

The SUMO protease Ulp2 modulates many of the SUMO-dependent processes in budding yeast. Ryu et al. discovered that cells lacking Ulp2 display a twofold increase in transcript levels across two particular chromosomes: chromosome I (ChrI) and ChrXII. Extra copies of ChrI and ChrXII can be eliminated following reintroduction of ULP2, suggesting that aneuploidy is a reversible adaptive mechanism to counteract loss of the SUMO protease.
Keywords: SUMO, aneuploidy, Ulp2, protease, Cln3, Ccr4
Abstract
Post-translational protein modification by the small ubiquitin-related modifier (SUMO) regulates numerous cellular pathways, including transcription, cell division, and genome maintenance. The SUMO protease Ulp2 modulates many of these SUMO-dependent processes in budding yeast. From whole-genome RNA sequencing (RNA-seq), we unexpectedly discovered that cells lacking Ulp2 display a twofold increase in transcript levels across two particular chromosomes: chromosome I (ChrI) and ChrXII. This is due to the two chromosomes being present at twice their normal copy number. An abnormal number of chromosomes, termed aneuploidy, is usually deleterious. However, development of specific aneuploidies allows rapid adaptation to cellular stresses, and aneuploidy characterizes most human tumors. Extra copies of ChrI and ChrXII appear quickly following loss of active Ulp2 and can be eliminated following reintroduction of ULP2, suggesting that aneuploidy is a reversible adaptive mechanism to counteract loss of the SUMO protease. Importantly, increased dosage of two genes on ChrI—CLN3 and CCR4, encoding a G1-phase cyclin and a subunit of the Ccr4–Not deadenylase complex, respectively—suppresses ulp2Δ aneuploidy, suggesting that increased levels of these genes underlie the aneuploidy induced by Ulp2 loss. Our results reveal a complex aneuploidy mechanism that adapts cells to loss of the SUMO protease Ulp2.
The basic proteome of an organism is greatly expanded and diversified through post-translational modifications (Beltrao et al. 2013). Covalent attachment of small molecules such as phosphate groups or small proteins such as ubiquitin alters the functional properties of target proteins in various ways. The small ubiquitin-related modifier (SUMO) protein is a conserved post-translational modification that modulates the binding properties, conformation, and/or localization of its substrates. Protein “sumoylation” is essential in most eukaryotes and regulates numerous cellular processes (Flotho and Melchior 2013).
Like other members of the ubiquitin-like protein (UBL) family, SUMO is covalently attached to target proteins in a process similar to ubiquitin–protein conjugation (Gareau and Lima 2010). SUMO is synthesized as an inactive precursor with a C-terminal peptide extension. A SUMO protease cleaves after a C-terminal-proximal Gly–Gly motif to form mature, conjugation-competent SUMO. In an ATP-dependent manner, the C terminus of mature SUMO is activated by the SUMO-activating enzyme (E1), forming a high-energy thioester bond with the E1. The SUMO is transferred to the active site cysteine of the SUMO-conjugating enzyme (E2) and then finally is attached to the lysine side chains of a target protein. Specificity of SUMO conjugation is often enhanced by a SUMO ligase (E3).
Sumoylation is a dynamic modification, with SUMO rapidly removed from substrates by SUMO proteases (Hickey et al. 2012). The two known SUMO proteases in budding yeast, Ulp1 and Ulp2, both localize primarily in the nucleus (Li and Hochstrasser 1999, 2000, 2003; Kroetz et al. 2009). While numerous functions and substrates of Ulp1 have been studied, most cellular roles of Ulp2 remain unclear. Ulp2 cleaves SUMO from specific substrates and efficiently depolymerizes polySUMO chains (Hickey et al. 2012).
SUMO modification has been shown to affect the transcriptional status of chromatin in both yeast and mammals (Shiio and Eisenman 2003; Nathan et al. 2006). Recent genetic studies from our laboratory implicate Ulp2 in the regulation of histone modification and transcriptional activation (HY Ryu, NR Wilson, D Su, A Lewis, and M Hochstrasser, unpubl.). However, the molecular mechanisms governing Ulp2-dependent regulation of gene expression are still poorly understood.
Aneuploidy usually results from unequal chromosome segregation during cell division. The imbalance in chromosome number resulting from such missegregation has paradoxical effects on cellular fitness (Pavelka et al. 2010a; Sheltzer and Amon 2011; Tang and Amon 2013). In humans, all monosomies, where only one copy of any chromosome is present, are embryonic-lethal (Hassold et al. 1996). Possessing an extra copy of a chromosome, termed trisomy, is almost always deleterious to humans. Indeed, 20 out of the 23 possible trisomies result in failure of the embryo to complete development (Brown 2008). However, under certain circumstances, aneuploidy enhances cell growth and provides a growth advantage as compared with euploid cells (those with the correct number of chromosomes) (Sheltzer and Amon 2011). Many cancers, including 90% of solid tumors and 75% of hematopoietic cancers, have an aberrant number of chromosomes (Weaver and Cleveland 2006).
To understand aneuploidy in greater depth, researchers have turned to the budding yeast Saccharomyces cerevisiae, where analysis of this phenomenon is more tractable. In one set of studies, stable disomic yeast strains (where one specific chromosome is duplicated in an otherwise haploid background) were generated (Torres et al. 2007; Sheltzer and Amon 2011). Transcriptomic studies using the disomic strains revealed a corresponding increase in mRNA transcript levels of the genes present on the duplicated chromosome (Pavelka et al. 2010b; Sheltzer et al. 2012). Therefore, yeast do not have a general dosage compensation mechanism to correct for an additional chromosome. Protein levels in these disomic strains are also nearly doubled (Pavelka et al. 2010b; Dephoure et al. 2014). These analyses show that, in budding yeast, an increase in chromosome copy number is accompanied by a parallel increase in both mRNA and protein levels.
As determined by numerous studies, the increase in gene, mRNA, and protein amounts negatively affects the growth of disomic yeast strains. Most disomic strains tested were sensitive to DNA-damaging reagents (Sheltzer et al. 2011). This may be due to genomic instability resulting from maintenance of an abnormal number of chromosomes. Interestingly, the severity of the sensitivity to genotoxic stress varied between disomic yeast strains, with the most sensitive strains being those with duplication of larger chromosomes (Sheltzer and Amon 2011).
Although aneuploidy generally reduces cell fitness, aneuploidy can provide a means for quickly adapting to cellular stress or mutation (Rancati et al. 2008; Mulla et al. 2014; Kaya et al. 2015). This selective aneuploidy is rapid compared with alternative adaptive mechanisms, such as accumulation of advantageous point mutations. In such adaptive aneuploidies, the duplicated chromosome frequently encodes a gene or genes that help mitigate the deleterious effects of the stress. For example, the pathogenic fungus Candida albicans develops a specific aneuploidy to counter the drug fluconazole (FLC) (Selmecki et al. 2006). Two genes encoded on the duplicated chromosome provide enhanced resistance to FLC; one is the direct drug target and the other regulates a drug efflux system that removes the drug from cells (Selmecki et al. 2008). Adaptive aneuploidies have also been observed in budding yeast and are triggered by various stressors, including DNA damage, gene deletions, and nutrient restrictions (Mulla et al. 2014). The resulting aneuploidy increases the dosage of a gene or genes that help to neutralize the stress. Therefore, the duplication of an entire chromosome, while at times deleterious, can often provide a selective growth advantage under specific stresses.
Here we provide evidence that the yeast SUMO protease mutant ulp2Δ rapidly develops a double disomy involving chromosome I (ChrI) and ChrXII; the duplicated chromosomes can be eliminated following reintroduction of ULP2 into the mutant. To our knowledge, this is the first example of a specific aneuploidy involving multiple chromosomes that occurs in response to deletion of a single, apparently nonessential gene. Using a tiled yeast genomic DNA library, we found that overexpression of either of two genes from ChrI—CLN3, encoding a G1 cyclin, or CCR4, encoding a component of the multifunctional Ccr4–Not regulatory complex—suppresses the development of aneuploidy in ulp2Δ cells. Moreover, loss of Whi5, a transcriptional repressor negatively regulated by the Cdc28–Cln3 kinase, or overexpression of NOT5, a gene encoding another subunit of the Ccr4–Not complex, also limits the aneuploidy generated by loss of Ulp2.
These findings suggest that the development of the ChrI and ChrXII double disomy is an essential adaptation that serves to amplify two genes, CLN3 and CCR4, in response to the potentially lethal effects inflicted by loss of the SUMO protease Ulp2. More generally, the data demonstrate that alterations of SUMO dynamics can be countered by selective aneuploidy. Given the evidence of SUMO dysregulation in cancer (Erfler and Vertegaal 2015) and the pervasiveness of aneuploidy in tumors, our results also suggest potential mechanistic links between these phenomena.
Results
ChrI and ChrXII are duplicated in ulp2Δ cells
To explore the relationship between Ulp2 and transcription, high-throughput RNA sequencing (RNA-seq) was used. In addition to wild-type and ulp2Δ cells, the transcriptomes of three other strains were analyzed. As a control, cells lacking the histone methyltransferase Set1 were tested, as the mRNA profile of this strain has been studied extensively (Martin et al. 2014). The SUMO pathway is linked to the ubiquitin–proteasome system through the Slx5–Slx8 heterodimer (Uzunova et al. 2007; Xie et al. 2007). Slx5–Slx8 is a SUMO targeted ubiquitin ligase (STUbL), and deletion of SLX5 in a ulp2Δ background suppresses the temperature sensitivity of ulp2Δ cells (Gillies et al. 2016). The mechanism behind this suppression is uncertain but could reflect restoration of proper transcriptional regulation in the ulp2Δ slx5Δ double mutant. To explore this possibility, the transcriptomes of the slx5Δ and the ulp2Δ slx5Δ mutants were also examined.
The RNA-seq data showed that overall transcription was not greatly altered in the SUMO pathway mutants (Supplemental Fig. S1A). However, there were many individual mRNAs whose levels were significantly different in the mutant strains compared with congenic wild-type cells (Supplemental Fig. S1B). By more stringent criteria, a limited set of genes was identified that were significantly differentially expressed in the mutant strains relative to wild type (Supplemental Fig. S1C). Selected RNA-seq data were validated by quantitative RT–PCR (qRT–PCR) and suggest that the RNA-seq data accurately represent the transcriptomes of ulp2Δ and wild-type cells (Supplemental Fig. S1D).
While the RNA-seq data did not yield any clear clues as to how Ulp2 modulates SUMO-dependent transcription globally, an unexpected result was observed when transcript levels relative to wild-type were mapped across each yeast chromosome (Fig. 1A,B). In ulp2Δ cells, the mRNA levels of most genes encoded on ChrI and ChrXII were roughly twice their level in wild-type cells. This is most clearly seen when comparing the profiles of similarly sized chromosomes (Fig. 1A [ChrI vs. ChrVI], B [ChrXII vs. ChrVII]). Such a chromosome-wide increase in mRNA transcript levels in ulp2Δ cells is prima facie evidence for the duplication of the affected chromosomes (see above). Notably, the doubling of transcript levels across these two chromosomes was not observed in any of the other tested strains, including the ulp2Δ slx5Δ double mutant. This suggests that the apparent chromosomal aneuploidy of the ulp2Δ single mutant is suppressed by loss of Slx5.
Figure 1.
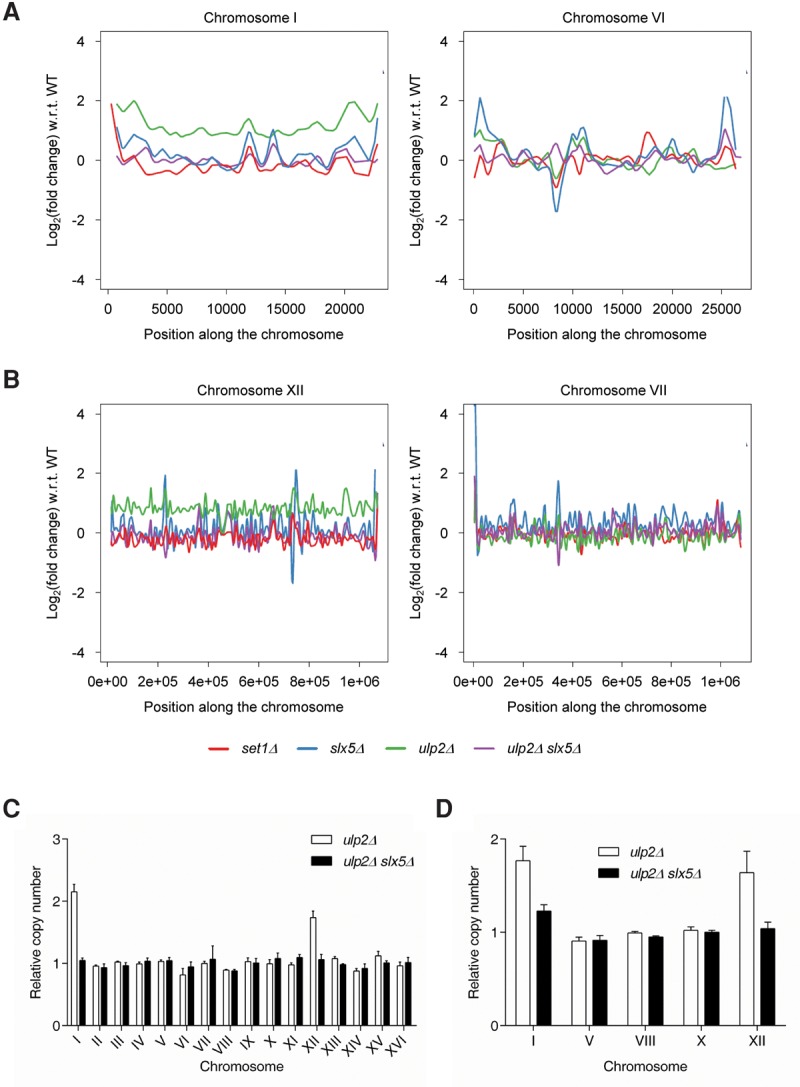
ChrI and ChrXII are duplicated in ulp2Δ cells. (A) Chromosome profiles of RNA-seq data (difference in mRNA level from mutant vs. wild type [WT]) mapped across ChrI and ChrVI, two of the smallest yeast chromosomes. (B) Chromosome profiles of RNA-seq data mapped across ChrXII and ChrVII, two of the largest chromosomes. Transcript levels from ChrI and ChrXII are twice as high in ulp2Δ cells (green line) with respect to (w.r.t) wild type. (C) Karyotype of ulp2Δ and ulp2Δ slx5Δ cells (MHY500 background) quantified by qPCR. Two primer pairs per chromosome (one per arm) were used to determine the chromosome copy number relative to wild-type (ULP2) cells. The qPCR reaction for each primer pair was performed in technical triplicate. The error bars represent the standard deviation (SD) of three PCR reactions performed using the genomic DNA from two independent colonies. (D) The chromosome copy number of ulp2Δ and ulp2Δ slx5Δ cells (BY4741 background) was determined by qPCR assay as described in C. The error bars indicate the SD of three PCR reactions performed using the genomic DNA from two independent clones.
To confirm directly, at the DNA level, the inferred disomies of ChrI and ChrXII in ulp2Δ cells, a qPCR-based aneuploidy assay was used (Pavelka et al. 2010b). Using one primer pair per yeast chromosome arm (32 primer pairs total), the copy number of each chromosome in ulp2Δ cells was determined in relation to the congenic wild-type euploid control (true haploid). Consistent with the RNA-seq data, ChrI was duplicated in ulp2Δ cells, whereas ChrXII was variably duplicated in the population (average copy number ∼1.6) (Fig. 1C). Notably, deletion of SLX5 in a ulp2Δ background suppressed the ChrI and ChrXII disomies. Duplication of ChrI and ChrXII was also observed in the BY4741 strain background used in the RNA-seq analysis (Fig. 1D). Taken together, our results reveal a previously unsuspected phenotype of ulp2Δ mutant cells. Because the Slx5–Slx8 STUbL is likely to process polySUMO-conjugated substrates of Ulp2 in ways that are toxic to cells (Gillies et al. 2016), the new data imply that aneuploidy is also a response to the action of this STUbL on Ulp2 substrates.
Aneuploidy develops rapidly in cells upon loss of ULP2
Several reports indicate that aneuploidy in yeast can be a rapid adaptation to specific types of stress (Hughes et al. 2000; Gresham et al. 2008; Rancati et al. 2008; Pavelka et al. 2010b; Yona et al. 2012; Mulla et al. 2014; Kaya et al. 2015). In all of these examples, one particular chromosome is duplicated. Our data above indicate that ChrI and, to a slightly more variable extent, ChrXII are both duplicated in otherwise haploid ulp2Δ cells; two types of measurement, RNA-seq (analysis at the RNA level) and qPCR (analysis at the DNA level), gave concordant results. Deletion of ULP2 is likely to impose a severe and potentially lethal cellular stress, which this specific aneuploidy may help to counter.
To assess the development of aneuploidy immediately following loss of the ULP2 gene, we used a strain carrying the ulp2Δ allele in the chromosome and a covering wild-type ULP2 plasmid that could be readily evicted, and ploidy was monitored by qPCR (Fig. 2A). The starting strain, MHY1379, was isolated from a complete tetrad and was therefore expected to be euploid, as was confirmed by the qPCR ploidy assay (Fig. 2B). The MHY1379 strain was streaked on FOA medium to evict the URA3-marked ULP2 plasmid; the URA3 gene product is toxic to cells on FOA. Single ulp2Δ colonies were then streaked onto FOA a second time to ensure complete elimination of the cover plasmid. Four clones from the second FOA plate were isolated and assayed for ploidy (Fig. 2C). Strikingly, all four independently derived ulp2Δ strains (A–D) had two copies of ChrI and an average of ∼1.6 copies of ChrXII. These results were similar to those obtained using the “old” ulp2Δ strains originally analyzed by RNA-seq and qPCR (Fig. 1C,D). The data indicate that the ChrI and ChrXII disomies develop quickly upon loss of ULP2.
Figure 2.

Selective aneuploidy develops rapidly upon loss of ULP2. (A) Plasmid eviction strategy for rapidly removing ULP2 from yeast. The ulp2Δ cells containing YCplac33-ULP2 were struck on SD + FOA twice to evict the YCplac33-ULP2 plasmid. (B) Karyotype of the parental MHY1379 strain (ulp2Δ + YCplac33-ULP2) determined by qPCR assay as described in Figure 1C. (C) Chromosome copy number determined by qPCR of four independent “young” ulp2Δ strains (A–D) obtained using the strategy outlined in A. Results were normalized to the chromosome copy number of the euploid wild-type strain shown in B. (D) Chromosome copy number of ulp2Δ cells expressing the indicated pRS315 (CEN, LEU2)-based plasmids was determined as in Figure 1C. The original MHY1379 was transformed with the indicated plasmids prior to eviction of the wild-type ULP2/URA3 plasmid. The error bars indicate the SD of three PCR reactions using genomic DNA from two independent clones. (E) Chromatin immunoprecipitation (ChIP) analysis using IgG-sepharose or anti-Flag agarose beads and strains expressing TAP-tagged Ndc10, Ubc9, and Ulp1 or Flag-tagged Ulp2. Real-time PCR signals at centromeric regions of ChrI, ChrIII, ChrXI, and ChrXII (CEN1, CEN3, CEN11, and CEN12) were quantitated and normalized to an internal background control and the input DNA. The error bars indicate the SD calculated from two independent chromatin preparations.
In each ulp2Δ strain, ChrXII was duplicated in many but not all cells in the population, possibly due to difficulties in faithful segregation during cell division of multiple copies of this chromosome, which carries the large rDNA gene cluster (D'Amours et al. 2004). Nevertheless, the highly reproducible and rapid duplication of ChrI and ChrXII suggests that this specific aneuploidy might promote viability of the ulp2Δ mutant. The results below also argue in favor of an advantage conferred by ChrXII disomy in ulp2Δ cells.
In an attempt to determine more precisely the timing of aneuploidy development following loss of Ulp2, an auxin-inducible degradation (AID) system was used (Nishimura et al. 2009). We constructed a yeast strain expressing a Ulp2 derivative tagged with an optimized AID* degron and a 9Myc epitope tag together with the Tir1-9Myc protein; the latter protein forms part of a ubiquitin ligase that drives ubiquitylation and degradation of AID*-tagged proteins (Morawska and Ulrich 2013). Upon addition of the synthetic auxin 1-naphthaleneacetic acid (NAA), levels of Ulp2 were monitored over short (∼4-h) and long (∼6-d) time periods (Supplemental Fig. S2A,C). High-molecular-mass SUMO conjugate levels increased as Ulp2 levels dropped, as observed previously in ulp2Δ cells (Bylebyl et al. 2003). Ploidy analysis of cells treated with NAA for 0.5 h to 6 d showed no evidence of ChrI or ChrXII duplication (Supplemental Fig. S2B,D). These results indicate either that the very small amount of Ulp2 remaining after sustained NAA treatment is sufficient to prevent aneuploidy or that an even longer period of time is needed for aneuploidy development. The latter seems unlikely given that the treatment time was similar to that used in the ULP2 plasmid eviction experiments (Fig. 2C).
Euploidy maintenance requires the catalytic activity of Ulp2
Ulp2, a cysteine protease, has an ∼200-residue catalytic domain flanked by large ∼400-residue extensions (Li and Hochstrasser 2000; Kroetz et al. 2009). To determine whether Ulp2 catalytic activity is required to prevent aneuploidy generation, a plasmid shuffle assay was used. A catalytic mutant, ulp2-C624A, was introduced on a LEU2-marked plasmid into cells that lacked the chromosomal copy of ULP2 but carried wild-type ULP2 on a URA3-marked plasmid. The plasmid-borne ULP2 was then evicted on FOA medium, leaving behind only the mutant ulp2-C624A allele. This shuffle experiment was also performed with an empty vector (negative control) or wild-type ULP2 (positive control). As expected, ulp2Δ cells transformed with the empty vector became disomic for both ChrI and ChrXII (Fig. 2D). Addition of wild-type ULP2 prevented the development of aneuploidy. In contrast, cells with the ulp2-C624A catalytic mutant, which express the inactive enzyme at levels similar to wild type (Kroetz et al. 2009), still duplicated ChrI and ChrXII, indicating that the SUMO protease activity of Ulp2 is required to maintain euploidy.
Given that Ulp2 is required for accurate chromosome segregation during cell division (Li and Hochstrasser 2000), it is possible that Ulp2 also has a structural role at centromeres. This might contribute to the ability of Ulp2 to maintain centromere cohesion (Baldwin et al. 2009). We therefore determined whether Ulp2 is stably localized at centromeres using a chromatin immunoprecipitation (ChIP) assay. A TAP-tagged version of the essential kinetochore protein Ndc10 was used as a positive control for centromere localization and binding. The ChIP assay revealed that Flag-tagged Ulp2 showed little if any binding to the centromere regions of ChrI, ChrIII, ChrXI, or ChrXII (Fig. 2E). Two other SUMO pathway proteins, Ubc9 and Ulp1, also failed to associate with centromeres. Thus, the ulp2Δ aneuploidy is unlikely to be due to a loss of stable Ulp2 association with centromeres.
Aneuploidy of ulp2Δ cells is reversed by reintroduction of ULP2
Loss of ULP2 quickly leads to ChrI and ChrXII aneuploidy development (Fig. 2). This state might be advantageous only in the absence of functional Ulp2. The expression imbalances caused by an abnormal number of chromosomes are generally deleterious to cell fitness. Hence, aneuploidy is often a temporary adaptation and is eventually supplanted by less disruptive evolutionary adaptations if the stress or mutation persists (Yona et al. 2012).
To test whether euploidy can be rapidly re-established upon reintroduction of the ULP2 gene into ulp2Δ cells, we analyzed chromosome copy number during continuous growth in liquid culture over many generations (Fig. 3A). Compared with ulp2Δ cells with an empty vector, ulp2Δ cells into which the wild-type ULP2 gene was reintroduced experienced a rapid reversal of ChrI and ChrXII aneuploidy. Of note, the extra copy of ChrXII was eliminated more rapidly than the additional copy of ChrI. Indeed, ChrXII disomy was eliminated within ∼50 cell divisions of growth after ULP2 reintroduction. Because ChrXII was not duplicated in all cells in the ulp2Δ population, it is conceivable that transfer of multiple ChrXII copies to daughter cells is difficult, possibly due to the large size of ChrXII or the structure of the large repetitive rDNA locus.
Figure 3.
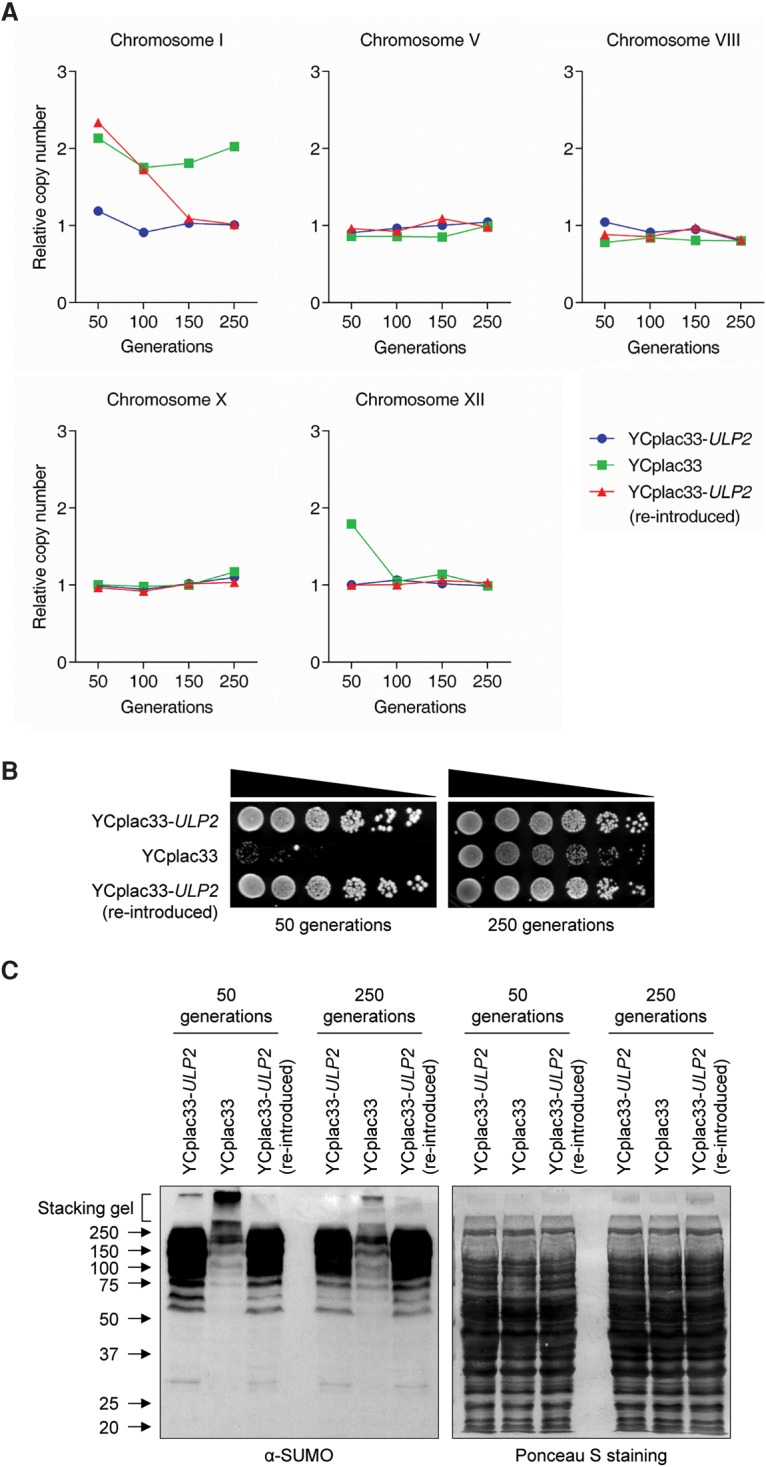
Aneuploidy of ulp2Δ cells can be reversed by reintroduction of ULP2. (A) The qPCR ploidy assay was performed using ulp2Δ strains with the indicated plasmids. After eviction of the YCplac33-ULP2 cover plasmid, ulp2Δ cells were retransformed with YCplac33-ULP2, and cells were monitored after the indicated number of generations. Chromosome copy number was analyzed as in Figure 1C. (B) Growth assay of the strains from A. After spotting cells in fivefold serial dilutions, YPD plates were incubated for 3 d at 30°C. (C) Immunoblot analysis of sumoylated proteins in extracts prepared from the strains in A. Equal loading was verified by staining the blot with Ponceau S.
We also observed that ChrXII, but not ChrI, disomy disappeared after ∼100 generations in ulp2Δ cells carrying an empty vector. Although ChrI disomy was not eliminated in ulp2Δ cells with an empty vector, the growth defect of these ulp2-null cells was significantly ameliorated after 250 generations (Fig. 3B). Furthermore, high-molecular-mass SUMO protein conjugates triggered by loss of Ulp2 had also diminished by ∼250 cell generations (Fig. 3C). We note that loss of aneuploidy was variable between experimental repetitions (cf. Fig. 3A and Supplemental Fig. S3), a result consistent with a previous report showing that the number of generations required to eliminate duplicated chromosomes varies among independent repetitions under the same stress conditions (Yona et al. 2012).
Segments of ChrI in high copy suppress ulp2Δ aneuploidy
If development of ChrI and ChrXII aneuploidy in ulp2Δ cells is adaptive, then there are likely genes on these two chromosomes whose increased dosage helps cells cope with loss of ULP2 (Mulla et al. 2014). Following this logic, providing cells with an additional exogenous source of these critical genes prior to eliminating ULP2 might prevent aneuploidy once ULP2 is evicted. ChrI is the smallest yeast chromosome and is fully represented in an available tiled yeast genomic DNA library (Jones et al. 2008). We transformed the “wild-type” ulp2Δ + ULP2 strain (MHY1379) with each of 22 different library plasmids containing individual ChrI fragments, and, after evicting the ULP2 cover plasmid, chromosome copy number was analyzed (Fig. 4A). Transformants expressing two different segments of ChrI reduced the ChrI/ChrIII ratio from ∼2 to 1, implying suppression of ulp2Δ aneuploidy (Fig. 4B). These results suggest that increased dosage of a gene or a noncoding DNA element present on these plasmids is sufficient to bypass the requirement for duplication of the entire ChrI in response to ULP2 loss.
Figure 4.

Overexpression of specific ChrI segments suppresses ulp2Δ aneuploidy. (A) Strategy used to identify specific regions of ChrI that suppress ulp2Δ aneuploidy. A ulp2Δ/YCplac33-ULP2 strain (MHY1379) was transformed with the indicated pGP564 (LEU2)-based ChrI library plasmids. Cells were struck on SD-Leu + FOA twice to evict YCplac33-ULP2. This strategy was used in subsequent ploidy assays as well. (B) Following YCplac33-ULP2 eviction, qPCR was used to assay chromosome copy number of ulp2Δ cells bearing the ChrI plasmids. Copy number was determined in relation to the ulp2Δ + ULP2 euploid strain. The ratio of ChrI to ChrIII copy number is plotted. (C,D) Schematic diagrams of plasmids pGP564-4 (C) and pGP564-10 (D). Locations of ORFs (gray arrows) and SpeI, HindIII, and NotI restriction sites are indicated. Subclones used in E and F are shown below the ChrI regions. “10-5” shows the ChrI segment that was inserted into plasmid pRS315-CCR4 and used in Figure 5, C and D. (E,F) qPCR ploidy assays of ulp2Δ cells with the high-copy plasmids shown in C and D. Chromosome copy number was determined as in Figure 1C. The error bars indicate the SD from three PCR reactions with two independent genomic DNA preparations.
The two suppressor plasmids, #4 and #10, encode a total of eight full-length ORFs. To determine the ChrI fragments or genes within these clones required for suppression of ulp2Δ aneuploidy, 10 smaller restriction fragments from the two ChrI plasmids were individually cloned into pGP564, a 2-µm vector with a LEU2 selectable marker (Fig. 4C,D), and tested for suppression (Fig. 4E,F). The average copy number of five chromosomes was compared with ulp2Δ cells expressing empty vector (negative control) and normalized to the euploid control strain (wild type + empty vector). Overexpression of two of the smaller fragments, one containing full-length CLN3 and the other containing full-length CCR4 and ATS1, prevented the development of ChrI but not ChrXII aneuploidy in nascent ulp2Δ cells. Because overexpressing the fragment containing only ATS1 (pGP564-10-2) did not affect ulp2Δ aneuploidy, it is likely that CCR4 is the gene relevant for preventing ChrI disomy. Our results therefore suggest that duplication of ChrI following loss of ULP2 may be circumvented by increased dosage of either of two specific ChrI genes: CLN3 or CCR4.
Additional copies of CLN3 or CCR4 are required for suppression of ulp2Δ aneuploidy
To confirm that Cln3, a G1-phase cyclin, and Ccr4, a subunit of the Ccr4–Not complex, were responsible for suppressing ulp2Δ-induced ChrI disomy, we measured chromosome copy numbers in ulp2Δ cells expressing CLN3, CCR4, ERV46, or ATS1 from the strong, constitutive GPD (TDH3) promoter (Fig. 5A). ChrI disomy, but not that of ChrXII, was indeed suppressed by CLN3 overexpression. Aneuploidy generally reduces cell fitness (Torres et al. 2007), but ulp2Δ strains that lost the ChrI disomy due to overexpression of CLN3 actually showed a more severe growth defect than the ulp2Δ mutant alone (Fig. 5B; Supplemental Fig. S4). This reduced growth is consistent with previous analyses showing that overexpression of CLN3 inhibits vegetative growth (Yoshikawa et al. 2011). Unlike CLN3 clones in overexpression vectors, a genomic fragment of CLN3 (4–6) (Fig. 4C) in a low-copy vector did not prevent ChrI disomy (Fig. 5C).
Figure 5.

Overexpression of CLN3 or CCR4 suppresses ulp2Δ aneuploidy. (A,C) Ploidy assays of ulp2Δ cells containing the indicated high-copy (A) or low-copy (C) plasmids. Chromosome copy number was analyzed as in Figure 1C. The error bars indicate the SD from three PCR reactions with two independent genomic DNA preparations. (B,D) Growth assays of the strains from A and C. After spotting cells in fivefold serial dilutions, SD-Leu plates were incubated for 3 d at 30°C. (E) qRT–PCR analysis of CLN3 and CCR4 mRNA in ulp2Δ cells with the indicated plasmids, relative to wild-type cells. Data were normalized to ACT1. The error bars indicate the SD from three PCR reactions using two independent RNA preparations. (F) Flow cytometry of DNA content in wild-type and ulp2Δ cells with vector p425-GPD or p425-GPD expressing CLN3 or CCR4. (1C) Unreplicated DNA; (2C) replicated DNA. (G,I) Ploidy analysis of ulp2Δ cells transformed with yeast genomic tiling library plasmids, including the indicated genes, as described in Figure 4B. The error bars indicate the SD from three PCR reactions with two genomic DNA preparations. (H,J) Ploidy analysis of indicated mutants (H) or ulp2Δ cells expressing the indicated plasmids (J). The error bars indicate the SD from two independent experiments. Statistical analyses were performed using a two-tailed t-test. (*) P < 0.05; (**) P < 0.01.
Intriguingly, strong overexpression of CCR4 suppressed both ChrI and ChrXII disomies along with the growth defect seen in cells lacking Ulp2 (Fig. 5A,B). Even when CCR4 (clone 10-5) (Fig. 4D) was introduced on a CEN vector, it was sufficient to suppress the aneuploidy caused by ULP2 loss and partially rescue the slow growth of the ulp2Δ mutant (Fig. 5C,D). These data suggest that Ccr4, presumably as part of the Ccr4–Not complex, is central to the two-chromosome aneuploidy of cells that have lost Ulp2.
It was reported previously that ccr4Δ cells display a G1-phase cyclin deficiency phenotype, and ccr4Δ cln3Δ double mutants are inviable, arresting as large unbudded G1 cells (Manukyan et al. 2008). We therefore analyzed the interactions of ulp2Δ with cln3Δ and ccr4Δ (Supplemental Fig. S5). Deletion of either CLN3 or CCR4 in a ulp2Δ background did not prevent ChrI or ChrXII duplication and did not alter ulp2Δ growth rates. Presumably, increased dosage of either CLN3 or CCR4 alone is sufficient to drive ChrI disomy. Testing this hypothesis would require simultaneously deleting both CLN3 and CCR4 in ulp2Δ cells, which is not possible due to the synthetic lethality of ccr4Δ cln3Δ (Manukyan et al. 2008).
Our results show that ulp2Δ aneuploidy is affected differently depending on the type of vector used to express CLN3 or CCR4. We therefore tested whether these differences depend on the mRNA levels of CLN3 or CCR4. For this, we performed qRT–PCR analysis on ulp2Δ cells expressing these genes on three different plasmids (Fig. 5E). CLN3 in a low-copy pRS315 plasmid in ulp2Δ cells caused only mildly increased mRNA levels compared with its expression in the pGP564 and p425-GPD vectors. Because ulp2Δ expressing the single-copy CLN3 plasmid did not prevent ChrI disomy, we suggest that a certain expression threshold of CLN3 is required to suppress the generation of ChrI aneuploidy.
Whereas the overexpression of CCR4 prevented the development of both ChrI and ChrXII aneuploidy upon loss of Ulp2, coexpression of CCR4 and ATS1 from pGP564 (pGP564-10-4) suppressed only the duplication of ChrI but not ChrXII (Fig. 4F). The expression level of CCR4 in the pGP564 plasmid in ulp2Δ cells was quite low compared with those from p425-GPD or pRS315 (Fig. 5E). It is therefore possible that ATS1 or other sequences upstream of CCR4 reduces CCR4 transcript levels. Taken together, our data reveal that, when highly expressed, the ChrI gene CCR4 fully suppresses the generation of aneuploidy triggered by loss of Ulp2.
ChrI aneuploidy is bypassed by rescuing the cell cycle defect of ulp2Δ cells
In budding yeast, accurate progression through the cell cycle is tightly regulated by the cyclin-dependent kinase Cdc28; kinase activity is controlled by various cyclins, such as the G1-phase cyclins (Cln1–3) and the B-type cyclins (Clb1–6). In addition, transcription of cell cycle factors is regulated by specific transcription factors: Swi4/Swi6/Mbp1 for G1/S genes, Mcm1/Fkh1/Fkh2/Ndd1 for G2/M genes, and Mcm1/Swi5/Ace2 for M/G1 genes (Bahler 2005). Most aneuploid yeast cells exhibit a delay in G1 phase (Torres et al. 2007; Thorburn et al. 2013), and ulp2Δ mutants have defects in cell cycle progression and chromosome segregation (Li and Hochstrasser 2000; Schwienhorst et al. 2000). Therefore, we asked whether the cell cycle defects found in cells lacking Ulp2 could be suppressed by increased dosage of CLN3 using flow cytometry measurements of propidium iodide-stained ulp2Δ cells transformed with p425-GPD-CLN3 (Fig. 5F). Overexpression of CLN3 caused a shift of the broad and irregular DNA distribution characteristic of ulp2Δ cells into two distinct peaks, roughly aligning with the haploid (1C) and diploid (2C) DNA peaks observed with wild-type cells. These results suggest that suppression of ChrI disomy by CLN3 overproduction in ulp2Δ cells may be caused by partial restoration of cell cycle progression.
To test whether other known cell cycle regulators prevent the ChrI disomy of ulp2Δ cells, we performed qPCR ploidy analysis using ulp2Δ cells expressing the corresponding genes in the pGP564 high-copy vector (Fig. 5G). None of the tested high-copy genes other than CLN3 prevented ChrI duplication. The Cdc28–Cln3 kinase is unique in its ability to hyperphosphorylate and inhibit Whi5, a repressor of G1/S transcription (Costanzo et al. 2004; de Bruin et al. 2004). Interestingly, deletion of WHI5 in ulp2Δ cells partially suppressed ChrI disomy (but not that of ChrXII) (Fig. 5H). Taken together, these results suggest that restoration of normal cell cycle progression by increased dosage of CLN3 or deletion of its target, the Whi5 transcriptional repressor, helps to prevent development of ChrI disomy in cells lacking ULP2.
The role of the Ccr4–Not complex in ulp2Δ aneuploidy
The Ccr4–Not complex is evolutionarily conserved from yeast to humans and consists of nine core subunits. The Ccr4–Not complex modulates gene expression at multiple steps, including transcription initiation and elongation, deadenylation and subsequent degradation of mRNA, translation, and even protein degradation (Miller and Reese 2012). Considering that overproduced Ccr4, a catalytic deadenylase subunit of the complex, thwarts the generation of both ChrI and ChrXII disomies upon loss of Ulp2, we asked whether this was a general feature of subunits of the complex. We tested high-copy expression of eight of the nine subunits; besides CCR4, only overexpression of a ChrXVI segment containing NOT5 suppressed duplication of ChrI upon loss of ULP2 (Fig. 5I). We confirmed that overexpression of the NOT5 gene by itself was sufficient to suppress ulp2Δ ChrI disomy, whereas ChrXII disomy was not strongly reduced (Fig. 5J). The slow growth of ulp2Δ was also suppressed by NOT5 overexpression (Supplemental Fig. S6B). These results suggest that the ChrI duplication triggered by ULP2 loss may serve to increase the expression of CCR4 in order to boost activity of the Ccr4–Not complex.
Discussion
Here, we identified a highly specific, two-chromosome aneuploidy as a recurring genetic alteration in cells suffering from inactivation of a supposedly nonessential component of the SUMO protein modification system. In particular, cells lacking an active Ulp2 SUMO protease become disomic for both ChrI and ChrXII. This occurs reproducibly in independent, mitotically growing cultures. Our findings suggest that the ChrI and ChrXII double disomy is an essential adaptation to loss of Ulp2, serving to amplify two ChrI genes (CLN3 and CCR4) and presumably one or more genes on ChrXII. Because the major function of Ulp2 is in disassembly of SUMO chains, these data imply not only that dysregulated accumulation of polySUMO-modified proteins is potentially lethal but that selective aneuploidy can mitigate the defect through several mechanisms. This is likely to be relevant to not only yeast and fungal growth but also human tumors where both aberrant SUMO dynamics and aneuploidy appear to play key roles in neoplastic development.
Aneuploidy has been found as an adaptation to various forms of stress in different yeast species (Mulla et al. 2014). FLC treatment of the pathogen C. albicans leads to specific single-chromosome duplications (Selmecki et al. 2006, 2008), while temperature stress (Yona et al. 2012) and gene deletions (Hughes et al. 2000; Rancati et al. 2008; Kaya et al. 2015) also can result in selective aneuploidies in S. cerevisiae. In these cases, the duplicated chromosome usually encodes a gene (or genes) that helps the organism cope with the mutation or environmental perturbation. Often there is a clear paralog of the deleted gene that is duplicated by disomy of the chromosome that carries the paralog (Hughes et al. 2000). The currently understood instances of aneuploidy as an evolutionary adaptation to genetic changes all vary in their causes and effects. For example, yeast cope with deletion of the essential MYO1 gene through several mechanisms, one being through development of ChrXVI trisomy and tetrasomy (Rancati et al. 2008). To our knowledge, a unique multichromosome aneuploidy consistently triggered by a genetic mutation, as observed in our study, is unprecedented (Liu et al. 2015).
While several examples of single-chromosome aneuploidies in response to mutation have been documented in yeast (Mulla et al. 2014), this compensatory mechanism may not be ideal because aneuploidy generally reduces cell fitness (Torres et al. 2007). Thus, aneuploidy can eventually be replaced by more subtle mutations that enhance cell fitness (Yona et al. 2012). We observed that ulp2Δ aneuploidy could be eliminated by reintroduction of ULP2 or, after longer times, could even be lost, in the case of ChrXII, in the continued absence of Ulp2.
The generation of aneuploidy is rapid compared with other evolutionary options and usually occurs through mitotic chromosome missegregation. Yeast lacking Ulp2 display increased chromosome instability (Li and Hochstrasser 2000), which might promote the usually rare combination of missegregation events needed to generate a specific double disomy. Aneuploidy in cells lacking Ulp2 can be suppressed by overexpression of CLN3 or CCR4, two genes located on ChrI. The cell cycle defects of ulp2Δ cells and ChrI aneuploidy are reduced by high levels of Cln3; elimination of the Whi5 transcriptional repressor target of Cdc28–Cln3 also reduces ulp2Δ ChrI aneuploidy. These results suggest that ChrI aneuploidy can ameliorate the cell cycle defects of ulp2Δ cells by restoring proper transcriptional regulation of G1/S cell cycle factors.
Most strikingly, both the ChrI and ChrXII disomies in ulp2Δ cells are repressed by overexpression of Ccr4, a catalytic subunit of the Ccr4–Not deadenylase complex. This places this multifunctional complex at the center of the adaptive aneuploidy caused by loss of Ulp2. Not5, another subunit of the complex, could also suppress at least the ChrI disomy. If elevated Ccr4–Not complex activity is required to relieve ulp2Δ-induced growth impairment, why does deletion of ULP2 not lead to duplication of ChrXVI, which encodes NOT5? Potentially, increased dosage of other genes on this chromosome is deleterious to ulp2Δ cells.
ChrXII is the largest yeast chromosome and includes the 1.2-Mb rDNA cluster. Mutant ulp2Δ cells overexpressing the rDNA locus still showed a twofold increase in ChrXII copy number (data not shown), suggesting that the benefit that comes from ChrXII aneuploidy in ulp2Δ cells is not due to increased dosage of the rDNA locus alone. ChrXII disomy in ulp2Δ cells might arise as a result of a consistent failure of ChrXII to properly segregate during cell division rather than conferring some adaptive advantage. However, the ability of excess CCR4 to suppress both ChrI and ChrXII disomies favors the adaptive model. Similarly, the finding that long-term culture of ulp2Δ cells leads to loss of ChrXII disomy is not expected if continual (nonadaptive) missegregation of this chromosome is the cause of its partial aneuploidy. Screens for suppression of ulp2Δ-induced aneuploidy by high-dosage ChrXII genes should clarify this issue.
While many phenotypic defects of ulp2 mutants have been documented, including sensitivity to a variety of stresses, genomic instability, and misregulation of the rDNA locus, much remains unknown regarding the physiological functions of Ulp2 (Li and Hochstrasser 2000; Bylebyl et al. 2003; Felberbaum et al. 2012). Few protein targets for desumoylation by Ulp2 have been documented. Identification of CLN3 and CCR4 as functionally relevant to the adaptive duplication of ChrI in ulp2Δ cells provides important initial insights into potentially essential functions of the Ulp2 protease. Cleavage of polySUMO-modified proteins by Ulp2-related SUMO proteases in other eukaryotes is also likely to have key roles in cell division and gene regulation.
Materials and methods
Yeast strains and growth conditions
Yeast strains used in this study are listed in Table 1. Standard media and experimental techniques were used for yeast growth and strain generation. To make MHY8915, the SET1 gene was deleted in a diploid strain by replacement with a HIS3MX6 cassette constructed from pFA6a-HIS3MX6 (Longtine et al. 1998), and haploid set1Δ segregants were identified following tetrad dissection. To generate MHY7863 and MHY9355 strains, the C-terminal insertion cassettes for ULP2-6xGly-3xFlag and ULP2-AID*-9Myc were constructed by PCR amplification from pFA6a-6xGly-3xFlag::HIS3MX6 (Funakoshi and Hochstrasser 2009) and pKan-AID*-9Myc (Morawska and Ulrich 2013) templates, respectively; diploid transformants were sporulated and dissected to isolate haploid cells with the tagged alleles. MHY5033 was derived by tetrad dissection of a ulp2Δ heterozygote purchased from the American Type Culture Collection. The CLN3, CCR4, and WHI5 deletion strains were generated by replacing each ORF with the kanMX6 module in ulp2Δ/ULP2 heterozygous diploid cells containing YCplac33-ULP2 (Longtine et al. 1998). Diploids were then sporulated, and the desired haploid cells were isolated. To create yeast strains newly null for ULP2, we evicted the YCplac33-ULP2 from MHY1379, a strain in which the chromosomal copy of ULP2 is deleted. In some cases, MHY1379 was first transformed with a LEU2-marked plasmid of interest. The transformed cells were streaked on FOA (or FOA-Leu) plates twice in succession to evict the YCplac33-ULP2 cover plasmid. All strains were verified by PCR and/or immunoblot analysis.
Table 1.
Yeast strains
Cells were grown at 30°C in YPD or synthetic complete medium with the appropriate amino acids and bases. For laboratory evolution experiments, cells were grown to stationary phase and then diluted 1:120 into fresh YPD medium, as described previously (Yona et al. 2012). This process was repeated daily (6.9 generations per day).
Plasmids
Plasmids used in this study are listed in Table 2. Wild-type ULP2, including 500 base pairs upstream of and downstream from the ORF, was PCR-amplified from yeast genomic DNA and cloned into pRS315 or YCplac33. QuikChange mutagenesis was used to mutate the ULP2 catalytic Cys (C624) to Ala (ulp2-C624A). To create high-copy plasmids containing subfragments of the ChrI #4 and #10 genomic library clones from the pGP564 vector, each HindIII or SpeI fragment from these clones was ligated into the HindIII or SpeI site of pGP564. After restriction digestion, the remaining vector-containing fragments were also religated. Overexpression constructs for ERV46, CLN3, CCR4, ATS1, and NOT5 were generated by PCR and cloned into p425-GPD. All constructs were confirmed by DNA sequencing.
Table 2.
Plasmids
RNA-seq analysis
Congenic wild-type (MHY2972=BY4741), ulp2Δ (MHY5033), slx5Δ (MHY7919), ulp2Δ slx5Δ (MHY9109), and set1Δ (MHY8915) yeast strains were grown in YPD overnight at 30°C and diluted to an OD600 of ∼0.2. Cells were grown for an additional ∼6 h at 30°C until reaching mid-log phase (OD600 of 0.8–1.0). Total RNA was isolated from 0.5 OD600 equivalents of cells using the RNeasy kit (Qiagen). RNA isolation was performed in triplicate for each yeast strain (15 total RNA samples). Contaminating DNA was removed from the samples using the DNA-free DNA removal kit according to the manufacturer's protocol (Ambion). RNA quality and concentration were determined using a NanoDrop spectrophotometer (Thermo Scientific). All RNA samples had an OD260/OD280 ratio of ∼2.0, indicative of high sample quality.
Purified RNA (8 µg per sample) was submitted to the Yale Center for Genome Analysis for RNA-seq analysis. Prior to sequencing, the samples were enriched for mRNA and depleted of rRNA by poly(A) tail selection using oligo(dT) columns. The mRNA samples were reverse-transcribed into cDNA, which was sequenced on an Illumina HiSeq2000 platform following the manufacturer's guidelines. Greater than 80-fold sequence coverage was obtained.
Sequences were aligned to S. cerevisiae Ensemble release R64-1-1. Reads were trimmed and filtered for quality using custom PERL scripts. The trimmed reads were aligned to the reference genome using TopHat (Trapnell et al. 2009). Aligned reads were processed with Cuffdiff (Trapnell et al. 2012) to estimate transcript abundance. The Cuffdiff results were processed in R using Cummerbund (Trapnell et al. 2012), and data were visualized using R.
qRT–PCR
Cells were grown, and total RNA was isolated as described above. Five-hundred nanograms of RNA was then used in reverse transcription reactions using the iScript cDNA synthesis kit (Bio-Rad). The cDNA was used in qPCR reactions using primers for the following genes: ACT1 (control), WSC4, RPL8A, KAP104, COA4, YPR078C, HSP12, CLN3, and CCR4. qPCR reactions were performed using the iQ SYBR Green Supermix kit from Bio-Rad on a CFX Connect thermal cycler (Bio-Rad). Each qPCR reaction was performed in technical triplicate. The ratio of mRNA in ulp2Δ over wild-type cells was calculated using the comparative CT method (ΔΔCT) (Schmittgen and Livak 2008). qRT–PCR reactions were performed in experimental quadruplicate using RNA prepared from independent yeast cultures.
qPCR ploidy assay
Genomic DNA was isolated from 1.0 OD600 equivalent of cells following a standard protocol (Funakoshi and Hochstrasser 2009). The qPCR ploidy assay was performed with the primers and according to the protocol of Pavelka et al. (2010b). The qPCR reactions were set up in 384-well plates and performed using the iQ SYBR Green Supermix kit in a Roche LightCycler 480 instrument. Each reaction was performed in technical triplicate. Analysis of the qPCR data was performed using a modified version of the classical comparative CT method (Schmittgen and Livak 2008), as developed by Pavelka et al. (2010b).
ChIP
ChIP experiments were performed as described previously (Ryu and Ahn 2014). Briefly, TAP-tagged or Flag-tagged proteins were precipitated with IgG-Sepharose beads (GE Healthcare) or anti-Flag agarose beads (Sigma), respectively. Quantitation of PCR signals was carried out with diluted template DNA (1:8 dilution for the immunoprecipitated DNA or 1:1000 dilution for the input DNA) following the method described in the preceding section and was normalized to the internal control (namely, PCR products amplified from an untranscribed region on ChrIV) and the input DNA.
Immunoblotting
Preparation of yeast whole-cell extracts and immunoblotting were done as described previously (Kroetz et al. 2009). To induce Ulp2 degradation via the AID*, cells were incubated in YPD with 500 µM NAA (Nishimura et al. 2009). Levels of the Ulp2-AID*-9Myc fusion protein and SUMO conjugate profiles were analyzed by immunoblotting with a 1:2000 dilution of 9E10 anti-Myc antibody (Covance) and anti-SUMO antibody (Li and Hochstrasser 1999), respectively.
Flow cytometry analysis
Analysis of yeast DNA content by flow cytometry was performed as previously described (Schober-Ditmore and Bachant 2000). Cells in log-phase culture (1 × 106 cells) were fixed in 70% ethanol, digested with RNase A, and stained with propidium iodide at 50 µg/mL final concentration. The analysis was performed using a FACS LSRII flow cytometer (BD Biosciences) with FACSDiva software.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health grant GM053756 to M.H., and in part by a predoctoral fellowship from the National Science Foundation and a Ruth L. Kirchstein National Research Service Award predoctoral fellowship (F31 AG046965) from the National Institutes of Health to N.R.W.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.282194.116.
References
- Bahler J. 2005. Cell-cycle control of gene expression in budding and fission yeast. Annu Rev Genet 39: 69–94. [DOI] [PubMed] [Google Scholar]
- Baldwin ML, Julius JA, Tang X, Wang Y, Bachant J. 2009. The yeast SUMO isopeptidase Smt4/Ulp2 and the polo kinase Cdc5 act in an opposing fashion to regulate sumoylation in mitosis and cohesion at centromeres. Cell Cycle 8: 3406–3419. [DOI] [PubMed] [Google Scholar]
- Beltrao P, Bork P, Krogan NJ, van Noort V. 2013. Evolution and functional cross-talk of protein post-translational modifications. Mol Syst Biol 9: 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S. 2008. Miscarriage and its associations. Semin Reprod Med 26: 391–400. [DOI] [PubMed] [Google Scholar]
- Bylebyl GR, Belichenko I, Johnson ES. 2003. The SUMO isopeptidase Ulp2 prevents accumulation of SUMO chains in yeast. J Biol Chem 278: 44113–44120. [DOI] [PubMed] [Google Scholar]
- Chen P, Johnson P, Sommer T, Jentsch S, Hochstrasser M. 1993. Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MATα2 repressor. Cell 74: 357–369. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Nishikawa JL, Tang XL, Millman JS, Schub O, Breitkreuz K, Dewar D, Rupes I, Andrews B, Tyers M. 2004. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 117: 899–913. [DOI] [PubMed] [Google Scholar]
- D’Amours D, Stegmeier F, Amon A. 2004. Cdc14 and condensin control the dissolution of cohesin-independent chromosome linkages at repeated DNA. Cell 117: 455–469. [DOI] [PubMed] [Google Scholar]
- de Bruin RAM, McDonald WH, Kalashnikova TI, Yates J, Wittenberg C. 2004. Cln3 activates G1-specific transcription via phosphorylation of the SBF transcription bound repressor Whi5. Cell 117: 887–898. [DOI] [PubMed] [Google Scholar]
- Dephoure N, Hwang S, O'Sullivan C, Dodgson SE, Gygi SP, Amon A, Torres EM. 2014. Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. Elife 3: e03023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erfler K, Vertegaal ACO. 2015. SUMOylation-mediated regulation of cell cycle progression and cancer. Trends Biochem Sci 40: 779–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felberbaum R, Wilson NR, Cheng D, Peng J, Hochstrasser M. 2012. Desumoylation of the endoplasmic reticulum membrane VAP family protein Scs2 by Ulp1 and SUMO regulation of the inositol synthesis pathway. Mol Cell Biol 32: 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotho A, Melchior F. 2013. Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem 82: 357–385. [DOI] [PubMed] [Google Scholar]
- Funakoshi M, Hochstrasser M. 2009. Small epitope-linker modules for PCR-based C-terminal tagging in Saccharomyces cerevisiae. Yeast 26: 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareau JR, Lima CD. 2010. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 11: 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74: 527–534. [DOI] [PubMed] [Google Scholar]
- Gillies J, Hickey CM, Su D, Wu ZP, Peng JM, Hochstrasser M. 2016. SUMO pathway modulation of regulatory protein binding at the ribosomal DNA locus in Saccharomyces cerevisiae. Genetics 202: 1377–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham D, Desai MM, Tucker CM, Jenq HT, Pai DA, Ward A, DeSevo CG, Botstein D, Dunham MJ. 2008. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet 4: e1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassold T, Abruzzo M, Adkins K, Griffin D, Merrill M, Millie E, Saker D, Shen J, Zaragoza M. 1996. Human aneuploidy: incidence, origin, and etiology. Environ Mol Mutagen 28: 167–175. [DOI] [PubMed] [Google Scholar]
- Hickey CM, Wilson NR, Hochstrasser M. 2012. Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol 13: 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Roberts CJ, Dai H, Jones AR, Meyer MR, Slade D, Burchard J, Dow S, Ward TR, Kidd MJ, et al. 2000. Widespread aneuploidy revealed by DNA microarray expression profiling. Nat Genet 25: 333–337. [DOI] [PubMed] [Google Scholar]
- Jones GM, Stalker J, Humphray S, West A, Cox T, Rogers J, Dunham I, Prelich G. 2008. A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nat Methods 5: 239–241. [DOI] [PubMed] [Google Scholar]
- Kaya A, Gerashchenko MV, Seim I, Labarre J, Toledano MB, Gladyshev VN. 2015. Adaptive aneuploidy protects against thiol peroxidase deficiency by increasing respiration via key mitochondrial proteins. Proc Natl Acad Sci 112: 10685–10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroetz MB, Su D, Hochstrasser M. 2009. Essential role of nuclear localization for yeast Ulp2 SUMO protease function. Mol Biol Cell 20: 2196–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SJ, Hochstrasser M. 1999. A new protease required for cell-cycle progression in yeast. Nature 398: 246–251. [DOI] [PubMed] [Google Scholar]
- Li SJ, Hochstrasser M. 2000. The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol Cell Biol 20: 2367–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SJ, Hochstrasser M. 2003. The Ulp1 SUMO isopeptidase: distinct domains required for viability, nuclear envelope localization, and substrate specificity. J Cell Biol 160: 1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GW, Yong MYJ, Yurieva M, Srinivasan KG, Liu J, Lim JSY, Poidinger M, Wright GD, Zolezzi F, Choi H, et al. 2015. Gene essentiality is a quantitative property linked to cellular evolvability. Cell 163: 1388–1399. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961. [DOI] [PubMed] [Google Scholar]
- Manukyan A, Zhang J, Thippeswamy U, Yang JY, Zavala N, Mudannayake MP, Asmussen M, Schneider C, Schneider BL. 2008. Ccr4 alters cell size in yeast by modulating the timing of CLN1 and CLN2 expression. Genetics 179: 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GM, King DA, Garcia-Nieto PE, Morrison AJ. 2014. Transcriptome profiling of Set5 and Set1 methyltransferases: tools for visualization of gene expression. Genom Data 2: 216–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JE, Reese JC. 2012. Ccr4–Not complex: the control freak of eukaryotic cells. Crit Rev Biochem Mol Biol 47: 315–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawska M, Ulrich HD. 2013. An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast 30: 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulla W, Zhu J, Li R. 2014. Yeast: a simple model system to study complex phenomena of aneuploidy. FEMS Microbiol Rev 38: 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119–122. [DOI] [PubMed] [Google Scholar]
- Nathan D, Ingvarsdottir K, Sterner DE, Bylebyl GR, Dokmanovic M, Dorsey JA, Whelan KA, Krsmanovic M, Lane WS, Meluh PB, et al. 2006. Histone sumoylation is a negative regulator in Saccharomyces cerevisiae and shows dynamic interplay with positive-acting histone modifications. Genes Dev 20: 966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. 2009. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6: 917–922. [DOI] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Li R. 2010a. Dr Jekyll and Mr Hyde: role of aneuploidy in cellular adaptation and cancer. Curr Opin Cell Biol 22: 809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. 2010b. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468: 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancati G, Pavelka N, Fleharty B, Noll A, Trimble R, Walton K, Perera A, Staehling-Hampton K, Seidel CW, Li R. 2008. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135: 879–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu HY, Ahn S. 2014. Yeast histone H3 lysine 4 demethylase Jhd2 regulates mitotic rDNA condensation. BMC Biol 12: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- Schober-Ditmore W, Bachant J. 2000. Yeast DNA flow cytometry. In In living color: protocols in flow cytometry and cell sorting (ed. Diamond RA, DeMaggio S), pp. 455–460. Springer Science and Business Media, New York. [Google Scholar]
- Schwienhorst I, Johnson ES, Dohmen RJ. 2000. SUMO conjugation and deconjugation. Mol Gen Genet 263: 771–786. [DOI] [PubMed] [Google Scholar]
- Selmecki A, Forche A, Berman J. 2006. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313: 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki A, Gerami-Nejad M, Paulson C, Forche A, Berman J. 2008. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol Microbiol 68: 624–641. [DOI] [PubMed] [Google Scholar]
- Sheltzer JM, Amon A. 2011. The aneuploidy paradox: costs and benefits of an incorrect karyotype. Trends Genet 27: 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, Brito IL, Hiraoka Y, Niwa O, Amon A. 2011. Aneuploidy drives genomic instability in yeast. Science 333: 1026–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Torres EM, Dunham MJ, Amon A. 2012. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci 109: 12644–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiio Y, Eisenman RN. 2003. Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci 100: 13225–13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YC, Amon A. 2013. Gene copy-number alterations: a cost–benefit analysis. Cell 152: 394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn RR, Gonzalez C, Brar GA, Christen S, Carlile TM, Ingolia NT, Sauer U, Weissman JS, Amon A. 2013. Aneuploid yeast strains exhibit defects in cell growth and passage through START. Mol Biol Cell 24: 1274–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AHY, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CWV, Bussey H, et al. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368. [DOI] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. 2007. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916–924. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzunova K, Gottsche K, Miteva M, Weisshaar SR, Glanemann C, Schnellhardt M, Niessen M, Scheel H, Hofmann K, Johnson ES, et al. 2007. Ubiquitin-dependent proteolytic control of SUMO conjugates. J Biol Chem 282: 34167–34175. [DOI] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW. 2006. Does aneuploidy cause cancer? Curr Opin Cell Biol 18: 658–667. [DOI] [PubMed] [Google Scholar]
- Xie Y, Kerscher O, Kroetz MB, McConchie HF, Sung P, Hochstrasser M. 2007. The yeast Hex3.Slx8 heterodimer is a ubiquitin ligase stimulated by substrate sumoylation. J Biol Chem 282: 34176–34184. [DOI] [PubMed] [Google Scholar]
- Yona AH, Manor YS, Herbst RH, Romano GH, Mitchell A, Kupiec M, Pilpel Y, Dahan O. 2012. Chromosomal duplication is a transient evolutionary solution to stress. Proc Natl Acad Sci 109: 21010–21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa K, Tanaka T, Ida Y, Furusawa C, Hirasawa T, Shimizu H. 2011. Comprehensive phenotypic analysis of single-gene deletion and overexpression strains of Saccharomyces cerevisiae. Yeast 28: 349–361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.