

Abstract
From the earliest Streptomyces genome sequences, the promise of natural product genome mining has been captivating: genomics and bioinformatics would transform compound discovery from an ad hoc pursuit to a high-throughput endeavor. Until recently, however, genome mining has advanced natural product discovery only modestly. Here, we argue that the development of algorithms to mine the continuously increasing amounts of (meta)genomic data will enable the promise of genome mining to be realized. We review computational strategies that have been developed to identify biosynthetic gene clusters in genome sequences and predict the chemical structures of their products. We then discuss networking strategies that can systematize large volumes of genetic and chemical data, and connect genomic information to metabolomic and phenotypic data. Finally, we provide a vision of what natural product discovery might look like in the future, specifically considering long-standing questions in microbial ecology regarding the roles of metabolites in interspecies interactions.
Introduction
Bacterial, fungal and plant natural products are a rich source of clinically used drugs including antibiotics, anticancer chemotherapeutics, immunosuppressants, cholesterol-lowering agents and anesthetics. Moreover, a wide range of food additives and crop protection agents originate from natural products. These molecules derive from large number of chemical classes such as polyketides, nonribosomal peptides, saccharides, alkaloids, and terpenoids, which together encompass a staggering diversity of chemical scaffolds. Intriguingly, most microbially derived (and some plant-derived) natural products are produced by metabolic pathways encoded by chromosomally adjacent genes: biosynthetic gene clusters (BGCs). These BGCs encode the enzymes, regulatory proteins and transporters that are necessary to produce, process and export a specialized metabolite. Importantly, this feature allows genomes to be mined for metabolites by the computational identification of BGCs.
Here, we will review the current and future impact of computational methodologies on the analysis of specialized metabolism and its applications in drug discovery from natural products. After outlining the recent evolution of (meta)genomic data, we will discuss algorithmic approaches for the identification, classification, dereplication and prioritization of BGCs in genomes and metagenomes. Subsequently, we examine how computational tools (for an overview, see Table 1) can be used to couple this genomic information to metabolite data and how networking approaches can be utilized to integrate multiple data sources (Fig. 1). We close by providing a perspective on how the use and further development of computational tools will change the field of natural product research in the next decade.
Table 1.
Overview of computational tools and databases for the analysis of secondary metabolites and their biosynthetic gene clusters. See reviews from Weber93 and Boddy94 for alternative overviews.
Figure 1. The role of computation in natural product discovery.
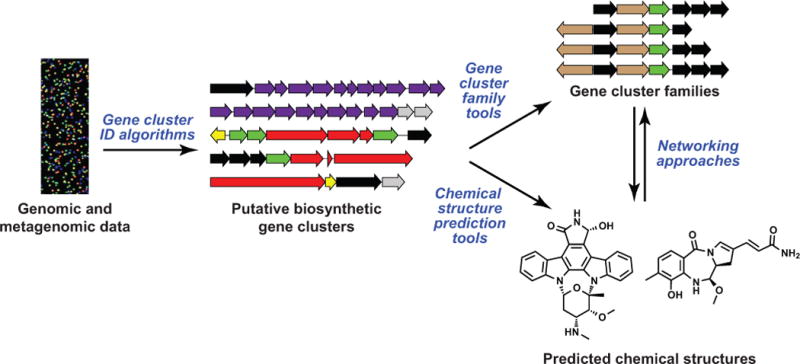
As shown in this overview schematic, which serves as an outline for the review, computational algorithms have been developed that enable or accelerate every key step in the natural product discovery pipeline: identifying BGCs from raw genomic and metagenomic sequence data, grouping BGCs into families, predicting the structure of a BGC’s small molecule product, and connecting gene cluster and molecular families using networking approaches.
The promise of genome mining
From the time the first Streptomyces genomes were sequenced more than a decade ago1,2, the promise of genome mining has been irresistible: the treasure trove of cryptic biosynthetic gene clusters they revealed would usher in a resurgence of natural product discovery. The key feature of this renaissance would be to turn the ad hoc, one-off process of discovering natural products into a high-throughput pipeline that would churn out many thousands of new small molecules from microbes, plus unnatural derivatives made possible by genetic engineering.
More than ten years later, this promise has not yet been realized. A skeptic could point out that not more than a few hundred molecules have been discovered over the last decade using genome mining, and that many of those molecules were so challenging to discover that the process would be difficult to generalize and automate.
However, we take a more optimistic view: that the promise of genome mining remains eminently realistic, and that its failure (to date) to yield thousands of new molecules is a kinetic, not a structural, problem. In this review, we argue that computational approaches to identifying biosynthetic gene clusters and predicting their small molecule products will finally equip scientists with the power to effectively explore the wide diversity of chemicals produced by organisms throughout the tree of life. Whereas recent and ongoing developments in synthetic biology and mass spectrometry (both recently reviewed elsewhere3–5) promise a massive increase in experimental throughput, computational developments will be the key to narrowing down the immense genomic diversity of extant biosynthetic pathways to a number that is feasible to evaluate using these approaches. Hence, the continued development of accurate and inventive algorithms to identify, classify, dereplicate and prioritize biosynthetic pathways will be of paramount importance to exploit the ever-increasing genomic data deluge to finally fulfill the promise of high-throughput natural product discovery.
An ongoing genomic revolution
In the near future, two important trends are likely to radically alter the landscape of genome mining. First, the number of genome sequences available will continue to rise exponentially: it is realistic to expect that within a decade, the nucleotide sequence databases will contain genome sequences of millions of bacterial and fungal strains. Research programmes to sequence entire culture collections, which together contain more than 1.5 million different bacterial and fungal strains (http://www.wfcc.info/ccinfo/), are already being initiated. And this just covers cultured microbes. Uncultured microbes appear to constitute the vast majority of biodiversity on earth: it has been estimated that about half of all identified bacterial phyla contain exclusively uncultured species6,7 and some reports predict that 99% of microbial strains are not readily culturable8. When single-cell genome sequencing of the uncultured majority of microbes9 increases in throughput and becomes less expensive, the number of genomes that can be sequenced will become almost unlimited. Moreover, as third-generation sequencing technologies (e.g., Pacific Biosciences and Oxford Nanopore) mature, it will become possible to cheaply obtain long read sequencing data sets with low error rates. This will make it feasible to rapidly obtain complete genome assemblies, without the many remaining gaps that currently plague genomic regions with repetitive sequences, including BGCs encoding NRPS (nonribosomal peptide synthetase) and PKS (polyketide synthase) enzymes10.
Second, the same technological developments will soon make it possible to close almost every genome captured within a metagenomic sample, eliminating the need for laborious and computationally intensive short-read assembly. This will lead to millions of additional genome sequences sampled directly from the environment, each with a rich set of data regarding the ecosystem and microbial community from which they derive.
These developments will have far-reaching repercussions for how natural product genome mining efforts should be developed in the coming years. Not only will synthetic biological refactoring of BGCs be paramount, as nearly all of these isolates will not exist in culture collections, but computational methods that compare enormous numbers of gene clusters within and between families and predict the structures and chemical diversity of their products will be essential. During this development, computational requirements of algorithms should be constantly optimized as well: already with the current data volumes, some of the bioinformatic tasks mentioned in this review take several months to finish on dedicated compute servers. Clever algorithmic innovation will be needed to allow these to scale up another thousand-fold.
Identifying biosynthetic gene clusters in genome sequences
Ever since the first bacterial genomes were sequenced, computational tools have been used to detect biosynthetic gene clusters in nucleotide sequences. At first, simple comparison techniques such as BLAST11 and HMMer12 were the methods of choice, using manually constructed lists of genes used as query sequences. Over the years, the methodologies have become increasingly sophisticated, however. Currently, a range of comprehensive software tools is available, which can be divided into two categories: high-confidence/low-novelty and low-confidence/high-novelty (Fig. 2).
Figure 2. Strategies for identifying BGCs.
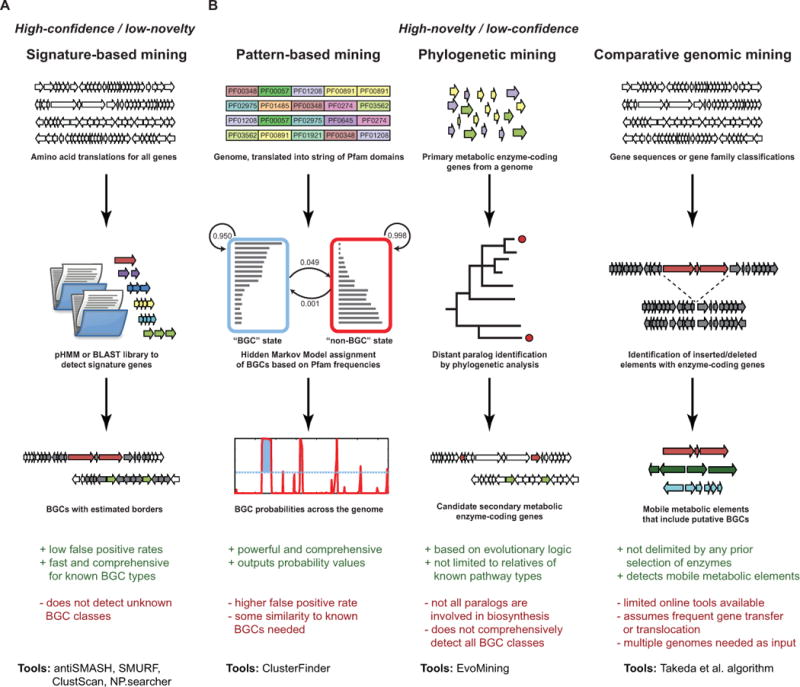
Several strategies have been designed for the genomic identification of BGCs. (a) The main high-confidence/low-novelty strategy is based on signature mining, using profile HMMs or BLAST searches to identify (combinations of) genes or protein domains that are specific for certain types of BGCs. (b) Recently, three high-novelty/low-confidence approaches have emerged that are focused on the identification of new BGC types: 1) pattern-based mining, based on the identification of genomic regions with protein domain frequencies that are generally indicative of involvement in specialized metabolism; 2) phylogenetic mining, based on the identification of functionally diverged paralogues of primary metabolic enzymes that have acquired functions in specialized metabolism during evolution; and 3) comparative genomic mining, which uses the identification of (horizontally or intra-chromosomally) transferred conserved syntenic blocks of enzyme-coding genes that belong to the accessory (pan) genome of a species to identify ‘mobile metabolic elements’ that are indicative of a role in specialized metabolism. Bullet points preceded by + and − at the bottom of the figure indicate advantages and disadvantages of a method, respectively. Tool(s) whose workflow corresponds to a column in the flowchart are listed at the bottom of each column.
High-confidence/low-novelty methods include tools such as CLUSEAN13, ClustScan14, np.searcher15, SMURF16 and antiSMASH17,18. The overall strategy for BGC detection shared among these tools is to utilize well-defined queries (usually implemented as profile HMMs19 generated from multiple sequence alignments) with manually curated cut-offs to identify signature genes or domains that are highly specific for known classes of biosynthetic pathways. The main advantage of this strategy is that it quickly and reliably yields an overview of the BGC repertoire of a single strain from its genome sequence, with a very low rate of false positives. Also, BGCs encoding known types of biosynthetic enzymes such as polyketide synthases and nonribosomal peptide synthetases are almost never missed, and a tool like antiSMASH can detect more than twenty classes of pathways (e.g., for the biosynthesis of aminocoumarins, oligosaccharides, thiopeptides, butyrolactones, etc.). Software tools based on these algorithms are therefore ideal for researchers who are, e.g., looking for gene clusters of a known biosynthetic class or who would like to get a quick overview of all detectable BGCs in one or more genomes for annotation purposes.
Low-confidence/high-novelty algorithms have only recently started to emerge to address an important shortcoming of the tools from the first category: their high specificity in detecting known gene cluster classes has the inherent consequence that they will not detect unknown types of gene clusters. Predicting gene clusters from unknown classes is a high priority, as they may encode molecules with entirely new chemical scaffolds20. New classes of gene clusters may be especially prevalent in the uncultivated majority of microorganisms often referred to as microbial ‘dark matter’21, but may even be hiding in plain sight in well-studied genomes from species like Escherichia coli22. Computationally detecting new classes of gene clusters requires more sophisticated algorithmic approaches. There are at least three possible strategies to solve this key challenge.
The first strategy, which is implemented in the recently published ClusterFinder algorithm22, is to look globally at the patterns of broad gene functions encoded in a genomic region instead of looking at the presence of specific individual signature genes. Similar to the way that gene identification algorithms detect stretches of nucleotides in a genome sequence that together look gene-like, ClusterFinder uses the Pfam database23 to translate a genome into a long string of protein domains and then looks for stretches within this string that look biosynthetic-gene-cluster-like based on their constituent broad gene families. The algorithm that is used for this is called a hidden Markov model (HMM), which is programmed to hover between two states (BGC and non-BGC) based on the frequencies of Pfam domains inside and outside known biosynthetic gene clusters of a wide range of types (provided by means of a training set). Due to the nature of the HMM, the probability of a Pfam domain to be part of a BGC is governed not only by its own frequency inside and outside BGCs from the training set, but also by the domains upstream and downstream of it. In this manner, ClusterFinder identifies genomic regions that are rich in Pfam domains that occur frequently in biosynthetic gene clusters. This strategy is capable of finding new gene cluster classes because biosynthetic pathways for entirely different molecules often utilize many of the same enzyme families common to secondary metabolism, such as oxidoreductases, methyltransferases, CoA-ligases, and cytochrome P450s24,25. Also, the operons are typically regulated by similar families of transcriptional regulators, and their small molecule products are often exported by similar families of transporters. Thus, genomic regions with high frequencies of these Pfam domains have a high likelihood of encoding a biosynthetic pathway, even in the total absence of signature genes for known biosynthetic gene clusters. Indeed, ClusterFinder enabled the identification of a large, previously unrecognized family of gene clusters that encode the biosynthesis of aryl polyenes in a wide range of bacteria from various phyla22.
The second strategy to identify new BGC classes is based on the idea that all secondary metabolic enzymes are distant paralogues of primary metabolic enzymes. The EvoMining approach26 exploits this conjecture by detecting the presence of ‘additional’ copies of primary metabolic enzymes in genomes (e.g., from amino acid metabolism), and then using phylogenetic analysis to identify outliers that have undergone significant sequence (and presumably also functional) divergence. These enzymes are subsequently visualized in their genomic context in order to identify new types of biosynthetic gene clusters.
The third way in which new types of biosynthetic pathways can be identified is by using large-scale comparative genomic alignment. According to this strategy, one would attempt to detect largely syntenic blocks of multiple orthologous genes that are not part of the core genome of a taxon and occur in different genomic contexts in different strains and species, or which otherwise display evolutionary hallmarks of specialized metabolic gene clusters. A dispersed taxonomic distribution and the presence of transposases at the borders (both pointing to horizontal gene transfer), as well as the detection of enzyme-associated Pfam domains would be additional indicators of a metabolic pathway with a specialized function. Takeda et al.27 have recently published an early version of such a motif-independent algorithm that identifies putative BGCs as small syntenic regions within the non-syntenic accessory genomes of Aspergillus species. The method successfully identified the kojic acid and oxylipin gene clusters, which do not have any signature genes specific to known pathways.
In the future, constructing a meta-algorithm combining scores from all the three of these strategies would probably be the most effective way to identify new classes of biosynthetic pathways with low false positive error rates, as it would take advantage of a broader range of characteristics.
Besides the challenge of identifying new classes of BGCs, one known class of BGCs is notoriously difficult to detect computationally: Ribosomally synthesized and Posttranslationally modified Peptides (RiPPs)28. RiPP BGCs have no shared signature genes and are so small that they are often missed by global pattern-matching algorithms like ClusterFinder. Tools like BAGEL29,30 and antiSMASH17,18 can detect a range of known subclasses of RiPPs based on the presence of shared tailoring or processing enzymes, but it is likely that these gene clusters are merely the tip of the iceberg. Advanced machine-learning techniques might be used in the future to detect RiPP prepeptide-encoding genes based on shared biases in amino acid frequencies and physicochemical properties.
Metagenome sequences: a special challenge
Compared to regular genome sequences, the analysis of metagenomic sequence data for BGCs presents several key challenges. As recently reviewed in more detail elsewhere31,32, there are two main approaches to identifying biosynthetic gene clusters in metagenomes: the PCR-based sequence tag approach and the shotgun assembly approach.
The sequence tag approach uses PCR amplification of known biosynthetic domains to identify clones in metagenomic libraries that harbor pathways of interest. This methodology, recently strengthened by full-fledged software automation33, is particularly powerful for identifying variants of known pathway types: it has been used to identify gene clusters that encode close relatives of molecules like rapamycin, teicoplanin and thiocoraline34. In this sense, it is an attractive alternative to synthetic chemical approaches to generating variants of known scaffolds, as it exploits the variation generated by evolution to alter moieties in a scaffold that would be difficult to access synthetically. However, the tag-based approach can also be used to find entirely new molecules that are produced by known BGC classes. Especially when coupled to phylogenomic analysis tools such as NaPDoS35, it can be used to identify domains that represent new areas of the extant biosynthetic diversity. Also, the recently released eSNAPD 2.033 specifically pinpoints gene clusters that are significantly different from all known gene clusters, which are visualized in its “New Clade Explorer” analysis module. In the future, such phylogenomic analysis of sequenced tags, combined with targeted cosmid sequencing, could be used to map the entire extant sequence diversity in known gene cluster families: after identifying a wide range of tag sequences in metagenomes obtained from a variety of environments, one could select an optimally diverse subset for cosmid sequencing based on approaches similar to the ones used in the Genomic Encyclopedia of Bacteria and Archaea project36.
In the shotgun assembly approach, metagenomic DNA is sequenced in bulk and then assembled en masse. For technical reasons related to the ability to assemble complete BGCs from short reads, this strategy has thus far largely been limited to the analysis of relatively low-complexity ecosystems or taxonomically enriched samples from more complex ecosystems. Even so, it has lead to the identification of a number of new pathways37,38. With spectacular recent developments in assembly algorithms39–41 and a notable increase in read lengths generated by new sequencing technologies, the time may now be ripe for a large-scale application of shotgun metagenomics to sequence and assemble BGCs directly from a wide variety of environments. With the cost of DNA synthesis dropping, synthetic biology will replace the need to isolate the cells from the source environment in order to access BGCs for heterologous expression. Adequate quality controls need to be in place, however, as the danger of chimeras in metagenomic sequence assemblies is ever-present, especially with fast-evolving and repetitive genes in BGCs that encode, e.g., modular polyketide synthases.
Notably, metagenomics in its current ‘short read-based’ form is a temporary research field, as the technical limitations that currently define it are likely to be solved soon. Given the technological developments anticipated in the coming years, the shotgun sequence approach may altogether replace tag sequencing for metagenomic BGC identification. And in the near future, the combination of fast-developing long-read technologies and new bioinformatic approaches42,43 may allow scientists to obtain full genome sequences of all but the least prominent members of a microbial community by parsing them out directly from the larger metagenomic dataset. In the near term, hybrid approaches that combine assembly and tag sequencing might also be of great help; in such a hybrid approach, assembled metagenomic contigs would be used to identify new candidate classes of BGCs for which primers are then designed for further exploration by tag sequencing, phylogenomic analysis and cosmid sequencing.
Dealing with thousands of gene clusters effectively
Although they are still being developed and improved, techniques for identifying gene clusters in genome and metagenome sequences have already identified tens of thousands of putative BGCs, creating a new challenge: how to make sense of and prioritize this growing list of BGCs. This challenge is particularly urgent since – as discussed above – the list of putative BGCs will likely soon number in the millions. In order to dereplicate and prioritize large numbers of gene clusters, it is essential to be able to classify them into gene cluster families (GCFs): in this way, it can easily be assessed whether newly identified BGCs are related to BGCs that produce known compounds, enabling gene cluster mining to supplement or replace the use of organic synthesis to generate semisynthetic analogs. Moreover, accurately determined GCFs allow the analysis of the taxonomic and environmental distribution within a family, which can provide clues about function. Finally, GCFs are ideal units to subject to multi-dimensional prioritization approaches to select BGCs for experimental characterization based on various indicators of potential for, e.g., pharmaceutical application44.
The challenge of classifying large numbers of BGCs into well-defined gene cluster families has already been taken on by several different research groups22,45,46, who each used different distance metrics and cutoffs to define relationships among BGCs (Box 1). The ways in which these strategies diverge reflect the differences in data sets that were analyzed as well as disparate goals of each metric. However, in order to provide a general solution and computational infrastructure for BGCs analogous to the Pfam domain classification for proteins, it would serve the broader community well to arrive at a common standard for family delineation that works robustly for a wide range of datasets and is implemented with user-friendly tools. One option would be a two-tier approach: first a categorization into broad classes (e.g., glycopeptides or ansamycins) using a metric measuring the overall similarity in gene content between each pair of clusters, followed by finer-scale delineation into GCFs (comprising one family for each molecule and its chemical variants) using sequence identity of domains shared between all BGCs.
Box 1. Defining BGC families.
Several efforts have recently been made to classify BGCs into gene cluster families (GCFs), based on their gene content and sequences (Fig. Box a).
In a recent manuscript reporting the comparative genomics of 75 Salinispora strains,45 BGCs with similar gene content were grouped into families termed ‘operational biosynthetic units’ (OBUs), based on sequence identity values of 90% and 85%, respectively, among homologous ketosynthase and condensation domains. While this method worked well to separate BGCs responsible for the production of different compounds in their dataset, the approach was not intended as a general solution for all organisms and all classes of BGCs. First of all, the approach only works for polyketide and nonribosomal peptide BGCs. Also, it is limited to the domains involved in the synthesis of their core scaffolds, not yet taking into account the complement of scaffold-tailoring enzymes found in a typical BGC. Finally, even recent hybridization of a PKS or NRPS BGC with other sub-clusters that encode the biosynthesis of other chemical moieties would not be detected by this method.
Recently, a more sophisticated strategy was developed to define GCFs: a distance metric composed of three parameters combined into a single overall score.46 The parameters used were the number of homologous genes shared between two BGCs with >50% sequence identity, the proportion of nucleotides involved in a PROmer74 pairwise alignment and the amino acid sequence identity between key signature domains involved in scaffold biosynthesis. The first two parameters were given a 25% weight in the final score, whereas the last parameter was given a 50% weight. When classifying 74 BGCs producing known compounds using this metric, the method performed robustly and consistently grouped BGCs for similar molecules into the same GCFs. One downside of the metric appears to be that the scores are calculated unilaterally, leading 3-amino-5-hydroxybenzoic acid (AHBA) BGCs to be grouped in a family with the BGC for rifamycin, which contains AHBA as just one of its chemical moieties (Fig. Box b); however, this could be fixed easily by, e.g., taking the average of each pair of two unilateral scores as a bilateral score. Another drawback of the metric is that its main component only works for pre-specified classes of BGCs for which appropriate signature domains have been defined.
A third approach to classify BGCs into families was recently developed.22 In this study, a distance metric devised for multidomain proteins75 was modified for the purpose of classifying BGCs, and uses the set of Pfam23 domains identified in each BGC as a basis. The modified metric consists of the Jaccard index (weighted 36%), which measures the number of unique Pfam domains shared between two BGCs, and the domain duplication index (weighted 64%), which measures the similarity in the number of domains for each Pfam type present in the BGCs. Additionally, sequence similarity information was (optionally) incorporated by weighting the score by the maximum bipartite matching of Pfam domain sequence identities. The main advantage of this metric is that it works for any type of BGC, regardless of what is known about the enzymes encoded in them. However, the approach has limited resolution when comparing BGCs that almost entirely consist of repetitions of the same Pfam domains, such as is the case for some large multimodular NRPS- or PKS-encoding gene clusters.
From genes to chemistry
Many GCFs that will be classified and prioritized using the approaches detailed in Box 1 will have no members with elucidated chemical structures. In order to further prioritize these orphan clusters for experimental characterization and connect them to high-throughput data generated by techniques such as mass spectrometry, it is essential that more accurate approaches are developed to predict chemical structures directly from genome sequences.
A range of algorithms have been developed to predict the substrate specificities of NRPS adenylation domains and PKS acyltransferase domains47–54. In tools like NP.searcher15 and antiSMASH17,18, individual monomer predictions are then combined to give a rough idea of the core scaffold of a polyketide or nonribosomal peptide. For some classes of RiPPs, intramolecular cross-links can also be predicted55. While ostensibly useful, such predictions currently provide limited detail and do not apply to important natural product classes such as terpenoids, alkaloids and saccharides. There are two possible solutions to improve chemical structure prediction, which are both worth pursuing in parallel: First, there is a need to devise more general strategies for connecting enzyme-encoding genes to predicted substrates and products, in order to be able to predict the final products of a wide range of pathways from scratch. Second, high-throughput experimental techniques can be used to connect actual molecules to computationally identified gene clusters. We discuss these solutions in more detail below.
Predicting entire chemical structures from scratch
Predicting the small molecule products of a wide range of biosynthetic pathways directly from genome sequence data is a daunting challenge. There exist an enormous variety of enzymes involved in synthesizing and tailoring natural product scaffolds, and innumerable variations on known chemical themes. From a computational perspective, the problem can largely be reduced to the question of how to acquire a sufficiently comprehensive training dataset to cover this diversity and complexity.
As a start, in order to effectively connect biosynthetic enzymes to the chemical transformations they catalyze, their BGCs should be meticulously and continuously catalogued. The Minimum Information about a Biosynthetic Gene cluster (MIBiG) standard56 takes a step in this direction, by allowing the scientific community to carefully annotate all enzyme functions and specificities for each BGC, as well as the level of evidence available for each observation. Also, systematically searching for correlations between natural product sub-structures and genes or sub-clusters in their BGCs may be helpful: e.g., sugar monomers could be linked to genes associated with their biosynthesis. By combinatorial permutation of scaffold biosynthesis predictions with predictions of tailoring reactions and additional chemical moieties, one might then be able to generate a library of molecular masses (or narrow mass ranges) for each BGC that can be used to identify which of the possible structures is truly produced by the organism in question15.
In the long run, it may also be necessary to enrich the training sets for, e.g., substrate specificity prediction algorithms, by systematically generating large amounts of training data experimentally. For example, a powerful dataset to train NRP structure predictors could be generated by expressing a diverse, carefully selected set of NRPS adenylation domains from synthetic genes and subjecting them to a high-throughput ATP-pyrophosphate exchange assay to determine their substrate selectivity. Similar approaches might soon be technically feasible for enzymes like glycosyltransferases, cytochrome P450s and terpene cyclases. Even if a subset of these enzymes displayed promiscuity in vitro that is not reflective of the reaction they catalyze in vivo, the combination of these results and existing data from known BGCs would yield a far more powerful and ‘fault tolerant’ predictive capability. A saying that is frequently applied to machine-learning problems is “garbage in, garbage out”, but for properly functioning algorithms the reverse will also be true: “great data in, great results out”.
Matching genes to molecules using mass spectrometry
Computational solutions alone will not be sufficient to realize the vision of high-throughput genome mining. In parallel, advances in analytical chemistry—particularly in mass spectrometry—are revolutionizing the ways in which the small molecule products of biosynthetic pathways are detected (reviewed in more detail elsewhere4,5). But when these experimental techniques are combined with computational algorithms directly, an even more powerful synergy arises.
For example, the peptidogenomics57 and glycogenomics58 methodologies combine the power of tandem mass spectrometry to profile the fragment composition of molecules with BGC predictions of chemical sub-structures that may correspond to these fragments. Recently, the computational coupling of mass spectrometric and genomic data for peptidogenomics has been entirely automated by a number of algorithms. This provides an unprecedentedly rapid method to connect gene clusters to molecules.
The RiPPQuest59 and NRPQuest60 algorithms both use a molecular networking approach61 to identify potential gene clusters for observed tandem mass spectra of lanthipeptides (a class of RiPPs) and nonribosomal peptides (NRPs), respectively. The search database for RiPPquest is compiled by finding all short open reading frames (ORFs) near each detected lanthionine synthetase-encoding gene in a genome, while NRPquest creates a database of possible NRPs by generating all possible orders of NRPS assembly-lines within each detected NRP BGC and then predicting the amino acids encoded by each NRPS module using NRPSPredictor251. A spectral networking approach enables multiple variants of a molecule to be assessed, which reduces the likelihood of a false negative result from unanticipated tailoring modifications. Also, it allows immediate identification of previously unknown variants of known peptides.
An alternative method, Pep2Path62, uses a probabilistic framework to predict the likelihood that each NRPS module selects every possible amino acid as a substrate, and then calculates combined probabilities for all possible NRPS assembly lines to match a mass spectrometry-derived mass shift sequence tag: a sequence of fragment molecular weight differences that is representative for the amino acid sequence of the peptide under study. Even though Pep2Path is based on the same algorithm for substrate specificity prediction as NRPquest (i.e., NRPSPredictor2), the advantage of this approach is that the algorithm will not fail to predicted a peptide-BGC link if a few modules are slightly mispredicted: e.g., if a module is specific for tyrosine, and a phenylalanine is observed, the probability of the module to be responsible for the observed amino acid will still be high. Pep2Path also has a tool for RiPP BGC identification, which searches all possible ORFs in a genome for hits to an observed mass shift sequence tag.
In the future, it would be powerful to combine the spectral networking approach of NRPquest with the probabilistic approach of Pep2Path: the strengths of both approaches could make it possible to rapidly annotate large and semi-automatically generated molecular networks from complex samples or sample collections. Also, developing similar algorithms for saccharides, terpenoids, alkaloids, and other classes of molecules, which is technically feasible, would make it possible to target a wide range of molecules using this technology. It might even be possible to use statistics to systematically connect the full set of spectrometrically observed fragment mass differences for a molecule—irrespective of its class—to all possible chemical transformations that can be predicted to occur in a pathway and all chemical moieties predicted to be synthesized based on the enzyme families that each BGC harbors.
In any case, the semi-automated identification of the small molecule products of BGCs that these strategies allow may soon have a profound effect on how natural product characterization operates: large numbers of ‘draft’ compound structures could be rapidly reconstructed and linked to their most probable biosynthetic gene clusters. Based on the characteristics of the molecules (e.g., the chemical moieties observed) and the gene clusters (e.g., their uniqueness compared to known gene clusters), a smaller number of compounds could then be prioritized for detailed structural characterization using methods such as NMR or X-ray crystallography. If this workflow becomes a reality, new computational tools will also be required to make the most of the large number of draft natural product structures for the purpose of dereplicating and classifying biosynthetic pathways.
Connecting multiple data types with networking approaches
Besides attempting to predict individual BGCs that correspond to individual molecules, large-scale genomic and molecular information could also be used more systematically to uncover links between genes and molecules in a wide range of organisms simultaneously. Using molecular networking of large numbers of mass spectra and BGC networking on large amounts of genomic data, comprehensive sets of molecular families (MFs) and GCFs can be reconstructed63. If, from a genetic perspective, GCFs are genotypes and MFs are their phenotypes, genome-wide association studies could be performed that statistically match these genotypes to phenotypes (Fig. 3), in a manner similar to how genetic diseases are matched to specific polymorphisms on the human chromosome. Given the extreme variability of natural product repertoires observed in many bacterial taxa22, only a relatively limited number of strains (thousands instead of millions) would then have to be assessed using the combination of both methods to be able to identify a large set of molecules that show a statistically significant correlation between the strains in which they are observed and the BGCs present in the genomes of these strains. When combined with even partial chemistry predictions, this method might enable scientists to discover large numbers of molecules and their corresponding gene clusters in the scope of a single large experiment. Even more intriguingly, all of these compounds could potentially be connected to specific biological activities in a similar manner: methods are currently emerging to classify molecules by their phenotypes in terms of the effect that they have on a range of different cells and cultures, which can be quantified and categorized using cytological profiling and functional signature ontologies64,65. In this way, combinatorial computational networking of genomic, metabolomic and phenotypic data could rapidly uncover families of molecules with phenotypes of interest in large strain collections, along with their corresponding gene cluster families that would allow heterologous or synthetic biology-driven expression and engineering of their biosynthetic pathways.
Figure 3. Big data challenges for biosynthesis.
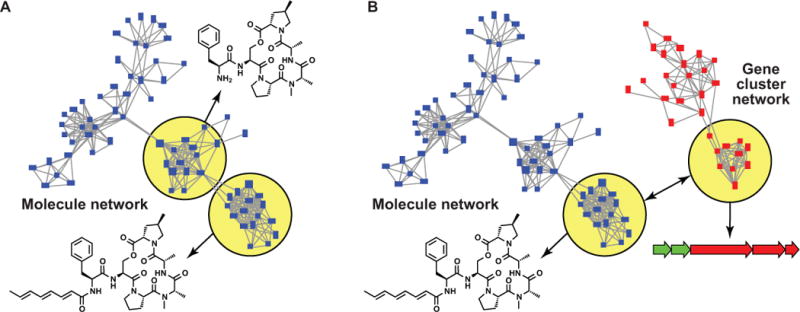
(a) In network-based algorithms that enable small molecule structure elucidation, networks are constructed in which each node is a mass ion, and edges are drawn between mass ions that are related by a mass difference that indicates a common chemical transformation. Sub-networks represent a molecular species of interest. (b) In an alternative approach, two distinct networks – one in which nodes are molecules, and the other in which nodes are BGCs – can be co-analyzed to connect BGCs to small molecules they encode and vice versa.
How computation will change natural product workflows in the future
The computational approaches discussed above hold great promise for improving individual steps in the natural product discovery process. However, the true promise of these approaches will be realized when they are connected in series, transforming natural product research from an ad hoc pursuit to a high-throughput endeavor.
As a result, ten years from now, a typical workflow will likely be a lot different than it is today (even though many approaches will likely extend efforts being pioneered now by various labs). One possibility: Starting with a molecule of interest, an MSn experiment will generate a fragmentation pattern revealing the set of possible chemical moieties represented by these fragments. These data are then matched probabilistically against a set of thousands BGCs detected in the complete genome sequences of more than a thousand bacterial species from the soil metagenomic sample from which the molecule was detected. This procedure pinpoints a family of around a dozen related candidate gene clusters which are near-perfect matches to the fragmentation data and appear to encode enzymatic pathways to synthesize AHBA-containing polyketides that are distant relatives to geldanamycin. Generating an in silico library of all the possible final products for each of these clusters based on detailed chemical structure predictions then identifies a single BGC that is predicted to be responsible for the compound with the observed total mass. NMR experiments on microgram quantities of the molecule confirm the correctness of this structure experimentally; all of this takes about one week. Querying a publicly available database of millions of putative BGCs reveals a few hundred non-redundant gene clusters, each of which appear to make an analog of the original molecule. Each of these BGCs is synthesized and expressed in a bacterial host that has been pre-optimized for producing AHBA-containing polyketides, and milligram quantities of several hundred analogs are obtained. High-throughput cytological profiling and toxicity screening then identify a handful of analogs with the most favorable pharmacological profiles.
Another possibility will be to start from several GCFs of interest. In this hypothetical case, the GCFs are chosen by a phenotypic/ecological criterion: they are overrepresented in the gut microbiomes of people predisposed to developing Crohn’s but without active disease; thus, they are candidate immunomodulatory factors that would serve a protective, tolerogenic role. Ten such GCFs are identified, each having a few hundred members on average. All gene clusters are ordered for synthesis, and expressed in one of several dozen hosts optimized for production of the predicted molecular class to which the gene cluster belongs. In the meantime, automated structure predictions for each of the clusters are refined by database matches to mass spectral networking data, generating high-quality structure predictions. Microgram quantities of around a thousand compounds are obtained and screened in cell-based immunological assays to narrow down the set to a few hundred high-confidence compounds—around one hundred molecular variants of each of 5 compounds that originated from 5 of the originally selected GCFs—that are produced in milligram quantities. Complete structure determination by MSn and NMR experiments reveals that the structure predictions were completely accurate for the majority of the molecules, and off by a simple chemical transformation (e.g., a hydroxylation or methylation) in the remaining cases. The structure prediction software automatically suggests enzymes from related BGCs that could be used to create unnatural derivatives of these molecules. A library of a few thousand derivatives is then utilized for detailed phenotypic screening, based on which a handful is prioritized for animal experiments.
At present, these examples are closer to science fiction than reality. But as natural product discovery continues its transformation from an ad hoc exercise to a systematic, computation-driven pursuit, science fiction might become reality with surprising speed.
Conclusion and future perspectives
In addition to changing the way natural products are discovered, advances in computation, along with increased deposition of metadata66, will help answer long-standing questions about the role of natural products in microbial and microbe-host ecology. This landmark challenge would address piecemeal observations that have raised tantalizing questions, but still no systematic study: Why do closely related organisms often have different gene clusters, but unrelated organisms have similar clusters? Are there certain environments or communities that select for the production of, e.g., a ribosome-targeted antibiotic or a rapamycin-family TOR inhibitor? Why does a single strain of Streptomyces or Sorangium harbor three to four dozen different biosynthetic gene clusters? What selective pressures, extremes in environment, or diverse competitors must a bacterium encounter in order to need an arsenal that large? A greater understanding of the role of small molecules in microbial ecology could be leveraged to advance natural product discovery by using earth-wide metagenomic sampling to pinpoint high-potential microbial ecosystems, and then targeting these for exhaustive single-cell sequencing.
Questions about the natural roles of natural products are particularly intriguing in their application to host-associated microbes. For example, can a computational analysis of the ecological distribution of biosynthetic gene clusters reveal a subset that have evolved to produce a molecule that targets eukaryotic or even mammalian enzymes? Can this process be refined to predict gene clusters whose small molecule products have a target that is expressed, e.g., in the mammalian gut or skin? Finally, can these predictions be merged with predictions of the chemical structure of a BGC’s product to produce a high-confidence list of candidate targets? These questions extend beyond the human host, as natural products from insect-, plant-, and sponge-associated bacteria have been an important source of natural products with human targets67–69; indeed, rapamycin and geldanamycin are exemplars of a larger class of bacterially produced molecules that may have evolved to target fungal enzymes that are conserved from fungi to humans70.
An important challenge will be to focus isolate and metagenome sequencing capacity on the most impactful samples in the near term. Although sequencing is becoming democratized and enormous latent sequence capacity exists in large centers, at companies, and in individual academic labs, samples still need to be collected and DNA needs to be extracted. Sequencing campaigns for natural product discovery should therefore be directed toward samples that are most likely to yield novelty worthy of experimental exploration. Although well-characterized clades of prolific natural product producers, including actinomycetes and myxobacteria, have been the focus of the vast majority of natural product discovery efforts, they are far from being exhausted. Recent taxonomically diverse genome sequencing efforts have also emphasized that understudied clades, including the host-associated Entotheonella71 and Photorhabdus/Xenorhabdus72,73 taxa, are also promising targets with many uncharacterized gene clusters. And although bacterial taxa that have many fewer gene clusters per Mb of genome sequence may seem less attractive as targets, the likelihood of finding a previously unknown molecular scaffold from a gene cluster in an entirely unmined taxon is high. In fact, the absolute number of distinct gene clusters in ‘low-producer’ taxa is likely to be at least as high as that in ‘high-producer’ taxa, simply because the ‘high-producers’ are a small minority. Taken together, these observations would suggest that a diversified portfolio of sequencing projects makes good sense in the coming decade. If the latest insights from ecology are thus effectively combined with state-of-the-art computational genomics and integrative omics analysis, a deep and quantitative understanding of specialized metabolism will be within grasp.
Figure for Box.
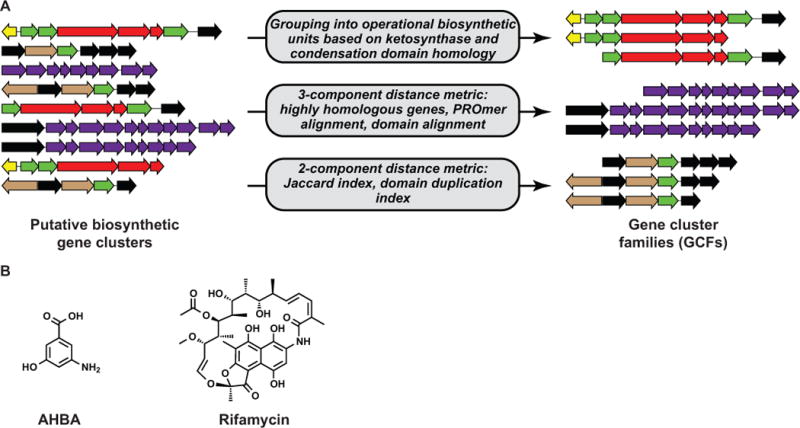
(a) Three algorithms have been developed recently to group biosynthetic gene clusters into families; see Box 1 for more details. (b) Chemical structures of 3-amino-5-hydroxybenzoic acid (AHBA) and rifamycin.
Acknowledgments
We are indebted to Peter Cimermancic, Mohamed Donia, and members of the Fischbach Group for helpful conversations. This work was supported by a Rubicon grant of the Netherlands Organization for Scientific Research (NWO; Rubicon 825.13.001) to M.H.M. and by grants from the W.M. Keck Foundation (M.A.F.), the David and Lucile Packard Foundation (M.A.F.), the Glenn Foundation (M.A.F.), the Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Disease program (M.A.F.), the Program for Breakthrough Biomedical Research (M.A.F.), DARPA award HR0011-12-C-0067 (M.A.F.), and NIH grants OD007290, AI101018, GM081879, and DK101674 (M.A.F.).
Footnotes
Competing financial interests
M.A.F. is on the scientific advisory boards of NGM Biopharmaceuticals and Warp Drive Bio.
Contributor Information
Marnix H. Medema, Email: marnix.medema@wur.nl.
Michael A. Fischbach, Email: fischbach@fischbachgroup.org.
References
- 1.Bentley SD, et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3 (2) Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 2.Ikeda H, et al. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat Biotechnol. 2003;21:526–531. doi: 10.1038/nbt820. [DOI] [PubMed] [Google Scholar]
- 3.Medema MH, Breitling R, Bovenberg R, Takano E. Exploiting plug-and-play synthetic biology for drug discovery and production in microorganisms. Nat Rev Microbiol. 2011;9:131–7. doi: 10.1038/nrmicro2478. [DOI] [PubMed] [Google Scholar]
- 4.Bouslimani A, Sanchez LM, Garg N, Dorrestein PC. Mass spectrometry of natural products: current, emerging and future technologies. Nat Prod Rep. 2014;31:718–29. doi: 10.1039/c4np00044g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krug D, Müller R. Secondary metabolomics: the impact of mass spectrometry-based approaches on the discovery and characterization of microbial natural products. Nat Prod Rep. 2014;31:768–83. doi: 10.1039/c3np70127a. [DOI] [PubMed] [Google Scholar]
- 6.Rappe MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol. 2003;57:369–394. doi: 10.1146/annurev.micro.57.030502.090759. [DOI] [PubMed] [Google Scholar]
- 7.Epstein SS. The phenomenon of microbial uncultivability. Curr Opin Microbiol. 2013;16:636–42. doi: 10.1016/j.mib.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Streit WR, Schmitz RA. Metagenomics–the key to the uncultured microbes. Curr Opin Microbiol. 2004;7:492–498. doi: 10.1016/j.mib.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Lasken RS. Genomic sequencing of uncultured microorganisms from single cells. Nat Rev. 2012;10:631–640. doi: 10.1038/nrmicro2857. [DOI] [PubMed] [Google Scholar]
- 10.Klassen JL, Currie CR. Gene fragmentation in bacterial draft genomes: extent, consequences and mitigation. BMC Genomics. 2012;13:14. doi: 10.1186/1471-2164-13-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eddy SR. Accelerated Profile HMM Searches. PLoS Comput Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weber T, et al. CLUSEAN: a computer-based framework for the automated analysis of bacterial secondary metabolite biosynthetic gene clusters. J Biotechnol. 2009;140:13–17. doi: 10.1016/j.jbiotec.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 14.Starcevic A, et al. ClustScan: an integrated program package for the semi-automatic annotation of modular biosynthetic gene clusters and in silico prediction of novel chemical structures. Nucleic Acids Res. 2008;36:6882–6892. doi: 10.1093/nar/gkn685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li MH, Ung PM, Zajkowski J, Garneau-Tsodikova S, Sherman DH. Automated genome mining for natural products. BMC Bioinforma [computer file] 2009;10:185. doi: 10.1186/1471-2105-10-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khaldi N, et al. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol. 2010;47:736–741. doi: 10.1016/j.fgb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medema MH, et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blin K, et al. antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–63. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 20.Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science (80-) 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rinke C, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–7. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 22.Cimermancic P, et al. Insights into Secondary Metabolism from a Global Analysis of Prokaryotic Biosynthetic Gene Clusters. Cell. 2014;158:412–421. doi: 10.1016/j.cell.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Punta M, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pelzer S, Wohlert SE, Vente A. Tool-box: tailoring enzymes for bio-combinatorial lead development and as markers for genome-based natural product lead discovery. Ernst Schering Res Found Workshop. 2005:233–59. doi: 10.1007/3-540-27055-8_11. at < http://www.ncbi.nlm.nih.gov/pubmed/15645724>. [DOI] [PubMed]
- 25.Weng J-K, Noel JP. The remarkable pliability and promiscuity of specialized metabolism. Cold Spring Harb Symp Quant Biol. 2012;77:309–20. doi: 10.1101/sqb.2012.77.014787. [DOI] [PubMed] [Google Scholar]
- 26.Cruz-Morales P, et al. Recapitulation of the evolution of biosynthetic gene clusters reveals hidden chemical diversity on bacterial genomes. bioRxiv. 2015 at < http://biorxiv.org/content/early/2015/06/08/020503.abstract>.
- 27.Takeda I, Umemura M, Koike H, Asai K, Machida M. Motif-independent prediction of a secondary metabolism gene cluster using comparative genomics: application to sequenced genomes of Aspergillus and ten other filamentous fungal species. DNA Res. 2014;21:447–57. doi: 10.1093/dnares/dsu010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnison PG, et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Jong A, van Hijum SA, Bijlsma JJ, Kok J, Kuipers OP. BAGEL: a web-based bacteriocin genome mining tool. Nucleic Acids Res. 2006;34:W273–9. doi: 10.1093/nar/gkl237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Jong A, van Heel AJ, Kok J, Kuipers OP. BAGEL2: mining for bacteriocins in genomic data. Nucleic Acids Res. 2010;38:W647–51. doi: 10.1093/nar/gkq365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson MC, Piel J. Metagenomic approaches for exploiting uncultivated bacteria as a resource for novel biosynthetic enzymology. Chem Biol. 2013;20:636–47. doi: 10.1016/j.chembiol.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 32.Charlop-Powers Z, Milshteyn A, Brady SF. Metagenomic small molecule discovery methods. Curr Opin Microbiol. 2014;19:70–5. doi: 10.1016/j.mib.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy BVB, Milshteyn A, Charlop-Powers Z, Brady SF. eSNaPD: a versatile, web-based bioinformatics platform for surveying and mining natural product biosynthetic diversity from metagenomes. Chem Biol. 2014;21:1023–33. doi: 10.1016/j.chembiol.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Owen JG, et al. Mapping gene clusters within arrayed metagenomic libraries to expand the structural diversity of biomedically relevant natural products. Proc Natl Acad Sci U S A. 2013;110:11797–802. doi: 10.1073/pnas.1222159110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziemert N, et al. The natural product domain seeker NaPDoS: a phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS One. 2012;7:e34064. doi: 10.1371/journal.pone.0034064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu D, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462:1056–1060. doi: 10.1038/nature08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kampa A, et al. Metagenomic natural product discovery in lichen provides evidence for a family of biosynthetic pathways in diverse symbioses. Proc Natl Acad Sci U S A. 2013;110:E3129–37. doi: 10.1073/pnas.1305867110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwan JC, et al. Genome streamlining and chemical defense in a coral reef symbiosis. Proc Natl Acad Sci U S A. 2012;109:20655–60. doi: 10.1073/pnas.1213820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boisvert S, Raymond F, Godzaridis E, Laviolette F, Corbeil J. Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol. 2012;13:R122. doi: 10.1186/gb-2012-13-12-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howe AC, et al. Tackling soil diversity with the assembly of large, complex metagenomes. Proc Natl Acad Sci U S A. 2014;111:4904–9. doi: 10.1073/pnas.1402564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. 2014:2. doi: 10.1093/bioinformatics/btv033. at < http://arxiv.org/abs/1409.7208>. [DOI] [PubMed]
- 42.Albertsen M, et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol. 2013;31:533–8. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 43.Nielsen HB, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol. 2014;32:822–8. doi: 10.1038/nbt.2939. [DOI] [PubMed] [Google Scholar]
- 44.Frasch H-J, Medema MH, Takano E, Breitling R. Design-based re-engineering of biosynthetic gene clusters: plug-and-play in practice. Curr Opin Biotechnol. 2013;24:1144–50. doi: 10.1016/j.copbio.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 45.Ziemert N, et al. Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. Proc Natl Acad Sci U S A. 2014;111:E1130–9. doi: 10.1073/pnas.1324161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doroghazi JR, et al. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol. 2014;10:963–8. doi: 10.1038/nchembio.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yadav G, Gokhale RS, Mohanty D. Computational approach for prediction of domain organization and substrate specificity of modular polyketide synthases. J Mol Biol. 2003;328:335–363. doi: 10.1016/s0022-2836(03)00232-8. [DOI] [PubMed] [Google Scholar]
- 48.Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson DH. Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMs) Nucleic Acids Res. 2005;33:5799–5808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Minowa Y, Araki M, Kanehisa M. Comprehensive analysis of distinctive polyketide and nonribosomal peptide structural motifs encoded in microbial genomes. J Mol Biol. 2007;368:1500–1517. doi: 10.1016/j.jmb.2007.02.099. [DOI] [PubMed] [Google Scholar]
- 50.Bachmann BO, Ravel J. Chapter 8. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 2009;458:181–217. doi: 10.1016/S0076-6879(09)04808-3. [DOI] [PubMed] [Google Scholar]
- 51.Röttig M, et al. NRPSpredictor2–a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011;39:W362–7. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prieto C, Garcia-Estrada C, Lorenzana D, Martin JF. NRPSsp: non-ribosomal peptide synthase substrate predictor. Bioinformatics. 2012;28:426–427. doi: 10.1093/bioinformatics/btr659. [DOI] [PubMed] [Google Scholar]
- 53.Khayatt BI, Overmars L, Siezen RJ, Francke C. Classification of the adenylation and acyl-transferase activity of NRPS and PKS systems using ensembles of substrate specific hidden Markov models. PLoS One. 2013;8:e62136. doi: 10.1371/journal.pone.0062136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baranašić D, et al. Predicting substrate specificity of adenylation domains of nonribosomal peptide synthetases and other protein properties by latent semantic indexing. J Ind Microbiol Biotechnol. 2014;41:461–7. doi: 10.1007/s10295-013-1322-2. [DOI] [PubMed] [Google Scholar]
- 55.Blin K, Kazempour D, Wohlleben W, Weber T. Improved lanthipeptide detection and prediction for antiSMASH. PLoS One. 2014;9:e89420. doi: 10.1371/journal.pone.0089420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Medema MH. The Minimum Information about a Biosynthetic Gene cluster (MIBiG) specification. Nat Chem Biol. 2015;X:XX–YY. doi: 10.1038/nchembio.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kersten RD, et al. A mass spectrometry-guided genome mining approach for natural product peptidogenomics. Nat Chem Biol. 2011;7:794–802. doi: 10.1038/nchembio.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kersten RD, et al. Glycogenomics as a mass spectrometry-guided genome-mining method for microbial glycosylated molecules. Proc Natl Acad Sci U S A. 2013;110:E4407–16. doi: 10.1073/pnas.1315492110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mohimani H, et al. Automated genome mining of ribosomal peptide natural products. ACS Chem Biol. 2014 doi: 10.1021/cb500199h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mohimani H, et al. NRPquest: Coupling Mass Spectrometry and Genome Mining for Nonribosomal Peptide Discovery. J Nat Prod. 2014 doi: 10.1021/np500370c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guthals A, Watrous JD, Dorrestein PC, Bandeira N. The spectral networks paradigm in high throughput mass spectrometry. Mol Biosyst. 2012;8:2535–2544. doi: 10.1039/c2mb25085c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Medema MH, et al. Pep2Path: Automated Mass Spectrometry-Guided Genome Mining of Peptidic Natural Products. PLoS Comput Biol. 2014;10:e1003822. doi: 10.1371/journal.pcbi.1003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nguyen DD, et al. MS/MS networking guided analysis of molecule and gene cluster families. Proc Natl Acad Sci U S A. 2013;110:E2611–20. doi: 10.1073/pnas.1303471110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schulze CJ, et al. ‘Function-first’ lead discovery: mode of action profiling of natural product libraries using image-based screening. Chem Biol. 2013;20:285–95. doi: 10.1016/j.chembiol.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Potts MB, et al. Using functional signature ontology (FUSION) to identify mechanisms of action for natural products. Sci Signal. 2013;6:ra90. doi: 10.1126/scisignal.2004657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yilmaz P, et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat Biotechnol. 2011;29:415–20. doi: 10.1038/nbt.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poulsen M, Oh D-C, Clardy J, Currie CR. Chemical analyses of wasp-associated streptomyces bacteria reveal a prolific potential for natural products discovery. PLoS One. 2011;6:e16763. doi: 10.1371/journal.pone.0016763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Piel J, et al. Exploring the chemistry of uncultivated bacterial symbionts: antitumor polyketides of the pederin family. J Nat Prod. 2005;68:472–9. doi: 10.1021/np049612d. [DOI] [PubMed] [Google Scholar]
- 69.Yu T-W, et al. The biosynthetic gene cluster of the maytansinoid antitumor agent ansamitocin from Actinosynnema pretiosum. Proc Natl Acad Sci U S A. 2002;99:7968–73. doi: 10.1073/pnas.092697199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cardenas ME, et al. Antifungal activities of antineoplastic agents: Saccharomyces cerevisiae as a model system to study drug action. Clin Microbiol Rev. 1999;12:583–611. doi: 10.1128/cmr.12.4.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wilson MC, et al. An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature. 2014;506:58–62. doi: 10.1038/nature12959. [DOI] [PubMed] [Google Scholar]
- 72.Crawford JM, Clardy J. Bacterial symbionts and natural products. Chem Commun (Camb) 2011;47:7559–66. doi: 10.1039/c1cc11574j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bode HB. Entomopathogenic bacteria as a source of secondary metabolites. Curr Opin Chem Biol. 2009;13:224–30. doi: 10.1016/j.cbpa.2009.02.037. [DOI] [PubMed] [Google Scholar]
- 74.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin K, Zhu L, Zhang DY. An initial strategy for comparing proteins at the domain architecture level. Bioinformatics. 2006;22:2081–2086. doi: 10.1093/bioinformatics/btl366. [DOI] [PubMed] [Google Scholar]
- 76.Van Heel AJ, de Jong A, Montalbán-López M, Kok J, Kuipers OP. BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013;41:W448–53. doi: 10.1093/nar/gkt391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anand S, et al. SBSPKS: structure based sequence analysis of polyketide synthases. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Medema MH, Takano E, Breitling R. Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol Biol Evol. 2013;30:1218–1223. doi: 10.1093/molbev/mst025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mohimani H, et al. Cycloquest: Identification of Cyclopeptides via Database Search of Their Mass Spectra against Genome Databases. J Proteome Res. 2011;10:4505–4512. doi: 10.1021/pr200323a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Markowitz VM, et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42:D560–7. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ichikawa N, et al. DoBISCUIT: a database of secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2013;41:D408–14. doi: 10.1093/nar/gks1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Conway KR, Boddy CN. ClusterMine360: a database of microbial PKS/NRPS biosynthesis. Nucleic Acids Res. 2013;41:D402–7. doi: 10.1093/nar/gks993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Diminic J, et al. Databases of the thiotemplate modular systems (CSDB) and their in silico recombinants (r-CSDB) J Ind Microbiol Biotechnol. 2013;40:653–659. doi: 10.1007/s10295-013-1252-z. [DOI] [PubMed] [Google Scholar]
- 84.Tae H, Sohng JK, Park K. MapsiDB: an integrated web database for type I polyketide synthases. Bioprocess Biosyst Eng. 2009;32:723–727. doi: 10.1007/s00449-008-0296-3. [DOI] [PubMed] [Google Scholar]
- 85.Hastings J, et al. The ChEBI reference database and ontology for biologically relevant chemistry: enhancements for 2013. Nucleic Acids Res. 2013;41:D456–63. doi: 10.1093/nar/gks1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bento AP, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42:D1083–90. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nakamura Y, et al. KNApSAcK Metabolite Activity Database for Retrieving the Relationships Between Metabolites and Biological Activities. Plant Cell Physiol. 2013;55:e7–e7. doi: 10.1093/pcp/pct176. [DOI] [PubMed] [Google Scholar]
- 88.Wang Y, et al. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37:W623–33. doi: 10.1093/nar/gkp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pence HE, Williams A. ChemSpider: An Online Chemical Information Resource. J Chem Educ. 2010;87:1123–1124. [Google Scholar]
- 90.Caboche S, et al. NORINE: a database of nonribosomal peptides. Nucleic Acids Res. 2008;36:D326–31. doi: 10.1093/nar/gkm792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lucas X, et al. StreptomeDB: a resource for natural compounds isolated from Streptomyces species. Nucleic Acids Res. 2013;41:D1130–6. doi: 10.1093/nar/gks1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Harborne JB. Dictionary of Natural Products. Phytochemistry. 1995;38:279. [Google Scholar]
- 93.Weber T. In silico tools for the analysis of antibiotic biosynthetic pathways. Int J Med Microbiol. 2014;304:230–5. doi: 10.1016/j.ijmm.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 94.Boddy CN. Bioinformatics tools for genome mining of polyketide and non-ribosomal peptides. J Ind Microbiol Biotechnol. 2014;41:443–450. doi: 10.1007/s10295-013-1368-1. [DOI] [PubMed] [Google Scholar]