

Abstract
Importance
Clearer delineation of the phenotypic heterogeneity within behavioral variant frontotemporal dementia (bvFTD) will help uncover underlying biological mechanisms, and will improve clinicians’ ability to predict disease course and design targeted management strategies.
Objective
To identify subtypes of bvFTD syndrome based on distinctive patterns of atrophy defined by selective vulnerability of specific functional networks targeted in bvFTD, using statistical classification approaches.
Design, Setting and Participants
In this retrospective observational study, 104 patients meeting the Frontotemporal Dementia Consortium consensus criteria for bvFTD were evaluated at the Memory and Aging Center of Department of Neurology at University of California, San Francisco. Patients underwent a multidisciplinary clinical evaluation, including clinical demographics, genetic testing, symptom evaluation, neurological exam, neuropsychological bedside testing, and socioemotional assessments. Ninety patients underwent structural Magnetic Resonance Imaging at their earliest evaluation at the memory clinic. From each patients’ structural imaging, the mean volumes of 18 regions of interest (ROI) comprising the functional networks specifically vulnerable in bvFTD, including the ‘salience network’ (SN), with key nodes in the frontoinsula and pregenual anterior cingulate, and the ‘semantic appraisal network’ (SAN) anchored in the anterior temporal lobe and subgenual cingulate, were estimated. Principal component and cluster analyses of ROI volumes were used to identify patient clusters with anatomically distinct atrophy patterns.
Main Outcome Measures
We evaluated brain morphology and other clinical features including presenting symptoms, neurologic exam signs, neuropsychological performance, rate of dementia progression, and socioemotional function in each patient cluster.
Results
We identified four subgroups of bvFTD patients with distinct anatomic patterns of network degeneration, including two separate salience network–predominant subgroups: frontal/temporal (SN-FT), and frontal (SN-F), and a semantic appraisal network–predominant group (SAN), and a subcortical–predominant group. Subgroups demonstrated distinct patterns of cognitive, socioemotional, and motor symptoms, as well as genetic compositions and estimated rates of disease progression.
Conclusions
Divergent patterns of vulnerability in specific functional network components make an important contribution to clinical heterogeneity of bvFTD. The data-driven anatomical classification identifies biologically meaningful phenotypes and provides a replicable approach to disambiguate the bvFTD syndrome.
INTRODUCTION
Behavioral variant frontotemporal dementia (bvFTD) is a clinical syndrome associated with frontal-predominant neurodegeneration.1-3 Despite numerous detailed accounts, no systematic understanding of the nature, frequency, and mechanisms of bvFTD clinical symptoms has arisen, in part due to tremendous diversity across patients.
The recent realization that unique functional neural networks are selectively vulnerable in bvFTD has fueled new insights to the syndrome’s clinical and pathological diversity.4-7 Evidence suggests that along with other networks, bvFTD targets intrinsic networks responsible for social-emotional-autonomic processing, i.e. the salience network (SN), and semantically-driven personal evaluation, the “limbic” or semantic appraisal network (SAN).8-10 The SN includes the frontoinsula and pregenual anterior cingulate structures and is closely allied with the SAN, which includes the temporal pole, ventral striatum, subgenual cingulate, and basolateral amygdala. We hypothesized that patterns of regional vulnerability of brain structures that undergo network-specific degeneration may reveal distinct subtypes within the bvFTD syndrome. A network layout will unfold clinical portfolios characterized by uniquely disrupted functional architecture and will provide specific therapeutic targets for pharmacological and behavioral treatments.
We examined the prevalence of early clinical symptoms in a large sample of bvFTD patients (n=104), and report the key features of their initial clinical presentation. We used statistical classification approaches to identify subgroups of patients with different patterns of grey matter loss in specific regions of interest in the SN and SAN, and report distinct symptom profiles of each subgroup.
METHODS
Subjects
One hundred and four patients, meeting the International bvFTD Criteria Consortium (FTDC) revised guidelines for the diagnosis of bvFTD,11 were evaluated at the University of California, San Francisco (UCSF) Memory and Aging Center (MAC). All patients underwent a complete clinical history, physical examination, neuropsychological evaluation and structural brain imaging. The diagnosis was made at a multidisciplinary consensus meeting for each patient individually. A retrospective chart review fulfilled the FTDC criteria for a subset of patients (n=16) who were initially diagnosed with Neary criteria.1 Eligibility criteria for age-matched healthy participants (n=44), who contributed as imaging controls included normal cognitive performance, normal structural brain image, and absence of neurological, psychiatric, or other major medical illnesses. Informed consent was obtained from all participants or their assigned surrogate decision makers. The study was approved by UCSF institutional review board for human research.
Clinical evaluation and detailed behavioral assessment
Clinical dementia rating (CDR), CDR Sum of Boxes (CDRSOB), and Frontotemporal Lobar Degeneration modified CDRSOB (FTLD-CDRSOB), were completed for each patient via a structured caregiver interview at the first visit to the memory clinic. Mini Mental State Exam (MMSE) and dementia rating scores were recorded at the first presentation to memory clinic. The National Alzheimer's Coordinating Center (NACC) symptom checklist to identify specific behavioral deficits was completed for 94 patients within 12 months of the first presentation to memory clinic. Patients included in the subsequent volumetric analyses (described below), completed their neuropsychological, socioemotional and neurological examinations within 90 days of structural brain imaging (eMethods). Socioemotional tests included The Awareness of Social Inference Test (TASIT),12 UCSF Cognitive Theory of Mind (cTOM),13 Interpersonal Reactivity Index (IRI)14,15 and Interpersonal Adjective Scale (IAS).16,17
Genetic analysis and autopsy
Genetic analysis identified the presence of FTD-causing mutations in C9ORF72, microtubule-associated protein tau (MAPT), and progranulin (GRN).18 Six patients chose to withhold from genetic testing. In the subset of bvFTD patients without an FTD-causing mutation, the frequency of GRN and MAPT allele distributions were compared to the normal population data from National Center for Biotechnology database. Twenty-four patients underwent autopsy and complete neuropathological assessment (eMethods).
Cross sectional model across stages of disease severity
To examine the effect of disease severity on symptomatology, utilizing the data from follow-up evaluations at the MAC, we generated a cross sectional model to categorize patients according to their FTLD-CDRSOB score.19 Based on previous evidence of ~3.5 annual gain of CDRSOB in bvFTD patients,20 we identified four levels of disease severity (FTLD-CDRSOB: 4–7.5, 8–11.5, 12–15.5, >16). We semi-randomly assigned patients into four roughly equal sets (n=29, n=27, n=28, and n=20, for FTLD-CDRSOB: 4–7.5, 8–11.5, 12–15.5, ≥ 16, respectively).
Cluster analysis and Principal Component Analysis
Ninety patients underwent a unified structural Magnetic Resonance Imaging (MRI) acquisition protocol (1.5, 3, or 4 Tesla), and were included in grey matter volume assessment (eMethods) using Voxel Based Morphometry (VBM) toolbox of Statistical Parametric Mapping (SPM8; http://www.fil.ion.ucl.ac.uk/spm/software/spm8/). We used the Neuromorphometric brain atlas (http://www.neuromorphometrics.com) to define regions of interest (ROI) uniquely contributing to 4 potentially bvFTD-affected networks, including SN, SAN, default-mode,21,22 and frontoparietal networks.23,24 Initial analyses showed no statistical benefit of default-mode and frontoparietal structures, hence the final models included only the 18 frontal and temporal ROIs comprising the SN and SAN networks. These included, bilaterally, the temporal poles, gyrus recti, subcallosal areas, anterior cingulate gyri, anterior insulae, basal forebrains, frontal operculae, posterior orbital gyri, and amygdalae. We estimated the mean grey matter volume of each ROI, for each patient, and used a cluster analysis based on Euclidean distance to derive four clusters of bvFTD patients (eMethods). Using the same ROIs, we performed a principal component analysis (PCA) to demonstrate the association between patient clusters and SN and SAN volume loss. We mapped each patient onto the Euclidean space based on the 1st and 2nd components of the PCA keeping their cluster identity. We modeled the analysis to identify the greatest possible number of distinct clusters of bvFTD patients that remained meaningfully separated when mapped onto dimensional space, with the result that 4-cluster solution was the best fit. We used a polytomous logistic regression model to determine the stability of the clusters (eMethods). VBM-derived grey matter volumes of the four bvFTD clusters were compared to an age-matched control population (n=44; age=62.7±1.5). PCA, cluster, and logistic regression analyses used MatLab statistical toolbox. Statistical comparisons of clinical-behavioral measures between subgroups were performed using SAS (SAS Institute, Cary, NC).
RESULTS
Presentation and progression of bvFTD syndrome
bvFTD patients presented to the memory clinic at an average age of 61 years (Table 1). Initial clinical assessment within 12 months of the first presentation revealed an MMSE of 22.5, a CDRSOB of 7.3, and an FTLD-CDRSOB of 9.9 (Table 1). 27.6% of patients who underwent genetic assessment (n=98) showed a genetic mutation. C9ORF72 accounted for more than 50% of mutations (Table 1). Mutation-negative bvFTD patients showed a trend towards a higher frequency of GRN-T-allele (rs5848; P=.054).
Table 1.
Patient Characteristics
| bvFTD patients (n=104) | |
|---|---|
| Age at disease onset– yr | 54.8 ± 9.5 |
| Age at first evaluation – yr | 60.8 ± 8.2 |
| Male sex – no. (%) | 63 (60.6) |
| White – no. (%) | 94 (90.4) |
| Right handedness – no. (%) | 93 (89.4) |
| Education – yr | 16.8 ± 9.0 |
| MMSE at initial evaluation | 22.5 ± 7.1 |
| CDR at initial evaluation | 1.4 ± 0.7 |
| CDRSOB at initial evaluation | 7.3 ± 3.5 |
| FTLD-CDRSOB at initial evaluationa | 9.9 ± 4.2 |
| Mutation carriers – no. (%)b | 27 (27.6) |
| C9ORF72 – no. (%) | 15 (55.6) |
| GRN – no. (%) | 6 (22.2) |
| MAPT – no. (%) | 6 (22.2) |
| GRN T allele frequency – no. (%) | 41 (31.1)c |
| Tau H1 allele frequency – no. (%) | 107 (81.1) |
Abbreviations: bvFTD = behavioral variant frontotemporal dementia; CDR = Clinical Dementia Rating; CDRSOB = CDR Sum of Boxes; C9ORF72 = chromosome 9 open reading frame 72 hexanucleotide expansions; FTLD-CDRSOB = Frontotemporal Lobar Degeneration - modified CDRSOB; GRN = progranulin; MAPT microtubule-associated protein tau; MMSE = Mini Mental State.
Age, MMSE, CDR, CDRSOB, and FTLD-CDRSOB, at initial evaluation represent patient characteristics measured within one year of first presentation. Values for age, education, MMSE, CDR, CDRSOB, and CDR-FTLD, are mean ± standard deviations. Race or ethnic group was self-reported. Scores on the MMSE range from 0 to 30, with higher scores denoting better cognitive function. Scores on the CDR, CDRSOB, and FTLD-CDRSOB range from 0 to 3, 0 to 18 and 0 to 24, respectively, with higher scores denoting more disability.
n=89 bvFTD patients had FTLD-CDRSOB recorded at their first evaluation.
percentages are calculated for the patients who underwent genetic testing (n=98).
P=.054, compared to allele frequency of normal population using Chi-square test.
Each core symptom of FTDC criteria showed > 70% prevalence, although no symptom was 100%, highlighting the clinical heterogeneity of bvFTD (Figure 1A). 73% of bvFTD patients presented with behavioral abnormalities as their first symptom (Figure 1B), while the rest presented with executive (16%), memory (6%), language (4%), or motor (1%) deficits as their first symptom. Visuospatial, sensory, or constitutional deficits were not reported as first symptoms. Based on NACC symptom categories, personality change was the most common early behavioral symptom (Figure 1C). Apathy, emotional blunting, disinhibition, obsessive behavior, and altered eating habits, were the next most common behavioral symptoms, respectively (Figure 1C). Violent behaviors and hallucinations were the least prevalent behavioral symptoms (data not shown). Prevalence of symptoms in all domains increased as the disease progressed (Figure 1D). 100% of patients reported both behavioral and executive deficits by the point of moderate severity, i.e.FTLD-CDRSOB 12-15.5. Although never reported as first symptoms, visuospatial and constitutional symptoms were reported in a subset of patients at all stages of severity (Figure 1D).
Figure 1. Prevalence of FTDC criteria and major symptom domains in bvFTD.

A: Prevalence of core diagnostic features of bvFTD patients. B: Percentage of patients with the initial symptoms in behavior, executive, memory, language, and motor domains. C: Rates of NACC behavioral symptom checklist categories in bvFTD patients during the first 12 months of presenting to memory clinic (n=94). Note that only the top six symptoms are shown in descending order from left to right. D: The rates of symptoms in each of the main domains, in bvFTD patients categorized according to their disease severity (i.e. FTLD-CDRSOB score). FTDC = Frontotemporal Dementia Criteria Consortium; FTLD-CDRSOB = Frontotemporal Lobar Degeneration modified Clinical Dementia Rating Sum of Boxes (CDRSOB); NACC = National Alzheimer Coordinating Center.
bvFTD subgroup analyses
We next tested the hypothesis that subsets of patients within the bvFTD syndrome would show distinct patterns of atrophy25-27 based on vulnerability of brain structures that undergo network-specific degeneration in bvFTD. More specifically, we predicted that unique subgroups of patients would be identified based on volumetric differences in SN and SAN structures.7-9,28 We identified 4 patient clusters based on 18 ROIs defining SN and SAN. A polytomous regression model predicted the assigned cluster with >0.80 probability for 99% of the patients and confirmed cluster stability. Keeping their cluster identity, we mapped each patient onto dimensional space derived from a PCA analysis based on the same ROIs (Figure 2A). Three of the four subgroups showed a distinct mapping along the 1st dimension (Figure 2A–top panel, yellow, blue and green subgroups). Both SN and SAN volumes contributed strongly to 1st dimension (Figure 2A–bottom panel, grey-scale gradient along x-axes), indicating that 1st dimension primarily predicts the overall degree of cortical atrophy. The 2nd dimension of the PCA more distinctly predicted the SAN volume (Figure 2A–bottom panel), and the cluster analysis identified a subset of patients with high SAN and intermediate SN volume loss (pink subgroup). Both yellow and blue subgroups show higher degree of SN volume loss (Figure 2A).
Figure 2. Anatomic subtypes of bvFTD.

bvFTD patients are clustered into four distinct subgroups based on the degree of atrophy in 18 ROIs defining SN and SAN functional networks. The four clusters consist of two distinct SN affected groups – frontotemporal (SN-FT, the yellow dots) and frontal (SN-F, the blue dots), one SAN affected group (the pink dots), and one subcortical group (green dots). A (top panel): Each patient is colored according to their cluster assignment and plotted onto the first two dimensions of a principal component analysis based on the same 18 ROIs. A (bottom panels): The grey scale indicates the normalized volume of the sum of each patient’s ROIs representing the SN and SAN networks respectively, where darker colors show greater degree of volume loss. x and y axes are the same as for the top panel. B: Voxel based morphometry-derived atrophy maps of each bvFTD subgroup. T maps show the atrophy patterns compared to age matched normal controls (n = 44), and are superimposed onto whole-brain template derived from elderly normal controls. Effects are corrected for age, gender and total intracranial volume of each individual, and are corrected for family-wise error at the whole brain level at P<.05. C: Percentage of mutation carriers for C9ORF72, MAPT, or GRN mutations in each subgroup. D: Percentage of patients found to have specific classes of neuropathology associated with each bvFTD subgroup, among the subset of patients with autopsy diagnosis. Patients who were diagnosed as Tau-other pathology included two SN-FT patients with FTLD-tau with MAPT mutation, one SAN patient with FTDP17, and one subcortical patient with argyrophilic grain disease. CBD = corticobasal degeneration; C9ORF72 = chromosome 9 open reading frame 72 hexanucleotide expansions; FTDP-17=Frontotemporal dementia with parkinsonism-17; FTLD=Frontotemporal lobar degeneration; GRN = progranulin; MAPT microtubule-associated protein tau; ROI = region of interest; SAN = sematic appraisal network; SN = salience network; TDP = transactive response DNA-binding protein 43.
VBM of each subgroup compared to an age-matched control group demonstrated distinct pattern of atrophy (Fig 2B). Three of the four clusters showed significant cortical atrophy (Figure 2A,B). One group showed both frontal and temporal grey matter loss with significant subcortical involvement (yellow dots, labeled SN frontal-temporal, or SN-FT), while the second group showed predominantly frontal and subcortical involvement (blue group, labeled SN frontal, or SN-F). The third cluster, which included patients with a high degree of atrophy to SAN structures, showed atrophy localized to right temporal lobe, with only modest subcortical involvement (pink group, labeled semantic appraisal network, or SAN). Finally, the fourth subset of patients showed minimal cortical and predominantly subcortical atrophy (green dots, labeled subcortical).
Global cognitive dysfunction, genetic, and neuropathologic features of bvFTD subgroups
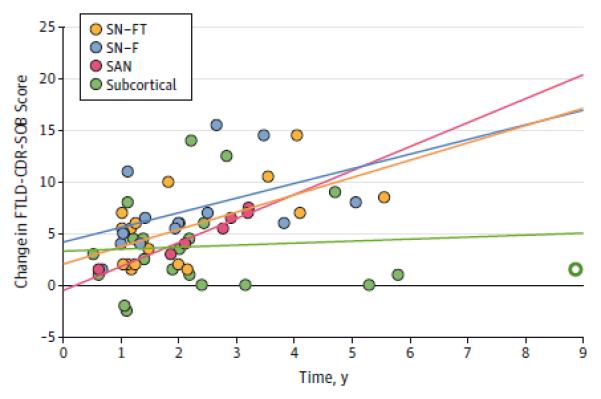
There were no differences between subgroups in age at evaluation, disease duration, and depression (Table 2). SN-FT had significantly higher dementia rating scores (i.e. CDR and FTLD-CDRSOB) and poorer MMSE scores compared to the subcortical group, while all other pairwise comparisons on symptom severity were equivalent. Subcortical patients, compared to other subgroups, showed a slower trend in disease progression (eFigure 1, P=.07).
Table 2.
Clinical characteristics of bvFTD subgroups
| SN–FT (n = 21) |
SN–F (n = 27) |
SAN (n = 8) |
Subcortical (n = 30) |
|
|---|---|---|---|---|
| Age at imaging – yr | 62.8 (1.8) | 61.7 (1.6) | 58.3 (2.7) | 60.9 (1.4) |
| Disease duration – yr | 6.6 (1.2) | 5.8 (1.1) | 5.1 (1.9) | 6.1 (1.0) |
|
| ||||
| Global cognitive measures and GDS | ||||
| MMSEh | 19.9 (1.4)a | 21.2 (1.2) | 25.6 (2.0) | 25.3 (1.1) |
| FTLD-CDRSOBi | 11.5 (0.9)a | 10.5 (0.7) | 8.4 (1.5) | 8.0 (0.7) |
| CDRj | 1.5 (0.1)a | 1.5 (0.1)a | 1.3 (0.2) | 0.9 (0.1) |
| GDSk | 4.6 (1.6) | 9.6 (1.3) | 6.3 (2.2) | 8.5 (1.2) |
|
| ||||
| Neurological Exam – no. (%) | ||||
| Reduced facial expression | 7 (33.3) | 10 (37.0) | 3 (37.5) | 9 (30.3) |
| Rigidity | 10 (47.6) | 14 (51.8) | 4 (50.0) | 12 (40.0) |
| Action or postural tremor | 7 (33.3) | 11 (40.7) | 3 (37.5) | 9 (30.0) |
| Incoordination | 6 (28.6) | 17 (63.0) | 2 (25.0) | 15 (50.0) |
| Motor neuron related signs | 2 (9.5) | 5 (18.5) | 1 (10.0) | 2 (6.7) |
| Parkinsonism related signs | 6 (28.57) | 14 (51.9) | 3 (37.5) | 11 (36.7) |
| Abnormal gait | 0 (0.0) | 10 (37.0)b | 2 (25.0)b | 5 (16.7)b |
|
| ||||
| Socioemotional functioning | ||||
| Emotion namingl | 6.5 (0.8) | 6.8 (0.6) | 5.6 (1.2) | 8.2 (0.5) |
| Paralinguistic sarcasm detectionm | 11.7 (1.5) | 11.6 (1.2) | 5.4 (2.4) c | 14.3 (1.0) |
| Complex social cognitionn | 35.6 (3.0) | 35.8 (2.5) | 36.1 (4.4) | 43.9 (1.7) |
| Cognitive perspective takingo | 12.3 (1.1) | 9.4 (0.9) | 10.0 (1.7) | 11.4 (0.7) |
| Empathic perspective takingp | 11.8 (2.3) | 12.9 (2.1) | 21.6 (3.7) | 12.7 (2.0) |
| Empathic concernq | 18.8 (3.0) | 21.9 (2.6) | 28.0 (4.7) | 19.5 (2.5) |
| Interpersonal warmthr | 28.6 (4.0) | 30.0 (4.3) | 46.4 (8.8) | 31.0 (3.5) |
| Interpersonal assertivenessr | 36.9 (3.9) | 32.0 (4.2) | 41.6 (8.5) | 38.4 (3.2) |
|
| ||||
| Neuropsychological performance t | ||||
| Executive | ||||
| Error insensitivity | 0.3 (0.2) | 0.6 (0.2) | 0.6 (0.3) | 0.6 (0.2) |
| Modified Trails | 0.3 (0.1) | 0.2 (0.0) | 0.2 (0.1) | 0.3 (0.0) |
| Stroop color naming | 67.0 (6.1) | 54.5 (5.0) | 65.2 (8.4) | 52.9 (4.2) |
| Stroop inhibition | 37.1 (4.6) | 23.8 (3.9) | 41.2 (5.4) | 26.9 (3.1) |
| Digit span forward | 5.9 (0.4) | 5.5 (0.3) | 6.5 (0.6) | 5.2 (0.3) |
| Digit span backward | 4.1 (0.3) | 3.1 (0.2) d | 4.2 (0.4) | 3.4 (0.2) |
| Design fluency | 4.8 (0.9) | 4.0 (0.7) | 5.2 (1.2) | 5.0 (0.6) |
| D-words (lexical fluency) | 7.6 (0.9) | 4.3 (0.9) e | 8.3 (1.2) | 7.6 (0.7) |
| Animals (Category fluency) | 10.3 (1.4) | 9.4 (1.1) | 11.0 (1.9) | 11.3 (1.0) |
| Visuospatial | ||||
| Benson copy | 15.1 (0.6) | 13.8 (0.5) | 14.8 (0.9) | 13.7 (0.5) |
| Face recognition | 10.0 (0.6) | 9.7 (0.5) | 10.0 (1.0) | 10.5 (0.4) |
| Number location | 8.8 (0.7) | 7.4 (0.5) | 6.9 (0.8) | 7.8 (0.4) |
| Memory | ||||
| Benson delayed recall | 6.9 (0.9) | 7.9 (0.7) | 4.2 (1.3) a | 8.8 (0.7) |
| CVLT short delay recall | 3.4 (0.4) a | 4.3 (0.3) | 3.9 (0.6) | 4.9 (0.3) |
| CVLT delayed recall | 2.2 (0.5) a | 3.8 (0.4) | 2.3 (0.7) | 4.2 (0.4) |
| Affect matching | 7.5 (0.8) | 7.9 (0.7) | 8.3 (1.4) | 9.6 (0.6) |
| Language | ||||
| Repetition | 4.3 (0.3) | 4.3 (0.2) | 3.9 (0.4) | 3.6 (0.2) |
| Reading irregular words | 5.0 (0.3) | 5.8 (0.2) | 5.0 (0.3) | 5.6 (0.2) |
| Syntax comprehension | 4.8 (0.3) | 3.3 (0.2)f | 3.9 (0.4) | 3.6 (0.2)b |
| Verbal agility | 5.5 (0.3) | 5.2 (0.3) | 5.0 (0.4) | 5.0 (0.2) |
| Boston Naming Test | 10.6 (0.7) | 12.6 (0.5) | 9.2 (1.0) g | 12.5 (0.5) |
| CVLT total score | 18.6 (1.2) | 18.3 (1.0) | 18.2 (1.8) | 19.3 (1.0) |
| Calculations | 3.8 (0.3) | 3.1 (0.3) | 3.6 (0.4) | 3.4 (0.2) |
Abbreviations: bvFTD = behavioral variant frontotemporal dementia; CDR = Clinical Dementia Rating; CVLT = California Verbal Learning Test – short form; FTLD-CDRSOB = Frontotemporal Lobar Degeneration - modified CDR Sum of Boxes; GDS = Geriatric Depression Scale. MMSE = Mini Mental State Exam; SAN indicates semantic appraisal network–predominant subgroup; SN-FT, salience network–predominant frontal/temporal subgroup; SN-F, salience network–predominant frontal subgroup.
All values, except for the neurological exam findings, indicate the least-square-means and standard errors, derived from a general linear model comparing the four subgroups. Pairwise comparisons that were statistically significant after correcting for multiple comparisons between groups (Tukey- Kramer) are indicated in the footnote. Socioemotional and neuropsychological test sores that appear in bold text indicate values that are <1.5 standard deviations below compared to an age-matched control group. Socioemotional and neuropsychological test performances were controlled for MMSE and sex in this model. Motor neuron related signs referred to the presence of one or more of the following: reduced muscle power, muscle atrophy, and fasciculations. Parkinsonism related signs referred to the presence of one or more of the following: rest tremors, body bradykinesia, and positive retropulsion test. Details of patient evaluations including, neurological examination, neuropsychiatric and socioemotional testing are provided in detail in the eMethods.
P < 0.05 compared to subcortical.
P < 0.05 compared to SN-FT.
P < 0.01 compared to subcortical.
P < 0.05 compared to SN-FT and SAN.
P < 0.05 compared to SN-FT, SAN and Subcortical.
P < 0.01 compared to SN-FT.
P < 0.05 compared to SN-F and subcortical.
scores range from 0-30 with higher scores indicating better cognition.
scores range from 0-24 with higher scores indicating more advanced dementia.
scores range from 0-3 with higher scores indicating more advanced dementia.
scores range from 0-30 with higher scores indicating greater depression.
Emotion naming was evaluated using the TASIT-EET (The Awareness of Social Inference Test-Emotion Evaluation Test), with scores ranging from 0-14 and higher scores indicating better performance. 1
Paralinguistic elements of sarcasm was evaluated using TASIT-SIM (TASIT-Social Inference Minimal), with scores ranging from 0-20 and higher scores showing better performance.1
Complex social cognition was evaluated using TASIT SIE (The Awareness of Social Inference Test-Social Inference Enriched), with scores ranging from 0-64 and higher scores indicating better performance.1
Cognitive perspective taking was evaluated using the UCSF-Theory of Mind test (TOM), with scores ranging from 0-16 and higher scores indicating better performance.2
Empathic perspective taking was evaluated using the Perspective Taking (PT) subscale of the Interpersonal Reactivity Index (IRI), with scores ranging from 7-35 and higher scores indicating better performance.3
Empathic concern was evaluated using the Empathic Concern (EC) subscale of the IRI, with scores ranging from 7-35 and higher scores indicating better performance.3
Interpersonal warmth and interpersonal assertiveness were evaluated using the Interpersonal Adjective Scale (IAS) and scored using the IAS computer scoring program which generates T-scores (mean=50, SD 10) by comparing subject scores to a gender-matched, community-based normative sample data.4
The subgroups showed differences in their distribution of mutation carriers and of primary neuropathology. C9ORF72 mutations were found in all subgroups, comprising the majority (88%) of mutations found in subcortical group, 40% of SN-FT and SN-F mutation carriers, and 33% of SAN mutation carriers (Figure 2C). The remaining 60% in the SN-FT group had exclusively MAPT mutations, while the remaining 60% in the SN-F group had exclusively GRN mutations (Figure 2C). All subgroups showed both Tau and TDP neuropathology (Figure 2D; n=24). 83% (5 of 6) of the subcortical group showed TDP, with the remaining patient having argyrophilic grain disease. All 3 patients with corticobasal degeneration pathology belonged to SN-F group. Pick’s disease was found in 2 patients, each classified as SN-FT and SAN. These results suggest that subcortical-type bvFTD patients have a higher likelihood of TDP pathology.
FTDC criteria and presenting symptoms of bvFTD subgroups
Core FTDC symptoms were found in the majority of patients in all 4 subgroups. The overall heterogeneity of symptom profiles across all bvFTD patients was partly explained by specific within-subgroup patterns; e.g. SAN patients showed 100% prevalence in disinhibition, with lower rates in other subgroups, and SN-FT patients showed 100% prevalence in hyperorality, with < 80% in other groups (Figure 3A). SN-F patients had the highest prevalence of altered neuropsychological performance (Figure 3A), and the highest rate of dyexecutive first symptoms (Figure 3B). 60% of SAN patients showed early obsessive behaviors, though it was <25% in other groups.
Figure 3. FTDC criteria, presenting symptoms and socioemotional dysfunction in bvFTD subgroups.

A: Prevalence of core diagnostic features in each bvFTD subgroup. B: Percentage of patients in each bvFTD subgroup with the initial symptoms in behavior, executive, memory, language, and motor domains. C: Rates of NACC behavioral symptom checklist categories in each bvFTD subgroup during the first 12 months of presenting to memory clinic. Note that only the top six symptoms are shown in descending order from left to right. D: Rates of specific socioemotional impairments observed in each bvFTD subgroup. Socioemotional function was considered impaired at a Z-score<1.35 based on published normative samples of age-matched healthy controls. Complex social cognition, emotion naming and sarcasm detection (paralinguistic, i.e. voice prosody and facial expression) abilities were measured with The Awareness of Social Inference Test. Cognitive perspective taking was assessed with the UCSF Cognitive theory of Mind (cTOM) test. Interpersonal assertiveness and warmth were measured with the Dominance and Warmth subscales of the Interpersonal Adjective Scales. Empathy was measured with the Interpersonal Reactivity Index. FTDC = Frontotemporal Dementia Criteria Consortium; NACC = National Alzheimer Coordinating Center.
Neurological exam, neuropsychological bedside testing and socioemotional evaluation of bvFTD subgroups
Neurological examination showed different rates of signs across subgroups, however, only abnormal gait reached statistical significance (Table 2; SN-FT 0%, other groups 16-37%, P<.010).
In neuropsychological bedside testing, all groups showed clinical impairments in aspects of executive, visuoconstruction, memory, and language testing. SN-F group showed worse executive function with significantly poorer digit-span-backward and lexical fluency abilities in pairwise comparisons (Table 2). SN-FT showed the most predominant memory dysfunction and significantly better syntax comprehension in pairwise comparisons (Table 2).
Socioemotional testing also revealed distinct clinical impairments across subgroups. 100% of SAN group was impaired at sarcasm detection, while only 28% were impaired in the subcortical group (Figure 3D). SAN showed significantly low scores in sarcasm detection compared to subcortical group in pairwise comparisons (Table 2). As a whole, the SAN group fell in the normal range for empathy and interpersonal and warmth (Table 2), while both SN-FT and subcortical groups showed diminished empathic perspective taking, empathic concern and, warmth. Only 59% of subcortical patients were impaired on complex social cognition, whereas 100% of patients in all other subgroups were impaired (Figure 3D).
DISCUSSION
We characterized a large cohort of bvFTD patients with detailed clinical and genetic evaluation and structural brain imaging. When patients were clustered into subgroups based on atrophy patterns, distinct profiles of neuropsychological, socioemotional, motor, and other clinical symptoms arose that provided greater precision within the striking heterogeneity seen across the group as a whole.
Our results underscored the diversity of the bvFTD syndrome. None of the FTDC criteria reached 100% prevalence, indicating that no one symptom is perfectly sensitive and specific to bvFTD. Nearly one quarter of our sample presented with first symptoms that were not behavioral (i.e. executive, memory, language, motor), suggesting that a non-behavioral first symptom cannot be taken as evidence that bvFTD will not emerge as the dominant clinical syndrome. Motor, visuospatial and constitutional symptoms, though considered atypical for bvFTD, were observed in a minority of patients. Despite the heterogeneity, however, personality, and socioemotional changes dominated throughout the disease, hence, reiterating the importance of standardized tools to quantify these deficits in bvFTD.
The discovery of intrinsic brain connectivity patterns has led to delineation of specific functional networks distributed in frontal and temporal lobes that are selectively vulnerable in bvFTD. Previous attempts have been made to identify subtypes of bvFTD based on clinical symptoms.2,29-31 Whitwell and colleagues25 performed one large study using statistical classification approaches based on regional anatomy, specifically 26 volumetric ROIs representing all structures in the frontal, temporal and parietal lobes, and suggested that bvFTD could be subdivided into different anatomic subtypes. Here, we extend these findings by demonstrating that volume loss in specific vulnerable functional networks alone accounts for the majority of meaningful anatomic variance in bvFTD. Though in our initial analytic approach we included additional network structures from frontoparietal and default-mode networks, we found that 18 volumetric ROIs from SN and SAN were adequate to classify patients into four subgroups. The SN-F and SN-FT subgroups paralleled the “frontal-predominant” and “frontotemporal-predominant” subgroups identified by Whitwell et.al, respectively.25 The SAN subgroup was comparable to Whitwell et.al’s “temporal dominant” group, although, as opposed to the largely bitemporal atrophy distribution they identified, our network-based approach identified a subset of predominantly right temporal patients.
We also identified a novel subgroup, representing 36% of our cohort, who have minimal cortical atrophy (i.e. subcortical). This finding is relevant to the debate around slowly progressive-bvFTD or “bvFTD-phenocopy”.31-33 Previous studies have reported a significant fraction of patients with minimal cortical atrophy and slow progression, yet otherwise having a clear bvFTD syndrome. Some have suggested that these patients may not have an underlying neurodegenerative disease.31 The current subcortical group showed ≥ 70 % prevalence in each core diagnostic symptom, while nearly 30% carried an FTD-related mutation, and the six autopsy diagnoses were 100% FTLD. This group also included patients with the slowest progression. These results confirm that the subcortical group is a true neurodegenerative phenotype, and the most likely degenerative pattern in patients with slowly progressive-bvFTD. The minimal cortical atrophy suggests that, in these patients, the typical bvFTD syndrome may result from disconnection of major network hubs from their striatal or thalamic partners.34
Identifying distinct subgroups within the bvFTD syndrome provides a neural substrate explaining much of the diversity reported in previous studies. The statistical division of patients with clear SN atrophy into two separate groups (SN-F and SN-FT) emphasizes that the presence of temporal atrophy in addition to frontal SN damage provides a meaningful anatomical differentiation of bvFTD patients. While one might initially presume that the SN-FT group simply represents the SN-F phenotype at a more advanced stage of disease severity, this hypothesis is contradicted by the clinical data, where SN-F patients showed worse executive abilities and more neurological exam abnormalities, particularly in gait and coordination, compared to SN-FT patients. The genetic and pathological probabilities were also distinct between SN-F and SN-FT groups. Given that both groups have similar degrees of structural damage to frontal SN structures, functional disruptions that require further investigation may likely underlie these divergent patterns.
The SAN subgroup, although consisting of fewer patients, showed very distinct behavioral characteristics. All SAN patients showed behavioral disinhibition, highlighting the centrality of right SAN/temporal lobe contributions to socioemotional sensitivity and maintenance of social decorum. The high prevalence of obsessive behaviors may reflect disruption in patients’ ability to value objects of interest, which is consistent with dysfunctional rostral caudate and medial orbotofrontal nodes of SAN.35,36 Consistent with the central role of right temporal structures in person perception, the SAN group also showed striking impairments in paralinguistic sarcasm detection (i.e., voice prosody, facial-expression).37 This suggests that SAN disruption may play a specific role in bvFTD patients’ difficulty in translating auditory and visual signals into meaningful social interactions. Preserved interpersonal warmth and empathic concern of SAN patients, indicates the critical role of their relatively intact frontal brain structures in these behaviors.38,39 Despite the focality of atrophy in the SAN group, they were neuropathologically diverse, comprising patients with TDP-B, Pick’s, and FTDP-17 tau pathology, and included patients with C9, GRN, and MAPT mutations.
Although the UCSF-MAC has a high diagnostic accuracy rate, with clinical diagnosis of bvFTD predicting an FTLD pathology in >95% of cases,11 future studies must corroborate these results in a clinicopathologically diagnosed cohort. Also, prospectively followed longitudinal studies will provide comprehensive phenotypic characteristics of each subgroup. This study suggests that classification of bvFTD patients by their patterns of volume loss in key networks may yield greater clarity in predicting patients’ specific cognitive, behavioral, and motor symptoms, as well as rate of progression. The identification of a subset of bvFTD patients with primarily subcortical atrophy, who are more likely to have TDP neuropathology and C9 mutations, suggests this approach also has modest utility for prediction of molecular pathology.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the patients, caregivers, and healthy controls for their participation in this research. The authors would like to thank Babu Adhimoolam MBBS for consultation on the VBM analysis and Jennifer Yokoyama PhD for consultation on the statistical analysis of genetic data. This study was supported by the NIH grants P01AG019724 (BLM), P50AG02350 (BLM), R01AG029577 (KPR), K23AG021606 (KPR), K23 AG038357 (KAV), K23AG040127 (VES), K23 AG042492-01 (BMB), K23 AG039414 (SEL), K23 AG045289 (DCP), a grant from the Alzheimer’s Association (PCTRB-13-288476) made possible by Part the CloudTM (KAV), grants from the Larry L. Hillblom Foundation, Inc. 2013-A-029-SUP, 2007/2I, 2015-A-034-FEL (KGR), and from the J.D. French Alzheimer’s Foundation (KAV).
KGR, KPR, PSP, DCP, IVL, WWS, GC, AMK, TS, SEL, GDR, HJR, MLG, ALB, ZAM, WC, MD, JHK, KLP, VES, BMB, LTG, MN, DDZ, LAN, RK, NB, TQW, AD, NR, AC, DHG, KAV, and BLM report no disclosures relevant to this work.
KGR and KPR would like to state here that we both have full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
AUTHOR CONTRIBUTIONS: KGR and KPR designed the study, performed the analyses including the statistical analyses. IVL gave expert guidance on statistical analyses. PSP and DCP did extensive patient chart reviews in addition to clinical evaluations of the patients. KAV, GDR, HJR, MLG, ALB, SEL, WWS, WC, ZAM, JHK, MD and BLM contributed to the clinical and diagnostic evaluation of patients. WWS and LTG contributed the neuropathological diagnoses. KPR, TS, JHK, VES, BMB, and KLP contributed the behavioral assessments and cognitive evaluations of the patients. All authors other than IVL contributed to consensus diagnostic panels at the time of patient assessments. GC, AMK, and DHG provided the genetic information for the patients. MN, DDZ, LAN, RK, NB, TQW, AD, NR and AC contributed to cognitive testing, dementia severity evaluations and caregiver interviews. KGR wrote the manuscript. KPR, KAV, PSP, WWS, DCP, SEL, and BLM, contributed in extensive editorial revisions of the manuscript.
AUTHOR DISCLOSURES: KGR, KPR, PSP, DCP, IVL, WWS, GC, AMK, TS, SEL, GDR, HJR, MLG, ALB, ZAM, WC, MD, JHK, KLP, VES, BMB, LTG, MN, DDZ, LAN, RK, NB, TQW, AD, NR, AC, DHG, KAV, and BLM report no disclosures relevant to this work.
REFERENCES
- 1.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 2.Snowden JS, Neary D, Mann DM. Frontotemporal dementia. Br J Psychiatry. 2002;180:140–143. doi: 10.1192/bjp.180.2.140. [DOI] [PubMed] [Google Scholar]
- 3.Hodges JR. Frontotemporal dementia (Pick's disease): clinical features and assessment. Neurology. 2001;56(11 Suppl 4):S6–10. doi: 10.1212/wnl.56.suppl_4.s6. [DOI] [PubMed] [Google Scholar]
- 4.Pievani M, de Haan W, Wu T, Seeley WW, Frisoni GB. Functional network disruption in the degenerative dementias. Lancet Neurol. 2011;10(9):829–843. doi: 10.1016/S1474-4422(11)70158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62(1):42–52. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou J, Gennatas ED, Kramer JH, Miller BL, Seeley WW. Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron. 2012;73(6):1216–1227. doi: 10.1016/j.neuron.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeo BT, Krienen FM, Sepulcre J, et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J Neurophysiol. 2011;106(3):1125–1165. doi: 10.1152/jn.00338.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seeley WW, Zhou J, Kim EJ. Frontotemporal dementia: what can the behavioral variant teach us about human brain organization? Neuroscientist. 2012;18(4):373–385. doi: 10.1177/1073858411410354. [DOI] [PubMed] [Google Scholar]
- 9.Zhou J, Seeley WW. Network dysfunction in Alzheimer's disease and frontotemporal dementia: implications for psychiatry. Biol Psychiatry. 2014;75(7):565–573. doi: 10.1016/j.biopsych.2014.01.020. [DOI] [PubMed] [Google Scholar]
- 10.Guo CC, Gorno-Tempini ML, Gesierich B, et al. Anterior temporal lobe degeneration produces widespread network-driven dysfunction. Brain. 2013;136:2979–2991. doi: 10.1093/brain/awt222. Pt 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. doi: 10.1093/brain/awr179. Pt 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDonald S, Bornhofen C, Shum D, Long E, Saunders C, Neulinger K. Reliability and validity of The Awareness of Social Inference Test (TASIT): a clinical test of social perception. Disabil Rehabil. 2006;28(24):1529–1542. doi: 10.1080/09638280600646185. [DOI] [PubMed] [Google Scholar]
- 13.Shany-Ur T, Poorzand P, Grossman SN, et al. Comprehension of insincere communication in neurodegenerative disease: lies, sarcasm, and theory of mind. Cortex. 2012;48(10):1329–1341. doi: 10.1016/j.cortex.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis MH. Measuring individual differences in empathy: Evidence for a multidimensional approach. Journal of Personality & Social Psychology. 1983;44(1):113–126. [Google Scholar]
- 15.Rankin KP, Gorno-Tempini ML, Allison SC, et al. Structural anatomy of empathy in neurodegenerative disease. Brain. 2006;129:2945–2956. doi: 10.1093/brain/awl254. Pt 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiggins JS, Trapnell P, Phillips N. Psychometric and geometric characteristics of the Revised Interpersonal Adjective Scales (IAS--R) Multivariate Behavioral Research. 1988;23(4) doi: 10.1207/s15327906mbr2304_8. [DOI] [PubMed] [Google Scholar]
- 17.Interpersonal Adjective Scales (IAS) Scoring Program [Computer Software] [computer program]. Version 1. Psychological Assessment Resources, Inc.; Odessa, FL: 1995. [Google Scholar]
- 18.Rohrer JD, Warren JD. Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol. 2011;24(6):542–549. doi: 10.1097/WCO.0b013e32834cd442. [DOI] [PubMed] [Google Scholar]
- 19.Borroni B, Agosti C, Premi E, et al. The FTLD-modified Clinical Dementia Rating scale is a reliable tool for defining disease severity in frontotemporal lobar degeneration: evidence from a brain SPECT study. Eur J Neurol. 2010;17(5):703–707. doi: 10.1111/j.1468-1331.2009.02911.x. [DOI] [PubMed] [Google Scholar]
- 20.O'Bryant SE, Waring SC, Cullum CM, et al. Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer's research consortium study. Arch Neurol. 2008;65(8):1091–1095. doi: 10.1001/archneur.65.8.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buckner RL, Andrews-Hanna JR, Schacter DL. The brain's default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- 22.Greicius MD, Krasnow B, Reiss AL, Menon V. Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proc Natl Acad Sci U S A. 2003;100(1):253–258. doi: 10.1073/pnas.0135058100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dosenbach NU, Fair DA, Miezin FM, et al. Distinct brain networks for adaptive and stable task control in humans. Proc Natl Acad Sci U S A. 2007;104(26):11073–11078. doi: 10.1073/pnas.0704320104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dosenbach NU, Fair DA, Cohen AL, Schlaggar BL, Petersen SE. A dual-networks architecture of top-down control. Trends Cogn Sci. 2008;12(3):99–105. doi: 10.1016/j.tics.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitwell JL, Przybelski SA, Weigand SD, et al. Distinct anatomical subtypes of the behavioural variant of frontotemporal dementia: a cluster analysis study. Brain. 2009;132:2932–2946. doi: 10.1093/brain/awp232. Pt 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitwell JL, Xu J, Mandrekar J, et al. Frontal asymmetry in behavioral variant frontotemporal dementia: clinicoimaging and pathogenetic correlates. Neurobiol Aging. 2013;34(2):636–639. doi: 10.1016/j.neurobiolaging.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whitwell JL, Avula R, Senjem ML, et al. Gray and white matter water diffusion in the syndromic variants of frontotemporal dementia. Neurology. 2010;74(16):1279–1287. doi: 10.1212/WNL.0b013e3181d9edde. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seeley WW, Allman JM, Carlin DA, et al. Divergent social functioning in behavioral variant frontotemporal dementia and Alzheimer disease: reciprocal networks and neuronal evolution. Alzheimer Dis Assoc Disord. 2007;21(4):S50–57. doi: 10.1097/WAD.0b013e31815c0f14. [DOI] [PubMed] [Google Scholar]
- 29.Josephs KA, Whitwell JL, Knopman DS, et al. Two distinct subtypes of right temporal variant frontotemporal dementia. Neurology. 2009;73(18):1443–1450. doi: 10.1212/WNL.0b013e3181bf9945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohrer JD. Behavioural variant frontotemporal dementia--defining genetic and pathological subtypes. J Mol Neurosci. 2011;45(3):583–588. doi: 10.1007/s12031-011-9542-2. [DOI] [PubMed] [Google Scholar]
- 31.Kipps CM, Hodges JR, Hornberger M. Nonprogressive behavioural frontotemporal dementia: recent developments and clinical implications of the 'bvFTD phenocopy syndrome'. Curr Opin Neurol. 2010;23(6):628–632. doi: 10.1097/WCO.0b013e3283404309. [DOI] [PubMed] [Google Scholar]
- 32.Khan BK, Yokoyama JS, Takada LT, et al. Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry. 2012;83(4):358–364. doi: 10.1136/jnnp-2011-301883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piguet O, Hornberger M, Mioshi E, Hodges JR. Behavioural-variant frontotemporal dementia: diagnosis, clinical staging, and management. Lancet Neurol. 2011;10(2):162–172. doi: 10.1016/S1474-4422(10)70299-4. [DOI] [PubMed] [Google Scholar]
- 34.Lee SE, Khazenzon AM, Trujillo AJ, et al. Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain. 2014;137:3047–3060. doi: 10.1093/brain/awu248. Pt 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Doherty J, Kringelbach ML, Rolls ET, Hornak J, Andrews C. Abstract reward and punishment representations in the human orbitofrontal cortex. Nat Neurosci. 2001;4(1):95–102. doi: 10.1038/82959. [DOI] [PubMed] [Google Scholar]
- 36.Roesch MR, Olson CR. Neuronal activity related to reward value and motivation in primate frontal cortex. Science. 2004;304(5668):307–310. doi: 10.1126/science.1093223. [DOI] [PubMed] [Google Scholar]
- 37.Rankin KP, Salazar A, Gorno-Tempini ML, et al. Detecting sarcasm from paralinguistic cues: anatomic and cognitive correlates in neurodegenerative disease. Neuroimage. 2009;47(4):2005–2015. doi: 10.1016/j.neuroimage.2009.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sollberger M, Neuhaus J, Ketelle R, et al. Interpersonal traits change as a function of disease type and severity in degenerative brain diseases. J Neurol Neurosurg Psychiatry. 2011;82(7):732–739. doi: 10.1136/jnnp.2010.205047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sollberger M, Rankin KP, Miller BL. Social cognition. Continuum (Minneap Minn) 2010;16(4):69–85. doi: 10.1212/01.CON.0000368261.15544.7c. Behavioral Neurology. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.