

Abstract
1-Amino-2,2-difluorocyclopropane-1-carboxylic acid (DFACC) is of interest in the study of 1-aminocyclopropane-1-carboxylic acid (ACC) deaminase due to the increased reactivity of its cyclopropyl functionality. It is shown that DFACC is unstable under near-physiological conditions where it primarily decomposes via specific-base catalysis to 3-fluoro-2-oxobut-3-enoic acid with a rate constant of 0.18 ± 0.01 min−1. Upon incubation with ACC deaminase, DFACC is found to be a slow-dissociating inhibitor of ACC deaminase with submicromolar affinity.
Authors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.
TOC image
1-Aminocyclopropane-1-carboxylic acid (ACC) deaminase is a pyridoxal-5′-phosphate (PLP)-dependent enzyme that catalyzes the decomposition of ACC (1) to α-ketobutyrate (2) and ammonia (see Scheme 1).1 It has been identified in a wide range of bacteria and fungi.1 The substrate of ACC deaminase is also the immediate biosynthetic precursor to ethylene, which is produced in plants via oxidation of ACC.2–4 While ethylene is an essential plant hormone that regulates fruit ripening, seed germination and leaf senescence,4,5 it can also be detrimental causing senescence, chlorosis and abscission when produced in excessive quantities under conditions of stress.4–6 The potential use of ACC deaminase to mitigate the deleterious effects of ethylene production has thus made it a focus of enzymological research to better understand its atypical mechanism of action.6b,7
Scheme 1.
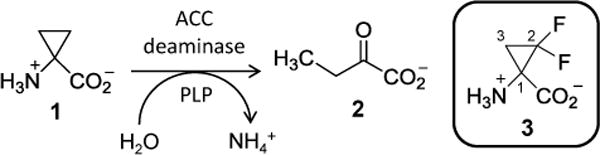
Reaction catalyzed by ACC deaminase (1→2) and the structure of DFACC (3).
The opening of the cyclopropane ring of ACC catalyzed by ACC deaminase is essentially a redox-neutral deamination reaction. The chemistry of ACC deaminase thus stands in contrast to that of typical PLP-dependent transaminases which instead couple the oxidative deamination of amino acids to the reductive amination of β-keto acids. The mechanism by which ACC is both deaminated and linearized by ACC deaminase along with the role played by PLP in this process remain an unresolved and intriguing question for which a number of possibilities have been proposed.7
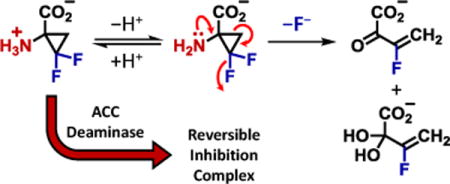
It is well documented that species containing a gem-difluorocyclopropane are susceptible to nucleophilic attack at both the CH2 and CF2 centers of the cyclopropane ring.8–11 Likewise, alkyl-substituted gem-difluorocyclopropanes have also been proposed to undergo 1,4-elimination reactions whereby fluoride is eliminated concomitant with ring-opening.12–14 Hence, the gem-difluoro analog of ACC, 1-amino-2,2-difluorocyclopropane-1-carboxylic acid (DFACC, 3), has been recognized for its potential to provide additional insight into the mechanism of ACC deaminase on account of the greatly enhanced reactivity of the gem-difluorocyclopropane moiety.14–16
Little is known, however, about the impact of introducing a basic amino functionality at the α-carbon of gem-difluorocyclopropyl amino acids. Such a modification may promote ring-opening in aqueous systems at near neutral pH. This would have consequences for the use of DFACC in both mechanistic studies of ACC deaminase and as a potential ligand15 for biological receptors. Herein we report the synthesis of racemic DFACC and show that it undergoes a pH-dependent ring-opening reaction that may involve a carbanionic intermediate. Interestingly, despite its instability, DFACC is still effective at reversibly inhibiting ACC deaminase with a Ki in the submicromolar range.
The chemical synthesis of rac-DFACC began with silylation of 2-(4′-methoxyphenyl)-2-propen-1-ol (4) and subsequent cyclization using difluorocarbene to generate the difluoro-substituted cyclopropane 6 (see Scheme 2). The silyl protecting group of 6 was removed, and the resulting alcohol (7) was oxidized to afford the carboxylic acid 8. A Curtius rearrangement of 8 was then performed to produce the Boc-protected amine 9. Next, the 4′-methoxyphenyl functionality of 9 was oxidatively cleaved using ruthenium tetroxide generated in situ to yield 10. After deprotection of the Boc group of 10, the crude product was purified using DOWEX 50WX8 cation exchange resin to afford DFACC (3) as the HCl salt.
Scheme 2.

Chemical synthesis of rac-DFACC (3).
Despite its stability as a HCl salt, when DFACC is incubated in D2O at neutral pD, it undergoes decomposition with a half-life of approximately 4 min (see below). This results in the production of two new species along with release of free fluoride ion. 1H NMR of the major (ca. 75%) product demonstrated an ABX spin system (X = F) with the AB protons appearing in the 5.65–5.85 ppm region and a JAB coupling constant of 4.8 Hz (see Figure S3.3). The 19F NMR likewise exhibited a doublet of doublets splitting pattern with JAF = 15.0 Hz and JBF = 46.0 Hz (see Figure S3.3). The magnitudes of JAF and JBF are most consistent with three-bond couplings between the fluorine and both the A and B protons rather than two-bond couplings, which can exceed 80 Hz.17–19 The 13C NMR of the major product also revealed the presence of two carbonyl carbons in the 165–195 ppm region and thus implicated an α-keto acid (see Supporting Information). Moreover, electrospray ionization mass spectrometry (ESI-MS) of the product mixture exhibited a signal at 117.00 m/z (negative ion mode) consistent with a composition of C4H2FO3− (calculated mass: 116.9993 Da). The major decomposition product was thus assigned as 3-fluoro-2-oxobut-3-enoic acid (11, see Scheme 2).
The NMR spectra of the minor decomposition product (ca. 25%) showed features very similar to those of 11. Again an ABX system was observed (X = F); however, the two AB quartets are found shifted upfield to the 4.70–4.95 ppm region. Likewise, the C2 13C resonance, which in the case of 11 is observed as a doublet at 189.4 ppm and split by the fluorine at C3, is replaced by a doublet 13C resonance at 91.4 ppm (see Supporting Information). This implies that the C2 carbonyl seen in 11 is absent and led to the assignment of the second product as the corresponding hydrate (12). The assignments for the two decomposition products were also consistent with additional measurements made using various two-dimensional NMR techniques (1H COSY, 1H-13C HSQC and 1H-13C HMBC), which are provided in the Supporting Information.
In addition to 11 and its hydrate (12), a new compound which makes up roughly 10% of the total product distribution (see Supporting Information) was also found when the reaction was run in H2O. This new product was identified as 3,3-difluoro-2-oxobutyric acid (15) by spectroscopic analysis (see Figure S3.5). Notably, 15 was not produced at a detectable level in the reactions run in D2O, and there was no evidence for deuterium incorporation at C4 of either 11 or its hydrate in D2O.
Based on these observations, four mechanisms describing the decomposition of DFACC in aqueous solution were considered (see Scheme 3). The first of these (A), which can account for the formation of 15 in buffered H2O, involves ring-opening of the conjugate base of DFACC (13) whereby a solvent-derived proton is transferred to the C3-carbon (13→14). The resulting intermediate would then undergo hydrolysis to produce the stable species 15. Mechanism B in Scheme 3 involves formation of a halohydrin intermediate (16). This mechanism begins with nucleophilic attack by hydroxide at C3 and is reminiscent of the reaction of 1-acetyl-2,2-difluoro-3-phenylcyclopropane with phenylthiolate8,9 and the addition of bromide to (2,2-difluorocyclopropyl)-phenylketone in N-pentylpyridinium bromide.10 While the α-amino group of DFACC is not directly involved in the ring-opening step of mechanism B (i.e., 3→16), it is important for the conversion of 17 to 18/11.
Scheme 3.
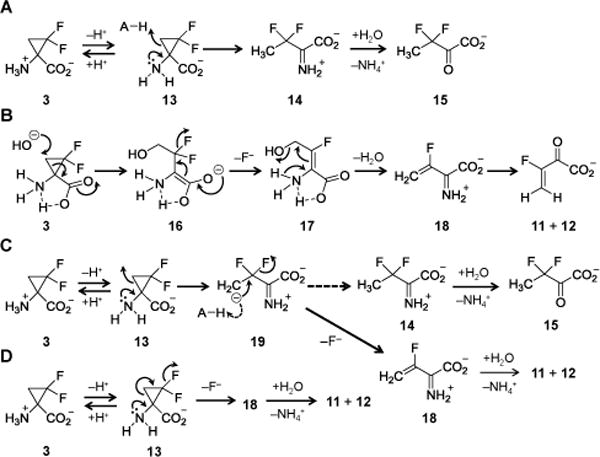
Hypotheses for the decomposition of DFACC (3).
Alternatively, the amino functionality could play a direct role in the DFACC ring-opening as shown in mechanisms C and D of Scheme 3. Both of these mechanisms, like A, also involve an initial rapid acid-base equilibrium; however, in the case of C, the proposed ring-opening results in a transient carbanion intermediate (19) that can undergo either protonation (19→ 14) or elimination (19→18). In contrast, mechanism D involves a one-step (i.e., (intra-DN)DN) elimination to produce 18 directly from 13. Neither mechanism B nor D, however, can explain the co-production of 15 in H2O-solvent systems, and would thus require the participation of a second, competing process such as 13→14 in mechanism A.
To examine these hypotheses, the role of the α-amino group in the decomposition of DFACC was investigated more closely. N-acetyl rac-DFACC was prepared from 9 (see Supporting Information) and found to be stable at all pH values considered. This result demonstrates the importance of a basic α-amino group for the observed decomposition of DFACC. The pH-dependence of the DFACC decomposition rate was then determined by following the disappearance of DFACC at different pH using 19F NMR. Triplicate reactions were run in 1.0 M succinate or phosphate (H2O) buffers at pH values ranging from 4.5 to 8.0 and a constant ionic strength of 3 M. DFACC was observed to decay via a first-order process with a pH-dependent rate constant (see Section S4 in the Supporting Information). This plot demonstrates that the decomposition of DFACC is slowed at low pH and reaches a plateau under alkaline conditions indicating base catalysis.
In order to obtain a measure of the apparent pKa and maximum first-order rate constant, k, for decomposition of DFACC, the data in Figure S4.3 were fit using Eqn. (1),
| (1) |
This provided values of 5.88 ± 0.06 for the pKa and 0.18 ± 0.01 min−1 for k. The observed pKa is too low to correspond to the second ionization constant of phosphoric acid, which is 6.8 at an ionic strength of 3 M.20 However, this pKa is consistent with those for the α-amino group of β,β-difluoroalanine and β,β-difluorophenylalanine, which have pKa values roughly 2 units lower than their nonfluorinated counterparts.21 Therefore, given the pKa of 8.15 for ACC,22 the corresponding pKa for DFACC is expected be ca. 6.0. This result suggests that decomposition of DFACC involves specific-base catalysis where the conjugate base of DFACC (i.e., 13) is the reactive species that formally undergoes ring-opening. This conclusion argues against formation of a halohydrin intermediate as shown in mechanism B. The above result along with the observation that decomposition of 3/13 is unaffected by the addition of a weak acid such as phenol (see Supporting Information) also casts doubt on the signifcance of pathway A, which involves general-acid catalysis.
Both mechanisms C and D in Scheme 3, however, are consistent with these results. The (intra-DN)DN ring-opening of mechanism D is analogous to an E1cB version of the 1,4-elimination reactions that have been proposed for the dehydrohalogenation of substituted gem-difluorocyclopropanes,12–14 and is consistent with the propensity of cyclopropanes to behave more as conjugated π-systems rather than sp3-hybridized carbocycles.23 However, mechanism D would require a competing process such as A in order to explain 15 and the product distributions in H2O versus D2O. In contrast, mechanism C can account for the formation of all observed products. More importantly, it possesses a product-determining step (i.e., 19→18/14) that would be susceptible to a normal solvent deuterium kinetic isotope effect and thus explain the formation of 15 in H2O as opposed to D2O buffers.
While mechanism C involves the intermediacy of a β-fluorocarbanion (i.e., RCF2CH2−), this species could be stabilized through negative hyperconjugation.24 Though a competing α-fluorinated carbanion (i.e., RCH2CF2−) could likewise be stabilized inductively, its stability may be offset by orbital repulsion between the lone pairs on the α-carbanion and the adjacent fluorines,25–27 resulting in preferential formation of the β-fluorocarbanion 19. Taken together, the available evidence seems to be most consistent with mechanism C.
In order to evaluate the potential of DFACC as a mechanistic probe of ACC deaminase, we first checked to see whether it would interact with the enzyme. The activity of ACC deaminase in the presence of DFACC (3) was thus assayed by following the deamination of ACC to α-ketobutyrate (2) at pH 7.0 using 1H NMR spectroscopy as shown in Figure 1. Each time trace exhibits a lag period during which the concentration of 2 increases to detectable levels. In the absence of rac-DFACC, this period is less than 1 h. In the presence of 5 and 50 μM DFACC, however, this period is extended to at least 2 and 3 h, respectively. Following the lag, each reaction reaches a roughly constant rate (ca. 16 μM/min with no DFACC) for the remainder of the observation period. This is consistent with a less than 10% drop in enzyme saturation assuming a Michaelis constant of 4 mM,7g and indicated an enzyme specific activity of approximately 1 μmol·min−1mg−1. Furthermore, when fresh ACC was added to each reaction after 48 h, by which time all of the original ACC had been consumed, equivalent rates of turnover were observed in each of the three reactions. These observations revealed that DFACC is a reversible instead of an irreversible inhibitor for ACC deaminase. Furthermore, no evidence of inhibition was observed when ACC deaminase was incubated in the presence of DFACC following its decomposition (see Figure S7.3 in the Supporting Information). This indicates that the decomposition product 11 does not react with the enzyme despite being a potential Michael acceptor and that DFACC itself is responsible for the inhibition.
Figure 1.
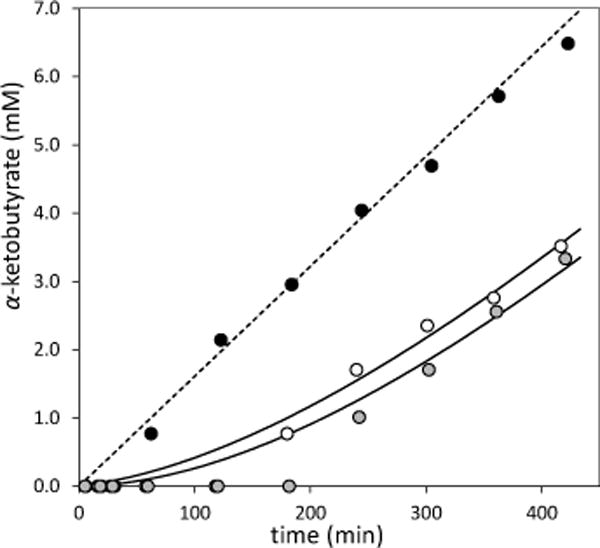
Inhibition of ACC deaminase with rac-DFACC (3). Reactions were run at room temperature with 0.5 μM enzyme in 500 μL of 100 mM NaPi buffer (pH 7.0) with 20 mM ACC and 0 (black), 5 (white) or 50 μM (grey) rac-DFACC. Reactions were followed by 1H NMR (see Supporting Information). The uninhibited time course was fit to a straight line to obtain the rate v0 (broken line), while the inhibition time courses were fit simultaneously using Eqns. (2) and (3). Fits exclude points where product is undetectable except at t = 0 min.
The rapid decomposition of DFACC in comparison to the lag times suggested a simplified kinetic model from which estimates of the DFACC dissociation constant Ki and dissociation rate constant kd could be made using the progress curves in Figure 1. This model assumes that a binding equilibrium between DFACC and enzyme is rapidly established such that the initial fraction f0 of enzyme being inhibited is given by
| (2) |
where x0 and s0 are the initial concentrations of DFACC and ACC, respectively, and KM is the Michaelis constant (4 mM). If dissociation of the enzyme-inhibitor complex is slow versus DFACC decomposition, then the observed time course of product formation will be given by
| (3) |
where v0 is approximately 16 μM/min (see Supporting Information for derivation). When Eqn. (3/2) was fit simultaneously to both inhibition time courses in Figure 1, values of Ki = 120 ± 40 nM and kd = 0.20 ± 0.01 h−1 were obtained. It should be emphasized that the true value of Ki may be significantly smaller if there is no rapid equilibration between the enzyme and inhibitor. The kinetic parameter estimates are nevertheless consistent with the conclusion that rac-DFACC is a reversible, slow-dissociating inhibitor of ACC deaminase that binds with submicromolar affinity.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (GM040541) and the Welch Foundation (F-1511).
Footnotes
Supporting Information
Details regarding the synthetic procedures, enzyme assays, kinetic analyses, and spectroscopic characterization of all compounds discussed. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
References
- 1.(a) Honma M, Shimomura T. Agric Biol Chem. 1978;42:1825. [Google Scholar]; (b) Nascimento FX, Rossi MJ, Soares CRFS, McConkey BJ, Glick BR. PLoS ONE. 2014;9:e99168. doi: 10.1371/journal.pone.0099168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kende H. Annu Rev Plant Physiol Plant Mol Biol. 1993;44:283. [Google Scholar]
- 3.Adams DO, Yang SF. Proc Natl Acad Sci USA. 1979;76:170. [Google Scholar]
- 4.Wang KLC, Li H, Ecker JR. The Plant Cell. 2002;14:S131. doi: 10.1105/tpc.001768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hyodo H. In: The plant hormone ethylene. Mattoo AK, Shuttle JC, editors. CRC Press; Boca Raton: 1991. pp. 65–80. [Google Scholar]
- 6.(a) Glick BR, Cheng Z, Czarny J, Duan J. Eur J Plant Pathol. 2007;119:329. [Google Scholar]; (b) Glick BR. Microbiol Res. 2014;169:30. doi: 10.1016/j.micres.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 7.(a) Walsh C, Pascal RA, Jr, Johnston M, Raines R, Dikshit D, Krantz A, Honma M. Biochemistry. 1981;20:7509. doi: 10.1021/bi00529a028. [DOI] [PubMed] [Google Scholar]; (b) Hill RK, Prakash SR, Wiesendanger R, Angst W, Martinoni B, Arigoni D, Liu HW, Walsh CT. J Am Chem Soc. 1984;106:795. [Google Scholar]; (c) Liu H-w, Auchus R, Walsh CT. J Am Chem Soc. 1984;106:5335. [Google Scholar]; (d) Li K, Du W, Que NLS, Liu H-w. J Am Chem Soc. 1996;118:8763. [Google Scholar]; (e) Yao M, Ose T, Sugimoto H, Horiuchi A, Nakagawa A, Wakatsuki S, Yokoi D, Murakami T, Honma M, Tanaka I. J Biol Chem. 2000;275:34557. doi: 10.1074/jbc.M004681200. [DOI] [PubMed] [Google Scholar]; (f) Ose T, Fujino A, Yao M, Watanabe N, Honma M, Tanaka I. J Biol Chem. 2003;278:41069. doi: 10.1074/jbc.M305865200. [DOI] [PubMed] [Google Scholar]; (g) Zhao Z, Chen H, Li K, Du W, He S, Liu H-w. Biochemistry. 2003;42:2089. doi: 10.1021/bi020567n. [DOI] [PubMed] [Google Scholar]; (h) Karthikeyan S, Zhao Z, Kao C-l, Zhou Q, Tao Z, Zhang H, Liu H-w. Angew Chem Int Ed. 2004;43:3425. doi: 10.1002/anie.200453353. [DOI] [PubMed] [Google Scholar]; (i) Karthikeyan S, Zhou Q, Zhao Z, Kao C-L, Tao Z, Robinson H, Liu H-w, Zhang H. Biochemistry. 2004;43:13328. doi: 10.1021/bi048878g. [DOI] [PubMed] [Google Scholar]; (j) Thibodeaux CJ, Liu H-w. Biochemistry. 2011;50:1950. doi: 10.1021/bi101927s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobayashi Y, Taguchi T, Morikawa T, Takase T, Takanashi H. Tetrahedron Lett. 1980;21:1047. [Google Scholar]
- 9.Kobayashi Y, Taguchi T, Morikawa T, Takase T, Takanashi H. J Org Chem. 1982;47:3232. [Google Scholar]
- 10.Xu W, Dolbier WR, Jr, Salazar J. J Org Chem. 2008;73:3535. doi: 10.1021/jo800337t. [DOI] [PubMed] [Google Scholar]
- 11.Dolbier WR, Jr, Cornett E, Martinez H, Xu W. J Org Chem. 2011;76:3450. doi: 10.1021/jo200423y. [DOI] [PubMed] [Google Scholar]
- 12.Osborn CL, Shields TC, Shoulders BA, Krause JF, Cortez HV, Gardner PD. J Am Chem Soc. 1965;87:3158. [Google Scholar]
- 13.Bessard Y, Kuhlmann L, Schlosser M. Tetrahedron. 1990;46:5230. [Google Scholar]
- 14.Dolbier WR, Jr, Battiste MA. Chem Rev. 2003;103:1071. doi: 10.1021/cr010023b. [DOI] [PubMed] [Google Scholar]
- 15.Sloan MJ, Kirk KL. Tetrahedron Lett. 1997;38:1677. [Google Scholar]
- 16.Kirihara M, Kawasaki M, Takuwa T, Kakuda H, Wakikawa T, Takeuchi Y, Kirk KL. Tetrahedron: Asymmetry. 2003;14:1753. [Google Scholar]
- 17.Dolbier WR., Jr . Guide to fluorine NMR for organic chemists. John Wiley & Sons; New Jersey: 2009. pp. 61–74. [Google Scholar]
- 18.Baklouti A, Chaabouni MM. J Fluorine Chem. 1981;19:181. [Google Scholar]
- 19.Nguyen T, Wakselman C. J Org Chem. 1989;54:5640. [Google Scholar]
- 20.Ellis KJ, Morrison JF. Method Enzymol. 1982;87:405. doi: 10.1016/s0076-6879(82)87025-0. [DOI] [PubMed] [Google Scholar]
- 21.Schlosser M, Brugger N, Schmidt W, Amrhein N. Tetrahedron. 2004;60:7731. [Google Scholar]
- 22.Shoukry MM, Hassan SS. Spectrochim Acta A Mol Biomol Spectrosc. 2014;118:146. doi: 10.1016/j.saa.2013.08.070. [DOI] [PubMed] [Google Scholar]
- 23.(a) Lowry TH, Richardson KS. Mechanism and Theory in Organic Chemistry. 3rd. HarperCollins Publishers; New York: 1989. pp. 31–33. [Google Scholar]; (b) Smith MB, March J. March’s Advanced Organic Chemistry. 5th. John Wiley & Sons; New York: 2001. pp. 181–182. [Google Scholar]
- 24.Dixon DA, Fukunaga T, Smart BE. J Am Chem Soc. 1986;108:4027. doi: 10.1021/ja00269a063. [DOI] [PubMed] [Google Scholar]
- 25.Andreades S. J Am Chem Soc. 1964;86:2003. [Google Scholar]
- 26.Castejon HJ, Wiberg KB. J Org Chem. 1998;63:3937. [Google Scholar]
- 27.Chambers RD. Fluorine in Organic Chemistry. CRC Press; Boca Raton: 2004. pp. 108–115. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.