

Abstract
Background:
Pulmonary arterial hypertension (PAH) is a rare and fatal disease. While many treatment options have been shown to improve quality of life, exercise tolerance, and hemodynamics in PAH, survival remains poor, in part due to the advanced stage at which patients present to PAH specialists.
Methods:
This perspective paper explores challenges related to the timing of referral, diagnosis, and initiation of therapy.
Results:
Multiple factors account for the delay in referral, including fallacies in physician education, commercial influence resulting in inappropriate prescribing practices, and barriers in access to care.
Conclusion:
Improving physician education, encouraging the prescription of PAH medications to be done predominantly by PAH specialists, overcoming barriers to care, and promoting screening for PAH will help ensure early referral, diagnosis, and treatment.
Keywords: Delayed diagnosis, early diagnosis, health services accessibility, hypertension–pulmonary, inappropriate prescribing, pulmonary medicine, referral and consultation
INTRODUCTION
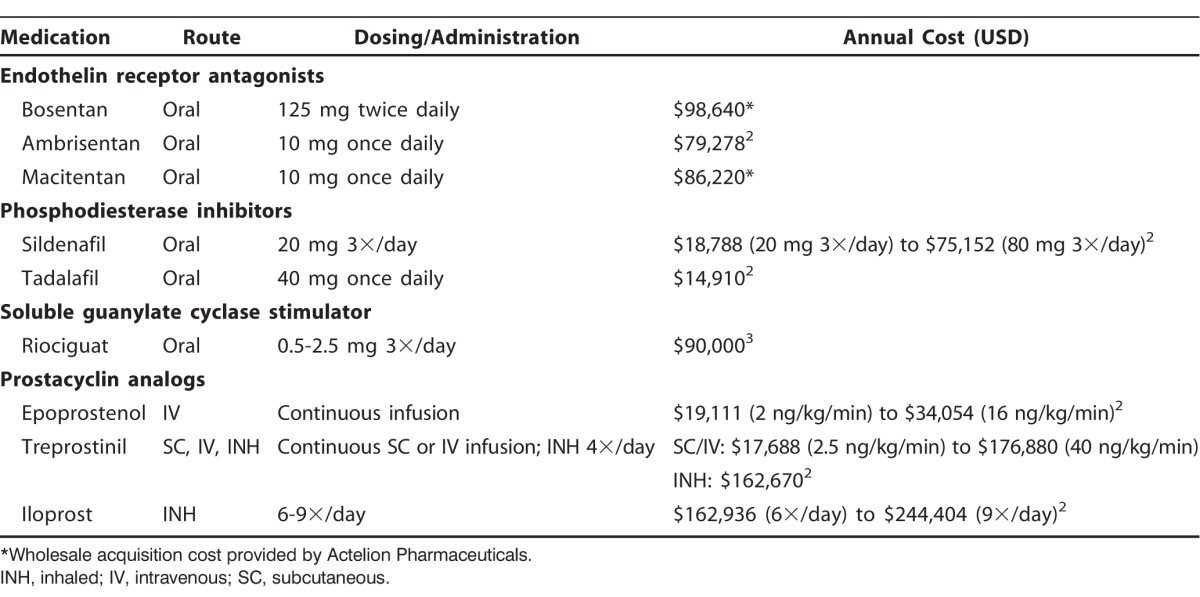
Pulmonary arterial hypertension (PAH) is a rare disease that leads to right-sided heart failure and death. National registries have demonstrated that the median survival in treatment-naïve PAH patients with advanced symptoms (World Health Organization [WHO] functional class [FC] III and IV) is 2.5 years and 6 months, respectively, compared with 6 years for patients with mild to moderate symptoms (WHO FC I-II).1 The 11 US Food and Drug Administration–approved treatment options for PAH are shown in Table 1.2,3 While only epoprostenol has been shown to improve survival, many of these therapies have been shown to improve exercise tolerance via increased 6-minute walk distance, hemodynamics, functional capacity, and time to clinical worsening.4,5
Table 1.
Pulmonary Arterial Hypertension Targeted Therapies
In 2013, the World Symposium on Pulmonary Hypertension was held in Nice, France, and the evidence-based guidelines for the diagnosis and management of PAH were revised.6,7 A practical approach for the treatment of PAH based on these guidelines is demonstrated in Figure 1 and Table 2,6,8 and Figure 27 contains a guideline-driven diagnostic algorithm for PAH.
Figure 1.

Treatment algorithm for pulmonary arterial hypertension.
*Refer to Table 2 for clinical determinants of risk.
Adapted from Galiè N, Corris PA, Frost A, et al.6 CCB, calcium channel blockers; INH, inhaled; IV, intravenous; RHC, right heart catheterization; SC, subcutaneous.
Table 2.
Determinants of Risk in Patients with Pulmonary Arterial Hypertension
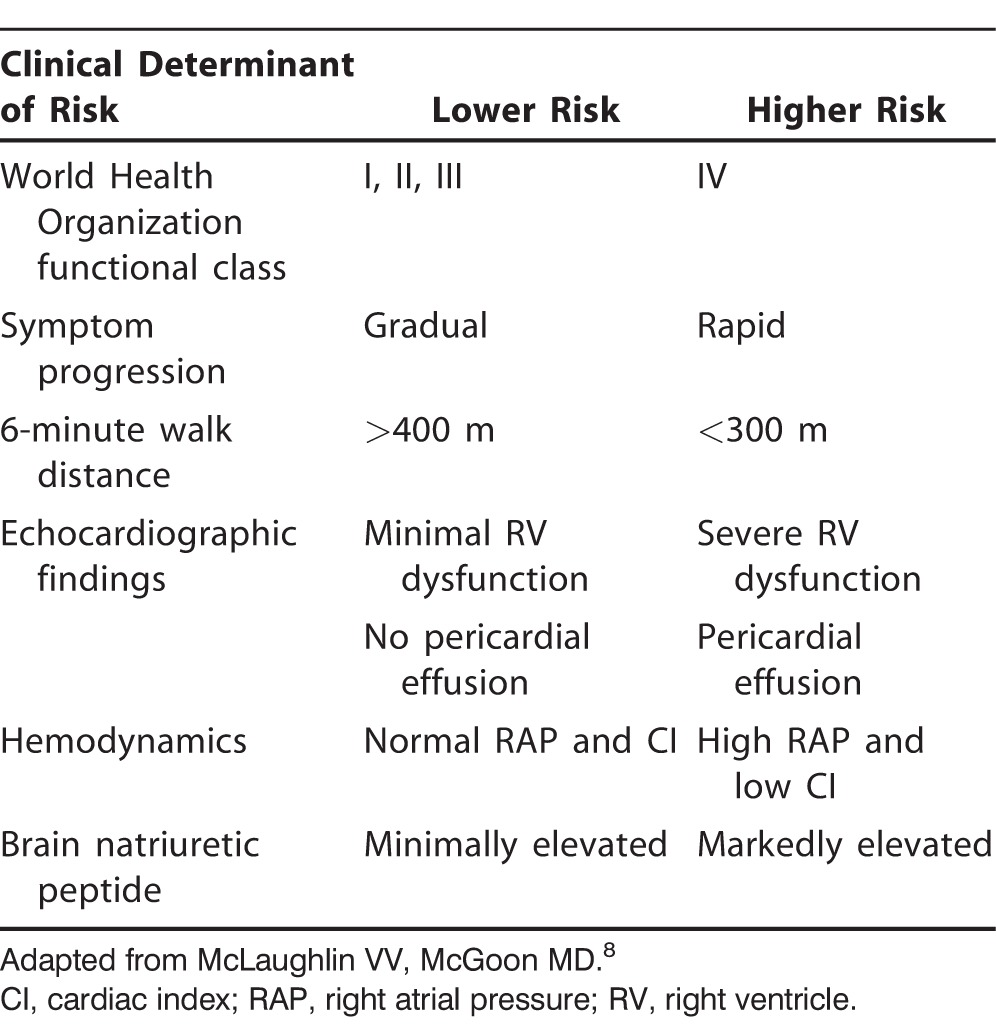
Figure 2.

Practical approach to diagnosis of pulmonary arterial hypertension.
Adapted from Hoeper MM, Bogaard HJ, Condliffe R, et al.7
ABG, arterial blood gas; CHD, congenital heart disease; CTD, connective tissue disease; CTEPH, chronic thromboembolic pulmonary hypertension; DLCO, diffusion capacity of the lung for carbon monoxide; ECG, electrocardiogram; HIV, human immunodeficiency virus; HR-CT, high-resolution computed tomography; mPAP, mean pulmonary arterial pressure; PAH, pulmonary arterial hypertension; PCWP, pulmonary capillary wedge pressure; PEA, pulmonary endarterectomy; PFTs, pulmonary function tests; PoPH, portopulmonary hypertension; PVOD, pulmonary veno-occlusive disease; PVR, pulmonary vascular resistance; RHC, right heart catheterization; RV, right ventricle; VQ, ventilation-perfusion.
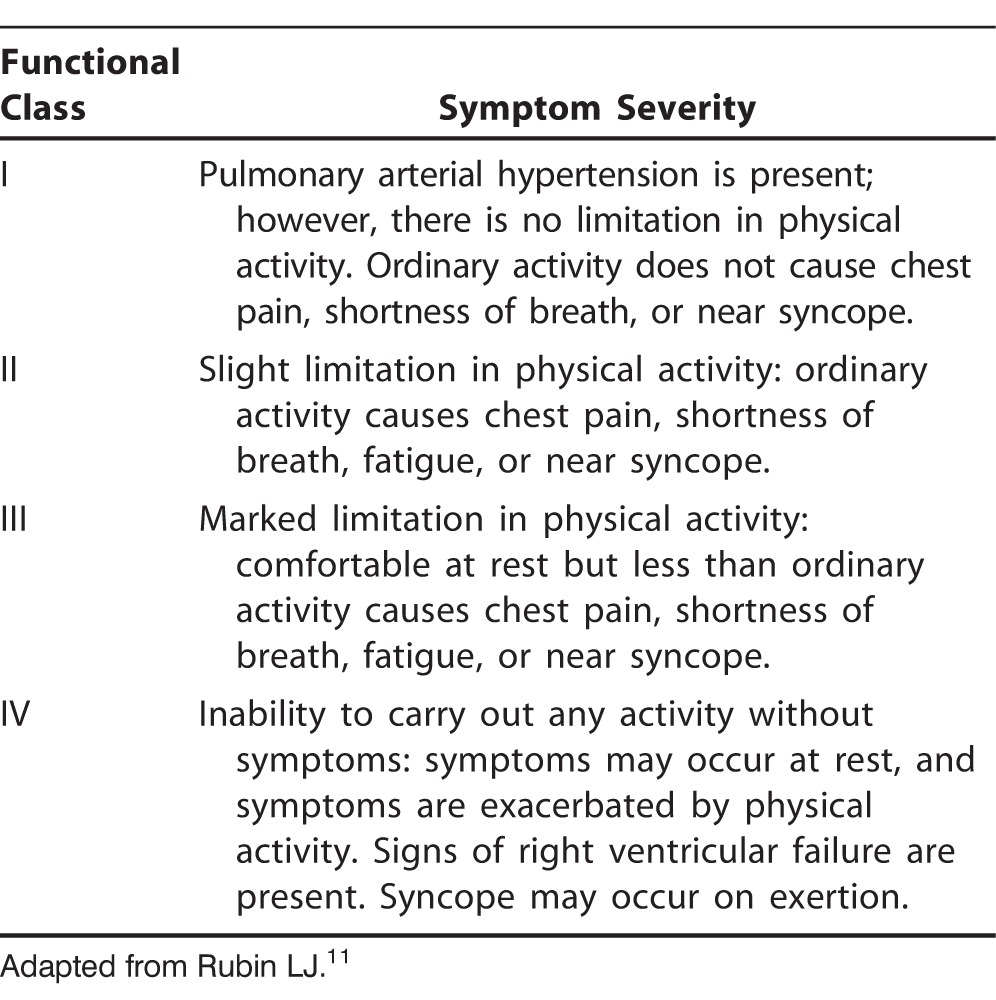
In a study investigating the accuracy of PAH diagnoses on referral to 3 large tertiary care PAH specialty centers, Deaño et al demonstrated that more than 60% of patients referred for evaluation and management of PAH presented with WHO FC III or IV symptoms at the time of diagnosis.9 Equally concerning was their finding that 33% of the 140 patients in the study were misdiagnosed, and 30% had been prescribed PAH medications prior to referral. Worse, 57% of these prescriptions were contrary to established PAH guidelines,6 in which the use of pulmonary vasodilator therapy is recommended only in patients with WHO groups 1 and 4 PAH. The WHO classification system by etiology of pulmonary hypertension (PH) is shown in Table 3,10 and the WHO FC system is presented in Table 4.11
Table 3.
World Health Organization Classification System for Pulmonary Arterial Hypertension
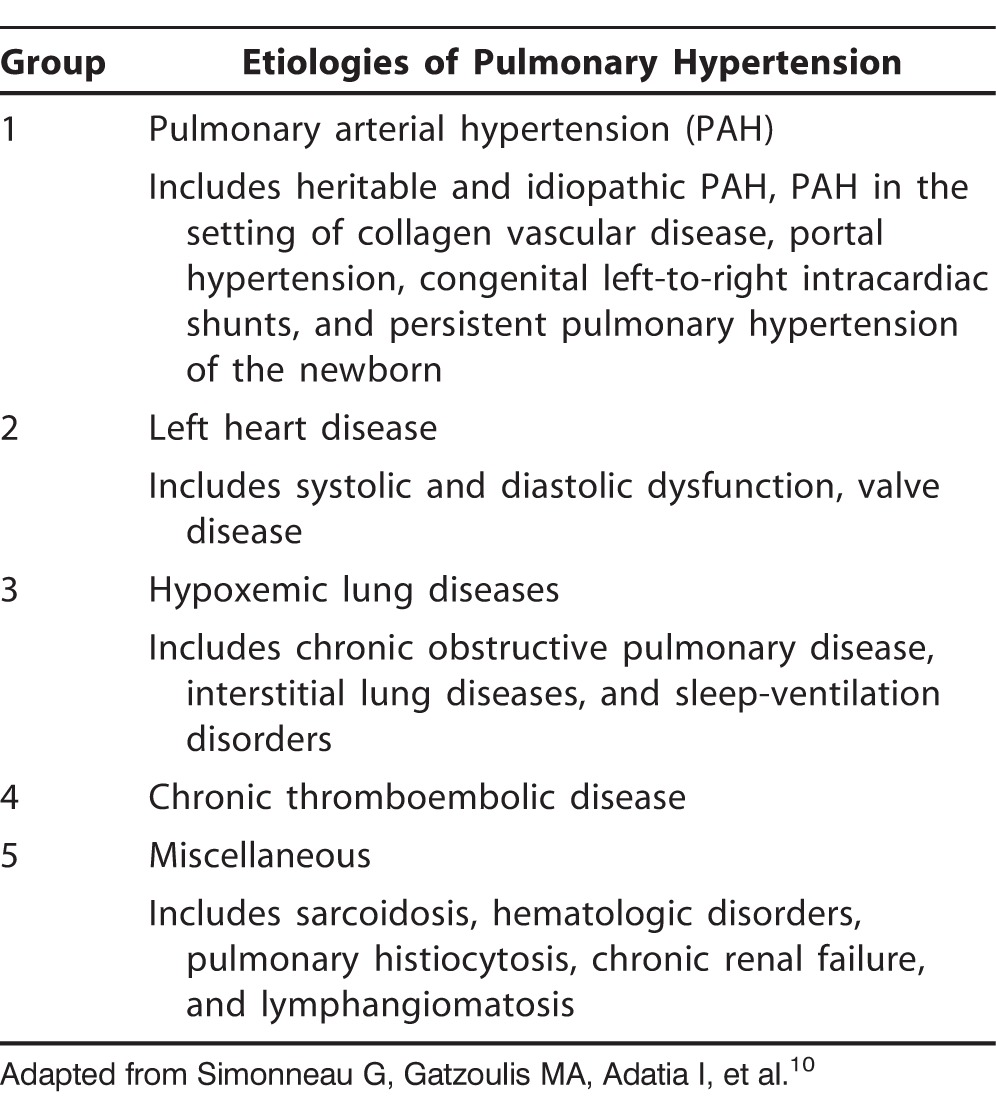
Table 4.
World Health Organization Functional Classes of Pulmonary Arterial Hypertension
The community of PAH physicians, those specifically trained in the diagnosis and management of PAH and dedicated to the care of patients with PAH, faces multiple challenges to improve the time to referral, diagnosis, and initiation of appropriate PAH therapy. This article explores these challenges and the controversies surrounding them.
PHYSICIAN EDUCATION
Despite the ongoing efforts of the PAH community, physician education on the topic of PAH remains problematic. Because of work-hour restrictions, internal medicine and pediatric residency programs have insufficient time to educate trainees about a relatively rare disease such as PAH.12
Specialty fellowship programs face similar time constraints. Although the Accreditation Council for Graduate Medical Education mandates PAH education, many pulmonary medicine fellows receive little or no exposure to echocardiography, right heart catheterization, or the advanced PAH therapies. In contrast, while cardiology fellows who train at programs with large PAH referral practices are more likely to be exposed to patients with PAH, the majority of training institutions do not have enough of a PAH presence to offer fellows significant exposure.
Education of general practitioners, general cardiologists, and pulmonologists is critical, as patients often present first to these physicians. Patients present with vague complaints of shortness of breath, chest pain, syncope, and fatigue, and these symptoms are often first mistaken for common conditions such as asthma.12 The failure to recognize the signs and symptoms of PAH may result in several visits and perhaps multiple subspecialty referrals before the correct diagnosis is made and treatment is initiated. This point is demonstrated by data from the REVEAL registry: 13.9% of patients were evaluated first by internists, 20.9% by pulmonologists, 24% by cardiologists, and 5.1% by rheumatologists.13 In this group of 2,493 patients, PAH was diagnosed >2 years after symptom onset in 526 (21.1%) patients.
PAH specialists and their governing societies have a key role to play in developing the curricula and core competencies for generalist and specialty fellowship programs. They should collaborate with generalist and general specialist societies to foster improved education via symposia at annual professional meetings and evidence-based guideline statements. Thus, cardiology and pulmonary medicine trainees, as well as internists and family practitioners, will develop at least a basic understanding of the signs and symptoms of PAH and maintain a high level of suspicion for the disease. In addition, physicians must learn that the combination of worsening dyspnea, chest pain on exertion, and syncope or near syncope likely represents advanced disease, and a sense of urgency should be instilled in them to refer such patients to a PAH specialty center.
COMMERCIAL INFLUENCE
With the increasing market availability of oral pulmonary vasodilator therapies, community physicians are frequently approached by pharmaceutical sales representatives from companies that manufacture PAH-specific therapies. These representatives distribute educational materials and medication samples or invite physicians to attend educational programs. In addition, the pharmaceutical companies fund investigational drug trials, support continuing medical education events, and raise awareness about PAH in the community. All of these initiatives are beneficial. However, the downside is that community physicians have developed an increased (and perhaps false) confidence in treating PAH.9
In a survey of 453 physicians, 92% believed that oral pulmonary vasodilator medications (sildenafil or bosentan) were effective treatment options for the management of PH in the setting of parenchymal lung disease, despite evidence-based guidelines.6 Furthermore, 30% started treating without confirming the diagnosis of PAH with a right heart catheterization.14
More worrisome is the delay in referral to tertiary care PAH centers that results from these physicians treating PAH. In the Deaño et al study, almost two-thirds of patients referred to PAH specialty centers had advanced symptoms on their initial evaluation by a PAH specialist. Six of these patients had WHO FC IV symptoms and were on oral pulmonary vasodilators alone at the time of referral.9 This treatment is contrary to accepted guidelines that recommend a parenteral prostanoid as first-line therapy for this patient population.6
The delay in initiation of parenteral prostanoid therapy can lead to treatment failure in patients who might have responded if the drug had been administered sooner and can also lead to a loss in functional capacity that may not recover. In addition, these patients can develop multisystem organ failure and cachexia and may present too late to be considered lung transplant candidates. The REVEAL registry demonstrated that half of patients who die with PAH have never been treated with a prostanoid. The consequence of late referral may therefore ultimately translate into a worse survival in these patients.
ACCESS TO CARE
Socioeconomic reasons result in delayed care in PAH centers. As an example, one of the major obstacles to referral for low-income patients is the distance from the PAH treatment center. Patients who live hours from the PAH center must make arrangements for transportation, gas money, a hotel stay, and meals, both for the initial visit and any subsequent visits.
Solutions are available for overcoming these barriers in access to care. Specialist outreach is an option but requires that the PAH specialist leave a busy practice to see only a few patients hours away. This problem can be avoided if the specialist has a focus clinic for PAH patients once a month in the outreach setting. Alternatively, telemedicine, in which the specialist has a virtual visit with the patient via telephone or video conference, is increasingly being used to assess and manage patients from a distance. Shared care between the primary provider and the PAH specialist is another option. Shared care allows for collaborative care in which a patient can be seen locally with input from the PAH specialist, thereby reducing the number of trips needed to the PAH center and providing for educational opportunities between the PAH specialist and the referring physician. The barrier in this case is that many community clinics and hospitals are unequipped to care for patients on parenteral prostanoid therapy and are uncomfortable about providing care because of lack of education and little or no previous exposure to this therapy. Education of these providers and emergency response teams in areas where patients on parental prostanoid therapy live is important to prevent life-threatening mistakes and to improve care.
Another major concern impacting access to care is the cost of therapy for PH. The mean retail 30-day cost of PAH therapies (including oral, inhaled, and parenteral treatment options) is $5,829,15 and the annual cost of each agent is shown in Table 1.2,3 Medicare plans pay 95% of this amount after the patient pays $4,350 out of pocket. Many patients not only pay substantial copayments for the medical therapy but also pay for oxygen and other non-PAH medications, resulting in a major hardship for both insured and uninsured patients.
Industry-sponsored patient assistance programs are available, but not all patients qualify. Nonprofit organizations also provide financial support to patients to help with copayments, appeal to insurance companies, and offer patients assistance in finding affordable insurance coverage. Clinical trials are another avenue through which patients may receive treatment for PAH without the burden of cost. These trials, however, are most often conducted only at PAH specialty centers.
For the provider, the use of PAH medications means resource-intensive prior authorizations and appeals if the medications are denied. Extensive nursing support and expertise are necessary to initiate and maintain patients on these therapies. This attention is best provided at a PAH center.
FUTURE DIRECTIONS
Although this article is based on experience in the United States, the problem of delayed referrals to a PAH specialist is of global concern.16 The number of deaths that result from PAH in the setting of congenital heart disease, rheumatic heart disease, and schistosomiasis is greater than the combined mortality from all other noncommunicable diseases worldwide.16 Physician education is a problem in the developing world, as is access to diagnostic technology, expensive medical therapies, and clinical trials.
However, one possible solution may lie in creating a model of care similar to that found in the United Kingdom (UK). In the UK, the National Health Service (NHS) of England commissions (plans, negotiates, purchases, and monitors) healthcare services to meet the needs of patients with PH.17 The PH Clinical Reference Group, made up of expert clinicians, commissioners, public health experts, patients, and caregivers, sets the standards of care and details the treatments and services the NHS buys for patients with PH. All patients suspected of having PAH or diagnosed with PAH are referred to 1 of 6 specialist adult PH centers in England or the Scottish Pulmonary Vascular Unit in Glasgow. As all care is delivered via these expert-designated centers, the inappropriate practice of initiating pulmonary vasodilator therapy without first performing a right heart catheterization does not occur. The prescription of pulmonary vasodilators is restricted to these centers. There is a national registry of incident cases, and quality is assured by audits of outcomes.
In the United States, the Pulmonary Hypertension Association has recently established a similar system, in which centers with expertise in PAH are accredited as Centers of Comprehensive Care and Regional Clinical Programs.18 The anticipated outcomes of this initiative are improved education for both patients and the medical community about standards of PAH care, reduction in time between symptom onset and diagnosis, creation of a network of centers for collaboration on clinical care and research, and appropriate use of therapies to improve long-term outcomes.
Another possible solution to improve time to diagnosis is to implement systematic screening in at-risk populations. Such populations would include patients with systemic sclerosis, portal hypertension, congenital heart disease, and human immunodeficiency virus (HIV), as well as patients with large, recurrent, or unprovoked pulmonary emboli (PE) because 0.5%-4% of patients with acute PE will develop chronic thromboembolic pulmonary hypertension (CTEPH).19 The DETECT algorithm, developed by collaborating rheumatologists and cardiologists, is an example of a sensitive, noninvasive, evidence-based screening tool that may be used to determine if patients with systemic sclerosis should be referred for echocardiography and right heart catheterization.20 The DETECT screening tool is widely available and has been shown to minimize missed diagnoses, detect mild PAH, and optimize utilization of resources. It is reasonable to imagine that collaboration between PAH specialists, infectious disease specialists, pulmonary embolism response teams (PERT), and hepatologists might result in similar screening tools for PAH in the setting of HIV, CTEPH, and portopulmonary hypertension, respectively.
Creating symptom-based clinics, such as breathlessness clinics, may have some value. The REVEAL registry showed that patients with other respiratory diseases (ie, obstructive airway disease and sleep apnea) were more likely to have >2 years elapse from symptom onset to diagnosis.13 Less severe impairment of right ventricular (RV) function shown by echocardiogram at the time of right heart catheterization was also found to be associated with delayed recognition of PAH.13 This finding is particularly true in young patients in whom RV failure occurs late and often long after initial symptom onset. Because 8% of children with congenital heart disease will eventually develop PAH, it is imperative that physicians caring for these patients have a high index of suspicion for PAH.16 In the future, cardiac magnetic resonance imaging, computed tomography, and novel biomarkers will likely play a greater role in evaluating RV function and detecting PAH at an earlier stage.
CONCLUSION
Recognizing the barriers to referral to PAH specialty centers is the first step toward overcoming them. By improving physician education, encouraging the use of PAH medications predominantly by PAH specialists, easing access to care, and promoting screening for PAH, patients may be referred earlier than they are at present, resulting in earlier diagnosis and initiation of therapy.
ACKNOWLEDGMENTS
The authors have no financial or proprietary interest in the subject matter of this article.
This article meets the Accreditation Council for Graduate Medical Education and the American Board of Medical Specialties Maintenance of Certification competencies for Patient Care and Medical Knowledge.
REFERENCES
- 1. D'Alonzo GE, Barst RJ, Ayres SW, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991. September 1; 115 5: 343- 349. [DOI] [PubMed] [Google Scholar]
- 2. Bishop D, Baker N, Gandhi R, Yen J, Shek A. Pulmonary arterial hypertension: bridging the gap between efficacy, quality of life, and cost-effectiveness. Formulary. 2010; 45: 190- 199. [Google Scholar]
- 3. Walker T. FDA approves first drug to treat two forms of pulmonary hypertension. Drug Topics . http://drugtopics.modernmedicine.com/drug-topics/content/clinical/clinical- pharmacology/fda-approves-first-drug-treat-two-forms- pulmonary. Published Oct 10, 2013. Accessed May 16, 2016. [Google Scholar]
- 4. Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996. February 1; 334 5: 296- 301. [DOI] [PubMed] [Google Scholar]
- 5. McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009. Apr 28;119(16);2250-2294. [DOI] [PubMed] [Google Scholar]
- 6. Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013. December 24; 62 25 Suppl: D60- D72. [DOI] [PubMed] [Google Scholar]
- 7. Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013. December 24; 62 25 Suppl: D42- D50. [DOI] [PubMed] [Google Scholar]
- 8. McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006. September 26; 114 13: 1417- 1431. [DOI] [PubMed] [Google Scholar]
- 9. Deaño RC, Glassner-Kolmin C, Rubenfire M, et al. Referral of patients with pulmonary hypertension diagnoses to tertiary pulmonary hypertension centers: the multicenter RePHerral study. JAMA Intern Med. 2013. May 27; 173 10: 887- 893. [DOI] [PubMed] [Google Scholar]
- 10. Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013. December 24; 62 25 Suppl: D34- D41. [DOI] [PubMed] [Google Scholar]
- 11. Rubin LJ. American College of Chest Physicians. Diagnosis and management of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004. July; 126 1 Suppl: 7S- 10S. [DOI] [PubMed] [Google Scholar]
- 12. Elliott CG, Barst RJ, Seeger W, et al. Worldwide physician education and training in pulmonary hypertension: pulmonary vascular disease: the global perspective. Chest. 2010. June; 137 6 Suppl: 85S- 94S. [DOI] [PubMed] [Google Scholar]
- 13. Brown LM, Chen H, Halpern S, et al. Delay in recognition of pulmonary arterial hypertension: factors identified from the REVEAL Registry. Chest. 2011. July; 140 1: 19- 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Minai OA, Nathan SD, Hill NS, Badesch DB, Stoller JK. Pulmonary hypertension in lung diseases: survey of beliefs and practice patterns. Respir Med. 2010. May; 104 5: 741- 748. [DOI] [PubMed] [Google Scholar]
- 15. Blankart CR, Stargardt T, Schreyögg J. Availability of and access to orphan drugs. an international comparison of pharmaceutical treatments for pulmonary arterial hypertension, Fabry disease, hereditary angioedema and chronic myeloid leukaemia. Pharmacoeconomics. 2011. January; 29 1: 63- 82. [DOI] [PubMed] [Google Scholar]
- 16. Rich S, Herskowitz A. Targeting pulmonary vascular disease to improve global health: pulmonary vascular disease: the global perspective. Chest. 2010. June; 137 6 Suppl: 1S- 5S. [DOI] [PubMed] [Google Scholar]
- 17. Understanding the management of pulmonary hypertension in adults in the UK . http://www.phauk.org/content/uploads/2016/05/PH-Service-Specification-V1.pdf. Accessed May 25, 2016. [Google Scholar]
- 18. Tenets of PH Care Centers. Pulmonary Hypertension Association Web site . http://www.phassociation.org/PHCareCenters. Accessed October 27, 2015. [Google Scholar]
- 19. Kiely DG, Elliot CA, Sabroe I, Condliffe R. Pulmonary hypertension: diagnosis and management. BMJ. 2013. Apr 16;346:f2028. [DOI] [PubMed] [Google Scholar]
- 20. Coghlan JG, Denton CP, Grünig E, et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis. 2014. July; 73 7: 1340- 1349. [DOI] [PMC free article] [PubMed] [Google Scholar]