

Abstract
Simultaneous spectral unmixing of excitation and emission spectra (ExEm unmixing) has the inherent ability to resolve donor emission, fluorescence resonance energy transfer (FRET)-sensitized acceptor emission and directly excited acceptor emission. We here develop an ExEm unmixing-based quantitative FRET measurement method (EES-FRET) independent of excitation intensity and detector parameter setting. The ratio factor (rK), predetermined using a donor-acceptor tandem construct, of total acceptor absorption to total donor absorption in excitation wavelengths used is introduced for determining the concentration ratio of acceptor to donor. We implemented EES-FRET method on a wide-field microscope to image living cells expressing tandem FRET constructs with different donor-acceptor stoichiometry.
OCIS codes: (170.2520) Fluorescence microscopy, (300.6170) Spectra
1. Introduction
Fluorescence resonance energy transfer (FRET) microscopy has become an invaluable tool for monitoring intracellular dynamic spatio-temporal activity of biochemical events during signal transduction. Improvements in the spectral characteristics of genetically encoded fluorescent proteins (FPs) and FPs-biosensors enable FRET-based visualization of dynamic signaling events to measure the protein-protein interaction of Bcl-2 family proteins during cell apoptosis [1,2] and the structural changes of fibroblast growth factors receptor dimerization on different ligands binding [3] as well as the dependence of mitochondrial calcium uptake from the cytosolic Ca2+ signals [4] within living cells. FRET microscopy based on multiple pairs of FP-fusions has been developed to simultaneously visualize multiple molecular events within single live cells [5–7].
Quantification of FRET needs to resolve three spectral components: direct donor fluorescence, direct acceptor fluorescence and FRET-sensitized acceptor fluorescence [5]. However, overlapping spectra between donor and acceptor, including donor spectral bleedthrough (donor emission into acceptor channel) and acceptor excitation cross (direct excitation of acceptor fluorophores by donor excitation), is inevitable for FP-FRET pairs [8]. The overlapping emission spectra of donor and acceptor can be easily resolved by spectral unmixing of emission spectra (Em unmixing) [9–13]. However, the FRET-sensitized and direct acceptor emission cannot be resolved by Em unmixing due to the same emission spectra. Therefore, donor spectral bleedthrough can be overcome by Em unmixing, but the acceptor excitation cross must be corrected using additional manner [13–16]. Simultaneous spectral unmixing of excitation and emission spectra (ExEm unmixing) has the inherent ability to resolve donor emission, FRET-sensitized acceptor emission and direct acceptor emission [5,6,17,18]. Thus ExEm unmixing is capable of simultaneously resolving the donor spectral bleedthrough and acceptor excitation cross without additional correction.
With the improvements of commercial confocal microscope with spectra detector, Em unmixing-based FRET quantification has been widely utilized to image biochemical events in single live cells [13,15,19]. Recently, Mustafa and associates demonstrated that ExEm unmixing with as few as two excitation wavelengths could be used for quantitative FRET measurement, and for the first time implemented ExEm unmixing-based quantitative FRET measurement on a laser scanning confocal microscope with two different excitations to image the FRET efficiency (E) of a genetically encoded FRET construct in living HEK cells [18]. Rigorous system calibration and signal correction are required for almost every quantitative FRET measurement on confocal microscope due to the mutable status of confocal microscope [20–22]. On the contrary, our and other studies demonstrated that the status and performance of wide-field microscopes were stable in quite a long time (at least three months) [13,20,22]. Recently, we for the first time developed a spectral wide-field microscope by integrating a liquid crystal tunable filter (LCTF) into a wide-field microscope, and successfully performed an Em unmixing-based FRET measurement on this spectral wide-field microscope [23].
In this report, we develop an ExEm unmixing-based quantitative FRET measurement method, termed as EES-FRET. In sharp contrast to the ExEm-FRET method proposed by Mustafa and associates [18], EES-FRET method adopts complete instrumental calibration including the excitation intensity spectrum (wavelength-dependent excitation intensity) and the emission-spectral response (optical transfer function of the emission light path) of system. In addition, we introduce a ratio factor (rK) of total acceptor absorption (KA) to total donor absorption (KD) in all excitation wavelengths used for the determination of concentration ratio (RC) of acceptor to donor. We implemented EES-FRET method on our spectral wide-field microscope to obtain the quantitative E and RC images of living HepG2 cells expressing FPs-based constructs with different acceptor-donor stoichiometry.
2. Theory
2.1 Basic equations for EES-FRET method
To keep the naming convention consistent throughout this manuscript, S without superscript represents the unit-area-normalized excitation-emission spectra, while S with superscript refers to the unit-area-normalized excitation or emission spectra of sample (ex denotes excitation spectra and em denotes emission spectra). For simplicity, we describe in Table 1 symbols and notations used throughout the report.
Table 1. Compilation of Symbols Used in the Article.
| Unit-area-normalized excitation spectrum of X (D for donor, A for acceptor) | ||
| Unit-area-normalized emission spectrum of X (D for donor, A for acceptor) | ||
| SX | Unit-area-normalized excitation-emission spectral signature of X (D for donor, A for acceptor, S for donor-acceptor sensitization): SD = S ex D⊗ S em D, SA = S ex A⊗ S em A, SS = S ex D⊗ S em A, ⊗: Outer product |
|
| SDA | Image-stack: Excitation-emission spectrum of FRET sample | |
| KX | Total X (D for donor, A for acceptor) absorption in all excitation wavelengths used: ∫Iex εX(λ)dλ | |
| (εX(λ) is the extinction coefficient of X at wavelength λ) | ||
| rK | The ratio of total acceptor absorption to total donor absorption in all excitation wavelengths used: KA/KD | |
| Cy | Concentration of y (d for free donor, a for free acceptor, da for the paired donor-acceptor) | |
| C t X | Concentration of total X (D for donor, A for acceptor) | |
| QX | Quantum yield of X (D for donor, A for acceptor) | |
| rQ | Quantum yield ratio of acceptor to donor: QA/QD | |
| WX | Weight of component X (D for donor, A for acceptor, S for donor-acceptor sensitization) | |
| E | FRET efficiency of the paired donor-acceptor | |
| Eapp | Apparent FRET efficiency (related to total donor): ECda/C t D | |
| RC | Concentration ratio of acceptor to donor: C t A/C t D | |
Considering a FRET sample containing free donor and free acceptor as well as FRET pairs, the net excitation-emission spectrum (SDA) can be linearly resolved into four excitation-emission components from free donor, the paired donor, the FRET-sensitized acceptor and the direct-excited acceptor as follow,
| (1) |
Where, E is FRET efficiency of the paired donor-acceptor in FRET sample; Cd is the concentration of free donor, Cda is the concentration of the paired donor-acceptor, and C t A is the concentration of total acceptor; KD and KA are the total donor absorption and total acceptor absorption respectively in all excitation wavelengths used; QD and QA are the quantum yield of donor and acceptor, respectively; SD, SA and SS are the unit-area-normalized excitation-emission spectral signatures of donor, acceptor and donor-acceptor sensitization, respectively, and they are the outer product of the excitation spectra (S ex X, X = D and A) and the emission spectra (S em X, X = D and A) measured from donor-only and acceptor-only sample, respectively [5,18]:
| (2) |
Sorting these terms in Eq. (1) according to the three spectral signatures introduced above:
| (3) |
where C t D (C t D = Cd + Cda) is the concentration of total donor, and
| (4) |
are the apparent concentrations (weights) of donor, donor-acceptor sensitization and acceptor, respectively, and they are extracted from SDA [Eq. (3)] by spectral unmixing, specifically by fitting the data using the least-squares method. From Eq. (4), we can obtain
| (5) |
| (6) |
| (7) |
Therefore, the formulae of apparent FRET efficiency (Eapp) normalized to donor concentration and total concentration ratio (RC) of acceptor to donor are
| (8) |
| (9) |
where rK = KA/KD and rQ = QA/QD.
2.2 Determination of rK factor using a donor-acceptor tandem sample
According to Eq. (9), we can predetermine rK factor using a donor-acceptor tandem construct with known RC as follow:
| (10) |
2.3 Process of quantitative EES-FRET measurement
As shown in Fig. 1 , the process of quantitative EES-FRET measurement can be broken into following three steps.
Fig. 1.
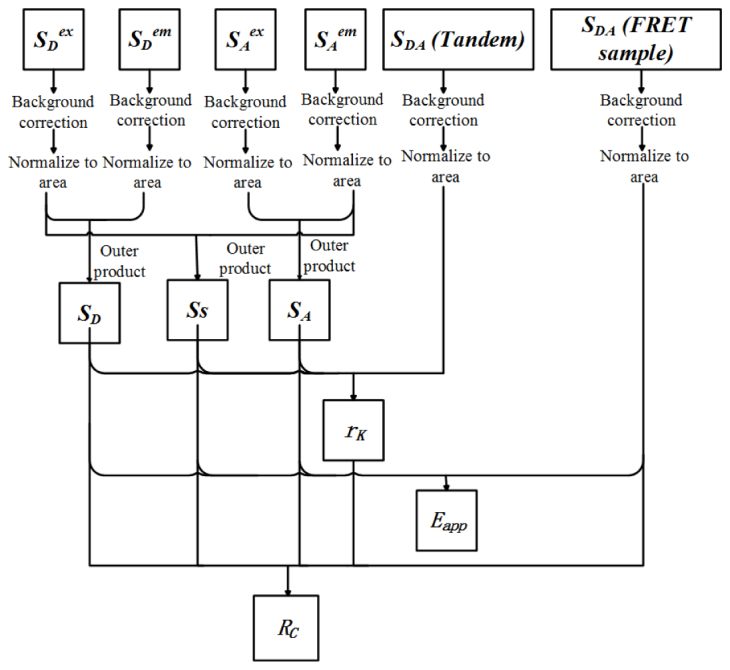
Scheme of quantitative EES-FRET measurement. Tandem: a tandem construct with known acceptor-donor concentration ratio.
Step 1: Measurement of excitation-emission spectral signatures (SD, SA and SS)
The excitation spectra for donor (Sex D) and acceptor (Sex A) are obtained by recording the emission of donor-only and acceptor-only sample respectively at a settled emission wavelength with different excitations. The emission spectra for donor (Sem D) and acceptor (Sem A) are obtained by measuring the emission spectral images of donor-only and acceptor-only sample respectively with a settled excitation wavelength. After background correction (background is removed from each spectrum), the excitation spectra should be corrected with excitation intensity spectrum and the emission spectra should be corrected with emission-spectral responses. All spectra should be normalized to unit area. Then, three unit-area-normalized excitation-emission spectral signatures (SD, SA and SS) are calculated according to Eq. (2).
Step 2: Determination of rK factor
The excitation-emission spectrum (image-stack: SDA (Tandem)) of a donor–acceptor tandem construct (a donor–acceptor pair connected by a polypeptide linker) with known acceptor-donor concentration ratio (RC) is firstly measured by recording the emission-spectral images with different excitations, and then is spectrally unmixed into the contributions of donor (weight: WD (Tandem)), donor-acceptor sensitization (weight: WS (Tandem)) and acceptor (weight: WA (Tandem)). The rK factor is calculated according to Eq. (10).
Step 3: Calculate Eapp and RC of FRET sample
The excitation-emission spectrum (image-stack: SDA (FRET sample)) of FRET sample is measured by recording the emission spectral images with different excitations. Three contributions (weights: WD, WS and WA) to the SDA (FRET sample) are obtained by spectral unmixing. Both Eapp and RC are then calculated according to Eq. (8) and Eq. (9), respectively.
3. Materials and methods
3.1 Spectral wide-field microscope for quantitative EES-FRET measurement
To perform EES-FRET method, we added two cubes to the filter-cube wheel in our recently developed spectral wide-field microscopic FRET imaging system with an E455Ipv2 dichroic mirror (455 nm dichroic mirror, D455) (Chroma, America) [23]. As depicted in Fig. 2 , one of the added cubes has a BP470/40 (Carl Zeiss, Germany) bandpass excitation filter and a FT495 (495 nm dichroic mirror, D495) (Carl Zeiss, Germany) dichroic mirror, and another one has a BP510/17 (Carl Zeiss, Germany) bandpass excitation filter and a DFT520 (520 nm dichroic mirror, D520) (Carl Zeiss, Germany) dichroic mirror. ET405/20x (Chroma, America) and BP436 (Carl Zeiss, Germany) bandpass excitation filters share the same 455 nm dichroic mirror (D455). The DD, AA and DA cubes are retained in the wide-field microscope for three-cube-based quantitative FRET measurement [24–26]. We can push or pull the secondary cube to alternatively implement quantitative EES-FRET measurement (a) using CCD1 or three-cube-based quantitative FRET measurement (b) using CCD2.
Fig. 2.
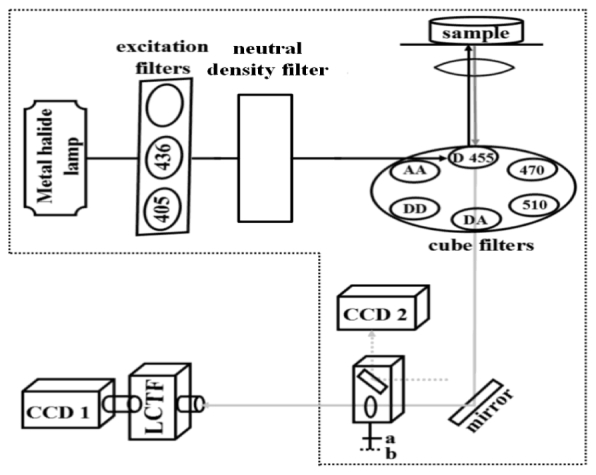
Illustration on the spectral wide-field microscope. 405: ET405/20x bandpass excitation filter (405 nm excitation); 436: BP436/20 bandpass excitation filter (436 nm excitation); D455: E455Ipv2 dichroic mirror (455 nm dichroic mirror, D455); 470: a cube contains a BP470/40 bandpass excitation filter (470 nm excitation) and a FT495 dichroic mirror (495 nm dichroic mirror, D495); 510: a cube contains a BP510/17 bandpass excitation filter (510 nm excitation) and a DFT520 dichroic mirror (520 nm dichroic mirror, D520); DD: a cube contains filters for donor excitation and donor emission; AA: a cube contains filters for acceptor excitation and acceptor emission; DA: a cube contains filters for donor excitation and acceptor emission; LCTF: liquid crystal tunable filter (Varispec LCTF, VIS-10-20-STD, CRI, Cambridge); CCD1: CCD camera for quantitative EES-FRET measurement; CCD2: CCD camera for three-cube-based quantitative FRET measurement; a: for CCD1 detection; b: for CCD2 detection.
3.2 Microscopic imaging
According to the expression level of FPs, excitation intensity was attenuated to 0.5% or 1% by regulating the intensity of metal halide lamp and/or choosing different neutral density filters. For EES-FRET method, bandpass excitation filters of ET405/20x, BP436/20, BP470/40 and BP510/17 were used to select four excitation wavelengths of 405 nm (405 nm excitation), 436 nm (436 nm excitation), 470 nm (470 nm excitation), and 510 nm (510 nm excitation), respectively, by pushing/pulling the mobilizable three-hole plan or/and shifting the filter-cube wheel [Fig. 2]. For emission spectral imaging of donor and acceptor, cells expressing Cerulean-only (donor-only) or Venus-only (acceptor-only) were excited with 436 nm excitation and emission wavelength range was from 464 to 607 nm by using LCTF. Emission at 503 nm was recorded for the detection of donor excitation spectrum, and emission at 529 nm was recorded for the detection of acceptor excitation spectrum. For excitation-emission spectral imaging of FRET samples, emission spectral range from 464 to 555 nm was spectrally scanned for the 405 nm and 436 nm excitations by using LCTF. Emission spectral range from 503 to 555 nm, and from 529 to 555 nm was spectrally scanned for the 470 nm excitation and 510 nm excitation, respectively. When emission was spectrally scanned with a step resolution of 13 nm using LCTF, the corresponding spectral images with each excitation were acquired with emCCD. For simplicity, we introduce image-stack (SDA) to denote the excitation-emission spectral image of FRET sample. In this report, an image-stack (SDA) contains 24 images including eight emission spectral images with 405 nm excitation, eight emission spectral images with 436 nm excitation, five emission spectral images with 470 nm excitation and three emission spectral images with 510 nm excitation. emCCD was typically run in 4 × 4 binning mode and background was removed based on the average reading in a non-fluorescent cell area of each image.
3.3 Cell culture, transfection and plasmids
Human hepatocellular carcinoma (HepG2) cells were obtained from the Department of Medicine, Jinan University, Guangzhou, China. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, New York) containing 10% fetal calf serum (Sijiqing, Hangzhou, China) at 37°C under 5% CO2 in a humidified incubator.
For transfection, cells were cultured in DMEM containing 10% FCS in a 30-mm glass dish at 37°C under 5% CO2 in a humidified incubator. After 24 h, when the cells reached 70% to 90% confluence, plasmid was transfected into the HepG2 cells for 24-48 h by using Lipofectamine 2000 (invitrogen, Carlsbad, American) in vitro transfection reagent.
Cerulean (C) and Venus(V)-kras plasmids were purchased from Addgene Company (Cambridge, Massachusetts). The FRET-standard constructs, including CTV (C-TRAF-Venus, the TRAF is a tumour necrosis factor receptor-associated factor domain including 229 amino acid), C32V (Cerulean-32-Venus, Addgene plasmid 29396), CVC (Cerulean-5-Venus-5-Cerulean, Addgene plasmid 27788) and VCV (Venus-5-Cerulean-5-Venus, Addgene plasmid 27788), were kindly provided by the Vogel lab (National Institutes of Health, Bethesda, Maryland) [11,27].
4. Results and discussion
4.1 Calibration of instrument
4.1.1 Excitation intensity spectrum
We calibrated the excitation intensity spectrum of our spectral wide-field microscope by recording the emission intensity at 630 nm of 7 g/l rhodamine B in ethylene glycol at different excitations. Background from ethylene glycol solvent without rhodamine B was subtracted. The measured excitation intensity spectrum of rhodamine B solution should be the excitation intensity spectrum of our instrument due to the constant quantum yield and emission maximum (≈630 nm) of this concentrated rhodamine B solution in the excitation wavelength range from 250 to 600 nm [28]. Figure 3(a) showed the relative excitation intensity at 405 nm, 436 nm, 470 nm and 510 nm excitations used in this study under different attenuation degrees (Transmission: T = 0.25%, 0.5% and 1%, respectively), and normalization of these relative excitation intensity showed the same excitation intensity spectrum (data not show) of our system at different attenuation degrees. We measured the excitation intensity spectra of our system under T = 0.25% for six times from 16 December to 20 December [Fig. 3(b)], and found that the excitation intensity spectra exhibited a modest shake for every starting up.
Fig. 3.

Excitation intensity spectra measured by recording the emission intensity at 630 nm of 7 g/l rhodamine B in ethylene glycol with 405, 436, 470 and 510 nm excitation, respectively. (a) Excitation intensity spectra (normalized to the maximum excitation intensity) measured under three attenuation degrees (Transmission: T = 0.25%, 0.5% and 1%). (b) Normalized excitation intensity spectra measured from 16 December to 20 December under T = 0.25%.
As the best-known quantum counter, the concentration of rhodamine B solution should be not less than 3 g/l [28] because the excitation spectrum from 250 to 600 nm of low concentrated rhodamine B solution is not constant. Considering the solubility of rhodamine B in ethylene glycol and fluorescence quenching in high concentration solution, the concentration of rhodamine B solution should not be too high. Rhodamine B should be dissolved in ethylene glycol completely to avoid influence of inhomogeneity in solution.
Regulating excitation intensity is usually necessary for obtaining good images of cells expressing different levels of FPs. Since EES-FRET method is dependent on the excitation intensity spectrum, we thus had better control excitation intensity by regulating the intensity of metal halide lamp rather than the neutral density filters according to the expression level of FPs in cells due to independence of the excitation intensity spectrum of our system on the intensity of metal halide lamp. Difference of excitation intensity spectra for every starting up the system [Fig. 3(b)] may be largely due to the instability of the power supply voltage. Therefore, we must calibrate the excitation intensity spectrum once the system is restarted. In reality, this calibration is very easy to be finished within 1 min.
4.1.2 Emission-spectral responses
Emission-spectral response, the optical transfer function of emission light path, indicates the spectral response of our system to different emissions. We calibrated the emission-spectral responses of our spectral wide-field microscope using a precalibrated light source (LS-1-CAL, Ocean Optics, Dunedin FL). The standard intensity spectrum of the LS-1-CAL was multiplied the nominal wavelength to transform them from energy units to values related to photon counts (photon spectrum) [Fig. 4(a) ], and the emission-spectral response of our system with D455 was obtained by comparing the ratio between the measured and standard spectra of CLS-1-CAL just as described previously [22,23]. Figure 4(b) showed the emission-spectral responses of our system with different dichroic mirrors: D455, D495 and D520. We repeated this calibration for our system with D455 for 15 days and obtained the same response [Fig. 4(c)], demonstrating the stability of our imaging system.
Fig. 4.

Emission-spectral responses of the spectral wide-field microscope. (a) Standard emission spectra of a precalibrated light source (LS-1-CAL). Open circles: intensity spectrum. Shaded circles: photon spectrum. (b) Emission-spectral responses (obtained by comparing the ratio between the measured and standard emission spectrum of the LS-1-CAL) of our system with different dichroic mirrors: D455, D495 and D520. (c) 13 emission-spectral responses of our system with D455 measured from 12 December to 28 December.
Different excitations share the same emCCD and LCTF, thus different emission-spectral responses of our system with different dichroic mirrors are only related to the different dichroic mirrors having different spectral transmittance. Since 405 nm and 436 nm excitations share the same D455, our system with the two excitations has the same emission-spectral response. Because ExEm method can be achieved with as few as two excitation wavelengths [18], 405 nm and 436 nm excitations may be enough for our EES-FRET method. In this case, we just need to measure the emission-spectral responses of our system with D455. Although we here demonstrated the stability of our system with D455 for only 15 days, our wide-field microscope had been proved to be stable at least 3 months [22]. Therefore, we need not measure the emission-spectral responses for subsequent measurement once our system is calibrated. However, periodical calibration for the unchanged device can ensure the precision of quantitative measurement.
4.2 Normalized excitation-emission spectral signatures
To obtain the excitation-emission spectra of donor and acceptor as well as donor-acceptor sensitization in living HepG2 cells, the cells were separately expressed with Cerulean (C) or Venus (V). We measured the excitation spectrum of donor by detecting the emission intensity at 503 nm of living HepG2 cells expressing Cerulean with different excitations, and also measured the excitation spectrum of acceptor by detecting the emission intensity at 529 nm of living HepG2 cells expressing Venus with different excitations [Fig. 5(a) , left]. The two excitation spectra calibrated by the excitation intensity spectrum [Fig. 3(a)] were normalized to unit area. We measured the emission spectra of both Cerulean and Venus in living HepG2 cells with 436 nm excitation [Fig. 5(a), right], and the two emission spectra calibrated by the emission-spectral response [Fig. 4(b)] were normalized to unit area. According to Eq. (2), we calculated the outer product of the excitation spectra and the emission spectra of donor and acceptor [Fig. 5(a)] to obtain the corresponding normalized excitation-emission spectral signatures of donor (SD) and acceptor (SA) as well as donor-acceptor sensitization (SS), respectively [Fig. 5(b)].
Fig. 5.

Three excitation-emission spectral signatures. (a) Excitation spectra of Cerulean (Sex D) and Venus (Sex A) (Left), and emission spectra of Cerulean (Sem D) and Venus (Sem A) (Right). These spectra were measured by recording the excitation and emission spectra of living HepG2 cells separately expressing donor (Cerulean) and acceptor (Venus). (b) Fitted pseudo-color images of three excitation-emission spectral signatures (SD, SA, SS) from the excitation and emission spectra (a) using Eq. (2).
Although two different excitations have been demonstrated to be enough for quantitative EES-FRET measurement [5], the fewer the excitations and emission wavelengths, the larger the error of the derived quantities due to the fitting procedure is going to be. However, the more the excitations, the more the measurement time should be required. We have previously performed a time-lapse images of living HepG2 cells expressing FPs with T = 1% of 436 nm excitation for 3 min, and found that this excitation did not induce significant photobleaching [22]. Considering rapidness and accuracy of measurement, we here chose emission wavelength range of 464 nm to 555 nm instead of total emission wavelength range, and four excitations of 405 nm, 436 nm, 470 nm and 510 nm for quantitative EES-FRET measurement. In reality, less time should be required when we only use 405 nm and 436 nm excitations to accomplish EES-FRET measurement on our system, because the two excitations share the same D455. In fact, the 405 nm and 436 nm excitations had been used to accomplish quantitative Iem-spFRET measurement on our system [23].
Although the excitation-emission spectral signatures (SD, SA and SS) are independent of the excitation intensity of our system and the parameter settings of detector as well as the expression levels of FPs due to the spectral correction using the excitation intensity spectrum and emission-spectral responses, they may be related to the physiological environments of a specific cell line. Therefore, once they are determined for a given FP-pair and cell line, we need not prepare donor-only and acceptor-only samples to measure them for subsequent FRET measurements. In reality, we found that the emission spectra of CFP/Cerulean and YFP/Venus in Huh-7 cells were very similar to the spectra of those FPs in human lung adenocarcinoma ASTC-a-1 cells [22,29].
Precise measurement of the excitation and emission spectra of both Cerulean and Venus is required for the accurate measurement of excitation-emission spectral signatures. Because of the emission peak at 503 nm for Cerulean and at 529 nm for Venus, we measured the excitation spectra of Cerulean and Venus by detecting the emission intensity at 503 nm and 529 nm respectively with different excitations. Considering the strong absorbability of Cerulean at 436 nm and the better absorbability of Venus at 436 nm than that at 405 nm, we measured the emission spectra of both Cerulean and Venus in living HepG2 cells with 436 nm excitation. To reduce the effect of background and autofluorescence on the fluorescence signal of FPs, we chose the cells expressing high levels of FPs to measure the above spectra. In addition, the signal from blank cells without FPs as background is subtracted from the fluorescence signal of cells expressing FPs. We recently evaluated the effect of autofluorescence in HepG2 cells on the spectra of FPs, and found that the autofluorescence of HepG2 cells is very low, and the concentration of FPs did not influence the measured spectra of FPs [16, 22].
4.3 Determination of rK factor
We used CTV plasmid as the donor-acceptor tandem sample to measure the ratio (rK) of total Venus absorption to total Cerulean absorption in all excitations used on our spectral wide-field microscope. A representative raw excitation-emission spectral image (image-stack, SDA (tandem)) of living HepG2 cells expressing CTV is shown in Fig. 6(a) . After correction with excitation intensity spectrum and emission-spectral responses, the SDA was linearly unmixed into the three contributions (weights: WD, WS and WA) of SD, SA and SS [Fig. 5(b)], and the rK factor was calculated according to Eq. (10), where the quantum yield ratio (rQ) of Venus (0.57) to Cerulean (0.62) is rQ = 0.57/0.62 = 0.919 [30,31]. Figure 6(b) showed the corresponding pixel-to-pixel pseudo-color rK image and Fig. 6(c) showed the corresponding histogram indicating a peak value about 1.96. The average peak rK value of Venus to Cerulean in living HepG2 cells was 1.9 ± 0.06 from 15 histograms including at least 60 cells.
Fig. 6.

Determination of rK factor using a donor-acceptor tandem sample (living HepG2 cells expressing CTV tandem construct with RC = 1). (a) A representative excitation-emission spectral image-stack of living cells expressing CTV construct. (b and c) Pixel-to-pixel rK image (b) and histograms (c) corresponding to (a).
Although KD and KA depend not only on the absorption spectra of donor and acceptor but also on the excitation intensity, rK is only related to the absorption spectra of donor and acceptor in all excitations used for a given cell line. Therefore, we can directly obtain the rK by measuring the excitation-emission spectrum of a donor-acceptor tandem construct instead of measuring the KD and KA. In contrast to the use of two extinction coefficient ratios of acceptor to donor at two excitations in emission-spectral unmixing-based FRET method [12,16], we here introduce rK in EES-FRET method for the determination of RC, which makes EES-FRET method more robust and accurate. Similar to the spectral signatures (SD, SA and SS), once rK is predetermined, we need not to measure it for subsequent FRET measurements for a given specific cell line and FP-pairs as well as excitation wavelengths used, even when we change emission optical path because it is independent of emission properties of imaging system [16].
4.4 Microscopic EES-FRET imaging of living cells expressing tandem constructs
To validate EES-FRET method, we implemented this method on our spectral wide-field microscope to obtain the FRET images of living HepG2 cells separately expressing C32V, CVC and VCV. Three representative spectral image-stacks of living cells separately expressing C32V, CVC and VCV construct were shown in Fig. 7(a) . Figure 7(b) exhibited the measured (upper panel) and fitted (lower panel) excitation-emission spectra for the brightest pixel in Fig. 7(a). Figures 7(c) and 7(d) showed the pixel-to-pixel pseudo-color E images [Fig. 7(c)] of Fig. 7(a) and the corresponding E histograms [Fig. 7(d)] exhibiting the peaks of ~37% for C32V and ~50% for CVC as well as ~70% for VCV. Figures 7(e) and 7(f) showed the pixel-to-pixel pseudo-color RC images [Fig. 7(e)] of Fig. 7(a) and the corresponding RC histograms [Fig. 7(f)] exhibiting the peaks of ~0.95 for C32V, ~0.48 for CVC and ~1.9 for VCV, in agreement with the corresponding theoretical values of 1, 0.5 and 2. The statistical E and RC values from 10 frames including at least 40 cells were 42.3% ± 3.1% and 0.97 ± 0.07 for C32V, 52.8% ± 5.4% and 0.49 ± 0.28 for CVC and 70.24 ± 6.7% and 1.95 ± 0.8 for VCV.
Fig. 7.

Microscopic EES-FRET imaging of living cells expressing constructs with different donor-acceptor stoichiometry. (a) Representative spectral image-stacks (SDA) for living HepG2 cells expressing C32V, CVC and VCV, respectively. (b) The measured and fitted excitation-emission spectra of the brightest pixel in (a). (c and d) Pixel-to-pixel E images (c) and histograms (d) corresponding to (a). (e and f) Pixel-to-pixel RC images (e) and histograms (f) corresponding to (a).
Considering the low transmittance of LCTF and the exact match between emCCD and LCTF, we chose a relative long expose time of 300 ms for emCCD imaging and time interval of 2000 ms for both emCCD imaging and LCTF operation. It thus took about 70 s to perform the EES-FRET measurement [Fig. 7]. During this period, the mobility of living cells may lead to a mismatch of spectral image and the nonuniform E image [Fig. 7(c), middle] just as described previously [23]. However, compared with the E and Rc images we recently obtained by using Em unmixing-based FRET measurement [23], EES-FRET method obtained more uniform and stable E and Rc images [Figs. 7(c)-7(f)], indicating that EES-FRET method had a better robustness. Fast measurement is especially important for quantitative live-cell FRET measurement. The theoretically shortest time to perform an EES-FRET measurement on our system is about 1 s. In reality, we can complete an EES-FRET measurement within 10 s on our spectral wide-field microscope for the cells expressing high levels of FPs.
5. Conclusions
We here developed an ExEm unmixing-based quantitative FRET measurement method (EES-FRET method). This method is independent of excitation intensity and parameter settings of detector including the exposure time and gain factor. Therefore, we can adjust the excitation intensity or the exposure time and gain factor of detector to obtain good images for quantitative EES-FRET measurement in the live cells expressing various levels of FPs. Implementation of EES-FRET method on our spectral wide-field microscope for live cells expressing FRET constructs with different stoichiometry exhibit stable FRET images. To our knowledge, this is the first report about the implementation of ExEm unmixing-based quantitative FRET measurement method on a wide-field microscope, which should expand the application range of live-cell FRET microscopy.
Acknowledgments
The authors thank Prof. S.S. Vogel (NIH/NIAAA) for providing C32V, CVC and VCV plasmids.
Funding
National Natural Science Foundation of China (NSFC) ( 81471699, 61527825); National Key Basic Research Program of China (2015CB352006); Science and Technology Plan Project of Guangdong Province (2014B090901060).
References and links
- 1.Düssmann H., Rehm M., Concannon C. G., Anguissola S., Würstle M., Kacmar S., Völler P., Huber H. J., Prehn J. H. M., “Single-cell quantification of Bax activation and mathematical modelling suggest pore formation on minimal mitochondrial Bax accumulation,” Cell Death Differ. 17(2), 278–290 (2010). 10.1038/cdd.2009.123 [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y., Xing D., Liu L., “PUMA promotes Bax translocation by both directly interacting with Bax and by competitive binding to Bcl-X L during UV-induced apoptosis,” Mol. Biol. Cell 20(13), 3077–3087 (2009). 10.1091/mbc.E08-11-1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sarabipour S., Hristova K., “Mechanism of FGF receptor dimerization and activation,” Nat. Commun. 7, 10262 (2016). 10.1038/ncomms10262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waldeck-Weiermair M., Malli R., Parichatikanond W., Gottschalk B., Madreiter-Sokolowski C. T., Klec C., Rost R., Graier W. F., “Rearrangement of MICU1 multimers for activation of MCU is solely controlled by cytosolic Ca(2.),” Sci. Rep. 5, 15602 (2015). 10.1038/srep15602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoppe A. D., Scott B. L., Welliver T. P., Straight S. W., Swanson J. A., “N-way FRET microscopy of multiple protein-protein interactions in live cells,” PLoS One 8(6), e64760 (2013). 10.1371/journal.pone.0064760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woehler A., “Simultaneous quantitative live cell imaging of multiple FRET-based biosensors,” PLoS One 8(4), e61096 (2013). 10.1371/journal.pone.0061096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galperin E., Verkhusha V. V., Sorkin A., “Three-chromophore FRET microscopy to analyze multiprotein interactions in living cells,” Nat. Methods 1(3), 209–217 (2004). 10.1038/nmeth720 [DOI] [PubMed] [Google Scholar]
- 8.Rizzo M. A., Springer G., Segawa K., Zipfel W. R., Piston D. W., “Optimization of pairings and detection conditions for measurement of FRET between cyan and yellow fluorescent proteins,” Microsc. Microanal. 12(3), 238–254 (2006). 10.1017/S1431927606060235 [DOI] [PubMed] [Google Scholar]
- 9.Clegg R. M., “Fluorescence resonance energy transfer and nucleic acids,” Methods Enzymol. 211, 353–388 (1992). 10.1016/0076-6879(92)11020-J [DOI] [PubMed] [Google Scholar]
- 10.Gu Y., Di W. L., Kelsell D. P., Zicha D., “Quantitative fluorescence resonance energy transfer (FRET) measurement with acceptor photobleaching and spectral unmixing,” J. Microsc. 215(2), 162–173 (2004). 10.1111/j.0022-2720.2004.01365.x [DOI] [PubMed] [Google Scholar]
- 11.Thaler C., Koushik S. V., Blank P. S., Vogel S. S., “Quantitative multiphoton spectral imaging and its use for measuring resonance energy transfer,” Biophys. J. 89(4), 2736–2749 (2005). 10.1529/biophysj.105.061853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wlodarczyk J., Woehler A., Kobe F., Ponimaskin E., Zeug A., Neher E., “Analysis of FRET signals in the presence of free donors and acceptors,” Biophys. J. 94(3), 986–1000 (2008). 10.1529/biophysj.107.111773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy S., Wilms C. D., Brumer E., Kahn J., Pnueli L., Arava Y., Eilers J., Gitler D., “SpRET: highly sensitive and reliable spectral measurement of absolute FRET efficiency,” Microsc. Microanal. 17(2), 176–190 (2011). 10.1017/S1431927610094493 [DOI] [PubMed] [Google Scholar]
- 14.Dinant C., van Royen M. E., Vermeulen W., Houtsmuller A. B., “Fluorescence resonance energy transfer of GFP and YFP by spectral imaging and quantitative acceptor photobleaching,” J. Microsc. 231(1), 97–104 (2008). 10.1111/j.1365-2818.2008.02020.x [DOI] [PubMed] [Google Scholar]
- 15.Megías D., Marrero R., Martínez Del Peso B., García M. Á., Bravo-Cordero J. J., García-Grande A., Santos A., Montoya M. C., “Novel lambda FRET spectral confocal microscopy imaging method,” Microsc. Res. Tech. 72(1), 1–11 (2009). 10.1002/jemt.20633 [DOI] [PubMed] [Google Scholar]
- 16.Zhang J., Li H., Chai L., Zhang L., Qu J., Chen T., “Quantitative FRET measurement using emission-spectral unmixing with independent excitation crosstalk correction,” J. Microsc. 257(2), 104–116 (2015). 10.1111/jmi.12189 [DOI] [PubMed] [Google Scholar]
- 17.Yuan J., Peng L., Bouma B. E., Tearney G. J., “Quantitative FRET measurement by high-speed fluorescence excitation and emission spectrometer,” Opt. Express 18(18), 18839–18851 (2010). 10.1364/OE.18.018839 [DOI] [PubMed] [Google Scholar]
- 18.Mustafa S., Hannagan J., Rigby P., Pfleger K., Corry B., “Quantitative Förster resonance energy transfer efficiency measurements using simultaneous spectral unmixing of excitation and emission spectra,” J. Biomed. Opt. 18(2), 026024 (2013). 10.1117/1.JBO.18.2.026024 [DOI] [PubMed] [Google Scholar]
- 19.Chen Y., Mauldin J. P., Day R. N., Periasamy A., “Characterization of spectral FRET imaging microscopy for monitoring nuclear protein interactions,” J. Microsc. 228(2), 139–152 (2007). 10.1111/j.1365-2818.2007.01838.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elangovan M., Wallrabe H., Chen Y., Day R. N., Barroso M., Periasamy A., “Characterization of one- and two-photon excitation fluorescence resonance energy transfer microscopy,” Methods 29(1), 58–73 (2003). 10.1016/S1046-2023(02)00283-9 [DOI] [PubMed] [Google Scholar]
- 21.Yu H., Chen T., Qu J., “Improving FRET efficiency measurement in confocal microscopy imaging,” Chin. Opt. Lett. 8(10), 947–949 (2010). 10.3788/COL20100810.0947 [DOI] [Google Scholar]
- 22.Chai L., Zhang J., Zhang L., Chen T., “Miniature fiber optic spectrometer-based quantitative fluorescence resonance energy transfer measurement in single living cells,” J. Biomed. Opt. 20(3), 037008 (2015). 10.1117/1.JBO.20.3.037008 [DOI] [PubMed] [Google Scholar]
- 23.Zhang L., Qin G., Chai L., Zhang J., Yang F., Yang H., Xie S., Chen T., “Spectral wide-field microscopic fluorescence resonance energy transfer imaging in live cells,” J. Biomed. Opt. 20(8), 086011 (2015). 10.1117/1.JBO.20.8.086011 [DOI] [PubMed] [Google Scholar]
- 24.Zhang L., Yu H., Zhang J., Chen T., “Binomial distribution-based quantitative measurement of multiple-acceptors fluorescence resonance energy transfer by partially photobleaching acceptor,” Appl. Phys. Lett. 104(24), 243706 (2014). 10.1063/1.4885084 [DOI] [Google Scholar]
- 25.Zal T., Gascoigne N. R. J., “Photobleaching-corrected FRET efficiency imaging of live cells,” Biophys. J. 86(6), 3923–3939 (2004). 10.1529/biophysj.103.022087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen H., Puhl H. L., 3rd, Koushik S. V., Vogel S. S., Ikeda S. R., “Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells,” Biophys. J. 91(5), L39–L41 (2006). 10.1529/biophysj.106.088773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koushik S. V., Blank P. S., Vogel S. S., “Anomalous surplus energy transfer observed with multiple FRET acceptors,” PLoS One 4(11), e8031 (2009). 10.1371/journal.pone.0008031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.J. R. Lakowicz, Principles of Fluorescence Spectroscopy (Springer, 2006), Chap. 2. [Google Scholar]
- 29.Wang L., Chen T., Qu J., Wei X., “Photobleaching-based quantitative analysis of fluorescence resonance energy transfer inside single living cell,” J. Fluoresc. 20(1), 27–35 (2010). 10.1007/s10895-009-0518-3 [DOI] [PubMed] [Google Scholar]
- 30.Rizzo M. A., Springer G. H., Granada B., Piston D. W., “An improved cyan fluorescent protein variant useful for FRET,” Nat. Biotechnol. 22(4), 445–449 (2004). 10.1038/nbt945 [DOI] [PubMed] [Google Scholar]
- 31.Nagai T., Ibata K., Park E. S., Kubota M., Mikoshiba K., Miyawaki A., “A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications,” Nat. Biotechnol. 20(1), 87–90 (2002). 10.1038/nbt0102-87 [DOI] [PubMed] [Google Scholar]