

Abstract
The “central dogma” of molecular biology describes how information contained in DNA is transformed into RNA and finally into proteins. In order for proteins to maintain their functionality in both the parent cell and subsequent generations, it is essential that the information encoded in DNA and RNA remains unaltered. DNA and RNA are constantly exposed to damaging agents, which can modify nucleic acids and change the information they encode. While much is known about how cells respond to damaged DNA, the importance of protecting RNA has only become appreciated over the past decade. Modification of the nucleobase through oxidation and alkylation has long been known to affect its base-pairing properties during DNA replication. Similarly, recent studies have begun to highlight some of the unwanted consequences of chemical damage on mRNA decoding during translation. Oxidation and alkylation of mRNA appear to have drastic effects on the speed and fidelity of protein synthesis. As some mRNAs can persist for days in certain tissues, it is not surprising that it has recently emerged that mRNA-surveillance and RNA-repair pathways have evolved to clear or correct damaged mRNA.
Keywords: 8-Oxoguanosine, O6-Methylguanosine, Oxidation, Alkylation, RNA surveillance, Translation, Ribosome, RNA damage
Introduction
Cellular fitness relies heavily on the ability of the cell to cope with mistakes resulting from biological processes being intrinsically imprecise and from exposure to a multitude of endogenous and exogenous insults. Damaging agents alter the chemical composition and, hence, the function of biomolecules, including DNA, RNA, protein, and lipids. Nucleic acids are specifically susceptible to chemical damage primarily due to the reactivity of the nitrogen and oxygen atoms of the nucleobase with a variety of chemicals. These chemical assaults include reactive oxygen species (ROS), ultraviolet light, and alkylating agents [1]. Curiously, certain species of damaged RNAs, typically oxidized, have been linked to a number of neurodegenerative disorders [2]. These observations suggest that the inability of the cell to clear damaged RNAs could contribute to disease or that certain diseases may interfere with cellular handling of damaged RNA.
Unlike naturally occurring modifications of specific nucleotides in rRNA and tRNA, unwanted modification resulting from chemical agents typically has deleterious effects on RNA’s function. Specifically, some modifications can prevent base pairing completely, while others alter the base-pairing properties of the modified nucleotide. Consistent with these ideas are recent discoveries showing that chemically damaged RNAs pose significant hurdles to translational fidelity and efficiency [3, 4]. Modifications interfere with the decoding process on the ribosome, whereby codon–anticodon interactions are disrupted and, depending on the type of damage, result in stalling or miscoding.
Accumulating evidence suggests that certain types of damaged RNA are selectively targeted for degradation. For instance, oxidized RNAs appear to turn over rapidly relative to intact RNA [5]. The exact details by which this selective degradation process operates are currently not fully understood, but it appears to involve the ribosome [4]. These models are largely based on the discovery of a number of ribosome-based-mRNA-surveillance mechanisms [6–8]. Recent studies have argued that certain quality control processes evolved to cope with chemically altered RNA [4, 9]. In addition, similar to some DNA repair machineries, certain RNA adducts appear to be repaired through direct-reversal strategies [10].
This review focuses on (1) the different types of chemical damage and their prevalence in health and disease; (2) how they impact the function of RNA; and (3) it describes the known and potential mechanisms that exist to handle modified RNA.
Types of damage and their prevalence in cells
Oxidative damage
Oxidative damage to RNA results from reactive oxygen species (ROS) reacting with the nucleobases. ROS is present under normal conditions as byproducts from metabolic reactions [11]. For instance, cellular respiration produces the superoxide radical O2− during electron transport as a result of molecular oxygen reduction by components of the electron transport chain [12]. O2− is also deliberately made by immune cells through the use of the enzyme NADPH oxidase to kill invading microbes [13]. Due to its toxic nature, organisms have evolved mechanisms to rid cells of O2−. Superoxide dismutase (SOD) catalyzes the metal-dependent dismutation of superoxide into O2 and hydrogen peroxide [14], which is then enzymatically reduced to water and molecular oxygen by catalase. If H2O2 is not eliminated immediately, it can react with intracellular iron through the Fenton and Haber–Weiss chemistry to form the highly reactive hydroxyl radical (·OH) [15]. In addition to superoxide and hydroxyl radicals, singlet-oxygen species 1O2 are also highly reactive and are known to oxidize nucleic acids. 1O2 is produced under photooxidative stresses and wounding [16]. Apart from these endogenous sources, multiple exogenous factors contribute to oxidative stress. These include ionizing and ultraviolet radiation [17] and toxic compounds, such as tobacco and certain drugs.
The reaction of ROS with nucleic acids results in a myriad of modifications [18]. Direct oxidation products include 8-oxo-7,8-dihydroguanosine (8-oxo-G), 8-oxo-7,8-dihydroadenosine (8-oxo-A), 5-hydroxyuridine (5-HO-U), and 5-hydroxycytidine (5-HO-C) (Fig. 1). In addition, during the course of 8-oxoG and 8-oxoA formation, intermediates can be modified to 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) and 4,6-diamino-5-formamidopyrimidine (FapyA) nucleosides, respectively [19]. Oxidation of cellular components can also lead to the production of other stably modified nucleobase products, for example, the etheno-adducts 1,N6-ethenoadenosine (ε-rA) and 3,N4-ethenocytidine (ε-rC) that form following lipid peroxidation [20]. Among these lesions, modification to the guanine base in the form of 8-oxo-G is notable. This is due to its high prevalence in DNA and RNA, presumably because of the intrinsic susceptibility of the guanine base to oxidation and, as importantly, a result of its drastic effect on base pairing [21]. In double-stranded DNA, unlike guanosine, which adopts the anti conformation, 8-oxoguanosine can adopt both the anti and syn conformations. In the syn conformation, 8-oxoguanosine base pairs with adenosine [22] (Fig. 2) and becomes mutagenic.
Fig. 1.
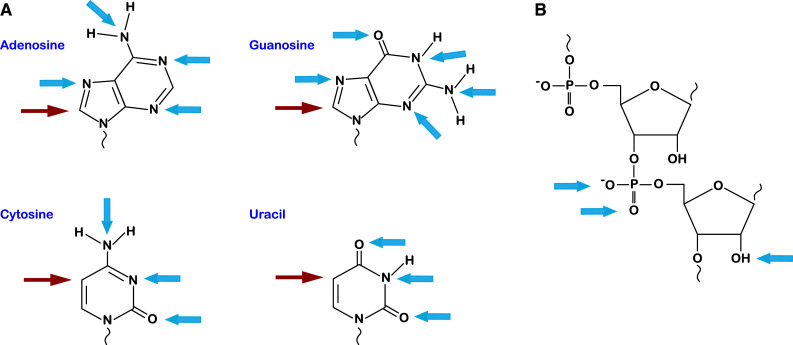
Targets of oxidative and alkylative damage on RNA. a Structures of the four RNA nucleobases with location of common oxidation sites marked (red arrows). Damage at these positions produces 8-oxo-7,8-dihydroadenosine, 8-oxo-7,8-dihydroguanosine, 5-hydroxycytosine, and 5-hydroxyuracil, respectively. Most of the nitrogen and oxygen atoms of the nucleobase are susceptible to alkylative damage (blue arrows). b The phosphodiester backbone and 2′-OH of the ribose are also targets for alkylation (blue arrows)
Fig. 2.

Damaged nucleobases exhibit altered base pairing. After oxidation, guanosine can still pair with cytosine, but is more likely to adopt a syn conformation and pair with adenosine (top). Aklylation of guanosine allows for efficient base pairing with uracil (bottom)
The quantification of modified nucleosides has traditionally relied on antibodies that specifically recognize the modified versus unmodified nucleoside. In the case of 8-oxoG, HPLC separation coupled with electrochemical detection has also been used successfully to quantify the modification in a number of samples [23]. More recently, HPLC/MS–MS has allowed for numerous modifications, including several damaged ones, to be catalogued [24]. Finally, genome-wide analysis is also becoming an option as affinity-based enrichment using antibodies together with next-generation sequencing can provide information about the location of modifications within hundreds of bases [25]. The ability to identify the specific context of the damaged site may provide some insight into how the modification affects gene regulation or other cellular responses.
Oxidized RNA in the form of 8-oxoG has been shown to accumulate, albeit to a low level, under normal physiological conditions, for which the modified nucleoside has been detected in human urine and red blood plasma [24, 26]. As would be predicted, measurements of 8-oxoG appear to correlate well with differences in metabolic rates. For instance, brains of old rats, which are characterized by mitochondrial dysfunction, show enhanced immunoreactivity with an antibody specific to 8-oxoG relative to young rats. The damage occurs predominantly in RNA and can be reversed by the addition of the antioxidants acetyl-l-carnitine and R-α-lipoic acid [27]. Furthermore, the presence of non-heme iron exacerbates mitochondrial dysfunction and contributes to oxidative damage through Fenton chemistry. Similar to aging brains, atrophic muscles resulting from aging or disuse accumulate oxidized RNA and not DNA, with the increase correlating to increased levels of free iron [28, 29]. Thus, the combination of reduced metabolic function and iron accumulation that occurs with age may have a profound effect on levels of RNA oxidation.
Some of the most convincing arguments about the prevalence and relevance of RNA damage in biology have come out of studies on neurodegenerative diseases. More than 15 years ago, several studies made the observation that brains of Alzheimer’s disease (AD) patients accumulate high levels of 8-oxoG in cytoplasmic and nuclear RNA, as detected by immunostaining after DNaseI treatment [30]. The level of 8-oxoG in the neuronal RNA of some of these patients is astonishingly high. In healthy individuals, less than 2 % of the total mRNA pool is immunoprecipitated by an anti 8-oxoG antibody; in AD patients, more than 50 % of the mRNA pool is immunoprecipitated [31]. These observations of elevated levels of 8-oxoG in cellular RNAs are not limited to AD patients. Indeed, in several neurodegenerative disorders [including Parkinson’s, dementia, and amyotrophic lateral sclerosis (ALS)], 8-oxoG levels in RNA are also significantly higher relative to healthy individuals [30–36]. What is clear from these studies is that neurons are especially prone to accumulation of oxidized RNA, which likely results from their high metabolic rate and in turn high levels of ROS. It is important to note that although the appearance of oxidized RNA appears to precede that of disease hallmarks, such as protein aggregation, whether it contributes directly to the pathogenicity of neurodegenerative disease states or is simply a consequence of disease is ambiguous. In some cases, the appearance of 8-oxoG in mouse models of disease can be delayed (through the addition of antioxidants, such as ascorbic acid) without affecting the onset of disease [35].
Alkylative damage
As described earlier, due to the nature of nucleic acids chemistry, RNA is also susceptible to alkylative damage (Fig. 1). Modifications have been documented to occur at nearly all of the nitrogen and oxygen atoms of the base, the phosphodiester backbone, and the 2′-OH of the ribose sugar [37]. Similar to oxidative damage, alkylation can result from endogenous agents; these include the universal methyl donor S-adenosyl methionine (SAM) and nitrosated bile acids. For instance, SAM has been shown to react with DNA in vitro to form a number of adducts [38]. Many of the prescribed chemotherapy agents, such as cyclophosphamide, streptozotocin, and Temodar, are alkylating agents that are known to target RNA in addition to DNA [39]. These agents selectively kill fast growing cells, because these cells do not have time to repair their DNA. Nevertheless, the observations that these agents also damage RNA suggest that RNA repair may play a role in cancer biology and is likely to be relevant to the survivability of healthy cells and tumors. Other exogenous agents include the highly mutagenic chemicals methylnitronitrosoguanidine (MNNG) and ethyl methanesulfonate (EMS) [40].
It should be noted that although some of the modifications to RNA do not alter the Watson–Crick face of the nucleobase, they could still have profound effects on the function of the RNA molecule by altering non-canonical base pairing or RNA–protein interaction. In purines, for example, N7 is especially reactive and is readily methylated. In contrast to DNA, for which the modification, beyond its accelerated rate of depurination, is not toxic [41], in RNA, N7 methylation interferes with Hoogsteen base pairing and how certain proteins recognize RNA. In addition to N7 methylation, modifications to the Watson–Crick face of the nucleobase are expected to be detrimental to RNA function [42, 43] similar to analogous studies on DNA replication. It is worth remembering that the list of possible modifications is extensive due to the diverse nature of the damaging agents (many groups can be added, from methyl to bulky aromatic groups) as well as the atom of the nucleobase on which these groups can be added. On adenosine (besides N7) N1, N3 and N6 can be modified; modifications to N1 and N3 are especially problematic, because they interrupt base pairing and/or the geometry of the minor groove [44, 45]. On guanosine (again besides N7) N1, N2, N3, and O6 can be modified and all in principle affect base pairing. Alkylation to O6 is especially notable, because it is potently mutagenic where O6-methylguanosine (O6MeG) readily base pairs with uracil instead of cytosine [46] (Fig. 2). On uracil, O2, N3, and O4 are readily modified; similar to O6MeG, O4MeU is highly mutagenic and base pairs with guanosine [47]. On cytosine, O2, N3, and N4 can be modified with modifications to O2 and N3 being potentially deleterious.
In contrast to oxidative damage, the extent of alkylated RNA accumulation under normal physiological conditions or in disease states has not been the subject of many studies. Nevertheless, more than 20 years ago, it was shown that rat hepatocyte cells treated with N-nitroso compounds, such as N-nitrosodimethylamine accumulate O6MeG in the cytoplasm as assessed by immunostaining [48]. As expected, the signal is sensitive to RNase treatment but not to DNase treatment. Furthermore, many of these adducts discussed earlier also accumulate in RNA when cells are treated with the mutagens methyl methanesulfonate (MMS) and 1-methyl-1-nitrosourea (MNU); some of these accumulate to levels that are more than fivefold higher than their counterparts in DNA [49]. What is even more interesting about these observations is the fact that alkylating agents are commonly used in chemotherapy, suggesting that the process by which cells cope with RNA damage might be relevant to the prognosis of cancer patients. Indeed, the chemotherapy drugs cisplatin, 5-fluorouracil, and doxorubicin (with the caveat that they follow diverse modes of action and do not alkylate RNA directly) to a certain extent appear to rely on RNA damage for their efficacy. Cisplatin forms cross-links on ribosomes, inhibiting translation [50]; 5-fluorouracil is incorporated into RNA, which appears to be one of the determinants for its cytotoxicity [51, 52]; and doxorubicin intercalates into RNA helices affecting the function of many RNAs [53]. Many of these agents, and in turn RNA damage, are known to elicit apoptosis through p53 activation [54], suggesting that alkylative damage to RNA is a real threat and cells have evolved systems to sense it.
RNA susceptibility to damage
In many of the studies described earlier, a particular damage to a nucleobase accumulates in RNA to levels that are much higher relative to the equivalent one in DNA [30, 55, 56]. This of course cannot be explained simply by inherent differences in chemical reactivities between the two polymers. Instead, the final structure, packaging, differential decay, and localization appear to be the main determinants for the observed disparity. For the most part, RNA exists in a single-stranded form, exposing the Watson–Crick face of the nucleobase, which is typically protected in double-stranded DNA. In addition, unlike DNA, which in eukaryotes is wrapped tightly in chromatin, RNA’s association with proteins is much less extensive. Consistent with these arguments, the extent of RNA damage varies greatly between different types of RNA in a manner that appears to correlate with the extent of protein association for the particular type of RNA [57]. For example, polyA-RNA was found to have levels of 8-oxoG that are almost fivefold higher than those measured in total RNA [4], which consists of mostly rRNA. That being said, even among different mRNAs, there are significant differences in the amount of damage [31]. Certain mRNAs are much more prone to oxidation relative to others, an effect that has no relation to transcript abundance [35, 58, 59]. In principle, this effect could be due to differences in sequence context; oxidation of guanosine depends critically on its neighbors [60]. Additional effects are likely due to differences in the structure, translation speed, and association with RNA binding proteins among different mRNAs.
In addition to structural and packaging differences between the polymers, it is reasonable to assume that cells repair damage from DNA much more rapidly than from the RNA pool. Finally, RNA appears to be especially susceptible to oxidation due to its localization in the cytoplasm in close proximity to the mitochondria, where ROS concentrations are much higher. In agreement with this proposal, mitochondrial DNA accumulates higher levels of oxidized nucleotide relative to nuclear DNA [61]. This is the very same argument that could be used to explain why exogenous ROS react more efficiently with cellular RNAs; these agents cannot enter the nucleus without passing first through the cytoplasm.
The effect of RNA damage on function
Damage to non-coding RNA
Non-coding RNAs account for the majority of RNA species in the cell and, therefore, may be expected to accumulate the bulk of oxidative damage. Ribosomal RNA (rRNA) is generally thought to be a poor target, given its complexity of folding and association with ribosomal proteins; however, under oxidative stress conditions, this may not be the case. In contrast to measurements under normal conditions, where 8-oxoG from rRNA is lower compared with that of total RNA, after treatment with H2O2, rRNA oxidation in E. coli correlated with the amount of 8-oxoG in the cell [62, 63]. The effects of oxidative stress on rRNA have also been observed in regions of the brain affected by AD. The total amount of rRNA is reduced in diseased tissue and AD patients’ cells carry higher amounts of 8-oxoG in rRNA than cells from healthy individuals [64, 65]. Interestingly, ribosomes collected from AD-affected neuronal cells are oxidized and are associated with higher levels of redox-active iron, suggesting that rRNA is particularly vulnerable to oxidation in the presence of Fe(II) [66].
One could predict that oxidative damage to functional domains of rRNA would affect translation in a variety of ways (Fig. 3). First, damage to residues that are required for folding could inhibit ribosome assembly, resulting in non-functional subunits and requiring a mechanism for their removal. Second, any modification to the decoding center on the ribosome could interfere with codon–anticodon interaction or with binding of aa-tRNA, causing expression of miscoded proteins or stalled ribosomes. Furthermore, if residues required for binding of EF-G or GTP are damaged, it would be expected to block elongation of the nascent peptide and potentially stall the ribosome. Similarly, modification of the peptidyl transferase center (PTC) could interfere with binding of P- or A-site tRNAs and stall translation, potentially creating truncated proteins that could accumulate or form aggregates detrimental to the cell.
Fig. 3.

RNA damage affects cellular fitness through multiple mechanisms. Damage to mRNA, rRNA, or tRNA could lead to failed peptide synthesis by miscoding, stalling, or mRNA turnover. Damage to rRNA could cause defects in ribosomal assembly or crosslinking with ribosomal proteins. tRNA damage may cause aminoacylation defects, potentially leading to production of miscoded proteins
Oxidative stress is also known to affect tRNAs. When tRNAs are treated with H2O2 in vitro, they accumulate 8-oxoG to the same extent when folded as when denatured, suggesting that their structure is not protective [62]. Damage to tRNA would potentially affect its function in a number of ways (Fig. 3); any modification to residues of the anticodon could interfere with codon–anticodon pairing and lead to miscoding or production of truncated proteins. Likewise, damage to other sites could affect amino-acylation and result in an accumulation of incompetent or misacylated tRNA that could then go on to add incorrect amino acids to the nascent polypeptide. In the event that a tRNA (or a rRNA) is damaged, such that it acquires a new activity or change in substrate preference, it could then promote miscoding on any newly synthesized protein and potentially promote the expression of a mutated protein that could be deleterious to the cell. tRNA cleavage has also been observed during oxidative stress, and a number of studies have indicated that the cleavage products have roles in promoting stress responses, including the formation of stress granules [67–69].
Recently, Wang et al. showed that even small RNAs can be damaged by ROS and identified a role for oxidative damage in modifying miRNA activity [70]. They reported that upon oxidative damage to miR-184, the sequence that it targets changes, leading to mismatched binding to Bcl-xL and Bcl-w 3′UTRs. This results in reduced expression of these genes and subsequently to apoptosis in rat heart cells. Apparently, small RNA species are not immune to ROS-generated damage, though how this relates to functional changes remains to be determined.
Damage to mRNA
A number of studies have looked at the effects of oxidative damage and the correlation between oxidized mRNA and translational efficiency. Several years ago Shan et al. showed that transcripts with higher levels of 8-oxoG result in reduced protein expression [58]. Studies by the same group also showed that in vitro oxidized reporter mRNAs, when introduced into cultured mammalian cells, produced less functional protein than intact mRNAs under conditions where the transcript levels did not change [59]. The resulting proteins were found to form aggregates, presumably due to misfolding, which could result from miscoding or premature termination on the oxidized transcript. Later studies by Stadtman et al. showed that oxidized mRNA associates with polysomes, but the yield of full-length protein product is decreased [71]. The same study also revealed that inhibition of the proteasome stabilizes short protein products that potentially result from premature termination. While these studies highlight the unwanted consequences of oxidation on mRNA function, it is worth nothing that due to the nature of the assays used, they reveal very little about the mechanism by which the damage affects translation. In addition to the multitude of adducts a crude oxidation treatment may introduce into the transcript, the precise location and identity of the adducts are not known. To examine the effect that 8-oxoG-containing mRNA has on translational efficiency, our laboratory recently took a reconstituted approach to assess decoding by the ribosome.
One might expect that, similar to DNA polymerization, 8-oxoG would cause miscoding due to its ability to pair with A. However, when an 8-oxoG is present in the A-site codon, rates of peptide-bond formation for cognate aminoacyl-tRNA (aa-tRNA) are about four orders of magnitude slower relative to those measured on a non-oxidized RNA; an event that would be expected to stall the ribosome [4]. Furthermore, the presence of the adduct resulted in a similar reduction in rate regardless of its position within the codon. These findings are reminiscent of those using in vitro oxidized RNA in extracts and cell culture [71]. Likewise, when we introduced a short mRNA containing a single 8-oxoG into eukaryotic extracts, shorter protein products were produced—of the size expected if the ribosome were stalled at the damaged base—compared with an intact mRNA. Interestingly, the response to a damaged base in a stop codon was only subtly different from the intact one—rates of peptide release were marginally affected, indicating that the interaction between the damaged codon and the proteinaceous release factor was insensitive to the change in chemistry.
What about other adducts? How might they affect the translational machinery? Recently, our laboratory explored the effects of the highly mutagenic base O6MeG on the decoding process. Similar to what has been observed with DNA polymerases, when an O6MeG residue is positioned in the A-site codon of an RNA, the ribosome readily incorporates incorrect aa-tRNAs by forming O6MeG-uridine codon–anticodon pairs. Conversely, at the second position of the codon, O6MeG was found not to promote miscoding, but instead slowed the observed rates of peptide-bond formation by >1000-fold for correct aa-tRNAs, without altering the rates for incorrect aa-tRNAs. It turns out that the effects of O6MeG are due to inhibition of the GTPase activation step by elongation factor EF-Tu, a key step in the early phase of tRNA selection. Interestingly, the related modified nucleotide N6-methyladenosine (m6A) has only a modest effect on decoding when placed at the second position of the codon, suggesting that the effects on tRNA selection are not merely due to the introduction of a methyl group but rather due to altered geometry of the base pair [3]. This strongly suggests that the decoding center of the ribosome is extremely sensitive to changes to the second position of the codon–anticodon. These studies are beginning to shed some important insights into the ribosomal response to damaged mRNAs and how different adducts and their position within the reading frame is deciphered by the translational machinery. These findings, in turn, are providing potential clues about the cellular handling of damaged mRNAs.
Quality control of damaged RNA
rRNA quality control
As mentioned earlier, the vast majority of cellular RNA is composed of rRNAs. These species of RNA tend to be long lived, and in some organisms, their half-life is several days [72]. Therefore, damaged ribosomes can persist if cells do not have the means to recognize and clear them. This becomes especially problematic if the modification profoundly affects the function of the RNA. While it is clear that chemical damage accumulates in ncRNAs, the extent to which cells recognize chemically damaged ribosomes and target them for degradation is currently unknown. Nevertheless, two pathways that degrade aberrant rRNAs have been described; these have been studied in the context of mutations to functional sites [73, 74].
Non-functional rRNA decay (NRD), originally described by the Moore lab, targets defective 25S rRNA of the large subunit. In contrast to bacteria, for which mutations in the peptidyl transferase center (PTC) of the ribosome are dominant lethal, yeast expressing PTC mutations in the background of wild-type endogenous rRNAs are viable [74, 75]. It turns out that eukaryotes possess a way of selectively ridding the cells of non-functional ribosomes, so that they do not initiate translation. Similarly, mutations that affect the decoding center of the ribosome in the 18S rRNA are subject to quality control [73]. It is worth noting that mutant 18S and 25S rRNAs are targeted for degradation post processing and assembly of the ribosomal subunits, as judged by sucrose-gradient fractionation [73]. However, defective rRNA turns over much faster than its wild-type counterpart, leading to almost an order of magnitude reduction in their steady-state levels [74]. Interestingly, although mutations in the decoding center or PTC both result in defective ribosomes, the manner by which cells appear to degrade the respective 18S and 25S rRNAs is distinct. In contrast to 25S NRD, 18S NRD is inhibited by cycloheximide, suggesting that it is dependent on translation. Furthermore, 18S NRD requires factors that are involved in mRNA-surveillance [73]. Defective 18S RNA is significantly stabilized in the absence of the yeast Dom34p, Hbs1p, and Ski7p, which are involved in no-go decay (NGD) and non-stop decay (NSD) of mRNAs (see below). These factors are responsible for the disassembly of stalled ribosomal complexes and likely lead to downstream events that degrade the rRNA [73]. 25S NRD does not require NGD or NSD factors, and is not inhibited by cycloheximide. In addition, whereas defective 18S rRNA is distributed throughout the cytoplasm, defective 25S rRNA localizes to perinuclear foci [74]. Hence, 25S NRD appears to follow 60S assembly but precedes the formation of the 80S ribosome. Furthermore, 25S NRD requires Mms1p and Rtt101p subunits of an E3 ligase complex that is likely to be involved in the ubiquitination of ribosomal proteins [76].
What are the physiologically relevant targets of NRD? So far, studies on this process have focused on mutants that disrupt functionally important sites of the ribosome. The extent to which these defective ribosomes are the real targets of this pathway is not likely to warrant its existence. Instead, many in the scientific community have successfully argued that NRD may have evolved to recognize chemically damaged ribosomes [77]. Consistent with these arguments, yeasts lacking 18S NRD factors Dom34p and Hbs1p are sensitive to oxidative stress and alkylative agents, such as MMS [78]. Interestingly, the 25S NRD factors Mms1p and Rtt101p are involved in DNA repair [79] and are sensitive to nucleic acid damaging agents [80], suggesting an overlap between RNA and DNA quality control processes. Indeed, recent reports have also implicated a number of base excision repair (BER) factors in RNA quality control, especially during ribosome biogenesis (for review see [81]). For example, the main eukaryotic apurinic/apyrimidinic endonuclease APE1, which is responsible for creating a nick in DNA at abasic sites, has an endonucleolytic activity on abasic RNA as well as different types of damaged RNA [82]. In addition to its potential role in RNA quality control, APE1 also appears to regulate gene expression by cleaving RNA targets, such as c-myc mRNA [83]. In the cell, APE1 has been shown to associate with rRNA species in the nucleolus, where it is thought to be involved in maintaining the integrity of ribosomes through rDNA repair and damaged-rRNA molecule removal [84]. Upon oxidation stress, the factor relocates from the nucleolus into the nucleoplasm. These observations suggest that under normal conditions, the factor is involved in rRNA quality control, whereas under stress conditions, it is involved in DNA repair [84]. In addition to APE1, the uracil glycosylase SMUG1 has been shown to interact with the pseudouridine synthase DKC1 [85]. DKC1 modifies uridines to pseudouridines in rRNA and is required for producing functional ribosomes. The depletion of SMUG1 is accompanied by a reduction in mature rRNA species, suggesting that the factor is involved in ribosome biogenesis perhaps through rRNA-quality control [85]. Consistent with these models, in vitro SMUG1 is active on RNA molecules harboring the modified base 5-hydroxymethyluracil [85]. How these enzymes switch substrates (from DNA or RNA) is not fully understood, but is likely to be regulated through protein partners. In the future, it will be interesting to explore the role of other DNA repair enzymes in RNA metabolism.
tRNA quality control
Similar to rRNA, tRNAs are long lived and play a central role in deciphering the genetic code. Furthermore, tRNAs have been documented to accumulate chemical damage (see above), which, in most instances, is likely to adversely affect their function. Much of our understanding of the ability of cells to recognize defective tRNAs and target them for degradation have come out of studies focusing on misprocessed, mutant, and hypomodified tRNAs. Pre-tRNAs undergo extensive maturation and processing following their synthesis by RNA polymerase III [86]. Defects in these processes are recognized by the TRAMP complex; Trf4p, a component of the TRAMP complex, polyadenylates the pre-tRNAs, targeting them for degradation by the nuclear exosome [87–90]. Mature tRNAs are also subject to quality control often referred to as rapid tRNA decay (RTD) [91]. For instance, tRNAs lacking post-transcriptional modifications are subject to 5′–3′ degradation in the nucleus by Rat1p and in the cytoplasm by Xrn1p [92]. The pathway involves the addition of a second CCA to the 3′-end of tRNAs with a weakened acceptor stem (as a result of hypomodification or mutation) by CCA-adding enzyme [93]. The additional extension on the 3′-end of the tRNAs is likely to facilitate 3′–5′ degradation by the exosome.
Interestingly, RTD is exquisitely sensitive to the modification status of the tRNA—an effect on the overall stability of the tertiary structure of the molecule [94]—suggesting that the process is likely to be responsible for also recognizing chemically damaged tRNAs. Many of the adducts described earlier, including 8-oxoG, which accumulates in tRNAs, have profound effects on the base pairing properties of RNA. By preventing base pairing or altering it, the overall structure of the damaged tRNA is likely to change, weakening the acceptor stem and, hence, allowing access for exonucleases to degrade the damaged tRNAs. Consistent with these ideas, deletion of some of the RTD factors discussed earlier renders yeast sensitive to nucleic acids damaging agents [4]. Furthermore, the TRAMP complex, which is involved in nuclear RNA-surveillance processes, is also required for maintenance of genome integrity [95], again providing a possible link between RNA and DNA metabolism as the cell responds to damaging agents.
Ribosome-independent quality control of damaged mRNA
Damaged mRNA appears to turn over rapidly relative to intact mRNA [5], suggesting that cells evolved pathways for the recognition and subsequent degradation of damaged mRNAs. Some of these processes are likely to involve the ribosome for initiating recognition (see below); this makes sense, as the ribosome is the only cellular complex that scans all mRNAs (at least through the coding region). Nevertheless, accumulating evidence suggests additional participation by ribosome-independent factors in mRNA quality control processes (reviewed in [1]). Of course, these pathways would involve direct recognition of adducts without assessing their effects on Watson–Crick base pairing. This is in contrast to how damage is typically recognized in DNA, for which most of the repair pathways rely heavily on the effect of the damage on base pairing and the geometry of the B helix. That being said, certain DNA repair enzymes, such as DNA-uracil glycosylase, remove damaged bases by having an active site tailored for the modified base [96]. Early studies aimed at the isolation of factors that might be involved in recognition of oxidized RNAs identified two factors: the human Y box-binding protein 1 (YB-1) and the bacterial polynucleotide phosphorylase (PNPase) [97, 98].
In vitro, YB-1 was shown to specifically bind 8-oxoG containing RNA suggesting a possible role for the factor in degrading oxidized RNA [98]. Consistent with these observations, overexpression of YB-1 in E. coli was found to confer resistance against paraquat, an inhibitor of electron transfer that increases ROS [98]. Curiously, under normal conditions, YB-1 stabilizes mRNAs by binding to the cap structure in the absence of protection by eIF4E and prevents decapping [99], which normally initiates the degradation process of mRNAs. Indeed depletion of YB-1 results in global destabilization of mRNAs [99]. Based on this, the perceived YB-1-induced protective phenotype against oxidative stress was explained through a sequestration mechanism [98], for which the protein prevents the translation of 8-oxoG-containing mRNAs. However, following arsenite-induced oxidative stress, YB-1 translocates from mRNA processing bodies (P-bodies) to stress granules [100]. These structures are proposed to provide a protecting environment for RNA during stress. From this, it is unclear whether YB-1 is directly involved in quality control of oxidized RNA. It is possible that YB-1 binding to oxidized mRNA is different from its typical role in RNA metabolism; for instance, upon binding to 8-oxoG-containing RNAs, it may recruit other factors to degrade the RNAs.
In E. coli, PNPase, a 3′–5′ exonuclease, has been reported to specifically target oxidized RNA over intact RNA through increased affinity toward 8-oxoG [101]. Its human counterpart has also been reported to preferentially bind oxidized RNA [102], and its depletion results in increased levels of 8-oxoG. However, a direct role for the factor in targeted degradation of oxidized RNA has yet to be documented [102]. Furthermore, in mammals, the factor is localized to the intermembrane space of the mitochondria, where it appears to function in maintaining mitochondrial homeostasis and has little to no role in RNA degradation in vivo [103].
Sanitation of the nucleotide pool
In addition to post-synthesis alteration, RNA can also modified through the incorporation of damaged free nucleotides. These monomers are susceptible to the same damaging agents as the RNA polymer, and if these are not cleared from the nucleotide pool, these can be incorporated during transcription. In addition to potentially interfering with the function of the RNA by affecting its folding, some of these modified NTPs are mutagenic and cause errors during transcription [104]. It is not surprising then that cells evolved strategies to clear damaged NTPs.
One of the best-studied NTP-sanitizing processes is the cellular handling of 8-oxoGTP. The presence of 8-oxoG in mRNA is detrimental to translation [4]. Furthermore, RNA polymerase incorporates 8-oxoGMP opposite to adenosine causing mutations in the RNA transcripts [104]. To deal with these deleterious effects of 8-oxoGTP, the E. coli MutT protein—a member of the Nudix (nucleoside diphosphate linked to X) family—catalyzes the hydrolysis of 8-oxoGTP to 8-oxoGMP [105]. The human MutT-like protein MTH1 serves a similar role, and the resulting 8-oxoGMP cannot be activated back to 8-oxoGTP, because gunaylate kinase (the enzyme responsible for these types of reactions) does not recognize the oxidized nucleotide monophosphate [105]. Humans have at least one more enzyme that degrades 8-oxoGTP; the NUDT5 protein hydrolyzes 8-oxoGTP to 8-oxoGDP [106]. The contributions of these MutT homologues in maintaining the integrity of RNA are best exemplified by the observation that deletion of MTH1 results in the accumulation of 8-oxoG in RNA and DNA in the hippocampal microglia of rats following excitotoxic-induced oxidative stress. In wild-type animals, excitotoxic-induced oxidative stress also results in elevated levels of MTH1 mRNA [107]. This suggests that oxidized NTPs are detrimental to cellular fitness due to their possible effect on RNA and, as a result, cells evolved specific processes to sanitize the nucleotide pool to prevent them from being incorporated during transcription. In the future, it will be interesting to explore the role of other NUDIX family members in the recognition of other damaged RNA precursors.
Damaged-RNA quality control through mRNA-surveillance pathways
The above pathways highlight some of the processes that may have evolved for the specific recognition and degradation of one type of RNA damage. However, cells utilize general mRNA-surveillance pathways to target aberrant mRNAs, typically with common features, for degradation [6–8]. Eukaryotic cells are known to have at least three cytoplasmic RNA-quality control processes: nonsense-mediated decay (NMD), no-go decay (NGD), and non-stop decay (NSD). It is worth noting that these processes have the commonality of taking advantage of the ribosome to initiate the recognition process. NMD targets transcripts with a premature stop codon for rapid degradation. The process is well conserved and utilizes translation factors as well as Upf proteins, which together recognize some poorly understood features of premature stop codons before the degradation process is initiated [7]. NGD targets transcripts that stall the ribosome, which classically included strong secondary structure containing transcripts, truncated ones and rare-codon-containing transcripts [108]. In a process dependent on translation, NGD transcripts are endonucleolytically cleaved before they are degraded by the general decay machinery: 5′–3′ Xrn1-dependent degradation and 3′–5′ exosome-dependent degradation. We note that, unless degraded, NGD targets are especially problematic for the cell, because they sequester valuable ribosomes from the translating pool. Dependent on the length of the transcript, tens of ribosomes could be removed from active translation. As a result, in addition to targeted-RNA degradation, NGD employs factors that rescue ribosomes through dissociation of the subunits and eventual recycling; in yeast, these factors are Dom34p, Hbs1p, and Rli1p [109–111]. Finally, NSD targets transcripts with no stop codon. The process resembles NGD in that transcripts are cleaved as a result of ribosomes stalling on the poly-Lys encoding polyA-tail [112, 113]. The similarities are reinforced by common requirements for similar factors [114], with one exception in yeast for which the process involves the non-conserved protein Ski7p [112, 113].
A potential role for mRNA-surveillance mechanisms in quality control of damaged mRNA (e.g., oxidized RNA) has not been studied in detail. Nonetheless, studies are beginning to uncover a direct link between mRNA-surveillance and chemical damage [4, 9]. Out of the three known processes, NGD is the most suitable for detecting chemical damage. While this process was initially described in the context of roadblocks to the translational machinery, such as long hairpins, many of these hurdles are artificial and are not likely to constitute a natural target. Indeed, the process appears to be triggered by a reduction in the rate of protein synthesis that results from certain types of chemical damage [4]. This is consistent with what we know about the effects of certain modifications on base pairing. Inhibition of base pairing has a dramatic effect on codon–anticodon interactions during tRNA selection by the ribosome, essentially stalling protein synthesis. The first clues about the role of NGD in clearing damaged RNA came out of studies on virus-mediated depurination of RNA. In these studies, enzymatic depurination of RNA decreased its half-life from ~3 to ~2 h in yeast, and the deletion of dom34 and xrn1 restored its half-life [9]. These studies suggest that NGD targets depurinated RNA. Chemical-induced depurination (through hydrolysis of the glycosidic linkage on adenosines and guanosines) is one of the most common damages in DNA [115] and is expected to occur in RNA. The resulting abasic sites cannot base pair with the anticodon, hence stalling translation. More recent work from our laboratory has also suggested that 8-oxoG-containing RNA is subject to NGD [4]. In vitro, 8-oxoG-containng RNAs stall translation making them likely targets for the process (Fig. 4). In vivo, 8-oxoG-contaning RNAs accumulate in the absence of Dom34p and Xrn1p. Therefore, emerging from these studies is the realization that NGD may have evolved to degrade chemicallydamaged RNA.
Fig. 4.

Model for No-go decay in response to stalling by a damaged mRNA. a Translating ribosome stalls with a damaged codon in the A-site. b Ribosome rescue factors, Dom34p and Hbs1p in yeast, are recruited to the stalled ribosome, and mRNA is endonucleolytically cleaved. c The resulting 3′-fragment is degraded by the 5′–3′ exonuclease Xrn1p, whereas the 5′-fragment is degraded through the action of the cytoplasmic exosome in a 3′–5′ direction
Direct repair of RNA
So far, our discussion has focused on processes that target defective RNAs and its precursors for degradation. Although these degradative processes are likely to constitute the most typical pathway for quality control of damaged mRNA, cells appear to have evolved at least one additional mechanism to cope with chemically modified RNA. Direct reversal of certain types of alkylation adducts is a strategy shared by common DNA- and RNA-repair mechanisms. As predicted, unlike most DNA-repair pathways, these mechanisms do not require a complementary strand for repair (for obvious reasons, these cannot be used to repair RNA). In vitro, certain homologues of the bacterial AlkB oxidative demethylase homologues repair RNA, or single-stranded DNA, almost exclusively [10]. These enzymes catalyze a molecular oxygen/Fe(II)/α-ketoglutarate-dependent oxidation reaction to hydroxylate the alkyl adducts (hydroxymethyl for methyl adducts) attached to a nitrogen atom to produce CO2 and succinate [10]. The reaction is completed by the dissociation of an aldehyde (formaldehyde, for example, from methyl adducts) from the unstable carbinoliminium, yielding an unmodified repaired base (reviewed in [116, 117]). Different members of the family have been shown to prefer diverse substrates, including tRNA, mRNA, and proteins, where they correct erroneous nucleobase substitutions or affect regulatory modifications on RNA and protein, although the details of their mechanism of action and its relevance to cellular function are not yet clear [118].
The bacterial AlkB protein, which is the most well-studied enzyme of the group, and its human homologue hABH3 have a preference for repairing RNA in vitro. Supporting a role for these enzymes in RNA repair in cells, both were shown to reactivate alkylated MS2 RNA phage in vivo [10]. In addition, the function of alkylated tRNA molecules is restored upon incubation with these enzymes [43]. It is worth noting that the real RNA targets for AlkB/hABH3 under normal conditions are currently unclear; certain species of RNAs are repaired much more efficiently than others. For example, AlkB repairs mRNAs much more efficiently than tRNAs [43]. We note that others in the field have argued that the RNA repair function of these enzymes may be irrelevant in vivo, where they associate with DNA-binding proteins, increasing their affinity to damaged DNA and suggesting that they are DNA-repair enzymes instead of RNA ones [119]. These latter arguments, however, cannot explain the observation that numerous single-stranded plant-infecting RNA viruses encode an AlkB homologue that has robust RNA-repair activity in vitro and in vivo [120]. Therefore, direct RNA-repair by an AlkB-like protein is likely to be a common strategy utilized by cells to protect them from alkylation damage. In RNA viruses, this process is likely to be critical for maintenance of genome integrity and stability, which may have been be relevant in early RNA-based organisms.
Concluding remarks
Recently, it has become evident that RNA damage occurs both through modification by exogenous agents as well as from byproducts of physiological processes. The effects of damage on RNA function have been observed in a number of settings, with a reduction in translational efficiency and potential for miscoding being a common outcome. Furthermore, the presence of accumulated oxidized RNA in degenerative diseases provides correlative evidence that these damages could be either a precursor to disease or an effect of disruption of cellular processes. It will be important to determine the details of how these effects may relate to RNA damage and aberrant protein synthesis. The cell has several mechanisms to rid itself of non-functional RNAs, and it appears likely that at least some of these may have evolved in response to a need to deal with RNA damage. Most of these surveillance mechanisms involve RNA degradation but, in the case of alkylative damage, repair may be an option as well. It will be interesting to see whether other types of damage elicit additional uncharacterized responses and what other factors may be involved.
The fields of RNA repair and quality control are relatively new, and we are only just starting to learn about the details of these pathways and their role in maintaining cellular homeostasis. For instance, it is currently not understood how the cell chooses which process to use to deal with certain adducts. Furthermore, little is known about how the ribosome-associated quality control machinery distinguishes between defective RNA molecules or programmed pausing that is used to control gene expression. The observation that certain AlkB homologues have unique substrate specificity for certain RNA species also begs the question of how the proteins recognize their RNA substrates. For example, analogous to transcription-coupled repair during DNA replication, are some of these factors ribosome associated and use the translation machinery to scan the mRNA pool? In addition to oxidative demethylation, are there are other mechanisms in place that are utilized to repair RNA? Finally, similar to cellular responses that are tightly coupled to DNA damage, it is feasible that cells have built in sensors, yet to be discovered, that are in place to detect environmental insults through RNA damage. Such a response system, for example, might be triggered by elevated ribosome stalling. This is likely to be an unappreciated pathway that is relevant to many of the chemotherapeutic agents that are designed to damage DNA but clearly have a profound effect on the integrity of RNA.
Acknowledgments
Research in the Zaher laboratory is supported by the National Institutes of Health (R01GM112641) and the Searle Scholars Program. We thank the members of the laboratory for comments and helpful discussions on the manuscript.
References
- 1.Wurtmann EJ, Wolin SL. RNA under attack: cellular handling of RNA damage. Crit Rev Biochem Mol Biol. 2009;44:34–49. doi: 10.1080/10409230802594043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nunomura A, Moreira PI, Takeda A, Smith MA, Perry G. Oxidative RNA damage and neurodegeneration. Curr Med Chem. 2007;14:2968–2975. doi: 10.2174/092986707782794078. [DOI] [PubMed] [Google Scholar]
- 3.Hudson BH, Zaher HS. O6-Methylguanosine leads to position-dependent effects on ribosome speed and fidelity. RNA. 2015;21:1648–1659. doi: 10.1261/rna.052464.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simms CL, Hudson BH, Mosior JW, Rangwala AS, Zaher HS. An active role for the ribosome in determining the fate of oxidized mRNA. Cell Rep. 2014;9:1256–1264. doi: 10.1016/j.celrep.2014.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hofer T, Badouard C, Bajak E, Ravanat JL, Mattsson A, Cotgreave IA. Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol Chem. 2005;386:333–337. doi: 10.1515/BC.2005.040. [DOI] [PubMed] [Google Scholar]
- 6.Graille M, Seraphin B. Surveillance pathways rescuing eukaryotic ribosomes lost in translation. Nat Rev Mol Cell Biol. 2012;13:727–735. doi: 10.1038/nrm3457. [DOI] [PubMed] [Google Scholar]
- 7.Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shoemaker CJ, Green R. Translation drives mRNA quality control. Nat Struct Mol Biol. 2012;19:594–601. doi: 10.1038/nsmb.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gandhi R, Manzoor M, Hudak KA. Depurination of Brome mosaic virus RNA3 in vivo results in translation-dependent accelerated degradation of the viral RNA. J Biol Chem. 2008;283:32218–32228. doi: 10.1074/jbc.M803785200. [DOI] [PubMed] [Google Scholar]
- 10.Aas PA, Otterlei M, Falnes PO, Vagbo CB, Skorpen F, Akbari M, Sundheim O, Bjoras M, Slupphaug G, Seeberg E, Krokan HE. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003;421:859–863. doi: 10.1038/nature01363. [DOI] [PubMed] [Google Scholar]
- 11.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 13.Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest. 1973;52:741–744. doi: 10.1172/JCI107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pick M, Rabani J, Yost F, Fridovich I. The catalytic mechanism of the manganese-containing superoxide dismutase of Escherichia coli studied by pulse radiolysis. J Am Chem Soc. 1974;96:7329–7333. doi: 10.1021/ja00830a026. [DOI] [PubMed] [Google Scholar]
- 15.Koppenol WH. The Haber–Weiss cycle—70 years later. Redox Rep. 2001;6:229–234. doi: 10.1179/135100001101536373. [DOI] [PubMed] [Google Scholar]
- 16.Clo E, Snyder JW, Ogilby PR, Gothelf KV. Control and selectivity of photosensitized singlet oxygen production: challenges in complex biological systems. ChemBioChem. 2007;8:475–481. doi: 10.1002/cbic.200600454. [DOI] [PubMed] [Google Scholar]
- 17.Ravanat JL, Douki T, Cadet J. Direct and indirect effects of UV radiation on DNA and its components. J Photochem Photobiol, B. 2001;63:88–102. doi: 10.1016/S1011-1344(01)00206-8. [DOI] [PubMed] [Google Scholar]
- 18.Barciszewski J, Barciszewska MZ, Siboska G, Rattan SI, Clark BF. Some unusual nucleic acid bases are products of hydroxyl radical oxidation of DNA and RNA. Mol Biol Rep. 1999;26:231–238. doi: 10.1023/A:1007058602594. [DOI] [PubMed] [Google Scholar]
- 19.Gajewski E, Rao G, Nackerdien Z, Dizdaroglu M. Modification of DNA bases in mammalian chromatin by radiation-generated free radicals. Biochemistry. 1990;29:7876–7882. doi: 10.1021/bi00486a014. [DOI] [PubMed] [Google Scholar]
- 20.Ku¨pfer PA, Leumann CJ. Oxidative Damage on RNA Nucleobases. In: Erdmann VA, Markiewicz WT, Barciszewski J, editors. Chemical biology of nucleic acids (fundamentals and clinical applications) Berlin: Springer; 2014. pp. 75–94. [Google Scholar]
- 21.Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G–T and A–C substitutions. J Biol Chem. 1992;267:166–172. [PubMed] [Google Scholar]
- 22.Hsu GW, Ober M, Carell T, Beese LS. Error-prone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature. 2004;431:217–221. doi: 10.1038/nature02908. [DOI] [PubMed] [Google Scholar]
- 23.Toyokuni S, Mori T, Dizdaroglu M. DNA base modifications in renal chromatin of Wistar rats treated with a renal carcinogen, ferric nitrilotriacetate. Int J Cancer. 1994;57:123–128. doi: 10.1002/ijc.2910570122. [DOI] [PubMed] [Google Scholar]
- 24.Weimann A, Belling D, Poulsen HE. Quantification of 8-oxo-guanine and guanine as the nucleobase, nucleoside and deoxynucleoside forms in human urine by high-performance liquid chromatography-electrospray tandem mass spectrometry. Nucleic Acids Res. 2002;30:E7. doi: 10.1093/nar/30.2.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKinlay A, Gerard W, Fields S. Global analysis of RNA oxidation in Saccharomyces cerevisiae . Biotechniques. 2012;52:109–111. doi: 10.2144/000113801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park EM, Shigenaga MK, Degan P, Korn TS, Kitzler JW, Wehr CM, Kolachana P, Ames BN. Assay of excised oxidative DNA lesions: isolation of 8-oxoguanine and its nucleoside derivatives from biological fluids with a monoclonal antibody column. Proc Natl Acad Sci USA. 1992;89:3375–3379. doi: 10.1073/pnas.89.8.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J, Head E, Gharib AM, Yuan W, Ingersoll RT, Hagen TM, Cotman CW, Ames BN. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-l-carnitine and/or R-alpha-lipoic acid. Proc Natl Acad Sci USA. 2002;99:2356–2361. doi: 10.1073/pnas.261709299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seo AY, Xu J, Servais S, Hofer T, Marzetti E, Wohlgemuth SE, Knutson MD, Chung HY, Leeuwenburgh C. Mitochondrial iron accumulation with age and functional consequences. Aging Cell. 2008;7:706–716. doi: 10.1111/j.1474-9726.2008.00418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hofer T, Marzetti E, Xu J, Seo AY, Gulec S, Knutson MD, Leeuwenburgh C, Dupont-Versteegden EE. Increased iron content and RNA oxidative damage in skeletal muscle with aging and disuse atrophy. Exp Gerontol. 2008;43:563–570. doi: 10.1016/j.exger.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shan X, Lin CL. Quantification of oxidized RNAs in Alzheimer’s disease. Neurobiol Aging. 2006;27:657–662. doi: 10.1016/j.neurobiolaging.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 32.Nunomura A, Chiba S, Kosaka K, Takeda A, Castellani RJ, Smith MA, Perry G. Neuronal RNA oxidation is a prominent feature of dementia with Lewy bodies. Neuroreport. 2002;13:2035–2039. doi: 10.1097/00001756-200211150-00009. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol. 1999;154:1423–1429. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding Q, Markesbery WR, Chen Q, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci. 2005;25:9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang Y, Kong Q, Shan X, Tian G, Ilieva H, Cleveland DW, Rothstein JD, Borchelt DR, Wong PC, Lin CL. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS One. 2008;3:e2849. doi: 10.1371/journal.pone.0002849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bradley-Whitman MA, Timmons MD, Beckett TL, Murphy MP, Lynn BC, Lovell MA. Nucleic acid oxidation: an early feature of Alzheimer’s disease. J Neurochem. 2014;128:294–304. doi: 10.1111/jnc.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sedgwick B. Repairing DNA-methylation damage. Nat Rev Mol Cell Biol. 2004;5:148–157. doi: 10.1038/nrm1312. [DOI] [PubMed] [Google Scholar]
- 38.Rydberg B, Lindahl T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-l-methionine is a potentially mutagenic reaction. EMBO J. 1982;1:211–216. doi: 10.1002/j.1460-2075.1982.tb01149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bellacosa A, Moss EG. RNA repair: damage control. Curr Biol. 2003;13:R482–R484. doi: 10.1016/S0960-9822(03)00408-1. [DOI] [PubMed] [Google Scholar]
- 40.Singer B, Bodell WJ, Cleaver JE, Thomas GH, Rajewsky MF, Thon W. Oxygens in DNA are main targets for ethylnitrosourea in normal and xeroderma pigmentosum fibroblasts and fetal rat brain cells. Nature. 1978;276:85–88. doi: 10.1038/276085a0. [DOI] [PubMed] [Google Scholar]
- 41.Lawrence CW, Borden A, Banerjee SK, LeClerc JE. Mutation frequency and spectrum resulting from a single abasic site in a single-stranded vector. Nucleic Acids Res. 1990;18:2153–2157. doi: 10.1093/nar/18.8.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu B, Fournier MJ. Interference probing of rRNA with snoRNPs: a novel approach for functional mapping of RNA in vivo. RNA. 2004;10:1130–1141. doi: 10.1261/rna.7190104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ougland R, Zhang CM, Liiv A, Johansen RF, Seeberg E, Hou YM, Remme J, Falnes PO. AlkB restores the biological function of mRNA and tRNA inactivated by chemical methylation. Mol Cell. 2004;16:107–116. doi: 10.1016/j.molcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Bodell WJ, Singer B. Influence of hydrogen bonding in DNA and polynucleotides on reaction of nitrogens and oxygens toward ethylnitrosourea. Biochemistry. 1979;18:2860–2863. doi: 10.1021/bi00580a029. [DOI] [PubMed] [Google Scholar]
- 45.Singer B, Pergolizzi RG, Grunberger D. Synthesis and coding properties of dinucleoside diphosphates containing alky pyrimidines which are formed by the action of carcinogens on nucleic acids. Nucleic Acids Res. 1979;6:1709–1719. doi: 10.1093/nar/6.4.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eadie JS, Conrad M, Toorchen D, Topal MD. Mechanism of mutagenesis by O6-methylguanine. Nature. 1984;308:201–203. doi: 10.1038/308201a0. [DOI] [PubMed] [Google Scholar]
- 47.Preston BD, Singer B, Loeb LA. Mutagenic potential of O4-methylthymine in vivo determined by an enzymatic approach to site-specific mutagenesis. Proc Natl Acad Sci USA. 1986;83:8501–8505. doi: 10.1073/pnas.83.22.8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lachapelle M, Fadlallah S, Krzystyniak K, Fournier M, Cooper S, Denizeau F. Colloidal gold ultraimmunocytochemical localization of DNA and RNA adducts in rat hepatocytes. Carcinogenesis. 1992;13:2335–2339. doi: 10.1093/carcin/13.12.2335. [DOI] [PubMed] [Google Scholar]
- 49.Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, Pena-Diaz J, Otterlei M, Slupphaug G, Krokan HE. Alkylation damage in DNA and RNA-repair mechanisms and medical significance. DNA Repair (Amst) 2004;3:1389–1407. doi: 10.1016/j.dnarep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 50.Heminger KA, Hartson SD, Rogers J, Matts RL. Cisplatin inhibits protein synthesis in rabbit reticulocyte lysate by causing an arrest in elongation. Arch Biochem Biophys. 1997;344:200–207. doi: 10.1006/abbi.1997.0198. [DOI] [PubMed] [Google Scholar]
- 51.Glazer RI, Lloyd LS. Association of cell lethality with incorporation of 5-fluorouracil and 5-fluorouridine into nuclear RNA in human colon carcinoma cells in culture. Mol Pharmacol. 1982;21:468–473. [PubMed] [Google Scholar]
- 52.Pettersen HS, Visnes T, Vagbo CB, Svaasand EK, Doseth B, Slupphaug G, Kavli B, Krokan HE. UNG-initiated base excision repair is the major repair route for 5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res. 2011;39:8430–8444. doi: 10.1093/nar/gkr563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu K, Henning D, Iwakuma T, Valdez BC, Busch H. Adriamycin inhibits human RH II/Gu RNA helicase activity by binding to its substrate. Biochem Biophys Res Commun. 1999;266:361–365. doi: 10.1006/bbrc.1999.1815. [DOI] [PubMed] [Google Scholar]
- 54.Chernova OB, Chernov MV, Agarwal ML, Taylor WR, Stark GR. The role of p53 in regulating genomic stability when DNA and RNA synthesis are inhibited. Trends Biochem Sci. 1995;20:431–434. doi: 10.1016/S0968-0004(00)89094-5. [DOI] [PubMed] [Google Scholar]
- 55.Fiala ES, Conaway CC, Mathis JE. Oxidative DNA and RNA damage in the livers of Sprague-Dawley rats treated with the hepatocarcinogen 2-nitropropane. Cancer Res. 1989;49:5518–5522. [PubMed] [Google Scholar]
- 56.Hofer T, Seo AY, Prudencio M, Leeuwenburgh C. A method to determine RNA and DNA oxidation simultaneously by HPLC-ECD: greater RNA than DNA oxidation in rat liver after doxorubicin administration. Biol Chem. 2006;387:103–111. doi: 10.1515/BC.2006.014. [DOI] [PubMed] [Google Scholar]
- 57.Gorg B, Qvartskhava N, Keitel V, Bidmon HJ, Selbach O, Schliess F, Haussinger D. Ammonia induces RNA oxidation in cultured astrocytes and brain in vivo. Hepatology. 2008;48:567–579. doi: 10.1002/hep.22345. [DOI] [PubMed] [Google Scholar]
- 58.Shan X, Chang Y, Lin CL. Messenger RNA oxidation is an early event preceding cell death and causes reduced protein expression. FASEB J. 2007;21:2753–2764. doi: 10.1096/fj.07-8200com. [DOI] [PubMed] [Google Scholar]
- 59.Shan X, Tashiro H, Lin CL. The identification and characterization of oxidized RNAs in Alzheimer’s disease. J Neurosci. 2003;23:4913–4921. doi: 10.1523/JNEUROSCI.23-12-04913.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nunez ME, Hall DB, Barton JK. Long-range oxidative damage to DNA: effects of distance and sequence. Chem Biol. 1999;6:85–97. doi: 10.1016/S1074-5521(99)80005-2. [DOI] [PubMed] [Google Scholar]
- 61.Shen Z, Wu W, Hazen SL. Activated leukocytes oxidatively damage DNA, RNA, and the nucleotide pool through halide-dependent formation of hydroxyl radical. Biochemistry. 2000;39:5474–5482. doi: 10.1021/bi992809y. [DOI] [PubMed] [Google Scholar]
- 62.Liu M, Gong X, Alluri RK, Wu J, Sablo T, Li Z. Characterization of RNA damage under oxidative stress in Escherichia coli . Biol Chem. 2012;393:123–132. doi: 10.1515/hsz-2011-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gong X, Tao R, Li Z. Quantification of RNA damage by reverse transcription polymerase chain reactions. Anal Biochem. 2006;357:58–67. doi: 10.1016/j.ab.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 64.Honda K, Smith MA, Zhu X, Baus D, Merrick WC, Tartakoff AM, Hattier T, Harris PL, Siedlak SL, Fujioka H, Liu Q, Moreira PI, Miller FP, Nunomura A, Shimohama S, Perry G. Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron. J Biol Chem. 2005;280:20978–20986. doi: 10.1074/jbc.M500526200. [DOI] [PubMed] [Google Scholar]
- 65.Ding Q, Markesbery WR, Cecarini V, Keller JN. Decreased RNA, and increased RNA oxidation, in ribosomes from early Alzheimer’s disease. Neurochem Res. 2006;31:705–710. doi: 10.1007/s11064-006-9071-5. [DOI] [PubMed] [Google Scholar]
- 66.Ding Q, Zhu H, Zhang B, Soriano A, Burns R, Markesbery WR. Increased 5S rRNA oxidation in Alzheimer’s disease. J Alzheimers Dis. 2012;29:201–209. doi: 10.3233/JAD-2012-111058. [DOI] [PubMed] [Google Scholar]
- 67.Thompson DM, Lu C, Green PJ, Parker R. tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA. 2008;14:2095–2103. doi: 10.1261/rna.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thompson DM, Parker R. The RNase Rny1p cleaves tRNAs and promotes cell death during oxidative stress in Saccharomyces cerevisiae . J Cell Biol. 2009;185:43–50. doi: 10.1083/jcb.200811119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emara MM, Ivanov P, Hickman T, Dawra N, Tisdale S, Kedersha N, Hu GF, Anderson P. Angiogenin-induced tRNA-derived stress-induced RNAs promote stress-induced stress granule assembly. J Biol Chem. 2010;285:10959–10968. doi: 10.1074/jbc.M109.077560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang JX, Gao J, Ding SL, Wang K, Jiao JQ, Wang Y, Sun T, Zhou LY, Long B, Zhang XJ, Li Q, Liu JP, Feng C, Liu J, Gong Y, Zhou Z, Li PF. Oxidative modification of miR-184 enables it to target Bcl-xL and Bcl-w. Mol Cell. 2015;59:50–61. doi: 10.1016/j.molcel.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 71.Tanaka M, Chock PB, Stadtman ER. Oxidized messenger RNA induces translation errors. Proc Natl Acad Sci USA. 2007;104:66–71. doi: 10.1073/pnas.0609737104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miller BG. The biological half-lives of ribosomal and transfer RNA in the mouse uterus. J Endocrinol. 1973;59:81–85. doi: 10.1677/joe.0.0590081. [DOI] [PubMed] [Google Scholar]
- 73.Cole SE, LaRiviere FJ, Merrikh CN, Moore MJ. A convergence of rRNA and mRNA quality control pathways revealed by mechanistic analysis of nonfunctional rRNA decay. Mol Cell. 2009;34:440–450. doi: 10.1016/j.molcel.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.LaRiviere FJ, Cole SE, Ferullo DJ, Moore MJ. A late-acting quality control process for mature eukaryotic rRNAs. Mol Cell. 2006;24:619–626. doi: 10.1016/j.molcel.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 75.Green R, Samaha RR, Noller HF. Mutations at nucleotides G2251 and U2585 of 23 S rRNA perturb the peptidyl transferase center of the ribosome. J Mol Biol. 1997;266:40–50. doi: 10.1006/jmbi.1996.0780. [DOI] [PubMed] [Google Scholar]
- 76.Fujii K, Kitabatake M, Sakata T, Miyata A, Ohno M. A role for ubiquitin in the clearance of nonfunctional rRNAs. Genes Dev. 2009;23:963–974. doi: 10.1101/gad.1775609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parker R. RNA degradation in Saccharomyces cerevisae . Genetics. 2012;191:671–702. doi: 10.1534/genetics.111.137265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Svensson JP, Pesudo LQ, Fry RC, Adeleye YA, Carmichael P, Samson LD. Genomic phenotyping of the essential and non-essential yeast genome detects novel pathways for alkylation resistance. BMC Syst Biol. 2011;5:157. doi: 10.1186/1752-0509-5-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zaidi IW, Rabut G, Poveda A, Scheel H, Malmstrom J, Ulrich H, Hofmann K, Pasero P, Peter M, Luke B. Rtt101 and Mms1 in budding yeast form a CUL4(DDB1)-like ubiquitin ligase that promotes replication through damaged DNA. EMBO Rep. 2008;9:1034–1040. doi: 10.1038/embor.2008.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Prakash L, Prakash S. Isolation and characterization of MMS-sensitive mutants of Saccharomyces cerevisiae. Genetics. 1977;86:33–55. doi: 10.1093/genetics/86.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jobert L, Nilsen H. Regulatory mechanisms of RNA function: emerging roles of DNA repair enzymes. Cell Mol Life Sci. 2014;71:2451–2465. doi: 10.1007/s00018-014-1562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Berquist BR, McNeill DR, Wilson DM., 3rd Characterization of abasic endonuclease activity of human Ape1 on alternative substrates, as well as effects of ATP and sequence context on AP site incision. J Mol Biol. 2008;379:17–27. doi: 10.1016/j.jmb.2008.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barnes T, Kim WC, Mantha AK, Kim SE, Izumi T, Mitra S, Lee CH. Identification of apurinic/apyrimidinic endonuclease 1 (APE1) as the endoribonuclease that cleaves c-myc mRNA. Nucleic Acids Res. 2009;37:3946–3958. doi: 10.1093/nar/gkp275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vascotto C, Fantini D, Romanello M, Cesaratto L, Deganuto M, Leonardi A, Radicella JP, Kelley MR, D’Ambrosio C, Scaloni A, Quadrifoglio F, Tell G. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol Cell Biol. 2009;29:1834–1854. doi: 10.1128/MCB.01337-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jobert L, Skjeldam HK, Dalhus B, Galashevskaya A, Vagbo CB, Bjoras M, Nilsen H. The human base excision repair enzyme SMUG1 directly interacts with DKC1 and contributes to RNA quality control. Mol Cell. 2013;49:339–345. doi: 10.1016/j.molcel.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 86.Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Copela LA, Fernandez CF, Sherrer RL, Wolin SL. Competition between the Rex1 exonuclease and the La protein affects both Trf4p-mediated RNA quality control and pre-tRNA maturation. RNA. 2008;14:1214–1227. doi: 10.1261/rna.1050408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kadaba S, Wang X, Anderson JT. Nuclear RNA surveillance in Saccharomyces cerevisiae: Trf4p-dependent polyadenylation of nascent hypomethylated tRNA and an aberrant form of 5S rRNA. RNA. 2006;12:508–521. doi: 10.1261/rna.2305406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ozanick SG, Wang X, Costanzo M, Brost RL, Boone C, Anderson JT. Rex1p deficiency leads to accumulation of precursor initiator tRNAMet and polyadenylation of substrate RNAs in Saccharomyces cerevisiae . Nucleic Acids Res. 2009;37:298–308. doi: 10.1093/nar/gkn925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vanacova S, Wolf J, Martin G, Blank D, Dettwiler S, Friedlein A, Langen H, Keith G, Keller W. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol. 2005;3:e189. doi: 10.1371/journal.pbio.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Alexandrov A, Chernyakov I, Gu W, Hiley SL, Hughes TR, Grayhack EJ, Phizicky EM. Rapid tRNA decay can result from lack of nonessential modifications. Mol Cell. 2006;21:87–96. doi: 10.1016/j.molcel.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 92.Chernyakov I, Whipple JM, Kotelawala L, Grayhack EJ, Phizicky EM. Degradation of several hypomodified mature tRNA species in Saccharomyces cerevisiae is mediated by Met22 and the 5′–3′ exonucleases Rat1 and Xrn1. Genes Dev. 2008;22:1369–1380. doi: 10.1101/gad.1654308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wilusz JE, Whipple JM, Phizicky EM, Sharp PA. tRNAs marked with CCACCA are targeted for degradation. Science. 2011;334:817–821. doi: 10.1126/science.1213671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Whipple JM, Lane EA, Chernyakov I, D’Silva S, Phizicky EM. The yeast rapid tRNA decay pathway primarily monitors the structural integrity of the acceptor and T-stems of mature tRNA. Genes Dev. 2011;25:1173–1184. doi: 10.1101/gad.2050711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Castano IB, Heath-Pagliuso S, Sadoff BU, Fitzhugh DJ, Christman MF. A novel family of TRF (DNA topoisomerase I-related function) genes required for proper nuclear segregation. Nucleic Acids Res. 1996;24:2404–2410. doi: 10.1093/nar/24.12.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mol CD, Arvai AS, Slupphaug G, Kavli B, Alseth I, Krokan HE, Tainer JA. Crystal structure and mutational analysis of human uracil-DNA glycosylase: structural basis for specificity and catalysis. Cell. 1995;80:869–878. doi: 10.1016/0092-8674(95)90290-2. [DOI] [PubMed] [Google Scholar]
- 97.Hayakawa H, Kuwano M, Sekiguchi M. Specific binding of 8-oxoguanine-containing RNA to polynucleotide phosphorylase protein. Biochemistry. 2001;40:9977–9982. doi: 10.1021/bi010595q. [DOI] [PubMed] [Google Scholar]
- 98.Hayakawa H, Uchiumi T, Fukuda T, Ashizuka M, Kohno K, Kuwano M, Sekiguchi M. Binding capacity of human YB-1 protein for RNA containing 8-oxoguanine. Biochemistry. 2002;41:12739–12744. doi: 10.1021/bi0201872. [DOI] [PubMed] [Google Scholar]
- 99.Evdokimova V, Ruzanov P, Imataka H, Raught B, Svitkin Y, Ovchinnikov LP, Sonenberg N. The major mRNA-associated protein YB-1 is a potent 5′ cap-dependent mRNA stabilizer. EMBO J. 2001;20:5491–5502. doi: 10.1093/emboj/20.19.5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tanaka T, Ohashi S, Kobayashi S. Roles of YB-1 under arsenite-induced stress: translational activation of HSP70 mRNA and control of the number of stress granules. Biochim Biophys Acta. 2014;1840:985–992. doi: 10.1016/j.bbagen.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 101.Wu J, Jiang Z, Liu M, Gong X, Wu S, Burns CM, Li Z. Polynucleotide phosphorylase protects Escherichia coli against oxidative stress. Biochemistry. 2009;48:2012–2020. doi: 10.1021/bi801752p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu J, Li Z. Human polynucleotide phosphorylase reduces oxidative RNA damage and protects HeLa cell against oxidative stress. Biochem Biophys Res Commun. 2008;372:288–292. doi: 10.1016/j.bbrc.2008.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen HW, Rainey RN, Balatoni CE, Dawson DW, Troke JJ, Wasiak S, Hong JS, McBride HM, Koehler CM, Teitell MA, French SW. Mammalian polynucleotide phosphorylase is an intermembrane space RNase that maintains mitochondrial homeostasis. Mol Cell Biol. 2006;26:8475–8487. doi: 10.1128/MCB.01002-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Taddei F, Hayakawa H, Bouton M, Cirinesi A, Matic I, Sekiguchi M, Radman M. Counteraction by MutT protein of transcriptional errors caused by oxidative damage. Science. 1997;278:128–130. doi: 10.1126/science.278.5335.128. [DOI] [PubMed] [Google Scholar]
- 105.Hayakawa H, Hofer A, Thelander L, Kitajima S, Cai Y, Oshiro S, Yakushiji H, Nakabeppu Y, Kuwano M, Sekiguchi M. Metabolic fate of oxidized guanine ribonucleotides in mammalian cells. Biochemistry. 1999;38:3610–3614. doi: 10.1021/bi982361l. [DOI] [PubMed] [Google Scholar]
- 106.Ishibashi T, Hayakawa H, Ito R, Miyazawa M, Yamagata Y, Sekiguchi M. Mammalian enzymes for preventing transcriptional errors caused by oxidative damage. Nucleic Acids Res. 2005;33:3779–3784. doi: 10.1093/nar/gki682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kajitani K, Yamaguchi H, Dan Y, Furuichi M, Kang D, Nakabeppu Y. MTH1, an oxidized purine nucleoside triphosphatase, suppresses the accumulation of oxidative damage of nucleic acids in the hippocampal microglia during kainate-induced excitotoxicity. J Neurosci. 2006;26:1688–1698. doi: 10.1523/JNEUROSCI.4948-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shoemaker CJ, Eyler DE, Green R. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science. 2010;330:369–372. doi: 10.1126/science.1192430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shoemaker CJ, Green R. Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc Natl Acad Sci USA. 2011;108:E1392–E1398. doi: 10.1073/pnas.1113956108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pisarev AV, Skabkin MA, Pisareva VP, Skabkina OV, Rakotondrafara AM, Hentze MW, Hellen CU, Pestova TV. The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol Cell. 2010;37:196–210. doi: 10.1016/j.molcel.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Frischmeyer PA, van Hoof A, O’Donnell K, Guerrerio AL, Parker R, Dietz HC. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–2261. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- 113.van Hoof A, Frischmeyer PA, Dietz HC, Parker R. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;295:2262–2264. doi: 10.1126/science.1067272. [DOI] [PubMed] [Google Scholar]
- 114.Saito S, Hosoda N, Hoshino S. The Hbs1-Dom34 protein complex functions in non-stop mRNA decay in mammalian cells. J Biol Chem. 2013;288:17832–17843. doi: 10.1074/jbc.M112.448977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 116.Shen L, Song CX, He C, Zhang Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu Rev Biochem. 2014;83:585–614. doi: 10.1146/annurev-biochem-060713-035513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fedeles BI, Singh V, Delaney JC, Li D, Essigmann JM. The AlkB family of Fe(II)/alpha-ketoglutarate-dependent dioxygenases: repairing nucleic acid alkylation damage and beyond. J Biol Chem. 2015;290:20734–20742. doi: 10.1074/jbc.R115.656462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ougland R, Rognes T, Klungland A, Larsen E. Non-homologous functions of the AlkB homologs. J Mol Cell Biol. 2015;7:494–504. doi: 10.1093/jmcb/mjv029. [DOI] [PubMed] [Google Scholar]
- 119.Yang CG, Yi C, Duguid EM, Sullivan CT, Jian X, Rice PA, He C. Crystal structures of DNA/RNA repair enzymes AlkB and ABH2 bound to dsDNA. Nature. 2008;452:961–965. doi: 10.1038/nature06889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.van den Born E, Omelchenko MV, Bekkelund A, Leihne V, Koonin EV, Dolja VV, Falnes PO. Viral AlkB proteins repair RNA damage by oxidative demethylation. Nucleic Acids Res. 2008;36:5451–5461. doi: 10.1093/nar/gkn519. [DOI] [PMC free article] [PubMed] [Google Scholar]