

Abstract
Younger siblings of children with autism spectrum disorder (ASD; high-risk siblings) exhibit lower levels of initiating joint attention (IJA; sharing an object or experience with a social partner through gaze and/or gesture) than low-risk siblings of children without ASD. However, high-risk siblings also exhibit substantial variability in this domain. The neurotransmitter dopamine is linked to brain areas associated with reward, motivation, and attention, and common dopaminergic variants have been associated with attention difficulties. We examined whether these common dopaminergic variants, DRD4 and DRD2, explain variability in IJA in high-risk (n = 55) and low-risk (n = 38) siblings. IJA was assessed in the first year during a semi-structured interaction with an examiner. DRD4 and DRD2 genotypes were coded according to associated dopaminergic functioning to create a gene score, with higher scores indicating more genotypes associated with less efficient dopaminergic functioning. Higher dopamine gene scores (indicative of less efficient dopaminergic functioning) were associated with lower levels of IJA in the first year for high-risk siblings, while the opposite pattern emerged in low-risk siblings. Findings suggest differential susceptibility—IJA was differentially associated with dopaminergic functioning depending on familial ASD risk. Understanding genes linked to ASD-relevant behaviors in high-risk siblings will aid in early identification of children at greatest risk for difficulties in these behavioral domains, facilitating targeted prevention and intervention.
Keywords: high-risk siblings, initiating joint attention, dopamine, differential susceptibility, autism spectrum disorder
Introduction
Autism spectrum disorder (ASD) is a complex neurodevelopmental condition characterized by a broad range of social and communication impairments and stereotyped patterns of behavior [American Psychiatric Association, 2013], with prevalence estimates of over 1 in 75 children [CDC, 2014]. The younger siblings of children with ASD (high-risk siblings) have high rates of ASD diagnosis, with recurrence rates of 4.5–18.7%, and exhibit substantial heterogeneity in behaviors associated with ASD [Grønborg, Schendel, & Parner, 2013; Messinger et al., 2013; Ozonoff et al., 2011; Risch et al., 2014], including initiating joint attention (IJA). Common genetic variants such as those seen in dopaminergic genes DRD4 and DRD2 may aid in understanding the variability of phenotypic presentation in high-risk siblings. The current study examined these dopaminergic variants in high-risk siblings and low-risk siblings (siblings with no family history of ASD) to better understand heterogeneity in a behavioral phenotype particularly relevant to ASD, initiating joint attention.
Genetics and ASD
Although recent estimates suggest substantial heritability for ASD [Colvert et al., 2015; Hallmayer et al., 2011], specific genes responsible for this heritability are not always clear [Geschwind, 2011]. Both rare and common variants contribute to understanding genetic susceptibility in ASD. Several rare variants (mutations with a minor allele frequency of less than 1%) associated with ASD have been identified [Betancur, 2011; Buxbaum, 2009; Geschwind & State, 2015]. Gene discovery efforts have yielded a number of ASD susceptibility genes, such as CHD8, CNTNAP2, NLGN4, NRXN1, and CNTN4 [e.g., Chen, Peñagarikano, Belgard, Swarup, & Geschwind, 2015; De Rubeis et al., 2014; Iossifov et al., 2014]. However, no specific gene accounts for a majority of ASD cases [Abrahams & Geschwind, 2008; Geschwind, 2011; Muhle, Trentacoste, & Rapin, 2004]. Even among siblings both diagnosed with ASD, most do not share the same ASD risk genes, underscoring the genetic heterogeneity of ASD [Yuen et al., 2015] and highlighting the potential difficulty of identifying replicable ASD susceptibility genes. Common variants (polymorphisms that occur in greater than 1–2% of the population) may comprise a substantial portion of the risk heritability of ASD [Gaugler et al., 2014; Klei et al., 2012]. However, identified common variants that in combination or alone influence ASD susceptibility have not been well-replicated [Anney et al., 2010; Devlin, Melhem, & Roeder, 2011; Muhle et al., 2004]. As genetic underpinnings of ASD are highly heterogeneous and a number of genes likely interact to influence susceptibility [Talkowski, Minikel, & Gusella, 2014], an approach focusing on the genetic basis of behaviors relevant to ASD may be productive in identifying genotypes associated with specific ASD-related traits [Muhle et al., 2004].
Heterogeneity Within ASD
In addition to ASD’s genetic variability, ASD is phenotypically heterogeneous, encompassing a broad spectrum of impairment. Those diagnosed can exhibit varied combinations of traits and symptoms [Rapin, 1991; Rutter & Schopler, 1987], resulting in a range of later outcomes [Howlin, Goode, Hutton, & Rutter, 2004]. ASD-relevant behaviors, which are characteristic of the disorder and its symptomatology, show substantial variability in both children with ASD and in their younger siblings. Even without an ASD diagnosis, high-risk siblings exhibit elevated ASD symptoms, lower levels of developmental functioning, and behavioral difficulties [Gangi, Ibañez, & Messinger, 2014; Georgiades et al., 2013; Messinger et al., 2013].
In low-risk children, common genetic variants have been linked to behavioral phenotypes [e.g., Bakermans-Kranenburg & van IJzendoorn, 2011; Lackner, Sabbagh, Hallinan, Liu, & Holden, 2012; Posner, Rothbart, & Sheese, 2007]. Here, we aimed to examine common genetic variants implicated in behavior in the context of familial risk for ASD, to determine whether these variants may play a role in the heterogeneity seen in behaviors that are relevant to and have implications for ASD. Though individual common genetic variants are unlikely to distinguish children with ASD from case controls, these variants may be related to phenotypic variability in ASD-relevant behaviors [Geschwind, 2011] among high-risk siblings. We examined the role of two common genetic variants (DRD4 and DRD2) in a sample including high-risk siblings to understand phenotypic, behavioral heterogeneity in the context of familial ASD risk.
Dopaminergic Variants and Behavior
While relationships between dopaminergic variants and behavior have been studied in typically developing children, there has been little examination in children at risk for ASD. Dopamine is a catecholamine that functions as a neurotransmitter in the brain, and it plays a role in several key domains including attention, reward-motivated behavior, and motor control. Dopamine is produced in brain areas including the substantia nigra and ventral tegmental area and then is transmitted through several main pathways, some of which are associated with the control of motivation-linked systems relevant to the current study. Specifically, the mesolimbic and mesocortical pathways begin in the ventral tegmental area and connect to the nucleus accumbens and cerebral cortex, respectively, and they are associated with response to reward and motivation.
Several common polymorphisms affect dopamine neurotransmission. The DRD4 gene encodes for dopamine receptor D4, which is expressed in areas including the frontal cortex, hippocampus, amygdala, and hypothalamus [Beaulieu & Gainetdinov, 2011]. Variants in a 48-base pair variable number tandem repeat of DRD4 can influence gene expression, and a “long” version (the 7-repeat allele) has been associated with suppressed receptor expression [Schoots & Van Tol, 2003]. The 7-repeat allele has been associated with varied attentional and behavioral difficulties in typically developing children and infants [Auerbach, Benjamin, Faroy, Geller, & Ebstein, 2001a, Auerbach, Faroy, Ebstein, Kahana, & Levine, 2001b; Gizer, Ficks, & Waldman, 2009; Schmidt, Fox, Perez-Edgar, Hu, & Hamer, 2001]. The DRD2 gene encodes for the dopamine receptor D2, which is expressed in areas including the striatum and nucleus accumbens [Beaulieu & Gainetdinov, 2011], and is associated with the Taq1A polymorphism on ANKK1. The A allele of the polymorphism (hereafter DRD2) is linked to a reduction in D2 receptor expression [Thompson et al., 1997] and is associated with risk for ASD and social interaction and communication difficulties [Hettinger et al., 2012; Salem et al., 2013].
ASD-Relevant Behavior
We focused on a specific ASD-relevant behavior, initiation of joint attention. Early deficits in initiating joint attention (IJA), a form of referential communication involving the use of gaze and gesture to coordinate attention between social partners and objects, are a core feature of ASD [Dawson et al., 2004; Mundy, Sigman, Ungerer, & Sherman, 1986]. Among high-risk siblings, early IJA is predictive of later ASD symptomatology [Ibañez, Grantz, & Messinger, 2012]. While some evidence suggests high-risk siblings tend to display fewer IJA behaviors than low-risk siblings [Cassel et al., 2007; Goldberg et al., 2005; Ibañez et al., 2012; Rozga et al., 2011], other investigations do not report differences [Toth, Dawson, Meltzoff, Greenson, & Fein, 2007; Yirmiya et al., 2006]. These mixed findings highlight the necessity for empirical work to explain phenotypic variability among high-risk siblings.
IJA is particularly relevant to high-risk siblings, as it may be predictive of later symptomatology. Given the role of the dopaminergic system in reward sensitivity and motivation, it may influence whether an infant finds social interaction through joint attention rewarding and is motivated to perform IJA behaviors. In addition, the dopaminergic system’s role in motor control and attention may play a role in an infant’s ability to shift attentional focus and execute such behaviors. Despite the relationship between dopaminergic variants and related functioning in typical development, similar associations have not been examined within children at risk for ASD. Investigating relations between behavioral phenotypes and dopaminergic genotypes in the context of familial risk for ASD may aid in understanding the manifestation of early ASD-relevant behaviors, enabling early identification of behavioral targets for early intervention.
Current Study
The current study examined dopaminergic genotypes DRD4 and DRD2 in relation to an ASD-relevant behavioral phenotype, initiating joint attention, in the context of familial autism risk.
Aim 1. Characterize dopaminergic genotype distributions in high-risk and low-risk siblings.
We examined distributions of genotype frequencies (DRD4, DRD2, and a dopamine gene score comprised of both genes) in high- and low-risk siblings. Due to the genetic heterogeneity of ASD and little evidence that dopaminergic genes confer ASD susceptibility, we did not expect genotype frequencies to differ between groups (i.e., risk alleles would not be overrepresented in high-risk siblings).
Aim 2. Examine the relationship between dopaminergic variants and ASD-relevant behavioral phenotype in high-risk and low-risk siblings.
Regression models tested the effect of dopaminergic genotype, as well as its interaction with risk group status, on IJA in the first year. Due to the potential role of dopamine in areas relevant to IJA (attentional control, social motivation and reward, motor control), we expected less efficient dopaminergic functioning to be associated with lower levels of IJA.
Methods
Participants
Participants were the infant siblings of children diagnosed with autism spectrum disorder (ASD; high-risk siblings, n = 55, 35 male) or the infant siblings of typically developing children with no history of ASD (low-risk siblings, n = 38, 16 male). High-risk siblings have at least one older sibling with a diagnosis of ASD, confirmed upon study enrollment by administration of the Autism Diagnostic Observation Schedule (ADOS) [Lord et al., 2000] and clinical diagnosis by a licensed clinical psychologist. Low-risk siblings have older siblings with no evidence of ASD, confirmed by a score lower than 9 on the Social Communication Questionnaire [Berument, Rutter, Lord, Pickles, & Bailey, 1999], a conservative cutoff score, and no family history of ASD in first degree relatives. All procedures were reviewed and approved by the University of Miami Institutional Review Board. Written informed consent was obtained from parents of all participants included in the study.
Measures
Early Social Communication Scales (ESCS)
Joint attention was assessed within the ESCS [Mundy et al., 2003] at 8, 10, and 12 months. The ESCS is a semi-structured assessment of infants’ nonverbal communication abilities, during which an examiner (seated across from the infant) presents and activates a series of toys, creating opportunities for the infant to initiate joint attention. After presenting and activating a toy, the examiner remains attentive and responds to the infant’s joint attention bids briefly. The current study focused on initiating joint attention (IJA) bids occurring during the ESCS (e.g., when infant gazed between the examiner and activated toy or showed an object to the examiner). Videotaped assessments were reliably coded by trained coders. Rates per minute of joint attention were calculated for each assessment age; a mean was calculated from the standardized values of each assessment age to provide a measure of IJA in the first year for analyses.
Dopamine genotypes
Genetic data was collected from saliva samples from participants using Oragene DNA collection kits. Genetic samples were sent for extraction and analysis to the John P. Hussman Institute for Human Genomics (HIHG) at the University of Miami Miller School of Medicine, where genotyping was conducted for DRD4 and DRD2. For analyses, genotypes for DRD4 (rs1805186) were grouped according to the presence or absence of the 7-repeat allele (“0” = no 7-repeat, “1” = at least one 7-repeat). For DRD2 (rs1800497), genotypes were grouped according the presence of the A allele (“0” = no A allele, “1” = at least one A allele).
A dopamine gene score was also created by coding DRD4 and DRD2 to reflect dopaminergic functioning. Higher scores indicated more “risk” genotypes (indexing less efficient dopaminergic functioning) and lower scores indicated fewer “risk” genotypes (indexing more efficient dopaminergic functioning). Gene scores served as an index of cumulative dopaminergic functioning for analyses of outcomes (for similar approaches, see Nikolova, Ferrell, Manuck, & Hariri [2011], Pearson-Fuhrhop et al. [2014], and Stice, Yokum, Burger, Epstein, & Smolen [2012]), with participants coded as having 0, 1, or 2 risk genotype sets.
Analytic Approach
For Aim 1, initial analyses examined distributions of genotype frequencies, testing whether allelic frequencies were consistent with Hardy–Weinberg equilibrium. Fisher’s exact tests determined whether genotype frequencies differed between high- and low-risk siblings or between ethnicities. For Aim 2, to first understand the roles of the DRD4 and DRD2 genotypes individually, we determined whether individual genotypes interacted with participants’ risk status to predict behavioral phenotype (IJA). A 2 (genotype) by 2 (risk group) ANOVA for each of the dopaminergic variants tested for main effects of genotype and risk status, as well as their interaction. To understand the effect of dopaminergic functioning across the two genotypes, this was followed by a regression in which dopamine score, status, and dopamine score*status interaction were entered as predictors to determine the cumulative effect of both dopaminergic genes. Interaction effects were followed up with individual models by risk group in which IJA was regressed on gene score.
Results
Dopaminergic Genotypes
Allelic distributions for DRD4, χ2(1) = 0.02, P = 0.85, and DRD2, χ2(1) = 0.00, P = 0.82, were consistent with Hardy–Weinberg equilibrium [Rodriguez, Gaunt, & Day, 2009]. Allele frequencies for DRD4, P = 0.82, and DRD2, P = 0.25, did not differ between high-risk and low-risk siblings (see Table 1). Allele frequencies for DRD4, P = 0.15, and DRD2, P = 0.14, did not differ by ethnicity (coded Non-Hispanic White/Caucasian, Hispanic/Latino, and Other; see supporting information Table 1 for genotypes by ethnicity).
Table 1.
DRD4 and DRD2 Allele Frequencies by Risk Group
| High-risk siblings |
Low-risk siblings |
|||
|---|---|---|---|---|
| Frequency | Percentage | Frequency | Percentage | |
| DRD4 | ||||
| – | 41 | 74.5% | 24 | 66.7% |
| 7/− | 13 | 23.6% | 11 | 30.6% |
| 7/7 | 1 | 1.8% | 1 | 2.8% |
| DRD2 | ||||
| G/G | 35 | 63.6% | 28 | 73.7% |
| A/G | 19 | 34.5% | 8 | 21.1% |
| A/A | 1 | 1.8% | 2 | 5.3% |
For analyses, genotypes for DRD4 and DRD2 were grouped according to the presence or absence of any alleles indicating less efficient dopaminergic functioning (7-repeat or A allele, respectively). Genotype frequencies for DRD4, P = 0.48, and DRD2, P = 0.37, did not differ between high-risk and low-risk siblings (see Table 2). Genotype frequencies for DRD4, P = 0.13, and DRD2, P = 0.14, did not differ by ethnicity. Dopamine composite scores also did not differ between high-risk and low-risk siblings, P = 0.69 (see Table 2), or by ethnicity, P = 0.23.
Table 2.
Genotype Frequencies by Risk Group
| High-risk siblings |
Low-risk siblings |
|||
|---|---|---|---|---|
| Frequency | Percentage | Frequency | Percentage | |
| DRD4 | ||||
| 7-repeat allele | 14 | 25.5% | 12 | 33.3% |
| No 7-repeat allele | 41 | 74.5% | 24 | 66.7% |
| DRD2 | ||||
| A allele | 20 | 36.4% | 10 | 26.3% |
| No A allele | 35 | 63.6% | 28 | 73.7% |
| Dopamine score | ||||
| 0 risk genotypes | 28 | 51.9% | 19 | 52.8% |
| 1 risk genotype | 18 | 33.3% | 14 | 38.9% |
| 2 risk genotypes | 8 | 14.8% | 3 | 8.3% |
Initiating Joint Attention
Preliminary analyses
Raw IJA scores (rates per minute) were examined using hierarchical linear modeling to determine whether there was a developmental change in IJA between 8 and 12 months (raw IJA scores by group are provided in supporting information Table 2). There were linear, β = 0.72, t(89) = 4.09, P < 0.001, and quadratic, β = −0.35, t(89) = −4.23, P < 0.001, effects of age. While high-risk and low-risk siblings differed in initial levels of IJA, β = −0.32, t(88) = −2.18, P = 0.032, they did not differ in their trajectories—high-risk siblings exhibited consistently lower levels of IJA than low-risk siblings from 8 to 12 months. For the purposes of subsequent analyses for this study, IJA scores were standardized at each age, and a mean was taken of these standardized scores for each infant in order to provide a measure of IJA in the first year.
DRD4
There was no main effect of genotype, F(1, 86) = 0.05, P = 0.82, partial η2 = 0.001, on IJA. There was a main effect of group status, F(1, 86) = 12.08, P = 0.001, partial η2 = 0.12, with high-risk siblings exhibiting lower levels of IJA than low-risk siblings. This main effect was modified by a genotype*status interaction effect, F(1, 86) = 8.80, P = 0.004, partial η2 = 0.09 (see supporting information Figure 1). Among children without the 7-repeat allele, levels of IJA did not differ between high-risk (M = 0.01, SD = 0.84) and low-risk (M = 0.11, SD = 0.73) siblings, t(62) = 0.48, P = 0.64. Among children with the 7-repeat allele, however, high-risk siblings (M = −0.52, SD = 0.75) exhibited lower levels of IJA than low-risk siblings (M = 0.73, SD = 1.01), t(24) = 3.62, P = 0.001.
DRD2
There was no main effect of genotype, F(1, 89) = 0.01, P = 0.91, partial η2 = 0.00, on IJA. There was a main effect of group status, F(1, 89) = 6.58, P = 0.01, partial η2 = 0.07, with high-risk siblings (M = −0.13, SD = 0.84) exhibiting lower levels of IJA than low-risk siblings (M = 0.29, SD = 0.86). There was no genotype*status interaction effect, F(1, 89) = 1.56, P = 0.22, partial η2 = 0.02 (see supporting information Figure 2).
Dopamine score
A regression model assessed effects of the dopamine score, risk group status, and their interaction on IJA, adjusted R2 = 0.13, F(3, 86) = 5.31, P = 0.002. There was no main effect of status, b = 0.03, t = 0.13, P = 0.90. There was a main effect of dopamine score, b = 0.50, t = 2.34, P = 0.02, such that children with higher dopamine scores tended to have higher IJA levels. There was also a dopamine score*status interaction effect, b = −0.81, t = −3.09, P = 0.003. Regression analyses by risk group indicated that in high-risk siblings, IJA levels decreased as dopamine scores increased (indicative of less efficient dopaminergic functioning), b = −0.31, t = −2.03, P = 0.047, while in low-risk siblings, IJA levels increased as dopamine scores increased, b = 0.50, t = 2.35, P = 0.03 (see Figure 1). Dopamine scores explained a significant proportion of variance in IJA in high-risk siblings, adjusted R2 = 0.06, F(1, 52) = 4.13, P = 0.047, and in low-risk siblings, adjusted R2 = 0.12, F(1, 34) = 5.53, P = 0.03.
Figure 1.
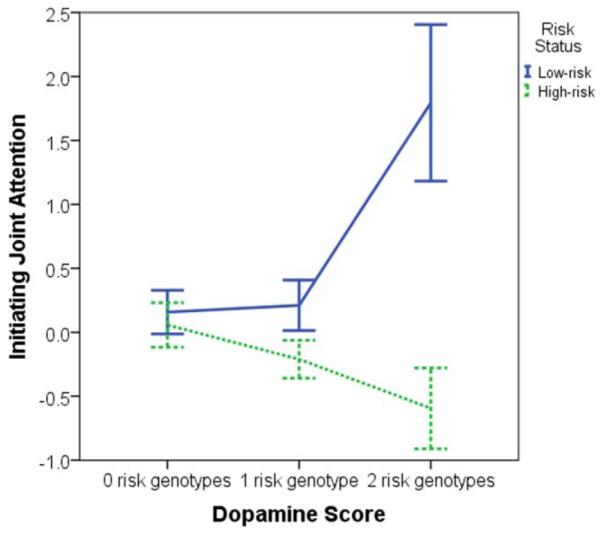
Mean levels of initiating joint attention (IJA) by group. Error bars reflect ± 1 SE. Initiating joint attention reflects a mean of standardized values. In high-risk siblings, 28 had a dopamine score of 0, 18 had a score of 1, and 8 had a score of 2. In low-risk siblings, 19 had a dopamine score of 0, 14 had a score of 1, and 3 had a score of 2.
Discussion
Children at elevated risk for ASD exhibit heterogeneity in symptomatology, other behaviors relevant to ASD, and outcomes. Among high-risk siblings, early behavior often predicts diagnosis, but these patterns of prediction are not clear. We aimed to refine our understanding of heterogeneity in early behavior relevant to ASD by examining the role of common genetic variants. We examined the association between common variants related to dopaminergic functioning and initiating joint attention (IJA). High-risk siblings with DRD4 and DRD2 genotypes linked to less efficient dopaminergic functioning exhibited lower levels of IJA than high-risk siblings with variants linked to more efficient dopaminergic functioning. To our knowledge, this is the first investigation of these genetic variants in relation to joint attention in siblings at risk for ASD.
For high-risk siblings, less efficient dopaminergic functioning (indexed by higher dopamine scores) was associated with less optimal behavior in the first year, i.e., with lower levels of IJA. IJA is important for the development of high-risk siblings—referential communication such as IJA is central to later language and social functioning in children at risk for ASD [Gangi et al., 2014; Ibañez et al., 2012; Malesa et al., 2012]. Early referential communication difficulties likely impact social functioning in children at risk for ASD, and these behaviors appear to be influenced by dopaminergic genotypes. Functioning of the dopaminergic system may be linked to IJA through its role in areas potentially important for the production of IJA behaviors, such as attentional control (relevant to the ability to shift attention between an object and social partner), social motivation and reward (relevant to whether an infant might find this social interaction rewarding and be motivated to initiate), and motor control (relevant to the ability to physically produce behaviors). This link may allow for early identification of high-risk siblings at greatest risk for behavioral difficulties in this area.
Although higher dopamine scores were associated with less optimal behavior among high-risk siblings, the opposite pattern emerged in low-risk siblings. Low-risk siblings with higher dopamine scores exhibited higher levels of IJA. This pattern suggests differential susceptibility, the hypothesis that children vary in their susceptibility to both adverse and beneficial effects of their environments [Belsky, 2005; Belsky, Bakermans-Kranenburg, & van IJzendoorn, 2007; Belsky & Pluess, 2009]. Common genetic variants have been identified as potential susceptibility factors that modify individuals’ susceptibility to influences affecting outcomes. The variants in the current study, DRD4 and DRD2, act as susceptibility genes in multiple contexts [e.g., Bakermans-Kranenburg & van IJzendoorn, 2006, 2011; Sheese, Voelker, Rothbart, & Posner, 2007; Van IJzendoorn & Bakermans-Kranenburg, 2006], but have not been examined in the context of familial risk for ASD.
Although the differential susceptibility hypothesis is often conceptualized as susceptibility to rearing, it may also encompass sensitivity to a broader range of influences [Belsky & Pluess, 2009]. For example, stronger associations between difficult child temperament and externalizing problems are found in children who have older siblings [Mesman et al., 2009]. Endogenous factors have also been conceptualized as internal environments that affect the relationship between genes and outcomes [Schmidt, Fox, Perez-Edgar, & Hamer, 2009]. That is, factors within an individual may play a role in moderating the association between genotype and developmental outcomes.
Familial risk for ASD confers increased risk for ASD and related sub-clinical deficits to younger siblings of children diagnosed with ASD. Within a differential susceptibility framework, we conceptualize familial ASD risk as a functional context. Familial ASD risk likely encompasses a combination of unknown factors that presumably are genetic and perhaps environmental, to which children may be more or less susceptible, such as CNVs linked to ASD susceptibility or exposure to environmental toxins [Hallmayer et al., 2011; Newschaffer et al., 2012; Yuen et al., 2015]. Here, high-risk siblings with alleles linked to less efficient dopaminergic functioning exhibited lower levels of IJA (the predicted pattern), while low-risk siblings did not exhibit this same pattern. In siblings with no alleles linked to less efficient dopaminergic functioning, high- and low-risk siblings exhibited similar levels of IJA. While this finding fits within the framework of the differential susceptibility hypothesis, and both dopaminergic genes have been found to act as susceptibility genes in other contexts, additional research will be necessary to determine the specific factors within familial risk for ASD (and lack of familial risk for ASD) that explain the mechanism through which dopaminergic functioning is differentially associated with levels of IJA in these groups. Future research could examine additional genetic factors, environmental factors, and parent qualities in high- and low-risk children.
Sample size limited analysis of high-risk siblings with ASD. Twelve high-risk siblings in the study sample were later diagnosed with ASD, a number insufficient for separate analyses. Findings from the current study should be interpreted with caution until replicated with larger sample sizes. Future research aimed at replicating our findings with larger sample sizes would strengthen our findings of a relationship between dopaminergic genotypes and ASD-relevant behavior and could profitably investigate this relationship among high-risk children with ASD outcomes.
Analyses of DRD4 and DRD2 genes individually suggest that DRD4 genotype may be more strongly associated with IJA than DRD2 genotype. In addition to the common dopaminergic variants in DRD4 and DRD2 examined in the current study, other dopaminergic variants might be examined in future investigations of behavioral characteristics of high-risk siblings and children with autism. For example, a VNTR in the DAT1 gene is associated with expression of the dopamine transporter [Fuke et al., 2001]. Together, these variants might provide a more comprehensive index of dopaminergic functioning. Additionally, information about copy number variations (CNVs) associated with ASD was not available for the participants in this study, and future research including known pathogenic CNVs may be able to examine their potential role in behavioral heterogeneity in high-risk siblings.
Genotypes outside the dopaminergic system may also impact the outcome of dopaminergic functioning and might further our understanding of dopamine’s role in behavioral outcomes. For example, catechol-O-methyl transferase (encoded by the COMT gene) is an enzyme that degrades catecholamines including dopamine, and a polymorphism in the COMT gene is associated with dopaminergic function [Chen et al., 2004]. Brain-derived neurotrophic factor (BDNF; coded for by the BDNF gene) may influence dopamine activity as well [Goggi, Pullar, Carney, & Bradford, 2003; Narita, Aoki, Takagi, Yajima, & Suzuki, 2003; Savitz, Solms, & Ramesar, 2006]. Serotonergic function may also interact with dopaminergic function to influence behavioral outcomes. Levels of dopaminergic functioning might influence sensitivity to reward, leading to either high or low motivation toward rewards. Dopaminergic functioning might then interact with levels of serotonergic functioning influencing effortful control, which could aid in regulation of approach tendencies related to reward sensitivity [Carver, Johnson, & Joormann, 2009].
In the current study, we found that dopaminergic risk genotypes were associated with lower levels of IJA in high-risk siblings. Given the systems in which dopamine plays a role, dopaminergic functioning could potentially affect children’s social motivation, reward sensitivity, attention coordination, and even motor control. High-risk siblings with lower dopaminergic functioning appear to exhibit less optimal behavior in early social interaction in the first year.
As the search for replicable genes associated with ASD risk is ongoing, an approach investigating genes that may be relevant to specific behaviors important for the development of children at risk for ASD may be a productive avenue of research. Genes that may not be associated with ASD itself may still be linked to particular behaviors. In addition to aiding identification of high-risk siblings at greatest risk for difficulties, findings may also aid in identifying resilient children. High-risk siblings with fewer genes associated with less efficient dopaminergic functioning were exhibiting fewer difficulties in IJA than high-risk siblings carrying more genotypes associated with less efficient dopaminergic functioning.
Referential communication is associated with ASD symptoms and outcome. Links between dopaminergic variants and behavioral phenotypes relevant to ASD, such as joint attention, can aid in understanding the developmental heterogeneity of high-risk siblings. Identification of common genetic variants—assessable at birth—that confer increased risk for ASD-relevant behaviors has the potential to aid in assessing risk and informing preventive interventions. If replicated, the current results suggest that genotype screening could aid in identifying siblings at the greatest risk for difficulties in areas relevant to later outcomes, even before the emergence of delays or difficulties. Developmental psychopathology could benefit from utilizing genetic markers with documented roles in healthy and problematic behaviors to assess risk and inform preventive interventions.
Supplementary Material
Acknowledgments
We would like to thank the families who have participated in our study. This research was supported by the National Institutes of Health (R01 HD047417 and R01 HD057284), National Institute of General Medical Sciences (R01 GM105004), and Autism Speaks.
References
- Abrahams BS, Geschwind DH. Advances in autism genetics: On the threshold of a new neurobiology. Nature Review Genetics. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anney R, et al. A genome-wide scan for common alleles affecting risk for autism. Human Molecular Genetics. 2010;19:4072–4082. doi: 10.1093/hmg/ddq307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association . Diagnostic and statistical manual of mental disorders: DSM-V. American Psychiatric Association; Washington, DC: 2013. [Google Scholar]
- Auerbach JG, Benjamin J, Faroy M, Geller V, Ebstein R. DRD4 related to infant attention and information processing: A developmental link to ADHD? Psychiatric Genetics. 2001a;11:31–35. doi: 10.1097/00041444-200103000-00006. [DOI] [PubMed] [Google Scholar]
- Auerbach JG, Faroy M, Ebstein R, Kahana M, Levine J. The association of the dopamine D4 receptor gene (DRD4) and the serotonin transporter promoter gene (5-HTTLPR) with temperament in 12-month-old infants. Journal of Child Psychology and Psychiatry. 2001b;42:777–783. doi: 10.1111/1469-7610.00774. [DOI] [PubMed] [Google Scholar]
- Bakermans-Kranenburg MJ, van IJzendoorn MH. Gene-environment interaction of the dopamine D4 receptor (DRD4) and observed maternal insensitivity predicting externalizing behavior in preschoolers. Developmental Psychobiology. 2006;48:406–409. doi: 10.1002/dev.20152. [DOI] [PubMed] [Google Scholar]
- Bakermans-Kranenburg MJ, van IJzendoorn MH. Differential susceptibility to rearing environment depending on dopamine-related genes: New evidence and a meta-analysis. Development and Psychopathology. 2011;23:39–52. doi: 10.1017/S0954579410000635. [DOI] [PubMed] [Google Scholar]
- Beaulieu J-M, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacological Reviews. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Belsky J. Differential susceptibility to rearing influence: An evolutionary hypothesis and some evidence. In: Ellis B, Bjorklund D, editors. Origins of the social mind: Evolutionary psychology and child development. Guilford; New York: 2005. pp. 139–163. [Google Scholar]
- Belsky J, Pluess M. Beyond diathesis stress: Differential susceptibility to environmental influences. Psychological Bulletin. 2009;135:885–908. doi: 10.1037/a0017376. [DOI] [PubMed] [Google Scholar]
- Belsky J, Bakermans-Kranenburg MJ, van IJzendoorn MH. For better and for worse: Differential susceptibility to environmental influences. Current Directions in Psychological Science. 2007;16:300–304. [Google Scholar]
- Berument SK, Rutter M, Lord C, Pickles A, Bailey A. Autism screening questionnaire: Diagnostic validity. The British Journal of Psychiatry. 1999;175:444–451. doi: 10.1192/bjp.175.5.444. [DOI] [PubMed] [Google Scholar]
- Betancur C. Etiological heterogeneity in autism spectrum disorders: More than 100 genetic and genomic disorders and still counting. Brain Research. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD. Multiple rare variants in the etiology of autism spectrum disorders. Dialogues in Clinical Neuro-science. 2009;11:35–43. doi: 10.31887/DCNS.2009.11.1/jdbuxbaum. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver CS, Johnson SL, Joormann J. Two-mode models of self-regulation as a tool for conceptualizing effects of the serotonin system in normal behavior and diverse disorders. Current Directions in Psychological Science. 2009;18:195–199. doi: 10.1111/j.1467-8721.2009.01635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassel TD, Messinger DS, Ibanez LV, Haltigan JD, Acosta SI, Buchman AC. Early social and emotional communication in the infant siblings of children with autism spectrum disorders: An examination of the broad phenotype. Journal of Autism and Developmental Disorders. 2007;37:122–132. doi: 10.1007/s10803-006-0337-1. [DOI] [PubMed] [Google Scholar]
- CDC Prevalence of autism spectrum disorder among children aged 8 years – Autism and developmental disabilities monitoring network, 11 sites, United States, 2010. Centers for disease control and prevention, morbidity and mortality weekly report, surveillance summaries. 2014;63:2. [PubMed] [Google Scholar]
- Chen JA, Peñagarikano O, Belgard TG, Swarup V, Geschwind DH. The emerging picture of autism spectrum disorder: Genetics and pathology. Annual Review of Pathology: Mechanisms of Disease. 2015;10:111–144. doi: 10.1146/annurev-pathol-012414-040405. [DOI] [PubMed] [Google Scholar]
- Chen J, et al. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): Effects on mRNA, protein, and enzyme activity in postmortem human brain. American Journal of Human Genetics. 2004;75:807–821. doi: 10.1086/425589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvert E, et al. Heritability of autism spectrum disorder in a UK population-based twin sample. JAMA Psychiatry. 2015;72:415–423. doi: 10.1001/jamapsychiatry.2014.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson G, Toth K, Abbott R, Osterling J, Munson J, Estes A, Liaw J. Early social attention impairments in autism: Social orienting, joint attention, and attention to distress. Developmental Psychology. 2004;40:271–283. doi: 10.1037/0012-1649.40.2.271. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Melhem N, Roeder K. Do common variants play a role in risk for autism? Evidence and theoretical musings. Brain Research. 2011;1380:78–84. doi: 10.1016/j.brainres.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuke S, Suo S, Takahashi N, Koike H, Sasagawa N, Ishiura S. The VNTR polymorphism of the human dopamine transporter (DAT1) gene affects gene expression. Pharmacogenomics Journal. 2001;1:152–156. doi: 10.1038/sj.tpj.6500026. [DOI] [PubMed] [Google Scholar]
- Gangi DN, Ibañez LV, Messinger DS. Joint attention initiation with and without positive affect: Risk group differences and associations with ASD symptoms. Journal of Autism and Developmental Disorders. 2014;44:1414–1424. doi: 10.1007/s10803-013-2002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaugler T, et al. Most genetic risk for autism resides with common variation. Nature Genetics. 2014;46:881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades S, et al. A prospective study of autistic-like traits in unaffected siblings of probands with autism spectrum disorder. JAMA Psychiatry. 2013;70:42–48. doi: 10.1001/2013.jamapsychiatry.1. [DOI] [PubMed] [Google Scholar]
- Geschwind DH. Genetics of autism spectrum disorders. Trends in Cognitive Sciences. 2011;15:409–416. doi: 10.1016/j.tics.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, State MW. Gene hunting in autism spectrum disorder: On the path to precision medicine. The Lancet Neurology. 2015:1109–1120. doi: 10.1016/S1474-4422(15)00044-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizer IR, Ficks C, Waldman ID. Candidate gene studies of ADHD: A meta-analytic review. Human Genetics. 2009;126:51–90. doi: 10.1007/s00439-009-0694-x. [DOI] [PubMed] [Google Scholar]
- Goggi J, Pullar IA, Carney SL, Bradford HF. Signalling pathways involved in the short-term potentiation of dopamine release by BDNF. Brain Research. 2003;968:156–161. doi: 10.1016/s0006-8993(03)02234-0. [DOI] [PubMed] [Google Scholar]
- Goldberg WA, et al. Brief report: Early social communication behaviors in the younger siblings of children with autism. Journal of Autism and Developmental Disorders. 2005;35:657–664. doi: 10.1007/s10803-005-0009-6. [DOI] [PubMed] [Google Scholar]
- Grønborg TK, Schendel DE, Parner ET. Recurrence of autism spectrum disorders in full- and half-siblings and trends over time: A population-based cohort study. JAMA Pediatrics. 2013 doi: 10.1001/jamapediatrics.2013.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmayer J, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Archives of General Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettinger JA, et al. DRD2 and PPP1R1B (DARPP-32) polymorphisms independently confer increased risk for autism spectrum disorders and additively predict affected status in male-only affected sib-pair families. Behavioral and Brain Functions. 2012;8(1):19. doi: 10.1186/1744-9081-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlin P, Goode S, Hutton J, Rutter M. Adult outcome for children with autism. Journal of Child Psychology and Psychiatry. 2004;45:212–229. doi: 10.1111/j.1469-7610.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- Ibañez LV, Grantz CJ, Messinger DS. The development of referential communication and autism symptomatology in high-risk infants. Infancy. 2012;18:687–707. doi: 10.1111/j.1532-7078.2012.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klei L, et al. Common genetic variants, acting additively, are a major source of risk for autism. Molecular Autism. 2012;3:9. doi: 10.1186/2040-2392-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner C, Sabbagh MA, Hallinan E, Liu X, Holden JJA. Dopamine receptor D4 gene variation predicts preschoolers’ developing theory of mind. Developmental Science. 2012;15:272–280. doi: 10.1111/j.1467-7687.2011.01124.x. [DOI] [PubMed] [Google Scholar]
- Lord C, et al. The autism diagnostic observation schedule—Generic: A standard measure of social and communication deficits associated with the spectrum of autism. Journal of Autism and Developmental Disorders. 2000;30:205–223. [PubMed] [Google Scholar]
- Malesa E, Foss-Feig J, Yoder P, Warren Z, Walden T, Stone W. Predicting language and social outcomes at age 5 for later-born siblings of children with autism spectrum disorders. Autism. 2012;17:558–570. doi: 10.1177/1362361312444628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesman J, Stoel R, Bakermans-Kranenburg MJ, IJzendoorn MH, Juffer F, Koot HM, Alink LRA. Predicting growth curves of early childhood externalizing problems: Differential susceptibility of children with difficult temperament. Journal of Abnormal Child Psychology. 2009;37:625–636. doi: 10.1007/s10802-009-9298-0. [DOI] [PubMed] [Google Scholar]
- Messinger D, et al. Beyond autism: A baby siblings research consortium study of high-risk children at three years of age. Journal of the American Academy of Child and Adolescent Psychiatry. 2013;52:300–308. doi: 10.1016/j.jaac.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhle R, Trentacoste SV, Rapin I. The genetics of autism. Pediatrics. 2004;113:e472–e486. doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- Mundy P, Sigman M, Ungerer J, Sherman T. Defining the social deficits of autism: The contribution of non-verbal communication measures. Journal of Child Psychology and Psychiatry. 1986;27:657–669. doi: 10.1111/j.1469-7610.1986.tb00190.x. [DOI] [PubMed] [Google Scholar]
- Mundy P, Delgado C, Block J, Venezia M, Hogan A, Siebert J. A manual for the abridged early social communication scales (ESCS) Department of Psychology, University of Miami; Coral Gables, Florida: 2003. [Google Scholar]
- Narita M, Aoki K, Takagi M, Yajima Y, Suzuki T. Implication of brain-derived neurotrophic factor in the release of dopamine and dopamine-related behaviors induced by methamphetamine. Neuroscience. 2003;119:767–775. doi: 10.1016/s0306-4522(03)00099-x. [DOI] [PubMed] [Google Scholar]
- Newschaffer CJ, et al. Infant siblings and the investigation of autism risk factors. Journal of Neurodevelopmental Disorders. 2012;4:7. doi: 10.1186/1866-1955-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova YS, Ferrell RE, Manuck SB, Hariri AR. Multilocus genetic profile for dopamine signaling predicts ventral striatum reactivity. Neuropsychopharmacology. 2011;36:1940–1947. doi: 10.1038/npp.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozonoff S, et al. Recurrence risk for autism spectrum disorders: A baby siblings research consortium study. Pediatrics. 2011;128:e488–e495. doi: 10.1542/peds.2010-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson-Fuhrhop KM, et al. Dopamine genetic risk score predicts depressive symptoms in healthy adults and adults with depression. PLoS ONE. 2014;9:e93772. doi: 10.1371/journal.pone.0093772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner MI, Rothbart MK, Sheese BE. Attention genes. Developmental Science. 2007;10:24–29. doi: 10.1111/j.1467-7687.2007.00559.x. [DOI] [PubMed] [Google Scholar]
- Rapin I. Autistic children: Diagnosis and clinical features. Pediatrics. 1991;87:751–760. [PubMed] [Google Scholar]
- Risch N, Hoffmann TJ, Anderson M, Croen LA, Grether JK, Windham GC. Familial recurrence of autism spectrum disorder: Evaluating genetic and environmental contributions. American Journal of Psychiatry. 2014;171:1206–1213. doi: 10.1176/appi.ajp.2014.13101359. [DOI] [PubMed] [Google Scholar]
- Rodriguez S, Gaunt TR, Day INM. Hardy-Weinberg equilibrium testing of biological ascertainment for mendelian randomization studies. American Journal of Epidemiology. 2009;169:505–514. doi: 10.1093/aje/kwn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozga A, Hutman T, Young GS, Rogers SJ, Ozonoff S, Dapretto M, Sigman M. Behavioral profiles of affected and unaffected siblings of children with autism: Contribution of measures of mother–infant interaction and nonverbal communication. Journal of Autism and Developmental Disorders. 2011;41:287–301. doi: 10.1007/s10803-010-1051-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter M, Schopler E. Autism and pervasive developmental disorders: Concepts and diagnostic issues. Journal of Autism and Developmental Disorders. 1987;17:159–186. doi: 10.1007/BF01495054. [DOI] [PubMed] [Google Scholar]
- Salem AM, Ismail S, Zarouk WA, Abdul B, Olwya S, Ahmed A, Abd El-Hamid S, Salem S. Genetic variants of neurotransmitter-related genes and miRNAs in Egyptian autistic patients. The Scientific World Journal. 2013;7 doi: 10.1155/2013/670621. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitz J, Solms M, Ramesar R. The molecular genetics of cognition: dopamine, COMT and BDNF. Genes, Brain and Behavior. 2006;5:311–328. doi: 10.1111/j.1601-183X.2005.00163.x. [DOI] [PubMed] [Google Scholar]
- Schmidt LA, Fox NA, Perez-Edgar K, Hu S, Hamer DH. Association of DRD4 with attention problems in normal childhood development. Psychiatric Genetics. 2001;11:25–29. doi: 10.1097/00041444-200103000-00005. [DOI] [PubMed] [Google Scholar]
- Schmidt LA, Fox NA, Perez-Edgar K, Hamer DH. Linking gene, brain, and behavior: DRD4, frontal asymmetry, and temperament. Psychological Science. 2009;20:831–837. doi: 10.1111/j.1467-9280.2009.02374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoots O, Van Tol HHM. The human dopamine D4 receptor repeat sequences modulate expression. Pharmacogenomics Journal. 2003;3:343–348. doi: 10.1038/sj.tpj.6500208. [DOI] [PubMed] [Google Scholar]
- Sheese BE, Voelker PM, Rothbart MK, Posner MI. Parenting quality interacts with genetic variation in dopamine receptor D4 to influence temperament in early childhood. Development and Psychopathology. 2007;19:1039–1046. doi: 10.1017/S0954579407000521. [DOI] [PubMed] [Google Scholar]
- Stice E, Yokum S, Burger K, Epstein L, Smolen A. Multilocus genetic composite reflecting dopamine signaling capacity predicts reward circuitry responsivity. The Journal of Neuroscience. 2012;32:10093–10100. doi: 10.1523/JNEUROSCI.1506-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Minikel EV, Gusella JF. Autism spectrum disorder genetics: Diverse genes with diverse clinical outcomes. Harvard Review of Psychiatry. 2014;22:65–75. doi: 10.1097/HRP.0000000000000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J, et al. D2 dopamine receptor gene (DRD2) Taql A polymorphism: Reduced dopamine D2 receptor binding in the human striatum associated with the A1 allele. Pharmacogenetics and Genomics. 1997;7:479–484. doi: 10.1097/00008571-199712000-00006. [DOI] [PubMed] [Google Scholar]
- Toth K, Dawson G, Meltzoff A, Greenson J, Fein D. Early social, imitation, play, and language abilities of young non-autistic siblings of children with autism. Journal of Autism and Developmental Disorders. 2007;37:145–157. doi: 10.1007/s10803-006-0336-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van IJzendoorn MH, Bakermans-Kranenburg MJ. DRD4 7-repeat polymorphism moderates the association between maternal unresolved loss or trauma and infant disorganization. Attachment & Human Development. 2006;8:291–307. doi: 10.1080/14616730601048159. [DOI] [PubMed] [Google Scholar]
- Yirmiya N, Gamliel I, Pilowsky T, Feldman R, Baron-Cohen S, Sigman M. The development of siblings of children with autism at 4 and 14 months: Social engagement, communication, and cognition. Journal of Child Psychology and Psychiatry. 2006;47:511–523. doi: 10.1111/j.1469-7610.2005.01528.x. [DOI] [PubMed] [Google Scholar]
- Yuen RKC, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nature Medicine. 2015;21:185–191. doi: 10.1038/nm.3792. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.