

Abstract
Background
Pre-clinical studies have demonstrated that natural and synthetic histone deacetylase (HDAC) inhibitors can impede the in vitro and in vivo growth of cell lines from a variety of gynecologic and other malignancies. We investigated the anti-tumor activity of panobinostat (LBH589) both in vitro and in vivo as either a single agent or in combination with conventional cytotoxic chemotherapy using patient-derived xenograft (PDX) models of primary serous ovarian tumors.
Methods
The ovarian cancer cell lines OVCAR8, SKOV3 and their paclitaxel-resistant derivatives OVCAR8-TR and SKOV3-TR were treated with increasing doses of LBH589. The effect of LBH589 on cell viability was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Serially transplanted primary human high-grade serous ovarian adenocarcinoma tissue was utilized to generate xenografts in 6-week old female NOD/SCID mice. The mice were then randomized into one of 4 treatment groups: (1) vehicle control; (2) paclitaxel and carboplatin (P/C); (3) LBH589; or (4) P/C + LBH589. Mice were treated for 21 days and tumor volumes and mouse weights were obtained every 3 days. These experiments were performed in triplicate with three different patient derived tumors. Wilcoxan rank-sum testing was utilized to assess tumor volume differences.
Results
In vitro treatment with LBH589 significantly reduced the viability of both taxol-sensitive and taxol-resistant ovarian cancer cell lines (p < 0.01). In vivo treatment with LBH589 alone appeared tumorstatic and reduced tumor growth when compared to vehicle treatment (p < 0.007) after 21 days. This single agent activity was confirmed in two additional experiments with other PDX tumors (p < 0.03, p < 0.05). A potential additive effect of LBH589 and P/C, manifested as enhanced tumor regression with the addition of LBH589 compared to vehicle (p < 0.02), in one of the three analyzed serous PDX models.
Conclusions
Our findings suggest that pan-HDAC inhibition with panobinostat precludes the growth of ovarian cancer cell lines in vitro and PDXs in vivo. Added benefit of LBH589 to standard P/C therapy was observed in one of three PDX models suggesting improved response in a subset of serous ovarian cancers.
Keywords: Ovarian cancer, Histone deacetylase inhibitor, Patient derived xenograft model, Conventional chemotherapy
Background
Epithelial ovarian cancer is the second most common, but most lethal gynecologic malignancy in the United States (US) and is estimated to affect approximately 22,280 women and lead to 14,240 deaths in 2016 [1]. Approximately 75 % of patients present with advanced stage disease, a factor largely attributed to the absence of effective screening strategies [2]. At the time of diagnosis, most women will undergo aggressive cytoreductive surgery with the subsequent delivery of platinum based chemotherapy [3]. The combination of carboplatin and paclitaxel is standard first line chemotherapy in the US and, while effective at generating responses in approximately 80 % of women, it is seldom curative [4]. Despite advances in therapy and delivery, recurrence and chemotherapy resistance are still formidable problems as the majority of patients with ovarian cancer who achieve a complete remission with first line platinum-based chemotherapy will ultimately develop recurrent disease that is less responsive to cytotoxic chemotherapy [5]. Finding new molecular targets and exploiting cellular pathways involved in the onset and progression of platinum resistant ovarian cancer will be essential to innovating the treatment of women with this lethal disease [6].
Epigenetic alterations have emerged as key factors in tumorigenesis and may have relevance in the therapy of women with ovarian cancer [7]. Specifically, interfering with gene transcription mechanisms through histone modification may lead to aberrant genetic changes responsible for the development of cancer [8, 9]. During mitosis, histones are modified through acetylation and deacetylation which serve to regulate chromosomal segregation and DNA access to transcription factors [10]. Acetylation causes a conformational change in the histone N-terminal tail resulting in displacement of the histone away from the DNA strand and increased transcriptional activation [11]. Histone deacetylases (HDAC) reverse this process and prevent interactions between transcription factors and DNA [12]. Multiple classes of HDACs have been described and have been shown to exert their effects in various tissues and cellular components [10].
Recognized as potent epigenetic inhibitors, HDAC inhibitors appear to exert anti-tumor effects through hyperacetylation of histones and demethylation of genomic DNA resulting in reactivation of genes that inhibit proliferation [10, 13, 14]. Both transcriptional and non-transcriptional mechanisms of action have been investigated [15]. Pre-clinical studies using cell lines from a variety of gynecologic [16–18] and other malignancies [19, 20] have demonstrated that natural and synthetic HDAC inhibitors can inhibit tumor cell growth in vitro and in vivo through cell cycle arrest as well as the induction of mitotic defects through histone mediated and histone independent interactions [15]. Investigations utilizing in vitro and in vivo models of ovarian cancer have demonstrated that HDAC inhibition synergizes with conventional chemotherapies to induce potent cytotoxic effects supporting the potential use of this combination in the clinic [18, 21–25]. Early clinical investigations determined that HDAC inhibitors were well tolerated therapeutics with single agent bioactivity against a variety of hematologic malignancies leading to the United States Food and Drug Administration approval of SAHA (Vorinistat) for the treatment of cutaneous T-cell lymphoma [26]. Although only modest single agent activity has been noted in solid tumors in Phase I and II human trials [7, 24, 27–31], mounting preclinical data suggest HDAC inhibition could be effective for patients with ovarian cancer [17, 32]. Relevant in vitro and in vivo models will be required to gain more insight into both the mechanisms of drug action and optimal combinations with conventional chemotherapy [22].
Investigators have observed that HDAC inhibition may be mediated in some cells by modulating expression of the Aurora A serine/threonine kinase [15, 33]. Aurora A is a key regulator of mitotic spindle cell formation and chromosomal segregation and is therefore critical to proper cell cycle progression [34, 35]. Located on chromosome 20q13.2, the AURKA gene has been noted to be amplified in several human epithelial tumors and has been an attractive target for developmental therapeutics [36–39]. In ovarian cancer, AURKA is amplified in several cell lines and amplification has been shown to correlate with poor prognosis [40, 41]. Additionally, investigators have shown that elevated Aurora A protein expression overrides the checkpoint mechanism that monitors mitotic spindle assembly and is involved in the development of resistance to paclitaxel [42]. Treatment of cancer cells with HDAC inhibitors resulted in a down regulation of Aurora A protein levels suggesting that the most robust responses to HDAC inhibition may be observed in those patients whose tumors exhibit heightened Aurora A expression [15]. Preliminary in vitro data suggests that HDAC inhibition potentiates the effects of Aurora A expression and can sensitize to Aurora kinase inhibitors in ovarian cancer cell line models [43].
In preliminary studies using the HDAC inhibitors trichostatin A (TSA) and SAHA, we demonstrated significant anti-tumor activity in PDX models of high grade serous ovarian cancer. We sought to explore the possibility that LBH589, a potent inhibitor of class I, II, and IV HDAC enzymes in clinical trial, may act to inhibit tumor cell growth through the degradation of Aurora A. Furthermore, we hypothesized that the administration of LBH589 in concert with conventional cytotoxic chemotherapy would manifest synergistic activity in a subset of serous ovarian cancer PDX models. This investigation sought to provide rationale for pursuing HDAC inhibition in a subset of women with serous ovarian cancer.
Methods
Cell culture and cell growth inhibition assay
The human ovarian cancer cell lines OVCAR-8, SKOV3, OVCAR8-TR and SKOV3-TR [44] were grown and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM 1X, 10 % fetal bovine serum [FBS], 1 % P/S). Cells were seeded in triplicate on 24-well plates and then treated with escalating doses of SAHA or LBH589 for 48 h at 37 °C. Cell viability was then assessed by MTT assay as previously described [45].
Tumor collection and propagation in vivo
Excess human serous ovarian tumor samples were obtained through an IRB approved centralized banking infrastructure at the Massachusetts General Hospital (MGH). Written informed consent was received from all participants. Tumor was enzymatically processed to achieve a single cell suspension and then depleted of hematologic components as described [46]. A specified number of cells were suspended in PBS:Matrigel® (1:1) and injected subcutaneously (s.c.) into 6–8 week old female NOD/SCID mice (Jackson Laboratory, Bar Harbor, ME). Animals were monitored continually to assess tumor formation and size, and euthanized when they became moribund or had excessive tumor burden. All animal experiments were approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee. For continued propagation in mice, the xenograft tumors were excised and enzymatically processed to a single cell suspension. The suspension was depleted of mouse H2Kd+ cells and the remaining tumor-derived cells were re-injected subcutaneously into new recipient NOD/SCID mice as described [46]. All of the primary human papillary serous ovarian tumors utilized in this study had undergone 4–5 passages in vivo and the serous histology of each was maintained over the serial transplantation process. Animals were housed and maintained in accordance with institutional guidelines.
Treatment with LBH589 alone and in combination
Mice bearing matched sized tumors (300–600 mm3) from three independent human papillary serous ovarian cancers (OV1, OV2, and OV3) were randomized into four cohorts of six mice each. The four groups were assigned to the following treatment regimens: (1) intraperitoneal (IP) injection of paclitaxel and carboplatin (P/C) vehicle one time per week + IP injection of LBH589 vehicle five times per week; 2) IP injection of paclitaxel (15 mg/kg) and carboplatin (50 mg/kg) one time per week + IP injection of LBH589 vehicle five times per week; (3) IP injection of P/C vehicle one time per week + IP injection of LBH589 (2.5 mg/kg) five times per week; or (4) IP injection of P/C one time per week + IP injection ofLBH589 five times per week. Mice were treated for 21 days with tumor volumes and mouse weights obtained every 3 days. At the end of the treatment period, animals were euthanized and portions of harvested tumors were snap frozen for protein analysis and embedded in paraffin for histological analysis.
Immunoblotting
For the in vitro cell line experiments, plated OVCAR8 and SKOV3 cells were treated with LBH586 (7.5 nM) or SAHA (2 μM) for 16 h and total cell lysates were prepared. A total of 10–20 μg of protein from each sample was electrophoresed on a precast Tris–Hepes–SDS polyacrylamide gel (Pierce, Thermo Scientific) and transferred onto a PVDF membrane. The membrane was blocked in 5 % non-fat dry milk prepared in TBST. Primary monoclonal antibodies directed against Aurora A kinase, acetylated tubulin and acetylated histone H3 were obtained from Cell Signaling Technology and used according to the manufacturer’s recommendations. Following incubation overnight at 4 °C, each blot was washed, incubated with the appropriate secondary antibody and developed using enhanced chemiluminescence.
Statistical analysis
Non-parametric Wilcoxan rank sum tests for unpaired samples were used to compare tumor sizes in the LBH589 treatment xenograft experiments. Two-way ANOVA analysis and Student’s t-tests were used to determine the statistical significance of results obtained in the MTT analyses of cell viability. Significance was set at p ≤ 0.05. STATA (College Station, TX) v10 software was used.
Results
LBH589 reduces ovarian cancer cell viability in vitro
Exposure to increasing doses of LBH589 decreased cell viability of both chemosensitive and chemoresistant ovarian cancer cell lines [44] (Fig. 1, p < 0.01). Treatment with 7.5 nM LBH589 led to a 50 % reduction in the viability of OVCAR8 cells while OVCAR8-TR cells were significantly less sensitive to LBH589 (50 % reduction around 7.5 nM, p < 0.01). A 50 % reduction in the viability of SKOV3 and SKOV3-TR cells was observed at LBH589 concentrations of 25 nM and 100 nM, respectively (p < 0.01). We compared the potency of LBH589 to the only established FDA approved HDAC inhibitor suberoylanilide hydroxamic acid (SAHA, Vorinistat) and observed that the concentrations required for inhibition of OVCAR8 and SKOV3 cell growth were approximately 2-fold less for LBH589. These results are summarized in Fig. 1.
Fig. 1.
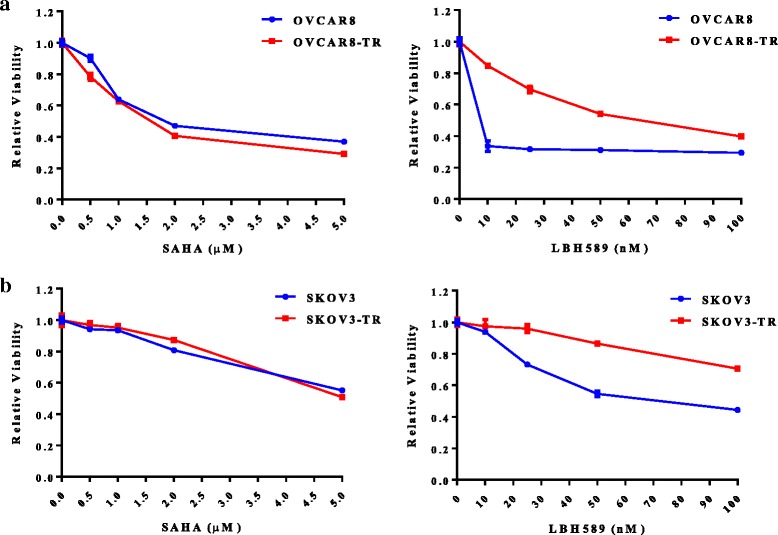
HDAC inhibition reduces the viability of ovarian cancer cell lines in vitro. The ovarian cancer cell lines OVCAR8 (a), SKOV3 (b) and their paclitaxel resistant derivatives OVCAR8-TR (a) and SKOV-3-TR (b) were treated in triplicate with the indicated concentrations of SAHA or LBH589 for 48 h. Relative viability was assessed by MTT assay. Data plotted are mean +/− SEM
Treatment with LBH589 and SAHA alters acetylated tubulin and Aurora A protein expression in vitro
Acetylated tubulin and acetylated histone H3 are known HDAC targets [47, 48]. We therefore analyzed the effect of LBH589 on acetylation of these proteins to validate that LBH behaves like other HDAC inhibitors and compared any observed effect to that obtained with SAHA. As shown in Fig. 2, treatment with both LBH586 and SAHA led to increased levels of acetylated tubulin and histone H3. We similarly analyzed Aurora A kinase levels following HDAC inhibition. Although Aurora-A kinase was detected in the OVCAR8 cell line, no significant change in its levels was observed following treatment with LBH589. In contrast, SAHA treatment led to a marked reduction in Aurora A kinase levels in OVCAR8. We detected no Aurora A kinase expression in SKOV3 cells.
Fig. 2.
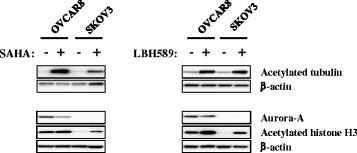
Effect of in vitro HDAC inhibition on acetylated histone and tubulin. OVCAR8 and SKOV3 cells were treated with 2 μM SAHA, 7.5 nM LBH589 or vehicle for 16 h. Total cellular protein was isolated and subjected to Western blot analysis to determine the relative levels of acetylated tubulin, acetylated histone H3 and Aurora-A kinase. β-actin was used as a protein loading control
HDAC inhibition blocks the growth of primary human serous ovarian tumor xenografts in vivo
We assessed the single agent activity of LBH589 as well as the potential therapeutic synergy of LBH589 and paclitaxel and carboplatin (P/C) combination therapy. These analyses were carried out with xenografts derived from three individual patient tumors. All of the patients were diagnosed with advanced stage, high grade serous ovarian cancer and had undergone primary upfront surgical cytoreduction (Table 1). To assess the single agent activity of LBH589, we treated mice harboring human serous ovarian cancer xenografts with either LBH589 or vehicle and regularly assessed the effect on tumor volume. Treatment with LBH589 alone appeared tumorstatic when compared to vehicle (OV1, p < 0.007; OV2, p < 0.03; OV3, p < 0.05) after 21 days (Fig. 3).
Table 1.
The clinical characteristics of the patients from whom the high grade serous ovarian carcinoma samples were obtained
| Patient | Age at diagnosis (Years) | Stage | Grade | Progression free survival (Months) | Overall survival (Months) | Current status |
|---|---|---|---|---|---|---|
| OV1 | 59 | IV | 3 | 22.8 | 74.4 | Deceased |
| OV2 | 64.5 | IIIC | 3 | N/A | N/A | N/A |
| OV3 | 38.3 | IV | 3 | 34.1 | 86.4 | Alive |
All patients underwent primary debulking surgery for advanced stage ovarian cancer and had optimal cytoreduction. One patient was lost to follow up and therefore recurrence and overall survival data are not available
Fig. 3.
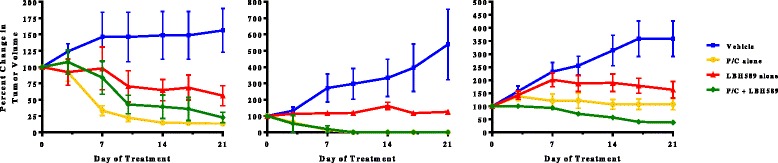
Pre-clinical analyses of LBH589 activity in vivo. Separate experiments utilizing PDXs derived from three individual patients are shown. Each evaluated the activity of LBH589 as a single agent and in combination with carboplatin and paclitaxel (P/C) chemotherapy. In all experiments, LBH589 induced statistically significant tumorstasis (p < 0.007, p < 0.03, p < 0.05). In one experiment (OV3), increased tumor regression was observed when LBH589 and P/C were combined (p < 0.02)
We then assessed whether LBH589 would have additional antitumor effects when combined with the standard of care chemotherapy regimen of paclitaxel and carboplatin (P/C). The OV3 PDXs demonstrated further tumor regression with the addition of LBH589 to P/C compared to P/C or LBH586 alone, (p < 0.02). This effect, however, was not confirmed with the OV1 and OV2 serous PDXs. In the two experiments where LBH589 did not show any additional benefit, treatment with P/C either alone or in combination with LBH589 resulted in complete tumor regression in all animals by day 21. In the majority of treatment arms, the animals lost an average of 10 % of their starting body weight with non-statistically significant weight loss in any arm receiving single agent P/C (p < 0.07) or LBH (p = 0.06). The most significant weight loss was observed in those animals receiving combination therapy (p < 0.005) (data not shown).
Discussion
These data suggest that single agent HDAC inhibition with LBH589 leads to tumorstatic effects in primary human xenografts derived from patients diagnosed with high-grade serous ovarian cancer. This pattern of tumor inhibition was demonstrated in triplicate across three separate high grade serous tumors obtained from patients who underwent optimal upfront surgical cytoreduction. Combination of LBH589 with conventional cytotoxic P/C, the current platform for the treatment of women with epithelial serous ovarian cancer, resulted in significant tumor regression in one of the analyzed PDX models. These data suggest that that HDAC inhibition may potentiate the activity of cytotoxic therapy in a subset of high grade serous ovarian tumors. These in vivo observations were confirmed by in vitro experiments with immortalized ovarian cancer cell lines OVCAR8 and SKOV3, as well as paclitaxel resistant derivatives of these lines [44] and echoes many recent reports that suggest HDAC inhibition can potentiate conventional and targeted therapeutics.
Numerous reports have implicated heightened HDAC activity, histone hypo-acetylation and subsequent silencing of tumor suppressor transcription with malignant transformation [49, 50]. Researchers have also proposed that in addition to histone mediated alterations, HDACs may induce DNA independent oncogenic changes to other proteins involved in cell cycle and apoptosis, such as p53, c-Myc, heat shock protein 90 (HSP90) and tubulin [15, 17, 47–49, 51, 52]. In ovarian cancer, heightened expression of many HDAC class proteins has been described in 60–90 % of tumors analyzed and many histone and non-histone mediated alterations have been described that alter the balance in favor of cellular growth and survival [17, 53]. Our data confirm that LBH589 treatment, like SAHA, leads to significant acetylation of both histone and a non-histone protein. While therapy with SAHA led to decreases in Aurora-A expression, LBH589 failed to modify the expression levels. LBH589 treatment led to significant decreases in cell viability at lower concentrations than that observed with SAHA suggesting a higher potency. This activity in a spectrum of immortalized ovarian cancer cell lines confirms the experience of other investigators using other HDAC inhibitors [16, 17, 22]. Though some investigators have suggested Aurora-A may modulate HDAC inhibition of cancer cell proliferation [15], our data suggest LBH589 mediates anti-tumor effects via alternative mechanisms. Collectively, we observed that LBH589 exerts potent cancer cell control mediated through histone and non-histone molecular modifications.
While the exact mechanism HDAC inhibition employs to induce cell cycle arrest and apoptosis of ovarian cancer cells remains elusive, much preclinical data supports that HDAC inhibition synergizes with many cytotoxic chemotherapies including paclitaxel, carboplatin and docetaxel [25, 47, 54–57]. Investigators have hypothesized that synergy may be due to both histone hyperacetylation fostering intercalation of platinum as well as the heightened acetylated tubulin stabilization reinforced by both taxanes and HDAC inhibitors [17]. Others have suggested that HDAC inhibition potentiates cytotoxic chemotherapies by increasing double stranded breaks, precluded homologous repair and blocking the phosphotidylinositol 3-kinase (PI3K) pathway in in vitro ovarian cancer cells [57]. Recent studies have also implicated modulation of the multidrug resistance protein (MDR1), epidermal growth factor receptor (EGFR) and tumor initiating cells in order to facilitate complementary action with both targeted and cytotoxic therapeutics [25, 58, 59].
These preclinical data provide context for our in vivo experiments utilizing a novel PDX model of serous ovarian cancer. Our studies confirmed that LBH589 precluded tumor growth compared to vehicle in three different tumors from women who underwent primary cytoreduction at our institution and suggest only a subset of high grade serous carcinomas will manifest heightened tumor regression if chemotherapy is administered in the setting of tonic HDAC inhibition. Importantly, we observed a tumorstatic response to P/C therapy in the OV3 PDXs only. In OV1 and OV2 PDXs P/C alone induced significant tumor regression. The observed toxicity of the treatments as manifested by mouse weight loss suggested that single agent LBH589 was well tolerated. These findings confirm numerous other studies that demonstrated that HDAC inhibitors were well tolerated by the animal models [17, 21, 22]. These observations provide rationale for the addition of HDAC inhibition in the setting of stable disease responses to conventional cytotoxic therapy. In the clinic, HDAC inhibition with SAHA (Vorinistat) has been combined with carboplatin and gemcitabine in a phase I trial that demonstrated partial responses in 6 out 7 women with recurrent ovarian cancer, though the trial was stopped early due to excessive toxicity [32].
Conclusions
To our knowledge, this is the first report testing HDAC inhibition in concert with conventional chemotherapy in a PDX model derived from patients with high grade serous ovarian cancer. We believe this model has noteworthy clinical relevance and lends support to previous investigations utilizing immortalized cell lines. Our data showed that LBH589 had significant potency as an HDAC inhibitor, and that it induced hyperacetylation in histone and tubulin proteins while decreasing cell viability. The in vivo experiments confirmed a single agent tumorstatic effect of LBH589, and demonstrated, in a subset of our PDX models, an enhanced effect on tumor regression when combined with P/C. This investigation highlights HDAC inhibition with LBH589 as a promising avenue for innovating the treatment of ovarian cancer. Future study of LBH589 as a sequential or maintenance therapy will likely be of merit to further define how best to utilize this specialized therapeutic.
Acknowledgements
We thank Ling Zhang, Lankai Guo and Minji Kim for expert technical assistance. We also thank Vanessa Therrien for patient sample collection. Funding provided by the Advanced Medical Research Fund (AMRF). LBH589 generously provided by Novartis Pharmaceuticals.
Funding
This study was funded by the American Medical Research Foundation which provided support for study design, data collection, analysis and interpretation and manuscript writing.
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article and its additional files.
Authors’ contributions
LAG: Experimental design, experiment execution, manuscript composition. WBG: Experimental design, data analysis and interpretation, manuscript composition. BRR: Experimental design, data analysis and interpretation, manuscript editing. RF: Experimental design, data analysis and interpretation, manuscript editing. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All patient tissues used in this study were obtained following informed consent of participating subjects. The tissue collection protocol was reviewed and approved by the Dana-Farber/Harvard Cancer Center IRB (protocol #07-049).
Abbreviations
- DMEM
Dulbecco’s Modified Eagle’s Medium
- HDAC
Histone deacetylase
- HSP90
Heat shock protein 90
- LBH589
Panobinostat
- P/C
Paclitaxel and carboplatin
Contributor Information
Leslie A. Garrett, Email: lgarret1@bidmc.harvard.edu
Whitfield B. Growdon, Email: wgrowdon@mgh.harvard.edu
Bo R. Rueda, Email: brueda@mgh.harvard.edu
Rosemary Foster, Email: Rfoster1@mgh.harvard.edu.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Schorge JO, Modesitt SC, Coleman RL, Cohn DE, Kauff ND, Duska LR, et al. SGO white paper on ovarian cancer: etiology, screening and surveillance. Gynecol Oncol. 2010;119:7–17. doi: 10.1016/j.ygyno.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Schorge JO, Bradford LS, del Carmen MG. Primary cytoreductive surgery for advanced ovarian cancer: is it the past, present, or future? Clin Adv Hematol Oncol. 2011;9:912–8. [PubMed] [Google Scholar]
- 4.Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2003;21:3194–200. doi: 10.1200/JCO.2003.02.153. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354:34–43. doi: 10.1056/NEJMoa052985. [DOI] [PubMed] [Google Scholar]
- 6.Growdon WB, Birrer MJ, Penson RT. Ovarian cancer: individualized and personalized care. Expert Rev Obstet Gynecol. 2010;5:409–19. doi: 10.1586/eog.10.35. [DOI] [Google Scholar]
- 7.Al-Janadi A, Chandana SR, Conley BA. Histone deacetylation: an attractive target for cancer therapy? Drugs R D. 2008;9:369–83. doi: 10.2165/0126839-200809060-00003. [DOI] [PubMed] [Google Scholar]
- 8.Kwon MJ, Shin YK. Epigenetic regulation of cancer-associated genes in ovarian cancer. Int J Mol Sci. 2011;12:983–1008. doi: 10.3390/ijms12020983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maradeo ME, Cairns P. Translational application of epigenetic alterations: ovarian cancer as a model. FEBS Lett. 2011;585:2112–20. doi: 10.1016/j.febslet.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–68. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 11.Gregory PD, Wagner K, Horz W. Histone acetylation and chromatin remodeling. Exp Cell Res. 2001;265:195–202. doi: 10.1006/excr.2001.5187. [DOI] [PubMed] [Google Scholar]
- 12.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 13.Frew AJ, Johnstone RW, Bolden JE. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett. 2009;280:125–33. doi: 10.1016/j.canlet.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 14.Camphausen K, Tofilon PJ. Inhibition of histone deacetylation: a strategy for tumor radiosensitization. J Clin Oncol. 2007;25:4051–6. doi: 10.1200/JCO.2007.11.6202. [DOI] [PubMed] [Google Scholar]
- 15.Park JH, Jong HS, Kim SG, Jung Y, Lee KW, Lee JH, et al. Inhibitors of histone deacetylases induce tumor-selective cytotoxicity through modulating Aurora-A kinase. J Mol Med (Berl) 2008;86:117–28. doi: 10.1007/s00109-007-0260-8. [DOI] [PubMed] [Google Scholar]
- 16.Takai N, Narahara H. Human endometrial and ovarian cancer cells: histone deacetylase inhibitors exhibit antiproliferative activity, potently induce cell cycle arrest, and stimulate apoptosis. Curr Med Chem. 2007;14:2548–53. doi: 10.2174/092986707782023299. [DOI] [PubMed] [Google Scholar]
- 17.Singh BN, Zhou H, Li J, Tipton T, Wang B, Shao G, et al. Preclinical studies on histone deacetylase inhibitors as therapeutic reagents for endometrial and ovarian cancers. Future Oncol. 2011;7:1415–28. doi: 10.2217/fon.11.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budman DR, Tai J, Calabro A, John V. The histone deacetylase inhibitor panobinostat demonstrates marked synergy with conventional chemotherapeutic agents in human ovarian cancer cell lines. Invest New Drugs. 2011;29:1224–9. doi: 10.1007/s10637-010-9467-6. [DOI] [PubMed] [Google Scholar]
- 19.Chen X, Wong P, Radany E, Wong JY. HDAC inhibitor, valproic acid, induces p53-dependent radiosensitization of colon cancer cells. Cancer Biother Radiopharm. 2009;24:689–99. doi: 10.1089/cbr.2009.0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LaBonte MJ, Wilson PM, Fazzone W, Groshen S, Lenz HJ, Ladner RD. DNA microarray profiling of genes differentially regulated by the histone deacetylase inhibitors vorinostat and LBH589 in colon cancer cell lines. BMC Med Genet. 2009;2:67. doi: 10.1186/1755-8794-2-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooper AL, Greenberg VL, Lancaster PS, van Nagell JR, Jr, Zimmer SG, Modesitt SC. In vitro and in vivo histone deacetylase inhibitor therapy with suberoylanilide hydroxamic acid (SAHA) and paclitaxel in ovarian cancer. Gynecol Oncol. 2007;104:596–601. doi: 10.1016/j.ygyno.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 22.Modesitt SC, Parsons SJ. In vitro and in vivo histone deacetylase inhibitor therapy with vorinostat and paclitaxel in ovarian cancer models: does timing matter? Gynecol Oncol. 2010;119:351–7. doi: 10.1016/j.ygyno.2010.06.030. [DOI] [PubMed] [Google Scholar]
- 23.Marchion D, Munster P. Development of histone deacetylase inhibitors for cancer treatment. Expert Rev Anticancer Ther. 2007;7:583–98. doi: 10.1586/14737140.7.4.583. [DOI] [PubMed] [Google Scholar]
- 24.Qiu T, Zhou L, Zhu W, Wang T, Wang J, Shu Y, et al. Effects of treatment with histone deacetylase inhibitors in solid tumors: a review based on 30 clinical trials. Future Oncol. 2013;9:255–69. doi: 10.2217/fon.12.173. [DOI] [PubMed] [Google Scholar]
- 25.Liu Z, Tong Y, Liu Y, Liu H, Li C, Zhao Y, et al. Effects of suberoylanilide hydroxamic acid (SAHA) combined with paclitaxel (PTX) on paclitaxel-resistant ovarian cancer cells and insights into the underlying mechanisms. Cancer Cell Int. 2014;14:112. doi: 10.1186/s12935-014-0112-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marks PA, Dokmanovic M. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs. 2005;14:1497–511. doi: 10.1517/13543784.14.12.1497. [DOI] [PubMed] [Google Scholar]
- 27.Fukutomi A, Hatake K, Matsui K, Sakajiri S, Hirashima T, Tanii H, et al. A phase I study of oral panobinostat (LBH589) in Japanese patients with advanced solid tumors. Invest New Drugs. 2012;30:1096–106. doi: 10.1007/s10637-011-9666-9. [DOI] [PubMed] [Google Scholar]
- 28.Hainsworth JD, Infante JR, Spigel DR, Arrowsmith ER, Boccia RV, Burris HA. A phase II trial of panobinostat, a histone deacetylase inhibitor, in the treatment of patients with refractory metastatic renal cell carcinoma. Cancer Invest. 2011;29:451–5. doi: 10.3109/07357907.2011.590568. [DOI] [PubMed] [Google Scholar]
- 29.Glaser KB. HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol. 2007;74:659–71. doi: 10.1016/j.bcp.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 30.Batty N, Malouf GG, Issa JP. Histone deacetylase inhibitors as anti-neoplastic agents. Cancer Lett. 2009;280:192–200. doi: 10.1016/j.canlet.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 31.Jones SF, Bendell JC, Infante JR, Spigel DR, Thompson DS, Yardley DA, et al. A phase I study of panobinostat in combination with gemcitabine in the treatment of solid tumors. Clin Adv Hematol Oncol. 2011;9:225–30. [PubMed] [Google Scholar]
- 32.Matulonis U, Berlin S, Lee H, Whalen C, Obermayer E, Penson R, et al. Phase I study of combination of vorinostat, carboplatin, and gemcitabine in women with recurrent, platinum-sensitive epithelial ovarian, fallopian tube, or peritoneal cancer. Cancer Chemother Pharmacol. 2015;76:417–23. doi: 10.1007/s00280-015-2813-9. [DOI] [PubMed] [Google Scholar]
- 33.Wang C, Chen J, Cao W, Sun L, Sun H, Liu Y. Aurora-B and HDAC synergistically regulate survival and proliferation of lymphoma cell via AKT, mTOR and Notch pathways. Eur J Pharmacol. 2016;779:1–7. [DOI] [PubMed]
- 34.Fu J, Bian M, Jiang Q, Zhang C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5:1–10. doi: 10.1158/1541-7786.MCR-06-0208. [DOI] [PubMed] [Google Scholar]
- 35.Warner SL, Bearss DJ, Han H, Von Hoff DD. Targeting Aurora-2 kinase in cancer. Mol Cancer Ther. 2003;2:589–95. doi: 10.4161/cbt.2.5.506. [DOI] [PubMed] [Google Scholar]
- 36.Doggrell SA. Dawn of Aurora kinase inhibitors as anticancer drugs. Expert Opin Investig Drugs. 2004;13:1199–201. doi: 10.1517/13543784.13.9.1199. [DOI] [PubMed] [Google Scholar]
- 37.Sen S, Zhou H, Zhang RD, Yoon DS, Vakar-Lopez F, Ito S, et al. Amplification/overexpression of a mitotic kinase gene in human bladder cancer. J Natl Cancer Inst. 2002;94:1320–9. doi: 10.1093/jnci/94.17.1320. [DOI] [PubMed] [Google Scholar]
- 38.Aust DE, Muders M, Kohler A, Schmidt M, Diebold J, Muller C, et al. Prognostic relevance of 20q13 gains in sporadic colorectal cancers: a FISH analysis. Scand J Gastroenterol. 2004;39:766–72. doi: 10.1080/00365520410003191. [DOI] [PubMed] [Google Scholar]
- 39.Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer. 2004;4:927–36. doi: 10.1038/nrc1502. [DOI] [PubMed] [Google Scholar]
- 40.Hu W, Kavanagh JJ, Deaver M, Johnston DA, Freedman RS, Verschraegen CF, et al. Frequent overexpression of STK15/Aurora-A/BTAK and chromosomal instability in tumorigenic cell cultures derived from human ovarian cancer. Oncol Res. 2005;15:49–57. doi: 10.3727/096504005775082101. [DOI] [PubMed] [Google Scholar]
- 41.Landen CN, Jr, Lin YG, Immaneni A, Deavers MT, Merritt WM, Spannuth WA, et al. Overexpression of the centrosomal protein Aurora-A kinase is associated with poor prognosis in epithelial ovarian cancer patients. Clin Cancer Res. 2007;13:4098–104. doi: 10.1158/1078-0432.CCR-07-0431. [DOI] [PubMed] [Google Scholar]
- 42.Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003;3:51–62. doi: 10.1016/S1535-6108(02)00235-0. [DOI] [PubMed] [Google Scholar]
- 43.Li Y, Liu T, Ivan C, Huang J, Shen DY, Kavanagh JJ, et al. Enhanced cytotoxic effects of combined valproic acid and the aurora kinase inhibitor VE465 on gynecologic cancer cells. Front Oncol. 2013;3:58. doi: 10.3389/fonc.2013.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duan Z, Lamendola DE, Duan Y, Yusuf RZ, Seiden MV. Description of paclitaxel resistance-associated genes in ovarian and breast cancer cell lines. Cancer Chemother Pharmacol. 2005;55:277–85. doi: 10.1007/s00280-004-0878-y. [DOI] [PubMed] [Google Scholar]
- 45.van Meerloo J, Kaspers GJ, Cloos J. Cell sensitivity assays: the MTT assay. Methods Mol Biol. 2011;731:237–45. doi: 10.1007/978-1-61779-080-5_20. [DOI] [PubMed] [Google Scholar]
- 46.Curley MD, Therrien VA, Cummings CL, Sergent PA, Koulouris CR, Friel AM, et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells. 2009;27:2875–83. doi: 10.1002/stem.236. [DOI] [PubMed] [Google Scholar]
- 47.Arts J, Angibaud P, Marien A, Floren W, Janssens B, King P, et al. R306465 is a novel potent inhibitor of class I histone deacetylases with broad-spectrum antitumoral activity against solid and haematological malignancies. Br J Cancer. 2007;97:1344–53. doi: 10.1038/sj.bjc.6604025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, et al. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–79. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18:769–74. [PubMed] [Google Scholar]
- 50.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 51.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/S0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 52.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–7. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 53.Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168–76. doi: 10.1016/j.canlet.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 54.Qian DZ, Kato Y, Shabbeer S, Wei Y, Verheul HM, Salumbides B, et al. Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res. 2006;12:634–42. doi: 10.1158/1078-0432.CCR-05-1132. [DOI] [PubMed] [Google Scholar]
- 55.Chobanian NH, Greenberg VL, Gass JM, Desimone CP, Van Nagell JR, Zimmer SG. Histone deacetylase inhibitors enhance paclitaxel-induced cell death in ovarian cancer cell lines independent of p53 status. Anticancer Res. 2004;24:539–45. [PubMed] [Google Scholar]
- 56.Ganai SA. Panobinostat: the small molecule metalloenzyme inhibitor with marvelous anticancer activity. Curr Top Med Chem. 2016;16:427–34. doi: 10.2174/1568026615666150813145800. [DOI] [PubMed] [Google Scholar]
- 57.Chao H, Wang L, Hao J, Ni J, Chang L, Graham PH, et al. Low dose histone deacetylase inhibitor, LBH589, potentiates anticancer effect of docetaxel in epithelial ovarian cancer via PI3K/Akt pathway in vitro. Cancer Lett. 2013;329:17–26. doi: 10.1016/j.canlet.2012.08.035. [DOI] [PubMed] [Google Scholar]
- 58.Liffers K, Kolbe K, Westphal M, Lamszus K, Schulte A. Histone deacetylase inhibitors resensitize EGFR/EGFRvIII-overexpressing, erlotinib-resistant glioblastoma cells to tyrosine kinase inhibition. Target Oncol. 2015. [Epub ahead of print]. [DOI] [PubMed]
- 59.Schech A, Kazi A, Yu S, Shah P, Sabnis G. Histone deacetylase inhibitor entinostat inhibits tumor-initiating cells in triple-negative breast cancer cells. Mol Cancer Ther. 2015;14:1848–57. doi: 10.1158/1535-7163.MCT-14-0778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset supporting the conclusions of this article is included within the article and its additional files.