

Abstract
Background
Silicone implants have been successfully used for breast augmentation and reconstruction in millions of women worldwide. The reaction to the silicone implant is highly variable; it can lead to local inflammatory symptoms, and sometimes to systemic symptoms and disease. Over 80 cases of anaplastic lymphoma kinase-negative anaplastic large cell lymphoma have been reported in patients with silicone breast implants and have been accepted as a new clinical entity. To the best of our knowledge, an intravascular large B-cell lymphoma associated with a silicone breast implant has not been reported previously.
Case presentation
A 48-year-old Caucasian woman who presented with high fever was found to have splenomegaly on physical examination. A laboratory diagnosis revealed pancytopenia, hypertriglyceridemia, and hyperferritinemia. She developed signs of altered sensorium, hemiparesis, aphasia, and cauda equina syndrome. On further evaluation, she fulfilled the necessary five out of eight criteria for diagnosis of macrophage activation syndrome/hemophagocytic lymphohistiocytosis. Dexamethasone administration was followed by prompt improvement; however, 3 days later she again manifested high fever, which persisted despite administration of immunoglobulin and cyclosporine A. Her silicone breast implant was considered a possible contributor to her macrophage activation syndrome and was therefore removed. A histological examination of the capsule tissue showed an extensive lymphohistiocytic/giant cell foreign body reaction suggestive of autoimmune/inflammatory syndrome induced by adjuvants. However, the histological examination unexpectedly also revealed an intravascular large B-cell lymphoma.
Conclusions
The genetic background of our patient with silicone breast implants might have predisposed her to three rare and difficult to diagnose syndromes/diseases: macrophage activation syndrome/hemophagocytic lymphohistiocytosis, autoimmune/inflammatory syndrome induced by adjuvants, and intravascular large B-cell lymphoma. The simultaneous manifestation of all three syndromes suggests causal interrelationships. Human leukocyte antigen testing in all women who undergo silicon breast implantation could in the future enable us to better evaluate the risk of potential side effects.
Electronic supplementary material
The online version of this article (doi:10.1186/s13256-016-0993-5) contains supplementary material, which is available to authorized users.
Keywords: Silicone breast implant, Siliconosis, Autoimmune inflammatory syndrome induced by adjuvants (ASIA), Macrophage activation syndrome (MAS), Hemophagocytic lymphohistiocytosis (HLH), Intravascular large B-cell lymphoma (IVLBCL)
Background
Hemophagocytic lymphohistiocytosis (HLH), also referred to as hemophagocytic syndrome, is a life-threatening hyperinflammatory syndrome that can occur in many underlying conditions: homozygous mutations affecting the cytotoxic function of natural killer (NK) cells and cytotoxic T cells (CTLs), infections, autoimmune diseases, malignancies, metabolic diseases, acquired immunodeficiency such as AIDS, iatrogenic immunosuppression, and solid organ or stem cell transplantation [1–3]. It is caused by an exaggerated or persistent immune stimulation and/or the failure to downregulate or to end an immune response [1]. It can be seen as a cytokine storm disorder, representing a variety of inflammatory etiologies with the final common result of overwhelming inflammation, hemodynamic instability, multiple organ failure, and potentially death [4]. HLH can be differentiated into primary (also called familial, hereditary, or genetic) and secondary (also called sporadic, reactive, or acquired) HLH [1, 2]. For the diagnostic criteria proposed by the Histiocyte Society in 2004, see Table 1 [5]. Macrophage activation syndrome (MAS) is an acquired form of HLH that occurs in autoimmune diseases, and it is mostly reported in patients with systemic juvenile idiopathic arthritis (sJIA), and less commonly in those with systemic lupus erythematosus (SLE), adult-onset Still’s disease (AOSD), rheumatoid arthritis (RA), spondyloarthropathy, and vasculitis [1, 6, 7]. There are no universally accepted criteria for MAS, thus many clinicians simply refer to the HLH criteria. Diagnosis of MAS can be difficult and it is hard to distinguish from flares of the underlying disease, and from sepsis-like syndromes, although it may also be associated with sepsis, especially in cases of intraphagocytic infections [6]. However, preliminary MAS diagnostic guidelines have shown a better capacity to identify MAS in sJIA and to differentiate it from systemic infections compared to the 2004-HLH diagnostic criteria [8].
Table 1.
Diagnostic criteria for hemophagocytic lymphohistiocytosis
| Diagnosis of HLH is based on the presence of 5 or more of the following: • Fever • Splenomegaly • Cytopenias (affecting 2 of 3 lineages in the peripheral blood) Hemoglobin <90 g/L Platelets <100 × 109/L Neutrophils <1.0 × 109/L • Hypertriglyceridemia and/or hypofibrinogenemia Fasting triglycerides >3.0 mmol/L (>263 mg/dL) Fibrinogen <1.5 g/L • Hemophagocytosis in bone marrow, spleen, or lymph nodes • Low or absent NK-cell activity • Ferritin >500 μg/L • Soluble IL-2 receptor >2400 U/mL |
Adapted from the Histiocyte Society HLH-2004 protocol [5]
HLH hemophagocytic lymphohistiocytosis, IL interleukin, NK natural killer
Autoimmune/inflammatory syndrome induced by adjuvants (ASIA) or Shoenfeld’s syndrome is a recently coined term for a spectrum of syndromes that includes macrophage myofascitis syndrome, Gulf war syndrome, post-vaccination phenomena, and siliconosis [9–11]. For the proposed criteria for the diagnosis of ASIA, see Table 2 [9, 12]. Silicone implants have been successfully used for breast augmentation and reconstruction in millions of women worldwide [13]. However, case reports and case series on side effects associated with silicone breast implants (SBIs) were published soon after their first application [14–24]. Anecdotal cases of breast cancer following SBI have been reported, too [25–27], however, epidemiological studies have not shown any evidence correlating malignancies with SBIs [28, 29]. Nevertheless, case reports of carcinomas and lymphomas continue to be published and no definitive consensus opinion has been obtained [30–33]. Meanwhile, over 80 cases of anaplastic lymphoma kinase (ALK)-negative anaplastic large cell lymphoma have been reported in patients with SBIs and it has been accepted as a new clinical entity [33]. Among the autoimmune connective tissue diseases diagnosed in patients with SBI, scleroderma has been reported most often; other diagnoses included SLE, RA, Sjögren’s syndrome, and mixed connective tissue diseases [16–18, 20, 21, 24]. Again, epidemiological studies and meta-analysis have rejected the presumed relationship between SBIs and autoimmune connective tissue diseases [13, 34] but, in seeming contradiction, case reports about autoimmune connective tissue diseases continue to be published [10, 23, 24]. However, a relationship between SBIs and a particular constellation of symptoms that did not fulfill diagnostic criteria for any recognized autoimmune connective tissue diseases has been documented in several studies [10, 13, 19, 22, 23, 34, 35]. Reported symptoms of these non-defined autoimmune phenomena are fatigue, muscular weakness, morning stiffness, arthralgia, myalgia, dry eyes, dry mouth, frequent sore throats, night sweats, rash, Raynaud’s phenomena, alopecia, adenopathy, poor sleep, headache, memory loss, and sensory loss [10, 13, 19, 20, 23, 34, 35]. In addition, the localized and very variable foreign body inflammatory reaction to the SBI can lead to the formation of an excessive fibrous capsule and capsular contracture, which occur in 2–50 % of individuals [36].
Table 2.
Suggested criteria for the diagnosis of autoimmune/inflammatory syndrome induced by adjuvants [9]
| Major criteria • Exposure to an external stimulus (infection, vaccine, silicone, adjuvant) prior to clinical manifestations • The appearance of ‘typical’ clinical manifestations: Myalgia, myositis, or muscle weakness Arthralgia and/or arthritis Chronic fatigue, un-refreshing sleep, or sleep disturbances Neurological manifestations (especially associated with demyelination) Cognitive impairment, memory loss Pyrexia, dry mouth • Removal of inciting agent induces improvement • Typical biopsy of involved organs Minor criteria • The appearance of autoantibodies or antibodies directed at the suspected adjuvant • Other clinical manifestations (e.g., irritable bowel syndrome) • Specific HLA (e.g., HLA DRB1…, HLA DQB1…) • Evolvement of an autoimmune disease (e.g., multiple sclerosis, systemic sclerosis) |
HLA human leukocyte antigen
An evaluation of these criteria in 93 cases of ASIA following hepatitis B vaccine revealed that fulfillment of either two major or of one major and two minor criteria is required to diagnose ASIA [12]
Intravascular large B-cell lymphoma (IVLBCL) is a rare subtype of diffuse large B-cell lymphoma [37–39]. It is characterized by proliferation and aggregation of clonal lymphocytes within the lumina of capillaries, arterioles, and venules. The surprising degree of sparing of the surrounding tissue and the absence of lymphoma cells in the lymph nodes is a hallmark of the disease [37–40]. This lymphoma is extremely heterogeneous in its clinical presentation and has been described in the small vessels of nearly every organ, leading to ischemia, organ dysfunction, and organ failure [37–40]. Therefore, it has also been called the oncologist’s “great imitator.” The majority of cases can be grouped into few discrete presentations: central nervous system involvement or cutaneous involvement (also called Western variant), fever of unknown origin, hemophagocytic syndrome/HLH, and multi-organ failure (also called Asian variant). An exhaustive search for an infectious etiology often contributes to the delay in diagnosis [39]. Up to two thirds of patients have neurologic manifestations [41]. The following syndromes have been found to be associated with IVLBCL: cerebrovascular events, encephalopathy, myelopathy and/or cauda equina syndrome, and peripheral or cranial neuropathies [41–44]. Beyond these presentations, there are single case reports of IVLBCL presenting primarily in other organ systems, for example, as interstitial lung disease, adrenal failure, pulmonary hypertension, nephrotic syndrome, myocardial infarction, and symmetric polyarthritis. Given the rarity of IVLBCL, its multiplicity of presentations, and its absence in lymph nodes (where lymphomas are usually localized), it is an extremely difficult diagnosis to make ante mortem [39]. IVLBCL can be fatal if diagnosis and treatment are delayed. It has emerged that 18Fluorodeoxy-glucose positron emission tomography/computed tomography (18FDG-PET/CT) may be useful to allow suspicion of a diagnosis of IVLBCL, to detect unexpected locations, to guide diagnostic biopsy, and to assess the response to treatment [45]. A random skin biopsy from normal-appearing skin has been shown to be highly sensitive in the diagnosis of ILVBCL and should be performed irrespective of the presence or absence of skin lesions in patients in whom IVLBCL is suspected [46]. Random transbronchial lung biopsies can be diagnostic in patients affected by IVLBCL either with negative 18FDG-PET/CT imaging or with no abnormal chest computed tomography [47, 48].
Case presentation
A 48-year-old Caucasian woman was referred to our hospital to exclude an unrecognized infection as the cause of three episodes of fever associated with splenomegaly. Her past medical history was unremarkable up to 4 years previously, when by routine mammography and subsequent biopsy an intraductal carcinoma was diagnosed. She underwent a skin-sparing mastectomy of her right breast combined with reconstruction and implantation of a silicone prosthesis (Polytech 445 g). No adjuvant chemotherapy or radiotherapy was provided or indicated. Three years later she underwent a reduction operation of her left breast combined with lipofilling of both breasts. She had experienced fever 8 and 5 months prior to hospitalization that lasted for about 3 weeks. She was treated at home with levofloxacin because of a known allergy to penicillin.
When fever appeared again together with sore throat, fatigue, sweating, cough (she smokes about 10 cigarettes daily), and an involuntary weight loss of 10 kg in the last year, she was hospitalized for 2 weeks in a peripheral hospital. Abnormal results from laboratory investigations included: hemoglobin 10.4 g/dL, leukocytes 3.6 × 109/L, serum total protein 5.3 g/dL, lactate dehydrogenase (LDH) 638 U/L, and ferritin 398 ng/mL. An abdominal ultrasound examination demonstrated her spleen was enlarged, with a diameter of 15 cm. X-ray examination of her chest showed bilateral apical pleural scarring, which was confirmed by computed tomography (CT). CT also revealed subpleural densities of uncertain significance, an enlarged liver and spleen, ovarian cysts, and a 2.3 cm large layer of fluid in the Douglas space. There was no evidence of enlarged mediastinal, abdominal, or retroperitoneal lymph nodes. A histologic examination of a bone marrow biopsy specimen was of no diagnostic help. 18FDG-PET/CT evidenced hypermetabolism of both adrenal glands. She was given azithromycin and her fever subsided but profound weakness remained. Extensive laboratory examinations did not indicate any infectious, autoimmune, or malignant diseases. She was referred to our Division of Infectious Diseases for a search for an unrecognized infectious disease.
On physical examination our patient was pale and weak. Her body temperature was 37.3 °C, blood pressure 110/90 mmHg, heart frequency 92 beats/minute, arterial blood oxygen saturation 98 %, body weight 59 kg, and high 168 cm. She complained of memory deficits and an unexplainable unstable gait. There were no signs of meningeal irritation. Since her breast operation 4 years previously she had needed sleeping tablets. During her first 6 days in hospital without any treatment her body temperature rose progressively from 37.3 to 39 °C. Because of the pleural scars and subpleural densities seen in the CT images, and the hypermetabolic adrenal glands shown by 18FDG-PET/CT [adrenocorticotropic hormone (ACTH) and cortisol concentrations were within the normal range], a tuberculosis (TB) infection was considered. Results from a repeated search by microscopy, culture and PCR analysis of Mycobacterium tuberculosis in her sputum and bronchial aspirate were unremarkable. A Mantoux skin test and QuantiFERON-TB assay were negative too. Leishmania infection was excluded by serology and microscopy of her bone marrow aspirate.
Despite repeated negative blood, urine, and stool cultures for pathogenic bacteria, among the many laboratory examinations, titers of 1:160 anti-Salmonella O and 1:2560 anti-Salmonella H antibodies were detected by Widal Wright’s reaction (Table 3). Thus empiric antibiotic therapy with ceftriaxone 2 g daily was started. There was no response to treatment and her high fever persisted. Therefore, indomethacin 50 mg three times daily was administered. This was followed by prompt defervescence; however, it lasted only for 3 days (Fig. 1). On the fourth day of indomethacin treatment, her fever of 39 °C recurred and was associated with increased weakness, malaise, and confusion. That night, two episodes of right hemiparesis with aphasia of about 30 min duration were observed. No areas of ischemia or hemorrhage were seen by magnetic resonance imaging (MRI)-angiography of her brain. An analysis of her cerebrospinal fluid found 282 mg/dL of protein, but there was no increase in cell number (Table 3). At this point, her symptoms were treated with dexamethasone 16 mg daily for 2 days followed by methylprednisolone 1 g daily for the next 4 days and then again dexamethasone 16 mg daily. Ceftriaxone was substituted by meropenem, and empiric anti-TB treatment and treatment with fluconazole were added. Corticosteroid therapy was followed by an impressive clinical improvement, including defervescence, regaining orientation, and regaining her ability to stand and to eat. However, on the fourth day of corticosteroid treatment, her high fever recurred together with extreme weakness, anorexia, and episodes of confusion.
Table 3.
Laboratory values
| Analyte | Reference | Day 1 | Day 12 | Day 22 | Day 29 |
|---|---|---|---|---|---|
| Leukocyte count (×103/μL) | 4.3–11.0 | 2.9 | 1.6 | 4.8 | 5.9 |
| Polymorphonucleocytes (×103/μL) | 1.9–8.0 | 1.3 | 1.0 | 3.7 | 5.1 |
| Lymphocytes (×103/μL) | 1.0–3.7 | 1.0 | 0.3 | 0.5 | 0.3 |
| Hemoglobin (g/dL) | 12–16 | 9.7 | 8.2 | 10.9 | 9.3 |
| Platelet count (×103/μL) | 140–450 | 151 | 77 | 68 | 58 |
| Triglycerides (mg/dL) | 30–150 | 128 | 296 | 301 | 442 |
| Fibrinogen (mg/dL) | 180–500 | - | 474 | 94 | 98 |
| Ferritin (ng/mL) | 13–150 | 309 | 673 | 1046 | 733 |
| sIL-2R(CD25) (kU/L) | 223–718 | - | 13,190 | 15,730 | 45,800 |
| sIL-2R/ferritin ratio | <2* | - | 20 | 15 | 62 |
| Lactate dehydrogenase (U/L) | 120–230 | 265 | 412 | 451 | 454 |
| Aspartate transaminase (IU/L) | <40 | 13 | 23 | 76 | 59 |
| Alanine transaminase (IU/L) | <40 | 12 | 17 | 59 | 68 |
| Total bilirubin (mg/dL) | <1.4 | 0.4 | 0.3 | 0.5 | 2.8 |
| Conjugated bilirubin (mg/dL) | <0.3 | 0.2 | 0.2 | 0.4 | 2.6 |
| Albumin (g/dL) | 3.5–5.2 | 2.8 | 2.8 | 2.1 | 1.7 |
| Erythrocyte sedimentation rate (mm/h) | <30 | 81 | 65 | - | - |
| C-reactive protein (mg/dL) | <0.50 | 0.62 | 9.9 | 16.8 | 8.2 |
| Lupus anticoagulant panel | |||||
| aPTT-low phospholipid | <1.15 | 0.85 | |||
| DRVVT ratio | <1.10 | 1.3 | |||
| Cerebrospinal fluid | |||||
| Cells (/μL) | ≤5 | 0 | 4 | ||
| Glucose (mg/dL) | 50–75 | 60 | 39 | ||
| Protein (mg/dL) | 15–45 | 282 | 152 | ||
| Chloride (mmol/L) | 120–132 | 113 | 107 | ||
| Lactate (mmol/L) | 1.1–2.2 | 2.9 | 6.6 | ||
| Link index | <0.7 | 0.5 | 0.5 | ||
| Widal Wright reaction to | |||||
| Salmonella O antigen | ≤1:160 | 1:160 | 1:160 | ||
| Salmonella H antigen | ≤1:160 | 1:2560 | 1:2560 | ||
High-resolution human leukocyte antigen (HLA) typing: HLA-A *32:01, HLA-B *40:02, *51:01, HLA-C *02:02,16:02, HLA-DRB1 *11:01,*11:04, HLA-DQB1 *03:01
Negative results: repeated blood cultures; QuantiFERON-TB test, search in blood for plasmodia, Plasmodium antigen, Leishmania immunoglobulin G (immunoblotting), hepatitis C virus antibody, Legionella pneumophila antibody, Mycoplasma pneumoniae antibody, Coxiella burnetii antibody, Bartonella henselae antibody, Brucella antibody, Borrelia burgdorferi antibody, Treponema pallidum antibody, HIV antibody, hepatitis B virus DNA, cytomegalovirus DNA, Epstein-Barr virus DNA, human herpesvirus6 DNA, parvovirus immunoglobulin M; urine cultures; search in urine for Legionella antigen; search in bone marrow aspirate for Leishmania, M. tuberculosis DNA; antinuclear antibodies
Values out of the reference range are in bold; normal routine laboratory values are not listed
*A ratio >2 is observed in most patients with lymphoma-associated hemophagocytic lymphohistiocytosis [50, 51]
aPPT activate partial thromboplastin time, DRVVT dilute Russell’s viper venom time, sIL-2R soluble interleukin-2 receptor
Fig. 1.
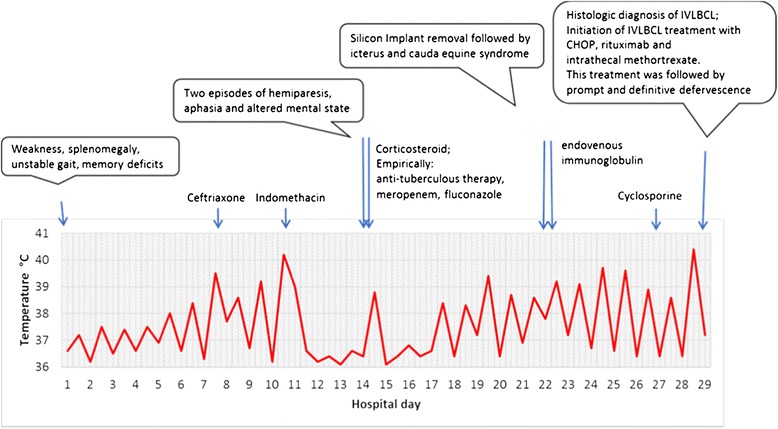
Chronological sequence of the main clinical features until the final diagnosis of intravascular large B-cell lymphoma (ILBCL) was made. CHOP cyclophosphamide, hydroxydaunorubicin (doxorubicin), vincristine, prednisone
The criteria for HLH/MAS were fulfilled (Table 1), but no underlying cause was evident. Because eight cases of AOSD associated with SBIs have been reported (see below), the silicone breast prosthesis of our patient was considered to have possibly contributed to her hyperinflammatory state, and consequently was explanted on the 22nd hospital day. A more detailed medical history revealed that the breast prosthesis had caused our patient many problems. Because of painful inflammation, the first prosthesis had to be explanted after 6 weeks and necrotic inflammatory tissue surrounding the prosthesis removed. Because an infection was suspected, the prosthesis was cleaned with antiseptics and then re-implanted, but no infectious agent could be isolated. Two months later, the prosthesis had to be removed definitively. Again, necrotic inflammatory tissue had to be removed and no infectious agent could be isolated. One year prior to her current hospitalization, a new silicone breast prosthesis was implanted. An inflammatory reaction with swelling of her breast was treated locally with infiltration of corticosteroids. Thereafter, our patient could tolerate the prosthesis and a mild pain was felt only when touching the prosthesis.
Significant worsening occurred after removal of the intact prosthesis. Her fever rose up to 40.4 °C, she became icteric (Table 3) and for the first time she experienced severe low back pain that radiated to her lower legs and required analgesic treatment with opium derivates. She complained of perianal anesthesia and fecal incontinence. The day of implant removal, 25 g of endovenous immunoglobulin was started and given daily for 3 days to treat the MAS. Finally, cyclosporine 100 mg twice daily was administered without any benefit. On the 29th hospital day, the results of the histologic examination of the tissue taken at implant removal were available and showed (a) normal pectoralis muscle with absence of neoplastic infiltration; (b) cutis and subcutis with dilated vessels showing an intraluminal proliferation of atypical large lymphocytes with a vesiculosus nucleus with several nucleoli (Fig. 2). A immunohistochemical analysis of the neoplastic cells revealed positivity for cluster of differentiation (CD) 20, B-cell lymphoma (BCL)-6, BCL-2, multiple myeloma oncogene 1 (MUM-1), and weaker staining for CD5. The proliferation index Ki-67 was over 90 %. Results for terminal deoxynucleotidyl transferase (TdT), CD10, CD34, CD3, and cytokeratin-CAM were negative. These results are consistent with the diagnosis of IVLBCL. The remaining parenchyma showed an extensive giant cell-lymphohistiocytic foreign body reaction, but no carcinomatous infiltration. Her familial medical history was notable for an uncle who has died from acute lymphatic leukemia at 43 years of age.
Fig. 2.
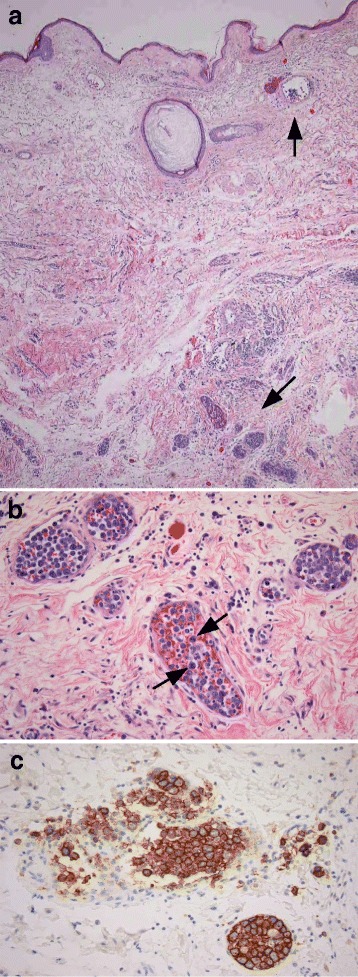
a Skin with enlarged vessels filled with tumor cells (arrows) mainly in the deep dermis (hematoxylin eosin stain, low magnification). b Intermediate vessels with tumor cells and some mitotic figure (arrows). c The tumor cells are strongly positive for CD20 by immunohistochemical reaction for CD20
Our patient was transferred to our Hematology Ward where she was treated with six cycles of R-CHOP [rituximab, cyclophosphamide, hydroxydaunorubicin (doxorubicin), vincristine, prednisone] chemotherapy. Intrathecal prophylaxis with methotrexate was also given. After 6 months her white blood cell count, platelet count, hemoglobin level, LDH level, and liver function tests were again within the reference ranges. After 2 years our patient’s clinical status is good, but sequelae of the cauda equina syndrome are still present, including low back pain, saddle anesthesia, difficulties in voiding the bladder and intestine, loss of sexual sensation, right plantar numbness, and gait instability. She continues to undergo 3-monthly follow-up visits (see Additional file 1 for supporting data).
Discussion
1. Diagnostic considerations
Elevated soluble interleukin-2 receptor (sIL-2R/CD25) has been observed in sera of patients with malignant lymphoma, and can therefore be used as a diagnostic and prognostic marker for malignant lymphoma [49]. sIL-2R was shown to be released from activated T cells mainly due to cleavage by matrix metalloprotease-9 (MMP-9) produced by tumor-associated macrophages [49]. Elevated sIL-2R is also seen in HLH and has been proposed as a diagnostic marker of the disease (Table 1). Secondary HLH can be caused by malignant lymphoma too [1–4]. HLH is considered characteristic of the so-called Asian variant of IVLBCL [38, 39]. A sIL-2R/ferritin ratio >2 has been observed in most patients with malignant lymphoma-associated HLH and only rarely in those with benign disease-associated HLH. Therefore, a high sIL-2R/ferritin ratio has been proposed as a useful marker for the diagnosis of a lymphoma-associated HLH [50, 51]. Our patient had sIL-2R/ferritin ratios of 20,15, and 62 (Table 3). Her serum had to be sent for sIL-2R testing to the Reference Center, Dipartimento Interaziendale e Medicina di Laboratorio, University of Padua, Italy, and the results were available 3 weeks later, when the histologic diagnosis of IVLBCL was already known. In order to be of diagnostic utility, sIL-2R testing results have to be available in a shorter period of time.
Autoptic or intra vitam diagnosis of IVLBCL of the adrenal glands in patients manifesting adrenal insufficiency has been reported [52–54]. Primary or secondary adrenal gland infiltration was detected in up to 67 % of patients diagnosed with IVLBCL [53]. Because the adrenal gland has a sufficient reserve, development of adrenal insufficiency requires destruction of 90 % of the adrenal gland tissue [53]. Indeed, a silent IVLBCL initially manifesting as a unilateral adrenal incidentaloma has been described [55]. In this case an abdominal CT scan performed on a regular follow-up appointment revealed asymptomatic bilateral enlargement of the adrenal glands, while the adrenal function remained normal, suggesting a relatively slow progress of the disease. An 18FDG-PET/CT showed an exclusive, strong uptake in both adrenal glands that was absent at a control one year after R-CHOP chemotherapy [55]. In our patient, the diagnostic significance of the exclusive hypermetabolic adrenals demonstrated by 18FDG-PET/CT carried out 7 weeks before the histologic diagnosis of IVLBCL was not recognized. Only adrenal tuberculosis was considered. Serum ACTH and cortisol levels were within the normal range. At that time, a biopsy of her adrenal glands [55], a random transbronchial biopsy of her lungs [47, 48], a random biopsy of her skin [46], or a biopsy of her liver probably could have facilitated an earlier diagnosis.
2. HLA-DRB1*11, HLA-DQB1*03, and siliconosis
Foreign body reaction to a silicone implant and capsule formation is a physiological process that protects the organism from a non-degradable foreign material that is too large to be engulfed by macrophages [11, 36]. However, this physiological process is highly variable and can lead to local inflammatory symptoms, an excessive capsular fibrosis, contracture in 2–50 % of patients, and sometimes systemic symptoms and disease [10–36]. Excessive capsular fibrosis and capsular contracture are well-recognized adverse events. However, harmful systemic effects of silicone, most notable connective tissue diseases (CTDs) or systemic autoimmune phenomena, remain controversial [13, 34]. A meta-analysis rejected the presumed relationship between SBIs and CTD [13, 34] but, in seeming contradiction, case reports about CTDs and systemic symptoms continue to be published [10, 23, 24]. As for other rare diseases or side effects [56], most epidemiological studies lack enough power to show significant associations [13]. In addition, most studies determined the risk of development of CTDs shortly after breast implantation, which may result in false-negative results, given that most patients complain only years after the breast implant operation as reported in other studies [10]. Another shortcoming of most epidemiological studies is that they focus on classic CTDs, ignoring other (systemic) autoimmune phenomena recently classed as ASIA syndrome [9–11, 13].
It seems obvious that a favorable genetic or predisposing background plays a key role in the development of rare diseases or side effects. Silicone gel was shown to enhance the development of autoimmune diseases in New Zealand black mice but failed to induce it in BALB7cAnPt mice [57]. Thus, it was hypothesized that silicone could play a similar role in the development of autoimmune diseases in a small percentage of women who are genetically susceptible to such diseases [57]. More than 200 genetic loci have been shown to be associated with one or more autoimmune disorders [58]. It has been suggested that most of these genetic associations reflect the immunoregulatory effect of the HLA molecules themselves [58]. For many of these disorders, genes within the major histocompatibility complex (MHC) have by far the strongest single genetic effect [58]. The human MHC is the most gene-dense region of the human genome and encodes the most polymorphic human proteins [59]. Its variants are associated with more than 100 different diseases, mostly autoimmune, infectious, or inflammatory diseases [60]. However, identification of causal variants within the MHC for the majority of these diseases has remained difficult due to the great variability and extensive linkage disequilibrium that exist among alleles throughout this locus [61]. In one study, 77 symptomatic patients with SBIs were reported to share important genetic characteristics (primarily HLA-DR53 = HLA-DRB4) that differentiate them from their asymptomatic controls [62]. Another study showed that all nine women with post-silicone implant systemic sclerosis had HLA-DQ5 or DQ7 (HLA-DQB1*0301) alleles [63]. Compared to 128 healthy Caucasian controls, these nine women had a significantly lower frequency of the hydrophobic leucine at position 26 of the first domain of the HLA-DQB1 allele [63]. Eleven women who had developed inflammatory myositis after they received silicone implants differed by increased frequency of HLA-DQA1*0102 from 76 women with myositis without silicone implants [64]. Two sisters who received SBIs developed polyarthritis and neurological symptoms but experienced dramatic improvement after implant removal. HLA typing revealed HLA-DRB1*0405, HLA-DQB1*0302, and HLA-DRB4*01 [21]. Three sisters who carried the BRCA-1 gene mutation had a preventive mastectomy and were reconstructed with SBIs. After the reconstruction, all three patients developed fatigue, arthralgia, myalgia, and sleep disturbances within a period of 4 years. Silicone implant replacement by non-silicone gel containing Monobloc Hydrogel breast implants was followed by improvement of all complaints in the three sisters [23]. These reports further indicate that the susceptibility to develop silicone implant incompatibility syndrome or ASIA may be genetically determined. HLA typing of all who receive silicone implants would enable physicians to better define the risk of adverse side effects of silicone implants.
3. Siliconosis and macrophage activation syndrome
In the animal model, silicone (dimethylpolysiloxane) exhibited potent adjuvant properties that varied according to its molecular weight and chemical composition [57, 65–68]. Silicone elastomer, pre-adsorbed with plasma proteins, activates human monocytes/macrophages in vitro to secrete IL-1ß, IL-6, and TNF-α [68]. In female Wistar rats it was shown that silicone induced persistent recruitment of leukocytes at the site of the injection and that macrophage activation was still present 45 days after injection. Activated macrophages exhibited an increased expression of adhesion and co-stimulatory molecules and an enhanced production of oxidant metabolites and nitric oxide (NO) [69]. In a study on biomaterial-induced macrophage activation it was shown that concentrations of markers of monocyte/macrophage activation, the chitinase-like proteins chitotriosidase and YKL-40, were significantly higher in patients with certain bio-implants and that these markers rose progressively as adverse reactions evolved [70]. A macrophage-specific PET imaging probe has recently been developed to image implant-surrounding activated macrophages and, thus, to evaluate in vivo medical device-associated inflammation [71].
Even if a silicone gel-filled breast implant does not rupture, small amounts of low molecular weight fluid dimethylpolysiloxane may permeate (bleed or sweat) out of the implant into the surrounding tissue [69]. Transcapsular migration of silicone particles from the mammary implant through the fibrous capsule of mammary prosthesis has been observed [72]. Silicone was identified within macrophages residing in the synovia and skin of patients with SBIs and CTD [73, 74]. One mechanism that has been proposed to explain an interaction between silicones and the immune system is the following. After silicones have been freed from the implant, they come into contact with native tissue, which may lead to denaturation of tissue macromolecules such as fibronectin, fibrinogen, and apolipoprotein B. The denatured macromolecules are sufficiently different from native tissue to be recognized as foreign, which may trigger activation of the immune system and lead to the generation of antibodies against them. These antibodies may cross react with native tissue, generating an autoimmune response [75].
Eight cases of AOSD associated with SBIs have been reported [76–80]. Improvement of AOSD was described in two of three patients in whom the SBI was removed [76]. AOSD is a systemic inflammatory disorder, a hyperferritinemic or cytokine storm syndrome that has similarities and pathophysiological overlap with MAS/secondary HLH [4, 7, 81]. AOSD has therefore also been defined as an autoimmune-associated, reactive, or secondary HLH [7, 81]. NK cytotoxicity was found to be reduced in AOSD [82]. Even if MAS has not been previously associated with SBIs, considering these studies it seems plausible that in our patient her SBI could have contributed to the development of MAS.
4. HLA-DRB1*11, HLA-DQB1*03, and lymphoma
There are a few studies that show a higher frequency of HLA-DRB1*11 and/or HLA-DQB1*03 in patients with hematologic malignancies [83], non-Hodgkin’s lymphoma, diffuse large B-cell lymphoma [84], mycosis fungoides (cutaneous T-cell non-Hodgkin’s lymphoma) [85–88], acute lymphoblastic leukemia [89], chronic lymphocytic leukemia [90], hairy cell leukemia (B-cell leukemia) [91], and HCV virus-associated lymphoma [92] compared to controls. These data indicate a probable genetic basis of the lymphomagenesis. Our patient’s uncle died at 43 years of age from acute lymphoblastic leukemia. The occurrence of more cases of hematologic malignancies in a family further suggest a genetic contribution to the pathogenesis [87, 88].
HLA-DRB1*11 or HLA-DQB1*03 have been associated with, among others, the autoimmune disease systemic sclerosis [93, 94]. Systemic sclerosis, like other autoimmune diseases, has been associated with lymphomas [95, 96]. Even the rarely occurring intravascular large B-cell lymphoma has been described in a woman with systemic sclerosis [97].
5. Siliconosis and lymphoma
Silicosis is a known risk factor for lung cancer [98]. Can siliconosis be considered a risk factor for cancer too? Although non-Hodgkin lymphomas of the breast are exceedingly rare, cases of breast implant-associated anaplastic large cell lymphoma (ALCL) continue to be reported [99–101]. The finding of more ALK-1-negative ALCL among breast implant-associated ALCLs compared to ALCL with breast involvement in women without breast prosthesis was considered as evidence in favor of an association between silicone breast prosthesis and ALK-1-negative ALCL [99]. Also, because the breast implant-associated ALK-1-negative ALCL has distinct clinical features, for example, a better prognosis, compared to non-implant-associated ALK-1-negative ALCL, it has been proposed as a new clinical entity [99–102]. The lack of strong epidemiological evidence for a causative role of breast implants most likely reflects the extreme rarity of the disease [102]. If siliconosis could have contributed to the pathogenesis of IVLBCL in our patient, one could speculate whether removal of the SBI and therefore removal of the chronic inflammatory stimulus could have improved disease prognosis.
Inflammation has been linked with tumorigenesis and tumor progression in many malignancies [98]. Chronic inflammation can promote tumor initiation and progression but can also protect against cancer through immune surveillance [103]. Chronic inflammation can increase the risk of cancer by providing bioactive molecules including cytokines, chemokines, and growth factors. These bioactive molecules maintain a sustained proliferation rate, interfere with apoptosis, promote angiogenesis, and induce the production of free radicals and reactive oxygen and nitrogen species (RONS) [98]. TNF-α, the prototypical proinflammatory cytokine, can exert pro-apoptotic and anti-apoptotic effects [98]. IL-6, another major mediator of inflammation, serves to block apoptosis in cells during the inflammatory process, keeping them alive, protecting them from cellular apoptotic deletion, and allowing them to progress towards neoplastic growth [104]. In cancers associated with chronic inflammation, either excessive free radical production or defective antioxidant mechanisms or both of these were observed [98]. Free radicals, RONS, can instigate carcinogenesis either directly by reacting with and damaging DNA and RNA or indirectly by interfering with transcription factors, for example, NF-kB [98, 105]. Free radicals and cytokines can both either induce or become induced by NF-kB [103]. As a consequence, a positive feedback loop exists between the ability of inflammatory cytokines to induce the synthesis of RONS and of RONS to elicit the generation of inflammatory cytokines [103].
Lymphoid neogenesis and lymphoma are associated with chronic inflammation in infections like chronic hepatitis C or Helicobacter pylori infection, and in chronic autoimmune diseases [98, 106]. Considering the key role of B- and T-cell activation in the pathogenesis of infections, autoimmune diseases, and lymphoma, it is perhaps not surprising that longstanding chronic inflammation and/or antigen stimulation have emerged as major predisposing factors of lymphoma in patients with these diseases [106].
6. Familial hemophagocytic lymphohistiocytosis- related gene mutations and macrophage activation syndrome
The complete cell-mediated cytotoxicity defect due to bi-allelic disruptive mutations in one of the familial HLH-related genes leads to full-blown familial HLH early in life, with a peak incidence between 1 and 6 months of age and with the typical and rapidly fatal course. However, the clinical impact of less complete or partial defects in this pathway remains to be clarified [2]. Mutations that lead to partial insufficiency of the cytotoxic machinery (missense mutations, single nucleotide polymorphisms, monoallelic mutations) have been found in patients with later-onset familial HLH [2, 107], in patients with secondary HLH including MAS [6, 108–111], and in those with other diseases, such as systemic juvenile idiopathic arthritis [112, 113] or lymphoma (see below). Therefore, distinctions between familial and secondary HLH are becoming increasingly blurred as new genetic causes are identified [6].
7. Familial hemophagocytic lymphohistiocytosis-related gene mutations and lymphoma
Mutations of the perforin gene and the UNC13D gene, both familial HLH-related genes, have been found in 27 % of children with ALCL [114, 115]. These data suggest that impaired cytotoxic machinery may represent a predisposing factor for ALCL. In one study, 11 out of 23 patients with bi-allelic perforin gene mutations, whose onset of familial HLH was delayed or abrogated, presented with B- or T-cell lymphoma or acute or chronic leukemia as the primary clinical illness [116]. In the mouse model of lymphomagenesis, perforin was shown to act as a suppressor of B-cell malignancies [117].
NK cells (innate immunity) and CTLs (adaptive immunity) are recognized for their role in the defense against viruses, in immune surveillance against a variety of malignancies, and in downregulating or ending an immune response [118–121]. These roles are performed by perforin initiating the apoptosis of dangerous cells, either those harboring an intracellular pathogen, those possessing the potential for uninhibited growth and spread, or those antigen-presenting cells and activated T cells that have just accomplished their immune response function. Perforin is a pore-forming protein stored in secretory granules of CTLs and NK cells that synergizes with the pro-apoptotic serine proteases (granzymes) to deliver a lethal hit to the target cell [118–121]. It is possible that in our patient polymorphisms of the familial HLH-related genes, the genes encoding the cytotoxic machinery, had a causal role for both the MAS and the IVLBCL. Unfortunately, we have not had the chance to sequence these genes.
Conclusions
To the best of our knowledge, this is the first reported case of IVLBCL associated with an SBI. MAS, siliconosis/ASIA, and IVLBCL are rare diseases and are difficult to diagnose. The combination of these three rare conditions suggests multifactorial interactions (Fig. 3). When IVLBCL is considered in the differential diagnosis of fever of unknown origin, then a diagnosis can be made by random skin biopsy or by biopsy of a suspected organ indicated by 18FDG-PET/CT. The chronic inflammation of siliconosis in our patient might have contributed to the development of her IVLBCL. MAS/secondary HLH is a known manifestation of IVLBCL. Siliconosis might also have predisposed our patient to MAS development, as there are case reports of siliconosis associated with AOSD, another cytokine storm syndrome like MAS. Hypothetical mutations of genes causing familial HLH could have favored both MAS and lymphoma development. Future expansion of genetic testing will further clarify the role of polymorphisms of genes encoding the cytotoxic machinery in the pathogenesis of hematologic malignancies, and possibly will discover polymorphisms of further genes involved in the pathogenesis of lymphoma, secondary HLH, and autoimmune diseases. HLA testing of all women who undergo silicon breast implantation could enable physicians to evaluate the risk of getting siliconosis/ASIA, a still-debated disease.
Fig. 3.
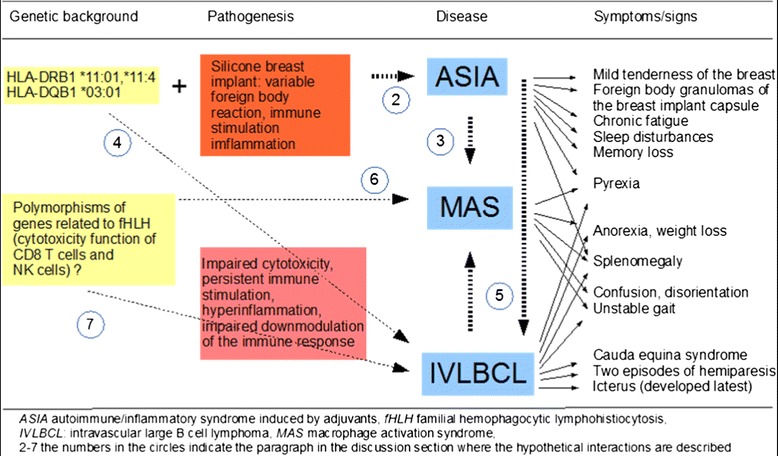
Hypothetical interactions between the different syndromes and diseases of the described patient
Abbreviations
18FDG-PET/CT, 18Fluorodeoxy-glucose positron emission tomography/computed tomography; ALCL, anaplastic large cell lymphoma; ALK-1, anaplastic lymphoma kinase-1; AOSD, adult-onset Still’s disease; ASIA, autoimmune inflammatory syndrome induced by adjuvants; BCL, B-cell lymphoma; CD, cluster of differentiation; CT, computer tomography; CTD, connective tissue diseases; CTL, cytotoxic T cell; HLH, hemophagocytic lymphohistiocytosis; IVLBCL, intravascular large B-cell lymphoma; LDH, lactate dehydrogenase; MAS, macrophage activation syndrome; MHC, major histocompatibility complex; MMP-9, matrix metalloprotease-9; MRI, magnetic resonance imaging; MUM-1, multiple myeloma oncogene 1; NK, natural killer; RA, rheumatoid arthritis; R-CHOP, rituximab, cyclophosphamide, hydroxydaunorubicin (doxorubicin), vincristine, prednisolone; RONS, reactive oxygen and nitrogen species; SBI, silicone breast implant; sJIA, systemic juvenile idiopathic arthritis; SLE, systemic lupus erythematosus; TB, tuberculosis; TdT, terminal deoxynucleotidyl transferase; TNF-α, tumor necrosis factor alpha
Acknowledgments
Not applicable.
Availability of data and materials
Supporting materials for the manuscript “Intravascular large B-cell lymphoma associated with silicone breast implant, HLA-DRB1*11:01, and HLA-DQB1*03:01 manifesting as macrophage activation syndrome and with severe neurological symptoms: a case report”.
Authors’ contributions
OM, AP, and MT were major contributors regarding the management of the patient, review of the articles, and manuscript preparation; MT and GN performed the histological examination; MCanova, PM, MCasini, BR, RB, LG, GR, and AB assisted in managing the patient. CV reviewed the genetic data. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Additional file
Availability of data and supporting materials. (DOCX 18.6 kb)
Contributor Information
Oswald Moling, Email: molosw@hotmail.com.
Andrea Piccin, Email: andrea.piccin@sabes.it.
Martina Tauber, Email: martina.tauber@sabes.it.
Peter Marinello, Email: peter.marinello@sabes.it.
Mariagrazia Canova, Email: mariagrazia.canova@sabes.it.
Marco Casini, Email: marco.casini@sabes.it.
Giovanni Negri, Email: giovanni.negri@sabes.it.
Bernd Raffeiner, Email: bernd.raffeiner@sabes.it.
Raffaella Binazzi, Email: raffaella.binazzi@sabes.it.
Latha Gandini, Email: latha.gandini@sabes.it.
Cinzia Vecchiato, Email: cinzia.vecchiato@sabes.it.
Giovanni Rimenti, Email: giovanni.rimenti@sabes.it.
Atto Billio, Email: atto.billio@sabes.it.
References
- 1.Janka GE, Lehmberg K. Hemophagocytic syndromes--an update. Blood Rev. 2014;28:135–42. doi: 10.1016/j.blre.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Sieni E, Cetica V, Hackmann Y, Coniglio ML, Da Ros M, Ciambotti B, et al. Familial hemophagocytic lymphohistiocytosis: when rare diseases shed light on immune system functioning. Front Immunol. 2014;5:167. doi: 10.3389/fimmu.2014.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Canna SW, Behrens EM. Not all hemophagocytes are created equally: appreciating the heterogeneity of the hemophagocytic syndromes. Curr Opin Rheumatol. 2012;24:113–8. doi: 10.1097/BOR.0b013e32834dd37e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canna SW, Behrens EM. Making sense of the cytokine storm: a conceptual framework for understanding, diagnosing, and treating hemophagocytic syndromes. Pediatr Clin North Am. 2012;59:329–44. doi: 10.1016/j.pcl.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 6.Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13:289–98. doi: 10.1038/gene.2012.3. [DOI] [PubMed] [Google Scholar]
- 7.Rosário C, Zandman-Goddard G, Meyron-Holtz EG, D’Cruz DP, Shoenfeld Y. The hyperferritinemic syndrome: macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. 2013;11:185. doi: 10.1186/1741-7015-11-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davì S, Minoia F, Pistorio A, Horne A, Consolaro A, Rosina S, et al. Performance of current guidelines for diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2014;66:2871–80. doi: 10.1002/art.38769. [DOI] [PubMed] [Google Scholar]
- 9.Shoenfeld Y, Agmon-Levin N. ‘ASIA’ - autoimmune/inflammatory syndrome induced by adjuvants. J Autoimmun. 2011;36:4–8. doi: 10.1016/j.jaut.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Cohen Tervaert JW, Kappel RM. Silicone implant incompatibility syndrome (SIIS): a frequent cause of ASIA (Shoenfeld’s syndrome) Immunol Res. 2013;56:293–8. doi: 10.1007/s12026-013-8401-3. [DOI] [PubMed] [Google Scholar]
- 11.Perricone C, Colafrancesco S, Mazor RD, Soriano A, Agmon-Levin N, Shoenfeld Y. Autoimmune/inflammatory syndrome induced by adjuvants (ASIA) 2013: unveiling the pathogenic, clinical and diagnostic aspects. J Autoimmun. 2013;47:1–16. doi: 10.1016/j.jaut.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Zafrir Y, Agmon-Levin N, Paz Z, Shilton T, Shoenfeld Y. Autoimmunity following hepatitis B vaccine as part of the spectrum of ‘Autoimmune (Auto-inflammatory) Syndrome induced by Adjuvants’ (ASIA): analysis of 93 cases. Lupus. 2012;21:146–52. doi: 10.1177/0961203311429318. [DOI] [PubMed] [Google Scholar]
- 13.Hajdu SD, Agmon-Levin N, Shoenfeld Y. Silicone and autoimmunity. Eur J Clin Invest. 2011;41:203–11. doi: 10.1111/j.1365-2362.2010.02389.x. [DOI] [PubMed] [Google Scholar]
- 14.Miyoshi K, Miyaoka T, Kotani Y, Saijo K. “Splenomegalia Hyperglobulinemica”. Proposal of a new clinical syndrome. Tokushima J Exp Med. 1963;10:185–94. [PubMed] [Google Scholar]
- 15.Uretsky BF, O’Brien JJ, Courtiss EH, Becker MD. Augmentation mammaplasty associated with a severe systemic illness. Ann Plast Surg. 1979;3:445–7. doi: 10.1097/00000637-197911000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Okano Y, Nishikai M, Sato A. Scleroderma, primary biliary cirrhosis, and Sjögren’s syndrome after cosmetic breast augmentation with silicone injection: a case report of possible human adjuvant disease. Ann Rheum Dis. 1984;43:520–2. doi: 10.1136/ard.43.3.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varga J, Schumacher HR, Jimenez SA. Systemic sclerosis after augmentation mammoplasty with silicone implants. Ann Intern Med. 1989;111:377–83. doi: 10.7326/0003-4819-111-5-377. [DOI] [PubMed] [Google Scholar]
- 18.Kaiser W, Biesenbach G, Stuby U, Grafinger P, Zazgornik J. Human adjuvant disease: remission of silicone induced autoimmune disease after explanation of breast augmentation. Ann Rheum Dis. 1990;49:937–8. doi: 10.1136/ard.49.11.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shoaib BO, Patten BM, Calkins DS. Adjuvant breast disease: an evaluation of 100 symptomatic women with breast implants or silicone fluid injections. Keio J Med. 1994;43:79–87. doi: 10.2302/kjm.43.79. [DOI] [PubMed] [Google Scholar]
- 20.Freundlich B, Altman C, Snadorfi N, Greenberg M, Tomaszewski J. A profile of symptomatic patients with silicone breast implants: a Sjögrens-like syndrome. Semin Arthritis Rheum. 1994;24(Suppl 1):44–53. doi: 10.1016/0049-0172(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 21.Meier LG, Barthel HR, Seidl C. Development of polyarthritis after insertion of silicone breast implants followed by remission after implant removal in 2 HLA-identical sisters bearing rheumatoid arthritis susceptibility genes. J Rheumatol. 1997;24:1838–41. [PubMed] [Google Scholar]
- 22.Laing TJ, Schottenfeld D, Lacey Jr JV, Gillespie BW, Garabrant DH, Cooper BC, Heeringa SG, Alcser KH, Mayes MD. Potential risk factors for undifferentiated connective tissue disease among women: implanted medical devices. Am J Epidemiol. 2001;154:610–7. [DOI] [PubMed]
- 23.Kappel RM, Cohen Tervaert JW, Pruijn GJ. Autoimmune/inflammatory syndrome induced by adjuvants (ASIA) due to silicone implant incompatibility syndrome in three sisters. Clin Exp Rheumatol. 2014;32:256–8. [PubMed] [Google Scholar]
- 24.Al Aranji G, White D, Solanki K. Scleroderma renal crisis following silicone breast implant rupture: a case report and review of the literature. Clin Exp Rheumatol. 2014;32:262–6. [PubMed] [Google Scholar]
- 25.Zaworski RE, DerHagopian RP. Locally recurrent carcinoma after breast reconstruction. Ann Plast Surg. 1979;3:326–9. doi: 10.1097/00000637-197910000-00005. [DOI] [PubMed] [Google Scholar]
- 26.Morgenstern L, Gleischman SH, Michel SL, Rosenberg JE, Knight I, Goodman D. Relation of free silicone to human breast carcinoma. Arch Surg. 1985;120:573. doi: 10.1001/archsurg.1985.01390290051008. [DOI] [PubMed] [Google Scholar]
- 27.Bingham HG, Copeland EM, Hackett R, Caffee HH. Breast cancer in a patient with silicone breast implants after 13 years. Ann Plast Surg. 1988;20:236–7. doi: 10.1097/00000637-198803000-00007. [DOI] [PubMed] [Google Scholar]
- 28.Bryant H, Brasher P. Breast implants and breast cancer--reanalysis of a linkage study. N Engl J Med. 1995;332:1535–9. doi: 10.1056/NEJM199506083322302. [DOI] [PubMed] [Google Scholar]
- 29.Nelson NJ. Silicone breast implants not linked to breast cancer risk. J Natl Cancer Inst. 2000;92:1714–5. doi: 10.1093/jnci/92.21.1714. [DOI] [PubMed] [Google Scholar]
- 30.McIntosh SA, Horgan K. Breast cancer following augmentation mammoplasty – a review of its impact on prognosis and management. J Plast Reconstr Aesthet Surg. 2007;60:1127–35. doi: 10.1016/j.bjps.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka Y, Morishima I, Kikuchi K. Invasive micropapillary carcinomas arising 42 years after augmentation mammoplasty: a case report and literature review. World J Surg Oncol. 2008;6:33. doi: 10.1186/1477-7819-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaudet G, Friedberg JW, Weng A, Pinkus GS, Freedman AS. Breast lymphoma associated with breast implants: two case-reports and a review of the literature. Leuk Lymphoma. 2002;43:115–9. doi: 10.1080/10428190210189. [DOI] [PubMed] [Google Scholar]
- 33.Lechner MG, Megiel C, Church CH, Angell TE, Russell SM, Sevell RB, et al. Survival signals and targets for therapy in breast implant-associated ALK--anaplastic large cell lymphoma. Clin Cancer Res. 2012;18:4549–59. doi: 10.1158/1078-0432.CCR-12-0101. [DOI] [PubMed] [Google Scholar]
- 34.Vasey FB, Zarabadi SA, Seleznick M, Ricca L. Where there’s smoke there’s fire: the silicone breast implant controversy continues to flicker: a new disease that needs to be defined. J Rheumatol. 2003;30:2092–4. [PubMed] [Google Scholar]
- 35.Solomon G, Espinoza L, Silverman S. Breast implants and connective-tissue diseases. N Engl J Med. 1994;331:1231. author reply 1233-4. Erratum in: N Engl J Med 1995;332:199. [DOI] [PubMed]
- 36.Steiert AE, Boyce M, Sorg H. Capsular contracture by silicone breast implants: possible causes, biocompatibility, and prophylactic strategies. Med Devices (Auckl) 2013;6:211–8. doi: 10.2147/MDER.S49522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura S, Ponzoni M, Campo E. Intravascular large B-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO classification of tumors of hematopoietic and lymphoid tissues. Lyon: IARC; 2008. p. 252–3.
- 38.Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136:333–8. doi: 10.5858/arpa.2010-0747-RS. [DOI] [PubMed] [Google Scholar]
- 39.Zuckerman D, Seliem R, Hochberg E. Intravascular lymphoma: the oncologist’s “great imitator”. Oncologist. 2006;11:496–502. doi: 10.1634/theoncologist.11-5-496. [DOI] [PubMed] [Google Scholar]
- 40.Fonkem E, Lok E, Robison D, Gautam S, Wong ET. The natural history of intravascular lymphomatosis. Cancer Med. 2014;3:1010–24. doi: 10.1002/cam4.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glass J, Hochberg FH, Miller DC. Intravascular lymphomatosis. A systemic disease with neurologic manifestations. Cancer. 1993;71:3156–64. doi: 10.1002/1097-0142(19930515)71:10<3156::AID-CNCR2820711043>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 42.Davis TS. Intravascular lymphoma presenting with cauda equina syndrome: treated with CHOP and rituxan. Leuk Lymphoma. 2003;44:887–8. doi: 10.1080/1042819021000055291. [DOI] [PubMed] [Google Scholar]
- 43.Abuzinadah A, Almalik Y, Shabani-Rad MT, Ho CH, George D, Alant J, Zochodne D. Cauda equina syndrome secondary to intravascular lymphoma. Neurol Clin Pract. 2012;2:158–61. [DOI] [PMC free article] [PubMed]
- 44.Colchester NT, Barker CS, Jogai S, Katifi HA. Cauda equina syndrome due to intravascular lymphoma: diagnosis by nasal biopsy. Pract Neurol. 2015;15:210–3. doi: 10.1136/practneurol-2014-001048. [DOI] [PubMed] [Google Scholar]
- 45.Colavolpe C, Ebbo M, Trousse D, Khibri H, Franques J, Chetaille B, et al. FDG-PET/CT is a pivotal imaging modality to diagnose rare intravascular large B-cell lymphoma: case report and review of literature. Hematol Oncol. 2014;33:99–109. doi: 10.1002/hon.2140. [DOI] [PubMed] [Google Scholar]
- 46.Matsue K, Asada N, Odawara J, Aoki T, Kimura S, Iwama K, et al. Random skin biopsy and bone marrow biopsy for diagnosis of intravascular large B cell lymphoma. Ann Hematol. 2011;90:417–21. doi: 10.1007/s00277-010-1101-3. [DOI] [PubMed] [Google Scholar]
- 47.Nakazato T, Sanada Y, Mihara A, Ito C, Aisa Y, Nakamura N. PET-negative pulmonary intravascular large B cell lymphoma diagnosed by a random transbronchial lung biopsy. Ann Hematol. 2012;91:811–2. doi: 10.1007/s00277-011-1333-x. [DOI] [PubMed] [Google Scholar]
- 48.Kaku N, Seki M, Doi S, Hayashi T, Imanishi D, Imamura Y, et al. A case of intravascular large B-cell lymphoma (IVLBCL) with no abnormal findings on chest computed tomography diagnosed by random transbronchial lung biopsy. Intern Med. 2010;49:2697–701. doi: 10.2169/internalmedicine.49.3986. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida N, Oda M, Kuroda Y, Katayama Y, Okikawa Y, Masunari T, et al. Clinical significance of sIL-2R levels in B-cell lymphomas. PLoS One. 2013;8:e78730. doi: 10.1371/journal.pone.0078730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabata C, Tabata R. Possible prediction of underlying lymphoma by high sIL-2R/ferritin ratio in hemophagocytic syndrome. Ann Hematol. 2012;91:63–71. doi: 10.1007/s00277-011-1239-7. [DOI] [PubMed] [Google Scholar]
- 51.Tsuji T, Hirano T, Yamasaki H, Tsuji M, Tsuda H. A high sIL-2R/ferritin ratio is a useful marker for the diagnosis of lymphoma-associated hemophagocytic syndrome. Ann Hematol. 2014;93:821–6. doi: 10.1007/s00277-013-1925-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chu P, Costa J, Lachman MF. Angiotropic large cell lymphoma presenting as primary adrenal insufficiency. Hum Pathol. 1996;27:209–11. doi: 10.1016/S0046-8177(96)90379-2. [DOI] [PubMed] [Google Scholar]
- 53.Fukushima A, Okada Y, Tanikawa T, Onaka T, Tanaka A, Higashi T, et al. Primary bilateral adrenal intravascular large B-cell lymphoma associated with adrenal failure. Intern Med. 2003;42:609–14. doi: 10.2169/internalmedicine.42.609. [DOI] [PubMed] [Google Scholar]
- 54.Askarian F, Xu D. Adrenal enlargement and insufficiency: a common presentation of intravascular large B-cell lymphoma. Am J Hematol. 2006;81:411–3. doi: 10.1002/ajh.20627. [DOI] [PubMed] [Google Scholar]
- 55.Takahashi Y, Iida K, Hino Y, Ohara T, Kurahashi T, Tashiro T, et al. Silent intravascular lymphoma initially manifesting as a unilateral adrenal incidentaloma. Case Rep Med. 2012;2012(849285):5. doi:10.1155/2012/849285. [DOI] [PMC free article] [PubMed]
- 56.Moling O, Cairon E, Rimenti G, Rizza F, Pristerá R, Mian P. Severe hepatotoxicity after therapeutic doses of acetaminophen. Clin Ther. 2006;28:755–60. doi: 10.1016/j.clinthera.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 57.McDonald AH, Weir K, Schneider M, Gudenkauf L, Sanger JR. Silicone gel enhances the development of autoimmune disease in New Zealand black mice but fails to induce it in BALB/cAnPt mice. Clin Immunol Immunopathol. 1998;87:248–55. doi: 10.1006/clin.1998.4532. [DOI] [PubMed] [Google Scholar]
- 58.Cho JH, Gregersen PK. Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med. 2011;365:1612–23. doi: 10.1056/NEJMra1100030. [DOI] [PubMed] [Google Scholar]
- 59.The MHC sequencing consortium Complete sequence and gene map of a human major histocompatibility complex. Nature. 1999;401:921–3. doi: 10.1038/44853. [DOI] [PubMed] [Google Scholar]
- 60.Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet. 2009;54:15–39. doi: 10.1038/jhg.2008.5. [DOI] [PubMed] [Google Scholar]
- 61.Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4(4):e1000024. doi:10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed]
- 62.Young VL, Nemecek JR, Schwartz BD, Phelan DL, Schorr MW. HLA typing in women with breast implants. Plast Reconstr Surg. 1995;96:1497–519. doi: 10.1097/00006534-199512000-00001. [DOI] [PubMed] [Google Scholar]
- 63.Morse JH, Fotino M, Zhang Y, Flaster ER, Peebles CL, Spiera H. Position 26 of the first domain of the HLA-DQB1 allele in post-silicone implant scleroderma. J Rheumatol. 1995;22:1872–5. [PubMed] [Google Scholar]
- 64.O’Hanlon T, Koneru B, Bayat E, Love L, Targoff I, Malley J, et al. Immunogenetic differences between Caucasian women with and those without silicone implants in whom myositis develops. Arthritis Rheum. 2004;50:3646–50. doi: 10.1002/art.20587. [DOI] [PubMed] [Google Scholar]
- 65.Chisholm CF, Nguyen BH, Soucie KR, Torres RM, Carpenter JF, Randolph TW. In vivo analysis of the potency of silicone oil microdroplets as immunological adjuvants in protein formulations. Pharm Sci. 2015;104:3681–90. doi: 10.1002/jps.24573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naim JO, Ippolito KM, Lanzafame RJ. The effect of molecular weight and gel preparation on humoral adjuvancy of silicone oils and silicone gels. Immunol Invest. 1995;24:537–47. doi: 10.3109/08820139509066849. [DOI] [PubMed] [Google Scholar]
- 67.Naim JO, Ippolito KM, van Oss CJ. Adjuvancy effect of different types of silicone gel. J Biomed Mater Res. 1997;37:534–8. doi: 10.1002/(SICI)1097-4636(19971215)37:4<534::AID-JBM12>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 68.Naim JO, Satoh M, Buehner NA, Ippolito KM, Yoshida H, Nusz D, et al. Induction of hypergammaglobulinemia and macrophage activation by silicone gels and oils in female A.SW mice. Clin Diagn Lab Immunol. 2000;7:366–70. doi: 10.1128/cdli.7.3.366-370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iribarren P, Correa SG, Sodero N, Riera CM. Activation of macrophages by silicones: phenotype and production of oxidant metabolites. BMC Immunol. 2002;3:6. doi: 10.1186/1471-2172-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sánchez O, Rodríguez-Sureda V, Domínguez C, Fernández-Figueras T, Vilches A, Llurba E, et al. Study of biomaterial-induced macrophage activation, cell-mediated immune response and molecular oxidative damage in patients with dermal bioimplants. Immunobiology. 2012;217:44–53. doi: 10.1016/j.imbio.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 71.Zhou J, Hao G, Weng H, Tsai YT, Baker DW, Sun X, Tang L. In vivo evaluation of medical device-associated inflammation using a macrophage-specific positron emission tomography (PET) imaging probe. Bioorg Med Chem Lett. 2013;23:2044–7. [DOI] [PMC free article] [PubMed]
- 72.Beekman WH, Feitz R, van Diest PJ, Hage JJ. Migration of silicone through the fibrous capsules of mammary prostheses. Ann Plast Surg. 1997;38:441–5. doi: 10.1097/00000637-199705000-00001. [DOI] [PubMed] [Google Scholar]
- 73.Silver RM, Sahn EE, Allen JA, Sahn S, Greene W, Maize JC, et al. Demonstration of silicon in sites of connective-tissue disease in patients with silicone-gel breast implants. Arch Dermatol. 1993;129:63–8. doi: 10.1001/archderm.1993.01680220075018. [DOI] [PubMed] [Google Scholar]
- 74.Greene WB, Raso DS, Walsh LG, Harley RA, Silver RM. Electron probe microanalysis of silicon and the role of the macrophage in proximal (capsule) and distant sites in augmentation mammaplasty patients. Plast Reconstr Surg. 1995;95:513–9. doi: 10.1097/00006534-199503000-00012. [DOI] [PubMed] [Google Scholar]
- 75.van Diest PJ, Beekman WH, Hage JJ. Pathology of silicone leakage from breast implants. J Clin Pathol. 1998;51:493–7. doi: 10.1136/jcp.51.7.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jara LJ, Medina G, Gómez-Bañuelos E, Saavedra MA, Vera-Lastra O. Still’s disease, lupus-like syndrome, and silicone breast implants. A case of ‘ASIA’ (Shoenfeld’s syndrome) Lupus. 2012;21:140–5. doi: 10.1177/0961203311430970. [DOI] [PubMed] [Google Scholar]
- 77.Katayama I, Umeda T, Nishioka K. Adult Still’s-disease-like illness in a patient with silicone breast implants. Clin Rheumatol. 1998;17:81–2. doi: 10.1007/BF01450969. [DOI] [PubMed] [Google Scholar]
- 78.Crétel E, Richard MA, Jean R, Durand JM. Still’s-like disease, breast prosthesis, and collagen implants. Rheumatol Int. 2001;20:129–31. doi: 10.1007/s002960000088. [DOI] [PubMed] [Google Scholar]
- 79.Montalto M, Vastola M, Santoro L, La Regina M, Curigliano V, Manna R, et al. Systemic inflammatory diseases and silicone breast prostheses: report of a case of adult still disease and review of the literature. Am J Med Sci. 2004;327:102–4. doi: 10.1097/00000441-200402000-00010. [DOI] [PubMed] [Google Scholar]
- 80.Błasiak A, Błachowicz A, Gietka A, Rell-Bakalarska M, Franek E. Still’s disease in patient with silicone breast implants: case report. Pol Arch Med Wewn. 2008;118:65–7. [PubMed] [Google Scholar]
- 81.Félix FH, Leal LK, Fontenele JB. Cloak and dagger: the case for adult onset Still disease and hemophagocytic lymphohistiocytosis. Rheumatol Int. 2009;29:973–4. doi: 10.1007/s00296-008-0825-z. [DOI] [PubMed] [Google Scholar]
- 82.Lee SJ, Cho YN, Kim TJ, Park SC, Park DJ, Jin HM, et al. Natural killer T cell deficiency in active adult-onset Still’s disease: correlation of deficiency of natural killer T cells with dysfunction of natural killer cells. Arthritis Rheum. 2012;64:2868–77. doi: 10.1002/art.34514. [DOI] [PubMed] [Google Scholar]
- 83.Khamaganova E, Aleschenko S, Murashova L, Zaretskaya Y. Immunogenetic factors of predisposition to blood malignancies in russian population. Russ J Immunol. 2001;6:265–70. [PubMed] [Google Scholar]
- 84.Choi HB, Roh SY, Choi EJ, Yoon HY, Kim SY, Hong YS, et al. Association of HLA alleles with non-Hodgkin’s lymphoma in Korean population. Int J Hematol. 2008;87:203–9. doi: 10.1007/s12185-008-0040-4. [DOI] [PubMed] [Google Scholar]
- 85.Jackow CM, McHam JB, Friss A, Alvear J, Reveille JR, Duvic M. HLA-DR5 and DQB1*03 class II alleles are associated with cutaneous T-cell lymphoma. J Invest Dermatol. 1996;107:373–6. doi: 10.1111/1523-1747.ep12363352. [DOI] [PubMed] [Google Scholar]
- 86.Hodak E, Lapidoth M, Kohn K, David D, Brautbar B, Kfir K, et al. Mycosis fungoides: HLA class II associations among Ashkenazi and non-Ashkenazi Jewish patients. Br J Dermatol. 2001;145:974–80. doi: 10.1046/j.1365-2133.2001.04496.x. [DOI] [PubMed] [Google Scholar]
- 87.Hodak E, Klein T, Gabay B, Ben-Amitai D, Bergman R, Gdalevich M, et al. Familial mycosis fungoides: report of 6 kindreds and a study of the HLA system. J Am Acad Dermatol. 2005;52:393–402. doi: 10.1016/j.jaad.2003.12.052. [DOI] [PubMed] [Google Scholar]
- 88.Vassallo C, Brazzelli V, Cestone E, Castello M, Ciocca O, Borroni RG, et al. Mycosis fungoides in childhood: description and study of two siblings. Acta Derm Venereol. 2007;87:529–32. doi: 10.2340/00015555-0254. [DOI] [PubMed] [Google Scholar]
- 89.Uçar F, Sönmez M, Erkut N, Balcı M, Yücel B, Yılmaz M, et al. Relation of HLA-A, -B, -DRB1 alleles and haplotypes in patients with acute leukemia: a case control study. Arch Med Res. 2011;42:305–10. doi: 10.1016/j.arcmed.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 90.Theodorou I, Abel L, Mauro F, Duprey B, Magnac C, Payelle-Brogard B, et al. High occurence of DRB1 11 in chronic lymphocytic leukaemia families. Br J Haematol. 2002;119:713–5. doi: 10.1046/j.1365-2141.2002.03926.x. [DOI] [PubMed] [Google Scholar]
- 91.Arons E, Adams S, Venzon DJ, Pastan I, Kreitman RJ. Class II human leucocyte antigen DRB1*11 in hairy cell leukaemia patients with and without haemolytic uraemic syndrome. Br J Haematol. 2014;166:729–38. doi: 10.1111/bjh.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.De Re V, Caggiari L, Talamini R, Crovatto M, De Vita S, Mazzaro C, et al. Hepatitis C virus-related hepatocellular carcinoma and B-cell lymphoma patients show a different profile of major histocompatibility complex class II alleles. Hum Immunol. 2004;65:1397–404. doi: 10.1016/j.humimm.2004.08.183. [DOI] [PubMed] [Google Scholar]
- 93.Beretta L, Rueda B, Marchini M, Santaniello A, Simeón CP, Fonollosa V, et al. Analysis of Class II human leucocyte antigens in Italian and Spanish systemic sclerosis. Rheumatology (Oxford) 2012;51:52–9. doi: 10.1093/rheumatology/ker335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Azzouz DF, Rak JM, Fajardy I, Allanore Y, Tiev KP, Farge-Bancel D, et al. Comparing HLA shared epitopes in French Caucasian patients with scleroderma. PLoS One. 2012;7(5):e36870. doi:10.1371/journal.pone.0036870. [DOI] [PMC free article] [PubMed]
- 95.Fallah M, Liu X, Ji J, Försti A, Sundquist K, Hemminki K. Autoimmune diseases associated with non-Hodgkin lymphoma: a nationwide cohort study. Ann Oncol. 2014;25:2025–30. doi: 10.1093/annonc/mdu365. [DOI] [PubMed] [Google Scholar]
- 96.Suvajdzic N, Djurdjevic P, Todorovic M, Perunicic M, Stojanović R, Novkovic A, et al. Clinical characteristics of patients with lymphoproliferative neoplasms in the setting of systemic autoimmune diseases. Med Oncol. 2012;29:2207–11. doi: 10.1007/s12032-011-0022-x. [DOI] [PubMed] [Google Scholar]
- 97.Takamine Y, Ikumi N, Onoe H, Hayase M, Nagasawa Y, Sakagami M, et al. Diagnostic value of brain biopsy in intravascular large B-cell lymphoma mimicking progressive multifocal leukoencephalopathy: case report. Nihon Rinsho Meneki Gakkai Kaishi. 2014;37:111–5. doi: 10.2177/jsci.37.111. [DOI] [PubMed] [Google Scholar]
- 98.Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185. doi:10.1155/2014/149185. [DOI] [PMC free article] [PubMed]
- 99.Lazzeri D, Agostini T, Bocci G, Giannotti G, Fanelli G, Naccarato AG, et al. ALK-1-negative anaplastic large cell lymphoma associated with breast implants: a new clinical entity. Clin Breast Cancer. 2011;11:283–96. doi: 10.1016/j.clbc.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 100.George EV, Pharm J, Houston C, Al-Quran S, Brian G, Dong H, et al. Breast implant-associated ALK-negative anaplastic large cell lymphoma: a case report and discussion of possible pathogenesis. Int J Clin Exp Pathol. 2013;6:1631–42. [PMC free article] [PubMed] [Google Scholar]
- 101.Hart AM, Lechowicz MJ, Peters KK, Holden J, Carlson GW. Breast implant-associated anaplastic large cell lymphoma: report of 2 cases and review of the literature. Aesthet Surg J. 2014;34:884–94. doi: 10.1177/1090820X14539503. [DOI] [PubMed] [Google Scholar]
- 102.Thompson PA, Lade S, Webster H, Ryan G, Prince HM. Effusion-associated anaplastic large cell lymphoma of the breast: time for it to be defined as a distinct clinico-pathological entity. Haematologica. 2010;95:1977–9. doi: 10.3324/haematol.2010.026237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Aivaliotis IL, Pateras IS, Papaioannou M, Glytsou C, Kontzoglou K, Johnson EO, et al. How do cytokines trigger genomic instability? J Biomed Biotechnol. 2012;2013:536761. doi:10.1155/2012/536761. [DOI] [PMC free article] [PubMed]
- 104.Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–12. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 105.Lowe DB, Storkus WJ. Chronic inflammation and immunologic-based constraints in malignant disease. Immunotherapy. 2011;3:1265–74. doi: 10.2217/imt.11.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Baecklund E, Smedby KE, Sutton LA, Askling J, Rosenquist R. Lymphoma development in patients with autoimmune and inflammatory disorders--what are the driving forces? Semin Cancer Biol. 2014;24:61–70. doi: 10.1016/j.semcancer.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 107.Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011;118:5794–8. doi: 10.1182/blood-2011-07-370148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Villanueva J, Lee S, Giannini EH, Graham TB, Passo MH, Filipovich A, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther. 2005;7:R30–7. doi: 10.1186/ar1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Grom AA, Villanueva J, Lee S, Goldmuntz EA, Passo MH, Filipovich A. Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. J Pediatr. 2003;142:292–6. doi: 10.1067/mpd.2003.110. [DOI] [PubMed] [Google Scholar]
- 110.Zhang K, Biroschak J, Glass DN, Thompson SD, Finkel T, Passo MH, et al. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis is associated with MUNC13-4 polymorphisms. Arthritis Rheum. 2008;58:2892–6. doi: 10.1002/art.23734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vastert SJ, van Wijk R, D’Urbano LE, de Vooght KM, de Jager W, Ravelli A, et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology (Oxford) 2010;49:441–9. doi: 10.1093/rheumatology/kep418. [DOI] [PubMed] [Google Scholar]
- 112.Wulffraat NM, Rijkers GT, Elst E, Brooimans R, Kuis W. Reduced perforin expression in systemic juvenile idiopathic arthritis is restored by autologous stem-cell transplantation. Rheumatology (Oxford) 2003;42:375–9. doi: 10.1093/rheumatology/keg074. [DOI] [PubMed] [Google Scholar]
- 113.Hazen MM, Woodward AL, Hofmann I, Degar BA, Grom A, Filipovich AH, et al. Mutations of the hemophagocytic lymphohistiocytosis-associated gene UNC13D in a patient with systemic juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:567–70. doi: 10.1002/art.23199. [DOI] [PubMed] [Google Scholar]
- 114.Cannella S, Santoro A, Bruno G, Pillon M, Mussolin L, Mangili G, et al. Germline mutations of the perforin gene are a frequent occurrence in childhood anaplastic large cell lymphoma. Cancer. 2007;109:2566–71. doi: 10.1002/cncr.22718. [DOI] [PubMed] [Google Scholar]
- 115.Ciambotti B, Mussolin L, d’Amore ES, Pillon M, Sieni E, Coniglio ML, et al. Monoallelic mutations of the perforin gene may represent a predisposing factor to childhood anaplastic large cell lymphoma. J Pediatr Hematol Oncol. 2014;36:e359–65. doi: 10.1097/MPH.0000000000000073. [DOI] [PubMed] [Google Scholar]
- 116.Chia J, Yeo KP, Whisstock JC, Dunstone MA, Trapani JA, Voskoboinik I. Temperature sensitivity of human perforin mutants unmasks subtotal loss of cytotoxicity, delayed FHL, and a predisposition to cancer. Proc Natl Acad Sci U S A. 2009;106:9809–14. doi: 10.1073/pnas.0903815106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bolitho P, Street SE, Westwood JA, Edelmann W, Macgregor D, Waring P, et al. Perforin-mediated suppression of B-cell lymphoma. Proc Natl Acad Sci U S A. 2009;106:2723–8. doi: 10.1073/pnas.0809008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Trapani JA, Thia KY, Andrews M, Davis ID, Gedye C, Parente P, et al. Human perforin mutations and susceptibility to multiple primary cancers. Oncoimmunology. 2013;2:e24185. doi: 10.4161/onci.24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Voskoboinik I, Sutton VR, Ciccone A, House CM, Chia J, Darcy PK, et al. Perforin activity and immune homeostasis: the common A91V polymorphism in perforin results in both presynaptic and postsynaptic defects in function. Blood. 2007;110:1184–90. doi: 10.1182/blood-2007-02-072850. [DOI] [PubMed] [Google Scholar]
- 120.Lykens JE, Terrell CE, Zoller EE, Risma K, Jordan MB. Perforin is a critical physiologic regulator of T-cell activation. Blood. 2011;118:618–26. doi: 10.1182/blood-2010-12-324533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Voskoboinik I, Trapani JA. Perforinopathy: a spectrum of human immune disease caused by defective perforin delivery or function. Front Immunol. 2013;4:441. doi:10.3389/fimmu.2013.00441. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Supporting materials for the manuscript “Intravascular large B-cell lymphoma associated with silicone breast implant, HLA-DRB1*11:01, and HLA-DQB1*03:01 manifesting as macrophage activation syndrome and with severe neurological symptoms: a case report”.