

Abstract
Human carcinoembryonic antigen-related cell adhesion molecule 1 (C?/Au: EACAM1) is a cell-surface signaling molecule involved in cell adhesion, proliferation, and immune response. It is also implicated in cancer angiogenesis, progression, and metastasis. This diverse set of effects likely arises as a result of the numerous homophilic and heterophilic interactions that CEACAM1 can have with itself and other molecules. Its N-terminal Ig variable (IgV) domain has been suggested to be a principal player in these interactions. Previous crystal structures of the β-sandwich-like IgV domain have been produced using Escherichia coli-expressed material, which lacks native glycosylation. These have led to distinctly different proposals for dimer interfaces, one involving interactions of ABED β-strands and the other involving GFCC′C″ β-strands, with the former burying one prominent glycosylation site. These structures raise questions as to which form may exist in solution and what the effect of glycosylation may have on this form. Here, we use NMR cross-correlation measurements to examine the effect of glycosylation on CEACAM1-IgV dimerization and use residual dipolar coupling (RDC) measurements to characterize the solution structure of the non-glycosylated form. Our findings demonstrate that even addition of a single N-linked GlcNAc at potential glycosylation sites inhibits dimer formation. Surprisingly, RDC data collected on E. coli expressed material in solution indicate that a dimer using the non-glycosylated GFCC′C″ interface is preferred even in the absence of glycosylation. The results open new questions about what other factors may facilitate dimerization of CEACAM1 in vivo, and what roles glycosylation may play in heterophylic interactions.
Keywords: cell adhesion, dimerization, glycosylation, nuclear magnetic resonance (NMR), structural model
Introduction
The human carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1)2 is involved in cell adhesion, proliferation, and immune response (1, 2). It also is implicated in cancer angiogenesis, progression, and metastasis (3). More specifically, it is known that CEACAM1 is a negative-regulator of cell proliferation and is down-regulated in some tumor cells (4–10). Yet, CEACAM1 expression is reported to protect tumor cells from killing by immune cells (11–14) and it has been found that the high expression level is associated with a number of other cancers (15–19). It is likely that this complex set of effects arises as a result of the numerous homophilic and heterophilic interactions that CEACAM1 can have with itself and other members of the CEACAM superfamily. The N-terminal Ig variable (IgV) domain of CEACAM1 has been suggested to be the basis of cis and trans homo-dimer formation (20), as well as interactions with other molecules (21–24). There are crystal structures of the IgV domain expressed in Escherichia coli, which have led to the suggestion of two distinct dimerization interfaces (24, 25). However, examination of the interfaces suggests that the extensive glycosylation found in native material would inhibit dimer formation in one of these cases. This raises questions about the actual type of dimer found in solution, with and without glycosylation. Here, we use NMR methods to examine dimer structures in solution using a non-glycosylated form expressed in E. coli, and several glycosylated variants expressed in HEK293 cells.
Dimerization of cell surface molecules is a well accepted mechanism for transmitting signals from the cell surface to the interior (26). In the case of CEACAM1 it is believed that modulation of intercellular adhesion as well as transmission of signals to the cell interior involves switching between cis and trans dimerization interactions. The full-length CEACAM1 molecule is large, sharing a topology with many other cell-surface signaling molecules; the extracellular domain is composed of one IgV-like domain and typically 3 IgC2-like domains; these are followed by a single transmembrane helix and cytoplasmic tails of different lengths (L and S forms) (20). Hence, subtle changes in the nature of dimerization, which may begin with the IgV domain, must be transmitted through the entire molecule to reach the cell interior and cytoplasmic tails that contain ITIM signaling motifs. Although it is possible that transmission of cis versus trans interactions may result from changes in inter-domain positions that propagate from the N terminus to the cytoplasmic elements, it is also possible that dimerization involves more direct interactions of other domains. IgC2-like domains of some family members are, in fact, believed to contribute to dimer stability in trans interactions (27) and the transmembrane domain of CEACAM1 has been found to promote the cis-dimerization (28, 29). To further complicate matters, the oligomeric states of CEACAM1 are believed to be diverse and dynamic (30). This makes it important to investigate in more detail the structural aspects of domain interactions, beginning with the IgV domain, before extending investigations to other constructs.
The existing crystal structures of the CEACAM1 IgV domain shows it exhibits a β sandwich topology with nine anti-parallel β strands, designated A, B, C, C′, C″, D, E, F, and G (Fig. 1A). Four of these are on one side of the molecule (ABED) and five (GFCC′C″) are on the other side. The asymmetric unit in an earlier crystal structure led to the suggestion that dimerization occurred via an interface involving the ABED face (25). More recent structures suggest dimerization occurs via the GFCC′C″ face (24). Supporting this more recent suggestion, interactions involving the GFCC′C″ side have been reported to be crucial for various types of biological response, including protection from killing by NK cells. More specifically mutagenesis of Arg-43 and Gln-44 on strand C′ indicate these residues are critical for protection through a process believed to involve dimer formation (31). Val-39 and Asp-40 mutations in the CC′ loop have also been shown to diminish homophilic dimer formation in full-length CEACAM1 (32), and this same loop region also seems to be required for intercellular interaction with certain other family members (33). The recent crystal structure, Protein Data Bank (PDB) code 4QXW, as well as 4WHD, provide direct evidence that contacts between the GF loop and adjacent strands, along with CC′ loop contacts, can form an interface (24). However, involvement of the ABED interface in dimer formation, as observed in the earlier crystal structure, PDB code 2GK2 (25), also has support. In particular, electron tomography data, supported by SPR data suggested both the ABED (parallel) and GFCC′C″ (antiparallel) interactions occur (30).
FIGURE 1.

Schematics depicting the structure of hCEACAM1-Igv and the constructs of hCEACAM1-Igv proteins used in this article. A, ribbon diagram of the crystal structure of the CEACAM1 N-terminal Igv domain (PDB code 4QXW). Nine β-sheets are labeled as A, B, C, C′, C″, D, E, F, and G, and they form two distinct faces, which can participate in dimer formation, ABED and GFCC′C″. The former makes dimerization contacts in the 2GK2 crystal structure and the latter makes dimerization contacts in the 4QXW crystal structure. B, CEACAM1-Igv constructs with approximate positions of glycosylation shown as high mannose glycans for the initial expression in HEK293S (GnT1−) cells, as a single GlcNAc after endoglycosidase F1 digestion of this product, and as a non-glycosylated asparagine (N) when produced in E. coli. The blue squares and green circles represent GlcNAc and mannose, respectively. Differences in the constructs at the N and C terminus are also denoted.
Some observations, particularly those based on the crystal structures, must be taken with caution because material was produced by E. coli expression where glycosylation does not occur. CEACAM1, like other members of this large family, is heavily glycosylated. The IgV domain alone has three putative N-glycosylation sites, 104, 111, and 115 based on the Uniprot sequence, Uniprot number P13688. We will use numbering that starts after a signal peptide and designate the sites as 70, 77, and 81. Site 70 is in the dimer interface of the x-ray crystal structure 2GK2 (25) and is highly conserved within the family (34). The authors of this crystal structure did note that the putative N-linked glycosylation site, Asn-70, is located in the dimer interface of 2GK2 and may suppress dimer formation in the presence of glycans. However, there is little site-specific data on the extent of glycosylation in the CEACAM1 preparations used in other studies. Thus, there is substantial reason to further investigate the structure of CEACAM1-IgV dimers under both well defined glycosylated and non-glycosylated conditions. It is also desirable to carry out these investigations in solution where dimer formation cannot be influenced by crystal contacts.
NMR offers an efficient way to assess dimer characteristics in solution, particularly when a crystal structure is available to provide a monomer structure that is likely conserved in solution. The presence of a dimer can be confirmed or discounted, and many times the actual structural form can be validated based on a limited set of NMR data. A non-glycosylated version of the IgV domain can be expressed using an E. coli host in media supplemented with [13C]glucose and [15N]ammonium chloride. The resulting uniform labeling allows assignment of most backbone resonances with a minimal set of triple resonance experiments (35). Glycosylated versions are usually expressed in eukaryotic cells that require media supplemented with a complete set of amino acids. If triple resonance assignment strategies are to be used, isotopically labeled versions of all required amino acids add considerable expense. However, when a pair of glycosylated and non-glycosylated proteins are examined, a limited set of less expensive amino acids labeled only with 15N can be used, and assignments can be transferred from non-glycosylated to glycosylated sets based on resonance overlap.
We, therefore, have used expression in E. coli to produce a uniformly labeled non-glycosylated protein and expression in mammalian HEK293 cells to produce glycosylated versions of the sparsely 15N-labeled protein (36). Three glycosylated forms have been prepared, one wild type, where glycans are large and complex, one expressed in GnT1− cells that carries predominantly (Man)5-(GlcNAc)2 glycans, and one in which these glycans are trimmed to a single GlcNAc residue. Once NMR resonance assignments were made on non-glycosylated material and transferred to the glycosylated material, 1H-15N HSQC spectra provided a structure-sensitive fingerprint that allowed comparison of the domain structure in the various constructs. They also provided a basis for measurement of rotational correlation times that are sensitive to dimer formation (37), and for the collection of residual dipolar coupling (RDC) data that are orientation dependent. The latter can more rigorously confirm the retention of the domain structure, as well as distinguish which of the dimer structures seen in crystal structures may be present in solution (38, 39). Our findings demonstrate that the non-glycosylated form exists as a dimer involving the GFCC′C″ interface in solution, but minimal glycosylation inhibits all types of dimerization. These observations open many questions about the roles that glycosylation may play in the presence of other domains, and in interactions of CEACAM1 with other cell-surface molecules.
Results
Aggregation State of Non-glycosylated CEACAM-IgV
A 15N-1H spectrum of the E. coli expressed, uniformly labeled, sample is shown in Fig. 2A. The cross-peaks are well dispersed and occur in numbers consistent with that expected for the 120-residue construct (112 of the expected 115, not counting prolines). The dispersal and peak count is typical of a well folded protein, and the significant proportion of cross-peaks in the low field region of the spectrum (8.5–11 ppm) is consistent with a substantial β-sheet content. Triple resonance experiments, as described under “Experimental Procedures,” allowed the assignment of 75/112 of the backbone resonances. Given the existence of a crystal structure and alternate means of assessing the preservation of this structure in solution, this level of backbone assignments is adequate, and full side chain assignments were not pursued.
FIGURE 2.
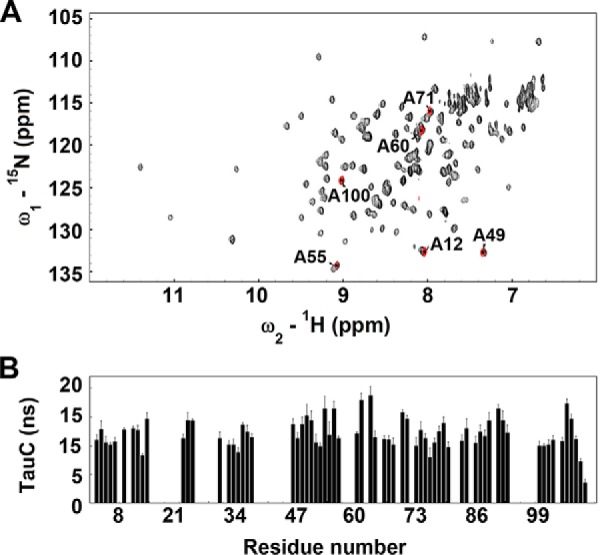
1H-15N HSQC spectra and rotational correlation times for CEACAM-IgV. A, the 1H-15N HSQC spectra of non-glycosylated CEACAM1-Igv (black) superimposed with (GlcNAc)1-CEACAM1-Igv (red). B, the cross-correlation time (τC) of assigned non-glycosylated CEACAM1-Igv residues. The τC were extracted from the SCT-CCR experiment. Data were collected at 25 °C with a protein concentration of 150 μm in 10 mm Tris, pH 8.0. Errors are standard deviations from fits to multipoint decay curves.
Rotational correlation times provide a convenient means of detecting the presence of a dimer under the conditions of NMR data collection. For overall protein rotation, one expects correlation times in nanoseconds to be about one-half the effective molecular mass in kDa when measured at 298 K in dilute aqueous solution. This relationship can be derived from the Stokes formula for rotational Brownian diffusion. For a CEACAM1-IgV monomer, therefore, a correlation time of about 6.5 ns is expected; for a CEACAM1-IgV dimer 13 ns is expected. There are several NMR methods available for measurement of correlation times. We have used a method based on interference between 15N relaxation contributions from chemical shift anisotropy and dipole-dipole mechanisms (37). This has an advantage over T1/T2 measurements in that it is less sensitive to chemical exchange effects. Residue-specific correlation times for 67 well resolved cross-peaks are presented in Fig. 2B. The average correlation time over this set is 12 ns, close to the value expected for a dimer. Correlation times can be reduced for residues with significant levels of internal motion and it is acceptable to exclude these when evaluating a correlation time for overall tumbling of a molecular complex. Excluding those below 9 ns gives an average of 12.5 ns. Hence, at the concentrations of our NMR experiment (150 μm) non-glycosylated versions of CEACAM-IgV appear to be dimers.
Aggregation State of Glycosylated CEACAM1-IgV
The glycosylated versions of CEACAM1-IgV were expressed in mammalian (HEK293) cell cultures producing large and complex wild type glycans (FreeStyleTM 293-F cells, Thermo Fisher Scientific) or cultures that lack the ability to extend to full complex type structures (HEK293S (GnT1−)) (40). The latter cell line produces glycans of predominantly the (Man)5-(GlcNAc)2 type. These high mannose glycans can also be trimmed to a single GlcNAc residue producing a third type of sample. Compared with the fully complex and high mannose forms of glycosylation, which are heterogeneous, we felt it important to initially work with a more homogeneous sample containing single GlcNAc residues at the three potential glycosylation sites. MS analysis indicated that all three sites were highly glycosylated. In particular, the Asn-70 site was more than 62% glycosylated with a single GlcNAc.
Uniform isotopic labeling of glycosylated proteins is extraordinarily expensive, and for our purposes only sufficient sites for rotational correlation time determination and verification of basic domain structure is required. Labeling of glycosylated samples, therefore, proceeded with a single labeled amino acid. [15N]Alanine was chosen because the alanine sites are fairly numerous (7) and they are well dispersed throughout the structure. 15N was chosen so that HSQC cross-peaks could be directly compared with cross-peaks from the uniformly labeled sample.
Alanine resonances of the minimally glycosylated sample are superposed (red cross-peaks) with cross-peaks of the uniformly 15N,13C-labeled CEACAM1-Igv expressed in E. coli in Fig. 2A. Only six alanine cross-peaks are observed in the minimally glycosylated sample. However, one alanine is near the N terminus of both species and amide protons in these largely disordered regions can exchange rapidly leading to peak broadening or intensity loss due to exchange with partially saturated water protons. All of the remaining 6 peaks superimpose well with alanine cross-peaks in the spectrum of the uniformly labeled sample.
HSQC spectra are frequently used as structural fingerprints, with shifts in cross-peak position of several tenths of a ppm in the proton domain and several ppms in the nitrogen domain indicating changes in secondary structure. The close superposition of cross-peaks in the HSQC spectra between the non-glycoylated E. coli expression product and the minimally glycosylated mammalian expression product argues strongly for preservation in solution of the domain structures seen in the crystal structures. Ala-71 is adjacent to the glycosylation site Asn-70, but even here the deviation is small. Ala-49 and Ala-55 are located in the GFCC′C″ dimer interface (more specifically the C′C″ motif). Ala-12 and Ala-71 are in the ABDE interface (more specifically AB and DE loops). Ala-12 shows the largest shift between glycosylated and non-glycosylated versions, but even this is small. Cross-peaks can move upon dimer formation, but changes tend to be smaller than any change in secondary structure, so differences in chemical shift provide no definitive evidence for a change in aggregation state.
Even though a small number of sites are labeled in the glycosylated versions, the number is adequate to determine a rotational correlation time for the protein. The individual correlation times for alanine residues in glycosylated and non-glycosylated versions are compared in Table 1. Excluding Ala-100, which appears to be affected by internal motion, the average correlation time for the sample with single GlcNAc residues at a concentration of 150 μm and 298 K, is 8.5 ns. The value of 8.5 ns is near that expected for a monomer. The fact that it is a little larger than expected may partly reflect the added mass of the monosaccharide residues and partly some dimerization. Also included is a correlation time measurement for this protein preparation at 300 μm. The fraction of dimer in the weak association limit is expected to increase as the square of the concentration and the effective correlation times should increase proportionately. The moderate increase (to an average of 10.3 ns) upon doubling the concentration supports a low dimerization constant. A sample with (Man)5-(GlcNAc)2 glycosylation at 150 μm gave an average correlation time of 9.3 ns. This may again reflect the bulk of the larger glycans, but could also suggest that larger glycans may promote some dimerization. This led to our testing of the wild type sample having complex N-glycans. The average correlation times observed are 14.2 and 15.4 ns, at concentrations of 150 μm and 1 mm, respectively. It is tempting to conclude that this increase in correlation time reflects increased mass resulting from dimerization. However, the mass also increases with the additional glycosylation. The most abundant wild type glycans, as assayed by mass spectrometry on a similarly expressed sample, have 11 or 12 sugar residues. If these are representative of all glycans, and all three sites are 100% occupied, glycosylation would add more than 8000 Da to the molecular mass. A linear dependence of correlation time on molecular weight would then lead to a prediction of a 11-ns correlation time if we use the 6.5-ns figure for a monomer and 14 ns if we use the 8.5-ns figure for a monomer. Our observations show only a slightly larger correlation time and little concentration dependence, both of which argue against dimer formation. Given that the linear dependence of the correlation time on molecular weight is only an approximation, we cannot definitely exclude restoration of dimerization in the presence of complex glycans, but the weight of the evidence argues against this.
TABLE 1.
Comparison of rotational correlation times (ns) of alanines in glycosylated and non-glycosylated CEACAM1-IgV
| 150 μm Non-glycosylated | 150 μm Non-glycosylated L18R | 150 μm Non-glycosylated I91R | 150 μm GlcNAc | 150 μm (Man)5-(GlcNAc)2 | 150 μm Complex glycan | 300 μm GlcNAc | 1 mm Complex glycan | |
|---|---|---|---|---|---|---|---|---|
| Ala-12 | 12.6 | 11.5 | 5.2 | 8.3 | 9.2 | 10.1 | 8.9 | 14.0 |
| Ala-49 | 15.1 | 12.1 | n/aa | 9.2 | 12.7 | 15.9 | 11.1 | 17.5 |
| Ala-55 | 15.4 | 12.4 | n/aa | 7.7 | 7.4 | 15.6 | 11.1 | 14.9 |
| Ala-60 | 12.1 | 12.3 | 5.9 | 9.0 | 8.1 | 13.9 | 9.2 | 16.2 |
| Ala-71 | 14.4 | 12.3 | 5.8 | 8.2 | NAa | 15.5 | 11.2 | 14.3 |
| Ala-100 | 10.2 | 12.0 | 4.7 | 6.0 | 7.7 | 12.7 | 7.6 | 13.0 |
a Peak not observable or errors >2.4 ns. Other errors average 1.3 ns.
The cross-correlation time measurements were also performed for mutants L18R and I91R of the non-glycosylated form to affirm the involvement of the GFCC′C″ interface in dimer formation. Leu-18 is in the middle of the ABED dimer interface as seen in the 2GK2 crystal structure and the residues on the respective monomers appear to participate in a favorable hydrophobic interaction. Replacement of this pair with a pair of positively charged arginines should result in strong repulsion and disruption of an ABED dimer if it formed in solution. Ile-91 is in the middle of the GFCC′C″ interface and appears to play a similar role in hydrophobic stabilization of the dimer as seen in the 4QXW crystal structure. Replacement of these residues with arginines should result in disruption of the GFCC′C″ dimer if it formed in solution. These mutations were generated for the non-glycosylated E. coli expression product and the average τC for the L18R and I91R mutants are 12.2 and 5.4 ns, respectively. The specific values for various alanines are listed in Table 1. According to Stokes law, the 5.4-ns correlation time suggests that mutant I91R is a monomer and L18R is a dimer, implying that GFCC′C″ is more likely the dimer interface.
Structural Characterization of the CEACAM1-IgV Dimer
RDCs between 15N-labeled amide nitrogens and directly bonded amide protons provide a good means of comparing structures found in solution to existing x-ray structures. These couplings measure the average of (3cos2θ-1), where θ is the angle between the inter-nuclear vector and the magnetic field in which data are collected. They do require partial ordering of the molecule, usually less than 0.1% departure from isotropy, and a minimum of 5 measurements is needed to assess the extent, anisotropy, and direction of this ordering. Adequate data can clearly be obtained for the uniformly labeled non-glycosylated protein. In this case, two media were used to help in identifying a symmetry axis for any symmetric dimers present. Bacteriophage and PEG C12E5 provided appropriate orientation and 59 and 55 measurements on assigned cross-peaks were obtained in the respective media as described under “Experimental Procedures.” Several of these measurements were excluded based on their association with regions of the protein showing high internal mobility (i.e. loop regions as identified from the crystal structures and low correlation time regions). The REDCAT software package was used, along with the 2GK2 and 4QXW monomer structures, to solve for the best set of order parameters describing the extent, anisotropy, and direction of alignment (41, 42). These parameters were used to back-calculate RDCs for comparison to experimental data. A correlation plot is shown in Fig. 3A with fits to both phage and PEG data. The Q factors of 0.30 and 0.29 are typical of agreement between solution NMR structures and x-ray structures that have root mean square deviations for backbone atom positions between 1.8 and 2.5 Å (43). Thus, at least the domain structure of CEACAM1-IgV seen in the 2GK2 and 4QXW structures is maintained (44).
FIGURE 3.
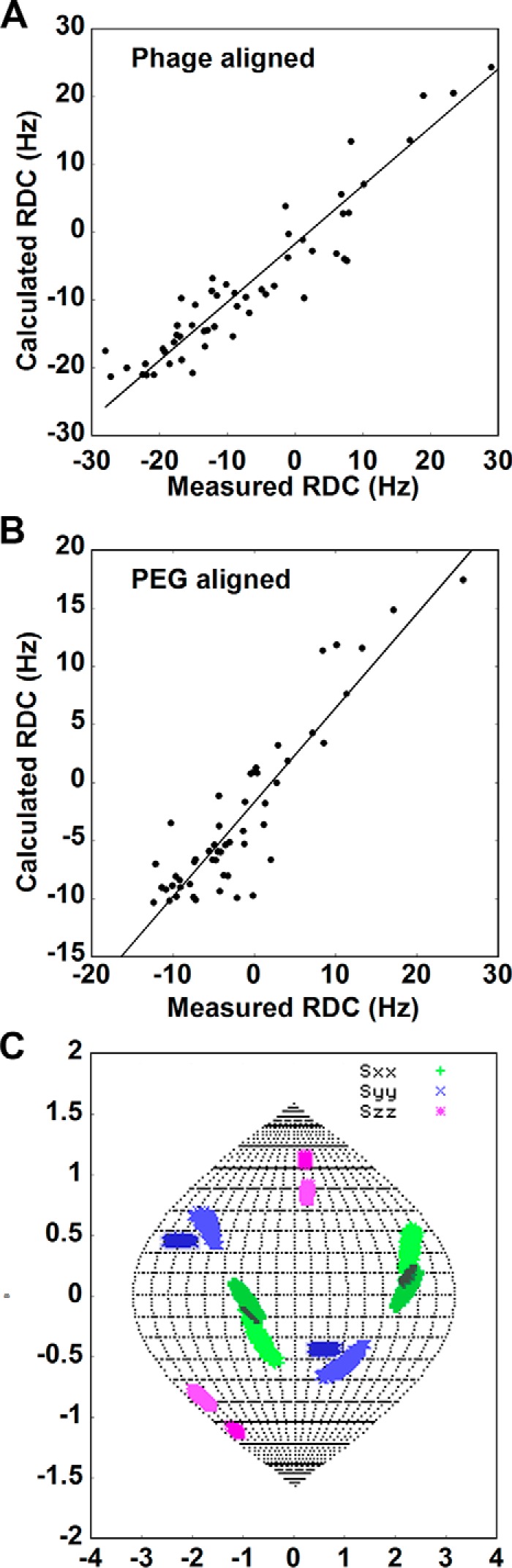
Using residual dipolar coupling data on non-glycosylated CEACAM1-IgV. Correlation plots between the experimental and back-calculated RDCs collected in (A) phage and (B) PEG alignment media using the crystal structure 4QXW chain A for back-calculations. C, Sauson-Flamsteed projections showing the orientation of phage (darker color) and PEG (lighter color) alignment tensor axes. The black area indicates overlap that identifies the symmetry axis. The plot was produced using the REDCAT program.
Beyond retention of domain structure, the orientation of the principle alignment frame that results from analysis of RDCs can provide information on the direction of the dimer symmetry axis. The orientations of alignment axes are conveniently displayed in the Sauson-Flamsteed plot shown in Fig. 3B. The clusters of points shown for each axis (x, green; y, blue; and z, red) are shown as light and dark shades of these colors for the two media. Note that the axis directions are different for the y and z axes in the two media reflecting differences in the types of interaction and nature of media particles (neutral disks and negatively charged rods for PEG and phage, respectively). The clusters for the x-axis overlap significantly. A symmetric dimer must have one of the alignment axes fall along the symmetry axis of the dimer, for any alignment medium used (38). The overlap of the x-axis identifies this as the symmetry axis. Once this axis is known, generating a model for a symmetric homodimer is relatively straightforward. Two copies of the monomer structure are loaded into appropriate molecular modeling/display software (Chimera (45)) with the symmetry axis along one of the display axes (usually z). One monomer is then rotated 180 degrees about this axis, and translated in a plane perpendicular to this axis, to produce a dimer structure. An appropriate scoring function, or some independent surface perturbation data, is usually used to identify the best dimer interface. However, in our case we have two potential dimers from the existing crystal structures and wish to identify the one that best fits our data. Therefore, a potential dimer (PDB codes 2GK2 or 4QXW) was superimposed on the rotated and un-rotated pair by matching Cα carbons of one-half of the dimer to the un-rotated monomer, and the rotated monomer was translated to make an interface as close as possible to that seen in the crystal structure. A grid search with step sizes of 1 Å in the plane perpendicular to the symmetry axis was then executed starting at this point and extending to 5 steps in each direction to generate a viable model. Models were first eliminated based on lack of a sufficient number of contact atom pairs (less than 3) and the existence of severe clashes (more than 1). For this purpose two atoms i and j with an inter-atom distance less than rvdw i + rvdw j + 1 Å are considered a contact pair, whereas they are considered to clash when the distance between them is smaller than rvdw i + rvdw j − 1 Å (45, 46). Energies for the remaining models were calculated using the minimize function in Chimera tools with the number of steps set to zero. Fig. 4 shows dimers with the lowest energy superimposed with dimer structures from PDB files 2GK2 (Fig. 4A) and 4QXW (Fig. 4B). Clearly the solution dimer built on 4QXW has the best fit. The contact pairs in the 4QXW dimer interface, Ile-91 and Leu-95, Gln-44 and Leu-95, are preserved in the modeled dimer and the calculated root mean square deviation for the backbone is 1.0 Å. However, the symmetry axis of 2GK2 is distinct from the one calculated from RDC and the backbone root mean square deviation of the lowest energy structure is 19.7 Å.
FIGURE 4.
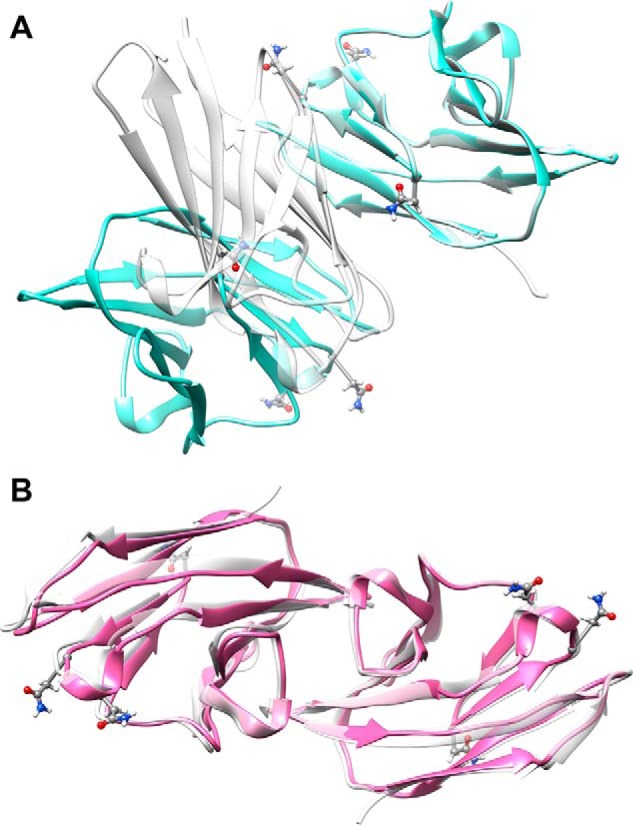
Non-glycosylated CEACAM1-IgV dimers constructed by rotation of monomers about the symmetry axis identified from RDC data. A, the rotated monomer has been translated to produce a model (shown in blue) with a best match to the dimer interface shown in the crystal structure 2GK2. B, the rotated monomer has been translated to produce a model (shown in red) with a best match to the dimer interface dimer shown in the crystal structure 4QXW. In each case the crystal structure dimers are shown in gray. One monomer of the model has been matched to a monomer in the crystal structure to depict the deviation in the position of the second monomer. The backbone root mean square deviation between the non-superimposed monomer is 19.7 Å in A and 1.0 Å in B. The glycosylation sites are represented in ball-and-stick mode for the asparagine residues.
RDCs have also been measured for the alanines in the single GlcNAc version, but 6 measurements are marginal for determination of the orientation of an alignment frame. However, the measurements for alanines in glycosylated and non-glycosylated versions can be compared directly. If the same dimer structure had been preserved in a molecule with a single GlcNAc at potential glycosylation sites, one would expect at most a simple scaling of RDC values between samples. RDCs are compared in Table 2. The deviations are larger than expected, supporting a change from dimer to monomer structure. Hence, glycosylation inhibits dimer formation, but the dimer structure that it inhibits is not the one with the obviously buried glycosylation site, but the one involving the GFCC′C″ interface.
TABLE 2.
RDC of alanines in CEACAM1 N-terminal IgV domain
| Non-glycosylated aligned in peg | Non-glycosylated aligned in phage | GlcNAc aligned in peg | |
|---|---|---|---|
| Hz | |||
| Ala-12 | −9.7 ± 1.7 | −12.9 ± 1.8 | −8.4 ± 3.5 |
| Ala-49 | 7.2 ± 2.8 | 7.1 ± 0.7 | 19.8 ± 4.1 |
| Ala-55 | 11.3 ± 6.1 | 23.4 ± 3.4 | 23.7 ± 3.8 |
| Ala-60 | NAa | NA | 15.8 ± 2.3 |
| Ala-71 | −5.5 ± 2.6 | −11.8 ± 1.8 | 0.8 ± 2.1 |
| Ala-100 | −1.3 ± 3.7 | −15.2 ± 1.8 | 2.8 ± 4.0 |
a Not available because of the fitting error >10 Hz.
Discussion
Our finding that in the absence of glycosylation the dimer formed uses the GFCC′C″ surface, rather than the ABED surface is at first surprising. For one thing, had we observed a dimer using the ABED surface, the inhibition of dimerization upon glycosylation would have been easy to rationalize. The ABED face hosts the Asn-70 glycosylation site and addition of a glycan could easily inhibit dimerization by steric hindrance. The dimer structure we find, the one from the 4QXW crystal structure that uses GFCC′C″ surface, places all three glycosylation sites well away from the interface, and it is more difficult to explain why the addition of single GlcNAc monosaccharides inhibits dimer formation.
Also the previous EM and SPR data suggesting the involvement of both ABDE and GFCC′C″ surfaces in dimerization needs to be explained (30). These prior studies used material derived from expression in HEK293 cells, which should be glycosylated, but we see no evidence of any strong dimer in the presence of glycosylation. However, the latter studies used constructs with multiple domains rather than the isolated IgV domains we used, and the additional domains could have contributed to dimer formation. Participation of additional domains in cis interactions of CEACAM1 is clearly supported (28, 32, 47).
There are pieces of evidence that do support the GFCC′C″ dimer as the major dimer form in solution. An examination of changes in solvent accessible surface (SAS) upon formation of the two dimers provides a rationale in favor the formation of a GFCC′C″ dimer. Solvent accessible surface values of 1192 versus 808 Å2 were observed for the GFCC′C″ and ABED dimer models, respectively. Also, upon further inspection of the crystal structure that led to the suggestion of an ABED dimer, a GFCC′C″ dimer interface can also be found when molecules in adjacent unit cells are examined. This, along with the observation of similar GFCC′C″ dimers for E. coli expressed material for CEACAM5 (48) and CEACAM6 (49), supports our observation of a GFCC′C″ dimer for CEACAM1 in solution.
Our observation that minimal glycosylation inhibits dimer formation of even the GFCC′C″ dimers is surprising, given that the glycosylation sites are well removed from the dimerization interface. In addition to the SPR and EM data cited above, which showed dimerization for HEK cell-expressed material that should have been heavily glycosylated, studies by Watt et al. (32), using mutagenesis and adhesion between cells overexpressing different CEACAM1 constructs and immobilized CEACAM1-Fc, identified the GFCC′C″ face of the terminal domain of CEACAM1 as important for interaction. These studies used CHO cells, which should have produced highly glycosylated forms of CEACAM1. However, these studies also used multiple domain constructs. Interestingly, in the same study employing a construct having just the N-terminal, transmembrane and short cytoplasmic domain showed negligible adhesion, opening the possibility that IgC2 domains participate or in some way facilitate dimerization.
There is also some direct support for the hypothesis that glycosylation inhibits dimerization. For example, in vitro adhesion assays reported by Watt et al. (32) show that a glycosylated N-terminal domain alone cannot form a tight dimer in vitro. More recent studies involving heterodimer formation between CEACAM6 and CEACAM8 show a decrease in binding affinity for isolated IgV domains when comparing glycosylated to non-glycosylated preparations (49). There was also a change in apparent stoichiometry when using glycosylated material, indicating that some glycoforms may more strongly inhibit dimerization.
We now must consider how minimally glycosylated sites that are far from the dimerization surface could inhibit dimerization. One possibility is that a local effect is propagated by protein structure. Glycosylation site Asn-81 is in a helical turn at the beginning of β-strand F and adjacent to Asp-82, which forms a salt bridge with Arg-64 and seems to be necessary for stability and dimerization (32). Also, Ala-71, which is adjacent to the Asn-70 glycosylation site and shows a small change in chemical shift on glycosylation, makes a strong hydrophobic contact with Tyr-31, a residue on β-strand C, an integral part of the GFCC′C″ dimer interface.
We cannot completely dismiss the possibility that dimerization may be restored in the presence of certain types of complex glycosylation. Our wild type sample carries a very heterogeneous set of glycans. Certain glycans are terminated with multiple negatively charged sialic acids, but these glycans are present in only a fraction of the structures. Hence, the possibility exists that electrostatic interactions of these glycans could stabilize the formation of a dimer. Asn-81 is sufficiently close to the dimer interface for the termini of an attached complex glycan to reach a positively charged patch composed of four arginines and two lysines, Lys-35, Arg-38, and Arg-43 that runs across the base of the dimer. Glycan-glycan interactions are also known to occur, particularly in sialic acid-terminated glycans in which bridging by divalent cations can be important (50). If such interactions exist, variation in glycosylation could be an important modulator of signaling the cis and trans homotypic interactions that appear to affect cellular function. It is now possible to engineer homogeneous glycans into glycoproteins so that some of these possibilities can be tested (51).
The potential involvement of glycosylation in homotypic interactions of CEACAMs, even if they are only inhibitory, suggests there may also be impacts of glycosylation on heterotypic CEACAM1 interactions. These interactions are quite numerous, including those that are physiologically important and those that involve pathogen proteins. Recently, it has been found that the IgV domain can interact with an immune regulator domain (T-cell immunoglobin domain and mucin domain 3, TIM-3), which shares a similar tertiary structure with CEACAM1-IgV (24). Although the initial structure of a heterodimer reported has been withdrawn, NMR and SPR data confirm the existence of an interaction (59). TIM-3 is a negative immune regulator involved in T-cell dysfunction occurring in cancer and chronic viral infection. Opacity (Opa) proteins from organisms such as Neisseria meningitidis and Neisseria gonorrhoeae use CEACAM1 as a receptor and appear to reduce antibody production as a consequence of this interaction (25). Mutagenesis suggests that Opa proteins target the interface that forms homo- and heterotypic dimers with other CEACAMs. Inhibition of normal interactions with TIM-3 may play a role here.
It will clearly be important to extend studies to multiple domain constructs. As discussed above, additional domains are believed to contribute to cis interactions. Also, understanding the mechanism of signaling is important. Both outside-in and inside-out cytoplasmic signaling are known (28). Propagating signals by domain-domain interactions may be essential (30, 54). Some of the NMR methods used here, RDCs, for example, are well suited to the investigation of domain-domain interactions (55). As all of these domains are heavily glycosylated, it will be important to study these constructs with defined glycosylation patterns.
Experimental Procedures
Protein Expression and Purification
A sample of the CEACAM1-IgV domain uniformly labeled in 13C and 15N was produced in E. coli to obtain a protein lacking glycosylation and allowing resonance assignment using triple resonance NMR methods. A pET28b expression vector that encodes a C-terminal His tag form of CEACAM1 was prepared using E. coli codon optimization for residues 34–141 of CEACAM1 (Genscript, China). This was used to transform E. coli BL21(DE3) in a 10-ml starter culture in LB media. When cell density A600 reached 0.8, isopropyl 1-thio-β-d-galactopyranoside was added to a final concentration of 0.5 mm to induce expression. Cells were grown at 25 °C in 1 liter of M9 minimal media containing 1 g of [15N]ammonium chloride and 2 g of [13C]glucose (Cambridge Isotope Laboratories). Cells were harvested after culture for 16 h. The protein was isolated from inclusion bodies and unfolded in 8 m urea. It was isolated using a Ni2+-NTA column and refolded by rapid dilution in 10 mm Tris, pH 8.0. The protein was further purified using a Superdex-75 gel filtration column and eluted with 10 mm Tris, pH 8.0. Protein from fractions with A280 greater than 100 milli-absorbance units was concentrated to ∼200 μm using Centricon tubes with a 3000 MWCO cut off. Total yield was ∼10 mg.
Samples of glycosylated CEACAM1-IgV, sparsely labeled with a selected 15N-labeled amino acid (alanine in the present case), were prepared by mammalian cell expression in either FreeStyleTM 293-F cells (Thermo Fisher Scientific, Waltham MA) for expression with wild type glycans or HEK293S GnTI− cells (ATCC catalog number CRL-3022) for expression with primarily (Man)5-(GlcNAc)2 glycans. HEK293S GnTI− cells were maintained using EX-CELL® 293 serum-free medium (Sigma) in a humidified CO2 platform shaker incubator at 37 °C. FreeStyleTM 293-F cells were maintained in a culture media mixture containing 9 parts FreeStyleTM 293 expression media (Thermo Fisher Scientific) and 1 part EX-CELL media in a humidified CO2 platform shaker incubator at 37 °C. An expression construct encoding the IgV domain 1 of human CEACAM1 (residues 34–141, UniProt P13688) was prepared essentially as described previously (56). Briefly, the coding region was codon optimized for human cells and synthesized by GeneArt AG (Regensburg, Germany) and subcloned into the pGEn2 expression vector (CEACAM1-pGEn2). The expression product from this construct contains an N-terminal sequence that signals secretion into the medium, followed by an His8 tag, AviTag, the “superfolder” GFP, the recognition sequence of the tobacco etch virus protease, and the protein of interest. A 500-ml suspension culture of HEK293S (GnT1−) cells, which produces predominantly (Man)5-(GlcNAc)2 glycosylation was transfected with CEACAM1-pGEn2 plasmid DNA using polyethyleneimine (Polysciences, Inc., Warrington, PA) as previously described (57). Transfection of FreeStyleTM 293-F cells with the CEACAM1-pGEn2 plasmid was performed under identical conditions to result in complex-type glycosylation of the recombinant product. For metabolic labeling of the HEK293 cell cultures with [15N]alanine, cells were transfected as described above, and 16 h after transfection the medium was exchanged for FreeStyleTM 293 expression medium (Thermo Fisher Scientific) depleted in GlutaMAXTM and supplemented with 200 mg/liter of l-[15N]alanine (l-[15N]alanine 98%, Cambridge Isotope Laboratories, Andover, MA), 4 mm glutamine, and 2.2 mm valproic acid (both from Sigma).
A protein production phase of 5 days at 37 °C was followed by centrifugation of the conditioned medium to remove cells. The culture supernatant was subjected to Ni-NTA Superflow chromatography (Qiagen, Valencia, CA) followed by elution in 25 mm HEPES, 300 mm NaCl, 300 mm imidazole, pH 7.0. Fractions containing fluorescence from GFP were pooled and concentrated to 1 mg/ml using an ultrafiltration pressure cell membrane (Millipore, Billerica, MA) with a 10-kDa molecular mass cutoff. The CEACAM1-GFP construct was digested with purified recombinant tobacco etch virus protease and re-applied to the Ni2+-NTA column to allow collection of the product in the flow-through fractions. The resulting peptide sequence is identical to the E. coli expressed version except for the linker GSGG left at the N terminus from the tobacco etch virus cleavage site and the absence of the His tag at the C terminus. Neither the N nor C terminius are near suggested dimer interfaces and should not contribute to differences in dimerization tendencies.
To produce a sample with minimal glycosylation the purified protein expressed in HEK293S (GnTI−) cells was treated with endoglycosidase F1, which cleaves between the two core GlcNAc residues and truncates glycans to a single GlcNAc residue. Fig. 1B shows the result of this processing. Both of the processing enzymes were generated in house, using standard E. coli expression procedures. The CEACAM1-IgV samples were further purified by Superdex 75 chromatography (GE Healthcare Life Sciences), with the bulk of the material eluting at a point consistent with a monomer of ∼12-kDa molecular mass. Peak fractions of CEACAM1 were collected and concentrated to 1 mg/ml using an ultrafiltration pressure cell membrane. The overall yield was 2 mg/liter for [15N]alanine-labeled CEACAM1 from HEK293S (GnTI−) cells and 26 mg/liter of [15N]alanine-labeled CEACAM1 from FreeStyleTM 293-F cells. Mass spectrometry analysis showed the product to be 81% 15N enriched at alanine sites. Additional glycan analysis was carried out in the CCRC Analytical Facility. MS/MS analysis of the tryptic peptide containing all three glycosylation sites showed a single GlcNAc residue at sites 70, 77, and 81 with percentage occupancy of 62.4, 65.1, and 63.2%, respectively. Assuming independence of sites 95% of proteins have glycosylation at least one site.
NMR Experiments
Three-dimensional NMR experiments including HNCO, HN(CA)CO, HNCA, HNCACB, and CBCA(CO)NH were used for sequence-specific backbone resonance assignment of the uniformly labeled E. coli expressed product. Data from these experiments were collected on a 200 μm sample in 10 mm Tris buffer at 30 °C, pH 8.0, containing 10% D2O using a 600 MHz Varian/Agilent spectrometer equipped with a 5-mm triple resonance cryo-probe. NMR data were processed by NMRPipe (58) and analyzed by SPARKY software (52). 67% of all backbone resonances and 48% of Cβ resonances were assigned in this manner. For the [15N]alanine-labeled glycosylated protein, assignments were made by overlap of cross-peaks with alanine cross-peaks in uniformly labeled material. In all cases shifts between the two preparations were small enough to allow definitive assignment. Assignments have been deposited in the BMRB with accession number 26803.
All RDC experiments were carried out at 25 °C on a 900 MHz Varian/Agilent spectrometer equipped with a cryo-probe. The 15N-H RDCs were measured from TROSY-based J-modulation experiments by varying a modulation delay from 0.5 to 14 ms (53). The protein was aligned in 4% alkyl polyethylene glycol detergent (PEG-C12E5, Sigma) or 13 mg/ml of bacteriophage Pf1 (ASLA Biotech). These two media, one with high surface charge and one with a neutral surface usually provide different orientations of alignment frames, and complement one another in determining symmetry axes for symmetric dimers (38). The deuterium splitting for the protein in these two media were 13 and 15.8 Hz, respectively, indicating a significant level of ordering. The order tensor solutions and the principle alignment frame were determined using the program REDCAT (42).
The backbone rotational correlation times were extracted for each of the assigned 15N backbone sites using shared constant-time cross-correlated relaxation experiments (SCT-CCR) (37). These experiments allow a direct measure of rotational correlation times for N-H bond vectors, relatively independent of effects from remote protons and chemical exchange. For rigid parts of the backbone they reflect correlation times for overall protein rotation. The SCT-CCR experiments were carried out using a Varian/Agilent 800 MHz spectrometer equipped with a triple resonance cryogenic probe at 25 °C.
Author Contributions
Y. Z. carried out the majority of the experiments and drafted the manuscript. J.-Y. Y. prepared proteins samples. K. M. helped design the experiments and edited the manuscript. J. H. P. helped design the experiments and edited the manuscript.
Acknowledgments
We thank Qi Gao and the CCRC MS analytical center for the analysis of mass spectrometry data and Annapoorani Ramiah for generation of site-directed mutants.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM033225 and P41GM103390. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
NMR assignments have been deposited in the BMRB with accession number 26803.
- CEACAM1
- carcinoembryonic antigen-related cell adhesion molecule 1
- PDB
- Protein Data Bank
- RDC
- residual dipolar coupling
- HSQC
- heteronuclear single quantum coherence
- Ni2+-NTA
- nickel-nitrilotriacetic acid
- SCT-CCR
- shared constant-time cross-correlated relaxation experiments.
References
- 1. Hammarström S. (1999) The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 9, 67–81 [DOI] [PubMed] [Google Scholar]
- 2. Najjar S. M. (2002) Regulation of insulin action by CEACAM1. Trends Endocrinol. Metab. 13, 240–245 [DOI] [PubMed] [Google Scholar]
- 3. Beauchemin N., and Arabzadeh A. (2013) Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 32, 643–671 [DOI] [PubMed] [Google Scholar]
- 4. Neumaier M., Paululat S., Chan A., Matthaes P., and Wagener C. (1993) Biliary glycoprotein, a potential human cell adhesion molecule, is down-regulated in colorectal carcinomas. Proc. Natl. Acad. Sci. U.S.A. 90, 10744–10748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hsieh J. T., Luo W., Song W., Wang Y., Kleinerman D. I., Van N. T., and Lin S. H. (1995) Tumor suppressive role of an androgen-regulated epithelial cell adhesion molecule (C-CAM) in prostate carcinoma cell revealed by sense and antisense approaches. Cancer Res. 55, 190–197 [PubMed] [Google Scholar]
- 6. Sappino A. P., Buser R., Seguin Q., Fernet M., Lesne L., Gumy-Pause F., Reith W., Favaudon V., and Mandriota S. J. (2012) The CEACAM1 tumor suppressor is an ATM and p53-regulated gene required for the induction of cellular senescence by DNA damage. Oncogenesis 1, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gray-Owen S. D., and Blumberg R. S. (2006) CEACAM1: contact-dependent control of immunity. Nat. Rev. Immunol. 6, 433–446 [DOI] [PubMed] [Google Scholar]
- 8. Leung N., Turbide C., Olson M., Marcus V., Jothy S., and Beauchemin N. (2006) Deletion of the carcinoembryonic antigen-related cell adhesion molecule 1 (Ceacam1) gene contributes to colon tumor progression in a murine model of carcinogenesis. Oncogene 25, 5527–5536 [DOI] [PubMed] [Google Scholar]
- 9. Volpert O., Luo W., Liu T. J., Estrera V. T., Logothetis C., and Lin S. H. (2002) Inhibition of prostate tumor angiogenesis by the tumor suppressor CEACAM1. J. Biol. Chem. 277, 35696–35702 [DOI] [PubMed] [Google Scholar]
- 10. Neumaier C., Nittka S., and Neumaier M. (2012) Loss of expression of the tumor suppressor CEACAM1 links different hereditary colorectal carcinoma subtypes to the genesis of sporadic colorectal carcinoma. Onkologie 35, 563–568 [DOI] [PubMed] [Google Scholar]
- 11. Iijima H., Neurath M. F., Nagaishi T., Glickman J. N., Nieuwenhuis E. E., Nakajima A., Chen D., Fuss I. J., Utku N., Lewicki D. N., Becker C., Gallagher T. M., Holmes K. V., and Blumberg R. S. (2004) Specific regulation of T helper cell 1-mediated murine colitis by CEACAM1. J. Exp. Med. 199, 471–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Markel G., Lieberman N., Katz G., Arnon T. I., Lotem M., Drize O., Blumberg R. S., Bar-Haim E., Mader R., Eisenbach L., and Mandelboim O. (2002) CD66a interactions between human melanoma and NK cells: a novel class I MHC-independent inhibitory mechanism of cytotoxicity. J. Immunol. 168, 2803–2810 [DOI] [PubMed] [Google Scholar]
- 13. Pantelic M., Kim Y. J., Bolland S., Chen I., Shively J., and Chen T. (2005) Neisseria gonorrhoeae kills carcinoembryonic antigen-related cellular adhesion molecule 1 (CD66a)-expressing human B cells and inhibits antibody production. Infect. Immun. 73, 4171–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen Z., Chen L., Baker K., Olszak T., Zeissig S., Huang Y. H., Kuo T. T., Mandelboim O., Beauchemin N., Lanier L. L., and Blumberg R. S. (2011) CEACAM1 dampens antitumor immunity by down-regulating NKG2D ligand expression on tumor cells. J. Exp. Med. 208, 2633–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Simeone D. M., Ji B., Banerjee M., Arumugam T., Li D., Anderson M. A., Bamberger A. M., Greenson J., Brand R. E., Ramachandran V., and Logsdon C. D. (2007) CEACAM1, a novel serum biomarker for pancreatic cancer. Pancreas 34, 436–443 [DOI] [PubMed] [Google Scholar]
- 16. Markel G., Ortenberg R., Seidman R., Sapoznik S., Koren-Morag N., Besser M. J., Bar J., Shapira R., Kubi A., Nardini G., Tessone A., Treves A. J., Winkler E., Orenstein A., and Schachter J. (2010) Systemic dysregulation of CEACAM1 in melanoma patients. Cancer Immunol. Immunother. 59, 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Obrink B. (2008) On the role of CEACAM1 in cancer. Lung Cancer 60, 309–312 [DOI] [PubMed] [Google Scholar]
- 18. Tilki D., Singer B. B., Shariat S. F., Behrend A., Fernando M., Irmak S., Buchner A., Hooper A. T., Stief C. G., Reich O., and Ergün S. (2010) CEACAM1: a novel urinary marker for bladder cancer detection. Eur. Urol. 57, 648–654 [DOI] [PubMed] [Google Scholar]
- 19. Kang W. Y., Chen W. T., Wu M. T., and Chai C. Y. (2007) The expression of CD66a and possible roles in colorectal adenoma and adenocarcinoma. Int. J. Colorectal Dis. 22, 869–874 [DOI] [PubMed] [Google Scholar]
- 20. Tchoupa A. K., Schuhmacher T., and Hauck C. R. (2014) Signaling by epithelial members of the CEACAM family: mucosal docking sites for pathogenic bacteria. Cell Commun. Signal. 12, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Virji M., Evans D., Griffith J., Hill D., Serino L., Hadfield A., and Watt S. M. (2000) Carcinoembryonic antigens are targeted by diverse strains of typable and non-typable Haemophilus influenzae. Mol. Microbiol. 36, 784–795 [DOI] [PubMed] [Google Scholar]
- 22. Virji M. (2000) The structural basis of CEACAM-receptor targeting by neisserial opa proteins: response. Trends Microbiol. 8, 260–261 [DOI] [PubMed] [Google Scholar]
- 23. Virji M., Watt S. M., Barker S., Makepeace K., and Doyonnas R. (1996) The N-domain of the human CD66a adhesion molecule is a target for Opa proteins of Neisseria meningitidis and Neisseria gonorrhoeae. Mol. Microbiol. 22, 929–939 [DOI] [PubMed] [Google Scholar]
- 24. Huang Y. H., Zhu C., Kondo Y., Anderson A. C., Gandhi A., Russell A., Dougan S. K., Petersen B. S., Melum E., Pertel T., Clayton K. L., Raab M., Chen Q., Beauchemin N., Yazaki P. J., et al. (2015) CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 517, 386–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fedarovich A., Tomberg J., Nicholas R. A., and Davies C. (2006) Structure of the N-terminal domain of human CEACAM1: binding target of the opacity proteins during invasion of Neisseria meningitidis and N. gonorrhoeae. Acta Crystallogr. D Biol. Crystallogr. 62, 971–979 [DOI] [PubMed] [Google Scholar]
- 26. Heldin C. H. (1995) Dimerization of cell surface receptors in signal transduction. Cell 80, 213–223 [DOI] [PubMed] [Google Scholar]
- 27. Zhou H., Fuks A., Alcaraz G., Bolling T. J., and Stanners C. P. (1993) Homophilic adhesion between Ig superfamily carcinoembryonic antigen molecules involves double reciprocal bonds. J. Cell Biol. 122, 951–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patel P. C., Lee H. S., Ming A. Y., Rath A., Deber C. M., Yip C. M., Rocheleau J. V., and Gray-Owen S. D. (2013) Inside-out signaling promotes dynamic changes in the carcinoembryonic antigen-related cellular adhesion molecule 1 (CEACAM1) oligomeric state to control its cell adhesion properties. J. Biol. Chem. 288, 29654–29669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lawson E. L., Mills D. R., Brilliant K. E., and Hixson D. C. (2012) The transmembrane domain of CEACAM1–4S is a determinant of anchorage independent growth and tumorigenicity. PLoS ONE 7, e29606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klaile E., Vorontsova O., Sigmundsson K., Müller M. M., Singer B. B., Ofverstedt L. G., Svensson S., Skoglund U., and Obrink B. (2009) The CEACAM1 N-terminal Ig domain mediates cis- and trans-binding and is essential for allosteric rearrangements of CEACAM1 microclusters. J. Cell Biol. 187, 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Markel G., Gruda R., Achdout H., Katz G., Nechama M., Blumberg R. S., Kammerer R., Zimmermann W., and Mandelboim O. (2004) The critical role of residues 43R and 44Q of carcinoembryonic antigen cell adhesion molecules-1 in the protection from killing by human NK cells. J. Immunol. 173, 3732–3739 [DOI] [PubMed] [Google Scholar]
- 32. Watt S. M., Teixeira A. M., Zhou G. Q., Doyonnas R., Zhang Y., Grunert F., Blumberg R. S., Kuroki M., Skubitz K. M., and Bates P. A. (2001) Homophilic adhesion of human CEACAM1 involves N-terminal domain interactions: structural analysis of the binding site. Blood 98, 1469–1479 [DOI] [PubMed] [Google Scholar]
- 33. Taheri M., Saragovi U., Fuks A., Makkerh J., Mort J., and Stanners C. P. (2000) Self recognition in the Ig superfamily. Identification of precise subdomains in carcinoembryonic antigen required for intercellular adhesion. J. Biol. Chem. 275, 26935–26943 [DOI] [PubMed] [Google Scholar]
- 34. Pavlopoulou A., and Scorilas A. (2014) A comprehensive phylogenetic and structural analysis of the carcinoembryonic antigen (CEA) gene family. Genome Biol. Evol. 6, 1314–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tugarinov V., Hwang P. M., and Kay L. E. (2004) Nuclear magnetic resonance spectroscopy of high-molecular-weight proteins. Annu. Rev. Biochem. 73, 107–146 [DOI] [PubMed] [Google Scholar]
- 36. Subedi G. P., Johnson R. W., Moniz H. A., Moremen K. W., and Barb A. (2015) High yield expression of recombinant human proteins with the transient transfection of HEK293 cells in suspension. J. Vis. Exp. e53568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu Y., and Prestegard J. H. (2008) Direct measurement of dipole-dipole/CSA cross-correlated relaxation by a constant-time experiment. J. Magn. Reson. 193, 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang X., Bansal S., Jiang M., and Prestegard J. H. (2008) RDC-assisted modeling of symmetric protein homo-oligomers. Protein Sci. 17, 899–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee H. W., Wylie G., Bansal S., Wang X., Barb A. W., Macnaughtan M. A., Ertekin A., Montelione G. T., and Prestegard J. H. (2010) Three-dimensional structure of the weakly associated protein homodimer SeR13 using RDCs and paramagnetic surface mapping. Protein Sci. 19, 1673–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reeves P. J., Callewaert N., Contreras R., and Khorana H. G. (2002) Structure and function in rhodopsin: high-level expression of rhodopin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Natl. Acad. Sci. U.S.A. 99, 13419–13424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schmidt C., Irausquin S. J., and Valafar H. (2013) Advances in the REDCAT software package. BMC Bioinformatics 14, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Valafar H., and Prestegard J. H. (2004) REDCAT: a residual dipolar coupling analysis tool. J. Magn. Reson. 167, 228–241 [DOI] [PubMed] [Google Scholar]
- 43. Bax A. (2003) Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Science 12, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bax A., and Grishaev A. (2005) Weak alignment NMR: a hawk-eyed view of biomolecular structure. Curr. Opin. Struct. Biol. 15, 563–570 [DOI] [PubMed] [Google Scholar]
- 45. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 46. Li A. J., and Nussinov R. (1998) A set of van der Waals and coulombic radii of protein atoms for molecular and solvent-accessible surface calculation, packing evaluation, and docking. Proteins 32, 111–127 [PubMed] [Google Scholar]
- 47. Stern N., Markel G., Arnon T. I., Gruda R., Wong H., Gray-Owen S. D., and Mandelboim O. (2005) Carcinoembryonic antigen (CEA) inhibits NK killing via interaction with CEA-related cell adhesion molecule 1. J. Immunol. 174, 6692–6701 [DOI] [PubMed] [Google Scholar]
- 48. Korotkova N., Yang Y., Le Trong I., Cota E., Demeler B., Marchant J., Thomas W. E., Stenkamp R. E., Moseley S. L., and Matthews S. (2008) Binding of Dr adhesins of Escherichia coli to carcinoembryonic antigen triggers receptor dissociation. Mol. Microbiol. 67, 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bonsor D. A., Günther S., Beadenkopf R., Beckett D., and Sundberg E. J. (2015) Diverse oligomeric states of CEACAM IgV domains. Proc. Natl. Acad. Sci. U.S.A. 112, 13561–13566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bucior I., Scheuring S., Engel A., and Burger M. M. (2004) Carbohydrate-carbohydrate interaction provides adhesion force and specificity for cellular recognition. J. Cell Biol. 165, 529–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barb A. W., Brady E. K., and Prestegard J. H. (2009) Branch-specific sialylation of IgG-Fc glycans by ST6Gal-I. Biochemistry 48, 9705–9707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Goddard T. D., and Kneller D. G. (2008) SPARKY 3. University of California, San Francisco, CA [Google Scholar]
- 53. Liu Y., and Prestegard J. H. (2009) Measurement of one and two bond N-C couplings in large proteins by TROSY-based J-modulation experiments. J. Magn. Reson. 200, 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Müller M. M., Klaile E., Vorontsova O., Singer B. B., and Obrink B. (2009) Homophilic adhesion and CEACAM1-S regulate dimerization of CEACAM1-L and recruitment of SHP-2 and c-Src. J. Cell Biol. 187, 569–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Esteban-Martín S., Fenwick R. B., Ådén J., Cossins B., Bertoncini C. W., Guallar V., Wolf-Watz M., and Salvatella X. (2014) Correlated inter-domain motions in adenylate kinase. PLoS Comput. Biol. 10, e1003721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Barb A. W., Meng L., Gao Z., Johnson R. W., Moremen K. W., and Prestegard J. H. (2012) NMR characterization of immunoglobulin G Fc glycan motion on enzymatic sialylation. Biochemistry 51, 4618–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Meng L., Forouhar F., Thieker D., Gao Z., Ramiah A., Moniz H., Xiang Y., Seetharaman J., Milaninia S., Su M., Bridger R., Veillon L., Azadi P., Kornhaber G., Wells L., et al. (2013) Enzymatic basis for N-glycan sialylation: structure of rat α2,6-sialyltransferase (ST6GAL1) reveals conserved and unique features for glycan sialylation. J. Biol. Chem. 288, 34680–34698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 59. Huang Y.-H., Zhu C., Kondo Y., Anderson A. C., Gandhi A., Russell A., Dougan S. K., Petersen B.-S., Melum E., Pertel T., Clayton K. L., Raab M., Chen Q., Beauchemin N., Yazaki P. J., et al. (2016) Corrigendum: CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 10.1038/nature17421 [DOI] [PMC free article] [PubMed] [Google Scholar]