

Abstract
The forward reaction of nicotinamide nucleotide transhydrogenase (NNT) reduces NADP+ at the expense of NADH oxidation and H+ movement down the electrochemical potential across the inner mitochondrial membrane, establishing an NADPH/NADP+ ratio severalfold higher than the NADH/NAD+ ratio in the matrix. In turn, NADPH drives processes, such as peroxide detoxification and reductive biosynthesis. In this study, we generated a congenic mouse model carrying a mutated NntC57BL/6J allele from the C57BL/6J substrain. Suspensions of isolated mitochondria from Nnt+/+, Nnt+/−, and Nnt−/− mouse liver were biochemically evaluated and challenged with exogenous peroxide under different respiratory states. The respiratory substrates were also varied, and the participation of concurrent NADPH sources (i.e. isocitrate dehydrogenase-2, malic enzymes, and glutamate dehydrogenase) was assessed. The principal findings include the following: Nnt+/− and Nnt−/− exhibit ∼50% and absent NNT activity, respectively, but the activities of concurrent NADPH sources are unchanged. The lack of NNT activity in Nnt−/− mice impairs peroxide metabolism in intact mitochondria. The contribution of NNT to peroxide metabolism is decreased during ADP phosphorylation compared with the non-phosphorylating state; however, it is accompanied by increased contributions of concurrent NADPH sources, especially glutamate dehydrogenase. NNT makes a major contribution to peroxide metabolism during the blockage of mitochondrial electron transport. Interestingly, peroxide metabolism in the Nnt+/− mitochondria matched that in the Nnt+/+ mitochondria. Overall, this study demonstrates that the respiratory state and/or substrates that sustain energy metabolism markedly influence the relative contribution of NNT (i.e. varies between nearly 0 and 100%) to NADPH-dependent mitochondrial peroxide metabolism.
Keywords: hydrogen peroxide, mitochondria, oxidation-reduction (redox), peroxidase, respiratory chain, tricarboxylic acid cycle (TCA cycle) (Krebs cycle), c57bl/6j, NADPH, transhydrogenase
Introduction
The coenzymes nicotinamide adenine dinucleotide (NAD) and NAD phosphate (NADP) are soluble electron carriers that are reduced in oxidative reactions during the catabolism of energy substrates in the cytosol and mitochondria. Despite their structural similarity, reduced NAD and NADP are used to drive rather different processes in the cells (1). Given their roles as specific electron donors in diverse metabolic pathways, it is interesting that the redox states of NAD and NADP are linked to each other in the mitochondria of many organisms because the enzyme nicotinamide nucleotide transhydrogenase (NNT)4 catalyzes the transfer of redox potential between these two coenzymes, reducing one at the expense of the oxidation of the other (2).
The academic and clinical interest in the understanding of the role of NNT in redox metabolism (3) has grown substantially because mutations in the gene (Nnt) encoding this protein are present in the most widely used experimental mouse substrain (i.e. C57BL/6J) (4) and in some humans (5). Homozygous mutations of Nnt that are linked to familiar glucocorticoid insufficiency have been documented in humans (5), whereas the C57BL/6J mouse substrains have been shown to exhibit metabolic abnormalities (6–8) with major implications regarding their use as experimental models in basic research (9–12). The phenotype of the Nnt mutation in heterozygotes is not clear in humans (13) or mice.
NADP+ reduction at the expense of NADH oxidation in the mitochondrial matrix is the well known primary role of NNT. Although mitochondria possess three other enzymatic oxidative reactions linked to NADP+ reduction (i.e. the enzymes isocitrate dehydrogenase (IDH2), malic enzymes (MEs), and glutamate dehydrogenase (GDH)), as highlighted in Fig. 1), NNT exhibits the following unique features. (i) The enzyme is assembled in the inner mitochondrial membrane (IMM) and is energy (i.e. mitochondrial protonmotive force, Δp)-dependent because it translocates H+ across the IMM (14, 15). (ii) It oxidizes NADH as an electron donor substrate and thus can couple the supply of NADPH to the substrate flux through the Krebs cycle NAD-dependent dehydrogenases. (iii) Because of this last property, NNT is a very high-capacity source of NADPH because it unites the reductive potentials of a variety of mitochondrial substrates (16, 17). The Δp maintained across the IMM by the activity of the respiratory chain thermodynamically drives the forward NNT reaction to establish an NADPH/NADP+ ratio that is at least 500-fold higher than the NADH/NAD+ ratio under the conditions present in respiring mitochondria (2). Canonically, the NADP redox potential drives the enzymatic degradation of organic and hydrogen peroxide via peroxidase and reductase systems involving glutathione or thioredoxin as substrates, and thus, NADPH can be considered an ultimate antioxidant against peroxides (1, 18). Additionally, the redox state of NADP in the mitochondrial matrix maintained by NNT can drive the otherwise unfavorable reductive carboxylation of α-ketoglutarate to isocitrate (i.e. the reverse of the IDH2 reaction) (3), which is an important pathway for some proliferative cells that rely on glutaminolysis.
FIGURE 1.
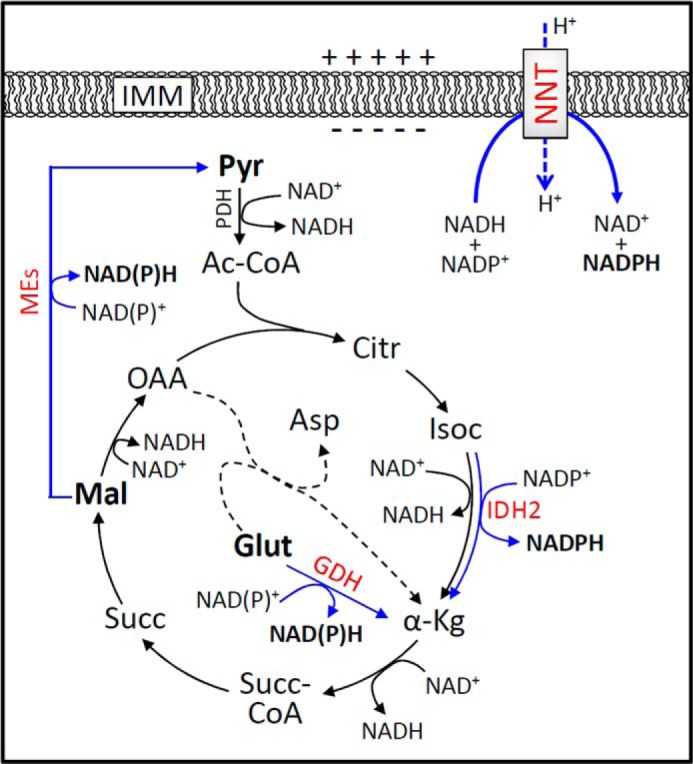
Schematic showing the sources of NADPH in the mitochondrial matrix. This schematic depicts the simplified intermediary oxidative catabolism of energy substrates via the mitochondrial Krebs cycle. NADP+ is reduced into NADPH during the oxidation of the substrates isocitrate (Isoc), malate (Mal), and glutamate (Glut) via IDH2, MEs, and GDH, respectively. In addition, NADPH is also a product of the forward reaction of NNT, an enzyme located in the IMM, whose activity is dependent on the NAD redox state and the electrochemical potential across the IMM. Enzymes that regenerate NADPH are shown in red, and their oxidative reactions are indicated by blue arrows. Two isoforms of ME occur in mitochondria and can reduce NADP+; one is specific for the NADP coenzyme only. The GDH reaction is coupled to either NAD+ or NADP+ reduction. Pyruvate (Pyr), malate, and glutamate, shown in bold, are exogenous substrates used to support mitochondrial energy metabolism during most of the experiments in this study. Dashed lines represent the aspartate transamination reaction that consumes glutamate to form and feed α-ketoglutarate (α-Kg) into the Krebs cycle, which is canonically a principal entry pathway for glutamate carbon skeleton. Other abbreviations used are as follows: Ac-CoA, acetyl-coenzyme A; Citr, citrate; Succ-CoA, succinyl-coenzyme A; Succ, succinate; OAA, oxaloacetate; Asp, aspartate. For simplicity, fluxes through other Krebs cycle enzymes, such as citrate synthase and aconitase, can be equally important to NADPH supply via a putative pathway but are not depicted.
In addition to supporting the forward reaction of NNT, NADH is mostly used as an electron donor to the mitochondrial electron transport chain at the level of respiratory complex I, which drives H+ ejection from the mitochondrial matrix and buildup of Δp. Because the energetic state of mitochondria, as represented by the NAD redox state and Δp, thermodynamically determines NNT activity, it is an experimental necessity that the function of NNT be evaluated with intact mitochondria respiring on substrates that support NAD+ reduction and respiratory chain buildup of Δp. Spectrophotofluorometric protocols have been long employed to evaluate changes in the redox states of NAD and NADP in intact and isolated mitochondria incubated under suitable conditions (16, 19–21). Nonetheless, discriminating between the NADPH supply via NNT and other concurrent NADPH sources in intact mitochondria (i.e. the relative contribution of NNT to the total NADPH supply) has been experimentally challenging but could be overcome via the molecular ablation of NNT expression/function (3, 16, 22, 23). However, few studies involving mammalian cells or isolated intact mitochondria have contributed to understanding NNT function in the context of integrated mitochondrial energy metabolism (17, 24).
Helping to advance the knowledge about NNT, a spontaneous loss-of-function Nnt mutation that arose in C57BL/6J mice (4) provides a potential genetic model to study many aspects of this enzyme in physiology and disease (25). Indeed, we recently described redox and permeability transition dysfunctions in liver mitochondria from Nnt-mutated C57BL/6J mice (which are homozygous for an allele named NntC57BL/6J) (16). This study provided novel findings that shed light on the theoretical scientific drawbacks of using widely spread Nnt-mutated mice substrains in biomedical research (9, 11, 12). In this study, we investigated the function of NNT by developing a congenic mouse model (derived from C57BL/6 substrains) bearing the three Nnt genotypes (i.e. Nnt+/+, Nnt+/−, and Nnt−/−) with the ultimate goal of evaluating the relative contribution of NNT to the NADPH supply during peroxide detoxification in isolated liver mitochondria under different respiratory states.
Results
The mating between C57BL/6/JUnib and C57BL/6J mice, which are, respectively, homozygous wild type and mutated for Nnt alleles (16), followed by successive backcrossing of the heterozygous mutated Nnt mice with C57BL/6/JUnib provided a congenic experimental model to study the relative contribution of NNT to mitochondrial peroxide metabolism at the expense of NADPH. It is noteworthy that this model allowed the study of liver mitochondria from mice that expressed mutated Nnt in heterozygosis, a genotype whose functional consequences are unknown but are probably relevant to the parents of patients with familiar glucocorticoid insufficiency linked to homozygous Nnt mutations (5, 13). The congenic mouse model nearly eliminates potential genetic modifiers between mouse substrains (26); thus, phenotypic differences between Nnt genotypes can be directly attributed to this gene (27). Congenic models improve the quality of quantitative and/or in vivo research with mutants (27).
The genotyping of the founders or the generated wild type (Nnt+/+), heterozygous mutant (Nnt+/−), and homozygous mutant (Nnt−/−) mice was performed as shown in Fig. 2. The generated congenic mice were designated as C57Unib.B6 and will hereafter be referred to only by their Nnt genotype.
FIGURE 2.

Nnt genotyping via DNA agarose gel following PCR amplification. Images representing the genotyping of the parental C57BL/6 substrains and congenic mice are shown on the left and right, respectively. DNA obtained from the tip of the mouse tail was subjected to a three-primer PCR amplification of fragments of wild type and mutated (i.e. the C57BL/6J mutated allele) Nnt alleles. In this assay, PCR products of 579 and of 743 bp were expected for the wild-type and mutant alleles, respectively. With heterozygous alleles, a third faint band of ∼1000 bp was also produced (as shown in the 6th and 7th lanes, right gel) and helped to differentiate the genotypes (6). The 1st lanes were loaded with DNA markers.
The maximal activities of the enzymes that catalyze the following four known reactions that regenerate NADPH in the mitochondrial matrix are shown in Fig. 3: NNT, IDH2, NADP-MEs, and GDH. The NNT activities in liver mitochondria from Nnt+/− and Nnt−/− mice were ∼50% and none, respectively, compared with wild-type mice (Fig. 3, A and B). Nonetheless, the activities of the remaining enzymes that serve as concurrent mitochondrial sources of NADPH (i.e. IDH2, NADP-MEs, and GDH) were similar across the three genotypes (Fig. 3B). Using the maximum activity of citrate synthase as a marker (28), the mitochondrial fractions isolated from the liver of Nnt+/+, Nnt+/−, and Nnt−/− mice were found to exhibit similar enrichments in the content of this organelle (Fig. 3C).
FIGURE 3.
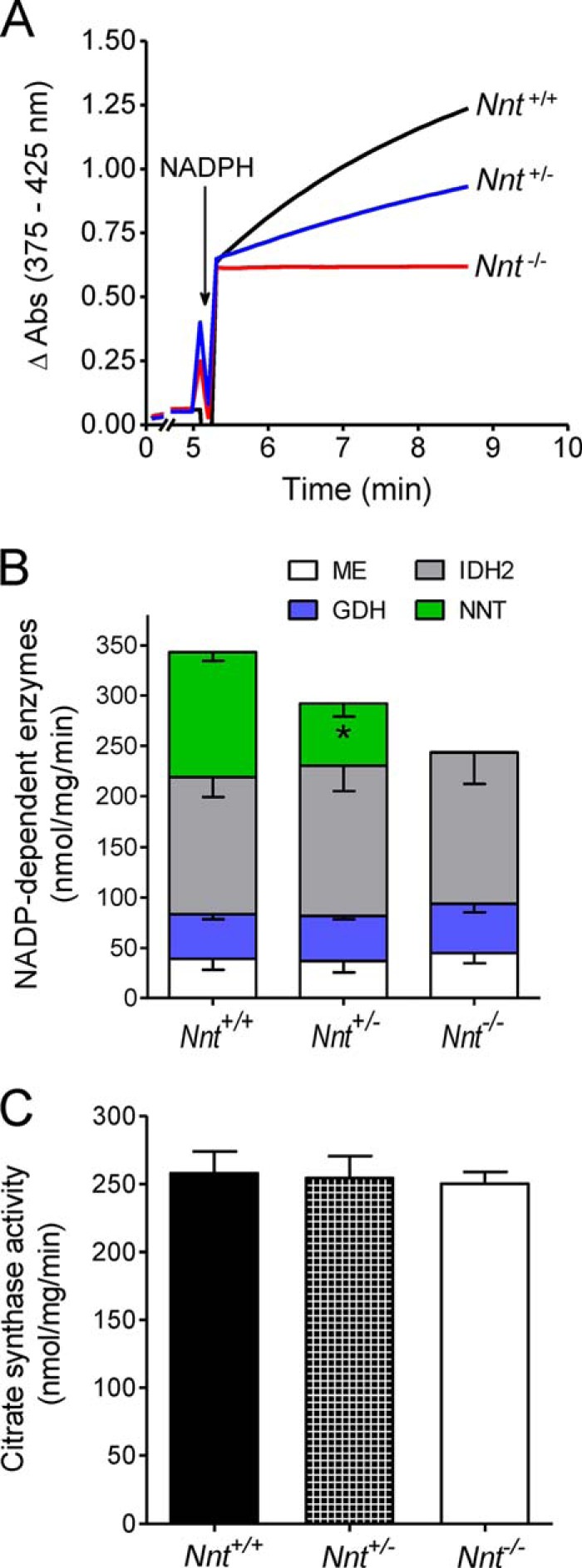
Activities of four NADP-dependent mitochondrial enzymes. The activities were determined spectrofluorometrically in suspensions of detergent-disrupted mitochondria from Nnt+/+, Nnt+/−, and Nnt−/− mice. A depicts representative traces of the activity of NNT. The stacked bars in B show the mean (−S.D.) of the activities of NADP-MEs (n = 5), GDH (n = 5), IDH2 (n = 8), and NNT (n = 5). *, significantly different (p ≤ 0.05) from the other two genotypes (one-way ANOVA and Fisher's LSD post hoc test). C depicts citrate synthase activity assessed as an index of mitochondrial enrichment in the isolated liver mitochondria of the three genotypes.
Below, we describe the results of a series of experiments in which NAD(P)H autofluorescence in suspended intact mitochondria respiring on exogenous substrates was continuously monitored and used to assess NADPH regeneration (i.e. NADP+ reduction) after challenging with an exogenous organic peroxide (t-BOOH). Using this method, we recently showed that the forward and reverse activities of NNT are completely absent in intact respiring liver mitochondria from Nnt-mutant C57BL/6J mice (16).
t-BOOH is metabolized via glutathione peroxidase/reductase and peroxiredoxin/thioredoxin reductase, with both systems operating at the expense of NADPH oxidation (29). Importantly, t-BOOH is not metabolized by catalase that could be present in isolated mitochondrial fractions from the liver (30). The time to recover the baseline fluorescence of NAD(P) following the addition of t-BOOH to mitochondrial suspensions varies depending on the t-BOOH concentration, as depicted in Fig. 4, A and B. Based on pilot studies and the data shown in Fig. 4, a bolus addition of 30 μm t-BOOH (a load of 30 nmol/mg) was used in most experiments. Using this concentration of t-BOOH, wild-type liver mitochondria generally recover the fully reduced state of NAD(P)H in ∼2.5 min and are capable of withstanding repeated t-BOOH challenges when respiring on malate/pyruvate as substrates. The time to metabolize the t-BOOH load (Fig. 4D) was used to calculate the rate of t-BOOH metabolism, as shown in Fig. 4E and in most of the other bar graphs in this paper. This rate of t-BOOH metabolism reflects NAD(P) re-reduction, and it can be accelerated in the presence of exogenous isocitrate (Fig. 4C), once it promotes direct NAD(P)+ reduction by IDH2. Although t-BOOH metabolism is slower in mitochondria from Nnt−/− mice compared with those from Nnt+/+ mice when energized by malate/pyruvate, mitochondria from both genotypes metabolize t-BOOH at similar high rates if isocitrate is also present in the medium (Fig. 4). The lack of difference between genotypes and the lower NAD(P)H fluorescence decay in Nnt−/− mitochondria is an indication that the rate of t-BOOH metabolism may not be limited by NAD(P) redox state when exogenous isocitrate is supporting a high NADPH supply via IDH2. In this condition, the peroxidase/reductase systems involved in t-BOOH metabolism could become a limiting factor. The larger drop in fluorescence in Nnt+/+ mitochondria after t-BOOH addition in Fig. 4C, which can be interpreted as a lower NAD(P) redox state, is likely due to NADH oxidation via the forward activity of NNT and does not necessarily mean a more oxidized state of NADP. Impaired t-BOOH metabolism in mice liver mitochondria in the absence of NNT activity was expected based on our previous study (16).
FIGURE 4.

Assessment of the NADPH-supported t-BOOH metabolism rate via the autofluorescence of NAD(P) in suspensions of isolated mitochondria. At the beginning (∼20 s) of the representative traces shown in A–C, mitochondria were added (1 mg/ml) to a standard reaction medium supplemented with 2.5 mm malate plus 5 mm pyruvate, followed by different bolus additions of t-BOOH (15, 30, or 45 μm). The fluorescence was continuously monitored. A and B, respectively, depict representative traces from the Nnt+/+ and Nnt−/− groups; C shows representative traces of Nnt+/+ and Nnt−/− mitochondria metabolizing t-BOOH in the presence of 1 mm isocitrate, which was added to fully support NADP+ reduction by IDH2. As illustrated by the dashed lines in A and B, the time elapsed between the t-BOOH addition and the recovery of the fluorescence to the baseline level (intersections of dashed lines) was taken as the time required to metabolize t-BOOH. D shows the time to metabolize 30 μm t-BOOH, which was used to calculate the specific rates shown in E (n = 4 for each group). Bars not sharing the same letter in E are different (p < 0.05) from each other. Bolus doses of 30 μm t-BOOH (i.e. a specific load of 30 nmol/mg) or less were used in subsequent experiments.
In the basal respiratory state shown in Fig. 5, the combination of exogenous substrates commonly used to sustain mitochondrial intermediate metabolism and respiration was varied to evaluate the dependence of the NADPH supply on the energy substrates fed to the mitochondria, which is arguably an issue in mitochondria without NNT activity. The non-phosphorylating and ADP-stimulated respiratory rates were also measured in the presence of these different combinations of substrates (Table 1). Nnt+/+ and Nnt+/− liver mitochondria equally metabolize t-BOOH irrespective of the added substrates, but mitochondria from Nnt−/− were only able to metabolize t-BOOH when energized by malate/pyruvate or malate/pyruvate/palmitoyl carnitine (Fig. 5, A and B); however, the rate was much slower compared with those of the other two genotypes, which exhibited 50% or normal NNT activity (Fig. 5F). With malate/glutamate or malate/pyruvate/glutamate/succinate as energy substrates, Nnt−/− mitochondria never recovered a reduced NADP state after t-BOOH addition. Although the autofluorescence of NADH and NADPH is indistinguishable, discrimination between NADH and NADPH oxidation was achieved by the sequential addition of 3-hydroxybutyrate and isocitrate to the system because the former only reduces NAD+. The results indicate that half of the normal NNT activity is sufficient to sustain its maximal relative contribution to mitochondrial t-BOOH metabolism. Malate/glutamate and malate/pyruvate/glutamate/succinate did not support a non-NNT supply of NADPH (Fig. 5, C–E) but did sustain the highest rates of ADP-stimulated mitochondrial respiration compared with malate/pyruvate and malate/pyruvate/palmitoyl carnitine (Table 1). The respiratory characteristics of liver mitochondria, which include the non-phosphorylating and ADP phosphorylation states and the ADP respiratory control ratio, did not differ among the genotypes. The ADP-stimulated respiration rates shown in Table 1 indicate that the best substrate combination for mitochondrial respiration (i.e. the highest NAD-reducing power) does not provide the best support for NADP+ reduction if the mitochondria lack NNT activity. These findings are important for the comprehensive discussion presented below regarding the non-NNT sources of NADPH in the mitochondria.
FIGURE 5.

t-BOOH metabolism supported by NADPH is dependent on the combination of substrates fed to Nnt−/− mitochondria: Nnt+/− mice mitochondria perform as Nnt+/+. The autofluorescence of reduced NAD(P) was continuously monitored in the representative experiments shown in A–E. Isolated mitochondria from the three mouse genotypes were added (1 mg/ml) at the beginning of the traces (∼20 s) to the standard reaction medium supplemented with 2.5 mm malate. Different respiratory substrates were combined with malate (Mal) in A (5 mm pyruvate (Pyr)), B (5 mm pyruvate plus 20 μm palmitoyl carnitine (PC)), C (5 mm glutamate (Glut)), and D (5 mm pyruvate plus 5 mm succinate (Succ) plus 5 mm glutamate). According to the arrows in A–E, t-BOOH (30 μm in A–D and 10 μm t-BOOH in E), 5 mm 3-hydroxybutyrate (3-OHB), and 1 mm isocitrate (Isoc) were sequentially added to the system. E shows representative traces of mitochondria from Nnt−/− in the incubation conditions of C and D but with a lower t-BOOH bolus addition (10 μm) and a longer duration to increase the limit of detection of t-BOOH metabolism rate to ∼0.6 nmol/mg/min. F shows the mean ± S.D. of the rate of t-BOOH metabolism under each experimental condition (n = 5). Bars not sharing the same letter are different (p < 0.05) from each other. # n.d. indicates that fluorescence recovery was not detected.
TABLE 1.
Oxygen consumption by isolated liver mitochondria from Nnt+/+, Nnt−/−, and Nnt+/−
Data are the mean ± S.D. (n = 5). Isolated liver mitochondria from the three Nnt genotypes (C57Unib.B6-Nnt+/+, C57Unib.B6-Nnt−/−, and C57Unib.B6-Nnt+/−) were incubated (0.5 mg/ml) in standard reaction medium containing 5 mm pyruvate and 2.5 mm malate; 5 mm pyruvate, 2.5 mm malate, and 20 μm l-palmitoylcarnitine; 2.5 mm malate and 5 mm glutamate; or 5 mm pyruvate, 2.5 mm malate, 5 mm glutamate, and 5 mm succinate. ADP (500 μm) followed by oligomycin (1 μg/ml) were added to achieve the ADP-stimulated and the non-phosphorylating respiratory states, respectively. The respiratory control is the ratio between ADP-stimulated and the non-phosphorylating respiratory rates.
| Substrates | Mice | Respiratory rate (ρmol O2/mg protein/s) |
Respiratory control ratio | |
|---|---|---|---|---|
| ADP-stimulated | Non-phosphorylating | |||
| Pyruvate/malate | Nnt+/+ | 518.3 ± 56.4 | 91.1 ± 11.0 | 5.7 ± 0.6 |
| Nnt−/− | 490.6 ± 90.9 | 93.5 ± 17.8 | 5.3 ± 0.4 | |
| Nnt+/− | 504.3 ± 53.6 | 90.6 ± 8.3 | 5.6 ± 0.5 | |
| Pyruvate/malate/palmitoyl carnitine | Nnt+/+ | 850.8 ± 124.5 | 141.1 ± 16.2 | 6.1 ± 0.7 |
| Nnt−/− | 903.0 ± 106.6 | 146.9 ± 32.1 | 6.3 ± 0.3 | |
| Nnt+/− | 915.6 ± 67.2 | 140.6 ± 19.5 | 6.6 ± 0.5 | |
| Malate/glutamate | Nnt+/+ | 1167.6 ± 149.0 | 115.3 ± 13.0 | 10.1 ± 0.8 |
| Nnt−/− | 1204.3 ± 226.4 | 123.9 ± 21.2 | 9.7 ± 0.4 | |
| Nnt+/− | 1210.0 ± 169.1 | 121.1 ± 15.7 | 10.0 ± 0.5 | |
| Pyruvate/malate/glutamate/succinate | Nnt+/+ | 1401.1 ± 200.9 | 259.5 ± 21.3 | 5.4 ± 0.4 |
| Nnt−/− | 1525.8 ± 313.3 | 269.2 ± 41.8 | 5.6 ± 0.5 | |
| Nnt+/− | 1426.2 ± 139.9 | 299.2 ± 25.0 | 5.4 ± 0.1 | |
To test whether a decreased mitochondrial energy state (i.e. partially dissipated Δp and a more oxidized NAD state) resulting from an increased respiratory rate affected NNT activity, we evaluated mitochondrial t-BOOH metabolism during non-phosphorylating and ADP phosphorylation respiratory states (Fig. 6). Malate/pyruvate were first used as energy substrates because they can sustain t-BOOH metabolism, although at a slow rate, in mitochondria devoid of NNT activity in the basal respiratory state (Fig. 5). Surprisingly, the rates of t-BOOH metabolism were similar in Nnt+/+ and Nnt−/− mitochondria during ADP phosphorylation (Fig. 6, A and C), indicating that the net flux through NNT is nearly null in this respiratory state. In contrast, stimulating oxidative phosphorylation with ADP increased t-BOOH metabolism in Nnt−/− mitochondria compared with the non-phosphorylating respiratory state. Thus, non-NNT sources of NADPH appear to be activated during ADP-stimulated respiration, and this effect of ADP is dependent on oxidative phosphorylation, i.e. higher respiration and substrate flux through Krebs cycle reactions. This conclusion can be drawn based on experiments in the presence of ADP plus the ATP synthase inhibitor oligomycin (Fig. 6C).
FIGURE 6.

Mitochondrial oxidative phosphorylation induced by ADP decreases the contribution of NNT to NADPH-supported t-BOOH metabolism. The autofluorescence of reduced NAD(P) was continuously monitored in the representative experiments shown in A and B. Isolated Nnt+/+ and Nnt−/− mitochondria were added (0.5 mg/ml) at the beginning of the traces (∼20 s) to the standard reaction medium. The substrates 2.5 mm malate plus 5 mm pyruvate or 2.5 mm malate plus 5 mm glutamate were present in the experiments shown, respectively, in A and B; in the experiments depicted in these panels, 2 mm ADP, 7.5 μm t-BOOH, and 1 μg/ml oligomycin (Oligo) were sequentially added as indicated by the arrows. C and D show the t-BOOH metabolism rates (mean ± S.D.) under each experimental condition differing with regard to the substrates and respiratory states. t-BOOH metabolism rates were determined in the presence of oligomycin with no exogenous ADP (i.e. the non-phosphorylating state), during ADP phosphorylation, and in the presence of oligomycin plus 2 mm ADP. n = 6 for each group and condition. *, different (p < 0.05) from Nnt+/+ in the same experimental condition; bars not sharing the same letter are different (p < 0.05) from each other within a given genotype. As in Fig. 5, # n.d. indicates fluorescence recovery was not detected during the non-phosphorylating state with malate plus glutamate.
As shown in Table 1, malate/glutamate sustains a higher rate of ADP-stimulated liver mitochondrial respiration than malate/pyruvate. This implies that the mitochondrial energy state and the mass action for the activity of the forward NNT reaction are also higher in the presence of malate/glutamate than malate/pyruvate. Therefore, we studied t-BOOH metabolism during ADP phosphorylation by mitochondria respiring on malate/glutamate (Fig. 6, B and D) and found that NNT still operates in the forward direction and contributes to t-BOOH metabolism, as indicated by the differences between Nnt+/+ and Nnt−/−. Curiously, Nnt−/− mitochondria are completely unable to metabolize t-BOOH when energized by malate/glutamate in the basal respiratory state (Fig. 5C) but could metabolize t-BOOH during ADP phosphorylation (Fig. 6B), although in a slower rate compared with Nnt+/+. Interestingly, experiments in the presence of ADP plus oligomycin revealed that ADP by itself, and/or AMP likely formed via adenylate kinase, can positively modulate sources of NADPH and sustain low rates of t-BOOH metabolism of ∼1 nmol/mg/min in mitochondria with no NNT activity respiring on malate/glutamate as substrates (Fig. 6D). Notably, GDH oxidizes glutamate and transfers electrons to either NAD+ or NADP+, and it is positively modulated by ADP and AMP, whereas ATP seems not regulate the activity of this enzyme (31). We have also tested the effects of 2 mm AMP or 2 mm ATP on t-BOOH metabolism rates in Nnt−/− mitochondria in the presence of oligomycin and malate/glutamate as substrates. These effects of adenine nucleotides on non-NNT sources of NADPH were studied in Nnt−/− mitochondria to rule out the contributions of NNT. In the presence of AMP, the t-BOOH metabolism rate was ∼0.6 nmol/mg/min (range from 0.4 to 0.8 nmol/mg/min, n = 7). Conversely, the effects of ATP (a creatine kinase/creatine phosphate-regenerating system was used to the maintain the adenine nucleotide pool in the form of ATP) on t-BOOH metabolism rates are less clear because it exhibited null effects in four out of six experiments (it was stimulated in two cases to produce rates of ∼0.3 and ∼0.5 nmol/mg/min). Putatively, the activity of adenylate kinase in liver mitochondria generates AMP when ADP is added. We also attempted to evaluate the effects of each of these adenine nucleotides in the presence of a classical adenylate kinase inhibitor (i.e. AP5A), but experimental controls revealed that AP5A alone marked influenced t-BOOH metabolism and biased results (data not shown).
GDH, the putative molecular target of ADP/AMP modulation, however, is not typically considered to be a relevant source of NADPH in mitochondria. A question remains regarding whether MEs were the source of or part of a pathway involving IDH2 (Fig. 1) to regenerate NADPH under the experimental conditions shown in Fig. 6B; if this is the case, then the contribution of GDH would be reciprocally lower. To improve the understanding about GDH as a source of NADPH, glutamate as the sole substrate plus malonate to inhibit succinate dehydrogenase were used to avoid the formation of malate via the Krebs cycle. Under this condition, we observed that Nnt−/− mitochondria metabolized t-BOOH fairly well in the presence of ADP (Fig. 7B), i.e. the mean rate of t-BOOH metabolism of ∼5 nmol/mg/min in Nnt−/− mitochondria. This finding provides strong evidence that GDH is an important mitochondrial source of NADPH when ADP is stimulating oxidative phosphorylation. Similarly, malate alone was tested as an NADP+ reductant during ADP-stimulated respiration (Fig. 7A) and was also found to be quite effective; the mean rate of t-BOOH metabolism was ∼2.5 nmol/mg/min in Nnt−/− mitochondria. Mitochondria possess two isoforms of ME (32) that can reduce NADP+, referred to here as NADP-MEs, and one of them exhibits dual specificity but has a greater affinity for NAD+ than NADP+ (33). The findings shown in Fig. 7A suggest that MEs form pyruvate and sustain NADPH-dependent t-BOOH metabolism, but it was not possible to ascertain whether the NAD- or NADP-ME supports also isocitrate formation and, thus, favors NADP+ reduction by IDH2 during this ADP-stimulated respiratory state.
FIGURE 7.

Independently of NNT, malate or glutamate alone can sustain NADPH-supported t-BOOH metabolism during oxidative phosphorylation: possible contributions of NADP-ME and GDH. The autofluorescence of reduced NAD(P) was continuously monitored in the representative experiments (n = 6 each group and condition) shown in A and B. Isolated Nnt+/+ and Nnt−/− mitochondria were added (0.5 mg/ml) at the beginning of the traces (∼20 s) to the standard reaction medium supplemented with 1 mm malonate (an inhibitor of succinate dehydrogenase). Malate (5 mm) was present in the experiments shown in A, whereas 5 mm glutamate was present in the experiments shown in B; in these experiments, 2 mm ADP, 7.5 μm t-BOOH, 1 μg/ml oligomycin (Oligo), 5 mm 3-hydroxybutyrate (3-OHB) and 1 mm isocitrate (Isoc) were sequentially added as indicated by arrows.
Next, we set a protocol to resemble features of mitochondrial energetics during anoxia by interrupting the electron transport chain via inhibiting respiratory complex III with antimycin A (Fig. 8). In this condition, the electrical membrane potential fully collapsed, and the rate of t-BOOH metabolism in Nnt+/+ mitochondria was nearly 3-fold higher than that in Nnt−/− mitochondria (Fig. 8), indicating that NNT operates in the forward direction and is the major source of NADPH. In addition to blocking electron transport distally at the level of complex III, we sequentially added the respiratory complex I inhibitor rotenone to block NADH oxidation by the mitochondrial respiratory chain (Fig. 8, A and B). Interestingly, mitochondria without NNT (i.e. from Nnt−/−) became completely unable to metabolize t-BOOH in the presence of rotenone, whereas the t-BOOH metabolism rate in Nnt+/+ mitochondria decreased by only 17%. To improve our understanding of this phenomenon and given the results of a recent study (34), we replaced rotenone with a high concentration of succinate (20 mm) to promote electron transfer from complex II to complex I and thus prevent NADH oxidation by the respiratory chain. Interestingly, similar to rotenone, succinate also completely abolished t-BOOH metabolism in Nnt−/− mitochondria (Fig. 8C). Mitochondrial NADH oxidation in the presence of antimycin A and the likely effect of NADH oxidation on the mitochondrial NADPH supply via concurrent NNT sources will be considered under the “Discussion.”
FIGURE 8.

NNT is the principal source of NADPH in depolarized mitochondria resulting from inhibition of electron transport and respiratory activity. As indicated in the representative traces (A–C), the addition of 0.5 mg/ml isolated mitochondria (Mito) was followed by blockage of electron transport induced by the addition of a respiratory complex III inhibitor (2 μm antimycin A, AA), whereas the autofluorescence of reduced NAD(P) and the mitochondrial membrane potential (Δψ, with the use of the dye safranine O) were monitored simultaneously. The NAD(P)H autofluorescence traces are exhibited only after the addition of antimycin A because this reagent causes optical drift. A bolus of t-BOOH was added as indicated before and after the addition of 2 μm rotenone (Rot) to block respiratory complex I (CI) (A and B); alternatively, rotenone was replaced by 20 mm succinate to block forward complex I electron transfer (C). Mitochondria from Nnt−/− were never able to recover the reduced state of NAD(P) when t-BOOH was added in the presence of antimycin A plus rotenone (B) or antimycin A plus succinate (C). Isocitrate (1 mm, Isoc) was added at the end of all traces to fully reduce NAD(P)+. The mean ± S.D. rates of t-BOOH metabolism under these two different experimental conditions are shown in D (n = 4); * indicates different (p < 0.05) from Nnt+/+ in the same condition; # indicates different (p < 0.05) from Nnt+/+ in the presence of antimycin A only; n.d., not detected.
The final questions considered in this study were related to a possible stimulation of mitochondrial respiration caused by H+ re-entry into the matrix (i.e. the utilization of Δp) as a result of the increased activity of the forward NNT reaction during peroxide metabolism. The rate of oxygen consumption by the mitochondria was monitored as t-BOOH was added during phosphorylating and non-phosphorylating respiratory states (Fig. 9). When respiring on malate/pyruvate substrates, both Nnt+/+ and Nnt−/− mitochondria exhibited a similar transient decrease in ADP-stimulated respiration upon t-BOOH addition, but t-BOOH addition did not change the non-phosphorylating respiration rate, regardless of genotype (Fig. 9, A and B). To briefly mention an issue that will be discussed below, complex I-independent respiration was also assessed (Fig. 9, C and D). With succinate as the substrate plus rotenone to inhibit complex I, t-BOOH addition did not elicit a detectable change in phosphorylation or the non-phosphorylating respiratory rate in mitochondria from mice of either genotype (Fig. 9, C and D). Although we used a high ADP load (i.e. 8 μmol/mg of mitochondrial protein) to stably stimulate oxidative phosphorylation, a constant spontaneous decline in oxygen consumption with time occurred during ADP phosphorylation when the mitochondria were energized by succinate plus rotenone compared with when malate/pyruvate was used.
FIGURE 9.

Independently of NNT function, t-BOOH inhibits ADP-stimulated respiration supported by NADH, whereas the activity of the forward NNT reaction does not significantly affect oxygen consumption. Isolated mitochondria from Nnt+/+ (A and C) and Nnt−/− (B and D) mice were suspended (0.25 mg/ml) in the standard reaction medium; pyruvate (5 mm) plus malate (2.5 mm) or succinate (5 mm) plus rotenone (1 μm) were present as respiratory substrates in A and B and in C and D, respectively. In these representative experiments, mitochondrial O2 consumption was measured, whereas 2 mm ADP, 7.5 μm t-BOOH, and 1 μg/ml oligomycin (Oligo) were sequentially added as indicated by the arrows.
Discussion
Various experimental approaches have established the notion that NNT is a physiologically relevant source of NADPH in mitochondria (3, 22, 35). It has often been stated in the literature that NNT has approximately a 50% contribution to the total NADPH flux in the mitochondrial matrix and that the other 50% includes contributions from IDH2 and NADP-MEs (2, 16, 22). However, the 50% contribution of NNT seems to stem from a study with bacteria (Escherichia coli) (2, 36), an organism that also contains the pentose phosphate pathway in the same cellular compartment as another major source of NADPH and lacks mitochondria (36). Correctly, a review by Rydstrom (2) pointed out that these estimates were from a study in prokaryotes. No experimental data from a higher organism with mitochondria were available to support a similar estimation. The uncertainty regarding the relative importance of NNT to NADP+ reduction in mammalian mitochondria is further complicated by the dependence of NNT activity on the mitochondrial energy state (i.e. Δp and the NAD redox state). Moreover, concurrent NADPH sources (i.e. the enzymes IDH2, NADP-MEs, and GDH) are presumably dependent on the energy substrates that fuel mitochondrial intermediary metabolism (i.e. the Krebs cycle) via different entry reactions.
In this study, we varied both the exogenous substrates supporting mitochondrial metabolism and the respiratory state in intact isolated mouse liver mitochondria to ascertain the relative contribution of NNT to NADPH-supported peroxide metabolism. The relative contributions of NNT under each condition studied were inferred by comparing Nnt+/+ and Nnt−/− mitochondria (the principal results are summarized in Fig. 10).
FIGURE 10.
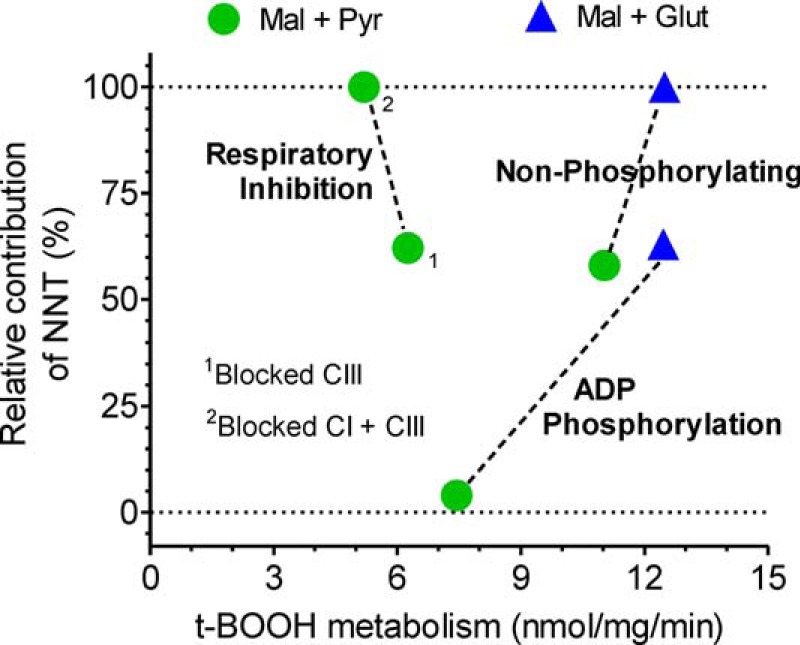
Graphical summary of the rate of t-BOOH metabolism and its relative dependence on NNT as the NADPH source under different experimental conditions. This figure summarizes the effects of the three mitochondrial respiratory states (non-phosphorylating, respiration stimulated by ADP phosphorylation, and respiratory inhibition induced by electron transfer blockage) and of two energy substrate conditions (2.5 mm malate plus 5 mm pyruvate or 2.5 mm malate plus 5 mm glutamate) on two variables shown on the abscissa and ordinate axes. Data represented on the abscissa are the mean values of the t-BOOH metabolism rate in Nnt+/+ mitochondria shown in Figs. 6 and 8, and the ordinate data are the respective relative (i.e. percentage) contributions from NNT; these values were calculated based on the mean differences between Nnt+/+ and Nnt−/− mitochondria divided by the rate in the Nnt+/+ mitochondria. 1, respiratory inhibition induced by 2 μm antimycin A blockage of the electron transfer at the level of respiratory complex III (CIII); 2, respiratory inhibition induced by antimycin A (blocked complex III) plus 2 μm rotenone-induced blockage of NADH oxidation at the level of respiratory complex I (CI). The intent of dashed lines is only to facilitate the visualization of a given respiratory state.
Novel findings regarding the NADPH supply to support peroxide metabolism in liver mitochondria were obtained, indicating that peroxide metabolism in the Nnt+/− mitochondria matched that in the Nnt+/+ mitochondria, that the respiratory state can dramatically modify the NNT contribution to NADPH regeneration, that non-NNT sources of NADPH reciprocally counterbalance the lower NADPH supply via NNT prevailing during the mitochondrial oxidative phosphorylation of ADP, and that GDH can comprise an important source of NADPH in phosphorylating mitochondria. One interesting finding was that net flux through NNT was nearly zero (i.e. did not contribute significantly to NADPH-supported peroxide metabolism) during oxidative phosphorylation when respiration was supported by malate/pyruvate substrates. As a result, the rate of peroxide metabolism was significantly decreased to 7.4 compared with 11 nmol/mg/min, as observed during the non-phosphorylating respiratory state in which the relative contribution of NNT was ∼60% (Fig. 10). Conversely, a rate of peroxide detoxification of ∼12.5 nmol/mg/min is 100% supported by NNT activity when malate/glutamate-energized mitochondria are in the non-phosphorylating respiratory state (Figs. 6D and 10). This 100% relative contribution of NNT also decreased but only to ∼63%, without impairing the peroxide metabolism rate, when the mitochondria were undergoing oxidative phosphorylation in the presence of malate/glutamate (Figs. 6D and 10). The oxidative phosphorylation of ADP leads to a partial dissipation of Δp and NADH oxidation, thereby decreasing the driving force for the forward NNT reaction. Depending on mass action, a reversal of NNT and the consequent NADPH oxidation are expected to occur and can be experimentally induced with chemical uncoupler in intact isolated liver mitochondria (16); however, a net reversal of NNT activity in liver mitochondria has not been directly demonstrated under biologically relevant conditions, including the anoxic conditions mimicked in this study (Fig. 8).
The stimulation of non-NNT NADPH sources during ADP-stimulated oxidative phosphorylation supported by malate plus pyruvate or by malate plus glutamate was further investigated in experiments using exogenous ADP plus the ATP synthase inhibitor oligomycin (Fig. 6). In parallel experiments, the effects of AMP or ATP were also evaluated under the same condition. The results indicated that ADP or AMP per se slightly activated concurrent sources of NADPH in the mitochondrial matrix only with malate plus glutamate as substrate. Therefore, most of the reciprocal compensation for NADPH-supported peroxide metabolism, when the relative contribution of NNT decreases during the oxidative phosphorylation of ADP supported by NAD-linked substrates, arises from enhanced respiratory rate and substrate flux through mitochondrial dehydrogenases. Because we also used glutamate alone as a substrate plus malonate to block the Krebs cycle at the level of succinate dehydrogenase, the data shown in Fig. 7 imply that GDH can be a direct source of NADPH during oxidative phosphorylation of ADP. It has long been known that GDH is positively modulated (allosteric) by ADP and AMP (31) and oxidizes glutamate while reducing either NAD+ or NADP+. Nonetheless, this enzyme is not typically considered to be an important NADPH source in intact respiring liver mitochondria, likely because the carbon skeleton of glutamate had been shown to enter the Krebs cycle mostly via transamination with oxaloacetate (i.e. the aspartate aminotransferase reaction) (37). Now, this view regarding Krebs cycle anaplerosis should include GDH activity linked to NADP+ reduction as a potentially important mitochondrion-feeding pathway when respiratory activity is stimulated by ADP and NADPH turnover enhanced by peroxide metabolism. Conversely, if respiratory rates and Krebs cycle flux are slow, α-ketoglutarate (generated from glutamate via aspartate aminotransferase) could accumulate and disfavor the activity of both IDH2 and GDH toward α-ketoglutarate formation and NADP+ reduction into NADPH, then explaining why non-phosphorylating mitochondria from Nnt−/− respiring on malate/glutamate metabolize t-BOOH slowly even when ADP is present to activate GDH (Fig. 6D).
According to Fig. 1, the flux through the depicted dehydrogenases IDH2, MEs, and pyruvate dehydrogenase (PDH) is directly and/or indirectly involved in NADPH regeneration in the mitochondrial matrix when malate alone or malate/pyruvate are the respiratory substrates. Higher rates of pyruvate oxidation via PDH during ADP-stimulated respiration, reminding that PDH is positively modulated by ADP (38), may also relieve a constraint (i.e. product inhibition and/or mass action) on the decarboxylation of malate to pyruvate via the NAD- or NADP-MEs. The fact that malate alone was able to sustain NADPH-dependent peroxide metabolism (Fig. 7A) indicates that the MEs can serve as both a direct source of NADPH (i.e. via flux through NADP-MEs) and part of a pathway that also includes PDH and malate dehydrogenase to replenish acetyl-CoA and oxaloacetate, respectively, thereby supporting citrate/isocitrate synthesis (via citrate synthase and aconitase) and flux through IDH2.
The reversal of NNT activity results in H+ ejection and NAD+ reduction and has been thought to occur under conditions that collapse mitochondrial energy transduction, such as the chemical uncoupling of oxidative phosphorylation (39) or anoxia in hepatocytes (40). This latter condition is of pathophysiological relevance because if NNT indeed oxidizes NADPH, it could contribute to the well known redox imbalance of tissue ischemia reperfusion. Although it has been proposed previously, the reversal of NNT activity during anoxia still needs well controlled experimental demonstrations because a lack of oxygen would have two opposing effects on NNT thermodynamics as follows: the dissipation of the mitochondrial membrane potential and the maintenance of NAD in a more highly reduced state (41). In this study, simultaneous measurements of the IMM potential and NAD(P) redox changes (Fig. 8) revealed that NNT still operates in the forward direction, and it comprises the main source of NADPH (i.e. a relative contribution of 62%) supporting peroxide metabolism when the energetic conditions of anoxic mitochondria are induced by the chemical inhibition of electron transport at the level of respiratory complex III. Surprisingly, mitochondria devoid of NNT and energized by malate/pyruvate were still able to metabolize peroxide at ∼2.1 nmol/mg/min when complex III was inhibited by antimycin A (Fig. 8B), suggesting that substrate fluxes via key mitochondrial dehydrogenases and that electron transfers paradoxically continue in this condition. The understanding of this phenomenon is aided by a recent demonstration that the metabolic signature of ischemic mitochondria was the reduction of fumarate to succinate via the reversal activity of the respiratory complex II (i.e. fumarate + reduced coenzyme Q → succinate + oxidized coenzyme Q) (34). Given that added malate is a ready source of fumarate via fumarase in isolated mitochondria, it can be speculated that this reductive reaction might be part of a short circuit in which NADH oxidation at complex I may continue, even during anoxia, because the donated electrons are transferred to oxidized coenzyme Q and then to fumarate, thus resulting in succinate formation (34). In fact, we observed that the subsequent addition of the complex I inhibitor rotenone (Fig. 8, A and B) or an excess of exogenous succinate (Fig. 8C) substantially reduced NAD when mitochondria were already poisoned with the complex III inhibitor antimycin A. Following this blockage of NADH oxidation via the rotenone inhibition of complex I or an unfavorable mass action because of a high concentration of exogenous succinate, t-BOOH metabolism only occurred in mitochondria containing functional NNT (Fig. 8A). This phenomenon may suggest that, in addition to NNT, a substrate flux through Krebs cycle dehydrogenases allows for non-NNT sources of NADPH to significantly contribute to peroxide detoxification despite the impaired oxidative metabolism prevailing in anoxic mitochondria (i.e. with distal inhibition of the respiratory chain).
Another interesting issue relates to whether the activity of the forward NNT reaction that causes H+ translocation back to the mitochondrial matrix substantially decreases mitochondrial coupling. This is important because in addition to supplying NADPH, the stimulation of respiration by decreased coupling could synergistically decrease the formation of reactive oxygen species from mitochondrial redox intermediates (18). The data shown in Figs. 6C and 10 offer insight into this issue. The peroxide metabolism supported by the NADPH supply from NNT was ∼6 nmol/mg/min (i.e. the difference between the genotypes in Fig. 6C) in mitochondria energized by malate/pyruvate and in the non-phosphorylating respiratory state. Stoichiometrically, this rate should be a 1:1 ratio in relation to the H+ influx into the mitochondrial matrix because of the activity of the forward NNT reaction (15). If complex I is the site of the electron entry, the respiratory chain would require an additional ∼0.3 nmol of O2/mg/min to maintain the Δp when NNT operates in the forward direction at 6 nmol/mg/min. Using a high resolution oxygraph and comparing Nnt+/+ and Nnt−/− mitochondria, the non-phosphorylating mitochondrial O2 consumption of ∼4 nmol of O2/mg/min was not found to increase to a detectable extent when NNT activity was elicited by t-BOOH addition (Fig. 9, A and B). During ADP-stimulated respiration, t-BOOH addition led to a transient decrease in the O2 consumption rate; it is important to remember that NNT does not contribute to peroxide metabolism under this condition with malate/pyruvate as respiratory substrates. Previous studies have shown that t-BOOH can inhibit complex I-supported but not complex II-supported mitochondrial respiration (42, 43). Because the inhibition of the mitochondrial respiratory chain at the level of complex I could counteract a potential increase in respiration as a result of NNT-mediated H+ reentry, we also studied the effects of t-BOOH on mitochondrial respiration relying on succinate as an electron donor to complex II (Fig. 9, C and D). Bypassing complex I, the H+ ejection by the respiratory chain per O2 consumed is decreased by 40%; thus, the calculated oxygen cost of NNT activity increases to 0.5 nmol of O2/mg/min. Even during succinate-supported respiration, which should favor the detection of the so-called uncoupling effect of NNT, t-BOOH addition and the resulting forward NNT reaction did not modify the measured O2-consumption rate in an NNT-dependent manner (i.e. Nnt+/+ versus Nnt−/−). Although it is predicted to occur to some extent (0.3–0.5 nmol of O2/mg/min, i.e. 5–8 pmol/mg/s), the stimulation of respiration by the forward NNT reaction was not detectable in liver mitochondria under the conditions studied, and arguably, such little uncoupling might not have an appreciable effect on the reactive oxygen species formation thermodynamically linked to the redox state of the mitochondrial electron carriers.
The technique used in this study and elsewhere allowed the continuous monitoring of changes in the autofluorescence of reduced NAD(P) as oxidants or reductant substrates were added to liver mitochondrial suspensions. Given the tissue specificity of mitochondrial characteristics (44), the results reported here may not be fully extended to mitochondria of other tissues. An organic peroxide such as t-BOOH is preferred over hydrogen peroxide, which is endogenously produced, for evaluating NADPH-supported peroxide metabolism in mitochondria because the catalase that is likely present in the isolated liver mitochondrial fraction (30) does not detoxify t-BOOH (45).
In summary, we have shown that the relative contribution of NNT to support the mitochondrial NADPH supply and peroxide detoxification is highly dynamic and can vary from nearly zero to 100% depending on the respiratory state and/or the substrates fueling mitochondrial intermediary metabolism. NNT exhibits a predominant (∼60% or higher) contribution during non-phosphorylating respiration or electron transport inhibition-induced respiratory failure. In contrast, the NNT contribution notably decreases during ADP-induced mitochondrial oxidative phosphorylation because the mass action for the forward NNT reaction is lower during this respiratory state. However, this decrease in the NNT contribution to NADPH-supported peroxide detoxification is counterbalanced by the fact that ADP stimulates substrate flux through mitochondrial dehydrogenases and favors NADPH supply by non-NNT sources in the mitochondrial matrix.
Experimental Procedures
Reagents
Malic acid, pyruvic acid sodium salt, succinic acid, glutamic acid, isocitric acid trisodium salt, palmitoyl carnitine, acetoacetic acid lithium salt, 3-hydroxybutyric acid, 3-acetylpyridine adenine dinucleotide (APAD), t-BOOH, rotenone, antimycin A, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone, NADPH, ADP, and most other chemicals were obtained from Sigma. Stock solutions of respiratory substrates and ADP, APAD, and NADPH were prepared in 20 mm HEPES with the pH adjusted to 7.2 using KOH.
Animals
C57BL/6J and C57BL/6/JUnib mice were obtained from the Campinas University Multidisciplinary Center for Biological Research in Laboratory Animals (CEMIB/UNICAMP, Campinas, Brazil). As described previously by our group (16), because of the historical origin of these two mice colonies from this provider, C57BL/6J and C57BL/6/JUnib mice are homozygous for mutated and wild-type Nnt alleles, respectively. A homozygous 17,814-bp deletion in the Nnt gene (absence of exons 7–11) arose spontaneously in the C57BL/6J mouse substrain (4, 9), and it was designated as NntC57BL/6J in the Mouse Genome Informatics Data Bank. The use of mice and the experimental protocols were approved by the local Committee for Ethics in Animal Research (CEUA-UNICAMP, approval number 3106-1). The animal procedures comply with national Brazilian guideline number 13 for “Control in Animal Experiments,” published on September 13, 2013.
Nnt Genotyping
Nnt genotyping was based on a three-primer two-allele PCR assay previously described (6). The Nnt wild-type and mutated alleles will give rise to 579- and 743-bp PCR products, respectively, detected by agarose gel electrophoresis. With heterozygous alleles, these two PCR products are present and an additional third faint band of ∼1000 bp unspecifically appears (6), likely because the two different PCR products give rise to an intermediate DNA template that is weakly amplified in the assay; this third band further helps to identify the heterozygous mouse.
Generation of Congenic Mice According to Conner (46) for Use in Experiments
Female C57BL/6J (homozygous for the Nnt mutation) mice were initially mated with male C57BL/6/JUnib mice (which are Nnt wild type) to generate male heterozygous mice (♂F1-Nnt+/−). Then, ♂F1-Nnt+/− mice were mated with female C57BL/6/JUnib mice to generate the second generation of male heterozygous mice (♂N2-Nnt+/−). This backcross was repeated for a further five generations, and then the mating between a heterozygous male (♂N7-Nnt+/−) and a female (♀N7-Nnt+/−) produced the Nnt+/+ and Nnt−/− genotypes. Their full designations are C57Unib.B6-Nnt+/+ and C57Unib.B6-Nnt−/−, but for simplicity, we refer to these N7 congenic mice by their Nnt genotype only (i.e. Nnt+/+ and Nnt−/−). Heterozygous mice (Nnt+/−) for experimental use were obtained by mating Nnt+/+ with Nnt−/− mice. The mouse colony was maintained homozygotic under identical housing conditions. Three-month-old female mice were used in the experiments shown here. The animals were kept under standard laboratory conditions (20–22 °C and 12/12-h light/dark cycle) with free access to a standard diet (NuvilabCR1, Nuvital, Colombo, PR, Brazil) and tap water in the local department's animal facility. The mice were euthanized by cervical dislocation prior to harvesting the liver.
Mitochondrial Isolation and Incubation Conditions
Intact and functional mitochondria were isolated from the livers of female adult mice by differential centrifugation, as described previously (44). Measurements of oxygen consumption, the redox state of NAD(P), and the membrane potential in suspensions of intact mitochondria were performed at 28 °C with continuous magnetic stirring in 2 ml of standard reaction medium (125 mm sucrose, 65 mm KCl, 2 mm KH2PO4, 1 mm MgCl2, 10 mm HEPES buffer, and 300 μm EGTA; pH was adjusted to 7.2 with KOH). When exogenous ATP was added for a specific purpose, a 10 mm creatine phosphate plus 5 units/ml creatine kinase system was used to prevent ADP accumulation. Supplementation with various respiratory substrates and the addition of other reagents are indicated in the figure legends. Assays with intact mitochondria were performed in either cuvettes with 1-cm sides or the oxygraph chamber described below.
NNT Activity
The medium contained 50 mm Tris (pH 8.0), 1 mg/ml palmitoyl-lysophosphatidylcholine, 0.1% Brij-35, 2 μm rotenone, 300 μm APAD, and 400 μg/ml mitochondrial protein. The reaction was started with 300 μm NADPH, and the differential absorbance (375–425 nm) was monitored (Shimadzu UV-1800, Kyoto, Japan) for the calculation of specific NNT activity as described elsewhere (16) at 37 °C.
NADP-linked GDH Activity
The medium contained 33 mm Tris, 0.33 mm EDTA, 1.33 mm MgCl2, 0.1% Triton X-100, 1 mm NADP+, 250 μm ADP, and 10 mm glutamate, and the pH was adjusted to 7.4. The development of NADPH fluorescence with time at 37 °C was followed in a microplate reader operating at excitation and emission wavelengths of 366 and 450 nm, respectively (SpectraMax M3, Molecular Devices). Reactions were performed with isolated mitochondria (150 μg/ml) in a final volume of 255 μl in a 96-well microplate (47). A blank without glutamate and known amounts of NADPH as standards were used to calculate the enzyme activity.
Mitochondrial NADP-dependent Malic Enzyme (NADP-ME) Activity
The medium (2 ml) contained 33 mm Tris, 0.33 mm EDTA, 5 mm MnCl2, 0.1% Triton X-100, 0.3 mm NADP+, and 3.3 mm malate, and the pH was adjusted to 7.4. This assay is based on the procedure of Geer et al. (48). Mitochondrial protein was present at 50 μg/ml, and a blank without malate was used. NADPH fluorescence was monitored at 37 °C (Hitachi F-4500, Tokyo, Japan), and the enzyme activity was calculated as described in our previous study (16) for IDH2.
NADP-dependent IDH (IDH2) Activity
IDH2 activity was determined as described previously (16) in a spectrofluorometer at 37 °C.
Citrate Synthase Activity
Activity was measured as described previously (49) in a microplate reader at 37 °C.
Oxygen Consumption
Mitochondrial respiration was measured in an oxygraph (OROBOROS Oxygraph-2k, Innsbruck, Austria) calibrated according to the manufacturer's instructions. After measuring the basal O2 consumption (i.e. the O2 consumption required to sustain mitochondrial Δp in the absence of exogenous ADP), the state of stimulated respiration linked to oxidative phosphorylation (also known as state 3) was elicited by the addition of saturating levels of ADP (500 μm or 2 mm); then 1 μg/ml oligomycin was added to inhibit the ADP phosphorylation by ATP synthase and to obtain the non-phosphorylating respiratory state. The final concentrations of mitochondrial proteins in the assay were either 0.25 or 0.5 mg/ml (the specific experimental conditions are described in the figure legends and in Table 1).
Redox State of Mitochondrial Nicotinamide Nucleotides (NAD(P))
Isolated mitochondria were suspended (0.5 or 1 mg/ml) in the standard reaction medium, and the changes in the redox state of NAD(P) were monitored in a spectrofluorometer (Hitachi F-7000 or Shimadzu RF-5301PC) as described in previous studies (16, 19). The mitochondrial peroxide-metabolizing system supported by NADPH was challenged with exogenous t-BOOH, an organic peroxide that is metabolized via the glutathione and thioredoxin peroxidase/reductase system at the expense of NADPH (45, 50). As a reference, known amounts of NADPH were added to the reaction medium in the absence of mitochondria.
Mitochondrial Membrane Potential (Δψ)
The mitochondrial Δψ was fluorometrically monitored with 5 μm safranine O dye added to standard reaction medium. The safranine fluorescence (excitation/emission wavelengths of 495/586 nm, respectively) was recorded simultaneously with the NAD(P) autofluorescence (366/450 nm) in a Hitachi F-7000 spectrofluorometer operating with slit widths of 5 nm. The relative safranine O fluorescence (ΔF/F) was calculated according to Figueira et al. (51), where F is the safranine fluorescence after the respiratory chain inhibitor antimycin A was added to the system to dissipate the IMM potential, and ΔF is any given fluorescence value minus F.
Statistical Analyses
The results are shown as representative traces or as the mean ± S.D. of independent experiments. The sample size is indicated in the figure legend. A one-way analysis of variance (ANOVA), a two-way repeated measures ANOVA with the Fisher's LSD as a post hoc test, or the Friedman's test followed by Dunn's post hoc test were used to assess differences between groups. p ≤ 0.05 was defined as significant.
Author Contributions
J. A. R. designed experiments, performed experiments, and analyzed results. A. F. performed experiments and analyzed results. L. A. C. P. assisted with establishment and maintenance of congenic mice. T. R. F. conceived the study, designed the experiments, performed the experiments, interpreted the data, and wrote the paper. R. F. C. conceived the study, designed the experiments, interpreted the data, and wrote the paper.
Acknowledgments
We thank Edilene S. Siqueira-Santos and Roberto C. Stahl for technical assistance.
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo Grant 2011/50400-0 (to R. F. C.), Fundação de Amparo à Pesquisa do Estado de São Paulo Scholarship Grant 2015/22063-0 (to A. F.), and by Conselho Nacional de Desenvolvimento Científico e Tecnológico Postdoctoral Fellowship Grant 403733/2014-6 (to T. R. F.). The authors declare that they have no conflicts of interest with the contents of this article.
- NNT
- nicotinamide nucleotide transhydrogenase
- GDH
- glutamate dehydrogenase
- IDH2
- isocitrate dehydrogenase 2
- IMM
- inner mitochondrial membrane
- ME
- malic enzyme
- PDH
- pyruvate dehydrogenase
- t-BOOH
- tert-butyl hydroperoxide
- ANOVA
- analysis of variance
- AP5A
- diadenosine-pentaphosphate
- APAD
- 3-acetylpyridine adenine dinucleotide.
References
- 1. Agledal L., Niere M., and Ziegler M. (2010) The phosphate makes a difference: cellular functions of NADP. Redox Rep. 15, 2–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rydström J. (2006) Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta 1757, 721–726 [DOI] [PubMed] [Google Scholar]
- 3. Gameiro P. A., Laviolette L. A., Kelleher J. K., Iliopoulos O., and Stephanopoulos G. (2013) Cofactor balance by nicotinamide nucleotide transhydrogenase (NNT) coordinates reductive carboxylation and glucose catabolism in the tricarboxylic acid (TCA) cycle. J. Biol. Chem. 288, 12967–12977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Toye A. A., Lippiat J. D., Proks P., Shimomura K., Bentley L., Hugill A., Mijat V., Goldsworthy M., Moir L., Haynes A., Quarterman J., Freeman H. C., Ashcroft F. M., and Cox R. D. (2005) A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 48, 675–686 [DOI] [PubMed] [Google Scholar]
- 5. Meimaridou E., Kowalczyk J., Guasti L., Hughes C. R., Wagner F., Frommolt P., Nürnberg P., Mann N. P., Banerjee R., Saka H. N., Chapple J. P., King P. J., Clark A. J., and Metherell L. A. (2012) Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat. Genet. 44, 740–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nicholson A., Reifsnyder P. C., Malcolm R. D., Lucas C. A., MacGregor G. R., Zhang W., and Leiter E. H. (2010) Diet-induced obesity in two C57BL/6 substrains with intact or mutant nicotinamide nucleotide transhydrogenase (Nnt) gene. Obesity 18, 1902–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Freeman H. C., Hugill A., Dear N. T., Ashcroft F. M., and Cox R. D. (2006) Deletion of nicotinamide nucleotide transhydrogenase: a new quantitative trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes 55, 2153–2156 [DOI] [PubMed] [Google Scholar]
- 8. Fergusson G., Ethier M., Guévremont M., Chrétien C., Attané C., Joly E., Fioramonti X., Prentki M., Poitout V., and Alquier T. (2014) Defective insulin secretory response to intravenous glucose in C57Bl/6J compared to C57Bl/6N mice. Mol. Metab. 3, 848–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kraev A. (2014) Parallel universes of black six biology. Biol. Direct 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Figueira T. R. (2013) A word of caution concerning the use of Nnt-mutated C57BL/6 mice substrains as experimental models to study metabolism and mitochondrial pathophysiology. Exp. Physiol. 98, 1643. [DOI] [PubMed] [Google Scholar]
- 11. Fontaine D. A., and Davis D. B. (2016) Attention to background strain is essential for metabolic research: C57BL/6 and the International Knockout Mouse Consortium. Diabetes 65, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bourdi M., Davies J. S., and Pohl L. R. (2011) Mispairing C57BL/6 substrains of genetically engineered mice and wild-type controls can lead to confounding results as it did in studies of JNK2 in acetaminophen and concanavalin A liver injury. Chem. Res. Toxicol. 24, 794–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fujisawa Y., Napoli E., Wong S., Song G., Yamaguchi R., Matsui T., Nagasaki K., Ogata T., and Giulivi C. (2015) Impact of a novel homozygous mutation in nicotinamide nucleotide transhydrogenase on mitochondrial DNA integrity in a case of familial glucocorticoid deficiency. BBA Clin. 3, 70–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leung J. H., Schurig-Briccio L. A., Yamaguchi M., Moeller A., Speir J. A., Gennis R. B., and Stout C. D. (2015) Structural biology. Division of labor in transhydrogenase by alternating proton translocation and hydride transfer. Science 347, 178–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bizouarn T., Sazanov L. A., Aubourg S., and Jackson J. B. (1996) Estimation of the H+/H− ratio of the reaction catalysed by the nicotinamide nucleotide transhydrogenase in chromatophores from over-expressing strains of Rhodospirillum rubrum and in liposomes inlaid with the purified bovine enzyme. Biochim. Biophys. Acta 1273, 4–12 [DOI] [PubMed] [Google Scholar]
- 16. Ronchi J. A., Figueira T. R., Ravagnani F. G., Oliveira H. C., Vercesi A. E., and Castilho R. F. (2013) A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic. Biol. Med. 63, 446–456 [DOI] [PubMed] [Google Scholar]
- 17. Lopert P., and Patel M. (2014) Nicotinamide nucleotide transhydrogenase (Nnt) links the substrate requirement in brain mitochondria for hydrogen peroxide removal to the thioredoxin/peroxiredoxin (Trx/Prx) system. J. Biol. Chem. 289, 15611–15620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Figueira T. R., Barros M. H., Camargo A. A., Castilho R. F., Ferreira J. C., Kowaltowski A. J., Sluse F. E., Souza-Pinto N. C., and Vercesi A. E. (2013) Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid. Redox Signal. 18, 2029–2074 [DOI] [PubMed] [Google Scholar]
- 19. Vercesi A. E. (1987) The participation of NADP, the transmembrane potential and the energy-linked NAD(P) transhydrogenase in the process of Ca2+ efflux from rat liver mitochondria. Arch. Biochem. Biophys. 252, 171–178 [DOI] [PubMed] [Google Scholar]
- 20. Lehninger A. L., Vercesi A., and Bababunmi E. A. (1978) Regulation of Ca2+ release from mitochondria by the oxidation-reduction state of pyridine nucleotides. Proc. Natl. Acad. Sci. U.S.A. 75, 1690–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Estabrook R. W. (1962) Fluorometric measurement of reduced pyridine nucleotide in cellular and subcellular particles. Anal. Biochem. 4, 231–245 [DOI] [PubMed] [Google Scholar]
- 22. Yin F., Sancheti H., and Cadenas E. (2012) Silencing of nicotinamide nucleotide transhydrogenase impairs cellular redox homeostasis and energy metabolism in PC12 cells. Biochim. Biophys. Acta 1817, 401–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ripoll V. M., Meadows N. A., Bangert M., Lee A. W., Kadioglu A., and Cox R. D. (2012) Nicotinamide nucleotide transhydrogenase (NNT) acts as a novel modulator of macrophage inflammatory responses. FASEB J. 26, 3550–3562 [DOI] [PubMed] [Google Scholar]
- 24. Vogel R., Wiesinger H., Hamprecht B., and Dringen R. (1999) The regeneration of reduced glutathione in rat forebrain mitochondria identifies metabolic pathways providing the NADPH required. Neurosci. Lett. 275, 97–100 [DOI] [PubMed] [Google Scholar]
- 25. Davisson M. T. (2005) Discovery genetics: serendipity in basic research. ILAR J. 46, 338–345 [DOI] [PubMed] [Google Scholar]
- 26. Conner D. A. (2002) Mouse colony management. Curr. Protoc. Mol. Biol. Chapter 23, Unit 23.28 [DOI] [PubMed] [Google Scholar]
- 27. Crusio W. E., Goldowitz D., Holmes A., and Wolfer D. (2009) Standards for the publication of mouse mutant studies. Genes Brain. Behav. 8, 1–4 [DOI] [PubMed] [Google Scholar]
- 28. Larsen S., Nielsen J., Hansen C. N., Nielsen L. B., Wibrand F., Stride N., Schroder H. D., Boushel R., Helge J. W., Dela F., and Hey-Mogensen M. (2012) Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 590, 3349–3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Flohé L., Toppo S., Cozza G., and Ursini F. (2011) A comparison of thiol peroxidase mechanisms. Antioxid. Redox Signal. 15, 763–780 [DOI] [PubMed] [Google Scholar]
- 30. Reinhart P. H., Taylor W. M., and Bygrave F. L. (1982) A procedure for the rapid preparation of mitochondria from rat liver. Biochem. J. 204, 731–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Godinot C., and Gautheron D. (1971) Regulation of pig heart mitochondrial glutamate dehydrogenase by nucleotides and phosphate: Comparison with pig heart and beef liver purified enzymes. FEBS Lett. 13, 235–240 [DOI] [PubMed] [Google Scholar]
- 32. Hsieh J. Y., Chen M. C., and Hung H. C. (2011) Determinants of nucleotide-binding selectivity of malic enzyme. PLoS ONE 6, e25312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin R. C., and Davis E. J. (1974) Malic enzymes of rabbit heart mitochondria. Separation and comparison of some characteristics of a nicotinamide adenine dinucleotide-preferring and a nicotinamide adenine dinucleotide phosphate-specific enzyme. J. Biol. Chem. 249, 3867–3875 [PubMed] [Google Scholar]
- 34. Chouchani E. T., Pell V. R., Gaude E., Aksentijević D., Sundier S. Y., Robb E. L., Logan A., Nadtochiy S. M., Ord E. N., Smith A. C., Eyassu F., Shirley R., Hu C. H., Dare A. J., James A. M., et al. (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arkblad E. L., Tuck S., Pestov N. B., Dmitriev R. I., Kostina M. B., Stenvall J., Tranberg M., and Rydström J. (2005) A Caenorhabditis elegans mutant lacking functional nicotinamide nucleotide transhydrogenase displays increased sensitivity to oxidative stress. Free Radic. Biol. Med. 38, 1518–1525 [DOI] [PubMed] [Google Scholar]
- 36. Sauer U., Canonaco F., Heri S., Perrenoud A., and Fischer E. (2004) The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli. J. Biol. Chem. 279, 6613–6619 [DOI] [PubMed] [Google Scholar]
- 37. Lenartowicz E., and Wojtczak A. B. (1988) Significance of the alanine aminotransferase reaction in the formation of α-ketoglutarate in rat liver mitochondria. Arch. Biochem. Biophys. 260, 309–319 [DOI] [PubMed] [Google Scholar]
- 38. Quinlan C. L., Goncalves R. L., Hey-Mogensen M., Yadava N., Bunik V. I., and Brand M. D. (2014) The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 289, 8312–8325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nicholls D. G., and Garland P. B. (1969) The control of isocitrate oxidation by rat liver mitochondria. Biochem. J. 114, 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoek J. B., and Rydström J. (1988) Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem. J. 254, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chandel N. S., Budinger G. R., Choe S. H., and Schumacker P. T. (1997) Cellular respiration during hypoxia. Role of cytochrome oxidase as the oxygen sensor in hepatocytes. J. Biol. Chem. 272, 18808–18816 [DOI] [PubMed] [Google Scholar]
- 42. Drahota Z., Kriváková P., Cervinková Z., Kmonícková E., Lotková H., Kucera O., and Houstek J. (2005) tert-Butyl hydroperoxide selectively inhibits mitochondrial respiratory-chain enzymes in isolated rat hepatocytes. Physiol. Res. 54, 67–72 [DOI] [PubMed] [Google Scholar]
- 43. Cervinková Z., Rauchová H., Kriváková P., and Drahota Z. (2008) Inhibition of palmityl carnitine oxidation in rat liver mitochondria by tert-butyl hydroperoxide. Physiol. Res. 57, 133–136 [DOI] [PubMed] [Google Scholar]
- 44. Chweih H., Castilho R. F., and Figueira T. R. (2015) Tissue and sex specificities in Ca2+ handling by isolated mitochondria in conditions avoiding the permeability transition. Exp. Physiol. 100, 1073–1092 [DOI] [PubMed] [Google Scholar]
- 45. Liu H., and Kehrer J. P. (1996) The reduction of glutathione disulfide produced by t-butyl hydroperoxide in respiring mitochondria. Free Radic. Biol. Med. 20, 433–442 [DOI] [PubMed] [Google Scholar]
- 46. Conner D. A. (2002) Mouse colony management. Curr. Protoc. Mol. Biol. Chapter 23, Unit 23.28 [DOI] [PubMed] [Google Scholar]
- 47. Zhang X., Vincent A. S., Halliwell B., and Wong K. P. (2004) A mechanism of sulfite neurotoxicity: direct inhibition of glutamate dehydrogenase. J. Biol. Chem. 279, 43035–43045 [DOI] [PubMed] [Google Scholar]
- 48. Geer B. W., Krochko D., Oliver M. J., Walker V. K., and Williamson J. H. (1980) A comparative-study of the NADP-malic enzymes from Drosophila and chick liver. Comp. Biochem. Physiol. B 65, 25–34 [Google Scholar]
- 49. Figueira T. R., Castilho R. F., Saito A., Oliveira H. C., and Vercesi A. E. (2011) The higher susceptibility of congenital analbuminemic rats to Ca2+-induced mitochondrial permeability transition is associated with the increased expression of cyclophilin D and nitrosothiol depletion. Mol. Genet. Metab. 104, 521–528 [DOI] [PubMed] [Google Scholar]
- 50. Chen Y., Cai J., and Jones D. P. (2006) Mitochondrial thioredoxin in regulation of oxidant-induced cell death. FEBS Lett. 580, 6596–6602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Figueira T. R., Melo D. R., Vercesi A. E., and Castilho R. F. (2012) Safranine as a fluorescent probe for the evaluation of mitochondrial membrane potential in isolated organelles and permeabilized cells. Methods Mol. Biol. 810, 103–117 [DOI] [PubMed] [Google Scholar]