

Abstract
Recent studies have shown that myocardial ischemia/reperfusion (I/R)-induced necrosis can be controlled by multiple genes. In this study, we observed that both strands (5p and 3p) of miR-223 were remarkably dysregulated in mouse hearts upon I/R. Precursor miR-223 (pre-miR-223) transgenic mouse hearts exhibited better recovery of contractile performance over reperfusion period and lesser degree of myocardial necrosis than wild type hearts upon ex vivo and in vivo myocardial ischemia. Conversely, pre-miR-223 knock-out (KO) mouse hearts displayed opposite effects. Furthermore, we found that the RIP1/RIP3/MLKL necroptotic pathway and inflammatory response were suppressed in transgenic hearts, whereas they were activated in pre-miR-223 KO hearts upon I/R compared with wild type controls. Accordingly, treatment of pre-miR-223 KO mice with necrostatin-1s, a potent necroptosis inhibitor, significantly decreased I/R-triggered cardiac necroptosis, infarction size, and dysfunction. Mechanistically, we identified two critical cell death receptors, TNFR1 and DR6, as direct targets of miR-223-5p, whereas miR-223-3p directly suppressed the expression of NLRP3 and IκB kinase α, two important mediators known to be involved in I/R-induced inflammation and cell necroptosis. Our findings indicate that miR-223-5p/-3p duplex works together and cooperatively inhibits I/R-induced cardiac necroptosis at multiple layers. Thus, pre-miR-223 may constitute a new therapeutic agent for the treatment of ischemic heart disease.
Keywords: inflammasome, ischemia, microRNA (miRNA), myocardial infarction, necrosis (necrotic death)
Introduction
Reperfusion of ischemic hearts by percutaneous coronary intervention, cardiac surgery, or thrombolytic therapy is commonly used in patients with myocardial infarction (1). However, it is well recognized that reperfusion could induce excessive oxidative stress and inflammation, leading to myocyte death (apoptosis and necrosis) (2). Over the past decades, tremendous efforts have been made to dissect mechanisms underlying the gene-regulated apoptosis in ischemia/reperfusion (I/R)13-induced cardiac injury, whereas gene-controlled necrosis has received less attention. In recent years, it has become clear that I/R-triggered cardiac necrosis can be regulated by multiple genes/mediators, such as cyclophilin D (CypD), NLRP3, receptor-interacting protein kinase 3 (RIP3) and its partners RIP1, mixed lineage kinase-like (MLKL), and Ca2+-calmodulin-dependent protein kinase (CaMKII) (3–7). Currently, the best characterized form of regulated necrosis (also referred to as necroptosis) is mediated by tumor necrosis factor-α (TNF-α)/TNFR1-induced protein complex RIP1-RIP3 (necrosome) (8). Importantly, several recent studies have demonstrated that inhibition of necroptosis by necrostatin-1, a small molecule capable of suppressing RIP1 kinase activity, alleviated reperfusion injury following acute myocardial infarction in mice, rats, and pigs (9–12). Nevertheless, given that I/R activates multiple necrosis-associated signals (5, 7, 13), making several necrotic pathways highly intertwined, blocking only one road to cell death may be unlikely to yield the desired outcome in a clinical setting. Therefore, it would be very significant to explore novel strategies aiming to target multiple cell death pathways in I/R hearts.
MicroRNAs (miRs; miRNAs), a highly conserved group of small non-protein-coding RNAs, possess the capacity to fine-tune expression of hundreds of genes (14). Our previous work and that of others have revealed that miRNAs are dysregulated in I/R hearts and actively involved in the modulation of cardiac cell death during I/R (14–19). Nonetheless, how such miRNAs regulate cell necrosis in I/R hearts remains poorly understood. Along this line, miR-223 has garnered special attention (20). Previously, only the 3′-arm of precursor miR-223 (pre-miR-223) (designated as miR-223 or the guide strand) was considered to maturate and become functional, whereas the complementary 5′-arm (referred as miR-223* or passenger strand) was destined to be degraded (20). However, we recently discovered that both arms of pre-miR-223 were co-expressed differently in septic mouse hearts and cooperatively modulated inflammatory response (21). (Note: to avoid confusion, miRNA/miRNA* nomenclature is now replaced by miRNA-3p and -5p, respectively) (22). Intriguingly, we and others have reported that the expression of miR-223-3p was up-regulated in mouse I/R hearts (14). Whether miR-223-5p is also dysregulated in mouse hearts upon I/R remains unclear. In addition, most targets of miR-223-3p validated so far are related to proinflammation (i.e. NLRP3, IKKα, interleukin (IL)-1β, IL-6, Roquin, and Pknox1) (20). Furthermore, miR-223-5p is predicted to directly interact with 3-untranslated regions (UTRs) of multiple death signal receptors (i.e. TNFR1, DR6, and TNFsf4) (mIRBase and miRDB). Hence, given that those targets of miR-223-5p/-3p are associated with cell necrosis/necroptosis, it would be significant to examine whether altered levels of miR-223-5p/-3p duplex impact the I/R-induced cardiac remodeling.
In this study, we utilized pre-miR-223 transgenic (TG) and knock-out (KO) mouse models that underwent I/R ex vivo and in vivo. Our results showed that pre-miR-223 TG hearts were resistant, whereas pre-miR-223-null hearts were sensitive, to I/R-triggered injury. Moreover, we observed that the RIP1/RIP3/MLKL necroptotic pathway and the NLRP3 inflammasome were suppressed in pre-miR-223 TG hearts, whereas they were activated in pre-miR-223 KO hearts upon I/R in vivo and ex vivo. Accordingly, inhibition of the necroptotic signaling in pre-miR-223 KO mice largely rescued I/R-triggered cardiac necrosis, infarction size, and dysfunction. Mechanistically, we identified that miR-223-5p/-3p worked together to inhibit multiple necrotic pathways and thereby protected hearts against I/R-induced damage.
Results
Alterations of MiR-223-5p and -3p Levels in Murine Hearts upon I/R in Vivo and ex Vivo
Previous work has shown that miR-223 is up-regulated in murine I/R hearts (14). However, the miR-223 examined in these studies was processed from the 3′-arm of pre-miR-223, which was named as the guide strand. Whether the 5′-arm of pre-miR-223 as a passenger strand is matured and dysregulated in mouse hearts upon I/R remains unknown. In this regard, we measured both arms of pre-miR-223 by a specific stem-loop real time polymerase chain reaction (PCR) in mouse hearts upon in vivo and ex vivo I/R (Fig. 1, A–D). We observed that, compared with a sham group, levels of miR-223-3p were significantly increased in mouse hearts during in vivo I/R and peaked in 4-h reperfused hearts (Fig. 1A). Importantly, mature miR-223-5p was detectable in mouse hearts rather than being degraded. Of interest, the miR-223-5p levels were remarkably increased in 30-min artery-occluded hearts and 1- and 4-h reperfused hearts but were greatly reduced by 33% in mouse hearts after 24-h reperfusion (Fig. 1B). In ex vivo 45-min ischemic hearts, miR-223-3p levels were elevated by 2-fold and further increased by 2.4-fold after 2-h reperfusion compared with sham samples (Fig. 1C). By contrast, the 5′-arm of pre-miR-223 was up-regulated by 2.2-fold in mouse hearts after 45-min ischemia, but such an elevation was attenuated at 2 h post-I/R ex vivo (Fig. 1D). These observations indicate that alterations of miR-223-5p/-3p are dynamic and dependent on the pathological conditions. Our results suggest that both arms of pre-miR-223 are processed to maturation, and both may play roles in I/R-induced cardiac injury.
FIGURE 1.

Expression levels of miR-223-3p and -5p were dynamically regulated in mouse ischemic/reperfused hearts. A and B, alteration of miR-223-3p and -5p levels in heart subjected in vivo to 30-min ischemia followed by reperfusion. C and D, both strands of pre-miR-223 were up-regulated in ex vivo hearts subjected to 45-min ischemia followed by 2-h reperfusion (error bars represent mean ± S.D.; n = 6 for in vivo sham and ischemic groups; n = 4 for in vivo 1-h, 4-h and n = 5 for 24-h reperfusion groups; n = 4 for ex vivo 45-min ischemia and 2-h reperfusion groups; *, p < 0.01 versus sham controls).
Overexpression of Pre-miR-223 Improves the Postischemic Recovery of Cardiac Function
Recently, we reported that a miR-223 TG mouse model in which a precursor miR-223 was driven by a cardiac-specific promoter, α-myosin heavy chain promoter (Fig. 2A), displayed physiological cardiac hypertrophy with no fibrosis (23). Notably, such miR-223 TG mouse model showed similar litter size and life expectancy as wild-type mice. In the present work, we focused on examining whether miR-223 plays any roles in I/R-induced cardiac injury and its underlying mechanisms. Hence, we first subjected miR-223 TG mice to 45 min of ex vivo global no-flow ischemia followed by 1 h of reperfusion using the Langendorff retrograde heart perfusion system. Stem-loop RT-PCR analysis confirmed that both miR-223-5p and -3p were markedly overexpressed in TG hearts compared with WTs (Fig. 2, B–D). Upon I/R, TG hearts exhibited significantly better functional recovery than wild-type (WT) hearts (Fig. 2, E–H) as measured by the rates of contraction (Fig. 2E), relaxation (Fig. 2F), and left ventricular developed pressure (LVDP) (Fig. 2G). In addition, the left ventricular end diastolic pressure (LVEDP) was significantly depressed in miR-223 hearts during reperfusion compared with WT controls (Fig. 2H). Together, these data reveal that elevation of miR-223-5p/-3p duplex is associated with improved cardiac function recovery during ex vivo ischemia/reperfusion.
FIGURE 2.

Overexpression of pre-miR-223 significantly improved cardiac function recovery during ex vivo I/R. A, diagram of pre-miR-223 TG vector. B–D, overexpression of miR-223-3p/-5p was determined in TG hearts by quantitative RT-PCR and 3% agarose gel electrophoresis. U6 was used as an internal control (*, p < 0.001; n = 3 for each group). E–H, pre-miR-223 TG hearts showed better functional recovery than WT hearts after 45-min global ischemia followed by 1-h reperfusion (n = 8 for WT; n = 7 for pre-miR-223 TG group; *, p < 0.005 versus WT). Error bars represent mean ± S.D. α-MHCp, α-myosin heavy chain promoter. hGHpA, human growth hormone poly A.
Overexpression of MiR-223-5p/-3p Duplex Dampens I/R-induced Cardiac Necroptosis but Not Apoptosis
It is well recognized that maintaining adequate numbers of living cardiomyocytes is critical to the overall preservation of cardiac function during I/R (24). Given the markedly improved recovery of myocardial function in miR-223 TG hearts, we then determined the effects of miR-223 on postischemic cellular damage. The extent of necrotic and apoptotic cell death was examined in mouse hearts upon ex vivo ischemia for 45 min followed by 1 h of reperfusion. Using fluorescent staining with propidium iodide (PI), a red dye that binds to necrotic cell nuclei (25), we observed that the number of PI-positive nuclei was reduced in pre-miR-223 TG hearts by ∼47% compared with WTs upon ex vivo I/R (Fig. 3, A and B). Consistently, during ex vivo I/R, cardiac release of lactate dehydrogenase (LDH), a biochemical marker for necrotic cell death, was significantly suppressed by ∼45% in TG hearts compared with WT samples (Fig. 3C). More intriguingly, ex vivo I/R-induced cardiac apoptosis, measured by TUNEL staining and Caspase-3 activity, did not differ between TGs and WT controls (Fig. 3, D–F). These results suggest that overexpression of miR-223-5p/-3p duplex suppresses I/R-initiated cellular necrosis but not apoptosis in the myocardium.
FIGURE 3.

Overexpression of miR-223-3p/-5p duplex attenuated I/R-induced myocardial necrosis and infarction size. A, PI was perfused into the mouse heart to label necrotic cells at the end of ex vivo I/R (45 min/1 h). Red, PI-positive nuclei; blue, 4′,6-diamidino-2-phenylindole-stained nuclei. Scale bars, 100 μm. B, quantitative analysis of myocardial necrotic cells in ex vivo I/R hearts (n = 8 for WT; n = 7 for TG group; *, p < 0.05 versus WTs). C, total LDH was measured in coronary effluent collected during the first 10 min of reperfusion (n = 8 for WT; n = 7 for TG group; *, p < 0.05 versus WTs). D and E, representative images (D) and quantitative results (E) of the TUNEL staining assay in WT and TG hearts after ex vivo I/R (45 min/1 h). Green, TUNEL-positive nuclei; red, α-actin; blue, DAPI. Scale bars, 100 μm. F, Caspase-3 activity was similar between TG hearts and WT hearts subjected to ex vivo I/R. p > 0.05 versus WTs (n = 6 hearts per group; p < 0.05 versus WTs). G–I, overexpression of miR-223-3p/-5p duplex greatly reduced in vivo myocardial infarct size after 30 min of left anterior descending artery occlusion followed by 24 h of reperfusion. The mouse hearts were stained by Evans Blue and triphenyltetrazolium chloride after in vivo I/R. White/gray, infarct size; red, area at risk; and blue, non-ischemic LV (n = 8 for WT; n = 10 for TG group; *, p < 0.01 versus WTs). Error bars represent mean ± S.D.
To further determine the consequence of increased miR-223-5p/-3p expression under in vivo conditions, we subjected WT and TG hearts to 30-min in vivo myocardial ischemia via coronary artery occlusion followed by 24-h reperfusion. We observed that the ratio of infarct-to-risk region was 18 ± 2.3% in TG hearts (n = 10) compared with 43 ± 3.7% in WT hearts (n = 8; p < 0.001) (Fig. 3, G and H), whereas the region at risk was not significantly different between the two groups (Fig. 3I). These results are consistent with our ex vivo findings that miR-223 duplex elicits cardioprotective effects on I/R-induced injury.
Knock-out of Pre-miR-223 Aggravates I/R-triggered Cardiac Dysfunction and Necrosis
Next, we examined the effects of pre-miR-223 deficiency on cardiac function recovery during ex vivo I/R using a global pre-miR-223 KO mouse model. Results of stem-loop RT-PCR analysis confirmed that both mature miR-223-5p and -3p were not expressed (Fig. 4A). Such pre-miR-223-null hearts were mounted onto the Langendorff apparatus and stabilized for 10 min with perfusion buffer followed by 30-min no-flow ischemia and 1-h reperfusion. During reperfusion, KO hearts (n = 8) exhibited significantly depressed functional recovery compared with WTs (n = 9) as evidenced by the decreased rates of contraction (+dP/dt) and relaxation (−dP/dt) in miR-223 KO hearts relative to WT hearts (Fig. 4, B and C). Similarly, the LVDP was recovered to 55 ± 4.7% of preischemic values after 1-h reperfusion in miR-223 KO hearts, whereas it was 67 ± 3.2% in WT hearts (p < 0.05) (Fig. 4D).
FIGURE 4.
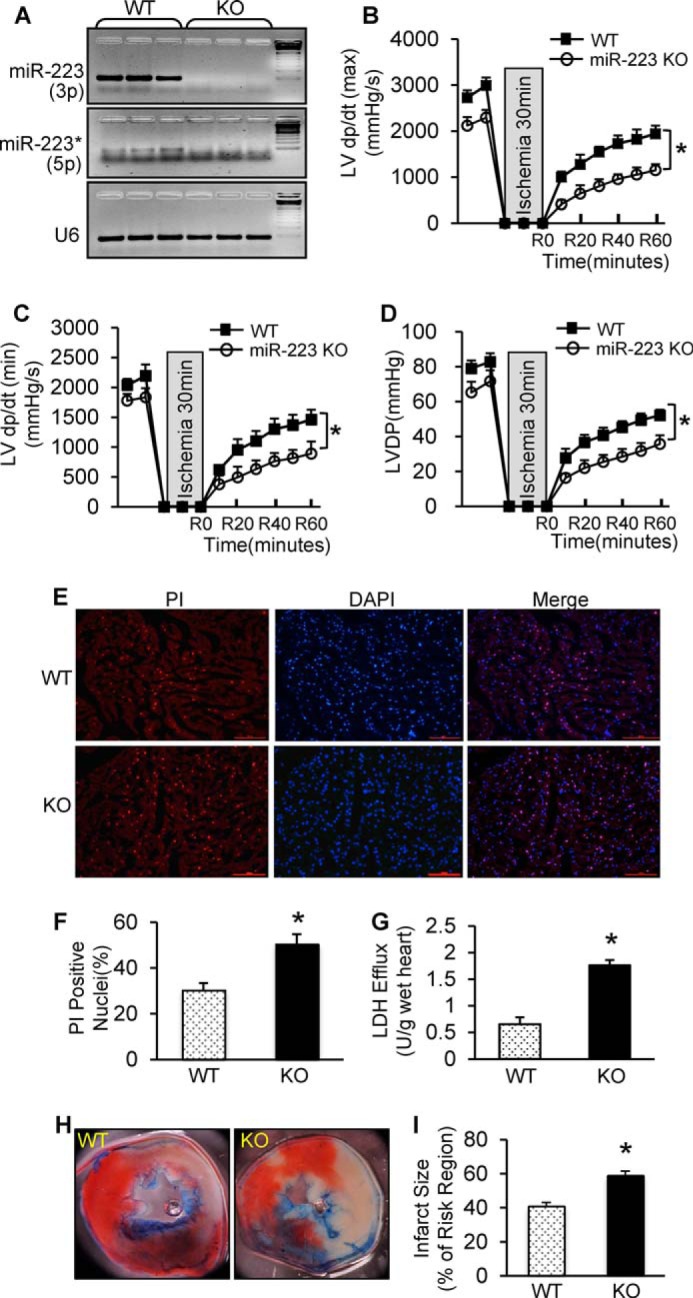
Ablation of pre-miR-223 aggravated ex vivo and in vivo I/R-induced cardiac injury. A, deletion of miR-223-3p/-5p expression was detected by semiquantitative RT-PCR. U6 was used as an internal control (n = 3 for each group). B–D, pre-miR-223 KO mice showed worse functional recovery than WTs during 1-h reperfusion. We performed 30-min ischemia for pre-miR-223 KO mice because KO mice could not bear 45-min ischemia (n = 8 for WT group; n = 9 for KO group; *, p < 0.005 versus WTs). E and F, pre-miR-223 KO hearts displayed a greater degree of myocardial necrosis than WTs as determined by PI staining (E) and quantitative analysis (F) (n = 8 for WT group; n = 9 for KO group; *, p < 0.05 versus WTs). G, total LDH in coronary effluent collected during the first 10 min of reperfusion was significantly increased in KO hearts compared with WTs (n = 8 for WT group; n = 9 for KO group; *, p < 0.05 versus WTs). H and I, pre-miR-223-null hearts exhibited a larger infarction size after 30 min of left anterior descending artery occlusion followed by 24 h of reperfusion. (n = 9 for WT group; n = 8 for KO group; *, p < 0.01 versus WTs). Error bars represent mean ± S.D.
We further assessed the degree of necrosis in these I/R hearts. Using PI staining, we observed that the number of PI-positive nuclei (red) in KO hearts was significantly increased by 1.7-fold compared with WTs (Fig. 4, E and F). Accordingly, the levels of LDH released from ischemic/reperfused KO hearts (n = 8) was 2.7-fold higher than that from ischemic/reperfused WTs (n = 9) (Fig. 4G). It is notable here that the degree of I/R-induced cardiac apoptosis, determined by three assays (TUNEL staining, DNA fragmentation, and Caspase-3 activity), was similar between the two groups (supplemental Fig. S1). Taken together, these results demonstrate that miR-223-5p/-3p deficiency promotes cellular necrosis in mouse hearts under ex vivo I/R conditions.
To elucidate the in vivo effects of pre-miR-223 ablation upon I/R, we subjected these KO mice to 30-min in vivo myocardial ischemia via coronary artery occlusion followed by 24-h reperfusion. We observed that the infarction size (ratio of infarct-to-risk region) was remarkably increased in KO hearts (59.2 ± 4.6%; n = 7) compared with WT hearts (41.1 ± 3.8%; n = 11; p < 0.05) (Fig. 4, H and I).
MiR-223 Negatively Regulates the RIP1/RIP3/MLKL Signaling Cascades and the Inflammatory Response in I/R Hearts
Currently, the complex RIP1-RIP3 and subsequently phosphorylated MLKL are well established to play an essential role in the regulation of necroptosis in various types of cells (8). Furthermore, recent studies have revealed that I/R injury significantly promoted RIP1 and RIP3 expression in animal hearts (6, 7). Given the remarkable reduction of cardiac necrosis in pre-miR-223 TG hearts upon I/R, we therefore examined whether overexpression of miR-223-5p/-3p altered the RIP1/RIP3/MLKL signaling pathway in mouse hearts in response to in vivo I/R (30 min/24 h). Results of Western blotting analysis showed that under pre-I/R conditions TG hearts displayed down-regulated effects of miR-223-5p/-3p on RIP1 and RIP3, but not MLKL, relative to WT controls (Fig. 5, A and B). Consistent with previous observations (4, 6, 7), after ischemia/reperfusion, protein levels of RIP1, RIP3, and MLKL were increased in WT hearts by 3.1-, 1.5-, and 1.7-fold, respectively, compared with pre-I/R WTs (Fig. 5, A and B). However, such I/R-induced elevations of the RIP1/RIP3/MLKL signaling cascades in WT hearts were significantly attenuated in pre-miR-223 TG hearts (Fig. 5, A and B). Conversely, ablation of pre-miR-223 caused up-regulation of RIP1 and RIP3 in mouse hearts under basal conditions and further aggravated the I/R-induced elevations of RIP1, RIP3, and MLKL protein levels compared with WT controls (n = 6; p < 0.05) (Fig. 5, C and D). Put together, these results indicate that miR-223-5p/-3p duplex could negatively modulate the RIP1/RIP3/MLKL axis in animal hearts upon I/R stress.
FIGURE 5.

MiR-223-3p/-5p duplex negatively regulated the RIP1/RIP3/MLKL necroptotic signaling cascades and the expression of inflammatory cytokines in I/R hearts. A and B, representative Western blots (A) and quantitative results (B) of RIP1, RIP3, and MLKL protein expression in WT hearts and pre-miR-223 TG hearts under in vivo pre-I/R and post-I/R (30 min/24 h) conditions (n = 6 hearts per experimental group; *, p < 0.05 versus pre-I/R WTs; #, p < 0.05 versus post-I/R WTs). C and D, representative Western blots (C) and quantitative results (D) of RIP1, RIP3, and MLKL protein expression in WT hearts and pre-miR-223 KO hearts under in vivo pre-I/R and post-I/R (30 min/24 h) conditions (n = 6 hearts per experimental group; *, p < 0.05 versus pre-I/R WTs; #, p < 0.05 versus post-I/R WTs). (Note: for the RIP1 immunoblotting, we initially used a rabbit anti-RIP1 antibody from Santa Cruz Biotechnology (in A), and the upper band was the real band of RIP1 (74 kDa); later on, we tried a rabbit anti-RIP1 from Cell Signaling Technology (in C)). E and F, the expression levels of IL-1β and TNF-α were analyzed by qRT-PCR in pre-miR-223 TG mouse hearts and WTs (E) and pre-miR-223 KO mouse hearts and WTs (F) subjected in vivo to 30-min ischemia followed by 4-h reperfusion. GAPDH mRNA was used as an internal control (n = 6; *, p < 0.05 versus pre-I/R WTs; #, p < 0.05 versus post-I/R WTs). Error bars represent mean ± S.D.
Given that 1) myocardial I/R could activate inflammatory response in vivo (26) and 2) necrotic cells also stimulate tissue inflammation (8), we therefore measured the expression levels of TNF-α and IL-1β, two prominent mediators for I/R-triggered inflammation, in miR-223 TG and KO hearts upon in vivo I/R. Results of RT-PCR analysis showed that, although the levels of TNF-α and IL-1β were too low to distinguish between groups under basal conditions, in vivo I/R (30 min/4 h) did dramatically stimulate the expression of TNF-α and IL-1β in WT hearts (Fig. 5E). However, such up-regulations of TNF-α and IL-1β were significantly reduced in ischemic/reperfused miR-223-overexpressing hearts compared with WT controls (n = 6; p < 0.05) (Fig. 5E). By contrast, ablation of pre-miR-223 remarkably aggravated the I/R-induced elevation of TNF-α and IL-1β relative to ischemic/reperfused WTs (n = 6; p < 0.05) (Fig. 5F). Collectively, these data indicate that up-regulation of miR-223-5p/-3p duplex could attenuate myocardial I/R-triggered inflammation.
Blocking RIP1 Signaling by Necrostatin-1s Counteracts Pre-miR-223-null-induced Detrimental Effects on Cardiac I/R Injury
Given that ablation of pre-miR-223 up-regulated the RIP1 necroptotic signaling (Fig. 5, C and D), we then asked whether blockade of the RIP1 signaling could rescue miR-223 KO-caused detrimental effects on cardiac I/R injury. To clarify this issue, we pretreated KO mice with necrostatin-1 (Nec-1) stable variant (Nec-1s), a newly developed specific inhibitor of RIP1 kinase (27), or the same volume of DMSO-PBS control buffer. 30 min later, hearts were isolated and mounted onto the Langendorff system to undergo ex vivo I/R (30 min/1 h). Consistent with previous reports using Nec-1 (6, 9, 11, 12), pretreatment of WT mice with Nec-1s significantly improved contractile functional recovery as evidenced by higher levels of left ventricle (LV) dP/dt (maximum/minimum) and LVDP and lower levels of LVEDP during reperfusion relative to buffer controls (n = 6; p < 0.05) (Fig. 6, A–D). Remarkably, Nec-1s rescued cardiac function recovery in the reperfused miR-223-null hearts (n = 6) to a greater degree than buffer-treated samples (n = 5; p < 0.05) (Fig. 6, A–D).
FIGURE 6.

Treatment of pre-miR-223 KO mice with Nec-1s largely rescued I/R-induced myocardial damage ex vivo and in vivo. A–D, cardiac function recovery was significantly improved in not only WT mice but also pre-miR-223 KO mice upon ex vivo 30-min ischemia followed by 1-h reperfusion (n = 5 for KO and n = 6 for WT in buffer controls; n = 6 for KO and WT in Nec-1s-treated group; *, p < 0.005). Error bars represent mean ± S.D.
In addition, we observed that Nec-1s-treated WT hearts exhibited significant reduction of necrotic nuclei as stained with the red PI dye (Fig. 7, A and B) and suppressed LDH release during reperfusion (Fig. 7C) relative to buffer-treated WTs. Similarly, I/R-caused aggravation of cardiac necrotic nuclei in miR-223 KO hearts was remarkably attenuated by pretreatment with Nec-1s compared with buffer-injected controls (Fig. 7, A and B). Accordingly, miR-223-null-induced elevation of LDH release was dramatically inhibited by Nec-1s treatment relative to buffer controls (Fig. 7C). Furthermore, the beneficial effects of Nec-1s on the in vivo myocardial I/R (30 min/24 h) was also validated in WT mice as evidenced by a smaller infarction size in Nec-1s-treated hearts (17 ± 1.9%; n = 9) than buffer samples (39 ± 4%; n = 10; p < 0.001) (Fig. 7, D and E), whereas the region at risk was not different between both groups (Fig. 7F). Likewise, pretreatment of miR-223 KO mice with Nec-1s resulted in a significant reduction of myocardial infarction size (29 ± 3.1%; n = 9) compared with the buffer-injected group (62 ± 5.7%; n = 8; p < 0.001) (Fig. 7, D and E). Collectively, these in vitro and in vivo data consistently demonstrate that inhibition of the RIP1-associated necroptotic pathway not only protects wild-type mice against I/R-induced cardiac damage but also largely offsets miR-223-null-mediated detrimental effects on myocardial I/R injury.
FIGURE 7.

A and B, representative images (A) and quantitative results (B) of the PI staining assay in KO and WT hearts after ex vivo I/R (30 min/1 h). Red, PI-positive nuclei; blue, 4′, 6-diamidino-2-phenylindole-stained nuclei. Scale bars, 100 μm. (n = 5 for KO and n = 6 for WT in buffer-injected samples; n = 6 KO and WT in Nec-1s-treated group; *, p < 0.05 versus WT controls.) C, total LDH in coronary effluent collected during the first 10 min of reperfusion was significantly decreased in Nec-1s-treated hearts compared with buffer-treated samples (n = 5 for KO and n = 6 for WT in I/R controls; n = 6 for KO and WT in Nec-1s-treated group; *, p < 0.05 versus WT controls). D–F, Nec-1s treatment greatly reduced in vivo myocardial infarct size after 30 min of left anterior descending artery occlusion followed by 24 h of reperfusion. Representative images (D) and quantitative analysis (E) of the ratio of infarct to risk region are shown (n = 8 for KO and n = 10 for WT in buffer controls; n = 9 for KO and WT in Nec-1s-treated group; *, p < 0.01 versus WT controls). Error bars represent mean ± S.D.
MiR-223 Protects Cardiomyocytes Against H2O2-induced Necrosis
Because 1) genetic mouse models (TG and KO) may have in vivo compensatory effects, 2) cardiac tissue contains other cell types (e.g. immune cells, fibroblasts, and endothelial cells) besides cardiomyocytes, and 3) miR-223 is ubiquitously expressed (20) and these may all influence the outcome of miR-223 TG/KO in the ischemic myocardium, we performed further studies using ex vivo cultured adult cardiomyocytes, a well controlled experimental setting. Cardiomyocytes were isolated from adult rats and cultured in laminin-precoated dishes. Subsequently, cardiomyocytes were infected with Ad.miR-223 and Ad.β-gal (Fig. 8A). Following infection for 36 h, we observed a close to 100% infection efficiency (Fig. 8B). Real time PCR revealed that both strands of miR-223 were overexpressed in Ad.miR-223-infected cardiomyocytes (Fig. 8C). We next treated these myocytes with H2O2 at a dose of 200 μm for 1 h. Cell viability analysis showed that ectopic expression of miR-223 increased cell survival by 2.6-fold relative to β-gal cells (Fig. 8, D and E). Accordingly, dead myocyte-released LDH was decreased by 38% in miR-223-cells compared with β-gal cells upon exposure to H2O2 (Fig. 8E). Furthermore, Western blotting analysis revealed that H2O2 could activate the RIP1/RIP3/MLKL necrotic pathway in β-gal myocytes; yet this pathway was attenuated in miR-223 myocytes (Fig. 8, F and G). Taken together, these data implicate miR-223 as a negative regulator of necrosis in cardiomyocytes.
FIGURE 8.

Acute overexpression of miR-223-3p/-5p in adult rat cardiomyocytes protected against H2O2-induced cell death. A, constructs of Ad.miR-223 and control Ad.β-gal. B, nearly 100% of cardiomyocytes were infected by Ad.miR-223 and Ad.β-gal. C, real time PCR showed overexpression of miR-223-5p and -3p in Ad.miR-223-infected cells. D and E, cardiomyocyte survival and LDH release were measured using MTS incorporation and an LDH analysis kit, respectively. F and G, representative Western blots and quantitative results of the necroptotic signaling cascades (RIP1/RIP3/MLKL) in Ad.miR-223- or Ad.β-gal-infected cardiomyocytes upon H2O2 treatment (n = 4; *, p < 0.05 versus Ad.β-gal control). Error bars represent mean ± S.D. Buff, buffer.
MiR-223-5p and -3p, Respectively, Target Death Receptors and the NLRP3 Inflammasome
To elucidate the potential mechanism(s) of miR-223-5p/-3p duplex-mediated inhibition of cardiac necroptosis during I/R, we dissected the possible targets of miR-223-5p/-3p and further examined their expression levels in TG and KO hearts. Currently, miR-223-3p targets, most of which are related to inflammation (e.g. IKKα, IL-1β, IL-6, Stat3, Roquin, Pknox1, CXCL2, CCL3, and NLRP3), have been well validated in variety of cell types (20). However, miR-223-5p targets have not been well identified thus far. According to the predictions by miRDB, most targets of miR-223-5p are associated with cell membrane receptors (e.g. Sema3a, TNFRsf1a (TNFR1), TNFrsf21 (DR6), and TNFsf4). Given the involvement of miR-223 in I/R-induced cardiac necroptosis, we then selected DR6 and TNFR1 for target validation of miR-223-5p. Based on the seed sequence pairing rule, 3-UTRs of DR6 and TNFR1 are assumed to contain a miR-223-5p-interacting region and are highly conserved among the species of human, mouse, and rat (Fig. 9, A and B). Using the luciferase reporter assay, we validated that miR-223-5p directly recognized both 3-UTRs of DR6 and TNFR1 in both HEK293 cells and rat cardiac H9c2 cells (Fig. 9, C and F). Co-transfection of miR-223-5p strongly inhibited luciferase activity from the reporter construct harboring a 3′-UTR segment of DR6 or TNFR1, whereas no effect was observed when a control miRNA was co-transfected with either reporter construct (Fig. 9, C–F).
FIGURE 9.
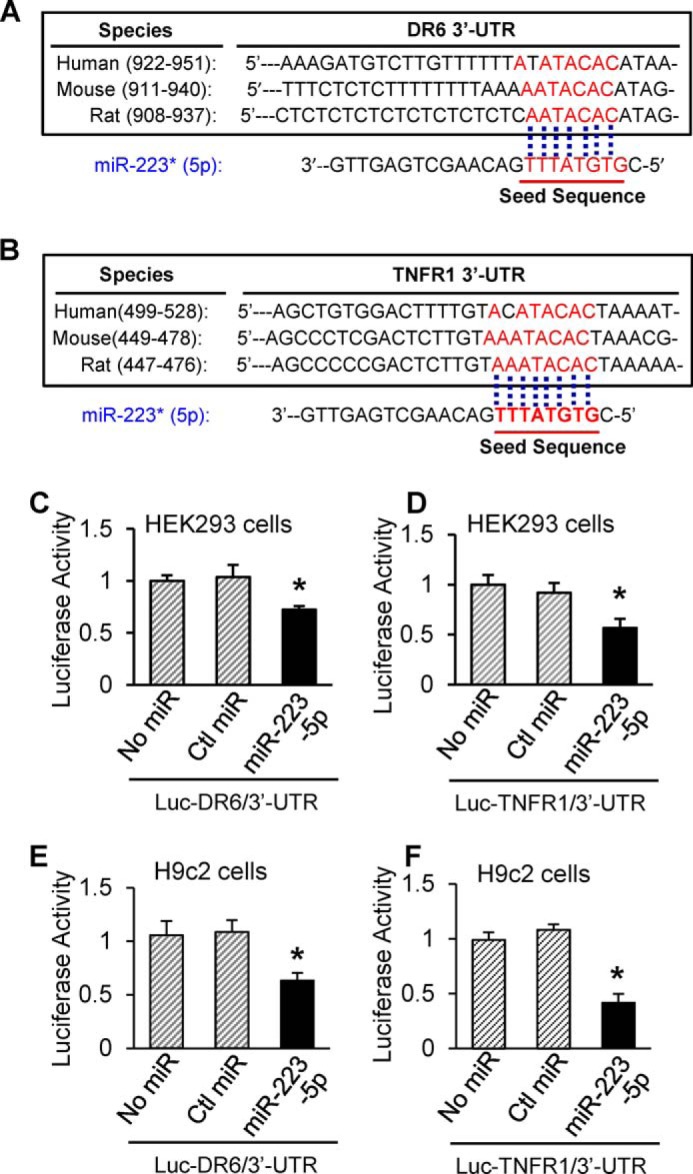
MiR-223-5p directly interacted with 3-UTRs of DR6 and TNFR1. A and B, the putative miR-223-5p binding sites in the 3′-UTR regions of DR6 and TNFR1 are conserved among mammalian species (mouse, human, and rat). C–F, luciferase (Luc) reporter assays showed that DR6 and TNFR1 were authentic targets of miR-223-5p. β-Gal was used as a transfection control. Similar results were observed in three additional, independent experiments (error bars represent mean ± S.D. of four independent experiments; *, p < 0.05 versus miR mimic controls). Ctl, control.
Remarkably, Ad.miR-223-infected cardiomyocytes displayed significant down-regulations of DR6 and TNFR1 compared with controls (Fig. 10A). Likewise, protein levels of DR6 and TNFR1 were significantly reduced by 28 and 42%, respectively, in miR-223-overexpressing hearts (Fig. 10B). By contrast, miR-223 KO hearts exhibited higher protein levels of DR6 and TNFR1 than WT hearts (Fig. 10C). Collectively, these data indicate that DR6 and TNFR1 represent two bona fide targets of miR-223-5p in the myocardium.
FIGURE 10.

A–C, representative Western blots and quantitative results of DR6 and TNFR1 protein expression in Ad.miR-223-infected rat cardiomyocytes (A), pre-miR-223 TG hearts (B), and pre-miR-223-null hearts (C) and their respective controls (n = 3 per group; *, p < 0.05 versus controls). D–F, protein levels of IKKα and NLRP3 were significantly reduced in Ad.miR-223-infected rat myocytes and pre-miR-223 TG hearts, whereas they were increased in pre-miR-223 KO hearts compared with the respective controls (n = 3 per group; *, p < 0.05 versus controls). Error bars represent mean ± S.D.
IKKα and NLRP3, two mediators known to participate in myocardial I/R injury (5, 28, 29), have been previously validated as genuine targets of miR-223-3p in macrophages (30, 31), but whether they are regulated by miR-223-3p in hearts remains unknown. We therefore examined protein levels of IKKα and NLRP3 in miR-223-overexpressing cardiomyocytes and miR-223 TG and KO hearts. Results of Western blotting analysis revealed that their protein levels negatively correlated with miR-223-3p levels as evidenced by reduced levels of IKKα and NLRP3 in miR-223-overexpressing myocytes and transgenic hearts (Fig. 10, D and E) but elevated levels in miR-223-null hearts (Fig. 10F) compared with WTs, respectively (n = 3; p < 0.05). Taken together, these results confirm that miR-223-3p negatively regulates the expression of IKKα and NLRP3 in animal hearts. Currently, it is well established that DR6/TNFR1 are upstream regulators of RIP1-dependent necroptotic signaling (32–34) and that the activation of NLRP3 inflammasome also promotes cell necroptosis (35, 36). In this regard, our data presented here suggest that miR-223-5p/-3p duplex may, respectively, modulate protein levels of death receptors (i.e. DR6 and TNFR1) and the NLRP3 inflammasome signaling cascades, thereby working in concert to inhibit myocardial I/R-induced necroptosis (Fig. 11).
FIGURE 11.
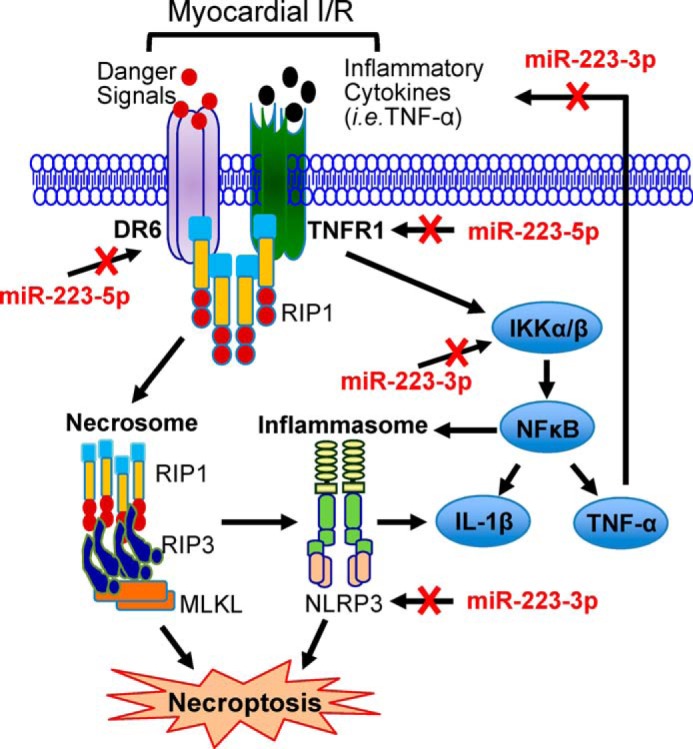
A working model showing that miR-223-5p and -3p work together to suppress the necroptotic cascades at multiple layers, which are marked in red.
Discussion
It is well recognized that a significant amount of myocyte death (apoptosis and necrosis) occurs during the onset of myocardial I/R (24). Given that apoptosis was thought to be the only regulated form of cell death for many years, genetic and pharmacological approaches targeting I/R-induced necrotic myocyte death are rarely studied. In this study, using gain- and loss-of-function approaches, we discovered that elevation of miR-223 could prevent I/R-caused myocyte necroptosis and consequently improved cardiac function recovery during reperfusion. By contrast, ablation of miR-223 aggravated I/R-triggered cardiac injury, and in particular, such detrimental effects could be rescued by necrostatin-1s-mediated inhibition of RIP1 kinase activity. Mechanistically, we identified for the first time that both 5′- and 3′-arms of pre-miR-223 could be processed to become mature miRNA, and they work together to suppress the RIP1-dependent necroptotic pathway at multiple layers in I/R hearts (Fig. 11).
Over the last decade, it has become clear that TNF-α can stimulate cells to form at least four distinctive protein complexes, including complex I, IIa, IIb, and IIc (37). Complex I usually controls NF-κB-associated cell survival signaling, complex IIa mediates Caspase-8/FADD (fas-associated protein with death domain)-related apoptosis, complex IIb favors RIP1-dependent apoptosis, and complex IIc (necrosome) induces cell necroptosis (37). As for which complex is formed in cells upon TNF-α activation, it depends on stress/disease conditions and cell types. In ischemic/reperfused hearts, previous studies have shown that the expression of TNF-α, together with its receptor TNFR1, was up-regulated in cardiomyocytes and consequently activated the NF-κB signaling pathway, resulting in myocardial inflammation and cell death (38, 39). Moreover, recent studies have indicated that myocardial I/R could promote the expression and formation of RIP1-RIP3 necrosome (complex IIc) in cardiomyocytes, leading to myocyte necroptosis and I/R injury (6, 7, 12). Actually, the RIP1-RIP3 necrosome is able to recruit MLKL, which creates a pore-forming complex at the plasma membrane and executes cell death (40). Importantly, sufficient expression of RIP3 and MLKL has been utilized as the only definite criterion for necroptosis to distinguish between different types of necrotic cell death as their levels correlate well with sensitivity to necroptosis (40–42). In the present study, we observed that miR-223-5p/-3p duplex negatively regulated the expression of RIP1 and RIP3, but not MLKL, in mouse hearts under pre-I/R condition. Furthermore, upon I/R, miR-223 TG hearts exhibited lower levels of RIP1, RIP3, and MLKL than those in WTs, suggesting that I/R-caused myocardial necroptosis is attenuated by miR-223. Consistently, myocardial infarction size was reduced in miR-223 TG mice compared with WTs. By contrast, miR-223 KO hearts exhibited opposite effects. Furthermore, miR-223-null-caused aggravation of cardiac necroptosis could be largely rescued by administration of necrostatin-1s, a newly developed specific inhibitor of necroptosis. Thus, these data presented in this study demonstrate that miR-223-5p/-3p duplex could inhibit I/R-triggered cardiac necroptosis via targeting the RIP1-associated necrotic pathway.
The miR-223-mediated inhibition of cardiac necroptosis could be interpreted by several possible mechanisms (Fig. 11). First, we identified that miR-223-5p directly interacted with 3-UTRs of TNFR1 and DR6 and thereby negatively regulated their protein levels. TNFR1 is well established to bind with the RIP1-RIP3 complex (42, 43), and DR6 has been identified to bind with TRADD (tumor necrosis factor receptor type 1-associated death domain protein), an adaptor of the RIP1-RIP3 complex (32–34). Therefore, it is plausible that reduced levels of TNFR1 and DR6 in miR-223-overexpressing hearts may result in the destabilization of the RIP1-RIP3 complex, leading to their degradation and, as a consequence, attenuation of necroptosis in I/R hearts. Second, although necroptosis is believed to be a potent inducer of inflammasomes, Cullen et al. (36) recently showed that all activators of the NLRP3 inflammasome promoted necroptosis in macrophages. In this study, we showed that the expression of NLRP3 could be directly down-regulated by miR-223-3p in the myocardium. In addition, I/R-triggered activation of the TNFR1-IKKα/β-NF-κB signaling cascades is well known to boost the NLRP3 inflammasome (42, 43). Thus, miR-223-5p-mediated inhibition of TNFR1 and miR-223-3p-caused reduction of IKKα would work in concert to indirectly suppress the NLRP3 inflammasome in I/R hearts (Fig. 11). Altogether, I/R-triggered elevation of the NLRP3 inflammasome could be dampened by the overexpression of miR-223-5p/-3p duplex, which may contribute to the reduction of cardiac necroptosis as presented in Fig. 3. Lastly, we observed that the expression of TNF-α and IL-1β was remarkably decreased in miR-223 TG hearts upon in vivo I/R compared with WT controls. Given that inflammatory cytokine (TNF-α and IFNα/β)-induced autocrine loops have been implicated as positive regulators of necroptosis in immune cells (43), reduced expression of TNF-α and IL-1β may also contribute to miR-223-mediated inhibition of cardiac necroptosis during I/R (Fig. 11).
There are several limitations in this work that should be acknowledged. First, although we determined the acute effects of miR-223 TG/KO on in vivo myocardial I/R injury, the long term consequence of miR-223 overexpression or deficiency in the ischemic/reperfused heart (e.g. the extent of fibrosis and cardiac function at different time points after I/R) was not characterized. Second, although the present work focused on miR-223-mediated effects in cardiac necroptosis, our study does not provide a detailed analysis of cardiac apoptosis in I/R hearts. In addition, two well known apoptotic markers (TUNEL staining and Caspase-3 activity) may also be positive for necrotic tissue. Hence, we cannot exclude the possible role of miR-223 in cardiac apoptosis and its contribution to I/R-induced cardiac damage. Finally, we utilized only one necrosis inhibitor (Nec-1s) in this study. Nec-1s is a newly developed analogue of Nec-1. Both Nec-1s and Nec-1 have been shown to inhibit RIP1 kinase; however, the original Nec-1 was recently found to inhibit two kinases other than RIP1 (27). Compared with necrostatin-1, this new compound showed a robust improvement in metabolic stability and displayed >1000-fold more selectivity toward inhibition of RIP1 than any other kinases (27). Thus, we selected only Nec-1s for the present study.
In summary, we have demonstrated that miR-223-5p and -3p both can be expressed in the myocardium and cooperatively dampen the I/R-induced cardiac necroptosis via targeting death receptors (i.e. TNFR1 and DR6) and inflammasome signaling (i.e. NLRP3 and IKKα) at multiple layers (Fig. 11). These findings uncover a novel role of miR-223-5p/-3p duplex in the regulation of cell necroptosis and may provide new and urgently needed therapeutic opportunities for myocardial ischemia/reperfusion injury.
Experimental Procedures
Mouse Models with Overexpression or Deletion of Pre-miR-223
A transgenic mouse model (FVB/N background) with cardiac-specific overexpression of pre-miR-223 was generated as described previously (23). Routine genotyping was performed by PCR. The endogenous mouse Oct2 (organic cation transporter, member 2) gene was used as an internal control. Expression levels of miR-223-5p/-3p in TG hearts were detected by stem-loop quantitative RT-PCR as described previously (16). U6 was used as a loading control to normalize the expression level. All primers used in this study are included in the supplemental material.
Pre-miR-223 knock-out mice (B6.Cg-Ptprca MiR223tm1Fcam/J) and wild-type mice (C57BL/6 background) were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were maintained and bred in the Division of Laboratory Animal Resources at the University of Cincinnati Medical Center. All animal protocols conformed to the Guidelines for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health and were approved by the University of Cincinnati Animal Care and Use Committee.
Myocardial Regional Ischemia/Reperfusion in Vivo
Male adult mice (10–12 weeks old) were anesthetized by intraperitoneal injection of a mixture of ketamine (100 mg/kg) and xylazine (5 mg/kg). Subsequently, these mice underwent occlusion of the left anterior descending coronary artery by tightening and tying the suture for 30 min followed by reperfusion (loosening the suture) as described previously (15). For measurement of miR-223-5p/-3p levels, heart samples were collected at the end of ischemia and 1, 4, and 24 h postreperfusion. For measurement of myocardial infarction size, we retied the occluder to reocclude the left anterior descending coronary artery and injected Evans Blue (0.5 ml of a 2% solution; Sigma-Aldrich) to the aorta to demarcate the non-ischemic myocardium 24 h after ischemia/reperfusion injury. The heart was rapidly excised and sliced into five slices. The heart slices were incubated in 1% 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich) for 15 min at 37 °C for demarcation of the viable and nonviable myocardium within the area at risk (AAR). The staining was stopped by ice-cold sterile saline, and the slices were fixed in 10% neutral buffered formaldehyde and individually weighed. Both sides of each slice were photographed. Each of the myocardial slices was weighed and the infarct size (IS), AAR, and non-ischemic LV were assessed with image analysis software. The AAR/LV, IS/AAR, and IS/LV ratios were calculated. Infarct sizes were determined and expressed as a percentage of the region at risk.
Myocardial Global No-flow Ischemia/Reperfusion ex Vivo
Isolation and perfusion of mouse hearts by the Langendorff method were carried out as described previously (15). In brief, hearts were retrogradely perfused with a phosphate-free Krebs-Henseleit buffer equilibrated at 37 °C with 5% CO2, 95% O2, pH 7.4. After a 10-min equilibration period, pre-miR-223 TG mouse hearts and WT controls underwent no-flow global ischemia for 45 min followed by 60-min reperfusion. Pre-miR-223 KO mice and controls were subjected to 30-min global ischemia followed by 60-min reperfusion. During the I/R, the parameters LVEDP, LVDP, maximum rate of contraction (+dP/dt), and minimum rate of relaxation (−dP/dt) were recorded with a DigiMed Heart Performance Analyzer (Micro-Med).
Pharmaceutical Inhibition of Necroptosis in I/R Mice
7-Cl-O-Nec-1 is a newly developed small molecule inhibitor of RIP1 kinase (27). Compared with necrostatin-1, a previously used RIP1 inhibitor, this new compound showed a robust improvement in metabolic stability and displayed exclusive selectivity toward inhibition of RIP1 (27). Accordingly, 7-Cl-O-Nec-1 is named as Nec-1s or RIP1 inhibitor II. In this study, pre-miR-223 KO mice and WT mice were administered with Nec-1s (Calbiochem, 502497) via intraperitoneal injection (1.65 μg/g) 30 min prior to in vivo or ex vivo myocardial ischemia/reperfusion. The control groups received the same volume (200 μl) of 0.002% DMSO, PBS solution.
Cardiac Necrosis and Apoptosis Assays
I/R-induced cardiac necrosis was assessed by 1) measuring LDH in the perfusion effluent and 2) PI staining of heart tissues. Perfusion effluent was collected every 10 min pre-ischemia and during reperfusion. Total LDH released from the I/R heart was determined using an in vitro Toxicology Assay kit (Sigma) and expressed as units/ml of wet heart weight (units/ml/g). Pyruvate kinase/LDH enzyme (Sigma, P-0294) was used as a standard control. PI is membrane-impermeant red fluorescent dye and that binds to nucleic acids (25). PI staining is generally excluded from viable cells and commonly used for identifying dead cells (25). For in situ detection of cardiac necrotic cells, at the end of reperfusion the I/R heart was immediately taken off from the Langendorff apparatus and then infused with PI solution (50 μg/ml in Krebs-Henseleit buffer) through the aorta using a 10-ml syringe followed by fixation by infusion with 4% paraformaldehyde and placed in prelabeled base molds filled with frozen tissue matrix. Subsequently, these hearts were snap frozen in liquid nitrogen. Frozen sections of 7 μm were cut and counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies). PI-positive (red) nuclei were counted from 10 randomly chosen microscope fields (200×) of the heart section and expressed as a percentage of total nuclei (blue DAPI-staining nuclei) from the same field.
I/R-induced cardiac apoptosis was assessed by 1) in situ TUNEL staining, 2) DNA fragmentation, and 3) Caspase-3 activity. The TUNEL staining was performed using the DeadEnd fluorometric TUNEL system (Promega) followed by staining with an anti-actin (α-sarcomeric) antibody (Sigma-Aldrich) and DAPI. TUNEL-positive (green) nuclei located in cardiomyocytes were counted from 10 randomly chosen microscope fields (200×) of the midventricular section and expressed as a percentage of total nuclei (both blue- and green-staining nuclei) from the same field. Two sections for each heart and five hearts for each group were used. For more accurate quantification of apoptosis in I/R hearts, DNA fragmentation was determined by a cell death detection ELISA kit (Roche Applied Science). Results were normalized to the standard provided in the kit and expressed as -fold increase over control. Caspase-3 activity was determined in cardiac lysates (100 μg) using the Caspase-3/CPP32 Fluorometric Assay kit (BioVision).
Preparation of Adult Rat Cardiomyocytes and Survival Assays upon H2O2 Treatment
Adult rat ventricular myocytes were isolated from Langendorff-perfused hearts of 6-week-old male Sprague-Dawley rats (Harlan Laboratories) as described before (15). Cardiomyocytes were then plated on laminin (10 μg/ml)-coated dishes for 1–2 h followed by infection with recombinant adenoviruses (Ad.miR-223 or Ad.β-gal) at a multiplicity of infection of 500. Thirty-six hours later, such cardiomyocytes were treated with H2O2 (200 μm) for 1 h. Subsequently, cardiomyocyte survival was assessed using the MTS incorporation assay kit (Promega) according to the manufacturer's instructions. Total LDH levels in culture supernatants were measured using an in vitro Toxicology Assay kit as described above.
Western Blotting Analysis
Protein samples were extracted from cultured cardiomyocytes or mouse hearts using procedures described in detail elsewhere (16). Heart samples were homogenated in Nonidet P-40 lysis buffer containing 0.1 mm PMSF and protease inhibitor (Roche Applied Science). Protein samples (50–75 μg) were fractionated by 10–12% SDS-PAGE and transferred to a 0.2-μm nitrocellulose blotting membrane. Binding of the primary antibody was detected by peroxidase-conjugated secondary antibodies and enhanced chemiluminescence (Thermo Fisher Scientific) or by infrared fluorescence secondary antibody and exposure on the Odyssey CLx Infrared Imaging System (LI-COR Biosciences). The corresponding bands were quantified by densitometry. The antibodies used in this study were as follows: rabbit anti-RIP1 (1:1000 dilution) (Santa Cruz Biotechnology, sc-7881 and Cell Signaling Technology, 3493), rabbit anti-RIP3 (1:1000 dilution) (Novus Biologicals, NBP1-77299), mouse anti-MLKL (1:1000 dilution) (Abcam Inc., ab194699), mouse anti-IKKα (1:500 dilution) (Novus Biologicals, NB100-56704), mouse anti-NLRP3 (1:330 dilution) (Adipogen AG-20B-0014), rabbit anti-DR6 (1:500 dilution) (Santa Cruz Biotechnology, sc-25772), and mouse anti-TNFR1 (1:250 dilution) (Santa Cruz Biotechnology, sc-8436). α-Actin (1:1000 dilution) (Sigma-Aldrich, A2172) was used as an internal control.
Luciferase Reporter Assay for MiR-223-5p Targets
For luciferase reporter experiments, a TNFR1 or DR6 3′-UTR segment of 120 bp and its respective mutant were amplified by footprint two-step PCR (16) and inserted into the pMIR-REPORT luciferase miR expression reporter vector (Ambion). HEK293 cells or rat cardiac H9c2 cells were co-transfected in 12-well plates with DharmaFECT Duo transfection reagent (Thermo Fisher Scientific) according to the manufacturer's instructions with 0.4 μg of the 3′-UTR luciferase reporter vector and 0.08 μg of the control vector pMIR-β-galactosidase (Ambion). For each well, 100 nm mimic miR-223-5p or mimic miR control (Thermo Fisher Scientific) was used. Cell lysates were prepared 48 h later, and luciferase activity was measured with a Monolight 3010 luminometer (BD Pharmingen) and expressed as relative light units with a luciferase assay kit (Promega). β-Galactosidase activity was measured with a commercially available kit (Promega). 3′-UTR activity of each construct was expressed as the ratio of luciferase to β-galactosidase activity. All transfections were performed in triplicate from three independent experiments.
Statistical Analysis
Data are expressed as mean ± S.D. Analysis of variance was conducted first across all investigated groups in measurements of myocardial function during ex vivo I/R in which we performed the Shapiro-Wilk test for normality, and there was no evidence of deviation from normality for all variables. Then post hoc pairwise tests were performed with assessment of statistical significance after Bonferroni correction of p values. p < 0.05 was considered statistically significant when Student's t test was used for two-group comparisons.
Author Contributions
D. Q. and X. W. both designed and performed experiments. Y. L. and L. Y. performed RT-PCR and genotyping. R. W., J. P., K. E., and X. M. were involved in data interpretation. Y. L., T. P., Q. H., and K.-J. Y. edited and reviewed the whole manuscript. G.-C. F. designed and conceived the study, provided financial and administrative support, wrote the main manuscript text, and gave final approval of the manuscript. All authors reviewed the manuscript.
Supplementary Material
This work was supported in part by National Institutes of Health Grants HL-087861 and GM-112930 (to G.-C. F.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Fig. S1 and a list of primers used.
- I/R
- ischemia/reperfusion
- IKK
- IκB kinase
- RIP
- receptor-interacting protein kinase
- MLKL
- mixed lineage kinase-like
- TNFR
- TNF receptor
- miR/miRNA
- microRNA
- TG
- transgenic
- LVDP
- left ventricular developed pressure
- LVEDP
- left ventricular end diastolic pressure
- PI
- propidium iodide
- LDH
- lactate dehydrogenase
- Nec-1s
- necrostatin-1 stable variant
- LV
- left ventricle
- Nec-1
- necrostatin-1
- AAR
- area at risk
- IS
- infarct size
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium.
References
- 1. Gershlick A. H., Banning A. P., Myat A., Verheugt F. W., and Gersh B. J. (2013) Reperfusion therapy for STEMI: is there still a role for thrombolysis in the era of primary percutaneous coronary intervention? Lancet 382, 624–632 [DOI] [PubMed] [Google Scholar]
- 2. Fröhlich G. M., Meier P., White S. K., Yellon D. M., and Hausenloy D. J. (2013) Myocardial reperfusion injury: looking beyond primary PCI. Eur. Heart J. 34, 1714–1722 [DOI] [PubMed] [Google Scholar]
- 3. Wang K., An T., Zhou L. Y., Liu C. Y., Zhang X. J., Feng C., and Li P. F. (2015) E2F1-regulated miR-30b suppresses Cyclophilin D and protects heart from ischemia/reperfusion injury and necrotic cell death. Cell Death Differ. 22, 743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oerlemans M. I., Koudstaal S., Chamuleau S. A., de Kleijn D. P., and Doevendans P. A., and Sluijter J. P. (2013) Targeting cell death in the reperfused heart: pharmacological approaches for cardioprotection. Int. J. Cardiol. 165, 410–422 [DOI] [PubMed] [Google Scholar]
- 5. Marchetti C., Toldo S., Chojnacki J., Mezzaroma E., Liu K., Salloum F. N., Nordio A., Carbone S., Mauro A. G., Das A., Zalavadia A. A., Halquist M. S., Federici M., Van Tassell B. W., Zhang S., et al. (2015) Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and nonischemic injury in the mouse. J. Cardiovasc. Pharmacol. 66, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oerlemans M. I., Liu J., Arslan F., den Ouden K., van Middelaar B. J., Doevendans P. A., and Sluijter J. P. (2012) Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res. Cardiol. 107, 270. [DOI] [PubMed] [Google Scholar]
- 7. Zhang T., Zhang Y., Cui M., Jin L., Wang Y., Lv F., Liu Y., Zheng W., Shang H., Zhang J., Zhang M., Wu H., Guo J., Zhang X., Hu X., et al. (2016) CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 22, 175–182 [DOI] [PubMed] [Google Scholar]
- 8. Silke J., Rickard J. A., and Gerlic M. (2015) The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 16, 689–697 [DOI] [PubMed] [Google Scholar]
- 9. Dmitriev Y. V., Minasian S. M., Demchenko E. A., and Galagudza M. M. (2013) Study of cardioprotective effects of necroptosis inhibitors on isolated rat heart subjected to global ischemia-reperfusion. Bull Exp. Biol. Med. 155, 245–248 [DOI] [PubMed] [Google Scholar]
- 10. Koshinuma S., Miyamae M., Kaneda K., Kotani J., and Figueredo V. M. (2014) Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury. J. Anesth. 28, 235–241 [DOI] [PubMed] [Google Scholar]
- 11. Zhang A., Mao X., Li L., Tong Y., Huang Y., Lan Y., and Jiang H. (2014) Necrostatin-1 inhibits Hmgb1-IL-23/IL-17 pathway and attenuates cardiac ischemia reperfusion injury. Transpl. Int. 27, 1077–1085 [DOI] [PubMed] [Google Scholar]
- 12. Koudstaal S., Oerlemans M. I., Van der Spoel T. I., Janssen A. W., Hoefer I. E., Doevendans P. A., Sluijter J. P., and Chamuleau S. A. (2015) Necrostatin-1 alleviates reperfusion injury following acute myocardial infarction in pigs. Eur. J. Clin. Invest. 45, 150–159 [DOI] [PubMed] [Google Scholar]
- 13. Linkermann A., Bräsen J. H., Darding M., Jin M. K., Sanz A. B., Heller J. O., De Zen F., Weinlich R., Ortiz A., Walczak H., Weinberg J. M., Green D. R., Kunzendorf U., and Krautwald S. (2013) Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 110, 12024–12029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu H., and Fan G. C. (2012) Role of microRNAs in the reperfused myocardium towards post-infarct remodelling. Cardiovasc .Res. 94, 284–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ren X. P., Wu J., Wang X., Sartor M. A., Qian J., Jones K., Nicolaou P., Pritchard T. J., and Fan G. C. (2009) MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation 119, 2357–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X., Zhang X., Ren X. P., Chen J., Liu H., Yang J., Medvedovic M., Hu Z., and Fan G. C. (2010) MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation 122, 1308–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ye Y., Hu Z., Lin Y., Zhang C., and Perez-Polo J. R. (2010) Downregulation of microRNA-29 by antisense inhibitors and a PPAR-γ agonist protects against myocardial ischaemia-reperfusion injury. Cardiovasc Res. 87, 535–544 [DOI] [PubMed] [Google Scholar]
- 18. Hinkel R., Penzkofer D., Zühlke S., Fischer A., Husada W., Xu Q. F., Baloch E., van Rooij E., Zeiher A. M., Kupatt C., and Dimmeler S. (2013) Inhibition of microRNA-92a protects against ischemia/reperfusion injury in a large-animal model. Circulation 128, 1066–1075 [DOI] [PubMed] [Google Scholar]
- 19. Wang J. X., Zhang X. J., Li Q., Wang K., Wang Y., Jiao J. Q., Feng C., Teng S., Zhou L. Y., Gong Y., Zhou Z. X., Liu J., Wang J. L., and Li P. F. (2015) MicroRNA-103/107 regulate programmed necrosis and myocardial ischemia/reperfusion injury through targeting FADD. Circ. Res. 117, 352–363 [DOI] [PubMed] [Google Scholar]
- 20. Taïbi F., Metzinger-Le Meuth V., Massy Z. A., and Metzinger L. (2014) miR-223: an inflammatory oncomiR enters the cardiovascular field. Biochim. Biophys. Acta 1842, 1001–1009 [DOI] [PubMed] [Google Scholar]
- 21. Wang X., Huang W., Yang Y., Wang Y., Peng T., Chang J., Caldwell C. C., Zingarelli B., and Fan G. C. (2014) Loss of duplex miR-223 (5p and 3p) aggravates myocardial depression and mortality in polymicrobial sepsis. Biochim. Biophys. Acta 1842, 701–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Essandoh K., Li Y., Huo J., and Fan G. C. (2016) MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock 46, 122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang L., Li Y., Wang X., Mu X., Qin D., Huang W., Alshahrani S., Nieman M., Peng J., Essandoh K., Peng T., Wang Y., Lorenz J., Soleimani M., Zhao Z. Q., et al. (2016) Overexpression of miR-223 tips the balance of pro- and anti-hypertrophic signaling cascades toward physiologic cardiac hypertrophy. J. Biol. Chem. 291, 15700–15713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Konstantinidis K., Whelan R. S., and Kitsis R. N. (2012) Mechanisms of cell death in heart disease. Arterioscler. Thromb. Vasc. Biol. 32, 1552–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Makarov R., Geserick P., Feoktistova M., and Leverkus M. (2013) Cell death in the skin: how to study its quality and quantity? Methods Mol. Biol. 961, 201–218 [DOI] [PubMed] [Google Scholar]
- 26. Vilahur G., and Badimon L. (2014) Ischemia/reperfusion activates myocardial innate immune response: the key role of the toll-like receptor. Front. Physiol. 5, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takahashi N., Duprez L., Grootjans S., Cauwels A., Nerinckx W., DuHadaway J. B., Goossens V., Roelandt R., Van Hauwermeiren F., Libert C., Declercq W., Callewaert N., Prendergast G. C., Degterev A., Yuan J., et al. (2012) Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 3, e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zingarelli B., Hake P. W., Denenberg A., and Wong H. R. (2002) Sesquiterpene lactone parthenolide, an inhibitor of IκB kinase complex and nuclear factor-κB, exerts beneficial effects in myocardial reperfusion injury. Shock 17, 127–134 [DOI] [PubMed] [Google Scholar]
- 29. Malik S., Sharma A. K., Bharti S., Nepal S., Bhatia J., Nag T. C., Narang R., and Arya D. S. (2011) In vivo cardioprotection by pitavastatin from ischemic-reperfusion injury through suppression of IKK/NF-κB and upregulation of pAkt-e-NOS. J. Cardiovasc. Pharmacol. 58, 199–206 [DOI] [PubMed] [Google Scholar]
- 30. Li T., Morgan M. J., Choksi S., Zhang Y., Kim Y. S., and Liu Z. G. (2010) MicroRNAs modulate the noncanonical transcription factor NF-κB pathway by regulating expression of the kinase IKKα during macrophage differentiation. Nat. Immunol. 11, 799–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bauernfeind F., Rieger A., Schildberg F. A., Knolle P. A., Schmid-Burgk J. L., and Hornung V. (2012) NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 189, 4175–4181 [DOI] [PubMed] [Google Scholar]
- 32. Pan G., Bauer J. H., Haridas V., Wang S., Liu D., Yu G., Vincenz C., Aggarwal B. B., Ni J., and Dixit V. M. (1998) Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett. 431, 351–356 [DOI] [PubMed] [Google Scholar]
- 33. Benschop R., Wei T., and Na S. (2009) Tumor necrosis factor receptor superfamily member 21: TNFR-related death receptor-6, DR6. Adv. Exp. Med. Biol. 647, 186–194 [DOI] [PubMed] [Google Scholar]
- 34. Hu R., Du Q., Yin X., Li J., Wang T., and Zhang L. (2014) Agonist antibody activates death receptor 6 downstream signaling involving TRADD recruitment. FEBS Lett. 588, 401–407 [DOI] [PubMed] [Google Scholar]
- 35. Kang T. B., Yang S. H., Toth B., Kovalenko A., and Wallach D. (2014) Activation of the NLRP3 inflammasome by proteins that signal for necroptosis. Methods Enzymol. 545, 67–81 [DOI] [PubMed] [Google Scholar]
- 36. Cullen S. P., Kearney C. J., Clancy D. M., and Martin S. J. (2015) Diverse activators of the NLRP3 inflammasome promote IL-1β secretion by triggering necrosis. Cell Rep. 11, 1535–1548 [DOI] [PubMed] [Google Scholar]
- 37. Conrad M., Angeli J. P., Vandenabeele P., and Stockwell B. R. (2016) Regulated necrosis: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 15, 348–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Flaherty M. P., Guo Y., Tiwari S., Rezazadeh A., Hunt G., Sanganalmath S. K., Tang X. L., Bolli R., and Dawn B. (2008) The role of TNF-α receptors p55 and p75 in acute myocardial ischemia/reperfusion injury and late preconditioning. J. Mol. Cell. Cardiol. 45, 735–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hamid T., Gu Y., Ortines R. V., Bhattacharya C., Wang G., Xuan Y. T., and Prabhu S. D. (2009) Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-κB and inflammatory activation. Circulation 119, 1386–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang H., Sun L., Su L., Rizo J., Liu L., Wang L. F., Wang F. S., and Wang X. (2014) Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146 [DOI] [PubMed] [Google Scholar]
- 41. Rickard J. A., O'Donnell J. A., Evans J. M., Lalaoui N., Poh A. R., Rogers T., Vince J. E., Lawlor K. E., Ninnis R. L., Anderton H., Hall C., Spall S. K., Phesse T. J., Abud H. E., Cengia L. H., et al. (2014) RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157, 1175–1188 [DOI] [PubMed] [Google Scholar]
- 42. Pasparakis M., and Vandenabeele P. (2015) Necroptosis and its role in inflammation. Nature 517, 311–320 [DOI] [PubMed] [Google Scholar]
- 43. Linkermann A., Stockwell B. R., Krautwald S., and Anders H. J. (2014) Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat. Rev. Immunol. 14, 759–767 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.