

Abstract
The use of photochemical transformations is a powerful strategy that allows for the formation of a high degree of molecular complexity from relatively simple building blocks in a single step. A central feature of all light-promoted transformations is the involvement of electronically excited states, generated upon absorption of photons. This produces transient reactive intermediates and significantly alters the reactivity of a chemical compound. The input of energy provided by light thus offers a means to produce strained and unique target compounds that cannot be assembled using thermal protocols. This review aims at highlighting photochemical transformations as a tool for rapidly accessing structurally and stereochemically diverse scaffolds. Synthetic designs based on photochemical transformations have the potential to afford complex polycyclic carbon skeletons with impressive efficiency, which are of high value in total synthesis.
1. Introduction
The synthesis of natural products defines the frontier of synthetic chemistry as it offers the practitioners the challenge of constructing complex and structurally diverse molecular frameworks. This wealth of synthetic challenges has been a valuable platform for expanding state-of-the-art synthetic methodology and discovering fundamentally new chemical protocols that can subsequently be implemented by the whole chemical community.1−12
Natural products that display biological activity often serve as vital targets for novel drug lead candidates.13−17 Access to these complex and structurally diverse assemblies constitutes a multifaceted challenge for chemists, which requires efficient and powerful synthetic strategies.18−20 The use of small-molecule libraries inspired by bioactive natural products is an essential part of drug discovery and is an attractive aspect of the early stages of drug development. Here, the transition from planar structures with a sp2-rich character to more structurally complex libraries that contain multiple sp3 centers may yield a higher probability of displaying selective biological activity.21
The increased attention to environmentally related issues has also led to the reassessment of several existing technologies, requiring the scientific community to devise novel and “green” methods. These processes should be energy-efficient, reduce the consumption of raw materials, and ultimately produce minimal amounts of waste.22−25 Ideal methods would provide the opportunity to transform simple feedstocks into highly functionalized and complex molecules. An attractive approach would be to explore the potential of photochemical reactions, as they only involve the absorption of photons. In this sense, photoinduced reactions offer powerful and efficient strategies for designing diverse organic frameworks that might otherwise be difficult to access.26,27
A central feature of all light-promoted transformations is the involvement of electronically excited states, formed upon the absorption of photons. This excitation leads to the generation of transient reactive intermediates and significantly alters the reactivity of a chemical compound (Figure 1), a process that can be controlled to generate the intended product in high yield and with excellent selectivity.28,29 Compared to thermal reactions, a majority of the prevailing photochemical reactions do not require additional reagents for activation, such as metal catalysts, Brønsted acids, or bases. The selective input of energy provided by light offers a means to produce strained and unique target molecules that cannot be assembled using thermal protocols, thus allowing for the production of immense molecular complexity in a single chemical step. Rational and efficient synthetic methodologies can thereby be designed as a rapid entry to diverse molecular scaffolds containing various functional groups, often in shorter synthetic sequences with respect to alternative multistep procedures.30,31
Figure 1.

Reaction pathways in (a) a thermal reaction with reagent R yielding product P catalyzed by a catalyst (cat.) via intermediate I′ and (b) in a photochemically induced reaction where the chemical reaction commences from the excited state of the reagent (R*).
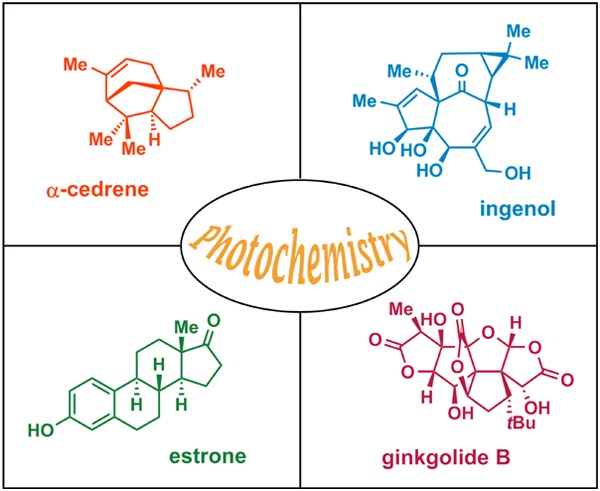
As previously mentioned, the use of photochemistry is appealing for generating molecular complexity that may not be accessible by conventional methods. As a result, a number of fascinating total syntheses of natural products have been achieved, which highlight the remarkable power of UV light for constructing advanced polycyclic carbon skeletons.27,32,33 The use of UV light for bond assembly has been known for a long period of time. Trommsdorff found in the early 19th century that crystals of the sesquiterpene santonin reacted upon exposure to sunlight, which may be considered the birth of photochemistry.34,35 From the mid-20th century, myriad examples have been reported where photochemistry has been exploited for remarkable rearrangements and construction of complex molecular scaffolds.27,36Figure 2 depicts some of the outstanding and now classic examples of natural product synthesis where photochemical reactions have been applied in key steps. These include the synthesis of α-cedrene (1),37 ingenol (2),38 estrone (3),39−41 and ginkgolide B (4),42 among others.
Figure 2.

Representative examples where photochemical reactions have been exploited for construction of complex natural products with polycyclic frameworks.
This review aims to demonstrate the importance of photochemical approaches for accessing complex chemotypes and its key role in the synthesis of advanced structures with relevance to biological systems. The review is organized by the different types of photochemical reactions, beginning with various [n + 2] photocycloadditions and then describing various photochemical rearrangements that can be achieved. The broader chemistry community has recently become well-aware of the virtues of photoredox catalysis to engage in an array of single-electron transfer (SET) events that have previously been elusive. The final chapter of this review therefore highlights the use of photoredox catalysis to trigger unique catalytic processes in natural product synthesis.
2. [2 + 2] Photocycloadditions
2.1. [2 + 2] Photocycloadditions of Olefins—A Versatile Method for Accessing Cyclobutanes
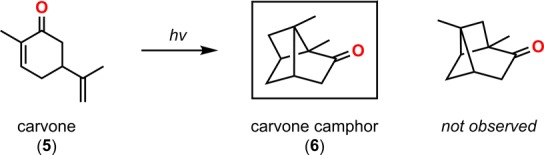
Ciamician and Silber reported on the [2 + 2] photocycloaddition in 1908 when they observed the formation of carvone camphor (6) when carvone (5) was exposed to light for 1 year (Scheme 1).43 In the [2 + 2] photocycloaddition of alkenes, α,β-unsaturated carbonyl compounds are generally employed, as they are more easily photoexcited. This produces an initial short-lived singlet state that decays by intersystem crossing (ISC) to produce a triplet state. The triplet exciplex that is formed with the ground state alkene moiety results in a triplet 1,4-biradical that undergoes spin inversion to the singlet biradical, thus allowing for generation of the desired cyclobutane (Scheme 2).44−46 The [2 + 2] photocycloaddition has the possibility of generating two different regioisomers, which are referred to as the head-to-tail and head-to-head products (Scheme 3, top). In general, head-to-tail products are formed when the R group is electron-donating, while head-to-head products are produced when the R group is electron-withdrawing (Scheme 3, bottom).47−50
Scheme 1. Ciamician and Silber’s early [2 + 2] Photocycloaddition of Carvone (5).
Scheme 2. Photoexcitation of Enones.

ISC = intersystem crossing.
Scheme 3. Regioselectivity in [2 + 2] Photocycloadditions.

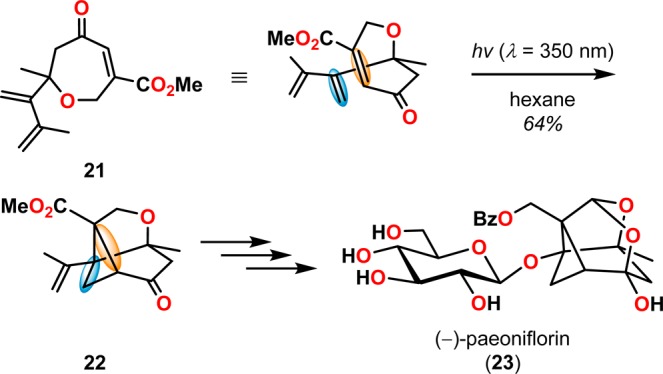
[2 + 2] Photocycloaddition affords cyclobutanes, an important and common structural motif in a variety of natural products (Figure 3). This makes the [2 + 2] photocycloaddition of two alkene units a powerful method in natural product synthesis for constructing precursors to either acyclic or cyclic systems, including carbo-, heterobi-, and oligocyclic structures. The utilization of the [2 + 2] photocycloaddition of olefins has, for example, been exploited in the total synthesis of (−)-biyouyanagin A (17, Scheme 4),51−53 (−)-littoralisone (20, Scheme 5),54 (−)-paeoniflorin (23, Scheme 6),55 (±)-punctaporonin C (26, Scheme 7),56,57 and (+)-solanascone (29, Scheme 8).58 Recent examples for use of the [2 + 2] photocycloadditions include the total syntheses of aquatolide (32, Scheme 9)59 and an intramolecular [2 + 2] photocycloaddition for construction of the tricyclic core of solanoeclepin A (14, Scheme 10).60
Figure 3.

Examples of natural products containing the cyclobutane scaffold.
Scheme 4. Intermolecular [2 + 2] Photocycloaddition in the Synthesis of (−)-Biyouyanagin A (17).

Scheme 5. Application of the Intramolecular [2 + 2] Photocycloaddition in the Total Synthesis of (−)-Littoralisone (20).

Scheme 6. [2 + 2] Photocycloaddition of Enone 21 in the Total Synthesis of (−)-Paeoniflorin (23).
Scheme 7. Intramolecular [2 + 2] Photocycloaddition for Synthesis of the (±)-Punctaporonin C (26) Core.

TIPS = triisopropylsilyl.
Scheme 8. Intramolecular [2 + 2] Photocycloaddition in the Synthesis of (+)-Solanascone (29).

Scheme 9. Photochemical [2 + 2] Cycloaddition in the Total Synthesis of Aquatolide (32).

Scheme 10. Intramolecular [2 + 2] Photocycloaddition for Construction of the Tricyclo[5.2.1.01,6]decane Core of Solanoeclepin A (14).

Bpin = (pinacolato)boron. TBS = tert-butyldimethylsilyl.
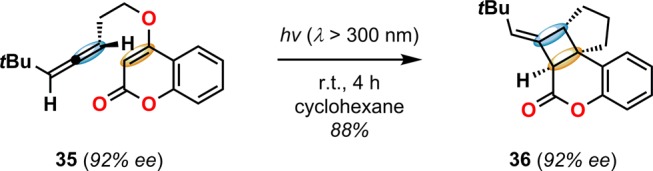
Optically active allenes appended to enones have also been shown to afford the cyclobutane photoadducts with high levels of asymmetric induction, thus providing access to optically active fused polycyclic structures that might otherwise be difficult to access (Scheme 11).61,62 A remarkable example for generation of molecular complexity from simple precursors can be encountered in the photocycloaddition/rearrangement sequence that converts pyrroles to aziridines (Scheme 12).63 Booker-Milburn and co-workers discovered that irradiation of pyrroles such as 37 could facilitate rearrangement from the initially produced [2 + 2] photocycloaddition adduct 38 to produce aziridine 39. This constitutes a novel photochemical sequence for conversion of substituted pyrroles into intricate tricyclic aziridines and was shown to be general for a variety of substituted pyrroles, ranging from mono- to tetrasubstituted.
Scheme 11. Intramolecular [2 + 2] Photocycloaddition of Optically Active Allene 35.
Scheme 12. Photocycloaddition/Rearrangement of Pyrroles to Aziridines.

From the aforementioned example it is obvious that the inherent ring strain in the cyclobutane ring system renders it amenable to strain-releasing reactions, making the cyclobutane products versatile substrates for further reactions. If the generated cyclobutane is fused to one or several rings, a tandem [2 + 2] photocycloaddition–fragmentation sequence offers powerful routes for accessing ring-expansion products and can thus be used as an intermediate for constructing more complex medium-sized ring systems (Scheme 13). Typical fragmentation pathways for the generated [2 + 2] photoadducts include Grob fragmentations (Figure 4, top), radical fragmentations (Figure 4, middle), or De Mayo reactions (Figure 4, bottom). The robust and sterically tolerant nature of the [2 + 2] photocycloaddition makes it well-suited for constructing C–C bonds in a plethora of contexts.64−66
Scheme 13. Formation of Cyclobutanes via [2 + 2] Photocycloaddition, Followed by Ring-Opening To Generate Medium-Sized Rings.

Figure 4.
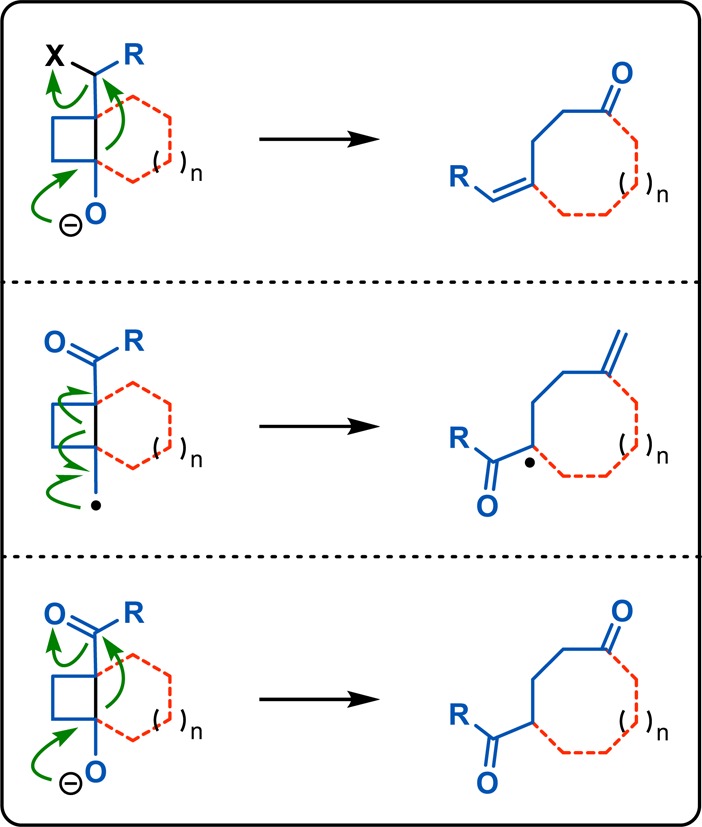
Fragmentation strategies for [2 + 2] photocycloaddition adducts: (top) Grob fragmentation, (middle) radical fragmentation, and (bottom) De Mayo reaction.
The use of the [2 + 2] photocycloaddition for the chemoselective annulation of larger ring systems is a powerful concept and was utilized in the total synthesis of (±)-gibberellic acid (46, Scheme 14),67 (±)-pentalenene (53, Scheme 15),68,69 and linderol A (56, Scheme 16).70−72 The [2 + 2] photocycloaddition in combination with a retro-aldol reaction is known as the De Mayo reaction and will be discussed in detail in section 2.3.
Scheme 14. Application of the [2 + 2] Photocycloaddition/Ozonolysis/Retro-Dieckmann Reaction Sequence in the Synthesis of (±)-Gibberellic Acid (46).

MOM = methoxymethyl. SEM = 2-(trimethylsilyl)ethoxymethyl.
Scheme 15. Allene–Enone [2 + 2] Photocycloaddition with Subsequent Lewis Acid-Catalyzed Rearrangement in the Synthesis of (±)-Pentalenene (53).

Scheme 16. Dimethylsulfoxonium Methylide Mediated Rearrangement of Cyclobutane 54 in the Synthesis of Linderol A (56).

A Grob fragmentation73 was employed in the synthesis of (±)-epikessane (62) to construct the hydroazulene skeleton via ring expansion.74 Cyclobutane 60, produced from [2 + 2] photocycloaddition of 4-acetoxy-2-cyclopentenone (57) and 1-acetoxy-2-carbo-methoxycyclopentene (58), treated with p-toluenesulfonyl chloride (p-TsCl) in pyridine, thereby triggering Grob fragmentation to afford ketone 61 bearing the hydroazulene core of epikessane (62) (Scheme 17). Epoxides can also be used as functional groups to achieve ring expansion through a Grob-type fragmentation and were utilized in the total synthesis of the hydroazulene sesquiterpene (+)-aphanamol I (see Scheme 50).75
Scheme 17. Grob Fragmentation for Generation of the Hydroazulene Skeleton in the Synthesis of (±)-Epikessane (62).

p-TsOH = p-toluenesulfonic acid; p-TsCl = p-toluenesulfonyl chloride.
Scheme 50. Synthesis of (+)-Aphanamol I (291) by Use of a Modified De Mayo Sequence.

Isocomene (63) is a sesquiterpene that belongs to a family of tricyclic angular triquinane sesquiterpenes, initially isolated from the plant Isocoma wrigthii.76,77 Tobe and co-workers used a [2 + 2] photocycloaddition approach using enone 66 to generate the tricyclic adduct 67.78 Subjecting enone 66 to an excess of 1,2-propadiene (43) in CH2Cl2 at −78 °C (λ = 300 nm) successfully gave the desired head-to-head product 67 in high yield and excellent selectivity (Scheme 18). Compound 67 was subsequently converted in two steps to epoxide 69, which under Lewis acid-mediated epoxide–carbonyl rearrangement conditions79 afforded the triquinane core. Further manipulations of triquinane 70 resulted in (±)-β-isocomene (64, Figure 5), which could undergo acid-catalyzed isomerization to furnish (±)-isocomene (63). A related ring expansion route by use of the epoxide-carbonyl rearrangement was also employed by Tobe and co-workers in the synthesis of (±)-modhephene (65).80
Scheme 18. Tobe’s Approach for Construction of the Tricyclic Core of (±)-Isocomene (63).

HMPA = hexamethylphosphoric triamide. m-CPBA = m-chloroperoxybenzoic acid.
Figure 5.

Structures of isocomene (63), β-isocomene (64), and modhephene (65).
Free radical fragmentations of the generated cyclobutanes to produce medium-sized ring systems represent a second class of essential cleavage reactions that have been widely exploited in organic synthesis.81−86 Shipe and Sorensen employed an intramolecular [2 + 2] photocycloaddition/radical SmI2-mediated fragmentation sequence in the convergent enantioselective syntheses of both natural (+)- and unnatural (−)-guanacastepene E (73) and formal total syntheses of (+)- and (−)-guanacastepene A (71, Figure 6).87,88 Initial π-allyl Stille cross-coupling afforded photosubstrate 76, which underwent intramolecular [2 + 2] photocycloaddition to furnish cyclobutane 77 in 82% yield. The selective ring opening of the cyclobutane (77) was accomplished using SmI2-mediated ketyl radical formation and radical fragmentation. Subsequent trapping of the samarium enolate with phenylselenenyl bromide gave organoselenide 78. Selenoxide elimination with mCPBA produced the tricyclic compound 79, which could be elaborated to (+)-guanacastepene A (71) and (+)-guanacastepene E (73) (Scheme 19).
Figure 6.

Structures of guanacastepenes A (71), C (72), and E (73).
Scheme 19. Use of the Intramolecular Enone–Olefin [2 + 2] Photocycloaddition and Stereoelectronically Controlled, Reductive, SmI2-Mediated Fragmentation in the Syntheses of (+)-Guanacastepene A (71) and (+)-Guanacastepene E (73).

mCPBA = m-chloroperoxybenzoic acid. DIPEA = diisopropylethylamine. HMPA = hexamethylphosphoric triamide. PMP = p-methoxyphenyl.
The radical fragmentation of [2 + 2] photocycloadducts involving Sn reagents has also been exploited in natural product synthesis. Lange and Gottardo utilized Bu3SnH in combination with azobis(isobutyronitrile) (AIBN) to affect the ring expansion of cyclobutylcarbinyl iodide 83 in the formal synthesis of pentalenene (53) (Scheme 20).89 A related radical fragmentation/elimination sequence has also been applied in the total synthesis of the sesquiterpenoid alismol (92) to afford the bicyclo[5.3.0]decane ring system 90 using a Bu3SnH/AIBN reagent combination (Scheme 21).90,91
Scheme 20. Bu3SnH-Mediated Fragmentation of Cyclobutane 83 in the Formal Synthesis of (±)-Pentalenene (53).

AIBN = azobis(isobutyronitrile).
Scheme 21. [2 + 2] Photocycloaddition with Subsequent Bu3SnH-Mediated Fragmentation in the Total Synthesis of Alismol (92).

AIBN = azobis(isobutyronitrile).
Laurenene (99) is a diterpene initially isolated by Corbett and co-workers in 1979 from Dacrydium cupressinum and contains the unique fenestrane motif (see Figure 30). The tetracyclic [5.5.5.7]fenestrane core was seen as a challenge that photocycloaddition chemistry was suited to address.92,93 To generate the sterically crowded quaternary carbon center, a [2 + 2] photocycloaddition of enone 94 furnished the essential cyclobutane intermediate 95 in 87% yield and established the three contiguous quaternary centers required for the central core of laurenene (99). Further manipulations transformed cyclobutane 95 to the unsaturated keto ester 96, where the cyclobutane underwent reductive ring opening under Birch reduction conditions (Na/NH3) at −33 °C, followed by hydrogenation using Pd/C to afford keto ester 97. A reduction–oxidation sequence generated the keto–aldehyde containing substrate, which underwent an intramolecular aldol condensation to give enone 98 containing the essential cycloheptane ring and core structure of (±)-laurenene (99) (Scheme 22).94 A related [2 + 2] photocycloaddition approach has also been reported for the synthesis of the sesquiterpene (±)-silphinene (105) (Scheme 23).95
Figure 30.
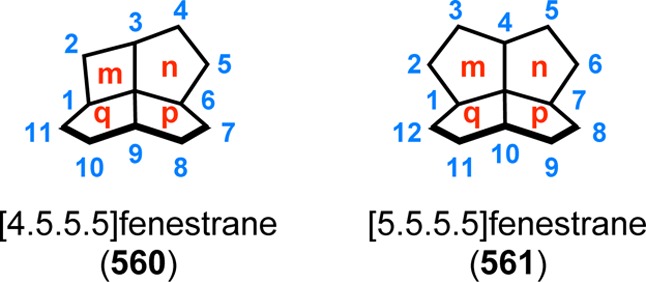
Numbering of the fenestrane framework.
Scheme 22. Synthesis of (±)-Laurenene (99) via Intramolecular [2 + 2] Photocycloaddition Followed by Reductive Cleavage.

p-TsOH = p-toluenesulfonic acid.
Scheme 23. Construction of the Core in Silphinene (105) By [2 + 2] Photocycloaddition.

LDA = lithium diisopropylamide.
Merrilactone A (106, Figure 7) is a sesquiterpene containing a bicyclo[3.3.0]octane core, two lactone moieties, an oxetane ring, and seven contiguous stereogenic centers, making it an alluring synthetic target.96,97 Originally isolated from the pericarps of Illicium merrillianum, merrilactone A (106) displays neurotrophic activity in cultures of fetal rat cortical neurons98,99 and is a promising potential therapeutic agent for the neurodegeneration associated with Alzheimer’s and Parkinson’s diseases.100−106 Several total syntheses of merrilactone A (106) have been reported following its isolation. The groups of Mehta107 and Inoue108 both employed a [2 + 2] photocycloaddition of the acetylene surrogate 1,2-dichloroethylene in the synthesis of (±)-merrilactone A and in the asymmetric synthesis of (−)-merrilactone A, respectively. In addition to the two syntheses by Mehta and Inoue, Greaney and co-workers have also reported on the total synthesis of (±)-merrilactone A, which relied on an initial [2 + 2] photocycloaddition of 4,5-dimethylmaleic anhydride and dimethylketene acetal.109 The photocycloaddition was chosen to produce the challenging syn angular methyl groups given the reactions robust nature in sterically encumbered environments.
Figure 7.
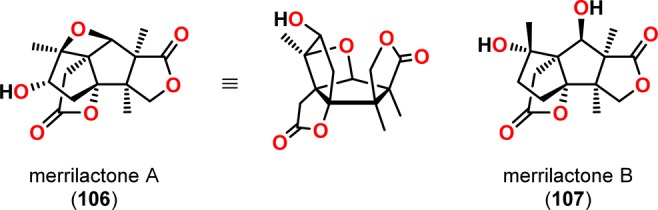
Structures of merrilactones A (106) and B (107).
In Mehta and Singh’s approach107 toward the synthesis of (±)-merrilactone A (Scheme 24), photochemical [2 + 2] cycloaddition of enone 109 and trans-1,2-dichloroethylene (110) afforded cyclobutane (111) in 43% yield, which was subsequently converted to enol ether 112 through a four-step procedure involving eliminative dehalogenation, acetonide deprotection, oxidation, and homologation. Acid-mediated intramolecular hemiacetal formation and oxidation furnished lactone 114. Ozonolysis and in situ reduction of the fused cyclobutene produced lactol 115, which was further elaborated into epoxide 116. Exposure of epoxide 116 to p-TsOH allowed for the homo-Payne rearrangement, yielding the target compound (±)-merrilactone A (106).
Scheme 24. Mehta and Singh’s Approach to (±)-Merrilactone A (106).

PCC = pyridinium chlorochromate. TBS = tert-butyldimethylsilyl. p-TsOH = p-toluenesulfonic acid.
For the asymmetric total synthesis of (−)-merrilactone A (106), Inoue and co-workers utilized the [2 + 2] photocycloaddition of the enantiomerically pure lactone 117 and cis-1,2-dichloroethylene (118) to afford cyclobutene 120 after Zn-promoted dehalogenation and LiAlH4 reduction. Cyclobutene 120 was subsequently converted into diene 121, which underwent ring-closing metathesis to yield a bicyclo[4.2.0]octane system that upon subjection to Pb(OAc)4 underwent oxidative ring expansion to give the cyclooctanedione 122. After some optimization, it was found that reacting cyclooctanedione 122 with NaN(TMS)2 allowed for site-selective deprotonation and diastereoselective C–C bond formation through a transannular aldol reaction, producing the desired bicyclo[3.3.0]octane system 124 with only small amounts of the other diastereomers. Further manipulations completed the asymmetric synthesis of (−)-merrilactone (106) in 31 steps with an overall yield of 1.1% (Scheme 25).
Scheme 25. Inoue and Co-Workers’ Approach toward (−)-Merrilactone A (106).

BTB = 2,6-bis(trifluoromethyl)benzyl.
The ginkgolides are a family of polycyclic oxygenated compounds obtained from the ginkgo tree (Ginkgo biloba). Extracts from the ginkgo tree have been used as herbal medicines for 5000 years for alleviating disorders such as coughs, bronchitis, and asthma.110−113 These therapeutic effects were shown to originate from five different compounds, called the ginkgolides, which differ only in the number and position of the hydroxyl groups (Figure 8). The ginkgolides were originally isolated by Furukawa114−116 in 1932, but their structures were not established until 1967, when X-ray crystallography studies confirmed the structure and absolute stereochemistry of the ginkgolides.117−121 Bilobalide (129)122 and ginkgolide J (127)123 were subsequently discovered and added as members of the ginkgolide family. The five ginkgolides (A, B, C, J, and M) all share an identical carbon skeleton, consisting of 6 rings, 11 stereogenic centers, and an uncommon tert-butyl moiety. The syntheses of ginkgolides A (125)124 and B (4)125 as well as bilobalide (129)126,127 were initially accomplished by Corey and co-workers.128
Figure 8.

Structures of ginkgolides A, B, C, M, and J (125, 4, 126–128) and bilobalide (129).
A remarkable example where [2 + 2] photocycloaddition has been employed to access complex polycyclic carbon skeletons is the total synthesis of ginkgolide B (4) reported by Crimmins and co-workers, where they made use of a [2 + 2] photocycloaddition in order to establish the two vicinal quaternary stereocenters.42,129 Previously, the same group reported the synthesis of the structurally related compound bilobalide (129) using a stereoselective, intramolecular [2 + 2] photocycloaddition as the key step for assembling the core of bilobalide (129) (Scheme 26).130,131 Here the [2 + 2] photocycloaddition of photosubstrate 130 gave the desired photoadduct 131 in 50% yield where both the (trimethylsilyl)oxy and the tert-butyl groups occupy pseudoequatorial positions on the generated five-membered ring. Hydroxylation of photoadduct 131 with MoOPH {(hexamethylphosphoric triamide)oxodiperoxy(pyridine)molybdenum(VI) [MoO5·pyr·HMPA]132,133} produced hydroxy ketone 132 in 80% yield. Oxidative cleavage of hydroxy ketone 132 afforded aldehyde 133 in 94% yield, which could subsequently be converted into acetal 134 in three steps. A regioselective Baeyer–Villiger oxidation of cyclobutanone 134 furnished lactone 135 in 95% yield. After construction of the basic skeleton of bilobalide, a three-step oxidation sequence involving Jones’ reagent134/dimethyldioxirane/Jones’ reagent furnished the target compound bilobalide (129) in excellent yield.
Scheme 26. Use of Intramolecular [2 + 2] Photocycloaddition in the Synthesis of Bilobalide (129).

m-CPBA = m-chloroperoxybenzoic acid. LDA = lithium diisopropylamide. MoOPH = (hexamethylphosphoric triamide)oxodiperoxy(pyridine)molybdenum(VI) (MoO5·pyr·HMPA). Piv = pivaloyl.
As previously shown for the ginkgolides, the introduction of the functional groups and the correct orchestration of the C5 and C9 quaternary carbon centers have been recognized as the crucial steps and certainly constitute a significant challenge for synthetic chemists. A resourceful synthetic approach was taken by Crimmins and co-workers, who made use of the enone–furan 138 as the key building block in the total synthesis of ginkgolide B (4, Scheme 27).42,129 The important photocycloaddition substrate 138 was prepared using a previously established homoenolate approach for the construction of carboalkoxycyclopentenones.135,136 Reacting the acetylenic ester 137 with the appropriate zinc–copper homoenolate gave the photosubstrate 138 in 82% yield. Subsequent irradiation of enone 138 in hexanes resulted in the formation of the tetracyclic photoadduct 139 in quantitative yield and in >98:2 diastereoselectivity, thus establishing the two quaternary centers of the ginkgolide skeleton. Conversion of cyclobutane 139 to the bridged lactone 140, accompanied by treatment with dimethyldioxirane, provided bis-hemiacetal 141, which was further transformed to lactone 142. Acid-catalyzed methanolysis of lactone 142 facilitated ring closure to construct the E ring of the ginkgolides, affording the pentacyclic lactone 143 in 88% yield. Finally, ring closure and additional functionalization of the F ring provided ginkgolide B (4).42,129
Scheme 27. Total Synthesis of Ginkgolide B (4) via Stereoselective Intramolecular [2 + 2] Photocycloaddition and Cyclobutane Ring-Opening Methodology.

CSA = camphorsulfonic acid. TES = triethylsilyl. p-TsOH = p-toluenesulfonic acid; HMPA = hexamethylphosphoric triamide.
Ring expansion by use of the Cargill rearrangement137−141 is an attractive way to produce bridged ketones from cyclobutenyl ketones (Scheme 28). Pirrung took advantage of this rearrangement in the synthesis of (±)-isocomene (63) (Scheme 29). Intramolecular [2 + 2] photocycloaddition of 144 generated tricyclo[6.3.0.01,6]undecanone 145. Subsequent Cargill rearrangement of cycloadduct 145 afforded the two bicyclo[3.3.0]octane and bicyclo[3.2.1]octane products 146 and 147 in a 1:5 ratio. Finally, bicyclo[3.2.1]octane 147 was transformed to (±)-isocomene via acid-catalyzed rearrangement.142,143 Additional routes to (±)-isocomene (63) include the use of the Paternò–Büchi reaction,144,145 the meta-photocycloaddition,146 and the oxa-di-π-rearrangement,147 and they will be discussed in more detail in the upcoming sections.
Scheme 28. Ring Expansion of Cyclobutanes via Cargill Rearrangement.
Scheme 29. Pirrung’s Approach to (±)-Isocomene (63).
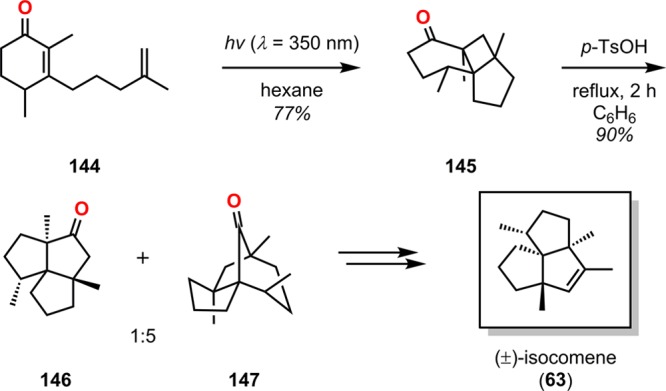
p-TsOH = p-toluenesulfonic acid.
The [2 + 2] photocycloaddition can also be accompanied by a thermally induced ring opening. This route was exploited by Schreiber and Santini in the synthesis of the sex pheromone periplanone-B (154, Scheme 30).148−150 The synthesis commenced with a [2 + 2] photocycloaddition of allene (43) and alkene 148 to afford a mixture of the syn and anti head-to-head photoproduct 149. It was revealed that the diastereomeric mixture converged to the same end product; thus, addition of vinylmagnesium bromide provided a mixture of allyl carbinols 150. Subsequent anion-accelerated oxy-Cope rearrangement followed by electrocyclic ring opening produced a mixture of s-cis diene 152 and s-trans diene 153. Cis/trans photoisomerization of the dienes showed a photostationary equilibrium in a 15:1 ratio with the s-trans isomer 153 being favored, which could be converted to periplanone B (154).
Scheme 30. [2 + 2] Photocycloaddition with Subsequent Thermal Ring Opening in the Synthesis of Periplanone B (154).

An intramolecular aza-Prins reaction with a guided fragmentation of a cyclobutylcarbinyl cation was used as the key step for assembling the bicyclo[3.3.1]nonene core of the nootropic alkaloid (−)-huperzine A (160).151 Here, the necessary photoadduct 156, generated from intramolecular [2 + 2] photocycloaddition in 58% yield from 155, was converted to cyclohexanone 157. Condensation of ketone 157 with NH2CO2Me in the presence of p-TsOH initiated the aza-Prins reation sequence, presumably via carbamate 158, to afford the desired fragmentation product 159, which is a known precursor to (−)-huperzine A (160) (Scheme 31).
Scheme 31. Synthesis of the Nootropic Alkaloid (−)-Huperzine A (160) Using a [2 + 2] Photocycloaddition/Cyclobutane Fragmentation Sequence through an Aza-Prins Reaction.

p-TsOH = p-toluenesulfonic acid.
It is clear that the wide range of natural products that can be accessed by the [2 + 2] photocycloaddition makes the reaction a highly useful tool in total synthesis. The prominent structures that have successfully been accessed utilizing a [2 + 2] photocycloaddition strategy are frameworks with multiple stereocenters and quaternary carbon centers. The ability to create several C–C bonds in hindered environments, in a single transformation, highlights the powerful features of the [2 + 2] photocycloaddition. The accessible fragmentation strategies that can subsequently be used allow for creative and effective construction of a multitude of complex natural products and their derivatives.
2.2. [2 + 2] Photocycloadditions between Olefins and Carbonyls—Oxetane Synthesis through the Paternò–Büchi Reaction
The [2 + 2] photocycloaddition of an alkene and a carbonyl compound—the Paternò–Büchi reaction—is a powerful method for construction of oxetanes, which are versatile intermediates in organic synthesis and occurring motifs in pharmaceutical compounds as well as in natural products (Figure 9).152,153 In these reactions the carbonyl motif usually serves as the light-absorbing species. Excitation of the nπ* state thus results in a singlet state that readily undergoes ISC to the triplet state, from which a majority of the Paternò–Büchi reactions occur. The diastereoselectivity is dictated by the lifetimes of the generated triplet 1,4-biradicals, as a consequence of the stepwise mechanism, and is connected with the mode of spin inversion events that allows for closed-shell products to be formed (Scheme 32). The regioselectivity can usually be predicted by formation of the most stable biradical upon addition of the carbonyl compound to the alkene moiety.154−159 A requirement for the Paternò–Büchi reaction to occur is that the alkene cannot have a lower triplet energy state than the carbonyl entity; if this would be the case, energy transfer is the major reaction pathway, with oxetane formation occurring slowly.160−165
Figure 9.
Compounds containing the oxetane motif.
Scheme 32. Illustration of the Paternò–Büchi Reaction.
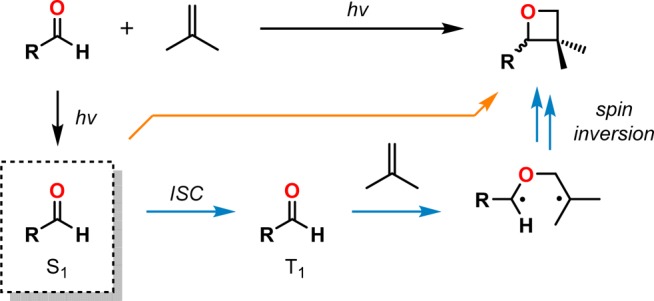
ISC = intersystem crossing.
Although the Paternò–Büchi reaction has been less frequently employed in natural product synthesis than the cyclobutane [2 + 2] photocycloaddition reaction, it has for example been utilized in the synthesis of (+)-preussin (167, Scheme 33),166 (±)-oxetanocin (171, Scheme 34),167 and (±)-1,13-herbertendiol (176, Scheme 35).168
Scheme 33. Total Synthesis of the Pyrrolidinol Alkaloid (+)-Preussin (167) by a Diastereoselective Paternò–Büchi Approach.

Scheme 34. Synthesis of (±)-Oxetanocin (171) Using the Paternò–Büchi Reaction.

Scheme 35. Synthesis of (±)-1,13-Herbertendiol (176).
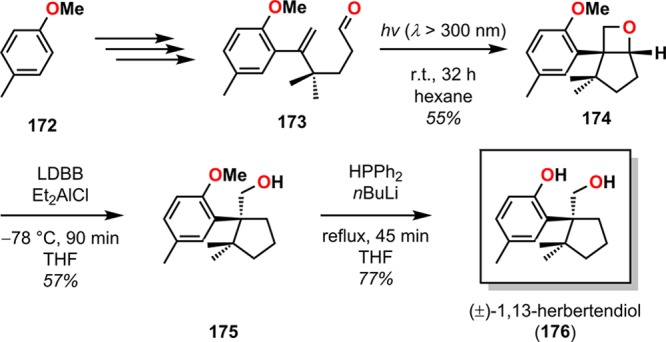
LDBB = lithium di-tert-butylbiphenylide.
An intramolecular version of the Paternò–Büchi reaction was applied by Greaney and co-workers in a six-step approach to the tetracyclic core intermediate 184 of merrilactone A (106).169 Starting from TBS-protected 3-hydroxycyclopentenone (177), nucleophilic addition of 178 afforded alcohol 179, which was subsequently desilylated and oxidized to generate hydroxy enone 180. A domino oxy/carbopalladation reaction with Pd(OAc)2 produced the bicyclic acetal 182 as a 1:1 mixture of diastereoisomers. Oxidative cleavage of alkene 182 gave ketone 183, which upon irradiation underwent [2 + 2] photocycloaddition to the tetracyclic oxetane 184 in 93% yield (Scheme 36). In addition to creating two new rings, the intramolecular Paternò–Büchi reaction also establishes three stereocenters, thus forming the oxa[3.3.3]propellane motif present in merrilactone A (106).
Scheme 36. Intramolecular Paternò–Büchi Reaction To Produce Oxetane 184 Bearing the Tetracyclic Core of Merrilactone A (106).

PDC = pyridinium dichromate. TBAF = tetra-n-butylammonium fluoride.
Rawal and co-workers also utilized an intramolecular Paternò–Büchi reaction as the key step in the stereocontrolled syntheses of the angular triquinane (±)-isocomene (63).144,145 It was envisioned that diquinane enone 192, which has three of the stereocenters correctly in place, would be a key intermediate in the synthesis. The synthesis of the desired methyl ketone substrate 188 for the Paternò–Büchi reaction was synthesized from norbornene 185 by alkylation with the methoxymethyl (MOM) ether of 3-iodopropan-1-ol (186) followed by reaction with dimsyl lithium and Zn. Irradiation of methyl ketone 188 produced the desired oxetane 189 in 92% yield, which was subsequently cleaved using an excess of iPr2NMgI. Oxidation of the resulting homoallylic alcohol under Swern conditions170 provided fragmentation precursor 190. Treatment of ketone 190 with lithium di-tert-butylbiphenylide (LDBB)171,172 as a one-electron reducing agent yielded the desired reductive fragmentation product 191, which was subsequently methylated at the bridgehead position and converted to diquinane 192. Treatment of iodide 192 with nBuLi allowed for anionic cyclization and in situ trapping of the resulting enolate to afford enol triflate 194, which upon treatment with Me2CuLi gave (±)-isocomene (63) (Scheme 37). The synthesis of (±)-isocomene (63) illustrates the advantages of the Paternò–Büchi photocycloaddition–reductive fragmentation strategy for access to intricate triquinane-based natural products.
Scheme 37. Rawal’s Synthesis of (±)-Isocomene (63) By an Intramolecular Paternò–Büchi Reaction.

LDA = lithium diisopropylamide. LDBB = lithium 4,4′-di-tert-butylbiphenylide. MOM = methoxymethyl. DMPU = N,N′-dimethylpropyleneurea.
(−)-Sarracenin (202) is a tricyclic highly oxygenated monoterpene originally isolated from Sarracenia flava.173 In the synthesis of (±)-sarracenin, Hoye and Richardson employed the Paternò–Büchi photocycloaddition of cyclopentadiene (195) and acetaldehyde (196) to access the exo diastereomeric oxetane 197. Acid-catalyzed methanolysis followed by exposure to TsCl in pyridine allowed for oxetane opening and formation of tosylate 198. Treating tosylate 198 with KOtBu and dimethyl β-styrenylmalonate (199) provided malonate 200, which underwent decarbomethoxylation followed by demethylation to yield alcohol 201. Methanolic ozonolysis followed by reductive workup with dimethyl sulfide (DMS) and subsequent acetic acid treatment produced (±)-sarracenin (202) in a nine-step sequence in an overall yield of 2% (Scheme 38).174
Scheme 38. Access to (±)-Sarracenin (202) Using a Paternò–Büchi Reaction.

CSA = camphorsulfonic acid. DMS = dimethyl sulfide. TsCl = p-toluenesulfonyl chloride.
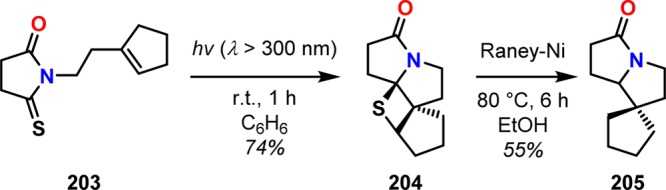
The Paternò–Büchi reaction can also be carried out with thiocarbonyl compounds.175 An intramolecular Paternò–Büchi reaction has for example been reported for the synthesis of spirocyclic pyrrolizinone 205.176 Here, irradiation of thioxasuccinimide system 203 afforded the tetracyclic thietane 204 as the major photoproduct in 74% yield. Reduction of photoproduct 204 with Raney-Ni gave the spiro pyrrolizinone 205 in 55% yield (Scheme 39), showing that carrying out the Paternò–Büchi with thiocarbonyl compounds can be used to access complex assemblies.
Scheme 39. Intramolecular Paternò–Büchi Reaction of Thiocarbonyl 203.
2.3. [2 + 2] Photocycloadditions Followed by Retro-Aldol Reaction—The De Mayo Reaction
An important contribution to the field of photochemistry was made in 1962 by De Mayo and co-workers when they observed that irradiation of alkenes with acetylacetone afforded 1,5-diketones in good yields. Subjecting the produced 1,5-diketones to catalytic amounts of acid or base subsequently led to cyclization to give cyclohexenones (Scheme 40).177 The first step in the De Mayo reaction involves tautomerization of the 1,3-dicarbonyl compound to the enol. For β-diketone 212 this affords the corresponding keto enol 213, which allows for [2 + 2] photocycloaddition with alkene 214 and thus generates a β-acylcyclobutanol (215). Subsequent retro-aldol condensation of the formed cyclobutanols produces the 1,5-dicarbonyl species 216 (Scheme 41), which can be further transformed into cyclooctadiones (see Scheme 42) and cyclohexenones.178,179 The poor regioselectivity that is sometimes observed in the De Mayo reaction can be explained by formation of a triplet 1,4-biradical from the keto enol reaction partner, which needs to undergo spin inversion to a singlet biradical for cyclobutane formation to occur (cf. Schemes 2 and 32).180,181
Scheme 40. Initial Photochemical Experiments Performed by De Mayo and Co-Workers.

Scheme 41. Depiction of the De Mayo Reaction.

Scheme 42. Formation of 1,5-Cyclooctadione 221 Using the De Mayo Reaction182.

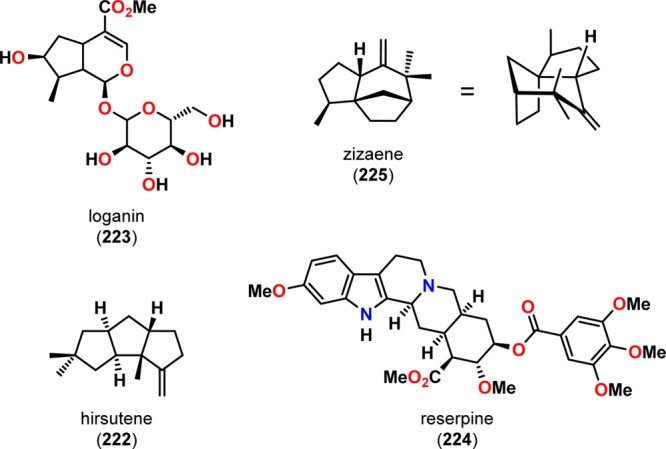
The synthetic utility of the De Mayo reaction in natural product synthesis has been readily explored and early examples include hirsutene (222),183,184 loganin (223),185−187 reserpine (224),188,189 and zizaene (225) (Figure 10).190,191 Another classical example employing an intramolecular [2 + 2] photocycloaddition followed by a retro-aldol reaction was reported in the total synthesis of (±)-longifolene (232),192,193 a tricyclic sesquiterpene containing a bicyclo[5.4.0]undecane scaffold.194−201 Here, the design of the complex carbon skeleton of this sesquiterpene was centered on a photoaddition/retro-aldolization sequence. Irradiation of benzyloxycarbonyl derivative 228 afforded cyclobutane 229 regioselectively, which upon hydrogenolysis underwent retro-aldol cleavage to give the bicyclo[5.4.0]undecane core. Subsequent functionalization of bicyclo[5.4.0]undecane 230 through a regioselective Wittig reaction/Simmons–Smith cyclopropanation sequence, followed by additional manipulations, produced (±)-longifolene (232) in ∼25% overall yield (Scheme 43).192,193 The expedient construction of the sesquiterpene (±)-longifolene (232) exemplifies the synthetic utility of the intramolecular De Mayo reaction in natural product synthesis.
Figure 10.
Examples of natural products accessed using the De Mayo reaction.
Scheme 43. Use of a Photocycloaddition/Retro-Aldol Sequence (De Mayo Reaction) in the Total Synthesis of (±)-Longifolene (232).

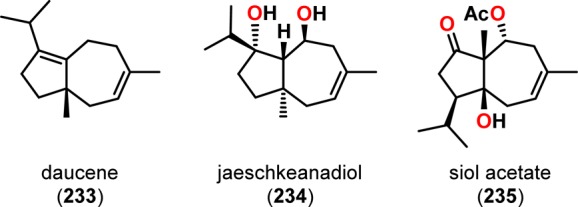
Daucene (233) is a member of the carotane-type sesquiterpenes and has an unusual hydroazulene skeleton. The daucane (carotane) sesquiterpenes all share a bicyclo[5.3.0]decane core with diverse functionality and varied degrees of oxidation (Figure 11) and have been shown to exhibit diverse biological activities.202−205 During the years, several total syntheses of daucene (233) have been reported.206−211 Seto and co-workers employed a highly regioselective intramolecular [2 + 2] photocycloaddition followed by a retro-aldol reaction to access a variety of carotane-type terpenes, including (±)-daucene.212 Irradiation of enone 239 resulted in efficient and regioselective photocyclization to afford photoproduct 240 in 88% yield (Scheme 44). Subsequent alkaline hydrolysis of the acetate group in photoproduct 240 resulted in retro-aldolization and ring expansion to provide the tricyclic ketol 242, which was subsequently transformed to monotosylate 243. Treating 243 with iPrLi initiated Grob fragmentation, followed by alkylation of the produced ketone to furnish the tertiary alcohol 244. Subjecting alcohol 244 to HCO2H at room temperature provided the dehydrated product (±)-daucene (233) in 28% yield together with two other regioisomeric products.
Figure 11.
Representative examples of daucane (carotane) sesquiterpenes.
Scheme 44. Synthesis of (±)-Daucene (233) by Use of the De Mayo Reaction.

The direct use of β-keto esters and β-keto acids in photocycloadditions can generate oxetanes through the Paternò–Büchi reaction (vide infra) instead of the desired cyclobutane. However, the use of dioxenones as β-keto ester and β-keto acid surrogates circumvents this problem by covalently locking the 1,3-dicarbonyl compound in the enol form and was originally reported by Baldwin and Wilkinson.213 Dioxenones have, after this observation, been applied to a number of natural product syntheses, such as the tricyclic skeleton of taxane diterpenes (Scheme 45).214,215
Scheme 45. Construction of the Taxane Skeleton (247) by Use of an Intramolecular Dioxenone Photocycloaddition–Fragmentation Strategy.

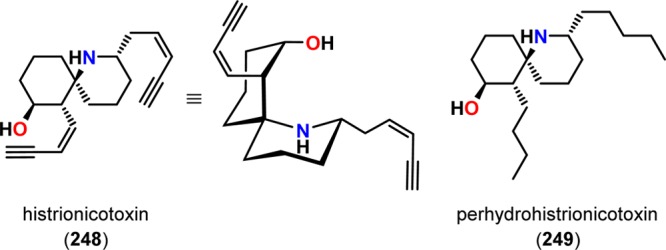
Another example of employing dioxenones can be found in the synthesis of perhydrohistrionicotoxin (249). Histrionicotoxin (248) and its derivatives (Figure 12) are powerful neurotoxic alkaloids initially isolated from the Columbian frog Dendrobates histrionicus.216−218 Initial attempts of synthesizing perhydrohistrionicotoxin (249) by Smith and Koft employed a [2 + 2] photocycloaddition methodology as the key step for assembling the core of perhydrohistrionicotoxin (249).219 Here, the cyclobutene fragment 251 was recognized as a potential intermediate in the synthesis of the natural product. Unfortunately, the outlined synthetic approach by the authors was not viable for converting the generated cyclobutene photoproduct 251 to perhydrohistrionicotoxin (249) (Scheme 46). However, a few years later, Winkler and co-workers reported the total synthesis of (−)-perhydrohistrionicotoxin, where the absolute configuration was derived from l-glutamic acid (253).220 For establishing the relative configuration, the authors employed an intramolecular De Mayo reaction between a vinylogous amide and a dioxenone to afford the necessary keto-lactone intermediate 257, which could be converted to (−)-perhydrohistrionicotoxin (249) (Scheme 47).
Figure 12.
Structures of histrionicotoxin (248) and perhydrohistrionicotoxin (249).
Scheme 46. Unsuccessful Approach toward the Synthesis of Perhydrohistrionicotoxin (249) via Cyclobutene Intermediate 251.

DMAP = 4-dimethylaminopyridine.
Scheme 47. Fragmentation of Photoproduct 255 en Route to the Total Synthesis of (−)-Perhydrohistrionicotoxin (249).

A related fragmentation strategy employing dioxenones has also been employed in the total synthesis of (±)-saudin (267, Scheme 48).221,222 Here, irradiation of dioxenone 261 afforded photoadduct 262 as a single diastereomer in 80% yield. Introduction of the furan motif was accomplished in a two-step sequence by first reacting 262 with nBuLi and Tf2O in the presence of TMEDA to produce enol triflate 263. Subsequent Stille coupling of enol triflate 263 with 3-furyltributylstannane (264) gave the furyl enol ether 265 in almost quantitative yield, which upon exposure to LiOH resulted in fragmentation. Cyclization to (±)-saudin (267) was carried out by treating the fragmented product with pyridinium tosylate. This 15-step route afforded (±)-saudin (267) in 5% overall yield and illustrates the potential of the intramolecular dioxenone photocycloaddition for the efficient construction of complex carbocyclic motifs.
Scheme 48. Access to (±)-Saudin (267) by Means of a [2 + 2] Photocycloaddition and Subsequent Retro-Aldol Reaction.

PPTS = pyridinium p-toluenesulfonate. Tf2O = trifluoromethanesulfonic anhydride. TMEDA = N,N,N′,N′-tetramethylethylenediamine.
A related retro-aldol approach was also utilized by Winkler and co-workers in the first total synthesis of (±)-ingenol (2, Figure 14).38 Ingenol (2) is a complex diterpenoid originally isolated from Euphorbia ingens in 1968 by Hecker.223 However, its structure was not determined until 1970, when the crystal structure of the triacetate compound was derived.224 The Euphorbia species contains a variety of diterpenoids with complex carbon skeletons, some of which are shown in Figure 13.225−229 Of the various diterpenoids, the ingenanes display several interesting biological activities, such as antileukemic and anti-HIV.230−233 In particular, ingenol 3-angelate (ingenol mebutate, 274, Figure 14) has received a great deal of attention since its approval by the Food and Drug Administration (FDA) in 2012 for treatment of actinic keratosis, a precancerous skin condition.234−236
Figure 14.
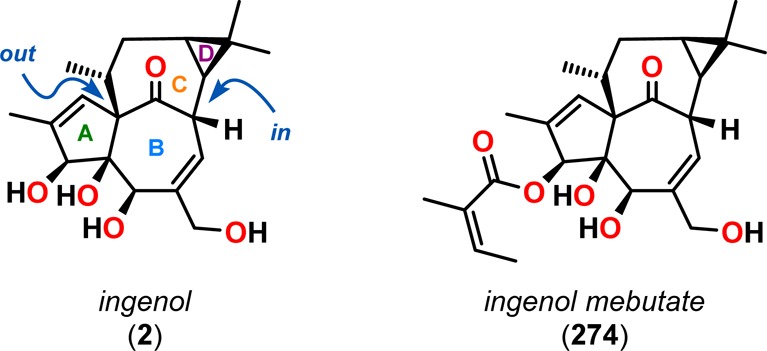
Structures of ingenol (2) and FDA approved ingenol mebutate (274).
Figure 13.
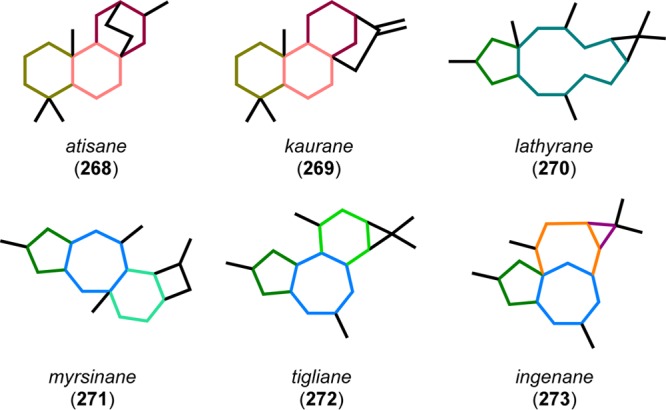
Skeletal types of Euphorbia diterpenes.
The biological activity and the structural complexity of ingenol (2) have motivated interest from synthetic organic chemists for several decades. The polyoxygenated tetracyclic core of ingenol (2) features several challenges, including the stereogenic triol unit of the A and B rings and a quaternary carbon stereocenter. However, the unusual in/out stereochemistry,237 or trans intrabridgehead stereochemical configuration, of the bicyclo[4.4.1]undecane motif represents a particularly daunting difficulty.238−250 In light of these challenges, a number of total syntheses of ingenol (2) have been reported.38,251−261
In the first total synthesis of ingenol (2),38 Winkler and co-workers established the rare and challenging trans intrabridgehead configuration of the BC ring system by employing inside–outside stereoisomerism methodologies previously developed in their laboratory.262 Initial attempts to use the De Mayo reaction on allylic alcohol 279, obtained from the bicyclic enone 275, resulted in a low yield (16%) of the desired photoadduct. However, irradiation of the corresponding allylic chloride (280) afforded photoadduct 281 with the important bicyclo[5.3.0]decane motif in 60% yield. Fragmentation of cyclobutane 281 with methanolic potassium carbonate followed by LiAlH4 reduction, chloride elimination, and silylation of the primary alcohol gave bicyclo[4.4.1]undecanone 282 in 35% yield over four steps. Cyclopropanation and reductive methylation subsequently produced cyclopropane 283, which allowed for further manipulations into (±)-ingenol (2) (Scheme 49).38
Scheme 49. Application of the De Mayo Reaction in the First Total Synthesis of (±)-Ingenol (2).

LDA = lithium diisopropylamide. p-MBOH = p-methoxybenzyl alcohol. TFA = trifluoroacetic acid. TFAA = trifluoroacetic anhydride. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
Modifications of the De Mayo reaction have also been developed and have for example been utilized in the synthesis of (+)-aphanamol I (291)75 and for constructing the 3,12-dioxatricyclo[8.2.1.06,13]tridecane skeleton of terpenoid natural products.263 Hansson and Wickberg’s strategy for accessing (+)-aphanamol I (291) depended upon a De Mayo-type sequence where the opening of the generated cyclobutane ring was achieved through a base-induced opening with concomitant β-elimination of a properly oriented oxirane.75 Here, the essential photosubstrate 286 was derived from (+)-(R)-limonene (284). Irradiation of photosubstrate 286 and cyclopentenone 287 afforded a 1:1 mixture of the regioisomeric photoadducts 288 and 289. Reacting photoadduct 289 with dimethyloxosulfonium methylide yielded the endo-epoxide 290, which upon alkaline hydrolysis by refluxing with LiOMe in MeOH produced (+)-aphanamol I (291) in 70% yield through fragmentation of the intermediate γ,δ-epoxy alcohol (Scheme 50).
Bach and co-workers explored an unusual fragmentation pathway for constructing 3,12-dioxatricyclo[8.2.1.06,13]tridecane skeletons.263 [2 + 2] Photocycloadditions of tetronate 292 gave photoproduct 293 in 65% yield as a single diastereoisomer. Facile ring expansion occurred upon treatment with KOH in aqueous MeOH to give the seven-membered ketolactone 295 via the intermediate tricyclic hemiacetal 294 (Scheme 51). The ring strain present in the tetracyclic compound 293 apparently facilitates nucleophilic substitution by hydroxide, thus resulting in cleavage of the cyclobutane ring.
Scheme 51. Hydroxide-Mediated Ring Expansion of [2 + 2] Photocycloaddition Product 293.

Recent work by Minter and Winslow involves the use of an unusual De Mayo approach for constructing the tetracyclic galanthan ring system (296, Figure 15),264 which is the core of the lycorine-type Amaryllidaceae alkaloids.265,266 Minter and Winslow utilized photosubstrate 305, synthesized in three steps from isocarbostyril (300), as the precursor to the galanthan skeleton. Irradiation of the De Mayo substrate 305 afforded a single product, which was shown to be photoproduct 307 with the correct cis stereochemistry of the vicinal tertiary ring protons. Subsequent ring closure was achieved under basic conditions using piperidine in refluxing benzene to afford the galanthan derivative 309 in five steps with 35% overall yield (Scheme 52).
Figure 15.

Structure of the galanthan skeleton (296) and examples of Amaryllidaceae alkaloids.
Scheme 52. Synthesis of Tetracyclic Compound 309 Bearing the Galanthan Skeleton.

p-TsOH = p-toluenesulfonic acid.
2.4. [2 + 2] Photocycloadditions of Vinylogous Amides—Formation of Nitrogen-Containing Ring Systems
In analogy to the De Mayo reaction, which utilizes 1,3-dicarbonyl compounds, [2 + 2] photocycloaddition adducts of vinylogous amides (enaminones) can undergo subsequent fragmentation. The fragmentation gives ketoimines or ketoiminium ions, which can be recyclized in a domino sequence. This photocycloaddition/retro-Mannich fragmentation sequence provides a powerful route for the construction of nitrogen-containing ring systems and has since its initial observation by Tamura and co-workers267 (Scheme 53) been exploited in natural product synthesis.268
Scheme 53. Tamura and Co-Workers’ Initial Observation of the Intramolecular Photocycloaddition/Retro-Mannich Fragmentation Sequence of Vinylogous Amides.

Similarly to Tamura and co-workers’ initial work, Schell and Cook investigated the use of secondary vinylogous amides. Irradiation of the secondary vinylogous amide 313 afforded ketoimine 315, which is presumably generated from cyclobutane intermediate 314 upon retro-Mannich fragmentation (Scheme 54).269 Pete and co-workers employed the intramolecular [2 + 2] photocycloaddition of N-alkenoyl β-enaminones to produce valuable synthons for synthesis of triquinanes or various sesquiterpenes (Scheme 55).270 Related photocycloaddition/retro-Mannich sequences have also been reported for 3-aminocyclopentenones271 and for the synthesis of 6-aza-bicyclo[3,2,1]octan-3-ones272 and pyrroles.273,274
Scheme 54. Intramolecular Photocycloaddition/Retro-Mannich Fragmentation of Secondary Vinylogous Amide 313.

Scheme 55. Intramolecular [2 + 2] Photocycloaddition of N-Alkenoyl β-Enaminones with Subsequent Acid- or Trimethylsilyl Iodide-Catalyzed Cyclobutane Ring Opening.

Examples of the application of the photocycloaddition/retro-Mannich reaction can be found in studies toward the synthesis of taxane skeletons by Swindell and co-workers.275−278 Photocycloaddition of vinylogous amide 321 generated photoproduct 322, which was converted to silyloxy ketone 323 through a Rubottom-type oxidation. Subsequent fragmentation and additional manipulations converted silyloxy ketone 323 to enone 324 having the basic BC subunit of the taxane diterpenoids (Scheme 56).
Scheme 56. Use of the Photocycloaddition/Retro-Mannich Sequence for the Construction of the BC Substructure of Taxanes.

TBS = tert-butyldimethylsilyl. m-CPBA = m-chloroperoxybenzoic acid.
The intramolecular photocycloaddition of vinylogous amides has also been exploited in the Sceletium alkaloid279 mesembrine (328) by Winkler and co-workers.280 Similarly to the alkaloids pretazettine (326) and sceletium A-4 (327), mesembrine (328) also features a functionalized cis-3a-aryloctahydroindole core (Figure 16). The desired photosubstrate 330 was accessible from veratrole (329) in four steps. Intramolecular photocycloaddition of 330 led to the photocycloaddition/reto-Mannich product 332 in 74% yield. Subsequent methylation using Me3OBF4 followed by treatment with DMAP facilitated ring closure, affording mesembrine (328) in 84% yield (Scheme 57).280 The efficient access to the alkaloid mesembrine (328) in merely seven steps with an overall yield of 33% from veratrole (329) illustrates the power of the synthetic utility of the photocycloaddition/retro-Mannich fragmentation/Mannich closure cascade sequence.
Figure 16.
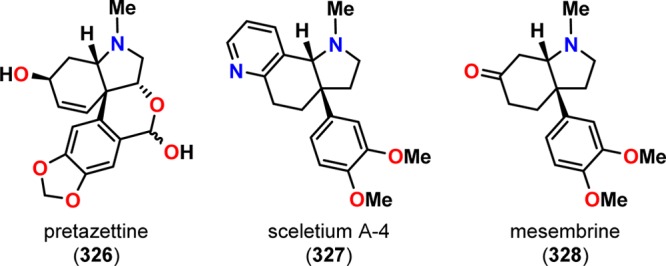
Examples of alkaloids containing the cis-3a-aryloctahydroindole motif.
Scheme 57. Synthesis of Mesembrine (328) via the Intramolecular Photocycloaddition of Vinylogous Amide 330.

DMAP = 4-dimethylaminopyridine.
White and Ihle have employed β-aminoalkylidene malonates for accessing spiropyrrolines and the tetracyclic core of the indolenine alkaloid koumine (339).281 The strategy for assembling the nonindolenine fragment 338 of koumine (339) was believed to be accessible through a photocycloaddition/retro-Mannich approach from β-aminoalkylidene malonate 333. Irradiation of malonate 333 indeed furnished cyclobutane 334, which upon removal of the Boc group caused spontaneous retro-Mannich fragmentation to spiroimine 335. Methylation of the imine nitrogen and subsequent treatment with MeNH2 triggered initial aminal formation, followed by intramolecular acylation to give lactam 337, which could be converted to ketone 338, a precursor for koumine (339) (Scheme 58). A related [2 + 2] photocycloaddition/retro-Mannich fission approach for tryptamine-based vinylogous amides was also employed by White and co-workers in the total syntheses of (±)-coerulescine (340), (±)-horsfiline (341), and (±)-elacomine (342) (Figure 17).282
Scheme 58. Photocycloaddition/Retro-Mannich Approach for Construction of the Koumine Core.

Boc = tert-butyloxycarbonyl. TFA = trifluoroacetic acid.
Figure 17.
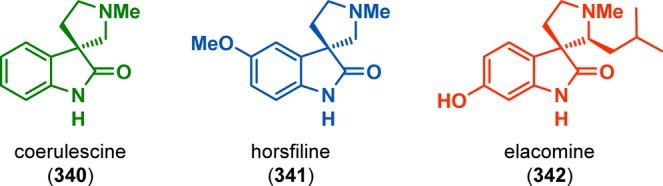
Synthesis of the spiro[pyrrolidine-3,3′-oxindole] alkaloids coerulescine (340), horsfiline (341), and elacomine (342) by use of the [2 + 2] photocycloaddition/retro-Mannich fragmentation process.
The formal synthesis of vindorosine (349), starting from l-tryptophan (343), is another example where the intramolecular vinylogous amide photocycloaddition/retro-Mannich fragmentation/Mannich closure sequence has been applied for construction of nitrogen-containing ring systems (Scheme 59).283 By starting from l-tryptophan (343), Winkler and co-workers showed that photosubstrate 344 afforded the highest stereoselectivity in photocycloaddition, providing photoproduct 346 in 91% yield as a single diastereomer. Treatment of keto imine 346 with LDA followed by TBSOTf and nBu4NF produced the desired tetracyclic ketone 347 in 51% yield. The latter intermediate could be converted to 348, a precursor to vindorosine (349), highlighting the efficiency of the vinylogous amide photocycloaddition approach to the synthesis of the aspidosperma alkaloids.
Scheme 59. Formal Synthesis of the Aspidosperma Alkaloid Vindorosine (349) by Photocycloaddition/Retro-Mannich Fragmentation.

Cbz = carboxybenzyl. LDA = lithium diisopropylamide. TBS = tert-butyldimethylsilyl.
The application of this methodology to the construction of more complex alkaloids is exemplified by Winkler and co-workers in the syntheses of the marine alkaloids ircinol (350), ircinal (351), manzamine A (352), and manzamine D (353) (Figure 18).284,285 [2 + 2] Photocycloaddition of vinylogous amide 354 afforded aminal 357. Exposure of aminal 357 to pyridinium acetate produced manzamine tetracycle 358 in 20% yield as a single stereoisomer from photosubstrate 354. Macrocyclization to produce the 13-membered D ring and subsequent manipulations allowed for conversion of precursor 358 to the four marine alkaloids ircinol (350), ircinal (351), manzamine A (352), and manzamine D (353) (Scheme 60). The establishment of all of the stereogenic centers in the manzamine skeleton from the single stereogenic center present in photosubstrate 354 validates the remarkable levels of stereochemical control that are possible using the photocycloaddition/fragmentation/Mannich closure cascade sequence in organic synthesis.
Figure 18.

Structures of the marine alkaloids ircinol A (350), ircinal A (351), manzamine A (352), and manzamine D (353).
Scheme 60. Photocycloaddition/Fragmentation/Mannich Closure to Ketone 358, a Precursor to Ircinol A (350), Ircinal A (351), Manzamine A (352), and Manzamine D (353).

Boc = tert-butyloxycarbonyl.
3. [3 + 2] Photocycloadditions
Apart from the meta-photocycloaddition reaction that is discussed in section 7, [3 + 2] photocycloadditions have been rarely applied in natural product synthesis. Several structurally interesting natural products have been isolated from the plant genus Aglaia (Figure 19).286−289 Porco and co-workers developed a biomimetic approach involving a [3 + 2] photocycloaddition/ketol shift rearrangement/reduction sequence using 3-hydroxyflavone and methyl cinnamate derivatives to access (−)-rocaglamide and related natural products.290−293 In the synthesis of methyl rocaglate (366), irradiation of 3-hydroxyflavone 362 produced oxidopyrylium intermediate 364, which undergoes a [3 + 2] photocycloaddition with methyl cinnamate (363) to furnish aglain core structure 365. Subsequent treatment with NaOMe/Me4NBH(OAc)3 was the basis for a ketol rearrangement/reduction sequence to provide methyl rocaglate (366) (Scheme 61).290,292 A related approach has also been employed to accomplish enantioselective photocycloaddition and was used in the asymmetric synthesis of rocaglamides.291,293
Figure 19.

Examples of natural products produced by the plant genus Aglaia.
Scheme 61. Synthesis of Methyl Rocaglate (366) Using a [3 + 2] Photocycloaddition/Ketol Shift Rearrangement/Reduction Sequence.

The groups of Porco and Stephenson have also reported a tandem dienone photorearrangement–cycloaddition reaction of cyclohexadienones tethered with various 2π and 4π functional groups that afforded polycyclic, bridged scaffolds.294 Photoirradiation of alkenyl ether-tethered cyclohexadienones, such as 367, resulted in the bridged tricyclic ketone 371, presumably via the photorearranged oxyallyl intermediate 368, which upon [3 + 2] photocycloaddition resulted in the observed product (Scheme 62). Using cyclohexadienone 372 containing a furfuryl alkyne ether tether led to the production of the polycyclic ketone 374 (Scheme 63). This tandem dienone photorearrangement/cycloaddition/[4 + 2] cycloaddition sequence generates four new bonds where the final hexacyclic framework 374 contains seven stereogenic centers, thus highlighting the complex architectures that can be created in this tandem process.
Scheme 62. Tandem Dienone Photorearrangement–Cycloaddition of Cyclohexadienone 367.

Scheme 63. Tandem Dienone Photorearrangement/Cycloaddition/[4 + 2] Cycloaddition To Afford Polycyclic Ketone 374.

4. Photochemical [4 + 2] Cycloadditions in Natural Product Synthesis
4.1. Photochemical Generation of Dienes—Combining Photoenolization with [4 + 2] Cycloadditions
o-Quinodimethanes are fundamental intermediates in total synthesis and are in general produced from o-alkyl-substituted aromatic carbonyl compounds.295−299 The high reactivity associated with the photochemically generated o-quinodimethanes can be utilized by subsequently trapping them in a Diels–Alder reaction (Figure 20).300−302
Figure 20.

Schematic depiction of the photoenolization/[4 + 2] cycloaddition sequence.
A classic example where the photochemically produced o-quinodimethanes subsequently underwent [4 + 2] cycloaddition can be found in the total synthesis of (+)-estrone (3) by Quinkert and co-workers. Here, photochemically triggered formation of dienol 380 from precursor 379 allowed for [4 + 2] cycloaddition to afford alcohol 381, which could subsequently be converted to estrone (3) via already known transformations (Scheme 64).303−305 Other early examples where the photoenolization/[4 + 2] cycloaddition sequence had been employed include the formal total synthesis of podophyllotoxin (385, Scheme 65)306 and the synthesis of 6-methylpretetramid (389, Scheme 66).307,308
Scheme 64. Application of the Photoenolization/[4 + 2] Cycloaddition Sequence in the Total Synthesis of Estrone (3).

Scheme 65. Tandem Photoenolization/Diels–Alder Reaction in the Synthesis of Podophyllotoxin (385).

Scheme 66. Photocyclization in the Total Synthesis of 6-Methylpretetramid (389).

The hamigerans are a family of natural products isolated from the poecilosclerid sponge Hamigera tarangaensis. The framework of hamigerans A–C (390–392) contains a substituted benzenoid core fused onto either a [4.3.0] or a [5.3.0] bicyclic ring system, three or four contiguous stereogenic centers, and an isopropyl group (Figure 21).309,310 In the total syntheses of hamigerans A (390) and B (391), Nicolaou and co-workers employed the photoinitiated [4 + 2] cycloaddition as the key step for assembling the hamigeran core.311−313 It was envisioned that incorporating an oxygen functionality at C6 would allow for the enantioselective synthesis of the targeted compounds. Irradiation of photosubstrate 394, derived from benzamide 393 as a 3:1 E/Z mixture, indeed triggered o-quinodimethane formation and afforded the tricyclic hydroxyl ester 396 in 92% yield as a mixture of C10 epimers (∼3:1; see Scheme 67). Subsequent epimerization at C5 and additional manipulations produced hamigeran A (390), which could be expediently converted to hamigeran B (391) in 82% yield through a Ba(OH)2-mediated cascade reaction involving saponification, decarboxylation, and auto-oxidation. A related photoenolization/Diels–Alder strategy was also applied by Nicolaou and Gray in the total synthesis of the lichen-derived antitumor agent hybocarpone (402) (Scheme 68) and analogues thereof.314
Figure 21.
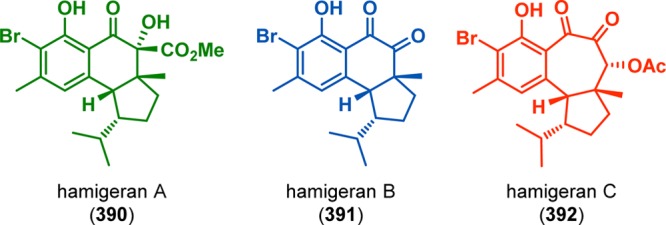
Structures of hamigerans A–C (390–392).
Scheme 67. Photochemical Generation and Diels–Alder Trapping of o-Quinodimethane 395 in the Total Synthesis of Hamigerans A (390) and B (391).

Scheme 68. Photoenolization/[4 + 2] Cycloaddition in the Total Synthesis of Hybocarpone (402).

A hetero-Diels–Alder reaction with the photochemically produced dienol 404 was utilized by Prabhakar and co-workers in the total synthesis of the alkaloid (±)-cis-alpinigenine (405).315,316 Here, photolysis of bis-aldehyde 403 led to the formation of at least seven photoproducts. The desired tetracyclic (±)-cis-alpinigenine (405) was presumably formed from o-quinodimethane intermediate 404, affording 405 in 20–30% yield (Scheme 69).
Scheme 69. Photolysis of Bis-aldehyde 403 for Generation of (±)-cis-Alpinigenine (405).

4.2. Photoisomerization Approaches for Triggering [4 + 2] Cycloadditions
The photoisomerization of cis-cycloalkenones to trans-cycloalkenones is a well-established procedure.317−323 An early example of this effect can be found in the photoisomerization of cis-2-cycloheptenone (406) to trans-2-cycloheptenone (407), where irradiation of a mixture of cis-2-cycloheptenone and excess cyclopentadiene (195) at −50 °C gave a single adduct (408) in 95% yield (Scheme 70).324 Intramolecular Diels–Alder trapping of photochemically generated trans-cyclic enones have also been reported as a rapid entry to complex polycyclic structures (Scheme 71).325
Scheme 70. Photoisomerization of cis-2-Cycloheptenone (406).
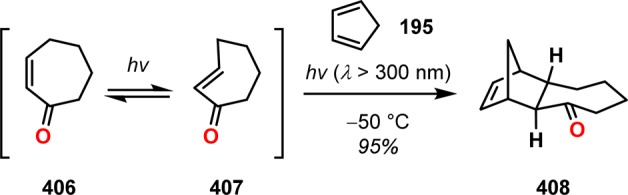
Scheme 71. Intramolecular Diels–Alder Reaction of Photochemically Generated trans-Cycloheptenone.
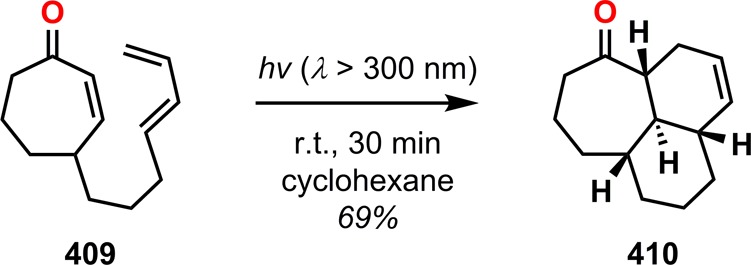
The diterpene vibsanin E (413) was initially isolated by Kawazu from the Japanese fish poison plant Viburnum odoratissimum and was among the first vibsane natural products to be isolated (Figure 22).326 Its highly oxygenated cycloheptane ring and the 3-oxatricyclo[6.3.2.05,10]tridecane core make vibsanin E (413) a considerable synthetic challenge.327,328 Davies and co-workers’ approach to (±)-5-epi-10-epivibsanin E (423) centered on three different cycloaddition steps: a Rh-catalyzed [4 + 3] cycloaddition, a heteronuclear [4 + 2] cycloaddition, and a photochemically induced [4 + 2] cycloaddition.329,330 The synthesis commenced with Rh-catalyzed [4 + 3] cycloaddition between diene 414 and vinyldiazoacetate 415 to afford cycloheptadiene 416 in 69% yield. The subsequent step involved reduction of ester 416 and oxidation of the resulting alcohol under Swern conditions to furnish aldehyde 417 in 90% yield. BF3·OEt2-assisted heteronuclear [4 + 2] cycloaddition of aldehyde 417 constructed the tricyclic vibsanin E core with the desired relative configuration. Further manipulations of tricycle 418 gave the conjugated ketone 419, which was shown to be resistant to a tandem conjugate addition/alkylation strategy that would have completed the synthesis of vibsanin E (413). Furthermore, thermal Diels–Alder reactions were also conducted but failed to give any product, highlighting the limited reactivity of 419 due to the sterically encumbered environment. An alternate strategy therefore had to be examined that increased the reactivity of enone 419. Intrigued by the possibility of exploiting a tandem photoisomerization/[4 + 2] cycloaddition sequence, the authors irradiated enone 419 in the presence of isoprene (421) to efficiently generate alkene 422 in 61% yield with the desired anti fusion across the enone moiety. This was further advanced to (±)-5-epi-10-epivibsanin E (423) (Scheme 72).
Figure 22.

Structures of vibsanin B (411), C (412), and E (413).
Scheme 72. Application of a Tandem Photochemical Isomerization/[4 + 2] Cycloaddition Sequence in the Total Synthesis of (±)-5-Epi-10-epivibsanin E (423).

DIBAL-H = diisobutylaluminum hydride.
Baldwin and co-workers employed the photoisomerization of (E,E,E,E)-tetraene 424 to produce the (E,E,E,Z)-tetraene intermediate 425. Tetraene 425 undergoes a radical cascade, ultimately revealing compound 427 (Scheme 73), which contains the crispatene core,331 similar to that reported by Padwa and co-workers in their studies of 1,3,5-hexatrienes.332,333 A recent example of the use of photoisomerization is encountered in Tang and co-workers’ synthesis of xanthanolide dimers.334 Xanthanolides are sesquiterpenoids with over a hundred members having been identified to date. A majority of the members in the xanthanolide family feature a butyrolactone moiety that is trans- or cis-fused to a seven-membered carbocycle, as in xanthatin (429, Figure 23).335−339 Although Tang and co-workers were not initially attempting to access dimeric products, they discovered that irradiation of xanthatin (429) under a N2 atmosphere led to a mixture of 3-epimogolide A (435) and mogolide A (436) in a combined yield of 74% (Scheme 74). The authors proposed that the reaction proceeded via photoinduced C1–C5 double bond isomerization to generate trans-cycloheptene 432, which rapidly undergoes Diels–Alder dimerization via 433 to produce the pentacyclic product 434. From intermediate 434, a subsequent [2 + 2] photocycloaddition gives the two dimerization products, 3-epimogolide A (435) and mogolide A (436).334 Although the photoisomerization strategy has not received significant attention in natural product synthesis, the examples discussed in this section highlight the synthetic utility of the photoisomerization of cis-cycloalkenes to trans-cycloalkenes to trigger subsequent reactions.
Scheme 73. Photoisomerization of (E,E,E,E)-Tetraene 424.

Figure 23.

Examples of monomeric and dimeric xanthanolides.
Scheme 74. Synthesis of 3-Epimogolide A (435) and Mogolide A (436).

5. Norrish-Type Photoreactions
The Norrish type I reaction refers to a photochemical reaction where the C–C bond between a carbonyl group and an α-carbon is homolytically cleaved. Several reaction modes are available for the generated 1,4-diradical and are dependent on the exact nature of the molecule (Scheme 75). One pathway involves decarbonylation with subsequent cyclization to furnish interesting cyclic products. Alternative pathways include formation of ketenes or aldehydes or a simple recombination of the 1,4-diradical, resulting in racemization of the α-carbon center.340−343
Scheme 75. Depiction of the Norrish Type I Reaction and the Possible Photoproducts That Can Be Generated.

The Norrish type II photoreaction involves an intramolecular γ-hydrogen abstraction of an excited carbonyl compound and was first reported by Ronald Norrish in the early 1930s.344−347 The primary 1,4-diradical that is formed can subsequently undergo further cleavage to produce alkenes and enols as the initial products (pathway A in Scheme 76). Alternatively, the produced 1,4-diradical can recombine to form cyclobutanols, a process that is called Norrish–Yang cyclization (pathway B in Scheme 76).348,349 The competition between fragmentation and cyclization is strongly dependent on the nature of the substituents.
Scheme 76. Depiction of the Norrish Type II Reaction Involving γ-Hydrogen Abstraction.

5.1. Norrish Type I Photoreactions
The utilization of the Norrish type I reaction can be found in the synthesis of α-cuparenone (439). Here, irradiation of diketone 438 produced α-cuparenone (439) in 85% yield via a decarbonylation/cyclization sequence (Scheme 77).350 A related approach was also used by the authors in the total synthesis of the sesquiterpene (±)-herbertenolide (442), where a Norrish type I reaction was employed to produce the essential cyclopentane core (Scheme 78).351
Scheme 77. Use of the Norrish Type I Reaction in the Synthesis of α-Cuparenone (439).

Scheme 78. Decarbonylation/Cyclization Sequence in the Total Synthesis of (±)-Herbertenolide (442).

In Nicolaou and co-workers’ studies on the hamigerans, a Norrish type I homolysis reaction was observed for hydroxy ketoester 443. Irradiation of a benzene solution containing hydroxy ketoester 443 produced an equilibrium mixture of C10 epimers. The authors speculated that this equilibration originates from a Norrish type I cleavage reaction of the C10–C11 bond, resulting in diradical 444. This diradical can either reclose, regenerating the hydroxyl ketoester 443, or undergo inversion prior to recombination to produce the C10 epimer 445 (Scheme 79). Furthermore, peculiar ring-contracted products were also derived from diketone 446. These ring-contraction products presumably proceed through a Norrish type I fragmentation, generating diradical 447. Subsequent expulsion of CO and intramolecular recombination of diradical 448 produces the observed ring-contracted product 449 (Scheme 80).312,313
Scheme 79. Photoinduced Epimerization of Hydroxy Ketoester 443 at C10 through Norrish Type I Fragmentation–Recombination.

Scheme 80. Norrish Type I Fragmentation of Diketone 446 Observed in the Total Syntheses of the Hamigerans.

Bicyclo[2.2.1]heptanones and related derivatives are prone to undergo Norrish type I cleavage. The resulting acyl radicals can subsequently abstract a hydrogen atom, leading to the formation of γ,δ-unsaturated aldehydes. Vandewalle and co-workers employed such an approach in the synthesis of (±)-boschnialactone (454).352 Irradiation of bicyclo[2.2.1]heptanone 450 promoted Norrish type I photoinduced α-cleavage to afford the 1,5-diradical 451. Subsequent hydrogen atom abstraction by the produced acyl radical gave the γ,δ-unsaturated aldehyde 452, which underwent facile ring closure to form lactol 453 in 90% yield. Oxidation with PCC followed by double bond hydrogenation using Pd/C gave (±)-boschnialactone 454 in 75% yield (Scheme 81). Additional applications of this photoinduced α-cleavage/hydrogen abstraction sequence can be found in the synthesis of (+)-juvabione (459, Scheme 82)353 and (±)-hop ether.354
Scheme 81. Norrish Type I Cleavage with Subsequent γ-Hydrogen Abstraction in Bicyclo[2.2.1]Heptanone 450.

PCC = pyridinium chlorochromate.
Scheme 82. Synthesis of (+)-Juvabione (459) through a Norrish Type I Fragmentation Methodology.

The formation of tetrahydrofuryl ethers upon irradiation of cyclobutanones in the presence of alcohols or other protic solvents is believed to proceed via initial formation of an oxacarbene species, which is subsequently inserted into the O–H bond.355−357 Molander and co-workers utilized this oxacarbene/insertion sequence in the synthesis of deacetoxyalcyonin acetate (465).358 The photochemical rearrangement of cyclobutanone 460 generated oxacarbene 461, which could be trapped with AcOH to afford the mixed acetal ring expansion product 462 in high yield and with complete retention of the stereocenter adjacent to the carbonyl. The mixed acetal 462 subsequently underwent a TiCl4-mediated [4 + 3] annulation at −80 °C to give the ester 464, which could be advanced to deacetoxyalcyonin acetate (465) (Scheme 83).
Scheme 83. Norrish Type I Cleavage with Subsequent Oxacarbene Trapping in the Total Synthesis of Deacetoxyalcyonin Acetate (465).

5.2. Norrish–Yang Cyclizations
(−)-Punctaporonin A (470), originally isolated from the fungus Poronia punctata, is a trihydroxylated tricyclic sesquiterpene containing a trans-cyclobutanol unit.359,360 For construction of the strained four-membered cyclobutanol scaffold, Sugimura and Paquette applied Norrish type II photochemistry.361 The synthesis commenced with diketone 466, which was converted to the essential photosubstrate 467. Irradiation of a benzene solution of ketone 467 generated diradical 468 via γ-hydrogen abstraction from the proximal isopropyl moiety, which produced the desired key Norrish–Yang cyclized product 469 in 49% yield along with approximately 20% of the Norrish β-fragmentation product (cf. Scheme 76). Removal of the SEM group and subsequent manipulations completed the synthesis and afforded (−)-punctaporonin A (470) (Scheme 84).
Scheme 84. Norrish–Yang Cyclization of Ketone 467 in the Total Synthesis of (−)-Punctaporonin A (470).

MOM = methoxymethyl. SEM = 2-(trimethylsilyl)ethoxymethyl.
Ouabain (471) and ouabagenin (472) belong to a class of steroids known as cardenolides and exhibit positive inotropic acitivity (Figure 24).362−364 The steroidal skeleton of ouabagenin (472), an aglycon of ouabain (471), possesses several characteristic features, such as cis-fused A/B and C/D rings, an angular hydroxyl unit at the C14 position, and a β-oriented butenolide group at C17, and poses a formidable synthetic challenge. In light of the need for a scalable and economically viable route to the cardenolides and derivatives thereof, Baran and co-workers’ synthesis commenced with cortisone acetate (473).365,366 Conversion of cortisone acetate (473) to adrenosterone with subsequent ketalization afforded ketone 474. Functionalization of the angular C19 methyl group was realized through a Norrish type II photochemical event (Scheme 85). Precedent for such a strategy can be found in the work of Jeger’s367 and Thomson’s368 groups on C19 functionalization of steroid scaffolds. Irradiation of ketone 474 efficiently provided the desired cyclobutanol 476 in 68% yield on a multigram scale after ring closure of diradical 475. Subsequent oxidative fragmentation employing N-iodosuccinimide as oxidant gave iodide 477. This process presumably proceeds via a transient hypoiodite species, which undergoes a chemoselective homolysis of the C11–C19 bond followed by recombination with an iodine radical to furnish the fragmentation product 477, which could be further transformed into ouabagenin (472).
Figure 24.
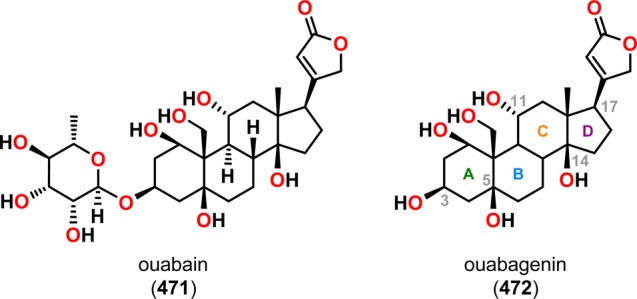
Structures of ouabain (471) and ouabagenin (472).
Scheme 85. Photoinduced Norrish–Yang Cyclization in the Synthesis of Ouabagenin (472).

NIS = N-iodosuccinimide. SDS = sodium dodecyl sulfate.
Booker-Milburn and co-workers recently applied the Norrish–Yang cyclization for the synthesis of aza-fuzed tricyclic lactones.369,370 It was realized that alkoxy-substituted maleimides such as 478 underwent a [5 + 2] cycloaddition/Norrish–Yang cyclization cascade sequence of the major initial photoadduct 479 to provide the complex azepine-fused alkylidene-oxetanol 482 in a single photochemical operation. The produced oxetane derivatives, such as 482, were shown to afford the tricyclic lactone 483, a result of acid-catalyzed rearrangement (Scheme 86). The rearrangement was proposed to proceed via initial oxetane ring-opening, followed by transannular amide cleavage. The resulting amine cyclizes to produce an aminol, which upon elimination of water forms the tricyclic lactone 483.
Scheme 86. Maleimide [5 + 2] Photocycloaddition/Norrish Type II Cascade for Synthesis of Tricyclic Lactone 483.

A recent application of the Norrish–Yang cyclization can be found in Inoue and co-workers’ synthesis of (+)-lactacystin (490).371 The C5–C9 core skeleton of (+)-lactacystin (490) was provided by commercially available (S)-pyroglutaminol (484), which was converted to diketone 485. Subjecting diketone 485 to UV light (λ < 360 nm) gave a complex product mixture, presumably due to unselective photoexcitation. As an alternative, the authors turned their attention to blue LEDs (λ ≈ 460 nm). Using the longer-wavelength-emitting blue LEDs led to smooth conversion to the cis-fused cyclobutanone 488. The observed chemoselective hydrogen abstraction in the Norrish type II reaction is presumed to originate from the more electron-rich nature of the ethereal C(sp3)–H bond in comparison to the aliphatic C–H bond. Subsequent oxidative ring-opening using Pb(OAc)4 generated ketoester 489 as a single isomer in 66% yield over two steps (Scheme 87), which could be converted to (+)-lactacystin (490) through homologation at C10, construction of C6 and C7 stereocenters, and attachment of the cysteine moiety.
Scheme 87. Application of the Norrish–Yang Cyclization in the Total Synthesis of (+)-Lactacystin (490).

Substrates lacking γ-hydrogens are also prone to undergo Norrish-Yang cyclization. Examples of this type have been applied in the synthesis of (±)-paulownin (493, Scheme 88)372 and the structurally related (+)-phrymarin I and (+)-phrymarin II.373
Scheme 88. Use of the Norrish–Yang Cyclization in the Total Synthesis of (±)-Paulownin (493).

6. The Oxa-di-π-methane Rearrangement—Access to Complex Molecular Frameworks
Substrates housing β,γ-unsaturated ketone motifs are generally competent in undergoing the oxa-di-π-methane rearrangement. The reaction involves triplet-sensitized irradiation and is the reason why the reaction is conducted in the presence of a sensitizer, usually by employing acetone as the solvent. The oxa-di-π-methane rearrangement ultimately furnishes an α-cyclopropyl ketone, where photochemical excitation brings about a 1,2-acyl migration, resulting in bond formation between what were formerly the α and γ atoms.374Scheme 89 depicts typical cyclic and acyclic substrates that undergo the oxa-di-π-methane rearrangement.
Scheme 89. Illustrative Examples of the Oxa-di-π-methane Rearrangement for (top) an Acyclic and (bottom) a Cyclic β,γ-Unsaturated Ketone.

The first reported example of the oxa-di-π-methane rearrangement dates back as far as 1966. Here, the authors discovered that a β,γ-unsaturated ketone underwent rearrangement to afford a cyclopropyl ketone.375 After this initial report, there have been a multitude of synthetic examples that address the photochemistry of substrates containing β,γ-unsaturated ketone scaffolds.376 The synthetic advantages of using the oxa-di-π-methane rearrangement as a tool in synthesis can be attributed to its feature of accessing molecular frameworks with significantly increased structural complexity that might be difficult to obtain via other routes. The relatively easily accessed starting materials, β,γ-unsaturated ketones, and the high tolerance of functional groups certainly increase the synthetic potential of the photochemical rearrangement.377
Stevens and Yates reported one of the initial applications of the oxa-di-π-methane rearrangement in total synthesis. The authors employed the oxa-di-π-methane rearrangement of bicyclo[2.2.2]octenone derivatives for the synthesis of cedrenoid sesquiterpenes.378 Acetophenone-sensitized irradiation of the Diels–Alder adduct 494 afforded the oxa-di-π-methane rearranged product 495. The strained tricyclic compound 495 was subsequently subjected to Me2CuLi to give compound 496, which underwent Krapcho decarbomethoxylation to ketoester 497 in 74% yield. The introduction of the functionalized two-carbon substituent was carried out by treating 497 with lithium acetylide (498). This afforded the propargylic alcohol 499, where attack of the acetylide occurs at the β-face of 497. Propargylic alcohol 499 was subsequently transformed into the Stork–Clarke β-diketone 500 (Scheme 90),378,379 which had previously been reported as a key intermediate in the synthesis of (±)-cedrol (501).436,380
Scheme 90. Use of the Oxa-di-π-methane Rearrangement in the Formal Total Synthesis of (±)-Cedrol (501).

Another example of employing the bicyclo[2.2.2]octenone skeleton for rearrangement into viable building blocks in total synthesis can be found in the synthesis of (±)-modhephene (65), which contains a carbocyclic [3.3.3]propellane framework (see Figure 25).381−394 Here, Mehta and Subrahmanyam applied a photochemical approach that involved the oxa-di-π-methane rearrangement for constructing the sesquiterpene hydrocarbon (±)-modhephene (65). In this route, diene 506 was converted to bicyclo[2.2.2]octenone 508 through a Diels–Alder cycloaddition. Irradiation of β,γ-unsaturated ketone 508, in the presence of acetone, afforded the tetracyclic strained cyclopropyl ketone 509 housing three quaternary carbon centers through a oxa-di-π-methane rearrangement. Conventional synthetic manipulations then converted the tetracyclic ketone 509 into the sesquiterpene (±)-modhephene (65) (Scheme 91).395,396
Figure 25.

Examples of carbocyclic propellanes.
Scheme 91. Construction of the Carbocyclic Framework of (±)-Modhephene (65) by Use of the Oxa-di-π-methane Rearrangement.

PCC = pyridinium chlorochromate.
Uyehara and co-workers have also utilized the oxa-di-π-methane rearrangement in the synthesis of (±)-modhephene (65) and (±)-isocomene (63).147 Here, oxa-di-π-rearrangement of bicyclo[2.2.2]oct-5-en-2-ones 512 and 514 afforded ketones 513 and 515, which could be further elaborated to (±)-modhephene (65) and (±)-isocomene (63), respectively (Scheme 92).
Scheme 92. Uyehara’s Approach to (±)-Modhephene (65) and (±)-Isocomene (63).

The structurally related magellanine (516),397−399 magellaninone (517),397−399 and paniculatine (518)400 are members of the Lycopodium alkaloids, which are a family of natural products that have attracted considerable interest from the synthetic community since their isolation in the 1970s due to their intriguing polycyclic skeleton and biological activity (Figure 26).401−404 These alkaloids share a diquinane core that is fused to a cyclohexenone or cyclohexanol and piperidine unit. Construction of the tetracyclic scaffolds of these alkaloids and also controlling the stereochemistry at a multitude of the carbon centers of the bicyclo[3.3.0]octane core certainly represent daunting synthetic challenges.405−409
Figure 26.
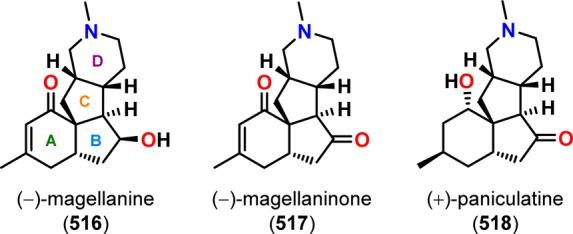
Structures of the Lycopodium alkaloids (−)-magellanine (516), (−)-magellaninone (517), and (+)-paniculatine (518).
The tetracyclic magellanine (516) with six contiguous stereogenic centers, one of which is a quaternary carbon center, was efficiently synthesized from commercially available acetovanillone 519 using the oxa-di-π-methane rearrangement as a key step.410 Liao and co-workers found that irradiation of Diels–Alder cycloadduct 520 yielded the tetracyclic diketone 521 through the oxa-di-π-methane rearrangement. Cyclopropyl ring-opening resulted in a linear triquinane (523), which was subsequently transformed into (±)-magellanine (516) (Scheme 93).
Scheme 93. Synthesis of the Tetracyclic (±)-Magellanine (516) by Use of the Oxa-di-π-methane Rearrangement.

AIBN = azobis(isobutyronitrile). TMSOTf = trimethylsilyl trifluoromethanesulfonate.
A similar approach by Liao and co-workers was also applied in the synthesis of (±)-Δ9(12)-capnellene (525), which was accessed in nine steps with 20% overall yield from 2-methoxy-4-methylphenol (524) (Scheme 94).411 Additional examples of linear triquinanes that have been assembled utilizing the oxa-di-π-methane rearrangement are (−)-complicatic acid (526),412,413 (−)-coriolin (527),414−418 (−)-hirsutene (222),419−422 (+)-hirsutic acid (528),412,413,423,424 and (−)-phellodonic acid (529) (Figure 27).425
Scheme 94. Synthesis of (±)-Δ9(12)-Capnellene (525) from 2-Methoxy-4-methylphenol (524).
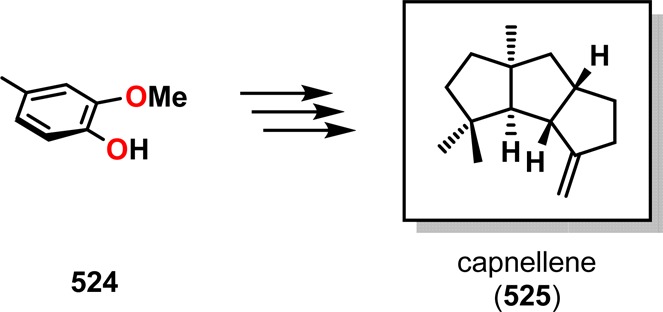
Figure 27.
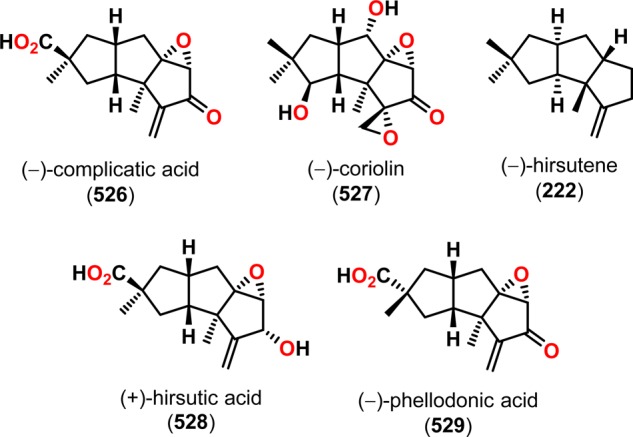
Examples of linear triquinanes that have been synthesized via the oxa-di-π-methane rearrangement.
The examples highlighted here illustrate the power of the oxa-di-π-methane rearrangement for the elaborate functionalization of simple scaffolds into highly condensed skeletons for use in the total synthesis of natural products.
7. The Meta-Photocycloaddition Reaction—A Powerful Method for Accessing Polycyclic Skeletons
Aromatic compounds are known for their robustness; however, upon electronic excitation the aromatic ring can be activated and engaged in reaction pathways that are generally not possible from the ground state. Scheme 95a depicts the cycloaddition between an alkene (ethylene) and an arene (benzene) and the three pathways, ortho-, meta-, and para-photocycloaddition, that can be observed.426 Ortho- and meta-photocycloadditions are the most frequently encountered outcomes, while para-photocycloaddition is rarely observed. The meta-photocycloaddition (Scheme 95b) is perhaps the most remarkable photochemical reaction, as it allows for the formation of three single bonds and up to six stereogenic centers in one step.427,428 The reaction proceeds via the formation of a tricyclic scaffold from which fragmentation of the three-membered ring can produce several products (Scheme 95c). Cleavage of the C2–C8 bond is considered as the most common pathway and generates a bicyclo[3.3.0]octane framework. On the other hand, cleavage of the C1–C2 bond or the bond between C1 and C8 furnishes a bicyclo[3.2.1]octane skeleton. The extensive bond reorganization associated with the meta-photocycloaddition reaction makes it a powerful method for generating significant molecular complexity in a single synthetic step, and it lacks a thermal counterpart. The versatility of the different fragmentation routes creates various exotic frameworks and it is this feature that has rendered the meta-photocycloaddition a widely used tool in a number of synthetic routes for accessing natural products.429,430
Scheme 95. (a) Three Modes of Photocycloadditions of an Alkene to a Benzene Ring, (b) Pathway for the Meta-Photocycloaddition, and (c) Key Bond Fragmentations for the Meta-Photocycloaddition Adduct for Accessing Complex Cyclic Frameworks.

Although Wilzbach and Kaplan reported the first example of a meta-photocycloaddition reaction in 1966,431 it was Wender’s successful implementation of the meta-photocycloaddition in the total synthesis of a variety of natural products that began to attract significant attention.432 In particular, Wender’s synthesis of α-cedrene (1) expanded the concept of the alkene–arene meta-photocycloaddition reaction and extended the chemical toolbox (Figure 28).37 The tricyclic α-cedrene (1) belongs to a family of naturally occurring sesquiterpenes. It was isolated433,434 from Juniperus cedrus and Juniperus thurifera in 1841; however, its structure and the tricycle undecane skeleton were not verified until 1953 by Stork and Breslow.435 The intriguing [5.3.1.01,5] tricyclic structure found in the cedranoids has attracted considerable interest among the synthetic community.436−447
Figure 28.
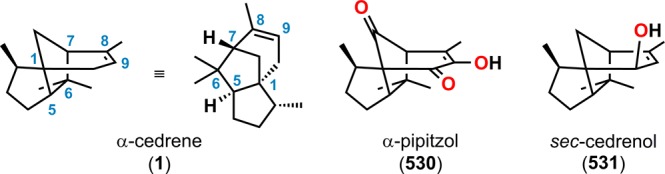
Examples of cedranoids.
Wender’s concise synthesis of (±)-α-cedrene (1) in merely four steps (Scheme 96) exemplified the ability of the meta-photocycloaddition reaction to provide the tricyclic sesquiterpene with the correct configuration at all four stereogenic centers. The high diastereo- and regioselectivity is dictated by 1,3-allylic (A1,3) strain between the aryl methoxy and the methyl group in 532. This favors a conformation for the ring closure step that gives the two products 533 and 534 in a 1:1 ratio. Subsequent fragmentation of the cyclopropane ring upon treatment with Br2, followed by reductive dehalogenation with nBu3SnH, provided convergent access to ketone 535 from the two cycloaddition products 533 and 534. Wolff–Kishner reduction of ketone 535 generated (±)-α-cedrene (1) in merely four steps.37
Scheme 96. Total Synthesis of (±)-α-Cedrene (1) Using Meta-Photocycloaddition.

As previously discussed in section 2.1, both (±)-isocomene (63)78 and silphinene (105)95 have been synthesized using the [2 + 2] photocycloaddition. However, the tricyclic cores of (±)-isocomene (63)146 and silphinene (105)448 can also be accessed by using a meta-photocycloaddition strategy and are depicted in Schemes 97 and 98, respectively.
Scheme 97. Photochemical Approach for Synthesis of (±)-Isocomene (63).

Scheme 98. Route to Silphinene (105) Using Meta-Photocycloaddition.

Pentacyclic (−)-retigeranic acid A (548), a structurally related natural product, was initially isolated from Himalayan lichen and is reported to have antibacterial activity.449,450 It is a structurally unique member of the class of sesterterpene natural products and houses a triquinane subunit and eight stereocenters.451,452 Due to retigeranic acid’s intriguing structure, several research groups have pursued its synthesis and related sesterterpenoids.453−455 Early examples of the total synthesis of retigeranic acid include reports by the groups of Corey,456 Hudlicky,457−460 and Paquette.461−463
Wender and Singh’s convergent approach to (−)-retigeranic acid A (Scheme 99) commenced with the preparation of the enantiomerically pure arene–alkene derivative 543 from commercially available 3-methylglutaric acid. Photolysis of the arene–alkene fragment 543 afforded a 1:2 mixture of photoadducts 544 and 545, which were found to be interconvertible vinylcyclopropane isomers, allowing them each to be converted to the Diels–Alder precursor 546. This precursor was then allowed to undergo an intramolecular Diels–Alder reaction to furnish the pentacyclic compound 547, which could be further transformed into (−)-retigeranic acid (548).464
Scheme 99. Synthesis of (−)-Retigeranic Acid A (548).

Subsequently, the Wender group reported on the total synthesis of rudmollin (554).465 The pseudoguaianolide rudmollin (554) was initially isolated from a coneflower by Herz and co-workers and exhibits antileukemic activity.466 It was realized by Wender and co-workers that the trans-fused perhydroazulene core of rudmollin (554) could be accessed through the use of an arene−olefin cycloaddition strategy. Their approach thus centered on accessing a seven-membered cyclic compound via intramolecular meta-photocycloaddition of substrate 549, which would establish the trans relationship between carbons C1 and C5. Furthermore, since these photocycloadditions generally preserve the geometry of the olefin moiety, this would also set the relative configuration between carbons C1 and C10 in the photocycloadduct. Performing the meta-photocycloaddition on substrate 549 afforded the two photocycloadducts 550 and 551 in a 7:3 ratio and hence established the relative configuration at the three carbon centers C1, C5, and C10. Here, the cyclopropane isomers 550 and 551 could be converted into a common product by employing a similar technique as with (±)-α-cedrene (1); see Scheme 96. However, instead of using Br2-induced cleavage of the cyclopropane bond, Hg(OAc)2-catalyzed hydrolysis was shown to convergently transform the cycloadducts 550 and 551 into compound 552, which could subsequently be used to access (±)-rudmollin (554) (see Scheme 100).465,467
Scheme 100. Total Synthesis of (±)-Rudmollin (554) Using an Intramolecular Meta-Photocycloaddition Approach.

Penifulvin A (555), an antiinsectan sesquiterpenoid, was originially isolated by Gloer and co-workers in 2006 from Penicillium griseofulvum.468,469 The penifulvins (Figure 29) contain an intricate dioxa[5.5.5.6]fenestrane skeleton where four rings are joined at a central quaternary carbon atom (Figure 30). In addition to the two vicinal quaternary stereocenters, a total of five stereogenic centers exist within the 15-carbon atom framework.470,471 The unique and complex molecular scaffold of penifulvins has made them an attractive target for total synthesis.472−474
Figure 29.
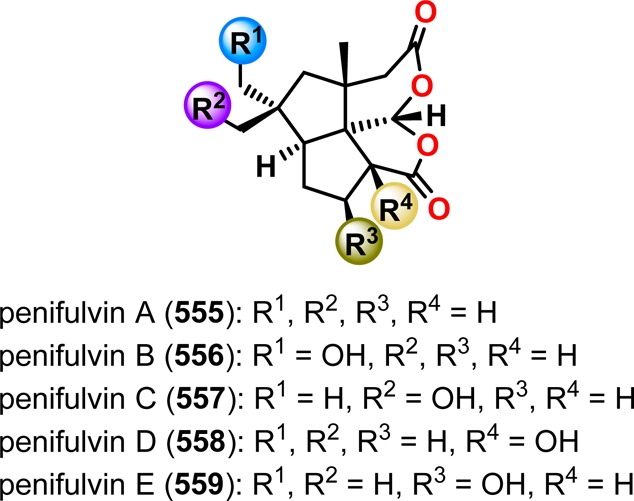
Structure of penifulvins A–E.
Recently, the Mulzer group used the arene olefin meta-photocycloaddition to access the sesquiterpene (−)-penifulvin A (555).475−477 The asymmetric synthesis of compound 566 was carried out by using Myers’ alkylation strategy (Scheme 101).478 Photoreaction of compound 566 resulted in a mixture of the two allylic regioisomers 569 and 570 in a 1:1 ratio. Subsequent cyclopropane ring opening using Birch-like reductive conditions afforded alcohol 571. This was followed by an oxidative cascade sequence to complete the concise synthesis of (−)-penifulvin A (555) (Scheme 102). In addition to the photocyclization event, which furnishes separable regioisomers, the synthesis did not require any protecting groups.475 The total syntheses of penifulvins B (556) and C (557) were subsequently accomplished using a similar meta-photocycloaddition approach as the key step to rapidly access the fenestrane-type carboskeletons.476 Wender and co-workers have also disclosed a method for synthesizing cis,cis,cis,trans-[5.5.5.5]fenestranes using the meta-photocycloaddition.479
Scheme 101. Synthesis of Enantiomerically Enriched Alcohol 566 Using Pseudoephedrine as a Chiral Auxiliary.

DIC = diisopropylcarbodiimide. DMAP = 4-dimethylaminopyridine. LDA = lithium diisopropylamide.
Scheme 102. Use of the Arene Olefin Meta-Photocycloaddition in the Total Synthesis of (−)-Penifulvin A (555).

IBX = 2-iodoxybenzoic acid. PCC = pyridinium chlorochromate.
Laurenene (99) is another natural product containing the fenestrane framework that has been synthesized using meta-photocycloaddition.480 Wender and co-workers started with a Diels–Alder reaction of commercially available 575 and cyclohepta-1,3-diene (576) to afford the tricyclic arene 577, which was subsequently elaborated to give lactone 579 upon Grob fragmentation of keto tosylate 578. Hydrogenation of lactone 579 and alkylation with homoprenyl iodide furnished the desired stereoisomer 581 with a 8:1 preference. Photolysis of lactol 582, prepared by LiAlH4 reduction of lactone 581, through a BiCl3 filter solution allowed for 80% conversion and isolation of a single hexacyclic product (583) in 51% yield. Completion of the synthesis was then accomplished in three steps to afford (±)-laurenene (99) in 5% overall yield (Scheme 103).
Scheme 103. Total Synthesis of (±)-Laurenene (99).

AIBN = azobis(isobutyronitrile). DMPU = N,N′-dimethylpropyleneurea. LDA = lithium diisopropylamide. NBS = N-bromosuccinimide.
Additional examples of natural products that have been accessed using the meta-photocycloaddition reaction as the key step are shown in Figure 31 and include (±)-coriolin (527),481 (±)-hirsutene (222),482 (±)-isoiridomyrmecin (586),483 (±)-modhephene (65),484 (±)-silphiperfol-6-ene (588),485 subergorgic acid (590),486 and crinipellin B (592).487
Figure 31.

Representative examples of natural products accessed by the meta-photocycloaddition reaction.
8. Visible-Light Photoredox Catalysis—Single-Electron Transfer Approaches in Natural Product Synthesis
A central theme in photoinduced reactions is the excitation of an electron from an occupied molecular orbital to an antibonding orbital to generate a neutral π–π* excited state. A fundamentally different approach would be to use photosensitizer compounds for activation of organic compounds. Upon absorption of visible light, these photosensitizers form excited states that can function both as powerful oxidants and reductants, which would allow for the generation of radical cations or radical anions under remarkably mild reaction conditions.488 Photoredox catalysis offers selective excitation of the organic compound, avoiding excitation of the whole organic network and decomposition, as can be the case with UV-light excitation. Visible-light photoredox catalysis is a rapidly emerging field that offers an attractive alternative to conventional methods of producing radical ion intermediates that are capable of engaging in a multitude of different pathways. This powerful method has enabled the development of novel reaction schemes and approaches for the controlled engineering of structurally complex and diverse frameworks through single-electron transfer (SET) reactions. In particular, transition-metal polypyridyl complexes based on Ru and Ir have proven to afford unique chemical reactivities when excited by visible light (Figure 32). These photocatalysts are chemically robust, afford long-lived excited state lifetimes, and possess favorable redox properties that can be conveniently fine-tuned by modifying the polypyridyl ancillary ligands (Figure 33).489−493 These properties allow overall redox-neutral reactions to be carried out as both reductants and oxidants that can be transiently generated during different stages in the catalytic process. This reactivity pattern is beneficial since it might allow for the exploration of alternative pathways under benign reaction conditions.494−504
Figure 32.

Examples of Ru- and Ir-based photosensitizers (bpy = 2,2′-bipyridine, bpz = 2,2′-bipyrazine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine, ppy = 2-phenylpyridine).
Figure 33.

Properties of the [Ru(bpy)3]2+ photocatalyst (594) (bpy = 2,2′-bipyridine, MLCT = metal-to-ligand charge transfer, A = sacrificial electron acceptor, D = sacrificial electron donor, S = substrate).
A variety of cyclobutane-containing natural products have been accessed via photoinduced SET. The use of visible-light photoredox catalysis can, for example, be found in the synthesis of magnosalin (599, Scheme 104),505ent-sceptrin (606, Scheme 105),506 (±)-epiraikovenal (614, Scheme 106),507 and (±)-cannabiorcicyclolic acid (619, Scheme 107).508 The synthesis of γ-butyrolactones from simple alkenes and unsaturated acids via polar radical crossover cycloaddition (PRCC) reactions was recently reported by Nicewicz and co-workers.509 The mechanism was proposed to involve single-electron oxidation of the alkene (621) by the excited acridinium photocatalyst (620+*), which produces an electrophilic alkene cation radical (622). The carboxylic acid (623) can subsequently add to the generated radical cation 622 to furnish radical 624, which undergoes 5-exo-trig radical cyclization to give 625. Hydrogen atom transfer (HAT) to radical 625 produces the desired γ-butyrolactone (626) and completes the catalytic cycle (Scheme 108, top). The authors subsequently applied the PRCC method to the synthesis of protolichesterinic acid (630). Irradiation of styrene 627 and β-haloacrylic acid (628) in the presence of 2.5 mol% of acridinium photocatalyst 620(BF4) and substoichiometric quantities (25 mol%) of the redox-active cocatalyst PhSSPh afforded lactone 629 in 69% yield, which upon treatment with RuCl3/NaIO4 followed by K2CO3 produced protolichesterinic acid (630) as a single isomer in 54% yield (Scheme 108, bottom).
Scheme 104. Synthesis of Magnosalin (599) via Photoinduced Electron Transfer.

p-OMeTPT = 2,4,6-tris(4-methoxyphenyl)pyrylium tetrafluoroborate.
Scheme 105. Visible-Light Photoredox Catalysis in the Synthesis of ent-Sceptrin (606).

BOM = benzyloxymethyl. TIPS = triisopropylsilyl. ppy = 2-phenylpyridine. fac = facial.
Scheme 106. Visible-Light-Promoted [2 + 2] Cycloaddition of 1,3-Diene 609 in the Synthesis of (±)-Epiraikovenal (614).

dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-trifluoromethylpyridine. dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine. HMPA = hexamethylphosphoric triamide.
Scheme 107. Visible-Light-Sensitized [2 + 2] Cycloaddition of Olefin 617 in the Synthesis of (±)-Cannabiorcicyclolic Acid (619).

CFL = compact fluorescent light.
Scheme 108. (Top) Proposed Mechanism for Polar Radical Crossover Cycloaddition (PRCC) of Alkenes and Unsaturated Acids and (Bottom) Its Application in the Synthesis of Protolichesterinic Acid (630).

HAT = hydrogen atom transfer.
The use of two electron-rich components in Diels–Alder cycloadditions typically requires harsher reaction conditions and prolonged reaction times as a result of the electronic mismatching. In contrast, radical cations generated from electron-rich olefins have been documented to react rapidly with electron-rich dienes in [4 + 2] cycloadditions, and the reactions typically occur with high chemo-, stereo-, and regioselectivity with rates that can be several orders of magnitude higher than the corresponding thermal process with the neutral species.510−513 The essential radical cations are usually produced using high loadings of one-electron oxidants, such as aminium salts,514 or photoinitiated electron transfer initiated by an organic photosensitizer.515
The Yoon group utilized visible-light photocatalysis as a means for the efficient generation of the vital radical cations.516−518 This allowed for an operationally simple protocol for radical cation Diels–Alder cycloadditions that employed low loadings (0.5 mol%) of [Ru(bpz)3]2+ (595519) as photosensitizer.516 Given the broad scope of this reaction, the authors became interested in heitziamide A (635), a natural product isolated as a racemate from the medicinal shrub Fagara heitzii.520 Irradiation of styrene 631 and myrcene (632) in the presence of 2 mol% [Ru(bpz)3]2+ (595) produced [4 + 2] adduct 633 in 80% yield. This process can be best described as an umpolung process that reverses the intrinsic electronic character of the electron-rich olefin, thus producing the cycloadduct with the natural regiochemistry of heitziamide A (635). As a comparison, the thermal Diels–Alder reaction furnished the unnatural isomeric cycloadduct without any trace of 633. Deprotection of the TBS protecting group and subsequent oxidation afforded carboxylic acid 634, which could be converted to heitziamide A (635) by EDC coupling with isobutylamine (Scheme 109).
Scheme 109. Visible-Light Photocatalytic Radical Cation Diels–Alder Cycloaddition in the Synthesis of Heitziamide A (635).

EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide. TBAF = tetra-n-butylammonium fluoride. TBS = tert-butyldimethylsilyl. NMO = N-methylmorpholine N-oxide. bpz = 2,2′-bipyrazine. DMAP = 4-dimethylaminopyridine.
Given the high oxidation potential of the [Ru(bpz)3]2+ photosensitizer (1.4 V vs SCE), the visible-light-mediated radical cation Diels–Alder reaction is believed to proceed via reductive quenching of the excited state of the photosensitizer {[Ru(bpz)3]2+•} by the electron-rich olefin moiety 631. This results in a radical cation (631+•) that undergoes the [4 + 2] cycloaddition to generate a radical cycloadduct, which can either be reduced by the reduced photosensitizer {[Ru(bpz)3]+} or abstract an electron from another olefin molecule to initiate radical chain processes.503,516,521 A related radical cation Diels–Alder approach has also been utilized in the synthesis of (±)-kingianic acid E (639) (Scheme 110).522
Scheme 110. Synthesis of (±)-Kingianic Acid (639) via a Radical Cation Diels–Alder Approach.

NMO = N-methylmorpholine N-oxide. TBAF = tetra-n-butylammonium fluoride. bpy = 2,2′-bipyridine. MV = methyl viologen.
Aplyviolene (646) belongs to a class of spongian diterpene natural products and contains a cis-perhydroazulene unit.523−525 For aplyviolene (646), formation of the bicyclic lactone subunit and the C8–C14 bond has been recognized as particularly challenging. In Schnermann and Overman’s synthesis of (−)-aplyviolene (646), the key step involved a photoredox-mediated fragmentation strategy, which allowed construction of the crucial C8–C14 bond.526 The synthesis commenced with (+)-fenchone (640), which was converted to N-(acyloxy)phthalimide 641. Irradiation of cyclopentenone 642 and N-(acyloxy)phthalimide 641 in the presence of 1 mol% [Ru(bpy)3]2+ (594), Hantsch ester 643, and DIPEA efficiently provided the coupled product 644 in 61% yield with <5% of the reductively dechlorinated analogue. Reductive enol silylation of coupled product 644 using Me2CuCNLi2 in the presence of TBS-Cl at −20 °C delivered the tricyclic enol ether 645, which had previously been converted to (−)-aplyviolene (646) (Scheme 111). The use of the photoredox-mediated fragmentation strategy thus allowed the construction of the C8–C14 bond and highlights the utility of using photoredox catalysis for generation of tertiary carbon radicals in the construction of quaternary stereocenters in a stereoselective fashion. A related strategy was also employed by Overman and co-workers in the synthesis of trans-clerodane natural products.527
Scheme 111. Photoredox-Mediated Tertiary Radical Generation in the Synthesis of (−)-Aplyviolene (646).

DIPEA = diisopropylethylamine. TBS = tert-butyldimethylsilyl. HMPA = hexamethylphosphoric triamide. bpy = 2,2′-bipyridine.
Pyrroloindoline alkaloids are a class of natural products that are formally derived from two molecules of tryptophan and have been shown to exhibit a broad range of biological activities. As a result of their structural complexity and broad biological activity, pyrroloindoline alkaloids have attracted attention from several research groups.528−530 In the synthesis of (+)-gliocladin C (652), Stephenson and co-workers utilized a photoredox-mediated indole coupling reaction as the key step to access the C3–C3′ bisindole motif. Irradiation of bromopyrrolindoline 648, derived from Boc-d-tryptophan methyl ester (647), and aldehyde 649 using 1 mol% [Ru(bpy)3]Cl2 and Bu3N as reductive quencher successfully provided the C3–C3′ coupling product 650 in 82% yield. Subsequent decarbonylation of the aldehyde at the C2′ position using a combination of Rh(CO)(PPh3)3Cl, dppp, and DPPA in xylenes afforded the decarbonylated product 651 in 85% yield, which could be further advanced to (+)-gliocladin C (652) in merely 10 steps from Boc-d-tryptophan methyl ester (647) and in 30% overall yield (Scheme 112).531
Scheme 112. Photoredox-Enabled Synthesis of (+)-Gliocladin (652).

Boc = tert-butyloxycarbonyl. Cbz = carboxybenzyl. DPPA = diphenylphosphoryl azide. dppp = 1,3-bis(diphenylphosphino)propane. bpy = 2,2′-bipyridine.
Beatty and Stephenson identified (+)-catharanthine (653) as an entry point for the synthesis of a number of structurally related alkaloids.532 Catharanthine has been reported to undergo a unique fragmentation of its C16–C21 bond533,534 and it was envisioned that such an oxidative fragmentation could be exploited to produce a common α-aminonitrile intermediate (654), which could subsequently be converted to a variety of analogous alkaloids. Visible-light irradiation of catharanthine (653) in the presence of the fluorinated photocatalyst 610 and TMSCN indeed produced an α-aminonitrile fragmentation product (654) in 93% yield. Performing the reaction in a photochemical flow reactor535−540 allowed for improved scalability, decreased reaction time, and the controlled generation of HCN, affording intermediate 654 in 96% yield. Refluxing a MeOH solution containing α-aminonitrile intermediate 654 and 1 equiv. of trifluoroacetic acid for 3 h gave (−)-pseudotabersonine (655) as the only observed product in 90% yield. Hydrogenation of the aminonitrile fragmentation product 654 followed by reflux in the presence of 1 equiv. of trifluoroacetic acid was anticipated to produce (−)-pseudovincadifformine (660). However, this yielded the natural product (+)-coronaridine (657) as the sole product in 48% yield over two steps and represents the highest yielding preparation of coronaridine from catharanthine reported thus far. An alternative approach to (−)-pseudovincadifformine (660) was therefore investigated and involved initial hydrogenation over Pd/C, followed by NaBH4 reduction to give 658. Exposing amine 658 to oxidative photoredox conditions employing malonate 659 as the terminal oxidant subsequently yielded (−)-pseudovincadifformine (660) in 58% yield (Scheme 113).532 This work demonstrates the synthetic utility of photoredox catalysis in natural product synthesis and the rapid access to structurally related alkaloid motifs from a common advanced intermediate.
Scheme 113. Synthesis of Alkaloid Natural Products Enabled by Photoredox Catalysis in Flow.

TFA = trifluoroacetic acid. dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-trifluoromethylpyridine. dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine. bpy = 2,2′-bipyridine. TMS = trimethylsilyl.
The examples described in this section highlight photoredox catalysis as a robust tool for the mild generation of radical species for potential access to a wide variety of complex molecular scaffolds, thus expanding the use of photochemistry en route to natural product targets.
9. Conclusions and Outlook
The purpose of this review has been to highlight some of the accomplishments that have been presented in natural product total synthesis using photochemistry. Photochemically mediated reactions have proven to be highly useful for construction of highly strained and complex molecular frameworks that can otherwise be challenging to access via conventional, thermal processes. However, the use of specialized equipment for performing these photochemical reactions could serve as a significant impediment for possible practitioners looking to implement these techniques. Although this may impede the advancement of photochemical transformations, continued triumphs in the total synthesis of natural products and biologically active compounds, along with further mechanistic studies to elucidate the general chemical reactivity of the electronically generated excited intermediates, will continue to inspire the development and application of photochemistry in organic synthesis.
The recent emergence of photoredox catalysis as a means to generate radical species in a controlled and mild fashion has further increased the ability of accessing complex molecular scaffolds. It is believed that such controlled redox manipulations will allow for chemoselective access to a wide array of novel structural motifs. Another important direction in modern photochemistry is its application to continuous flow processing. Compared to normal batch reactors, carrying out photochemical reactions in flow reactors can greatly improve the yields and selectivities due to the avoidance of unproductive reaction pathways.
On the whole, photochemistry serves as a useful tool for mastering new and environmentally benign transformations. In the future, it can also be used to expand our classical chemical toolbox by offering improved reaction conditions and can facilitate the discovery of new transformations for efficient bond construction. Thus, in combination with flow chemistry and other advanced techniques, photochemistry will continue to serve as an attractive option for accessing intricate chemotypes of biological and medicinal importance.
Acknowledgments
Financial support from the NSF (CHE-1440118) and the NIH (GM096129), the Camille and Henry Dreyfus Foundation and University of Michigan to C.R.J.S., and from the NIH (GM073855) and Boston University to J.A.P., Jr., is gratefully acknowledged. M.D.K. gratefully acknowledges financial support from the Swedish Research Council (637-2013-7314) and the Royal Swedish Academy of Agriculture and Forestry (Kungliga Skogs- och Lantbruksakademien) for a postdoctoral fellowship.
Glossary
Abbreviations Used
- AIBN
azobis(isobutyronitrile)
- Boc
tert-butyloxycarbonyl
- BOM
benzyloxymethyl
- Bpin
(pinacolato)boron
- bpy
2,2′-bipyridine
- bpz
2,2′-bipyrazine
- BTB
2,6-bis(trifluoromethyl)benzyl
- Cbz
carboxybenzyl
- m-CPBA
m-chloroperoxybenzoic acid
- CFL
compact fluorescent light
- CSA
camphorsulfonic acid
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- dF(CF3)ppy
2-(2,4-difluorophenyl)-5-trifluoromethylpyridine
- DIBAL-H
diisobutylaluminum hydride
- DIC
diisopropylcarbodiimide
- DIPEA
diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DMPU
N,N′-dimethylpropyleneurea
- DMS
dimethyl sulfide
- DPPA
diphenylphosphoryl azide
- dppp
1,3-bis(diphenylphosphino)propane
- dtbbpy
4,4′-di-tert-butyl-2,2′-bipyridine
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- fac
facial
- FDA
Food and Drug Administration
- HAT
hydrogen atom transfer
- HIV
human immunodeficiency virus
- HMPA
hexamethylphosphoric triamide
- IBX
2-iodoxybenzoic acid
- ISC
intersystem crossing
- LDA
lithium diisopropylamide
- LDBB
lithium 4,4′-di-tert-butylbiphenylide
- p-MBOH
p-methoxybenzyl alcohol
- MLCT
metal-to-ligand charge transfer
- MOM
methoxymethyl
- MoOPH
(hexamethylphosphoric triamide)oxodiperoxy(pyridine)molybdenum(VI) (MoO5·pyr·HMPA)
- MV
methyl viologen
- NBS
N-bromosuccinimide
- NIS
N-iodosuccinimide
- NMO
N-methylmorpholine N-oxide
- PCC
pyridinium chlorochromate
- PDC
pyridinium dichromate
- Piv
pivaloyl
- PMP
p-methoxyphenyl
- PPTS
pyridinium p-toluenesulfonate
- ppy
2-phenylpyridine
- PRCC
polar radical crossover cycloaddition
- SCE
saturated calomel electrode
- SDS
sodium dodecyl sulfate
- SEM
2-(trimethylsilyl)ethoxymethyl
- SET
single-electron transfer
- TBAF
tetra-n-butylammonium fluoride
- TBS
tert-butyldimethylsilyl
- TES
triethylsilyl
- Tf2O
trifluoromethanesulfonic anhydride
- TFA
trifluoroacetic acid
- TFAA
trifluoroacetic anhydride
- TIPS
triisopropylsilyl
- TMEDA
N,N,N′,N′-tetramethylethylenediamine
- TMSOTf
trimethylsilyl trifluoromethanesulfonate
- p-OMeTPT
2,4,6-tris(4-methoxyphenyl)pyrylium tetrafluoroborate
- p-TsCl
p-toluenesulfonyl chloride
- p-TsOH
p-toluenesulfonic acid
Biographies
Markus D. Kärkäs obtained his Ph.D. degree in 2013 from Stockholm University, under the supervision of Prof. Björn Åkermark. His research focused on the development and mechanistic insight of artificial water oxidation catalysts. After completing his Ph.D. studies he continued to work as a researcher in the group of Prof. Åkermark and was involved in studying and developing homogeneous as well as heterogeneous water oxidation catalysts. He is currently a Swedish Research Council (Vetenskapsrådet) postdoctoral fellow in the research group of Prof. Corey Stephenson at the University of Michigan, where his research is directed toward the development of novel synthetic transformations involving visible-light photoredox catalysis.
John A. Porco, Jr. received his Ph.D. degree in 1992 from Harvard University under the direction of Prof. Stuart Schreiber. He did postdoctoral studies with Chi-Huey Wong at the Scripps Research Institute. After a period in industry, John joined the Department of Chemistry at Boston University in 1999 as assistant professor and was promoted to professor of chemistry in September 2004. From 2002 to 2013 he was director of the NIH-funded Center for Chemical Methodology and Library Development (CMLD-BU) and since 2013 has directed the BU Center for Molecular Discovery (BU-CMD). His research is focused in two major areas: the development of new synthetic methodologies for efficient chemical synthesis of complex molecules and the synthesis of complex chemical libraries.
Corey R. J. Stephenson received an Honours B.Sc. degree in 1998 from the University of Waterloo and a Ph.D. degree in 2005 from the University of Pittsburgh, where he worked under the direction of Prof. Peter Wipf. After carrying out postdoctoral research in the laboratory of Prof. Erick M. Carreira at ETH Zürich, Switzerland, he joined the Department of Chemistry at Boston University in 2007 as assistant professor and co-principal investigator in the Center for Chemical Methodology and Library Development (CMLD-BU). He was granted tenure and promoted to associate professor in February 2013. In July 2013, he joined the Department of Chemistry at the University of Michigan, Ann Arbor, as an associate professor and was promoted to professor in September 2015. His research interests include photoredox catalysis, natural product synthesis, and continuous flow chemistry.
The authors declare no competing financial interest.
References
- Nicolaou K. C.; Chen J. S.. Classics in Total Synthesis III; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Keasling J. D.; Mendoza A.; Baran P. S. Synthesis: A Constructive Debate. Nature 2012, 492, 188–189. 10.1038/492188a. [DOI] [PubMed] [Google Scholar]
- Mulzer J. Trying to Rationalize Total Synthesis. Nat. Prod. Rep. 2014, 31, 595–603. 10.1039/c3np70105k. [DOI] [PubMed] [Google Scholar]
- Trost B. M. The Atom Economy—A Search for Synthetic Efficiency. Science 1991, 254, 1471–1477. 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- Broadwater S. J.; Roth S. L.; Price K. E.; Kobašlija M.; McQuade D. T. One-Pot Multi-Step Synthesis: A Challenge Spawning Innovation. Org. Biomol. Chem. 2005, 3, 2899–2906. 10.1039/b506621m. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Chen J. S. The Art of Total Synthesis through Cascade Reactions. Chem. Soc. Rev. 2009, 38, 2993–3009. 10.1039/b903290h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey E. J. The Logic of Chemical Synthesis: Multistep Synthesis of Complex Carbogenic Molecules (Nobel Lecture). Angew. Chem., Int. Ed. Engl. 1991, 30, 455–465. 10.1002/anie.199104553. [DOI] [Google Scholar]
- Young I. S.; Baran P. S. Protecting-Group-Free Synthesis as an Opportunity for Invention. Nat. Chem. 2009, 1, 193–205. 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]
- Hoffmann R. W. Natural Product Synthesis: Changes over Time. Angew. Chem., Int. Ed. 2013, 52, 123–130. 10.1002/anie.201203319. [DOI] [PubMed] [Google Scholar]
- Fürstner A. Catalysis for Total Synthesis: A Personal Account. Angew. Chem., Int. Ed. 2014, 53, 8587–8598. 10.1002/anie.201402719. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C. Inspirations, Discoveries, and Future Perspectives in Total Synthesis. J. Org. Chem. 2009, 74, 951–972. 10.1021/jo802351b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttruff C. A.; Eastgate M. D.; Baran P. S. Natural Product Synthesis in the Age of Scalability. Nat. Prod. Rep. 2014, 31, 419–432. 10.1039/C3NP70090A. [DOI] [PubMed] [Google Scholar]
- Paterson I.; Anderson E. A. The Renaissance of Natural Products as Drug Candidates. Science 2005, 310, 451–453. 10.1126/science.1116364. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Hale C. R. H.; Nilewski C.; Ioannidou H. A. Constructing Molecular Complexity and Diversity: Total Synthesis of Natural Products of Biological and Medicinal Importance. Chem. Soc. Rev. 2012, 41, 5185–5238. 10.1039/c2cs35116a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M.; Snader K. M. The Influence of Natural Products upon Drug Discovery. Nat. Prod. Rep. 2000, 17, 215–234. 10.1039/a902202c. [DOI] [PubMed] [Google Scholar]
- Carey J. S.; Laffan D.; Thomson C.; Williams M. T. Analysis of the Reactions Used for the Preparation of Drug Candidate Molecules. Org. Biomol. Chem. 2006, 4, 2337–2347. 10.1039/b602413k. [DOI] [PubMed] [Google Scholar]
- Butler M. S. Natural Products to Drugs: Natural Product-Derived Compounds in Clinical Trials. Nat. Prod. Rep. 2008, 25, 475–516. 10.1039/b514294f. [DOI] [PubMed] [Google Scholar]
- Newhouse T.; Baran P. S.; Hoffmann R. W. The Economies of Synthesis. Chem. Soc. Rev. 2009, 38, 3010–3021. 10.1039/b821200g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grondal C.; Jeanty M.; Enders D. Organocatalytic Cascade Reactions as a New Tool in Total Synthesis. Nat. Chem. 2010, 2, 167–178. 10.1038/nchem.539. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Edmonds D. J.; Bulger P. G. Cascade Reactions in Total Synthesis. Angew. Chem., Int. Ed. 2006, 45, 7134–7186. 10.1002/anie.200601872. [DOI] [PubMed] [Google Scholar]
- Morton D.; Leach S.; Cordier C.; Warriner S.; Nelson A. Synthesis of Natural-Product-Like Molecules with Over Eighty Distinct Scaffolds. Angew. Chem., Int. Ed. 2009, 48, 104–109. 10.1002/anie.200804486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon R. A. Catalysis: The Key to Waste Minimization. J. Chem. Technol. Biotechnol. 1997, 68, 381–388. 10.1002/(SICI)1097-4660(199704)68:4<381::AID-JCTB620>3.3.CO;2-V. [DOI] [Google Scholar]
- Tucker J. L. Green Chemistry, a Pharmaceutical Perspective. Org. Process Res. Dev. 2006, 10, 315–319. 10.1021/op050227k. [DOI] [Google Scholar]
- Lipshutz B. H.; Ghorai S. Transitioning Organic Synthesis from Organic Solvents to Water. What’s Your E Factor?. Green Chem. 2014, 16, 3660–3679. 10.1039/C4GC00503A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns N. Z.; Baran P. S.; Hoffmann R. W. Redox Economy in Organic Synthesis. Angew. Chem., Int. Ed. 2009, 48, 2854–2867. 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Sorensen E. J. Rapid Complexity Generation in Natural Product Total Synthesis. Chem. Soc. Rev. 2009, 38, 2981–2982. 10.1039/b918568m. [DOI] [Google Scholar]
- Bach T.; Hehn J. P. Photochemical Reactions as Key Steps in Natural Product Synthesis. Angew. Chem., Int. Ed. 2011, 50, 1000–1045. 10.1002/anie.201002845. [DOI] [PubMed] [Google Scholar]
- Olivucci M.; Santoro F. Chemical Selectivity through Control of Excited-State Dynamics. Angew. Chem., Int. Ed. 2008, 47, 6322–6325. 10.1002/anie.200800898. [DOI] [PubMed] [Google Scholar]
- Schapiro I.; Melaccio F.; Laricheva E. N.; Olivucci M. Using the Computer to Understand the Chemistry of Conical Intersections. Photochem. Photobiol. Sci. 2011, 10, 867–886. 10.1039/c0pp00290a. [DOI] [PubMed] [Google Scholar]
- Hoffmann N. Photochemical Reactions of Aromatic Compounds and the Concept of the Photon as a Traceless Reagent. Photochem. Photobiol. Sci. 2012, 11, 1613–1641. 10.1039/c2pp25074h. [DOI] [PubMed] [Google Scholar]
- Turro N. J. Fun with Photons, Reactive Intermediates, and Friends. Skating on the Edge of the Paradigms of Physical Organic Chemistry, Organic Supramolecular Photochemistry, and Spin Chemistry. J. Org. Chem. 2011, 76, 9863–9890. 10.1021/jo201786a. [DOI] [PubMed] [Google Scholar]
- De Keukeleire D.; He S.-L. Photochemical Strategies for the Construction of Polycyclic Molecules. Chem. Rev. 1993, 93, 359–380. 10.1021/cr00017a017. [DOI] [Google Scholar]
- For a review on pharmaceuticals containing polycyclic scaffolds, see the following:Stockdale T. P.; Williams C. M. Pharmaceuticals that Contain Polycyclic Hydrocarbon Scaffolds. Chem. Soc. Rev. 2015, 44, 7737–7763. 10.1039/C4CS00477A. [DOI] [PubMed] [Google Scholar]
- Trommsdorff H. Ueber Santonin. Ann. Chem. Pharm. 1834, 11, 190–207. 10.1002/jlac.18340110207. [DOI] [Google Scholar]
- For a discussion on the beginnings of organic photochemistry, see the following:Roth H. D. The Beginnings of Organic Photochemistry. Angew. Chem., Int. Ed. Engl. 1989, 28, 1193–1207. 10.1002/anie.198911931. [DOI] [Google Scholar]
- Hoffmann N. Photochemical Reactions as Key Steps in Organic Synthesis. Chem. Rev. 2008, 108, 1052–1103. 10.1021/cr0680336. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Howbert J. J. Synthetic Studies on Arene-Olefin Cycloadditions: Total Synthesis of (±)-α-Cedrene. J. Am. Chem. Soc. 1981, 103, 688–690. 10.1021/ja00393a041. [DOI] [Google Scholar]
- Winkler J. D.; Rouse M. G.; Greaney M. F.; Harrison S. J.; Jeon Y. T. The First Total Synthesis of (±)-Ingenol. J. Am. Chem. Soc. 2002, 124, 9726–9728. 10.1021/ja026600a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinkert G.; Weber W.-D.; Schwartz U.; Dürner G. A Photochemical Route to (±)-Estrone. Angew. Chem., Int. Ed. Engl. 1980, 19, 1027–1029. 10.1002/anie.198010271. [DOI] [Google Scholar]
- Quinkert G.; Schwartz U.; Stark H.; Weber W.-D.; Baier H.; Adam F.; Dürner G. Asymmetric Total Synthesis of (+)-Estrone. Angew. Chem., Int. Ed. Engl. 1980, 19, 1029–1030. 10.1002/anie.198010291. [DOI] [Google Scholar]
- Quinkert G.; Stark H. Stereoselective Synthesis of Enantiomerically Pure Natural Products—Estrone as Example. Angew. Chem., Int. Ed. Engl. 1983, 22, 637–655. 10.1002/anie.198306373. [DOI] [Google Scholar]
- Crimmins M. T.; Pace J. M.; Nantermet P. G.; Kim-Meade A. S.; Thomas J. B.; Watterson S. H.; Wagman A. S. The Total Synthesis of (±)-Ginkgolide B. J. Am. Chem. Soc. 2000, 122, 8453–8463. 10.1021/ja001747s. [DOI] [Google Scholar]
- Ciamician G.; Silber P. Chemische Lichtwirkungen. Ber. Dtsch. Chem. Ges. 1908, 41, 1928–1935. 10.1002/cber.19080410272. [DOI] [Google Scholar]
- Crimmins M. T.; Reinhold T. L. Enone Olefin [2 + 2] Photochemical Cycloadditions. Org. React. 1993, 44, 297–588. 10.1002/0471264180.or044.02. [DOI] [Google Scholar]
- Abe M.; Ye J.; Mishima M. The Chemistry of Localized Singlet 1,3-Diradicals (Biradicals): From Putative Intermediates to Persistent Species and Unusual Molecules with a π-Single Bonded Character. Chem. Soc. Rev. 2012, 41, 3808–3820. 10.1039/c2cs00005a. [DOI] [PubMed] [Google Scholar]
- Abe M. Diradicals. Chem. Rev. 2013, 113, 7011–7088. 10.1021/cr400056a. [DOI] [PubMed] [Google Scholar]
- Suishu T.; Shimo T.; Somekawa K. Substituent Effects on the Regiochemistry of Enone-Alkene 2 + 2-Photocycloadditions. Experimental Results and FMO Analysis. Tetrahedron 1997, 53, 3545–3556. 10.1016/S0040-4020(97)00105-1. [DOI] [Google Scholar]
- Corey E. J.; Bass J. D.; LeMahieu R.; Mitra R. B. A Study of the Photochemical Reactions of 2-Cyclohexenones with Substituted Olefins. J. Am. Chem. Soc. 1964, 86, 5570–5583. 10.1021/ja01078a034. [DOI] [Google Scholar]
- Hastings D. J.; Weedon A. C. Origin of the Regioselectivity in the Photochemical Cycloaddition Reactions of Cyclic Enones with Alkenes: Chemical Trapping Evidence for the Structures, Mechanism of Formation, and Fates of 1,4-Biradical Intermediates. J. Am. Chem. Soc. 1991, 113, 8525–8527. 10.1021/ja00022a052. [DOI] [Google Scholar]
- Andrew D.; Hastings D. J.; Weedon A. C. The Mechanism of the Photochemical Cycloaddition Reaction between 2-Cyclopentenone and Polar Alkenes: Trapping of Triplet 1,4-Biradical Intermediates with Hydrogen Selenide. J. Am. Chem. Soc. 1994, 116, 10870–10882. 10.1021/ja00103a003. [DOI] [Google Scholar]
- Nicolaou K. C.; Sarlah D.; Shaw D. M. Total Synthesis and Revised Structure of Biyouyanagin A. Angew. Chem., Int. Ed. 2007, 46, 4708–4711. 10.1002/anie.200701552. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Wu T. R.; Sarlah D.; Shaw D. M.; Rowcliffe E.; Burton D. R. Total Synthesis, Revised Structure, and Biological Evaluation of Biyouyanagin A and Analogues Thereof. J. Am. Chem. Soc. 2008, 130, 11114–11121. 10.1021/ja802805c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C.; Li L.; Li Y.; Xie Z. Construction of Two Vicinal Quaternary Carbons by Asymmetric Allylic Alkylation: Total Synthesis of Hyperolactone C and (−)-Biyouyanagin A. Angew. Chem., Int. Ed. 2009, 48, 7853–7856. 10.1002/anie.200902908. [DOI] [PubMed] [Google Scholar]
- Mangion I. K.; MacMillan D. W. C. Total Synthesis of Brasoside and Littoralisone. J. Am. Chem. Soc. 2005, 127, 3696–3697. 10.1021/ja050064f. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S.; Kawamura M.; Takano S. Total Synthesis of (−)-Paeoniflorin. J. Am. Chem. Soc. 1994, 116, 4081–4082. 10.1021/ja00088a055. [DOI] [Google Scholar]
- Fleck M.; Bach T. Total Synthesis of the Tetracyclic Sesquiterpene (±)-Punctaporonin C. Angew. Chem., Int. Ed. 2008, 47, 6189–6191. 10.1002/anie.200801534. [DOI] [PubMed] [Google Scholar]
- Fleck M.; Bach T. Total Synthesis of Punctaporonin C by a Regio- and Stereoselective [2 + 2]-Photocycloaddition. Chem. - Eur. J. 2010, 16, 6015–6032. 10.1002/chem.201000036. [DOI] [PubMed] [Google Scholar]
- Srikrishna A.; Ramasastry S. S. V. Enantiospecific Total Synthesis of Phytoalexins, (+)-Solanascone, (+)-Dehydrosolanascone, and (+)-Anhydro-β-rotunol. Tetrahedron Lett. 2005, 46, 7373–7376. 10.1016/j.tetlet.2005.08.124. [DOI] [Google Scholar]
- Saya J. M.; Vos K.; Kleinnijenhuis R. A.; van Maarseveen J. H.; Ingemann S.; Hiemstra H. Total Synthesis of Aquatolide. Org. Lett. 2015, 17, 3892–3894. 10.1021/acs.orglett.5b01888. [DOI] [PubMed] [Google Scholar]
- Kleinnijenhuis R. A.; Timmer B. J. J.; Lutteke G.; De Gelder R.; Van Maarseveen J. H.; Smits J. M. M.; Hiemstra H. Formal Total Synthesis of Solanoeclepin A: Enantioselective Allene Diboration and Intramolecular [2 + 2]-Photocycloaddition for the Construction of the Tricyclic Core. Chem. - Eur. J. 2016, 22, 1266–1269. 10.1002/chem.201504894. [DOI] [PubMed] [Google Scholar]
- Carreira E. M.; Hastings C. A.; Shepard M. S.; Yerkey L. A.; Millward D. B. Asymmetric Induction in Intramolecular [2 + 2]-Photocycloadditions of 1,3-Disubstituted Allenes with Enones and Enoates. J. Am. Chem. Soc. 1994, 116, 6622–6630. 10.1021/ja00094a017. [DOI] [Google Scholar]
- For a review on enantioselective [2 + 2] cycloadditions, see the following:Xu Y.; Conner M. L.; Brown M. K. Cyclobutane and Cyclobutene Synthesis: Catalytic Enantioselective [2 + 2] Cycloadditions. Angew. Chem., Int. Ed. 2015, 54, 11918–11928. 10.1002/anie.201502815. [DOI] [PubMed] [Google Scholar]
- Maskill K. G.; Knowles J. P.; Elliott L. D.; Alder R. W.; Booker-Milburn K. I. Complexity from Simplicity: Tricyclic Aziridines from the Rearrangement of Pyrroles by Batch and Flow Photochemistry. Angew. Chem., Int. Ed. 2013, 52, 1499–1502. 10.1002/anie.201208892. [DOI] [PubMed] [Google Scholar]
- Lee-Ruff E.; Mladenova G. Enantiomerically Pure Cyclobutane Derivatives and Their Use in Organic Synthesis. Chem. Rev. 2003, 103, 1449–1484. 10.1021/cr010013a. [DOI] [PubMed] [Google Scholar]
- Namyslo J. C.; Kaufmann D. E. The Application of Cyclobutane Derivatives in Organic Synthesis. Chem. Rev. 2003, 103, 1485–1537. 10.1021/cr010010y. [DOI] [PubMed] [Google Scholar]
- Iriondo-Alberdi J.; Greaney M. F. Photocycloaddition in Natural Product Synthesis. Eur. J. Org. Chem. 2007, 2007, 4801–4815. 10.1002/ejoc.200700239. [DOI] [Google Scholar]
- Nagaoka H.; Shimano M.; Yamada Y. Total Synthesis of (±)-Gibberellic Acid. Tetrahedron Lett. 1989, 30, 971–974. 10.1016/S0040-4039(00)95293-6. [DOI] [Google Scholar]
- Morimoto T.; Horiguchi T.; Yamada K.; Tsutsumi K.; Kurosawa H.; Kakiuchi K. Acid-Catalyzed Rearrangement of an Allene-Cyclohexenone Photoadduct and its Application in the Synthesis of (±)-Pentalenene. Synthesis 2004, 2004, 753–756. 10.1055/s-2004-815988. [DOI] [Google Scholar]
- Paquette L. A.; Annis G. D. Total Synthesis of (±)-Pentalenene, the Least Oxidized Neutral Triquinane Metabolite of Streptomyces griseochromogenes. J. Am. Chem. Soc. 1983, 105, 7358–7363. 10.1021/ja00363a025. [DOI] [Google Scholar]
- Yamashita M.; Inaba T.; Shimizu T.; Kawasaki I.; Ohta S. Stereoconvergent Transformation of 1,2a-Disubstituted Benzo[b]cyclobuta[d]pyrans to 1,3-Disubstituted Tetrahydrodibenzofuran-4-ols and its Application to the Second-Generation Synthesis of (±)-Linderol A. Synlett 2004, 1897–1900. 10.1055/s-2004-830875. [DOI] [PubMed] [Google Scholar]
- Yamashita M.; Inaba T.; Nagahama M.; Shimizu T.; Kosaka S.; Kawasaki I.; Ohta S. Novel Stereoconvergent Transformation of 1,2a-Disubstituted 1,2,2a,8b-Tetrahydro-3H-benzo[b]cyclobuta[d]pyran-3-ones to 1,3-Disubstituted 1,2,4a,9b-Tetrahydrodibenzofuran-4-ols and its Application to the Second-Generation Synthesis of (±)-Linderol A. Org. Biomol. Chem. 2005, 3, 2296–2304. 10.1039/b503890a. [DOI] [PubMed] [Google Scholar]
- Yamashita M.; Yadav N. D.; Sawaki T.; Takao I.; Kawasaki I.; Sugimoto Y.; Miyatake A.; Murai K.; Takahara A.; Kurume A.; Ohta S. Asymmetric Total Synthesis of (−)-Linderol A. J. Org. Chem. 2007, 72, 5697–5703. 10.1021/jo070682+. [DOI] [PubMed] [Google Scholar]
- For a review on the synthetic applications of Grob fragmentations, see the following:Prantz K.; Mulzer J. Synthetic Applications of the Carbonyl Generating Grob Fragmentation. Chem. Rev. 2010, 110, 3741–3766. 10.1021/cr900386h. [DOI] [PubMed] [Google Scholar]
- Liu H.-J.; Lee S. P. Total Synthesis of 5-Epikessane and Dehydrokessane. Tetrahedron Lett. 1977, 18, 3699–3702. 10.1016/S0040-4039(01)83330-X. [DOI] [Google Scholar]
- Hansson T.; Wickberg B. A Short Enantiospecific Route to Isodaucane Sesquiterpenes from Limonene. On the Absolute Configuration of (+)-Aphanamol I and II. J. Org. Chem. 1992, 57, 5370–5376. 10.1021/jo00046a018. [DOI] [Google Scholar]
- Zalkow L. H.; Harris R. N. III.; Van Derveer D.; Bertrand J. A. Isocomene: A Novel Sesquiterpene from Isocoma Wrightii. X-Ray Crystal Structure of the Corresponding Diol. J. Chem. Soc., Chem. Commun. 1977, 456–457. 10.1039/c39770000456. [DOI] [Google Scholar]
- Bohlmann F.; Van N. L.; Pickardt J. Terpen-Derivate. 108. Über ein anomales Sesquiterpen aus Berkheya radula (Harv.) De Willd. Chem. Ber. 1977, 110, 3777–3781. 10.1002/cber.19771101207. [DOI] [Google Scholar]
- Tobe Y.; Yamashita T.; Kakiuchi K.; Odaira Y. Stereocontrolled Total Synthesis of (±)-Isocomene and (±)-β-Isocomene via Ring Enlargement. J. Chem. Soc., Chem. Commun. 1985, 898–899. 10.1039/C39850000898. [DOI] [Google Scholar]
- Rickborn B.; Gerkin R. M. The Lithium Salt-Catalyzed Epoxide-Carbonyl Rearrangement. J. Am. Chem. Soc. 1968, 90, 4193–4194. 10.1021/ja01017a071. [DOI] [Google Scholar]
- Tobe Y.; Yamashita S.; Yamashita T.; Kakiuchi K.; Odaira Y. Chelation-Controlled Regioselective Epoxide-Carbonyl Rearrangement: A Ring Enlargement Route to (±)-Modhephene. J. Chem. Soc., Chem. Commun. 1984, 0, 1259–1260. 10.1039/C39840001259. [DOI] [Google Scholar]
- Lange G. L.; Gottardo C. Free Radical Fragmentation of Derivatives of [2 + 2] Photoadducts. Tetrahedron Lett. 1990, 31, 5985–5988. 10.1016/S0040-4039(00)98009-2. [DOI] [Google Scholar]
- Crimmins M. T.; Dudek C. M.; Cheung A. W.-H. A Fragmentation-Rearrangement Sequence of Cyclobutylcarbinyl Radicals. Tetrahedron Lett. 1992, 33, 181–184. 10.1016/0040-4039(92)88044-6. [DOI] [Google Scholar]
- Crimmins M. T.; Huang S.; Guise-Zawacki L. E. Radical Fragmentation of Cyclobutanes: An Approach to Medium Ring Fused Carbocycles. Tetrahedron Lett. 1996, 37, 6519–6522. 10.1016/0040-4039(96)01433-5. [DOI] [Google Scholar]
- Lange G. L.; Merica A.; Chimanikire M. Total Synthesis of (±)-Dictamnol by a Free Radical Fragmentation/Elimination Sequence. Confirmation of its Revised Structure. Tetrahedron Lett. 1997, 38, 6371–6374. 10.1016/S0040-4039(97)01466-4. [DOI] [Google Scholar]
- Crimmins M. T.; Wang Z.; McKerlie L. A. Double Diastereoselection in Intramolecular Photocycloadditions: A Radical Rearrangement Approach to the Total Synthesis of the Spirovetivane Phytoalexin (±)-Lubiminol. J. Am. Chem. Soc. 1998, 120, 1747–1756. 10.1021/ja973824y. [DOI] [Google Scholar]
- For a review on SmI2-mediated reactions in total synthesis, see the following:Nicolaou K. C.; Ellery S. P.; Chen J. S. Samarium Diiodide Mediated Reactions in Total Synthesis. Angew. Chem., Int. Ed. 2009, 48, 7140–7165. 10.1002/anie.200902151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipe W. D.; Sorensen E. J. A Convergent Synthesis of the Tricyclic Architecture of the Guanacastepenes Featuring a Selective Ring Fragmentation. Org. Lett. 2002, 4, 2063–2066. 10.1021/ol0259342. [DOI] [PubMed] [Google Scholar]
- Shipe W. D.; Sorensen E. J. Convergent, Enantioselective Syntheses of Guanacastepenes A and E Featuring a Selective Cyclobutane Fragmentation. J. Am. Chem. Soc. 2006, 128, 7025–7035. 10.1021/ja060847g. [DOI] [PubMed] [Google Scholar]
- Lange G. L.; Gottardo C. Free Radical Fragmentation of [2 + 2] Photoadduct Derivatives. Formal Synthesis of Pentalenene. J. Org. Chem. 1995, 60, 2183–2187. 10.1021/jo00112a043. [DOI] [Google Scholar]
- Lange G. L.; Gottardo C. Synthesis of the Guaiane (±)-Alismol Using a Free Radical Fragmentation/Elimination Sequence. Tetrahedron Lett. 1994, 35, 8513–8516. 10.1016/S0040-4039(00)78424-3. [DOI] [Google Scholar]
- Lange G. L.; Gottardo C.; Merica A. Synthesis of Terpenoids Using a Free Radical Fragmentation/Elimination Sequence. J. Org. Chem. 1999, 64, 6738–6744. 10.1021/jo990307k. [DOI] [PubMed] [Google Scholar]
- Corbett R. E.; Lauren D. R.; Weavers R. T. The Structure of Laurenene, a New Diterpene from the Essential Oil of Dacrydium cupressinum. Part 1. J. Chem. Soc., Perkin Trans. 1 1979, 1774–1790. 10.1039/p19790001774. [DOI] [Google Scholar]
- Corbett R. E.; Couldwell C. M.; Lauren D. R.; Weavers R. T. The Structure of Laurenene, a New Diterpene from the Essential Oil of Dacrydium cupressinum. Part 2. Crystal Structure. J. Chem. Soc., Perkin Trans. 1 1979, 1791–1794. 10.1039/p19790001791. [DOI] [Google Scholar]
- Crimmins M. T.; Gould L. D. Intramolecular Photocycloaddition-Cyclobutane Fragmentation: Total Synthesis of (+)-Laurenene. J. Am. Chem. Soc. 1987, 109, 6199–6200. 10.1021/ja00254a058. [DOI] [Google Scholar]
- Crimmins M. T.; Mascarella S. W. Intramolecular Photocycloaddition-Cyclobutane Fragmentation: Total Synthesis of (+)-Silphinene. J. Am. Chem. Soc. 1986, 108, 3435–3438. 10.1021/ja00272a044. [DOI] [Google Scholar]
- Birman V. B.; Danishefsky S. J. The Total Synthesis of (±)-Merrilactone A. J. Am. Chem. Soc. 2002, 124, 2080–2081. 10.1021/ja012495d. [DOI] [PubMed] [Google Scholar]
- Inoue M.; Sato T.; Hirama M. Total Synthesis of Merrilactone A. J. Am. Chem. Soc. 2003, 125, 10772–10773. 10.1021/ja036587+. [DOI] [PubMed] [Google Scholar]
- Huang J.-m.; Yokoyama R.; Yang C.-s.; Fukuyama Y. Merrilactone A, a Novel Neurotrophic Sesquiterpene Dilactone from Illicium merrillianum. Tetrahedron Lett. 2000, 41, 6111–6114. 10.1016/S0040-4039(00)01023-6. [DOI] [Google Scholar]
- Huang J.-M.; Yang C.-S.; Tanaka M.; Fukuyama Y. Structures of Merrilactones B and C, Novel Anislactone-Type Sesquiterpenes from Illicium merrillianum, and Chemical Conversion of Anislactone B to Merrilactone A. Tetrahedron 2001, 57, 4691–4698. 10.1016/S0040-4020(01)00418-5. [DOI] [Google Scholar]
- Hefti F. Neurotrophic Factor Therapy for Nervous System Degenerative Diseases. J. Neurobiol. 1994, 25, 1418–1435. 10.1002/neu.480251109. [DOI] [PubMed] [Google Scholar]
- Hefti F. Pharmacology of Neurotrophic Factors. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 239–267. 10.1146/annurev.pharmtox.37.1.239. [DOI] [PubMed] [Google Scholar]
- Sofroniew M. V.; Howe C. L.; Mobley W. C. Nerve Growth Factor Signaling, Neuroprotection, and Neural Repair. Annu. Rev. Neurosci. 2001, 24, 1217–1281. 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- Lu B.; Pang P. T.; Woo N. H. The Yin and Yang of Neurotrophin Action. Nat. Rev. Neurosci. 2005, 6, 603–614. 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- Park H.; Poo M.-m. Neurotrophin Regulation of Neural Circuit Development and Function. Nat. Rev. Neurosci. 2013, 14, 7–23. 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- Huang T.; Larsen K. T.; Ried-Larsen M.; Møller N. C.; Andersen L. B. The Effects of Physical Activity and Exercise on Brain-Derived Neurotrophic Factor in Healthy Humans: A Review. Scand. J. Med. Sci. Sports 2014, 24, 1–10. 10.1111/sms.12069. [DOI] [PubMed] [Google Scholar]
- For a review on the chemistry and biology of neurotrophic natural products, see the following:Xu J.; Lacoske M. H.; Theodorakis E. A. Neurotrophic Natural Products: Chemistry and Biology. Angew. Chem., Int. Ed. 2014, 53, 956–987. 10.1002/anie.201302268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta G.; Singh S. R. Total Synthesis of (±)-Merrilactone A. Angew. Chem., Int. Ed. 2006, 45, 953–955. 10.1002/anie.200503618. [DOI] [PubMed] [Google Scholar]
- Inoue M.; Sato T.; Hirama M. Asymmetric Total Synthesis of (−)-Merrilactone A: Use of a Bulky Protecting Group as Long-Range Stereocontrolling Element. Angew. Chem., Int. Ed. 2006, 45, 4843–4848. 10.1002/anie.200601358. [DOI] [PubMed] [Google Scholar]
- Shi L.; Meyer K.; Greaney M. F. Synthesis of (±)-Merrilactone A and (±)-Anislactone A. Angew. Chem., Int. Ed. 2010, 49, 9250–9253. 10.1002/anie.201005156. [DOI] [PubMed] [Google Scholar]
- Strømgaard K.; Nakanishi K. Chemistry and Biology of Terpene Trilactones from Ginkgo Biloba. Angew. Chem., Int. Ed. 2004, 43, 1640–1658. 10.1002/anie.200300601. [DOI] [PubMed] [Google Scholar]
- van Beek T. A. Ginkgolides and Bilobalide: Their Physical, Chromatographic and Spectroscopic Properties. Bioorg. Med. Chem. 2005, 13, 5001–5012. 10.1016/j.bmc.2005.05.056. [DOI] [PubMed] [Google Scholar]
- Nakanishi K. Terpene Trilactones from Gingko Biloba: From Ancient Times to the 21st Century. Bioorg. Med. Chem. 2005, 13, 4987–5000. 10.1016/j.bmc.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Leistner E.; Drewke C. Ginkgo Biloba and Ginkgotoxin. J. Nat. Prod. 2010, 73, 86–92. 10.1021/np9005019. [DOI] [PubMed] [Google Scholar]
- Furukawa S. Constituents of Ginkgo Biloba L. Leaves. I. Sci. Papers Inst. Phys. Chem. Res. 1932, 19, 27–38. [Google Scholar]
- Furukawa S. Constituents of Ginkgo Biloba L. Leaves. II. Sci. Papers Inst. Phys. Chem. Res. 1932, 19, 39–42. [Google Scholar]
- Furukawa S. Constituents of Ginkgo Biloba L. Leaves. IV. Sci. Papers Inst. Phys. Chem. Res. 1933, 21, 278–285. [Google Scholar]
- Maruyama M.; Terahara A.; Itagaki Y.; Nakanishi K. The Ginkgolides. I. Isolation and Characterization of the Various Groups. Tetrahedron Lett. 1967, 8, 299–302. 10.1016/S0040-4039(00)71538-3. [DOI] [Google Scholar]
- Maruyama M.; Terahara A.; Nakadaira Y.; Woods M. C.; Nakanishi K. The Ginkgolides. III. The Structure of the Ginkgolides. Tetrahedron Lett. 1967, 8, 309–313. 10.1016/S0040-4039(00)71540-1. [DOI] [Google Scholar]
- Maruyama M.; Terahara A.; Nakadaira Y.; Woods M. C.; Takagi Y.; Nakanishi K. The Ginkgolides. IV. Stereochemistry of the Ginkgolides. Tetrahedron Lett. 1967, 8, 315–319. 10.1016/S0040-4039(00)71541-3. [DOI] [Google Scholar]
- Sakabe N.; Takada S.; Okabe K. The Structure of Ginkgolide A, a Novel Diterpenoid Trilactone. Chem. Commun. 1967, 259–261. 10.1039/c19670000259. [DOI] [Google Scholar]
- Nakanishi K. The Ginkgolides. Pure Appl. Chem. 1967, 14, 89–114. 10.1351/pac196714010089. [DOI] [PubMed] [Google Scholar]
- Nakanishi K.; Habaguchi K.; Nakadaira Y.; Woods M. C.; Maruyama M.; Major R. T.; Alauddin M.; Patel A. R.; Weinges K.; Baehr W. Structure of Bilobalide, a Rare tert-Butyl Containing Sesquiterpenoid Related to the C20-Ginkgolides. J. Am. Chem. Soc. 1971, 93, 3544–3546. 10.1021/ja00743a051. [DOI] [Google Scholar]
- Weinges K.; Hepp M.; Jaggy H. Chemie der Ginkgolide, II. Isolierung und Strukturaufklärung eines neuen Ginkgolids. Liebigs Ann. Chem. 1987, 1987, 521–526. 10.1002/jlac.198719870368. [DOI] [Google Scholar]
- Corey E. J.; Ghosh A. K. Total Synthesis of Ginkgolide A. Tetrahedron Lett. 1988, 29, 3205–3206. 10.1016/0040-4039(88)85122-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey E. J.; Kang M.-C.; Desai M. C.; Ghosh A. K.; Houpis I. N. Total Synthesis of (±)-Ginkgolide B. J. Am. Chem. Soc. 1988, 110, 649–651. 10.1021/ja00210a083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey E. J.; Su W.-G. Total Synthesis of a C15 Ginkgolide, (±)-Bilobalide. J. Am. Chem. Soc. 1987, 109, 7534–7536. 10.1021/ja00258a050. [DOI] [Google Scholar]
- Corey E. J.; Su W.-G. Enantioselective Total Synthesis of Bilobalide, a C15 Ginkgolide. Tetrahedron Lett. 1988, 29, 3423–3426. 10.1016/0040-4039(88)85179-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on the synthesis of ginkgolides, see the following:Corey E. J. Robert Robinson Lecture. Retrosynthetic Thinking—Essentials and Examples. Chem. Soc. Rev. 1988, 17, 111–133. 10.1039/CS9881700111. [DOI] [Google Scholar]
- Crimmins M. T.; Pace J. M.; Nantermet P. G.; Kim-Meade A. S.; Thomas J. B.; Watterson S. H.; Wagman A. S. Total Synthesis of (±)-Ginkgolide B. J. Am. Chem. Soc. 1999, 121, 10249–10250. 10.1021/ja993013p. [DOI] [Google Scholar]
- Crimmins M. T.; Jung D. K.; Gray J. L. A Total Synthesis of (±)-Bilobalide. J. Am. Chem. Soc. 1992, 114, 5445–5447. 10.1021/ja00039a077. [DOI] [Google Scholar]
- Crimmins M. T.; Jung D. K.; Gray J. L. Synthetic Studies on the Ginkgolides: Total Synthesis of (±)-Bilobalide. J. Am. Chem. Soc. 1993, 115, 3146–3155. 10.1021/ja00061a014. [DOI] [Google Scholar]
- Vedejs E. Method for Direct Hydroxylation of Enolates. Transition Metal Peroxide Reactions. J. Am. Chem. Soc. 1974, 96, 5944–5946. 10.1021/ja00825a047. [DOI] [Google Scholar]
- Vedejs E.; Larsen S. Hydoxylation of Enolates with Oxodiperoxymolybdenum(pyridine)(hexamethylphosphoric triamide), MoO5 · Py · HMPA (MoOPH): 3-Hydroxy-1,7,7-trimethylbicyclo[2.2.1]heptan-2-one. Org. Synth. 2003, 127. 10.1002/0471264180.os064.20. [DOI] [Google Scholar]
- Heilbron I.; Jones E. R. H.; Sondheimer F. Researches on Acetylenic Compounds. Part XV. The Oxidation of Primary Acetylenic Carbinols and Glycols. J. Chem. Soc. 1949, 604–607. 10.1039/jr9490000604. [DOI] [Google Scholar]
- Crimmins M. T.; Nantermet P. G. Addition of Zinc Homoenolates to Acetylenic Esters: A Formal [3 + 2] Cycloaddition. J. Org. Chem. 1990, 55, 4235–4237. 10.1021/jo00301a003. [DOI] [Google Scholar]
- Crimmins M. T.; Nantermet P. G.; Trotter B. W.; Vallin I. M.; Watson P. S.; McKerlie L. A.; Reinhold T. L.; Cheung A. W.-H.; Stetson K. A. Addition of Zinc Homoenolates to Acetylenic Esters and Amides: A Formal [3 + 2] Cycloaddition. J. Org. Chem. 1993, 58, 1038–1047. 10.1021/jo00057a013. [DOI] [Google Scholar]
- Cargill R. L.; Beckham M. E.; Siebert A. E.; Dorn J. Synthesis and Characterization of Bridged Tricyclic Ketones. J. Org. Chem. 1965, 30, 3647–3650. 10.1021/jo01022a014. [DOI] [Google Scholar]
- Cargill R. L.; Damewood J. R.; Cooper M. M. Photoreduction of a β,γ-Unsaturated Ketone. J. Am. Chem. Soc. 1966, 88, 1330–1331. 10.1021/ja00958a052. [DOI] [Google Scholar]
- Cargill R. L.; Crawford J. W. Synthesis and Rearrangement of Tricyclo[4.3.2.01,6]undec-10-en-2-one. Tetrahedron Lett. 1967, 8, 169–171. 10.1016/S0040-4039(00)90510-0. [DOI] [Google Scholar]
- Peet N. P.; Cargill R. L.; Bushey D. F. Synthesis and Acid-Catalyzed Rearrangements of Tricyclo[4.3.2.0]undecanones. J. Org. Chem. 1973, 38, 1218–1221. 10.1021/jo00946a034. [DOI] [Google Scholar]
- Cargill R. L.; Jackson T. E.; Peet N. P.; Pond D. M. Acid-Catalyzed Rearrangements of β,γ-Unsaturated Ketones. Acc. Chem. Res. 1974, 7, 106–113. 10.1021/ar50076a002. [DOI] [Google Scholar]
- Pirrung M. C. Total Synthesis of (±)-Isocomene. J. Am. Chem. Soc. 1979, 101, 7130–7131. 10.1021/ja00517a087. [DOI] [Google Scholar]
- Pirrung M. C. Total Synthesis of (±)-Isocomene and Related Studies. J. Am. Chem. Soc. 1981, 103, 82–87. 10.1021/ja00391a016. [DOI] [Google Scholar]
- Rawal V. H.; Dufour C.; Eschbach A. Stereocontrolled Synthesis of Isocomene by a Novel Photocycloaddition-Fragmentation Strategy. J. Chem. Soc., Chem. Commun. 1994, 1797–1798. 10.1039/C39940001797. [DOI] [Google Scholar]
- Rawal V. H.; Eschbach A.; Dufour C.; Iwasa S. Photocyclization-Fragmentation Route to Di- and Triquinanes: Stereocontrolled Asymmetric Synthesis of (−)-Isocomene. Pure Appl. Chem. 1996, 68, 675–678. 10.1351/pac199668030675. [DOI] [Google Scholar]
- Wender P. A.; Dreyer G. B. Synthetic Studies on Arene-Olefin Cycloadditions. II. Total Synthesis of (±)-Isocomene. Tetrahedron 1981, 37, 4445–4450. 10.1016/0040-4020(81)80011-7. [DOI] [Google Scholar]
- Uyehara T.; Murayama T.; Sakai K.; Onda K.; Ueno M.; Sato T. Rearrangement Approaches to Cyclic Skeletons. XIII. Total Synthesis of Triquinane Sesquiterpenes, (±)-Modhephene, and (±)-Isocomene, on the Basis of Formal Substitution at Both Bridgeheads of a Bicyclo[2.2.2]oct-5-en-2-one. Bull. Chem. Soc. Jpn. 1998, 71, 231–242. 10.1246/bcsj.71.231. [DOI] [Google Scholar]
- Schreiber S. L.; Santini C. A Cyclobutene Bridgehead Approach to the Germacranes. Tetrahedron Lett. 1981, 22, 4651–4654. 10.1016/S0040-4039(01)83004-5. [DOI] [Google Scholar]
- Schreiber S. L.; Santini C. Cyclobutene Bridgehead Olefin Route to the American Cockroach Sex Pheromone, Periplanone-B. J. Am. Chem. Soc. 1984, 106, 4038–4039. 10.1021/ja00326a028. [DOI] [Google Scholar]
- Schreiber S. L. [2 + 2] Photocycloadditions in the Synthesis of Chiral Molecules. Science 1985, 227, 857–863. 10.1126/science.4038558. [DOI] [PubMed] [Google Scholar]
- White J. D.; Li Y.; Kim J.; Terinek M. Cyclobutane Synthesis and Fragmentation. A Cascade Route to the Lycopodium Alkaloid (−)-Huperzine A. J. Org. Chem. 2015, 80, 11806–11817. 10.1021/acs.joc.5b01619. [DOI] [PubMed] [Google Scholar]
- Paternò E.; Chieffi G. Synthesis in Organic Chemistry Using Light. Note II. Compounds of Unsaturated Hydrocarbons with Aldehydes and Ketones. Gazz. Chim. Ital. 1909, 39, 341–361. [Google Scholar]
- Büchi G.; Inman C. G.; Lipinsky E. S. Light-Catalyzed Organic Reactions. I. The Reaction of Carbonyl Compounds with 2-Methyl-2-butene in the Presence of Ultraviolet Light. J. Am. Chem. Soc. 1954, 76, 4327–4331. 10.1021/ja01646a024. [DOI] [Google Scholar]
- Freilich S. C.; Peters K. S. Observation of the 1,4-Biradical in the Paterno-Buchi Reaction. J. Am. Chem. Soc. 1981, 103, 6255–6257. 10.1021/ja00410a062. [DOI] [Google Scholar]
- Caldwell R. A.; Majima T.; Pac C. Some Structural Effects on Triplet Biradical Lifetimes. Norrish II and Paterno-Buchi Biradicals. J. Am. Chem. Soc. 1982, 104, 629–630. 10.1021/ja00366a050. [DOI] [Google Scholar]
- Freilich S. C.; Peters K. S. Picosecond Dynamics of the Paterno-Buchi Reaction. J. Am. Chem. Soc. 1985, 107, 3819–3822. 10.1021/ja00299a011. [DOI] [Google Scholar]
- Abe M.; Kawakami T.; Ohata S.; Nozaki K.; Nojima M. Mechanism of Stereo- and Regioselectivity in the Paternò-Büchi Reaction of Furan Derivatives with Aromatic Carbonyl Compounds: Importance of the Conformational Distribution in the Intermediary Triplet 1,4-Diradicals. J. Am. Chem. Soc. 2004, 126, 2838–2846. 10.1021/ja039491o. [DOI] [PubMed] [Google Scholar]
- Brogaard R. Y.; Schalk O.; Boguslavskiy A. E.; Enright G. D.; Hopf H.; Raev V.; Tarcoveanu E.; Sølling T. I.; Stolow A. The Paternò-Büchi Reaction: Importance of Triplet States in the Excited-State Reaction Pathway. Phys. Chem. Chem. Phys. 2012, 14, 8572–8580. 10.1039/c2cp40819h. [DOI] [PubMed] [Google Scholar]
- Griesbeck A. G.; Abe M.; Bondock S. Selectivity Control in Electron Spin Inversion Processes: Regio- and Stereochemistry of Paternò-Büchi Photocycloadditions as a Powerful Tool for Mapping Intersystem Crossing Processes. Acc. Chem. Res. 2004, 37, 919–928. 10.1021/ar040081u. [DOI] [PubMed] [Google Scholar]
- Bach T. Stereoselective Intermolecular [2 + 2]-Photocycloaddition Reactions and Their Application in Synthesis. Synthesis 1998, 1998, 683–703. 10.1055/s-1998-2054. [DOI] [Google Scholar]
- Abe M. Recent Progress Regarding Regio-, Site-, and Stereoselective Formation of Oxetanes in Paternò-Büchi Reactions. J. Chin. Chem. Soc. 2008, 55, 479–486. 10.1002/jccs.200800072. [DOI] [Google Scholar]
- D’Auria M.; Racioppi R. Oxetane Synthesis through the Paternò-Büchi Reaction. Molecules 2013, 18, 11384–11428. 10.3390/molecules180911384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porco J. A. Jr.; Schreiber S. L.. The Paterno–Büchi Reaction. In Comprehensive Organic Synthesis; Trost B. M., Fleming I., Paquette L. A., Eds.; Pergamon Press: Oxford, 1991; Vol. 5, pp 151–192. [Google Scholar]
- Griesbeck A. G.Photocycloadditions of Alkenes to Excited Carbonyls. In Synthetic Organic Photochemistry; Griesbeck A. G., Mattay J., Eds.; Marcel Dekker: New York, 2005; pp 89–139. [Google Scholar]
- Abe M.Formation of a Four-Membered Ring: Oxetanes. In Handbook of Synthetic Photochemistry; Albini A., Fagnoni M., Eds.; Wiley-VCH: Weinheim, Germany, 2010; pp 217–239. [Google Scholar]
- Bach T.; Brummerhop H.; Harms K. The Synthesis of (+)-Preussin and Related Pyrrolidinols by Diastereoselective Paternò-Büchi Reactions of Chiral 2-Substituted 2,3-Dihydropyrroles. Chem. - Eur. J. 2000, 6, 3838–3848. 10.1002/1521-3765(20001016)6:20<3838::AID-CHEM3838>3.3.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Hambalek R.; Just G. A Short Synthesis of (±)-Oxetanocin. Tetrahedron Lett. 1990, 31, 5445–5448. 10.1016/S0040-4039(00)97868-7. [DOI] [Google Scholar]
- Boxall R. J.; Ferris L.; Grainger R. S. Synthesis of C-13 Oxidised Cuparene and Herbertane Sesquiterpenes via a Paternò-Büchi Photocyclisation-Oxetane Fragmentation Strategy: Total Synthesis of 1,13-Herbertenediol. Synlett 2004, 2379–2381. 10.1055/s-2004-832813. [DOI] [Google Scholar]
- Iriondo-Alberdi J.; Perea-Buceta J. E.; Greaney M. F. A Paternò-Büchi Approach to the Synthesis of Merrilactone A. Org. Lett. 2005, 7, 3969–3971. 10.1021/ol0514496. [DOI] [PubMed] [Google Scholar]
- Omura K.; Swern D. Oxidation of Alcohols by “Activated” Dimethyl Sulfoxide. A Preparative, Steric and Mechanistic Study. Tetrahedron 1978, 34, 1651–1660. 10.1016/0040-4020(78)80197-5. [DOI] [Google Scholar]
- Freeman P. K.; Hutchinson L. L. Organolithium Reagents from Alkyl Halides and Lithium Di-tert-butylbiphenyl. Tetrahedron Lett. 1976, 17, 1849–1852. 10.1016/S0040-4039(00)92942-3. [DOI] [Google Scholar]
- Freeman P. K.; Hutchinson L. L. Alkyllithium Reagents from Alkyl Halides and Lithium Radical Anions. J. Org. Chem. 1980, 45, 1924–1930. 10.1021/jo01298a034. [DOI] [Google Scholar]
- Miles D. H.; Kokpol U.; Bhattacharyya J.; Atwood J. L.; Stone K. E.; Bryson T. A.; Wilson C. Structure of Sarracenin. An Unusual Enol Diacetal Monoterpene from the Insectivorous Plant Sarracenia flava. J. Am. Chem. Soc. 1976, 98, 1569–1573. 10.1021/ja00422a048. [DOI] [Google Scholar]
- Hoye T. R.; Richardson W. S. A Short, Oxetane-Based Synthesis of (±)-Sarracenin. J. Org. Chem. 1989, 54, 688–693. 10.1021/jo00264a034. [DOI] [Google Scholar]
- For a review on the photochemistry of thiocarbonyl compounds, see the following:Coyle J. D. The Photochemistry of Thiocarbonyl Compounds. Tetrahedron 1985, 41, 5393–5425. 10.1016/S0040-4020(01)91341-9. [DOI] [Google Scholar]
- Padwa A.; Jacquez M. N.; Schmidt A. An Approach toward Azacycles Using Photochemical and Radical Cyclizations of N-Alkenyl Substituted 5-Thioxopyrrolidin-2-ones. J. Org. Chem. 2004, 69, 33–45. 10.1021/jo035127w. [DOI] [PubMed] [Google Scholar]
- De Mayo P.; Takeshita H.; Satta A. B. M. A. The Photochemical Synthesis of 1,5-Diketones and their Cyclisation: A New Annulation Process. Proc. Chem. Soc. 1962, 119. [Google Scholar]
- De Mayo P.; Takeshita H. Photochemical Syntheses: 6. The Formation of Heptandiones from Acetylacetone and Alkenes. Can. J. Chem. 1963, 41, 440–449. 10.1139/v63-061. [DOI] [Google Scholar]
- Hikino H.; De Mayo P. Photochemical Cycloaddition as a Device for General Annelation. J. Am. Chem. Soc. 1964, 86, 3582–3583. 10.1021/ja01071a052. [DOI] [Google Scholar]
- De Mayo P. Photochemical Syntheses. 37. Enone Photoannelation. Acc. Chem. Res. 1971, 4, 41–47. 10.1021/ar50038a001. [DOI] [Google Scholar]
- Oppolzer W. The Intramolecular [2 + 2] Photoaddition/Cyclobutane-Fragmentation Sequence in Organic Synthesis. Acc. Chem. Res. 1982, 15, 135–141. 10.1021/ar00077a002. [DOI] [Google Scholar]
- Benchikh le-Hocine M.; Do Khac D.; Fetizon M. Model Studies in Taxane Diterpene Synthesis. Part III. Synth. Commun. 1992, 22, 245–255. 10.1080/00397919208021299. [DOI] [Google Scholar]
- Disanayaka B. W.; Weedon A. C. A Short Synthesis of Hirsutene Using the de Mayo Reaction. J. Chem. Soc., Chem. Commun. 1985, 1282–1283. 10.1039/c39850001282. [DOI] [Google Scholar]
- Disanayaka B. W.; Weedon A. C. Application of the de Mayo Reaction to the Preparation of Tricyclo[6.3.0.02,6]undecanes: A Photochemical Synthesis of (±)-Hirsutene. J. Org. Chem. 1987, 52, 2905–2910. 10.1021/jo00389a046. [DOI] [Google Scholar]
- Büchi G.; Carlson J. A.; Powell J. E. Jr.; Tietze L. F. The Total Synthesis of Loganin. J. Am. Chem. Soc. 1970, 92, 2165–2167. 10.1021/ja00710a078. [DOI] [Google Scholar]
- Büchi G.; Carlson J. A.; Powell J. E.; Tietze L. F. Total Synthesis of Loganin. J. Am. Chem. Soc. 1973, 95, 540–545. 10.1021/ja00783a038. [DOI] [Google Scholar]
- Partridge J. J.; Chadha N. K.; Uskokovic M. R. Asymmetric Synthesis of Loganin. Stereospecific Formation of (1R,2R)- and (1S,2S)-2-Methyl-3-cyclopenten-1-ol and (2R)- and (2S)-2-Methylcyclopentanone. J. Am. Chem. Soc. 1973, 95, 532–540. 10.1021/ja00783a037. [DOI] [PubMed] [Google Scholar]
- Pearlman B. A. A Method for Effecting the Equivalent of a de Mayo Reaction with Formyl Acetic Ester. J. Am. Chem. Soc. 1979, 101, 6398–6404. 10.1021/ja00515a039. [DOI] [Google Scholar]
- Pearlman B. A. A Total Synthesis of Reserpine. J. Am. Chem. Soc. 1979, 101, 6404–6408. 10.1021/ja00515a040. [DOI] [Google Scholar]
- Barker A. J.; Pattenden G. Zizaane Sesquiterpenes. Synthesis of the Coates-Sowerby Tricyclic Ketone. Tetrahedron Lett. 1981, 22, 2599–2600. 10.1016/S0040-4039(01)90530-1. [DOI] [Google Scholar]
- Barker A. J.; Pattenden G. Synthetic photochemistry. A New Synthesis of (±)-Zizaene via an Intramolecular Variant of the De Mayo Reaction. J. Chem. Soc., Perkin Trans. 1 1983, 1901–1904. 10.1039/P19830001901. [DOI] [Google Scholar]
- Oppolzer W.; Godel T. A New and Efficient Total Synthesis of (±)-Longifolene. J. Am. Chem. Soc. 1978, 100, 2583–2584. 10.1021/ja00476a071. [DOI] [Google Scholar]
- Oppolzer W.; Godel T. Syntheses of (±)- and Enantiomerically Pure (+)-Longifolene and of (±)- and Enantiomerically Pure (+)-Sativene by an Intramolecular de Mayo Reaction. Helv. Chim. Acta 1984, 67, 1154–1167. 10.1002/hlca.19840670429. [DOI] [Google Scholar]
- For the first isolation of longifolene, see the following:Simonsen J. L. The Constituents of Indian Turpentine from Pinus longifolia, Roxb. Part I. J. Chem. Soc., Trans. 1920, 117, 570–578. 10.1039/CT9201700570. [DOI] [Google Scholar]
- Moffett R. H.; Rogers D. The Molecular Configuration of Longifolene Hydrochloride. Chem. Ind. 1953, 916–917. [Google Scholar]
- Ourisson G.; Longifolene V. Absolute Configuration of Longifolene, Stereochemistry of its Derivatives. Stereochemistry of the Santalenes. Bull. Soc. Chim. Fr. 1955, 895–902. [Google Scholar]
- Corey E. J.; Ohno M.; Vatakencherry P. A.; Mitra R. B. Total Synthesis of d,l-Longifolene. J. Am. Chem. Soc. 1961, 83, 1251–1253. 10.1021/ja01466a056. [DOI] [Google Scholar]
- Corey E. J.; Ohno M.; Mitra R. B.; Vatakencherry P. A. Total Synthesis of Longifolene. J. Am. Chem. Soc. 1964, 86, 478–485. 10.1021/ja01057a039. [DOI] [Google Scholar]
- Volkmann R. A.; Andrews G. C.; Johnson W. S. Novel Synthesis of Longifolene. J. Am. Chem. Soc. 1975, 97, 4777–4779. 10.1021/ja00849a062. [DOI] [Google Scholar]
- Ho G. A.; Nouri D. H.; Tantillo D. J. The Cationic Cascade Route to Longifolene. J. Org. Chem. 2005, 70, 5139–5143. 10.1021/jo0503658. [DOI] [PubMed] [Google Scholar]
- For a review on longifolene and its derivatives, see the following:Dev S. Aspects of Longifolene Chemistry. An Example of Another Facet of Natural Products Chemistry. Acc. Chem. Res. 1981, 14, 82–88. 10.1021/ar00063a004. [DOI] [Google Scholar]
- Galal A. M.; Abourashed E. A.; Ross S. A.; ElSohly M. A.; Al-Said M. S.; El-Feraly F. S. Daucane Sesquiterpenes from Ferula hermonis. J. Nat. Prod. 2001, 64, 399–400. 10.1021/np000526x. [DOI] [PubMed] [Google Scholar]
- Yang S.-P.; Cai Y.-J.; Zhang B.-L.; Tong L.-J.; Xie H.; Wu Y.; Lin L.-P.; Ding J.; Yue J.-M. Structural Modification of an Angiogenesis Inhibitor Discovered from Traditional Chinese Medicine and a Structure-Activity Relationship Study. J. Med. Chem. 2008, 51, 77–85. 10.1021/jm070906g. [DOI] [PubMed] [Google Scholar]
- Chen H.-D.; He X.-F.; Ai J.; Geng M.-Y.; Yue J.-M. Trigochilides A and B, Two Highly Modified Daphnane-Type Diterpenoids from Trigonostemon chinensis. Org. Lett. 2009, 11, 4080–4083. 10.1021/ol901506x. [DOI] [PubMed] [Google Scholar]
- For a review on the biogenesis, isolation and chemistry of the daucane (carotane) sesquiterpenes, see the following:Ghisalberti E. L. The Daucane (Carotane) Class of Sesquiterpenes. Phytochemistry 1994, 37, 597–623. 10.1016/S0031-9422(00)90327-3. [DOI] [Google Scholar]
- Yamasaki M. Total Synthesis of the Sesquiterpene (−)-Daucene. J. Chem. Soc., Chem. Commun. 1972, 606b–607. 10.1039/C3972000606B. [DOI] [Google Scholar]
- De Broissia H.; Levisalles J.; Rudler H. Total Synthesis of the Sesquiterpenes (+)-Daucene, (+)-Carotol, and (−)-Daucol. J. Chem. Soc., Chem. Commun. 1972, 855. 10.1039/C39720000855. [DOI] [Google Scholar]
- Mehta G.; Krishnamurthy N. New Approach towards Carotane Sesquiterpenes: A Short Synthesis of (−)-Daucene. Synth. Commun. 1988, 18, 1267–1274. 10.1080/00397918808060920. [DOI] [Google Scholar]
- Park Y. S.; Little R. D. Redox Electron-Transfer Reactions: Electrochemically Mediated Rearrangement, Mechanism, and a Total Synthesis of Daucene. J. Org. Chem. 2008, 73, 6807–6815. 10.1021/jo801199s. [DOI] [PubMed] [Google Scholar]
- Bennett N. B.; Stoltz B. M. A Unified Approach to the Daucane and Sphenolobane Bicyclo[5.3.0]decane Core: Enantioselective Total Syntheses of Daucene, Daucenal, Epoxydaucenal B, and 14-para-Anisoyloxydauc-4,8-diene. Chem. - Eur. J. 2013, 19, 17745–17750. 10.1002/chem.201302353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an intermolecular [2 + 2] photocycloaddition approach to synthesize daucene, see the following:Audenaert F.; De Keukeleire D.; Vandewalle M. A New and Facile Perhydroazulene Formation: The Total Synthesis of the Carotane (+)-Daucene. Tetrahedron 1987, 43, 5593–5604. 10.1016/S0040-4020(01)87740-1. [DOI] [Google Scholar]
- Seto H.; Fujimoto Y.; Tatsuno T.; Yoshioka H. Synthetic Studies on Carotane and Dolastane Type Terpenes: A New Entry to the Total Synthesis of (±)-Daucene via Intramolecular [2 + 2] Photocycloaddition. Synth. Commun. 1985, 15, 1217–1224. 10.1080/00397918508077268. [DOI] [Google Scholar]
- Baldwin S. W.; Wilkinson J. M. Photochemical Annelations. 6. Four-Carbon Photochemical Annelation of Alkenes with 2,2,6-Trimethyl-1,3-dioxolenone. J. Am. Chem. Soc. 1980, 102, 3634–3635. 10.1021/ja00530a059. [DOI] [Google Scholar]
- Winkler J. D.; Lee C. S.; Rubo L.; Muller C. L.; Squattrito P. J. Stereoselective Synthesis of the Tricyclic Skeleton of the Taxane Diterpenes. The First C-Silylation of a Ketone Enolate. J. Org. Chem. 1989, 54, 4491–4493. 10.1021/jo00280a007. [DOI] [Google Scholar]
- Winkler J. D.; Subrahmanyam D. Studies Directed Towards the Synthesis of Taxol: Preparation of C-13 Oxygenated Taxane Congeners. Tetrahedron 1992, 48, 7049–7056. 10.1016/S0040-4020(01)91213-X. [DOI] [Google Scholar]
- Witkop B. New Directions in the Chemistry of Natural Products: The Organic Chemist as a Pathfinder for Biochemistry and Medicine. Experientia 1971, 27, 1121–1138. 10.1007/BF02286882. [DOI] [PubMed] [Google Scholar]
- Daly J. W.; Karle I.; Myers C. W.; Tokuyama T.; Waters J. A.; Witkop B. Histrionicotoxins: Roentgen-Ray Analysis of the Novel Allenic and Acetylenic Spiroalkaloids Isolated from a Colombian Frog, Dendrobates Histrionicus. Proc. Natl. Acad. Sci. U. S. A. 1971, 68, 1870–1875. 10.1073/pnas.68.8.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on synthesis of histrionicotoxin alkaloids, see the following:Sinclair A.; Stockman R. A. Thirty-Five Years of Synthetic Studies Directed towards the Histrionicotoxin Family of Alkaloids. Nat. Prod. Rep. 2007, 24, 298–326. 10.1039/b604203c. [DOI] [PubMed] [Google Scholar]
- Koft E. R.; Smith A. B. III. Intramolecular [2 + 2] Photochemical Cycloadditions. 3. Perhydrohistrionicotoxin Synthetic Studies: Synthesis of Spiro[4.5]decanones via Intramolecular [2 + 2] Photocycloaddition. J. Org. Chem. 1984, 49, 832–836. 10.1021/jo00179a016. [DOI] [Google Scholar]
- Winkler J. D.; Hershberger P. M. A Stereoselective Synthesis of (−)-Perhydrohistrionicotoxin. J. Am. Chem. Soc. 1989, 111, 4852–4856. 10.1021/ja00195a042. [DOI] [Google Scholar]
- Winkler J. D.; Doherty E. M. The First Total Synthesis of (±)-Saudin. J. Am. Chem. Soc. 1999, 121, 7425–7426. 10.1021/ja9916198. [DOI] [Google Scholar]
- Winkler J. D.; Doherty E. M. Control of Relative Stereochemistry of Quaternary Carbon Centers via the Intramolecular Dioxenone Photocycloaddition: An Approach to the Synthesis of Saudin. Tetrahedron Lett. 1998, 39, 2253–2256. 10.1016/S0040-4039(98)00285-8. [DOI] [Google Scholar]
- Hecker E. Cocarcinogenic Principles from the Seed Oil of Croton Tiglium and from Other Euphorbiaceae. Cancer Res. 1968, 28, 2338–2348. [PubMed] [Google Scholar]
- Zechmeister K.; Brandl F.; Hoppe W.; Hecker E.; Opferkuch H. J.; Adolf W. Structure Determination of the New Tetracyclic Diterpene Ingenol-Triacetate with Triple Product Methods. Tetrahedron Lett. 1970, 11, 4075–4078. 10.1016/S0040-4039(01)98669-1. [DOI] [Google Scholar]
- Hecker E. New Toxic, Irritant and Cocarcinogenic Diterpene Esters from Euphorbiaceae and from Thymelaeaceae. Pure Appl. Chem. 1977, 49, 1423–1431. 10.1351/pac197749091423. [DOI] [Google Scholar]
- Liao S.-G.; Chen H.-D.; Yue J.-M. Plant Orthoesters. Chem. Rev. 2009, 109, 1092–1140. 10.1021/cr0782832. [DOI] [PubMed] [Google Scholar]
- Vasas A.; Hohmann J. Euphorbia Diterpenes: Isolation, Structure, Biological Activity, and Synthesis (2008–2012). Chem. Rev. 2014, 114, 8579–8612. 10.1021/cr400541j. [DOI] [PubMed] [Google Scholar]
- Durán-Peña M. J.; Ares J. M. B.; Collado I. G.; Hernández-Galán R. Biologically Active Diterpenes Containing a gem-Dimethylcyclopropane Subunit: An Intriguing Source of PKC Modulators. Nat. Prod. Rep. 2014, 31, 940–952. 10.1039/C4NP00008K. [DOI] [PubMed] [Google Scholar]
- Wang H.-B.; Wang X.-Y.; Liu L.-P.; Qin G.-W.; Kang T.-G. Tigliane Diterpenoids from the Euphorbiaceae and Thymelaeaceae Families. Chem. Rev. 2015, 115, 2975–3011. 10.1021/cr200397n. [DOI] [PubMed] [Google Scholar]
- Kupchan S. M.; Uchida I.; Branfman A. R.; Dailey R. G. Jr.; Fei B. Y. Antileukemic Principles Isolated from Euphorbiaceae Plants. Science 1976, 191, 571–572. 10.1126/science.1251193. [DOI] [PubMed] [Google Scholar]
- Blanco-Molina M.; Tron G. C.; Macho A.; Lucena C.; Calzado M. A.; Muñoz E.; Appendino G. Ingenol Esters Induce Apoptosis in Jurkat Cells through an AP-1 and NF-κB Independent Pathway. Chem. Biol. 2001, 8, 767–778. 10.1016/S1074-5521(01)00048-5. [DOI] [PubMed] [Google Scholar]
- Ogbourne S. M.; Suhrbier A.; Jones B.; Cozzi S.-J.; Boyle G. M.; Morris M.; McAlpine D.; Johns J.; Scott T. M.; Sutherland K. P.; Gardner J. M.; Le T. T. T.; Lenarczyk A.; Aylward J. H.; Parsons P. G. Antitumor Activity of 3-Ingenyl Angelate: Plasma Membrane and Mitochondrial Disruption and Necrotic Cell Death. Cancer Res. 2004, 64, 2833–2839. 10.1158/0008-5472.CAN-03-2837. [DOI] [PubMed] [Google Scholar]
- Fujiwara M.; Ijichi K.; Tokuhisa K.; Katsuura K.; Shigeta S.; Konno K.; Wang G. Y.; Uemura D.; Yokota T.; Baba M. Mechanism of Selective Inhibition of Human Immunodeficiency Virus by Ingenol Triacetate. Antimicrob. Agents Chemother. 1996, 40, 271–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration. 2012 Novel New Drugs Summary; FDA: Washington, DC, 2013; www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugInnovation/UCM337830.pdf.
- For a review on ingenol mebutate, see the following:Vasas A.; Rédei D.; Csupor D.; Molnár J.; Hohmann J. Diterpenes from European Euphorbia Species Serving as Prototypes for Natural-Product-Based Drug Discovery. Eur. J. Org. Chem. 2012, 2012, 5115–5130. 10.1002/ejoc.201200733. [DOI] [Google Scholar]
- Liang X.; Grue-Sørensen G.; Månsson K.; Vedsø P.; Soor A.; Stahlhut M.; Bertelsen M.; Engell K. M.; Högberg T. Syntheses, Biological Evaluation and SAR of Ingenol Mebutate Analogues for Treatment of Actinic Keratosis and Non-Melanoma Skin Cancer. Bioorg. Med. Chem. Lett. 2013, 23, 5624–5629. 10.1016/j.bmcl.2013.08.038. [DOI] [PubMed] [Google Scholar]
- For a review on in/out isomerism, see the following:Alder R. W.; East S. P. In/Out Isomerism. Chem. Rev. 1996, 96, 2097–2112. 10.1021/cr940246k. [DOI] [PubMed] [Google Scholar]
- Rigby J. H.; Moore T. L.; Rege S. Synthetic Studies on the Ingenane Diterpenes. Inter- and Intramolecular [6 + 4] Tropone-Diene Cycloaddition Reactions. J. Org. Chem. 1986, 51, 2398–2400. 10.1021/jo00362a051. [DOI] [Google Scholar]
- Funk R. L.; Olmstead T. A.; Parvez M. A Solution to the in,out-Bicyclo[4.4.1]undecan-7-one Problem Inherent in Ingenane Total Synthesis. J. Am. Chem. Soc. 1988, 110, 3298–3300. 10.1021/ja00218a049. [DOI] [Google Scholar]
- Funk R. L.; Olmstead T. A.; Parvez M.; Stallman J. B. Stereoselective Construction of the Complete Ingenane Ring System. J. Org. Chem. 1993, 58, 5873–5875. 10.1021/jo00074a003. [DOI] [Google Scholar]
- Winkler J. D.; Hong B.-C.; Bahador A.; Kazanietz M. G.; Blumberg P. M. Inside-outside stereoisomerism. VII. Methodology for the Synthesis of 3-Oxygenated Ingenanes. The First Ingenol Analogs with High Affinity for Protein Kinase C. J. Org. Chem. 1995, 60, 1381–1390. 10.1021/jo00110a048. [DOI] [Google Scholar]
- Rigby J. H.; Rege S. D.; Sandanayaka V. P.; Kirova M. Studies on Intramolecular Higher-Order Cycloaddition Reactions. J. Org. Chem. 1996, 61, 842–850. 10.1021/jo9518009. [DOI] [Google Scholar]
- Nakamura T.; Matsui T.; Tanino K.; Kuwajima I. A New Approach for Ingenol Synthesis. J. Org. Chem. 1997, 62, 3032–3033. 10.1021/jo970172n. [DOI] [PubMed] [Google Scholar]
- Rigby J. H.; Hu J.; Heeg M. J. Synthetic Studies on the Ingenane Diterpenes. Construction of an ABC Tricycle Exhibiting trans-Intrabridgehead Stereochemistry. Tetrahedron Lett. 1998, 39, 2265–2268. 10.1016/S0040-4039(98)00243-3. [DOI] [Google Scholar]
- Winkler J. D.; Kim S.; Harrison S.; Lewin N. E.; Blumberg P. M. Synthesis and Biological Evaluation of Highly Functionalized Analogues of Ingenol. J. Am. Chem. Soc. 1999, 121, 296–300. 10.1021/ja9741842. [DOI] [Google Scholar]
- Kigoshi H.; Suzuki Y.; Aoki K.; Uemura D. Synthetic Studies of Ingenol: Synthesis of in,out-Tricyclo[7.4.1.01,5]tetradecan-14-one. Tetrahedron Lett. 2000, 41, 3927–3930. 10.1016/S0040-4039(00)00519-0. [DOI] [Google Scholar]
- Tang H.; Yusuff N.; Wood J. L. Progress toward the Total Synthesis of Ingenol: Construction of the Complete Carbocyclic Skeleton. Org. Lett. 2001, 3, 1563–1566. 10.1021/ol015855a. [DOI] [PubMed] [Google Scholar]
- Rigby J. H.; Bazin B.; Meyer J. H.; Mohammadi F. Synthetic Studies on the Ingenane Diterpenes. An Improved Entry into a trans-Intrabridgehead System. Org. Lett. 2002, 4, 799–801. 10.1021/ol017280n. [DOI] [PubMed] [Google Scholar]
- Watanabe K.; Suzuki Y.; Aoki K.; Sakakura A.; Suenaga K.; Kigoshi H. Formal Synthesis of Optically Active Ingenol via Ring-Closing Olefin Metathesis. J. Org. Chem. 2004, 69, 7802–7808. 10.1021/jo048833l. [DOI] [PubMed] [Google Scholar]
- Grainger R. S.; Owoare R. B. Selective 1,5-Alkylidenecarbene Insertion Reactions on [3.2.1] Oxabicyclic Ethers: A New Approach toward the AB Ring System of Ingenol. Org. Lett. 2004, 6, 2961–2964. 10.1021/ol048911r. [DOI] [PubMed] [Google Scholar]
- Jørgensen L.; McKerrall S. J.; Kuttruff C. A.; Ungeheuer F.; Felding J.; Baran P. S. 14-Step Synthesis of (+)-Ingenol from (+)-3-Carene. Science 2013, 341, 878–882. 10.1126/science.1241606. [DOI] [PubMed] [Google Scholar]
- McKerrall S. J.; Jørgensen L.; Kuttruff C. A.; Ungeheuer F.; Baran P. S. Development of a Concise Synthesis of (+)-Ingenol. J. Am. Chem. Soc. 2014, 136, 5799–5810. 10.1021/ja501881p. [DOI] [PubMed] [Google Scholar]
- Tanino K.; Onuki K.; Asano K.; Miyashita M.; Nakamura T.; Takahashi Y.; Kuwajima I. Total Synthesis of Ingenol. J. Am. Chem. Soc. 2003, 125, 1498–1500. 10.1021/ja029226n. [DOI] [PubMed] [Google Scholar]
- Nickel A.; Maruyama T.; Tang H.; Murphy P. D.; Greene B.; Yusuff N.; Wood J. L. Total Synthesis of Ingenol. J. Am. Chem. Soc. 2004, 126, 16300–16301. 10.1021/ja044123l. [DOI] [PubMed] [Google Scholar]
- Kim S.; Winkler J. D. Approaches to the Synthesis of Ingenol. Chem. Soc. Rev. 1997, 26, 387–399. 10.1039/cs9972600387. [DOI] [Google Scholar]
- Kuwajima I.; Tanino K. Total Synthesis of Ingenol. Chem. Rev. 2005, 105, 4661–4670. 10.1021/cr040636z. [DOI] [PubMed] [Google Scholar]
- Cha J. K.; Epstein O. L. Synthetic Approaches to Ingenol. Tetrahedron 2006, 62, 1329–1343. 10.1016/j.tet.2005.10.035. [DOI] [Google Scholar]
- Busch T.; Kirschning A. Recent Advances in the Total Synthesis of Pharmaceutically Relevant Diterpenes. Nat. Prod. Rep. 2008, 25, 318–341. 10.1039/b705652b. [DOI] [PubMed] [Google Scholar]
- Hayakawa I.; Asuma Y.; Ohyoshi T.; Aoki K.; Kigoshi H. Synthetic Study on 13-Oxyingenol: Construction of the Full Carbon Framework. Tetrahedron Lett. 2007, 48, 6221–6224. 10.1016/j.tetlet.2007.06.135. [DOI] [Google Scholar]
- Ohyoshi T.; Miyazawa Y.; Aoki K.; Ohmura S.; Asuma Y.; Hayakawa I.; Kigoshi H. Synthetic Studies toward 13-Oxyingenol: Construction of the Fully Substituted Tetracyclic Compound. Org. Lett. 2011, 13, 2160–2163. 10.1021/ol103151k. [DOI] [PubMed] [Google Scholar]
- Ohyoshi T.; Funakubo S.; Miyazawa Y.; Niida K.; Hayakawa I.; Kigoshi H. Total Synthesis of (−)-13-Oxyingenol and its Natural Derivative. Angew. Chem., Int. Ed. 2012, 51, 4972–4975. 10.1002/anie.201201383. [DOI] [PubMed] [Google Scholar]
- Winkler J. D.; Henegar K. E.; Williard P. G. Inside-Outside Stereoisomerism. II. Synthesis of the Carbocyclic Ring System of the Ingenane Diterpenes via the Intramolecular Dioxolenone Photocycloaddition. J. Am. Chem. Soc. 1987, 109, 2850–2851. 10.1021/ja00243a062. [DOI] [Google Scholar]
- Hehn J. P.; Herdtweck E.; Bach T. A Photocycloaddition/Fragmentation Approach toward the 3,12-Dioxatricyclo[8.2.1.06,13]tridecane Skeleton of Terpenoid Natural Products. Org. Lett. 2011, 13, 1892–1895. 10.1021/ol2004462. [DOI] [PubMed] [Google Scholar]
- Minter D. E.; Winslow C. D. A Photochemical Approach to the Galanthan Ring System. J. Org. Chem. 2004, 69, 1603–1606. 10.1021/jo0356560. [DOI] [PubMed] [Google Scholar]
- Lewis J. R. Amaryllidacae alkaloids. Nat. Prod. Rep. 1997, 14, 303–308. 10.1039/np9971400303. [DOI] [Google Scholar]
- Jin Z. Amaryllidaceae and Sceletium Alkaloids. Nat. Prod. Rep. 2013, 30, 849–868. 10.1039/c3np70005d. [DOI] [PubMed] [Google Scholar]
- Tamura Y.; Ishibashi H.; Hirai M.; Kita Y.; Ikeda M. Photochemical Syntheses of 2-Aza- and 2-Oxabicyclo[2.1.1]hexane Ring Systems. J. Org. Chem. 1975, 40, 2702–2710. 10.1021/jo00907a002. [DOI] [Google Scholar]
- Winkler J. D.; Mazur Bowen C.; Liotta F. [2 + 2] Photocycloaddition/Fragmentation Strategies for the Synthesis of Natural and Unnatural Products. Chem. Rev. 1995, 95, 2003–2020. 10.1021/cr00038a010. [DOI] [Google Scholar]
- Schell F. M.; Cook P. M. Intramolecular Photochemistry of a Vinylogous Amide and some Transformations of the Photoproduct. J. Org. Chem. 1984, 49, 4067–4070. 10.1021/jo00195a041. [DOI] [Google Scholar]
- Amougay A.; Pete J.-P.; Piva O. Intramolecular [2 + 2] Photocycloaddition of N-Alkenoyl β-Enaminones. Tetrahedron Lett. 1992, 33, 7347–7350. 10.1016/S0040-4039(00)60184-3. [DOI] [Google Scholar]
- Roupany A. J. A.; Baker J. R. Diverse Products Accessible via [2 + 2] Photocycloadditions of 3-Aminocyclopentenones. RSC Adv. 2013, 3, 10650–10653. 10.1039/c3ra42106f. [DOI] [Google Scholar]
- Kwak Y.-S.; Winkler J. D. Synthesis of 6-Aza-bicyclo[3,2,1]octan-3-ones via Vinylogous Imide Photochemistry: An Approach to the Synthesis of the Hetisine Alkaloids. J. Am. Chem. Soc. 2001, 123, 7429–7430. 10.1021/ja010542w. [DOI] [PubMed] [Google Scholar]
- Winkler J. D.; Ragains J. R. Intramolecular Photoaddition of Vinylogous Amides with Allenes: A Novel Approach to the Synthesis of Pyrroles. Org. Lett. 2006, 8, 4031–4033. 10.1021/ol061453x. [DOI] [PubMed] [Google Scholar]
- Lutteke G.; AlHussainy R.; Wrigstedt P. J.; Hue B. T. B.; de Gelder R.; van Maarseveen J. H.; Hiemstra H. Formation of Bicyclic Pyrroles and Furans Through an Enone Allene Photocycloaddition and Fragmentation Sequence. Eur. J. Org. Chem. 2008, 2008, 925–933. 10.1002/ejoc.200701017. [DOI] [Google Scholar]
- Swindell C. S.; deSolms S. J. Synthesis of the Taxane Diterpenes: Construction of a BC Ring Intermediate for Taxane Synthesis. Tetrahedron Lett. 1984, 25, 3801–3804. 10.1016/S0040-4039(01)91171-2. [DOI] [Google Scholar]
- Swindell C. S.; Patel B. P. Synthesis and Stereochemistry of a Saturated Tricyclic Taxane Model. Tetrahedron Lett. 1987, 28, 5275–5278. 10.1016/S0040-4039(00)96706-6. [DOI] [Google Scholar]
- Swindell C. S.; Patel B. P.; DeSolms S. J.; Springer J. P. A Route for the Construction of the Taxane BC Substructure. J. Org. Chem. 1987, 52, 2346–2355. 10.1021/jo00388a002. [DOI] [Google Scholar]
- Swindell C. S.; Patel B. P. Stereoselective Construction of the Taxinine AB System through a Novel Tandem Aldol-Payne Rearrangement Annulation. J. Org. Chem. 1990, 55, 3–5. 10.1021/jo00288a002. [DOI] [Google Scholar]
- For a review on Sceletium alkaloids, see the following:Jin Z. Amaryllidaceae and Sceletium Alkaloids. Nat. Prod. Rep. 2011, 28, 1126–1142. 10.1039/c0np00073f. [DOI] [PubMed] [Google Scholar]
- Winkler J. D.; Muller C. L.; Scott R. D. A New Method for the Formation of Nitrogen-Containing Ring Systems via the Intramolecular Photocycloaddition of Vinylogous Amides. A Synthesis of Mesembrine. J. Am. Chem. Soc. 1988, 110, 4831–4832. 10.1021/ja00222a053. [DOI] [Google Scholar]
- White J. D.; Ihle D. C. Tandem Photocycloaddition-Retro-Mannich Fragmentation of Enaminones. A Route to Spiropyrrolines and the Tetracyclic Core of Koumine. Org. Lett. 2006, 8, 1081–1084. 10.1021/ol052955y. [DOI] [PubMed] [Google Scholar]
- White J. D.; Li Y.; Ihle D. C. Tandem Intramolecular Photocycloaddition-Retro-Mannich Fragmentation as a Route to Spiro[pyrrolidine-3,3′-oxindoles]. Total Synthesis of (±)-Coerulescine, (±)-Horsfiline, (±)-Elacomine, and (±)-6-Deoxyelacomine. J. Org. Chem. 2010, 75, 3569–3577. 10.1021/jo1002714. [DOI] [PubMed] [Google Scholar]
- Winkler J. D.; Scott R. D.; Williard P. G. Asymmetric Induction in the Vinylogous Amide Photocycloaddition Reaction. A Formal Synthesis of Vindorosine. J. Am. Chem. Soc. 1990, 112, 8971–8975. 10.1021/ja00180a049. [DOI] [Google Scholar]
- Winkler J. D.; Axten J. M. The First Total Syntheses of Ircinol A, Ircinal A, and Manzamines A and D. J. Am. Chem. Soc. 1998, 120, 6425–6426. 10.1021/ja981303k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. D.; Stelmach J. E.; Siegel M. G.; Haddad N.; Axten J.; Dailey W. P. III. An Approach to the Synthesis of the Manzamine Alkaloids via the Vinylogous Amide PHotocycloAddition/Retro-Mannich Fragmentation/Mannich Closure Cascade (pharM). Isr. J. Chem. 1997, 37, 47–67. 10.1002/ijch.199700008. [DOI] [Google Scholar]
- Wang B.-G.; Ebel R.; Nugroho B. W.; Prijono D.; Frank W.; Steube K. G.; Hao X.-J. Aglacins A–D, First Representatives of a New Class of Aryltetralin Cyclic Ether Lignans from Aglaia cordata. J. Nat. Prod. 2001, 64, 1521–1526. 10.1021/np0102962. [DOI] [PubMed] [Google Scholar]
- Wang S.-K.; Cheng Y.-J.; Duh C.-Y. Cytotoxic Constituents from Leaves of Aglaia elliptifolia. J. Nat. Prod. 2001, 64, 92–94. 10.1021/np000341q. [DOI] [PubMed] [Google Scholar]
- Proksch P.; Edrada R. A.; Ebel R.; Bohnenstengel F. I.; Nugroho B. W. Chemistry and Biological Activity of Rocaglamide Derivatives and Related Compounds in Aglaia Species (Meliaceae). Curr. Org. Chem. 2001, 5, 923–938. 10.2174/1385272013375049. [DOI] [Google Scholar]
- Hwang B. Y.; Su B.-N.; Chai H.; Mi Q.; Kardono L. B. S.; Afriastini J. J.; Riswan S.; Santarsiero B. D.; Mesecar A. D.; Wild R.; Fairchild C. R.; Vite G. D.; Rose W. C.; Farnsworth N. R.; Cordell G. A.; Pezzuto J. M.; Swanson S. M.; Kinghorn A. D. Silvestrol and Episilvestrol, Potential Anticancer Rocaglate Derivatives from Aglaia silvestris. J. Org. Chem. 2004, 69, 3350–3358. 10.1021/jo040120f. [DOI] [PubMed] [Google Scholar]
- Gerard B.; Jones G. II; Porco J. A. Jr. A Biomimetic Approach to the Rocaglamides Employing Photogeneration of Oxidopyryliums Derived from 3-Hydroxyflavones. J. Am. Chem. Soc. 2004, 126, 13620–13621. 10.1021/ja044798o. [DOI] [PubMed] [Google Scholar]
- Gerard B.; Sangji S.; O’Leary D. J.; Porco J. A. Jr. Enantioselective Photocycloaddition Mediated by Chiral Brønsted Acids: Asymmetric Synthesis of the Rocaglamides. J. Am. Chem. Soc. 2006, 128, 7754–7755. 10.1021/ja062621j. [DOI] [PubMed] [Google Scholar]
- Roche S. P.; Cencic R.; Pelletier J.; Porco J. A. Jr. Biomimetic Photocycloaddition of 3-Hydroxyflavones: Synthesis and Evaluation of Rocaglate Derivatives as Inhibitors of Eukaryotic Translation. Angew. Chem., Int. Ed. 2010, 49, 6533–6538. 10.1002/anie.201003212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajkiewicz N. J.; Roche S. P.; Gerard B.; Porco J. A. Jr. Enantioselective Photocycloaddition of 3-Hydroxyflavones: Total Syntheses and Absolute Configuration Assignments of (+)-Ponapensin and (+)-Elliptifoline. J. Am. Chem. Soc. 2012, 134, 13108–13113. 10.1021/ja305342f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos P. H.; Antalek M. T.; Porco J. A. Jr.; Stephenson C. R. J. Tandem Dienone Photorearrangement-Cycloaddition for the Rapid Generation of Molecular Complexity. J. Am. Chem. Soc. 2013, 135, 17978–17982. 10.1021/ja409992m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cava M. P.; Napier D. R. Condensed Cyclobutane Aromatic Systems. II. Dihalo Derivatives of Benzocyclobutene and Benzocyclobutadiene Dimer. J. Am. Chem. Soc. 1957, 79, 1701–1705. 10.1021/ja01564a048. [DOI] [Google Scholar]
- Oppolzer W. Intramolecular Cycloaddition Reactions of ortho-Quinodimethanes in Organic Synthesis. Synthesis 1978, 1978, 793–802. 10.1055/s-1978-24895. [DOI] [Google Scholar]
- Charlton J. L.; Alauddin M. M. Orthoquinodimethanes. Tetrahedron 1987, 43, 2873–2889. 10.1016/S0040-4020(01)86825-3. [DOI] [Google Scholar]
- Michellys P.-Y.; Pellissier H.; Santelli M. Cycloadditions of ortho-Quinodimethanes Derived from Benzocyclobutenes in Organic Synthesis. A Review. Org. Prep. Proced. Int. 1996, 28, 545–608. 10.1080/00304949609458572. [DOI] [Google Scholar]
- Segura J. L.; Martín N. o-Quinodimethanes: Efficient Intermediates in Organic Synthesis. Chem. Rev. 1999, 99, 3199–3246. 10.1021/cr990011e. [DOI] [PubMed] [Google Scholar]
- Diels O.; Alder K. Synthesen in der Hydroaromatischen Reihe. Justus Liebigs Ann. Chem. 1928, 460, 98–122. 10.1002/jlac.19284600106. [DOI] [Google Scholar]
- For a review on the use of the Diels–Alder reaction in total synthesis, see the following:Nicolaou K. C.; Snyder S. A.; Montagnon T.; Vassilikogiannakis G. The Diels-Alder Reaction in Total Synthesis. Angew. Chem., Int. Ed. 2002, 41, 1668–1698. 10.1002/1521-3773(20020517)41:10<1668::AID-ANIE1668>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- For a review on photoenolization, see the following:Sammes P. G. Photoenolisation. Tetrahedron 1976, 32, 405–422. 10.1016/0040-4020(76)80055-5. [DOI] [Google Scholar]
- Quinkert G.; Weber W.-D.; Schwartz U.; Dürner G. A Photochemical Route to (±)-Estrone. Angew. Chem., Int. Ed. Engl. 1980, 19, 1027–1029. 10.1002/anie.198010271. [DOI] [Google Scholar]
- Quinkert G.; Schwartz U.; Stark H.; Weber W.-D.; Baier H.; Adam F.; Dürner G. Asymmetric Total Synthesis of (+)-Estrone. Angew. Chem., Int. Ed. Engl. 1980, 19, 1029–1030. 10.1002/anie.198010291. [DOI] [Google Scholar]
- For a review on the synthesis of estrone, see the following:Quinkert G.; Stark H. Stereoselective Synthesis of Enantiomerically Pure Natural Products—Estrone as Example. Angew. Chem., Int. Ed. Engl. 1983, 22, 637–740. 10.1002/anie.198306373. [DOI] [Google Scholar]
- Kraus G. A.; Wu Y. Hydrogen Atom Abstraction Reactions in Organic Synthesis. A Formal Total Synthesis of Racemic Podophyllotoxin. J. Org. Chem. 1992, 57, 2922–2925. 10.1021/jo00036a030. [DOI] [Google Scholar]
- Barton D. H. R.; Clive D. L. J.; Magnus P. D.; Smith G. Experiments of the Synthesis of Tetracycline. Part V. Photocyclisation of Ring B. J. Chem. Soc. C 1971, 2193–2203. 10.1039/j39710002193. [DOI] [PubMed] [Google Scholar]
- Barton D. H. R.; Magnus P. D.; Hase T. Experiments on the Synthesis of Tetracycline. Part VII. The Total Synthesis of 6-Methylpretetramid. J. Chem. Soc. C 1971, 2215–2225. 10.1039/j39710002215. [DOI] [PubMed] [Google Scholar]
- Cambie R. C.; Lal A. R.; Kernan M. R.; Bergquist P. R. Chemistry of Sponges, 17. A Novel Brominated Benzocylooctane Derivative from Hamigera taragensis. J. Nat. Prod. 1995, 58, 940–942. 10.1021/np50120a020. [DOI] [PubMed] [Google Scholar]
- Wellington K. D.; Cambie R. C.; Rutledge P. S.; Bergquist P. R. Chemistry of Sponges. 19. Novel Bioactive Metabolites from Hamigera tarangaensis. J. Nat. Prod. 2000, 63, 79–85. 10.1021/np9903494. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Gray D.; Tae J. Total Synthesis of Hamigerans: Part 1. Development of Synthetic Technology for the Construction of Benzannulated Polycyclic Systems by the Intramolecular Trapping of Photogenerated Hydroxy-o-quinodimethanes and Synthesis of Key Building Blocks. Angew. Chem., Int. Ed. 2001, 40, 3675–3678. 10.1002/1521-3773(20011001)40:19<3675::AID-ANIE3675>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Gray D.; Tae J. Total Synthesis of Hamigerans: Part 2. Implementation of the Intramolecular Diels-Alder Trapping of Photochemically Generated Hydroxy-o-quinodimethanes; Strategy and Completion of the Synthesis. Angew. Chem., Int. Ed. 2001, 40, 3679–3683. 10.1002/1521-3773(20011001)40:19<3679::AID-ANIE3679>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Gray D. L. F.; Tae J. Total Synthesis of Hamigerans and Analogues Thereof. Photochemical Generation and Diels-Alder Trapping of Hydroxy-o-quinodimethanes. J. Am. Chem. Soc. 2004, 126, 613–627. 10.1021/ja030498f. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Gray D. L. F. Total Synthesis of Hybocarpone and Analogues Thereof. A Facile Dimerization of Naphthazarins to Pentacyclic Systems. J. Am. Chem. Soc. 2004, 126, 607–612. 10.1021/ja030497n. [DOI] [PubMed] [Google Scholar]
- Prabhakar S.; Lobo A. M.; Oliveira I. M. C. Photochemical Approach to the Rhoeadine Alkaloid Skeleton: Synthesis of (±)-cis-Alpinigenine. J. Chem. Soc., Chem. Commun. 1977, 419–420. 10.1039/C39770000419. [DOI] [Google Scholar]
- Prabhakar S.; Lobo A. M.; Tavares M. R.; Oliveira I. M. C. Total Synthesis of the Alkaloids (±)-Alpinigenine and (±)-cis-Alpinigenine. J. Chem. Soc., Perkin Trans. 1 1981, 1273–1277. 10.1039/P19810001273. [DOI] [Google Scholar]
- Eaton P. E.; Lin K. trans-2-Cyclooctenone. J. Am. Chem. Soc. 1964, 86, 2087–2088. 10.1021/ja01064a049. [DOI] [Google Scholar]
- Eaton P. E.; Lin K. trans-2-Cycloheptenone. J. Am. Chem. Soc. 1965, 87, 2052–2054. 10.1021/ja01087a038. [DOI] [Google Scholar]
- Eaton P. E. Photochemical Reactions of Simple Alicyclic Enones. Acc. Chem. Res. 1968, 1, 50–57. 10.1021/ar50002a003. [DOI] [Google Scholar]
- Dunkelblum E.; Hart H. Stereochemistry and Mechanism of the Photochemical Addition of Methanol to Cycloheptenones. J. Am. Chem. Soc. 1977, 99, 644–646. 10.1021/ja00444a072. [DOI] [Google Scholar]
- Hart H.; Dunkelblum E. Stereochemistry of the Photoinduced and Michael Addition of Methanol to Seven- and Eight-Membered 2-Cycloalkenones. J. Am. Chem. Soc. 1978, 100, 5141–5147. 10.1021/ja00484a038. [DOI] [Google Scholar]
- Ghosh S.; Saha S. Photo-Induced Diels-Alder Reaction. A Novel Route to Trans Fused Benzobicyclo[5.3.0]decanes and [5.4.0]Undecanes. Tetrahedron Lett. 1985, 26, 5325–5326. 10.1016/S0040-4039(00)95029-9. [DOI] [Google Scholar]
- Mintas M.; Schuster D. I.; Williard P. G. Stereochemistry and Mechanism of [4 + 2] Photocycloaddition of Pummerer’s Ketone to Furan. J. Am. Chem. Soc. 1988, 110, 2305–2306. 10.1021/ja00215a053. [DOI] [Google Scholar]
- Corey E. J.; Tada M.; LaMahieu R.; Libit L. trans-2-Cycloheptenone. J. Am. Chem. Soc. 1965, 87, 2051–2052. 10.1021/ja01087a037. [DOI] [Google Scholar]
- Dorr H.; Rawal V. H. The Intramolecular Diels-Alder Reactions of Photochemically Generated trans-Cycloalkenones. J. Am. Chem. Soc. 1999, 121, 10229–10230. 10.1021/ja992287+. [DOI] [Google Scholar]
- Kawazu K. Isolation of Vibsanines A, B, C, D, E and F from Viburnum odoratissimum. Agric. Biol. Chem. 1980, 44, 1367–1372. 10.1271/bbb1961.44.1367. [DOI] [Google Scholar]
- Fukuyama K.; Katsube Y.; Kawazu K. Structure and Absolute Configuration of Vibsanine E Isolated from Leaves of Viburnum odoratissimum Ker. J. Chem. Soc., Perkin Trans. 2 1980, 1701–1703. 10.1039/p29800001701. [DOI] [Google Scholar]
- For a review on the synthesis of vibsane-type diterpenoids, see the following:Mak J. Y. W.; Williams C. M. Key Achievements in the Total Synthesis of Vibsane-Type Diterpenoids. Nat. Prod. Rep. 2012, 29, 440–448. 10.1039/c2np00067a. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Loe Ø.; Stafford D. G. Sequential Cycloaddition Approach to the Tricyclic Core of Vibsanin E. Total Synthesis of (±)-5-epi-10-epi-Vibsanin E. Org. Lett. 2005, 7, 5561–5563. 10.1021/ol052005c. [DOI] [PubMed] [Google Scholar]
- Nikolai J.; Loe Ø.; Dominiak P. M.; Gerlitz O. O.; Autschbach J.; Davies H. M. L. Mechanistic Studies of UV Assisted [4 + 2] Cycloadditions in Synthetic Efforts toward Vibsanin E. J. Am. Chem. Soc. 2007, 129, 10763–10772. 10.1021/ja072090e. [DOI] [PubMed] [Google Scholar]
- Moses J. E.; Baldwin J. E.; Marquez R.; Adlington R. M.; Claridge T. D. W.; Odell B. Biomimetic Synthesis of the Crispatene Core. Org. Lett. 2003, 5, 661–663. 10.1021/ol020242z. [DOI] [PubMed] [Google Scholar]
- Padwa A.; Brodsky L.; Clough S. Orbital Symmetry and Steric Control in the Photorearrangement of 1,3,5-Hexatrienes to Bicyclo[3.1.0]hex-2-enes. J. Am. Chem. Soc. 1972, 94, 6767–6775. 10.1021/ja00774a033. [DOI] [Google Scholar]
- For a review on photochemical transformations of hetero-1,3,5-hexatrienes into five-membered rings, see the following:George M. V.; Mitra A.; Sukumaran K. B. Thermal and Photochemical Transformations of Hetero-1,3,5-hexatrienes into Five-Membered Rings—Possible Pericyclic Reactions. Angew. Chem., Int. Ed. Engl. 1980, 19, 973–983. 10.1002/anie.198009731. [DOI] [Google Scholar]
- Shang H.; Liu J.; Bao R.; Cao Y.; Zhao K.; Xiao C.; Zhou B.; Hu L.; Tang Y. Biomimetic Synthesis: Discovery of Xanthanolide Dimers. Angew. Chem., Int. Ed. 2014, 53, 14494–14498. 10.1002/anie.201406461. [DOI] [PubMed] [Google Scholar]
- Bohlmann F.; Knoll K.-H.; El-Emary N. A. Neuartige Sesquiterpenlactone aus Pulicaria crispa. Phytochemistry 1979, 18, 1231–1233. 10.1016/0031-9422(79)80146-6. [DOI] [Google Scholar]
- Ahmed A. A.; Jakupovic J.; Bohlmann F.; Regaila H. A.; Ahmed A. M. Sesquiterpene Lactones from Xanthium pungens. Phytochemistry 1990, 29, 2211–2215. 10.1016/0031-9422(90)83040-8. [DOI] [Google Scholar]
- Ahmed A. A.; Mahmoud A. A.; El-Gamal A. A. A Xanthanolide Diol and a Dimeric Xanthanolide from Xanthium Species. Planta Med. 1999, 65, 470–472. 10.1055/s-2006-960817. [DOI] [PubMed] [Google Scholar]
- Shishido K. Recent Advances in the Total Synthesis of Xanthanolide Sesquiterpenoids. Heterocycles 2009, 78, 873–889. 10.3987/REV-08-647. [DOI] [Google Scholar]
- Vasas A.; Hohmann J. Xanthane Sesquiterpenoids: Structure, Synthesis and Biological Activity. Nat. Prod. Rep. 2011, 28, 824–842. 10.1039/c0np00011f. [DOI] [PubMed] [Google Scholar]
- For a review on Norrish type I reactions, see the following:Weiss D. S. The Norrish Type I Reaction in Cycloalkanone Photochemistry. Org. Photochem. 1981, 5, 347–420. [Google Scholar]
- Barltrop J. A.; Coyle J. D. Evidence for a Discrete Biradical Intermediate in the Photolysis of 2,3-Dimethylcyclohexanone. J. Chem. Soc. D 1969, 1081–1082. 10.1039/c29690001081. [DOI] [Google Scholar]
- Diau E. W.-G.; Kötting C.; Zewail A. H. ChemPhysChem 2001, 2, 273–293. 10.1002/1439-7641(20010518)2:5<273::AID-CPHC273>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Diau E. W.-G.; Kötting C.; Zewail A. H. ChemPhysChem 2001, 2, 294–309. 10.1002/1439-7641(20010518)2:5<294::AID-CPHC294>3.3.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Norrish R. G. W.; Appleyard M. E. S. Primary Photochemical Reactions. Part IV. Decomposition of Methyl Ethyl Ketone and Methyl Butyl Ketone. J. Chem. Soc. 1934, 874–880. 10.1039/jr9340000874. [DOI] [Google Scholar]
- Norrish R. G. W.; Bamford C. H. Photodecomposition of Aldehydes and Ketones. Nature 1936, 138, 1016. 10.1038/1381016a0. [DOI] [Google Scholar]
- Norrish R. G. W.; Bamford C. H. Photo-decomposition of Aldehydes and Ketones. Nature 1937, 140, 195–196. 10.1038/140195b0. [DOI] [Google Scholar]
- For a review on Norrish type II reactions, see the following:Scaiano J. C.; Lissi E. A.; Encina M. V. Chemistry of the Biradicals Produced in the Norrish Type II Reaction. Rev. Chem. Intermed. 1978, 2, 139–196. 10.1007/BF03155998. [DOI] [Google Scholar]
- Yang N. C.; Yang D.-D. H. Photochemical Reactions of Ketones in Solution. J. Am. Chem. Soc. 1958, 80, 2913–2914. 10.1021/ja01544a092. [DOI] [Google Scholar]
- For a review on Norrish–Yang cyclizations, see the following:Wagner P.; Park B. S. Photoinduced Hydrogen Atom Abstraction by Carbonyl Compounds. Org. Photochem. 1991, 11, 227–366. [Google Scholar]
- Natarajan A.; Ng D.; Yang Z.; Garcia-Garibay M. A. Parallel Syntheses of (+)- and (−)-α-Cuparenone by Radical Combination in Crystalline Solids. Angew. Chem., Int. Ed. 2007, 46, 6485–6487. 10.1002/anie.200700679. [DOI] [PubMed] [Google Scholar]
- Ng D.; Yang Z.; Garcia-Garibay M. A. Total Synthesis of (±)-Herbertenolide by Stereospecific Formation of Vicinal Quaternary Centers in a Crystalline Ketone. Org. Lett. 2004, 6, 645–647. 10.1021/ol0499250. [DOI] [PubMed] [Google Scholar]
- Callant P.; Storme P.; Van der Eycken E.; Vandewalle M. Iridoids: Stereospecific Synthesis of Functionalized Cyclopentanoid Intermediates via Bicyclo[2.2.1]heptanones. Tetrahedron Lett. 1983, 24, 5797–5800. 10.1016/S0040-4039(00)94204-7. [DOI] [Google Scholar]
- Itagaki N.; Iwabuchi Y. Enantio- and Diastereocontrolled Synthesis of (+)-Juvabione Employing Organocatalytic Desymmetrisation and Photoinduced Fragmentation. Chem. Commun. 2007, 1175–1176. 10.1039/b616641e. [DOI] [PubMed] [Google Scholar]
- Lin C.-H.; Su Y.-L.; Tai H.-M. A Total Synthesis of (±)-Hop Ether. Heterocycles 2006, 68, 771–777. 10.3987/COM-05-10646. [DOI] [Google Scholar]
- Morton D. R.; Lee-Ruff E.; Southam R. M.; Turro N. J. Molecular Photochemistry. XXVII. Photochemical Ring Expansion of Cyclobutanone, Substituted Cyclobutanones, and Related Cyclic Ketones. J. Am. Chem. Soc. 1970, 92, 4349–4357. 10.1021/ja00717a036. [DOI] [Google Scholar]
- Pirrung M. C.; Chang V. K.; DeAmicis C. V. Mechanism and Synthetic Applications of the Photochemical Generation and X–H Insertion Reactions of Oxacarbenes. J. Am. Chem. Soc. 1989, 111, 5824–5831. 10.1021/ja00197a049. [DOI] [Google Scholar]
- Umbricht G.; Hellman M. D.; Hegedus L. S. Photochemical Ring Expansion of α-Alkoxycyclobutanones to 2-Acetoxy-5-alkoxytetrahydrofurans: Nucleophilic Reactions at the 2-Position. J. Org. Chem. 1998, 63, 5173–5178. 10.1021/jo980429x. [DOI] [Google Scholar]
- Molander G. A.; St. Jean D. J. Jr.; Haas J. Toward a General Route to the Eunicellin Diterpenes: The Asymmetric Total Synthesis of Deacetoxyalcyonin Acetate. J. Am. Chem. Soc. 2004, 126, 1642–1643. 10.1021/ja0398464. [DOI] [PubMed] [Google Scholar]
- Anderson J. R.; Briant C. E.; Edwards R. L.; Mabelis R. P.; Poyser J. P.; Spencer H.; Whalley A. J. S. Punctatin A (Antibiotic M95464): X-ray Crystal Structure of a Sesquiterpene Alcohol with a New Carbon Skeleton from the Fungus, Poronia punctata. J. Chem. Soc., Chem. Commun. 1984, 405–406. 10.1039/c39840000405. [DOI] [Google Scholar]
- The metabolites isolated from the fungus Poronia punctata were initially named punctatins. To avoid confusion, the name punctatins was modified to punctaporonins; see the following:Anderson J. R.; Briant C. E.; Edwards R. L.; Mabelis R. P.; Poyser J. P.; Spencer H.; Whalley A. J. S.; Freer A. A.; Gravel D.; Murray S.; Ladouceur G. Corrigenda to “Punctatin A (Antibiotic M95464): X-Ray Crystal Structure of a Sesquiterpene Alcohol with a New Carbon Skeleton from the Fungus, Poronia punctata” and “Punctatins B and C (Antibiotics M95154 and M95155): Further Sesquiterpene Alcohols from the Fungus Poronia punctata. J. Chem. Soc., Chem. Commun. 1986, 984. 10.1039/c39860000984. [DOI] [Google Scholar]
- Sugimura T.; Paquette L. A. Enantiospecific Total Synthesis of the Sesquiterpene Antibiotics (−)-Punctatin A and (+)-Punctatin D. J. Am. Chem. Soc. 1987, 109, 3017–3024. 10.1021/ja00244a025. [DOI] [Google Scholar]
- Rahimtoola S. H.; Tak T. The Use of Digitalis in Heart Failure. Curr. Probl. Cardiol. 1996, 21, 781–853. 10.1016/S0146-2806(96)80001-6. [DOI] [PubMed] [Google Scholar]
- Bagrov A. Y.; Shapiro J. I.; Fedorova O. V. Endogenous Cardiotonic Steroids: Physiology, Pharmacology, and Novel Therapeutic Targets. Pharmacol. Rev. 2009, 61, 9–38. 10.1124/pr.108.000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero C. P.; Medarde M.; San Feliciano A. A Short Review on Cardiotonic Steroids and Their Aminoguanidine Analogues. Molecules 2000, 5, 51–81. 10.3390/50100051. [DOI] [Google Scholar]
- Renata H.; Zhou Q.; Baran P. S. Strategic Redox Relay Enables A Scalable Synthesis of Ouabagenin, A Bioactive Cardenolide. Science 2013, 339, 59–63. 10.1126/science.1230631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renata H.; Zhou Q.; Dünstl G.; Felding J.; Merchant R. R.; Yeh C.-H.; Baran P. S. Development of a Concise Synthesis of Ouabagenin and Hydroxylated Corticosteroid Analogues. J. Am. Chem. Soc. 2015, 137, 1330–1340. 10.1021/ja512022r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrli H.; Heller M. S.; Schaffner K.; Jeger O. UV-Bestrahlung von 11-Oxo-Steroiden I Die Darstellung von 11β, 19-Cyclo-, 9β, 19-Cyclo- und 19-Hydroxy-5α-pregnan-Verbindungen. Helv. Chim. Acta 1961, 44, 2162–2173. 10.1002/hlca.19610440745. [DOI] [Google Scholar]
- Finucane B. W.; Thomson J. B. The Photoisomerisation of an 11-Oxo-olean-12-ene. J. Chem. Soc. D 1969, 380. 10.1039/c29690000380. [DOI] [Google Scholar]
- Booker-Milburn K. I.; Baker J. R.; Bruce I. Rapid Access to Azepine-Fused Oxetanols from Alkoxy-Substituted Maleimides. Org. Lett. 2004, 6, 1481–1484. 10.1021/ol049645k. [DOI] [PubMed] [Google Scholar]
- Hickford P. J.; Baker J. R.; Bruce I.; Booker-Milburn K. I. Acid-Catalyzed Rearrangement of Fused Alkylideneoxetanols. Org. Lett. 2007, 9, 4681–4684. 10.1021/ol701625q. [DOI] [PubMed] [Google Scholar]
- Yoshioka S.; Nagatomo M.; Inoue M. Application of Two Direct C(sp3)–H Functionalizations for Total Synthesis of (+)-Lactacystin. Org. Lett. 2015, 17, 90–93. 10.1021/ol503291s. [DOI] [PubMed] [Google Scholar]
- Kraus G. A.; Chen L. A Total Synthesis of Racemic Paulownin Using a Type II Photocyclization Reaction. J. Am. Chem. Soc. 1990, 112, 3464–3466. 10.1021/ja00165a033. [DOI] [Google Scholar]
- Ishibashi F.; Hayashita M.; Okazaki M.; Shuto Y. Improved Procedure for the Enantiometric Synthesis of 1-Hydroxy/acetoxy-2,6-diaryl-3,7-dioxabicyclo[3.3.0]octane Lignans: Total Syntheses of (+)-Paulownin, (+)-Phrymarin I and (+)-Phrymarin II. Biosci., Biotechnol., Biochem. 2001, 65, 29–34. 10.1271/bbb.65.29. [DOI] [PubMed] [Google Scholar]
- For a review on the oxa-di-π-methane rearrangement, see the following:Zimmerman H. E.; Armesto D. Synthetic Aspects of the Di-π-methane Rearrangement. Chem. Rev. 1996, 96, 3065–3112. 10.1021/cr910109c. [DOI] [PubMed] [Google Scholar]
- Tenney L. P.; Boykin D. W. Jr.; Lutz R. E. Novel Photocyclization of a Highly Phenylated β,γ-Unsaturated Ketone to a Cyclopropyl Ketone, Involving Benzoyl Group Migration. J. Am. Chem. Soc. 1966, 88, 1835–1836. 10.1021/ja00960a059. [DOI] [Google Scholar]
- Hixson S. S.; Mariano P. S.; Zimmerman H. E. Di-π-Methane and Oxa-Di-π-Methane Rearrangements. Chem. Rev. 1973, 73, 531–551. 10.1021/cr60285a005. [DOI] [Google Scholar]
- Banwell M. G.; Bon D. J.-Y. D.. Applications of Di-π-Methane and Related Rearrangement Reactions in Chemical Synthesis. In Molecular Rearrangements in Organic Synthesis; Rojas C. M., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2015. [Google Scholar]
- Stevens K. E.; Yates P. Cedrenoid sesquiterpenes. Synthesis of the (±) Stork–Clarke β-Diketone. J. Chem. Soc., Chem. Commun. 1980, 990–991. 10.1039/C39800000990. [DOI] [Google Scholar]
- Yates P.; Stevens K. E. Bicyclopentanoid Sesquiterpenes. The Synthesis of Cedranoid Sesquiterpenes via the Photo-Rearrangement of Bicyclo[2.2.2]octenones. Tetrahedron 1981, 37, 4401–4410. 10.1016/0040-4020(81)80006-3. [DOI] [Google Scholar]
- Stork G.; Clarke F. H. Jr. Cedrol: Stereochemistry and Total Synthesis. J. Am. Chem. Soc. 1961, 83, 3114–3125. 10.1021/ja01475a030. [DOI] [Google Scholar]
- Wiberg K. B. Small Ring Propellanes. Chem. Rev. 1989, 89, 975–983. 10.1021/cr00095a001. [DOI] [Google Scholar]
- Levin M. D.; Kaszynski P.; Michl J. Bicyclo[1.1.1]pentanes, [n]Staffanes, [1.1.1]Propellanes, and Tricyclo[2.1.0.02,5]pentanes. Chem. Rev. 2000, 100, 169–234. 10.1021/cr990094z. [DOI] [PubMed] [Google Scholar]
- For a review on propellane-containing natural products, see the following:Pihko A. J.; Koskinen A. M. P. Synthesis of Propellane-Containing Natural Products. Tetrahedron 2005, 61, 8769–8807. 10.1016/j.tet.2005.06.013. [DOI] [Google Scholar]
- For a review on synthesis of propellanes, see the following:Delia E. W.; Lochert I. J. Synthesis of Bridgehead-Substituted Bicyclo[1.1.1]pentanes. A Review. Org. Prep. Proced. Int. 1996, 28, 411–441. 10.1080/00304949609356550. [DOI] [Google Scholar]
- Oppolzer W.; Bättig K. A Short and Efficient Synthesis of (±)-Modhephene by a Stereoelectronically-Controlled Ene-Reaction. Helv. Chim. Acta 1981, 64, 2489–2491. 10.1002/hlca.19810640756. [DOI] [Google Scholar]
- Wilkening D.; Mundy B. P. A Formal Synthesis of Modhephene. Tetrahedron Lett. 1984, 25, 4619–4622. 10.1016/S0040-4039(01)91215-8. [DOI] [Google Scholar]
- Mundy B. P.; Wilkening D.; Lipkowitz K. B. Total Synthesis of Modhephene. J. Org. Chem. 1985, 50, 5727–5731. 10.1021/jo00350a057. [DOI] [Google Scholar]
- Fitjer L.; Majewski M.; Kanschik A. Towards a Synthesis of (±)-Modhephene via Cascade Rearrangement: Synthesis and Rearrangement of Dispiro[3.0.4.2]undecanes to [3.3.3]Propellanes. Tetrahedron Lett. 1988, 29, 1263–1264. 10.1016/S0040-4039(00)80271-3. [DOI] [Google Scholar]
- Mash E. A.; Math S. K.; Flann C. J. Homochiral Ketals in Organic Synthesis. Enantioselective Synthesis and Absolute Configuration of (−)-Modhephene. Tetrahedron 1989, 45, 4945–4950. 10.1016/S0040-4020(01)81075-9. [DOI] [Google Scholar]
- Curran D. P.; Shen W. Tandem Transannular Radical Cyclizations. Total Syntheses of (±)-Modhephene and (±)-epi-Modhephene. Tetrahedron 1993, 49, 755–770. 10.1016/S0040-4020(01)80321-5. [DOI] [Google Scholar]
- Suri S. C. A Formal Total Synthesis of Modhephene. Tetrahedron Lett. 1993, 34, 8321–8324. 10.1016/S0040-4039(00)61421-1. [DOI] [Google Scholar]
- Fitjer L.; Majewski M.; Monzó-Oltra H. Synthesis of Tricyclopentanoid Sesquiterpenes via Rearrangement Routes: (±)-Modhephene, (±)-Epimodhephene and (±)-Isocomene. Tetrahedron 1995, 51, 8835–8852. 10.1016/0040-4020(95)00486-R. [DOI] [Google Scholar]
- Uyehara T.; Murayama T.; Sakai K.; Ueno M.; Sato T. Formal Substitution at Both Bridgeheads of a Bicyclo[2.2.2]oct-5-en-2-one and Its Application to a Synthesis of (±)-Modhephene. Tetrahedron Lett. 1996, 37, 7295–7298. 10.1016/0040-4039(96)01652-8. [DOI] [Google Scholar]
- De Boeck B.; Pattenden G. α-Ketenyl Radical Intermediates in the Synthesis of Propellanes. A Formal Synthesis of Modhephene. Tetrahedron Lett. 1998, 39, 6975–6978. 10.1016/S0040-4039(98)01480-4. [DOI] [Google Scholar]
- Mehta G.; Subrahmanyam D. A Total Synthesis of (±)-Modhephene. J. Chem. Soc., Chem. Commun. 1985, 768–769. 10.1039/C39850000768. [DOI] [Google Scholar]
- Mehta G.; Subrahmanyam D. Photochemical Oxa-Di-π-Methane Rearrangement Approach to [3.3.3]Propellanes. Total Synthesis of Sesquiterpene Hydrocarbon (±)-Modhephene. J. Chem. Soc., Perkin Trans. 1 1991, 395–401. 10.1039/P19910000395. [DOI] [Google Scholar]
- Hirst G. C.; Johnson T. O. Jr.; Overman L. E. First Total Synthesis of Lycopodium Alkaloids of the Magellanane Group. Enantioselective Total Syntheses of (−)-Magellanine and (+)-Magellaninone. J. Am. Chem. Soc. 1993, 115, 2992–2993. 10.1021/ja00060a064. [DOI] [Google Scholar]
- Paquette L. A.; Friedrich D.; Pinard E.; Williams J. P.; St. Laurent D.; Roden B. A. Total Synthesis of the Tetracyclic Diquinane Lycopodium Alkaloids Magellanine and Magellaninone. J. Am. Chem. Soc. 1993, 115, 4377–4378. 10.1021/ja00063a072. [DOI] [Google Scholar]
- Williams J. P.; St. Laurent D. R.; Friedrich D.; Pinard E.; Roden B. A.; Paquette L. A. Total Synthesis of the Lycopodium Alkaloids Magellanine and Magellaninone by Three-fold Annulation of 2-Cyclopentenone. J. Am. Chem. Soc. 1994, 116, 4689–4696. 10.1021/ja00090a017. [DOI] [Google Scholar]
- For the first total synthesis of (+)-paniculatine, see the following:Sha C.-K.; Lee F.-K.; Chang C.-J. Tandem Radical Cyclizations Initiated with α-Carbonyl Radicals: First Total Synthesis of (+)-Paniculatine. J. Am. Chem. Soc. 1999, 121, 9875–9876. 10.1021/ja992315o. [DOI] [Google Scholar]
- Castillo M.; Morales G.; Loyola L. A.; Singh I.; Calvo C.; Holland H. L.; MacLean D. B. Paniculatine. A New Alkaloid from L. paniculatum Desvaux. Can. J. Chem. 1975, 53, 2513–2514. 10.1139/v75-357. [DOI] [Google Scholar]
- Castillo M.; Loyola L. A.; Morales G.; Singh I.; Calvo C.; Rolland H. L.; MacLean D. B. The Alkaloids of L. magellanicum and the Structure of Magellanine. Can. J. Chem. 1976, 54, 2893–2899. 10.1139/v76-409. [DOI] [Google Scholar]
- Castillo M.; Morales G.; Loyola L. A.; Singh I.; Calvo C.; Holland H. L.; MacLean D. B. The Alkaloids of L. paniculatum and the Structure of Paniculatine. Can. J. Chem. 1976, 54, 2900–2908. 10.1139/v76-410. [DOI] [Google Scholar]
- Loyola L. A.; Morales G.; Castillo M. Alkaloids of Lycopodium magellanicum. Phytochemistry 1979, 18, 1721–1723. 10.1016/0031-9422(79)80193-4. [DOI] [Google Scholar]
- Ishizaki M.; Niimi Y.; Hoshino O.; Hara H.; Takahashi T. A Formal Total Synthesis of Lycopodium alkaloid, (±)-Magellanine, by Using the Intramolecular Pauson Khand Reaction. Tetrahedron 2005, 61, 4053–4065. 10.1016/j.tet.2005.02.044. [DOI] [Google Scholar]
- Kozaka T.; Miyakoshi N.; Mukai C. Stereoselective Total Syntheses of Three Lycopodium Alkaloids, (−)-Magellanine, (+)-Magellaninone, and (+)-Paniculatine, Based on Two Pauson-Khand Reactions. J. Org. Chem. 2007, 72, 10147–10154. 10.1021/jo702136b. [DOI] [PubMed] [Google Scholar]
- Chandra A.; Pigza J. A.; Han J.-S.; Mutnick D.; Johnston J. N. Total Synthesis of the Lycopodium Alkaloid (+)-Serratezomine A. J. Am. Chem. Soc. 2009, 131, 3470–3471. 10.1021/ja900536d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S.-Z.; Lei T.; Wei K.; Yang Y.-R. Collective Total Synthesis of Tetracyclic Diquinane Lycopodium Alkaloids (+)-Paniculatine, (−)-Magellanine, (+)-Magellaninone and Analogues Thereof. Org. Lett. 2014, 16, 5612–5615. 10.1021/ol502679v. [DOI] [PubMed] [Google Scholar]
- Lin K.-W.; Ananthan B.; Tseng S.-F.; Yan T.-H. Exceedingly Concise and Elegant Synthesis of (+)-Paniculatine, (−)-Magellanine, and (+)-Magellaninone. Org. Lett. 2015, 17, 3938–3940. 10.1021/acs.orglett.5b01975. [DOI] [PubMed] [Google Scholar]
- Yen C.-F.; Liao C.-C. Concise and Efficient Total Synthesis of Lycopodium Alkaloid Magellanine. Angew. Chem., Int. Ed. 2002, 41, 4090–4093. 10.1002/1521-3773(20021104)41:21<4090::AID-ANIE4090>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Hsu D.-S.; Chou Y.-Y.; Tung Y.-S.; Liao C.-C. Photochemistry of Tricyclo[5.2.2.02,6]undeca-4,10-dien-8-ones: An Efficient General Route to Substituted Linear Triquinanes from 2-Methoxyphenols. Total Synthesis of (±)-Δ9(12)-Capnellene. Chem. - Eur. J. 2010, 16, 3121–3131. 10.1002/chem.200902752. [DOI] [PubMed] [Google Scholar]
- Austin K. A. B.; Banwell M. G.; Harfoot G. J.; Willis A. C. Chemoenzymatic Syntheses of the Linear Triquinane-type Sesquiterpenes (+)-Hirsutic Acid and (−)-Complicatic Acid. Tetrahedron Lett. 2006, 47, 7381–7384. 10.1016/j.tetlet.2006.07.145. [DOI] [Google Scholar]
- Banwell M. G.; Austin K. A. B.; Willis A. C. Chemoenzymatic Total Syntheses of the Linear Triquinane-type Natural Products (+)-Hirsutic Acid and (−)-Complicatic Acid from Toluene. Tetrahedron 2007, 63, 6388–6403. 10.1016/j.tet.2007.03.073. [DOI] [Google Scholar]
- Demuth M.; Ritterskamp P.; Weigt E.; Schaffner K. Total Synthesis of (−)-Coriolin. J. Am. Chem. Soc. 1986, 108, 4149–4154. 10.1021/ja00274a050. [DOI] [Google Scholar]
- Singh V.; Samanta B. Generation of Molecular Complexity from Aromatics: A Formal Total Synthesis of Coriolin from 6-Methylsaligenin. Tetrahedron Lett. 1999, 40, 383–386. 10.1016/S0040-4039(98)02318-1. [DOI] [Google Scholar]
- Singh V.; Alam S. Q. Intramolecular Diels-Alder Reaction in 1-Oxaspiro[2.5]octa-5,7-dien-4-one and Sigmatropic Shifts in Excited States: Novel Route to Sterpuranes and Linear Triquinanes: Formal Total Synthesis of (±)-Coriolin. Chem. Commun. 1999, 2519–2520. 10.1039/a908223i. [DOI] [Google Scholar]
- Singh V.; Samanta B.; Kane V. V. Molecular Complexity from Aromatics: Synthesis and Photoreaction of endo-Tricyclo[5.2.2.02,6]undecanes. Formal Total Syntheses of (±)-Coriolin. Tetrahedron 2000, 56, 7785–7795. 10.1016/S0040-4020(00)00674-8. [DOI] [Google Scholar]
- For the total synthesis of (±)-coriolin, see the following:Demuth M.; Ritterskamp P.; Schaffner K. Novel Variant of the Tricyclooctanone Approach to Cyclopentanoid Natural Products. Synthesis of (±)-Coriolin. Helv. Chim. Acta 1984, 67, 2023–2027. 10.1002/hlca.19840670741. [DOI] [Google Scholar]
- Banwell M. G.; Edwards A. J.; Harfoot G. J.; Jolliffe K. A. A Chemoenzymatic Synthesis of (−)-Hirsutene from Toluene. J. Chem. Soc. Perkin Trans. 1 2002, 2439–2441. 10.1039/B208778B. [DOI] [Google Scholar]
- Banwell M. G.; Edwards A. J.; Harfoot G. J.; Jolliffe K. A. A Chemoenzymatic Synthesis of the Linear Triquinane (−)-Hirsutene and Identification of Possible Precursors to the Naturally Occurring (+)-Enantiomer. Tetrahedron 2004, 60, 535–547. 10.1016/j.tet.2003.10.122. [DOI] [Google Scholar]
- Singh V.; Vedantham P.; Sahu P. K. A Novel Stereoselective Total Synthesis of (±)-Hirsutene from Saligenin. Tetrahedron Lett. 2002, 43, 519–522. 10.1016/S0040-4039(01)02183-9. [DOI] [Google Scholar]
- Singh V.; Vedantham P.; Sahu P. K. Reactive Species from Aromatics and Oxa-di-π-methane Rearrangement: A Stereoselective Synthesis of (±)-Hirsutene from Salicyl Alcohol. Tetrahedron 2004, 60, 8161–8169. 10.1016/j.tet.2004.06.096. [DOI] [Google Scholar]
- Singh V.; Tosh D. K.; Mobin S. M. Synthesis of Embellished Bicyclo[2.2.2]octenones and a Sigmatropic 1,2-Acyl Shift in an Excited State: A Novel and Stereoselective Route to (±)-Hirsutic Acid C and Complicatic Acid. Tetrahedron Lett. 2004, 45, 1729–1732. 10.1016/j.tetlet.2003.12.095. [DOI] [Google Scholar]
- Singh V.; Pal S.; Tosh D. K.; Mobin S. M. Molecular Complexity from Aromatics. Cycloaddition of Cyclohexa-2,4-dienones, Sigmatropic 1,2-Acyl Shift and Ring-Closing Metathesis: A New, Efficient, and Stereoselective Synthesis of (±)-Hirsutic Acid C and Medium Ring Carbocycles. Tetrahedron 2007, 63, 2446–2454. 10.1016/j.tet.2007.01.006. [DOI] [Google Scholar]
- Reekie T. A.; Austin K. A. B.; Banwell M. G.; Willis A. C. The Chemoenzymatic Total Synthesis of Phellodonic Acid, a Biologically Active and Highly Functionalized Hirsutane Derivative Isolated from the Tasmanian Fungus Phellodon melaleucus. Aust. J. Chem. 2008, 61, 94–106. 10.1071/CH07403. [DOI] [Google Scholar]
- For a recent review on arene–alkene photocycloadditions, see the following:Streit U.; Bochet C. G. The Arene-Alkene Photocycloaddition. Beilstein J. Org. Chem. 2011, 7, 525–542. 10.3762/bjoc.7.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit U.; Birbaum F.; Quattropani A.; Bochet C. G. Photocycloaddition of Arenes and Allenes. J. Org. Chem. 2013, 78, 6890–6910. 10.1021/jo4002307. [DOI] [PubMed] [Google Scholar]
- Wegmann M.; Bach T. Influence of the -CH2X Substituent on the Regioselectivity of Intramolecular meta-Photocycloaddition Reactions. J. Org. Chem. 2015, 80, 2017–2023. 10.1021/jo5028613. [DOI] [PubMed] [Google Scholar]
- Chappell D.; Russell A. T. From α-Cedrene to Crinipellin B and Onward: 25 Years of the Alkene-Arene meta-Photocycloaddition Reaction in Natural Product Synthesis. Org. Biomol. Chem. 2006, 4, 4409–4430. 10.1039/B614011B. [DOI] [PubMed] [Google Scholar]
- Cornelisse J. The meta Photocycloaddition of Arenes to Alkenes. Chem. Rev. 1993, 93, 615–669. 10.1021/cr00018a002. [DOI] [Google Scholar]
- Wilzbach K. E.; Kaplan L. A Photochemical 1,3 Cycloaddition of Olefins to Benzene. J. Am. Chem. Soc. 1966, 88, 2066–2067. 10.1021/ja00961a052. [DOI] [Google Scholar]
- Wender P. A.; Ternansky R.; deLong M.; Singh S.; Olivero A.; Rice K. Arene-Alkene Cycloadditions and Organic Synthesis. Pure Appl. Chem. 1990, 62, 1597–1602. 10.1351/pac199062081597. [DOI] [Google Scholar]
- Walter P. Ueber das Feste und Flüssige Cedernöl. Justus Liebigs Ann. Chem. 1841, 39, 247–251. 10.1002/jlac.18410390213. [DOI] [Google Scholar]
- Walter P. Ueber das Krystallisirte und über das Flüssige Cedernöl. Justus Liebigs Ann. Chem. 1843, 48, 35–38. 10.1002/jlac.18430480105. [DOI] [Google Scholar]
- Stork G.; Breslow R. The Structure of Cedrene. J. Am. Chem. Soc. 1953, 75, 3291–3291. 10.1021/ja01109a524. [DOI] [Google Scholar]
- Stork G.; Clarke F. H. Jr. The Total Synthesis of Cedrol and Cedrene. J. Am. Chem. Soc. 1955, 77, 1072–1073. 10.1021/ja01609a104. [DOI] [Google Scholar]
- Corey E. J.; Girotra N. N.; Mathew C. T. Total Synthesis of dl-Cedrene and dl-Cedrol. J. Am. Chem. Soc. 1969, 91, 1557–1559. 10.1021/ja01034a062. [DOI] [Google Scholar]
- Crandall T. G.; Lawton R. G. Biogenetic-Type Synthesis of Cedrene. J. Am. Chem. Soc. 1969, 91, 2127–2129. 10.1021/ja01036a055. [DOI] [Google Scholar]
- Demole E.; Enggist P.; Borer C. Applications Synthétiques de la Cyclisation d’Alcools Tertiaires γ-Éthyléniques en α-Bromotétrahydrofurannes sous l’Action du N-Bromosuccinimide. II. Cyclisation du (±)-Nérolidol en Diméthyl-2,5-(méthyl-4-pentène-3-yl)-2-cycloheptène-4-one, Tétraméthyl-3,3,7,10-oxa-2-tricyclo[5.5.0.01,4]-dodécène-9, β-Acoratriène, Cédradiène-2,8, épi-2-α-Cédrène et α-Cédrène. Helv. Chim. Acta 1971, 54, 1845–1864. 10.1002/hlca.19710540712. [DOI] [Google Scholar]
- Ohta Y.; Hirose Y. Electrophile Induced Cyclization of Farnesol. Chem. Lett. 1972, 1, 263–266. 10.1246/cl.1972.263. [DOI] [Google Scholar]
- Andersen N. H.; Syrdal D. D. Chemical Simulation of the Biogenesis of Cedrene. Tetrahedron Lett. 1972, 13, 2455–2458. 10.1016/S0040-4039(01)84845-0. [DOI] [Google Scholar]
- Kuehne M. E.; Damon R. E. Thiyl Radical Induced Cyclizations of Dienes. Cyclization of α-Acoradiene, α-Bulnesene, and Geranyl Acetate to Cedrane, Patchulane, and Cyclogeranyl Acetate Products. J. Org. Chem. 1977, 42, 1825–1832. 10.1021/jo00431a001. [DOI] [Google Scholar]
- Breitholle E. G.; Fallis A. G. Total Synthesis of (±)-Cedrol and (±)-Cedrene via an Intramolecular Diels-Alder Reaction. J. Org. Chem. 1978, 43, 1964–1968. 10.1021/jo00404a025. [DOI] [Google Scholar]
- Rigby J. H.; Kirova-Snover M. Studies on Intramolecular Cr(0)-promoted [6π+2π] Cycloaddition Reactions. Synthesis of β-Cedrene. Tetrahedron Lett. 1997, 38, 8153–8156. 10.1016/S0040-4039(97)10221-0. [DOI] [Google Scholar]
- Lee H.-Y.; Lee S.; Kim D.; Kim B. K.; Bahn J. S.; Kim S. Total Synthesis of α-Cedrene: A New Strategy Utilizing N-Aziridinylimine Radical Chemistry. Tetrahedron Lett. 1998, 39, 7713–7716. 10.1016/S0040-4039(98)01680-3. [DOI] [Google Scholar]
- Kerr W. J.; McLaughlin M.; Morrison A. J.; Pauson P. L. Formal Total Synthesis of (±)-α- and β-Cedrene by Preparation of Cedrone. Construction of the Tricyclic Carbon Skeleton by the Use of a Highly Efficient Intramolecular Khand Annulation. Org. Lett. 2001, 3, 2945–2948. 10.1021/ol016054a. [DOI] [PubMed] [Google Scholar]
- For a recent report on the synthesis of α-cedrene through the use of an oxidative-dearomatization-induced [5 + 2] cycloaddition approach, see the following:Green J. C.; Pettus T. R. R. An Oxidative Dearomatization-Induced [5 + 2] Cascade Enabling the Syntheses of α-Cedrene, α-Pipitzol, and sec-Cedrenol. J. Am. Chem. Soc. 2011, 133, 1603–1608. 10.1021/ja109925g. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Ternansky R. J. Synthetic Studies on Arene-Olefin Cycloadditions—VII: A Three-Step Total Synthesis of (±)-Silphinene. Tetrahedron Lett. 1985, 26, 2625–2628. 10.1016/S0040-4039(00)98120-6. [DOI] [Google Scholar]
- Rao P. S.; Sarma K. G.; Seshadri T. R. Chemical Components of the Lobaria Lichens from the Western Himalayas. Curr. Sci. 1965, 34, 9–11. [Google Scholar]
- Kaneda M.; Takahashi R.; Iitaka Y.; Shibata S. Retigeranic Acid, a Novel Sesterterpene Isolated from the Lichens of Lobaria Retigera Group. Tetrahedron Lett. 1972, 13, 4609–4611. 10.1016/S0040-4039(01)94378-3. [DOI] [Google Scholar]
- Kaneda M.; Iitaka Y.; Shibata S. X-ray Studies of C25 Terpenoids. IV. The Crystal Structure of Retigeranic Acid p-Bromoanilide. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1974, 30, 358–364. 10.1107/S0567740874002822. [DOI] [Google Scholar]
- Sugawara H.; Kasuya A.; Iitaka Y.; Shibata S. Further Studies on the Structure of Retigeranic Acid. Chem. Pharm. Bull. 1991, 39, 3051–3054. 10.1248/cpb.39.3051. [DOI] [Google Scholar]
- Gybin A. S.; Smit V. A.; Caple R.; Veretenov A. L.; Shashkov A. S.; Vorontsova L. G.; Kurella M. G.; Chertkov V. S.; Karapetyan A. A. The Sequence of a Stepwise AdE Reaction and Intramolecular Pauson-Khand Cycloaddition as an Entry into the Synthesis of Polycyclic Compounds. J. Am. Chem. Soc. 1992, 114, 5555–5566. 10.1021/ja00040a012. [DOI] [Google Scholar]
- Hog D. T.; Mayer P.; Trauner D. A Unified Approach to trans-Hydrindane Sesterterpenoids. J. Org. Chem. 2012, 77, 5838–5843. 10.1021/jo300726z. [DOI] [PubMed] [Google Scholar]
- Hog D. T.; Huber F. M. E.; Jiménez-Osés G.; Mayer P.; Houk K. N.; Trauner D. Evolution of a Unified Strategy for Complex Sesterterpenoids: Progress toward Astellatol and the Total Synthesis of (−)-Nitidasin. Chem. - Eur. J. 2015, 21, 13646–13665. 10.1002/chem.201501423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey E. J.; Desai M. C.; Engler T. A. Total Synthesis of (±)-Retigeranic Acid. J. Am. Chem. Soc. 1985, 107, 4339–4341. 10.1021/ja00300a049. [DOI] [Google Scholar]
- Hudlicky T.; Short R. P. Terpenic Acids by Cyclopentene Annulation of Exocyclic Dienes. Synthesis of the Triquinane Portion of Retigeranic Acid. J. Org. Chem. 1982, 47, 1522–1527. 10.1021/jo00347a031. [DOI] [Google Scholar]
- Hudlicky T.; Radesca-Kwart L.; Li L.-q.; Bryant T. Short, Enantioselective Synthesis of (−)-Retigeranic Acid via [2 + 3] Annulation. Tetrahedron Lett. 1988, 29, 3283–3286. 10.1016/0040-4039(88)85141-4. [DOI] [Google Scholar]
- Hudlicky T.; Fleming A.; Radesca L. The [2 + 3] and [3 + 4] Annulation of Enones. Enantiocontrolled Total Synthesis of (−)-Retigeranic Acid. J. Am. Chem. Soc. 1989, 111, 6691–6707. 10.1021/ja00199a032. [DOI] [Google Scholar]
- Hudlicky T.; Fleming A.; Radesca L. The [2 + 3] and [3 + 4] Annulation of Enones. Enantiocontrolled Total Synthesis of (−)-Retigeranic Acid [Erratum to Document Cited in CA111(11):97589v]. J. Am. Chem. Soc. 1989, 111, 9136–9136. 10.1021/ja00207a040. [DOI] [Google Scholar]
- Paquette L. A.; Wright J.; Drtina G. J.; Roberts R. A. Enantiospecific Total Synthesis of Natural (−)-Retigeranic Acid A and Two (−)-Retigeranic Acid B Candidates. J. Org. Chem. 1987, 52, 2960–2962. 10.1021/jo00389a070. [DOI] [Google Scholar]
- Wright J.; Drtina G. J.; Roberts R. A.; Paquette L. A. A Convergent Synthesis of Triquinane Sesterterpenes. Enantioselective Synthesis of (−)-Retigeranic Acid A. J. Am. Chem. Soc. 1988, 110, 5806–5817. 10.1021/ja00225a036. [DOI] [Google Scholar]
- For a recent example of the construction of the tetracyclic core of retigeranic acid A, see the following:Zhang J.; Wang X.; Li S.; Li D.; Liu S.; Lan Y.; Gong J.; Yang Z. Diastereoselective Synthesis of Cyclopentanoids: Applications to the Construction of the ABCD Tetracyclic Core of Retigeranic Acid A. Chem. - Eur. J. 2015, 21, 12596–12600. 10.1002/chem.201502423. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Singh S. K. Synthetic Studies on Arene-Olefin Cycloadditions. 11. Total Synthesis of (−)-Retigeranic Acid. Tetrahedron Lett. 1990, 31, 2517–2520. 10.1016/0040-4039(90)80114-2. [DOI] [Google Scholar]
- Wender P. A.; Fisher K. Seven-Membered Ring Synthesis Based on Arene Olefin Cycloadditions: The Total Synthesis of (±)-Rudmollin. Tetrahedron Lett. 1986, 27, 1857–1860. 10.1016/S0040-4039(00)84394-4. [DOI] [Google Scholar]
- Herz W.; Kumar N.; Blount J. F. Antileukemic C-15-Functionalized Ambrosanolides from Rudbeckia Mollis. J. Org. Chem. 1981, 46, 1356–1361. 10.1021/jo00320a026. [DOI] [Google Scholar]
- For a recent report on the synthesis of rudmollin, see the following:Carroll G. L.; Allan A. K.; Schwaebe M. K.; Little R. D. Atom Transfer Reactions of TMM Diyls Directed toward the Synthesis of Rudmollin. Org. Lett. 2000, 2, 2531–2534. 10.1021/ol006210y. [DOI] [PubMed] [Google Scholar]
- Shim S. H.; Swenson D. C.; Gloer J. B.; Dowd P. F.; Wicklow D. T. Penifulvin A: A Sesquiterpenoid-Derived Metabolite Containing a Novel Dioxa[5,5,5,6]fenestrane Ring System from a Fungicolous Isolate of Penicillium griseofulvum. Org. Lett. 2006, 8, 1225–1228. 10.1021/ol060107c. [DOI] [PubMed] [Google Scholar]
- Shim S. H.; Gloer J. B.; Wicklow D. T. Penifulvins B–E and a Silphinene Analogue: Sesquiterpenoids from a Fungicolous Isolate of Penicillium griseofulvum. J. Nat. Prod. 2006, 69, 1601–1605. 10.1021/np060327z. [DOI] [PubMed] [Google Scholar]
- Keese R. Carbon Flatland: Planar Tetracoordinate Carbon and Fenestranes. Chem. Rev. 2006, 106, 4787–4808. 10.1021/cr050545h. [DOI] [PubMed] [Google Scholar]
- Boudhar A.; Charpenay M.; Blond G.; Suffert J. Fenestranes in Synthesis: Unique and Highly Inspiring Scaffolds. Angew. Chem., Int. Ed. 2013, 52, 12786–12798. 10.1002/anie.201304555. [DOI] [PubMed] [Google Scholar]
- Chakraborty T. K.; Chattopadhyay A. K.; Samanta R.; Ampapathi R. S. Stereoselective Construction of Quaternary Chiral Centers Using Ti(III)-Mediated Opening of 2,3-Epoxy Alcohols: Studies Directed toward the Synthesis of Penifulvins. Tetrahedron Lett. 2010, 51, 4425–4428. 10.1016/j.tetlet.2010.06.076. [DOI] [Google Scholar]
- Mehta G.; Khan T. B. Model Studies toward a Synthesis of Asperaculin A: Exploration of Iterative Intramolecular Pauson–Khand Reaction Based Strategies to Access the Dioxa[5.5.5.6]fenestrane Framework. Tetrahedron Lett. 2012, 53, 4558–4561. 10.1016/j.tetlet.2012.06.059. [DOI] [Google Scholar]
- Das D.; Kant R.; Chakraborty T. K. An Approach to a Bislactone Skeleton: A Scalable Total Synthesis of (±)-Penifulvin A. Org. Lett. 2014, 16, 2618–2621. 10.1021/ol500768w. [DOI] [PubMed] [Google Scholar]
- Gaich T.; Mulzer J. Total Synthesis of (−)-Penifulvin A, an Insecticide with a Dioxafenestrane Skeleton. J. Am. Chem. Soc. 2009, 131, 452–453. 10.1021/ja8083048. [DOI] [PubMed] [Google Scholar]
- Gaich T.; Mulzer J. From Silphinenes to Penifulvins: A Biomimetic Approach to Penifulvins B and C. Org. Lett. 2010, 12, 272–275. 10.1021/ol902594b. [DOI] [PubMed] [Google Scholar]
- Gaich T.; Mulzer J.. Biomimetic Total Synthesis of the Penifulvin Family. In Asymmetric Synthesis II: More Methods and Applications; Christmann M., Bräse S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. [Google Scholar]
- Myers A. G.; Yang B. H.; Chen H.; McKinstry L.; Kopecky D. J.; Gleason J. L. Pseudoephedrine as a Practical Chiral Auxiliary for the Synthesis of Highly Enantiomerically Enriched Carboxylic Acids, Alcohols, Aldehydes, and Ketones. J. Am. Chem. Soc. 1997, 119, 6496–6511. 10.1021/ja970402f. [DOI] [Google Scholar]
- Wender P. A.; Dore T. M.; deLong M. A. An Arene-Alkene Photocycloaddition-Radical Cyclization Cascade: The First Syntheses of cis,cis,cis,trans-[5.5.5.5 ]-Fenestranes. Tetrahedron Lett. 1996, 37, 7687–7690. 10.1016/0040-4039(96)01740-6. [DOI] [Google Scholar]
- Wender P. A.; Von Geldern T. W.; Levine B. H. Synthetic Studies on Arene-Olefin Cycloadditions. 10. A Concise, Stereocontrolled Total Synthesis of (±)-Laurenene. J. Am. Chem. Soc. 1988, 110, 4858–4860. 10.1021/ja00222a072. [DOI] [Google Scholar]
- Wender P. A.; Howbert J. J. Synthetic Studies on Areneolefin Cycloadditions. VI. Two Syntheses of (±)-Coriolin. Tetrahedron Lett. 1983, 24, 5325–5328. 10.1016/S0040-4039(00)87859-4. [DOI] [Google Scholar]
- Wender P. A.; Howbert J. J. Synthetic Studies on Arene-Olefin Cycloadditions. III. Total Synthesis of (±)-Hirsutene. Tetrahedron Lett. 1982, 23, 3983–3986. 10.1016/S0040-4039(00)88675-X. [DOI] [Google Scholar]
- Wender P. A.; Dreyer G. B. Synthetic Studies on Arene-Olefin Cycloadditions. V. Total Synthesis of (±)-Isoiridomyrmecin. Tetrahedron Lett. 1983, 24, 4543–4546. 10.1016/S0040-4039(00)85950-X. [DOI] [Google Scholar]
- Wender P. A.; Dreyer G. B. Synthetic Studies on Arene-Olefin Cycloadditions. 4. Total Synthesis of (±)-Modhephene. J. Am. Chem. Soc. 1982, 104, 5805–5807. 10.1021/ja00385a051. [DOI] [Google Scholar]
- Wender P. A.; Singh S. K. Synthetic Studies on Arene-Olefin Cycloadditions. VIII. Total Syntheses of (±)-Silphiperfol-6-ene, (±)-7-α-H-Silphiperfol-5-ene and (±)-7-β-H-Silphiperfol-5-ene. Tetrahedron Lett. 1985, 26, 5987–5990. 10.1016/S0040-4039(00)98278-9. [DOI] [Google Scholar]
- Wender P. A.; deLong M. A. Synthetic Studies on Arene-Olefin Cycloadditions. XII. Total Synthesis of (±)-Subergorgic Acid. Tetrahedron Lett. 1990, 31, 5429–5432. 10.1016/S0040-4039(00)97864-X. [DOI] [Google Scholar]
- Wender P. A.; Dore T. M. A Formal Synthesis of Crinipellin B Based on the Arene-Alkene meta-Photocycloaddition Reaction. Tetrahedron Lett. 1998, 39, 8589–8592. 10.1016/S0040-4039(98)01965-0. [DOI] [Google Scholar]
- For synthesis of fine chemicals using sunlight, see the following:Esser P.; Pohlmann B.; Scharf H.-D. The Photochemical Synthesis of Fine Chemicals with Sunlight. Angew. Chem., Int. Ed. Engl. 1994, 33, 2009–2023. 10.1002/anie.199420091. [DOI] [Google Scholar]
- Kalyanasundaram K. Photophysics, Photochemistry and Solar Energy Conversion with Tris(bipyridyl)ruthenium(II) and Its Analogues. Coord. Chem. Rev. 1982, 46, 159–244. 10.1016/0010-8545(82)85003-0. [DOI] [Google Scholar]
- Juris A.; Balzani V.; Barigelletti F.; Campagna S.; Belser P.; von Zelewsky A. Ru(II) Polypyridine Complexes: Photophysics, Photochemistry, Eletrochemistry, and Chemiluminescence. Coord. Chem. Rev. 1988, 84, 85–277. 10.1016/0010-8545(88)80032-8. [DOI] [Google Scholar]
- Teplý F. Photoredox Catalysis by [Ru(bpy)3]2+ to Trigger Transformations of Organic Molecules. Organic Synthesis Using Visible-Light Photocatalysis and Its 20th Century Roots. Collect. Czech. Chem. Commun. 2011, 76, 859–917. 10.1135/cccc2011078. [DOI] [Google Scholar]
- Campagna S.; Puntoriero F.; Nastasi F.; Bergamini G.; Balzani V. Photochemistry and Photophysics of Coordination Compounds: Ruthenium. Top. Curr. Chem. 2007, 280, 117–214. 10.1007/128_2007_133. [DOI] [Google Scholar]
- Tucker J. W.; Stephenson C. R. J. Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. J. Org. Chem. 2012, 77, 1617–1622. 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]
- Narayanam J. M. R.; Stephenson C. R. J. Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev. 2011, 40, 102–113. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]
- Xuan J.; Xiao W.-J. Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. 10.1002/anie.201200223. [DOI] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y.; Yi H.; Lei A. Synthetic Applications of Photoredox Catalysis with Visible Light. Org. Biomol. Chem. 2013, 11, 2387–2403. 10.1039/c3ob40137e. [DOI] [PubMed] [Google Scholar]
- Schultz D. M.; Yoon T. P. Solar Synthesis: Prospects in Visible Light Photocatalysis. Science 2014, 343, 1239176. 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas J. J.; Nguyen J. D.; Cole K. P.; Stephenson C. R. J. Enabling Novel Photoredox Reactivity via Photocatalyst Selection. Aldrichimica Acta 2014, 47, 15–25. [Google Scholar]
- Hopkinson M. N.; Sahoo B.; Li J.-L.; Glorius F. Dual Catalysis Sees the Light: Combining Photoredox with Organo-, Acid, and Transition-Metal Catalysis. Chem. - Eur. J. 2014, 20, 3874–3886. 10.1002/chem.201304823. [DOI] [PubMed] [Google Scholar]
- Zeitler K. Photoredox Catalysis with Visible Light. Angew. Chem., Int. Ed. 2009, 48, 9785–9789. 10.1002/anie.200904056. [DOI] [PubMed] [Google Scholar]
- Yoon T. P.; Ischay M. A.; Du J. Visible Light Photocatalysis as a Greener Approach to Photochemical Synthesis. Nat. Chem. 2010, 2, 527–532. 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]
- Kärkäs M. D.; Matsuura B. S.; Stephenson C. R. J. Enchained by Visible Light–Mediated Photoredox Catalysis. Science 2015, 349, 1285–1286. 10.1126/science.aad0193. [DOI] [PubMed] [Google Scholar]
- For a review on the utilization of photosensitizers in artificial photosynthesis, see the following:Kärkäs M. D.; Verho O.; Johnston E. V.; Åkermark B. Artificial Photosynthesis: Molecular Systems for Catalytic Water Oxidation. Chem. Rev. 2014, 114, 11863–12001. 10.1021/cr400572f. [DOI] [PubMed] [Google Scholar]
- Riener M.; Nicewicz D. A. Synthesis of Cyclobutane Lignans via an Organic Single Electron Oxidant–Electron Relay System. Chem. Sci. 2013, 4, 2625–2629. 10.1039/c3sc50643f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z.; Wang X.; Wang X.; Rodriguez R. A.; Moore C. E.; Gao S.; Tan X.; Ma Y.; Rheingold A. L.; Baran P. S.; Chen C. Asymmetric Syntheses of Sceptrin and Massadine and Evidence for Biosynthetic Enantiodivergence. Science 2014, 346, 219–224. 10.1126/science.1255677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtley A. E.; Lu Z.; Yoon T. P. [2 + 2] Cycloaddition of 1,3-Dienes by Visible Light Photocatalysis. Angew. Chem., Int. Ed. 2014, 53, 8991–8994. 10.1002/anie.201405359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z.; Yoon T. P. Visible Light Photocatalysis of [2 + 2] Styrene Cycloadditions by Energy Transfer. Angew. Chem., Int. Ed. 2012, 51, 10329–10332. 10.1002/anie.201204835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller M. A.; Riener M.; Nicewicz D. A. Butyrolactone Synthesis via Polar Radical Crossover Cycloaddition Reactions: Diastereoselective Syntheses of Methylenolactocin and Protolichesterinic Acid. Org. Lett. 2014, 16, 4810–4813. 10.1021/ol5022993. [DOI] [PubMed] [Google Scholar]
- Bellville D. J.; Wirth D. W.; Bauld N. L. Cation-Radical Catalyzed Diels-Alder Reaction. J. Am. Chem. Soc. 1981, 103, 718–720. 10.1021/ja00393a061. [DOI] [Google Scholar]
- Bauld N. L.; Bellville D. J.; Pabon R.; Chelsky R.; Green G. Theory of Cation-Radical Pericyclic Reactions. J. Am. Chem. Soc. 1983, 105, 2378–2382. 10.1021/ja00346a045. [DOI] [Google Scholar]
- Bellville D. J.; Bauld N. L.; Pabon R.; Gardner S. A. Theoretical Analysis of Selectivity in the Cation Radical Diels-Alder. J. Am. Chem. Soc. 1983, 105, 3584–3588. 10.1021/ja00349a038. [DOI] [Google Scholar]
- For a review on cation radical cycloadditions, see the following:Bauld N. L. Cation Radical Cycloadditions and Related Sigmatropic Reactions. Tetrahedron 1989, 45, 5307–5363. 10.1016/S0040-4020(01)89486-2. [DOI] [Google Scholar]
- Yueh W.; Bauld N. L. Mechanistic Aspects of Aminium Salt-Catalyzed Diels-Alder Reactions: The Substrate Ionization Step. J. Phys. Org. Chem. 1996, 9, 529–538. 10.1002/(SICI)1099-1395(199608)9:8<529::AID-POC813>3.0.CO;2-1. [DOI] [Google Scholar]
- Gieseler A.; Steckhan E.; Wiest O. Photoinduced, Electron-Transfer-Catalyzed Diels-Alder Reaction Between Indole and 1,3-Cyclohexadienes. Synlett 1990, 1990, 275–277. 10.1055/s-1990-21063. [DOI] [Google Scholar]
- Lin S.; Ischay M. A.; Fry C. G.; Yoon T. P. Radical Cation Diels-Alder Cycloadditions by Visible Light Photocatalysis. J. Am. Chem. Soc. 2011, 133, 19350–19353. 10.1021/ja2093579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Padilla C. E.; Ischay M. A.; Yoon T. P. Visible Light Photocatalysis of Intramolecular Radical Cation Diels-Alder Cycloadditions. Tetrahedron Lett. 2012, 53, 3073–3076. 10.1016/j.tetlet.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtley A. E.; Cismesia M. A.; Ischay M. A.; Yoon T. P. Visible Light Photocatalysis of Radical Anion Hetero-Diels-Alder Cycloadditions. Tetrahedron 2011, 67, 4442–4448. 10.1016/j.tet.2011.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutchley R. J.; Lever A. B. P. Ruthenium(II) Tris(bipyrazyl) Dication—A New Photocatalyst. J. Am. Chem. Soc. 1980, 102, 7128–7129. 10.1021/ja00543a053. [DOI] [Google Scholar]
- Mbaze L. M.; Lado J. A.; Wansi J. D.; Shiao T. C.; Chiozem D. D.; Mesaik M. A.; Choudhary M. I.; Lacaille-Dubois M.-A.; Wandji J.; Roy R.; Sewald N. Oxidative Burst Inhibitory and Cytotoxic Amides and Lignans from the Stem Bark of Fagara heitzii (Rutaceae). Phytochemistry 2009, 70, 1442–1447. 10.1016/j.phytochem.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Cismesia M. A.; Yoon T. P. Characterizing Chain Processes in Visible Light Photoredox Catalysis. Chem. Sci. 2015, 6, 5426–5434. 10.1039/C5SC02185E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew S. L.; Lawrence A. L.; Sherburn M. S. Unified Total Synthesis of the Natural Products Endiandric acid A, Kingianic acid E, and Kingianins A, D, and F. Chem. Sci. 2015, 6, 3886–3890. 10.1039/C5SC00794A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambley T. W.; Poiner A.; Taylor W. C. Diterpene Metabolites of the Marine Sponge Chelonaplysilla violacea: Aplyviolene and Aplyviolacene. Tetrahedron Lett. 1986, 27, 3281–3282. 10.1016/S0040-4039(00)84775-9. [DOI] [Google Scholar]
- Buckleton J. S.; Bergquist P. R.; Cambie R. C.; Clark G. R.; Karuso P.; Rickard C. E. F. Structure Determination of Aplyviolene from Chelonaplysilla violacea. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1986, 42, 1846–1848. 10.1107/S0108270186090315. [DOI] [Google Scholar]
- For a review on diterpenoids from marine sponges, see the following:Keyzers R. A.; Northcote P. T.; Davies-Coleman M. T. Spongian Diterpenoids from Marine Sponges. Nat. Prod. Rep. 2006, 23, 321–334. 10.1039/b503531g. [DOI] [PubMed] [Google Scholar]
- Schnermann M. J.; Overman L. E. A Concise Synthesis of (−)-Aplyviolene Facilitated by a Strategic Tertiary Radical Conjugate Addition. Angew. Chem., Int. Ed. 2012, 51, 9576–9580. 10.1002/anie.201204977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller D. S.; Untiedt N. L.; Dieskau A. P.; Lackner G. L.; Overman L. E. Constructing Quaternary Stereogenic Centers Using Tertiary Organocuprates and Tertiary Radicals. Total Synthesis of trans-Clerodane Natural Products. J. Am. Chem. Soc. 2015, 137, 660–663. 10.1021/ja512527s. [DOI] [PubMed] [Google Scholar]
- Depew K. M.; Marsden S. P.; Zatorska D.; Zatorski A.; Bornmann W. G.; Danishefsky S. J. Total Synthesis of 5-N-Acetylardeemin and Amauromine: Practical Routes to Potential MDR Reversal Agents. J. Am. Chem. Soc. 1999, 121, 11953–11963. 10.1021/ja991558d. [DOI] [Google Scholar]
- Kim J.; Ashenhurst J. A.; Movassaghi M. Total Synthesis of (+)-11,11′-Dideoxyverticillin A. Science 2009, 324, 238–241. 10.1126/science.1170777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo K.; Newhouse T.; Mori I.; Takayama H.; Baran P. S. Total Synthesis Guided Structure Elucidation of (+)-Psychotetramine. Angew. Chem., Int. Ed. 2011, 50, 2716–2719. 10.1002/anie.201008048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furst L.; Narayanam J. M. R.; Stephenson C. R. J. Total Synthesis of (+)-Gliocladin C Enabled by Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed. 2011, 50, 9655–9659. 10.1002/anie.201103145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty J. W.; Stephenson C. R. J. Synthesis of (−)-Pseudotabersonine, (−)-Pseudovincadifformine, and (+)-Coronaridine Enabled by Photoredox Catalysis in Flow. J. Am. Chem. Soc. 2014, 136, 10270–10273. 10.1021/ja506170g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukovic J.; Goodbody A. E.; Kutney J. P.; Misawa M. Production of 3′,4′-Anhydrovinblastine: A Unique Chemical Synthesis. Tetrahedron 1988, 44, 325–331. 10.1016/S0040-4020(01)85824-5. [DOI] [Google Scholar]
- Sundberg R. J.; Hunt P. J.; Desos P.; Gadamasetti K. G. Oxidative Fragmentation of Catharanthine by Dichlorodicyanoquinone. J. Org. Chem. 1991, 56, 1689–1692. 10.1021/jo00005a007. [DOI] [Google Scholar]
- Tucker J. W.; Zhang Y.; Jamison T. F.; Stephenson C. R. J. Visible-Light Photoredox Catalysis in Flow. Angew. Chem., Int. Ed. 2012, 51, 4144–4147. 10.1002/anie.201200961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen J. D.; Reiß B.; Dai C.; Stephenson C. R. J. Batch to Flow Deoxygenation Using Visible Light Photoredox Catalysis. Chem. Commun. 2013, 49, 4352–4354. 10.1039/C2CC37206A. [DOI] [PubMed] [Google Scholar]
- Knowles J. P.; Elliott L. D.; Booker-Milburn K. I. Flow Photochemistry: Old Light through New Windows. Beilstein J. Org. Chem. 2012, 8, 2025–2052. 10.3762/bjoc.8.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.; Straathof N. J. W.; Hessel V.; Noël T. Photochemical Transformations Accelerated in Continuous-Flow Reactors: Basic Concepts and Applications. Chem. - Eur. J. 2014, 20, 10562–10589. 10.1002/chem.201400283. [DOI] [PubMed] [Google Scholar]
- Garlets Z. J.; Nguyen J. D.; Stephenson C. R. J. The Development of Visible-Light Photoredox Catalysis in Flow. Isr. J. Chem. 2014, 54, 351–360. 10.1002/ijch.201300136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore K.; Seeberger P. H. Continuous Flow Photochemistry. Chem. Rec. 2014, 14, 410–418. 10.1002/tcr.201402035. [DOI] [PubMed] [Google Scholar]