

Abstract
Background:
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors. This cancer may be due to a multistep process with an accumulation of epigenetic alterations in tumor suppressor genes (TSGs), leading to hypermethylation of the genes. Hypermethylation of TSGs is associated with silencing and inactivation of them. It is well-known that DNA hypomethylation is the initial epigenetic abnormality recognized in human tumors. Estrogen receptor alpha (ERα) is one of the TSGs which modulates gene transcription and its hypermethylation is because of overactivity of DNA methyltransferases. Fortunately, epigenetic changes especially hypermethylation can be reversed by pharmacological compounds such as genistein (GE) and 17-beta estradiol (E2) which involve in preventing the development of certain cancers by maintaining a protective DNA methylation. The aim of the present study was to analyze the effects of GE on ERα and DNMT1 genes expression and also apoptotic and antiproliferative effects of GE and E2 on HCC.
Materials and Methods:
Cells were treated with various concentrations of GE and E2 and the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay was used. Furthermore, cells were treated with single dose of GE and E2 (25 μM) and flow cytometry assay was performed. The expression level of the genes was determined by quantitative real-time reverse transcription polymerase chain reaction.
Results:
GE increased ERα and decreased DNMT1 genes expression, GE and E2 inhibited cell viability and induced apoptosis significantly.
Conclusion:
GE can epigenetically increase ERα expression by inhibition of DNMT1 expression which in turn increases apoptotic effect of E2. Furthermore, a combination of GE and E2 can induce apoptosis more significantly.
Keywords: DNMT1, E2, epigenetic, estrogen receptor alpha, genistein, hepatocellular carcinoma
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors in the world and also is the second among cancers of digestive tract after stomach cancer.[1,2] The disease has a wide geographical variation.[3] The major risk factors of HCC are chronic viral hepatitis B and C.[4] Other factors include nutritional factors, toxins, and metabolic diseases.[5,6] Besides, HCC may be due to a multistep process with an accumulation of genetic and epigenetic alterations in regulatory genes, leading to activation of oncogenes and inactivation of tumor suppressor genes (TSGs). Epigenetics refers to heritable changes that play an important role in the control of gene expression.
In contrast to genetic events, the epigenetic pathway is a reversible alteration and characterized by three main mechanisms:
DNA hypermethylation leading to inactivation of genes,
DNA hypomethylation causing genomic instability,
Histone modifications affecting chromatin conformation.
It should be noted that DNA methylation does not change the genetic information. In fact, it alters the readability of the DNA and results in inactivation of genes by subsequent messenger RNA transcript repression.[7,8] Epigenetic silenced TSGs genes are involved in important molecular pathways of carcinogenesis example, cell cycle regulation, apoptosis, DNA repair or cell adhesion, and hypermethylation have been widely reported in all types of tumors.[9] It is well-known that DNA hypomethylation is the initial epigenetic abnormality recognized in human tumors such as ovarian epithelial carcinomas,[10,11] prostate metastatic tumors,[12] HCC[13] and Wilms' tumors.[14] Collectively, cancer is an epigenetic disease. Hypermethylation at the CpG islands found in estrogen response element (ERE) promoters occurs in conjunction with ligand bonded alpha subunit estrogen receptor alpha (ERα) dimers wherein the ligand ERα dimer complex acts as a transcription factor and binds to the ERE promoter.[15] ERα signaling plays a key role in hormonal cancer progression.[16] It has been reported that hypermethylation of ERα gene is a marker for HCC.[15] Furthermore, the relation between ERα and malignant disease has been discussed in a variety of tissues including breast, colon, blood, bladder, and liver.[17,18,19,20,21,22,23,24,25]
In many cancers, hypermethylation of CpG islands results from overactivity of DNA methyltransferases (DNMTs). In humans, the primary DNMTs are DNMT1, DNMT3a, and DNMT3b. DNMT1 is the most abundant in human.
Fortunately, epigenetic changes can be reversed by pharmacological intervention. Several active compounds in the food can decrease the risk of cancers by epigenetic mechanisms. Dietary phytoestrogens like genistein (GE) have shown to possess multiple cell regulatory activities within cancer cells.[26] Besides, it has been reported that phytoestrogens like GE involve in preventing the development of certain cancers such as prostate and mammary cancers by maintaining a protective DNA methylation profile.[27] Some of the dietary phytoestrogens exert their chemopreventive effects by modulating various components of the epigenetic mechanism in humans and have potentially beneficial effects on DNA methylation pattern.[26]
Isoflavones are structurally similar to 17-beta estradiol (E2) and have the ability to bind to ERs.[28] They act as agonists or antagonists of E2[29] and exert weak estrogenic activity in some tissues and antiestrogenic activity in others. They exert dual actions (both inhibitory and stimulatory effects) depending on their concentration.[30,31,32] Because of antiprolifratory effect, they protect against some cancer such as uterine, breast, prostate, lung and colon cancer.[33,34,35,36]
In previous study, we indicated that E2 can inhibit proliferation and induce apoptosis in PLC/PRF5 HCC cell line[28] and in this study, we investigated whether GE could alter the ERα and DNMT1 expression and also investigated apoptotic and proliferative effects of GE combined with E2 on PLC/PRF5 HCC cell line.
MATERIALS AND METHODS
Materials
Human HCC cell line (PLC/PRF5) was purchased from the National Cell Bank of Iran-Pasteur Institute. GE, E2 were purchased from Sigma (Sigma, St. Louis, MO). Total RNA extraction Kit (TRIZOL reagent), real-time polymerase chain reaction (PCR) kits (qPCR MasterMix Plus for SYBR Green I dNTP), DMEM (Dulbecco’s modified Eagle’s medium nutrient mixture F-12 Ham), and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were purchased from Qiagen. All other chemicals were obtained from the best available sources.
Cell culture
The cells were cultured and grown in DMEM supplemented with 10% fetal bovine serum. The cultures were incubated at 37°C in a humidified incubator containing 5% CO2, 95% ambient air. When cells became >80% confluent, 5 × 105 cells (PLC/PRF5) were seeded into 24-well plates (Becton, Dickinson) for 24 h in DMEM culture medium before they were incubated with certain concentrations of GE (1, 5, 10, 25, 50, 75, and 100 μM/L), which was dissolved in dimethyl sulfoxide (DMSO); DMSO was present at 0.01–0.3% in the medium. After 24 h, culture medium was changed with culture medium contains various concentrations of GE. On days 2, 3, and 4 after treatment with GE, MTT assay was done. The MTT assay for determination of IC50 value for E2 was done as done for GE with certain concentrations of E2 (1, 5, 10, 25, 50, 75, and 100 μM/L) which was dissolved in DMSO; DMSO was present at 0.01–0.3% in the medium. Photography was done for cultures before and after treatment with GE and E2 at different times using inverted microscope (Nikon, TE 2000-U, Japan).
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay for cell viability and IC50
After 24, 48, and 72 h of the treatment, the IC50 value for GE and E2 in PLC/PRF5 group were determined. Briefly, 5 × 105 Cells (PLC/PRF5) were counted and placed into each well of a 24-well culture plates. After 24 h of seeding, various concentrations of GE and E2 were added to the cells except in the control groups and after 24, 48, and 72 h of drug exposure, the MTT survival assay was then carried out for the evaluation of the cell viability with different drug concentration. The cells measured spectrophotometrically at 570 nm. All experiments were repeated 3 times with at least three measurements (triplicates).
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide solution was added to the medium to assess cell proliferation and viability by measuring the reduction of yellow MTT by mitochondrial dehydrogenases in viable cells. This yields purple formazan crystals that detected colorimetrically at 570 nm.
Determination of apoptotic cells by flow cytometry assay
The cells were cultured in 24-well culture plates and divided into 11 groups after 24 h. Three groups received a single dose of GE at the concentration of 25 μM and also 3 groups received a single dose of E2 at the concentration of 25 μM for 24, 48, and 72 h, respectively. One group received GE (25 μM) for 24 h and followed by E2 (25 μM) for 24 h (total treatment time 48 h) and other group received same dose of GE (GE) for 48 h and followed by E2 (25 μM) for 24 h (total treatment time 72 h). Final 3 groups received DMSO as control groups. In the GE treated groups (3 groups), E2 treated groups (3 groups) after 24, 48, and 72 h and GE-E2 groups (2 groups) after 24 h of E2 treatment and also control groups, all the adherent cells were collected with 0.05% trypsin, washed with cold phosphate-buffered saline and re suspended in binding buffer (1x). After addition of Annexin V-FITC and propidium iodide (PI, Becton, Dickinson, San Diego, CA), analysis was carried out according to the manufacturer’s protocol (BMS500F1/100CE Annexin V-FITC, eBiscience, USA). Finally, the apoptotic cells were counted by FACScan flow cytometry (Becton, Dickinson, Heidelberg, Germany). All experiments were processed independently 3 times. A minimum of 5 × 105 cell/ml were analyzed for each sample.
Determination of gene expression by real-time quantitative reverse transcription polymerase chain reaction
Real-time quantitative reverse transcription (RT)-PCR amplification and analysis were achieved to quantitatively estimate the expression of ERα and DENMT1 in GE (25 μM)-treated PLC/PRF5 cells at different times. Total RNA was isolated by RNeasy mini kit (Qiagen) according to the manufacturer’s protocol and then treated by RNase-free DNase (Qiagen) to eliminate the genomic DNA. The RNA concentration was determined using a BioPhotometer (Biowave II Germany). Total RNA (100 ng) was reverse transcribed to complementary DNA (cDNA) by using the RevertAid™First Strand cDNA Synthesis Kit (Fermentas, K1622 for 100 reactions) according to the manufacturer’s instructions. Real-time RT-PCR was performed by the Maxima™ SYBR Green/ROX qPCR Master Mix (2x1.25 ml, K0221). ERα and DNMT1 primers were obtained from articles[37,38,39] which their sequences are shown in Table 1. Real-time PCR reactions were performed using the Steponeplus (BD facscalibur StepOne plus v2.2). Thermal cycling conditions for ERα was: An initial denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95° C for 20 s, annealing at 58°C for 15 s and extension at 72°C for 15 s. Thermal cycling condition for DNMT1 was: An initial denaturation at 95°C for 10 min followed by 40 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 20 s and extension at 72°C for 20 s. Data were analyzed using the comparative Ct (ΔΔct) method, the relative expression level of ERα and DNMT1 was calculated by determining a ratio between the amount of these genes and that of endogenous control. Melting curve was used to determine melting temperature of specific amplification products and primer dimmers. These experiments were carried out in triplicate and independently repeated at least 3 times. GAPDH was used as a reference gene for internal control.
Table 1.
Real-time polymerase chain reaction primers used in the study

RESULTS
Result of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay for cell viability and IC50
The effects of the various concentrations of GE and E2 (as mentioned) on the cell viability were assessed by MTT assay. The dose- and time-dependent antiproliferative effects were observed with IC50s for GE and E2 [Figures 1 and 2]. Reduction of cell viability by 50% (IC50) required 25 μM GE for GE-treatment groups and same dose of E2 for E2-treatment groups at different time periods (24, 48, and 72 h). Each experiment was repeated 3 times for consistency of the result. The percentage of cell viability for GE (25 μM)-treatment groups were 52% (P < 0.001), 48% (P < 0.001), and 45% (P < 0.001) and for E2 (25 μM)-treatment groups were 55% (P < 0.001), 51% (P < 0.001), and 48% (P < 0.001) at different time periods (24, 48, and 72 h), respectively [Figures 3 and 4].
Figure 1.

Effect of genistein (GE) on the viability of hepatocellular carcinoma cell line determined by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay. The cells were treated without and with different concentrations of GE for 24, 48, and 72 h. Each experiment was conducted in triplicate. Mean values from the three experiments ± standard error of mean are shown. Asterisks (*) indicate significant differences between treated cells and the control group (*P < 0.001)
Figure 2.
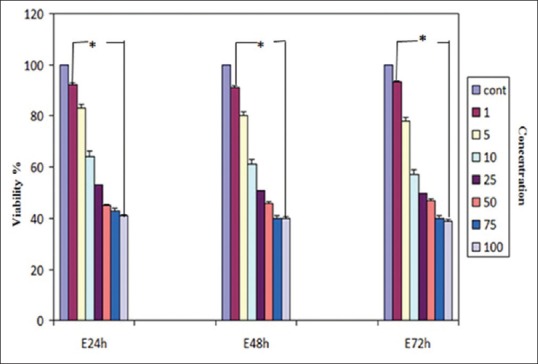
Effect of E2 on the viability of hepatocellular carcinoma cell line determined by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay. The cells were treated without and with different concentrations of E2 for 24, 48, and 72 h. Each experiment was conducted in triplicate. Mean values from the three experiments ± standard error of mean are shown. Asterisks (*) indicate significant differences between treated cells and the control group (*P < 0.001)
Figure 3.
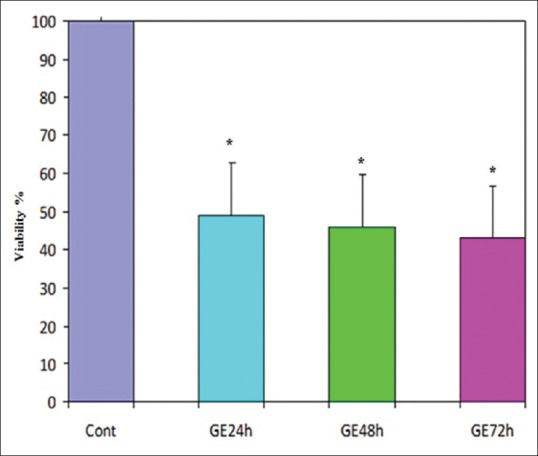
The cell vitality in the cells which treated with genistein (GE) at a concentration of 25 μM in different times was analyzed using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. The amounts of reduced MTT in all groups treated with GE were significantly lower than that of the control group. Mean values from the three experiments ± standard error of mean are shown. Asterisks (*) indicate significant differences between treated cells and the control group (*P < 0.001)
Figure 4.
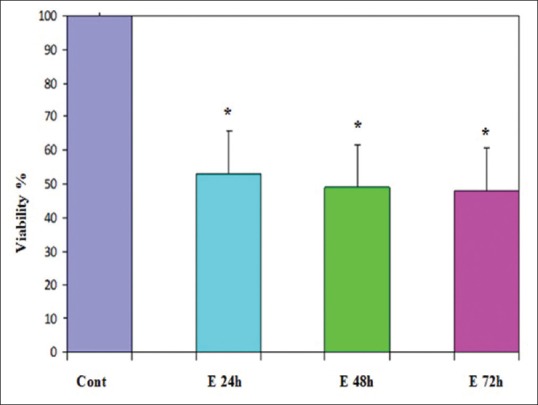
Effect of E2 at a concentration of 25 μM on cell viability of PLC/PRF5 cells. The effect of E2 on the viability of PLC/PRF5 cells was determined by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay at different time periods (24, 48, and 72 h). Mean values from the three experiments ± standard error of mean are shown. Asterisks (*) indicate significant differences between treated cells and the control group (*P < 0.001)
Result of determination of apoptosis by flow cytometry assay
The apoptosis-inducing effect of GE and E2 was investigated by flow cytometric analysis of PLC/PRF5 cells stained with Annexin V and propidium iodide. We observed via flow cytometry that these compounds induce apoptosis in this cell line significantly. The percentage of apoptotic cells in the GE (25 μM)-treatment groups at different times (24, 48, and 72 h) were 30, 36, 42% (P < 0.001) [Figure 5] and in the E2 (25 μM)-treatment groups at different times (24, 48, and 72 h) were 22, 30, 37% (P < 0.001) [Figure 6], respectively. The percentage of apoptotic cells in the group that was treated with GE (25 μM) for 24 h and followed by E2 (25 μM) for 24 h was 44% and in the group that was treated with GE (25 μ) for 48 h and followed by E2 (25 μM) for 24 h was 60% (P < 0.001) [Figure 7]. Relative analysis between GE treatment groups and E2 treatment groups at different times indicated that GE induces apoptosis more significantly and the percentage of apoptotic cells in the groups that treated with combined compound were significantly higher than that of the experimental groups that treated with GE or E2 alone, with 44% and 60% apoptotic cells respectively as shown in the [Figure 8] (*P < 0.001). The apoptotic effect was not observed in DMSO control group. A minimum of 5 × 105 cells/ml were analyzed for each sample. Results were obtained from three independent experiments and were expressed as mean ± standard error of mean.
Figure 5.

The apoptosis-inducing effect of genistein (GE) was investigated by flow cytometric analysis of PLC/PRF5 cells stained with Annexin V and propidium iodide. Result of flow cytometry indicated that GE induces cell apoptosis in PLC/PRF5 cells significantly. Asterisks (*) indicate significant differences between treated cells and the control group. Results were obtained from three independent experiments and were expressed as mean ± standard error of mean n = 3. (a) 24 h. (b) 48 h. (c) 72 h (*P < 0.001)
Figure 6.

Effects of E2 on PLC/PRF5 cell apoptosis. The cells were treated with E2 (25 μM) for 24, 48, and 72 h and the apoptosis-inducing effect of E2 was investigated by flow cytometric analysis of PLC/PRF5 cells stained with Annexin V and propidium iodide. Asterisks (*) indicate significant differences between treated cells and the control group. Results were obtained from three independent experiments and were expressed as mean ± standard error of mean. P <0.001, n = 3. (a) 24 h. (b) 48 h. (c) 72 h
Figure 7.

The apoptosis-inducing effect of genistein (GE) and E2 combination (as described in the methods) were investigated by flow cytometric analysis of PLC/PRF5 cells stained with Annexin V and propidium iodide. The combination of GE and E2 induced cell apoptosis in PLC/PRF5 cells significantly. Asterisks (*) indicate significant differences between treated cells and the control group. Results were obtained from three independent experiments and were expressed as mean ± standard error of mean n = 3. (a) GE 24 h/E2 24 h. (b) GE 48 h/E2 24 h. 48 h. (c) control
Figure 8.
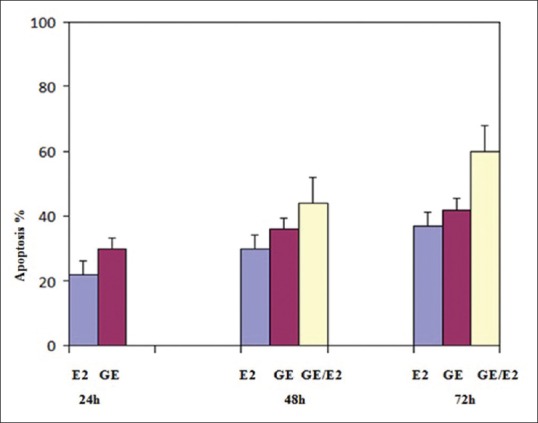
Relative analysis between genistein (GE) treatment groups, E2 treatment groups and combined compound treatment groups at different times indicated that GE induces apoptosis more significantly than E2 and also percentage of apoptotic cells in the groups that treated with combined compound were significantly higher than that of the experimental groups that treated with GE or E2 alone (P < 0.001)
Result of determination of gene expression by real-time polymerase-chain-reaction
Using quantitative RT-PCR, GE (25 μM) was shown to significantly increase ERα expression [Figure 9] and decrease DNMT1 expression [Figure 10] in PLC/PRF5 cell line at different time periods (24, 48, and 72 h). The relative expression of ERα was 2, 2.5, and 3.1 (P < 0.001) and that of DNMT1 were 0.33, 0.26, and 0.21 (P < 0.001) in different time periods, respectively.
Figure 9.
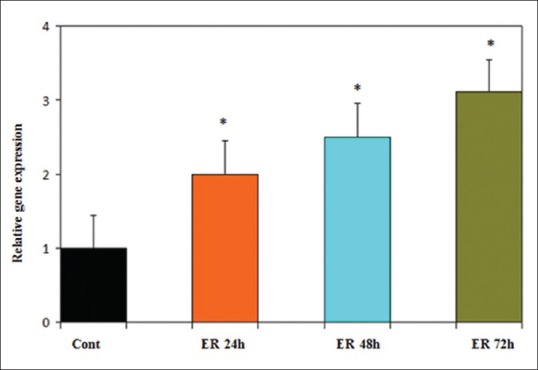
Time course of estrogen receptor alpha (ERα) expression in PLC/PRF5 cells in response to genistein (GE) (25 μM). Quantitative reverse transcription polymerase chain reaction analysis demonstrated that GE increased ERα expression significantly. Asterisks (*) indicate significant differences between treated cells and the control group. Data are presented as means ± standard error of mean P < 0.006, n = 3
Figure 10.
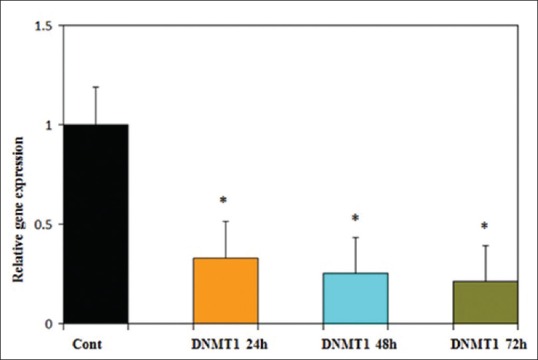
Time course of DNMT1 expression in PLC/PRF5 cells in response to genistein (GE) (25 μM). Quantitative reverse transcription polymerase chain reaction analysis demonstrated that GE decreased DNMT1 expression significantly. Asterisks (*) indicate significant differences between treated cells and the control group. Data are presented as means ± standard error of mean. P <0.001, n = 3
DISCUSSION
Our study clearly demonstrated that GE (25 μM) can down-regulate the expression of DNMT1, up-regulate the expression of ERα, inhibit cell proliferation, and induce cell apoptosis in PLC/PRF5 cell line with a dose- and time-dependent manner. Similarly, it has been indicated that dietary GE affects ERα expression via modulating epigenetic pathways such as DNMT-involved transcription regulation. It should be noted that GE induces a maximal ERα increment at 25 μM in a time-dependent manner.[40] Gu et al. demonstrated that GE inhibits the growth of MHCC97-H HCC cells in vitro in a concentration-dependent fashion which is more potent in the 10 μg/ml and 20 μg/ml GE-treated groups.[41] In other cancers such as stomach cancer, breast cancer, prostate cancer, colon cancer, leukemia and melanoma, GE strongly inhibits cell proliferation and plays an important role in the prevention and inhibition of tumor.[42,43,44] Similarly, it has been reported that GE induces and increases the apoptotic population in ovarian cancer cells (Choi et al., 2007). In rodent models, GE can protect against mammary[45,46,47,48] and prostate cancers.[49,50] A relationship between the intake of soy foods and reduced breast or prostate cancer has been reported in several epidemiological studies.[51,52,53,54,55]
These findings about effect of GE appear to lend support for our current finding, but many studies have shown that GE has proliferative or biphasic effect that is not consistent with our result;[56] Hsieh et al. reported that GE increases MCF-7 cell proliferation at a concentration of 0.01–1 μM, and maximal growth stimulation is observed at 1 μM and this level of growth is sustained up to 10 μM.[57] At relatively low concentrations, GE is full agonists for ERα as well as for the proliferation of ER-dependent breast cancer cells.[58]
Many mechanisms and different pathways have been reported for GE, although the exact mechanisms of GE await further elucidation; Hsieh et al. reported that GE can act via an ER-mediated mechanism.[57] Other observations reported that GE binds to the ER with an affinity approximately 100-fold less than that of estradiol.[59,60]
Finally, it has been shown that GE induces cell cycle arrest in the G0/G1 and G2/M phases[61,62,63,64,65,66,67,68] and the number of S phase cells are decreased in a progressive way as the GE incubation time is increased.[69,70,71] It has previously been reported that structurally distinct phytoestrogens, including the GE, exert their estrogenic effects through direct binding and activation of the ERs (Kuiper et al., 1997; Barkhem et al., 1998). GE has biphasic effect with cytotoxic effect at concentrations >10 μM in breast cancer cell lines and cell death becomes apparent by about 72 h whereas exposure to 10 μM for only 24 h is tolerated.[58]
Our data clearly shown that E2 has a significant inhibitory effect on the growth of liver cancer cells and induces apoptosis in this cell line with a dose- and time-dependent manner. Similar results have been obtained by other studies; Huang et al. reported that estrogen and the estrogen-like compounds (E2) induce anti-proliferative and apoptotic effects in Hep3B cells, and the E2 and the E2-like compounds mediated apoptotic effect is ER dependent. Among the estrogen-like compounds, E2 and GE show the stronger anti-tumor potential.[72] It has been reported that 17 β-estradiol at a concentration of 1 nM significantly increase apoptosis of MDA-MB-231 breast cancer cells.[73] Besides, dietary phytoestrogens play a protective role against prostate and colon cancer and the formation of polyps[74,75] and reduce colorectal cancer development[76] and also decrease colorectal cancer risk.[77] Over the past decades, epidemiological studies have indicated that consumption of diets rich in phytoestrogens is associated with low risk of breast cancer.[78]
All reports mentioned above confirm our finding about E2, but many studies have reported that phytoestrogen have stimulatory and prolifratory effects.[79] It has been reported that estrogen can stimulate the growth of stromal cells derived from the hyperplastic prostate (Collins et al., 1994). Hong et al. (2004) found that estrogen can stimulate the growth of prostatic stromal cells and increase smooth muscle cell markers (Hong et al., 2004).[80]
Furthermore, it has been shown that 17 β-estradiol administration exerts a growth-inhibitory effect on ER-positive cell lines (human gastric carcinoma-27, AGS).[81,82,83]
It should be noted that phytoestrogens act through different mechanisms and pathways. They exert anticancer activity by two mechanisms including an anti-estrogenic mechanism which is due to structural similarity with estradiol and anti-aromatase activity. Many studies have reported that phytoestrogens exert antiproliferative effects by inhibition of tyrosine kinase activity, DNA topoisomerase II, and angiogenesis.[58] Other studies have reported that estrogen acts through four molecular pathways: Ligand-independent, ligand-dependent, cell-surface (nongenomic) signaling, and DNA binding-independent.[84] Phytoestrogens reduce cancer risk by binding to ERs or interacting with enzymes involved in sex steroid biosynthesis and metabolism.[85]
Our finding demonstrated that combination of GE and E2 induces apoptosis and inhibits proliferation more significant than that of these compounds alone. Other studies reported that the expression of ER and apoptosis-induction in MCF-7 cells treated with a combination of estradiol (5 μM) and GE (5 μM) are more significant than that of these compound alone.[86] Rajah et al. reported that 1 μM GE plus 1 nM 17 β-estradiol significantly increase apoptosis with a concomitant decrease in ERK1/2 phosphorylation. High concentrations of GE (100 μM) both in the presence and absence of 17 β-estradiol also increases apoptosis.[73]
In summary, a combination of GE and E2 has been used synergistically in other studies but we first used these compounds separately and then used GE following by E2 (as mentioned in the material and method) and this is the advantage of our research compared to other researches using these compounds. Considering the results of our research, a combination of GE and E2 may be good candidate for HCC treatment.
We did not perform enzyme activity assays related to methylation and histone modifications and also enzyme immunoassay related to protein levels, but we will perform in the next researches and also further researches are needed to determine the clinical applications of GE.
CONCLUSIONS
Our study clearly demonstrated that GE increases ERα expression and decreases DNMT1 expression and also inhibits proliferation and induces apoptosis in human HCC cell line through epigenetic mechanism which can provide a new strategy for HCC treatment. It should be noted that when GE (25 μM) treatment followed by E2 (25 μM) treatment, apoptosis was increased more significantly.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
This article was supported by adjutancy of research of Isfahan medical university-Iran with code number 393005.
REFERENCES
- 1.Bosch FX, Ribes J, Borràs J. Epidemiology of primary liver cancer. Semin Liver Dis. 1999;19:271–85. doi: 10.1055/s-2007-1007117. [DOI] [PubMed] [Google Scholar]
- 2.Michielsen PP, Francque SM, van Dongen JL. Viral hepatitis and hepatocellular carcinoma. World J Surg Oncol. 2005;3:27. doi: 10.1186/1477-7819-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruix J, Sherman M, Llovet JM, Beaugrand M, Lencioni R, Burroughs AK, et al. Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona. 2000 EASL conference. European Association for the Study of the Liver. J Hepatol. 2001;35:421–30. doi: 10.1016/s0168-8278(01)00130-1. [DOI] [PubMed] [Google Scholar]
- 4.Tischoff I, Tannapfel A. DNA methylation in hepatocellular carcinoma. World J Gastroenterol. 2008;14:1741–8. doi: 10.3748/wjg.14.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet. 1981;2:1129–33. doi: 10.1016/s0140-6736(81)90585-7. [DOI] [PubMed] [Google Scholar]
- 6.Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int J Cancer. 2001;94:153–6. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 7.Riggs AD, Pfeifer GP. X-chromosome inactivation and cell memory. Trends Genet. 1992;8:169–74. doi: 10.1016/0168-9525(92)90219-t. [DOI] [PubMed] [Google Scholar]
- 8.Razin A, Cedar H. DNA methylation and genomic imprinting. Cell. 1994;77:473–6. doi: 10.1016/0092-8674(94)90208-9. [DOI] [PubMed] [Google Scholar]
- 9.De Zhu J. The altered DNA methylation pattern and its implications in liver cancer. Cell Res. 2005;15:272–80. doi: 10.1038/sj.cr.7290296. [DOI] [PubMed] [Google Scholar]
- 10.Cheng P, Schmutte C, Cofer KF, Felix JC, Yu MC, Dubeau L. Alterations in DNA methylation are early, but not initial, events in ovarian tumorigenesis. Br J Cancer. 1997;75:396–402. doi: 10.1038/bjc.1997.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehrlich M, Woods CB, Yu MC, Dubeau L, Yang F, Campan M, et al. Quantitative analysis of associations between DNA hypermethylation, hypomethylation, and DNMT RNA levels in ovarian tumors. Oncogene. 2006;25:2636–45. doi: 10.1038/sj.onc.1209145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bedford MT, van Helden PD. Hypomethylation of DNA in pathological conditions of the human prostate. Cancer Res. 1987;47:5274–6. [PubMed] [Google Scholar]
- 13.Lin CH, Hsieh SY, Sheen IS, Lee WC, Chen TC, Shyu WC, et al. Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res. 2001;61:4238–43. [PubMed] [Google Scholar]
- 14.Ehrlich M, Jiang G, Fiala E, Dome JS, Yu MC, Long TI, et al. Hypomethylation and hypermethylation of DNA in Wilms tumors. Oncogene. 2002;21:6694–702. doi: 10.1038/sj.onc.1205890. [DOI] [PubMed] [Google Scholar]
- 15.Vo AT, Millis RM. Epigenetics and breast cancers. Obstet Gynecol Int. 2012;17:10–7. doi: 10.1155/2012/602720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mann M, Cortez V, Vadlamudi RK. Epigenetics of estrogen receptor signaling: Role in hormonal cancer progression and therapy. Cancers (Basel) 2011;3:1691–707. doi: 10.3390/cancers3021691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teng J, Wang ZY, Jarrard DF, Bjorling DE. Roles of estrogen receptor alpha and beta in modulating urothelial cell proliferation. Endocr Relat Cancer. 2008;15:351–64. doi: 10.1677/erc.1.01255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Danel L, Cordier G, Revillard JP, Saez S. Presence of estrogen binding sites and growth-stimulating effect of estradiol in the human myelogenous cell line HL60. Cancer Res. 1982;42:4701–5. [PubMed] [Google Scholar]
- 19.Hatfill SJ, Brusnicky J, Fester E. Immunocytochemical identification of nuclear estrogen-receptors in human acute myeloid leukemia. Leuk Res. 1991;15:315–20. doi: 10.1016/0145-2126(91)90006-f. [DOI] [PubMed] [Google Scholar]
- 20.Holst F, Stahl PR, Ruiz C, Hellwinkel O, Jehan Z, Wendland M, et al. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat Genet. 2007;39:655–60. doi: 10.1038/ng2006. [DOI] [PubMed] [Google Scholar]
- 21.Pancholi S, Lykkesfeldt AE, Hilmi C, Banerjee S, Leary A, Drury S, et al. ERBB2 influences the subcellular localization of the estrogen receptor in tamoxifen-resistant MCF-7 cells leading to the activation of AKT and RPS6KA2. Endocr Relat Cancer. 2008;15:985–1002. doi: 10.1677/ERC-07-0240. [DOI] [PubMed] [Google Scholar]
- 22.Boix L, Bruix J, Castells A, Fuster J, Bru C, Visa J, et al. Sex hormone receptors in hepatocellular carcinoma. Is there a rationale for hormonal treatment? J Hepatol. 1993;17:187–91. doi: 10.1016/s0168-8278(05)80036-4. [DOI] [PubMed] [Google Scholar]
- 23.van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 24.van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Bernards R, et al. Expression profiling predicts outcome in breast cancer. Breast Cancer Res. 2003;5:57–8. doi: 10.1186/bcr562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Archer KJ, Mas VR, Maluf DG, Fisher RA. High-throughput assessment of CpG site methylation for distinguishing between HCV-cirrhosis and HCV-associated hepatocellular carcinoma. Mol Genet Genomics. 2010;283:341–9. doi: 10.1007/s00438-010-0522-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hishida M, Nomoto S, Inokawa Y, Hayashi M, Kanda M, Okamura Y, et al. Estrogen receptor 1 gene as a tumor suppressor gene in hepatocellular carcinoma detected by triple-combination array analysis. Int J Oncol. 2013;43:88–94. doi: 10.3892/ijo.2013.1951. [DOI] [PubMed] [Google Scholar]
- 27.Day JK, Bauer AM, DesBordes C, Zhuang Y, Kim BE, Newton LG, et al. Genistein alters methylation patterns in mice. J Nutr. 2002;132:2419S–23. doi: 10.1093/jn/132.8.2419S. [DOI] [PubMed] [Google Scholar]
- 28.Nikbakht Dastjerdi M, Kavoosi F. Inhibitory effect of 17-beta estradiol on hepatocellular carcinoma cell line. Indian J Appl Res. 2015;5:435–40. [Google Scholar]
- 29.Brownson DM, Azios NG, Fuqua BK, Dharmawardhane SF, Mabry TJ. Flavonoid effects relevant to cancer. Int Res Conf Food Nutr Cancer J Nutr. 2002;132:3482–9. doi: 10.1093/jn/132.11.3482S. [DOI] [PubMed] [Google Scholar]
- 30.Kyselova Z. Toxicological aspects of the use of phenolic compounds in disease prevention. Interdiscip Toxicol. 2011;4:173–83. doi: 10.2478/v10102-011-0027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patisaul HB, Jefferson W. The pros and cons of phytoestrogens. Front Neuroendocrinol. 2010;31:400–19. doi: 10.1016/j.yfrne.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ziegler RG. Phytoestrogens and breast cancer. Am J Clin Nutr. 2004;79:183–4. doi: 10.1093/ajcn/79.2.183. [DOI] [PubMed] [Google Scholar]
- 33.Breithofer A, Graumann K, Scicchitano MS, Karathanasis SK, Butt TR, Jungbauer A. Regulation of human estrogen receptor by phytoestrogens in yeast and human cells. J Steroid Biochem Mol Biol. 1998;67:421–9. doi: 10.1016/s0960-0760(98)00139-3. [DOI] [PubMed] [Google Scholar]
- 34.Setchell KD, Cassidy A. Dietary isoflavones: Biological effects and relevance to human health. J Nutr. 1999;129:758S–67. doi: 10.1093/jn/129.3.758S. [DOI] [PubMed] [Google Scholar]
- 35.Tikkanen MJ, Adlercreutz H. Dietary soy-derived isoflavone phytoestrogens. Could they have a role in coronary heart disease prevention? Biochem Pharmacol. 2000;60:1–5. doi: 10.1016/s0006-2952(99)00409-8. [DOI] [PubMed] [Google Scholar]
- 36.Wiseman H. The therapeutic potential of phytoestrogens. Expert Opin Investig Drugs. 2000;9:1829–40. doi: 10.1517/13543784.9.8.1829. [DOI] [PubMed] [Google Scholar]
- 37.Mirza S, Sharma G, Parshad R, Gupta SD, Pandya P, Ralhan R. Expression of DNA methyltransferases in breast cancer patients and to analyze the effect of natural compounds on DNA methyltransferases and associated proteins. J Breast Cancer. 2013;16:23–31. doi: 10.4048/jbc.2013.16.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurebayashi J, Otsuki T, Kunisue H, Tanaka K, Yamamoto S, Sonoo H. Expression levels of estrogen receptor-alpha, estrogen receptor-beta, coactivators, and corepressors in breast cancer. Clin Cancer Res. 2000;6:512–8. [PubMed] [Google Scholar]
- 39.Otsuki T, Yamada O, Kurebayashi J, Moriya T, Sakaguchi H, Kunisue H, et al. Estrogen receptors in human myeloma cells. Cancer Res. 2000;60:1434–41. [PubMed] [Google Scholar]
- 40.Li Y, Meeran SM, Patel SN, Chen H, Hardy TM, Tollefsbol TO. Epigenetic reactivation of estrogen receptor-a (ERa) by genistein enhances hormonal therapy sensitivity in ERa-negative breast cancer. Mol Cancer. 2013;12:9. doi: 10.1186/1476-4598-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gu Y, Zhu CF, Dai YL, Zhong Q, Sun B. Inhibitory effects of genistein on metastasis of human hepatocellular carcinoma. World J Gastroenterol. 2009;15:4952–7. doi: 10.3748/wjg.15.4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yanagihara K, Ito A, Toge T, Numoto M. Antiproliferative effects of isoflavones on human cancer cell lines established from the gastrointestinal tract. Cancer Res. 1993;53:5815–21. [PubMed] [Google Scholar]
- 43.Entschladen F, Drell TL, 4th, Lang K, Joseph J, Zaenker KS. Tumour-cell migration, invasion, and metastasis: Navigation by neurotransmitters. Lancet Oncol. 2004;5:254–8. doi: 10.1016/S1470-2045(04)01431-7. [DOI] [PubMed] [Google Scholar]
- 44.Gu Y, Zhu CF, Iwamoto H, Chen JS. Genistein inhibits invasive potential of human hepatocellular carcinoma by altering cell cycle, apoptosis, and angiogenesis. World J Gastroenterol. 2005;11:6512–7. doi: 10.3748/wjg.v11.i41.6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fritz WA, Coward L, Wang J, Lamartiniere CA. Dietary genistein: Perinatal mammary cancer prevention, bioavailability and toxicity testing in the rat. Carcinogenesis. 1998;19:2151–8. doi: 10.1093/carcin/19.12.2151. [DOI] [PubMed] [Google Scholar]
- 46.Lamartiniere CA, Zhao YX, Fritz WA. Genistein: Mammary cancer chemoprevention, in vivo mechanisms of action, potential for toxicity, and bioavailability in rats. J Womens Cancer. 2000;2:11–9. [Google Scholar]
- 47.Lamartiniere CA, Moore JB, Brown NM, Thompson R, Hardin MJ, Barnes S. Genistein suppresses mammary cancer in rats. Carcinogenesis. 1995;16:2833–40. doi: 10.1093/carcin/16.11.2833. [DOI] [PubMed] [Google Scholar]
- 48.Murrill WB, Brown NM, Zhang JX, Manzolillo PA, Barnes S, Lamartiniere CA. Prepubertal genistein exposure suppresses mammary cancer and enhances gland differentiation in rats. Carcinogenesis. 1996;17:1451–7. doi: 10.1093/carcin/17.7.1451. [DOI] [PubMed] [Google Scholar]
- 49.Wang J, Eltoum IE, Lamartiniere CA. Dietary genistein suppresses chemically-induced prostate cancer in Lobund-Wistar rats. Cancer Lett. 2001;12:321–9. doi: 10.1016/s0304-3835(01)00811-4. [DOI] [PubMed] [Google Scholar]
- 50.Lee JY, Kim HS, Song YS. Genistein as a potential anticancer agent against ovarian cancer. J Tradit Complement Med. 2012;2:96–104. doi: 10.1016/s2225-4110(16)30082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mäkelä SI, Pylkkänen LH, Santti RS, Adlercreutz H. Dietary soybean may be antiestrogenic in male mice. J Nutr. 1995;125:437–45. doi: 10.1093/jn/125.3.437. [DOI] [PubMed] [Google Scholar]
- 52.Messina M, Barnes S, Setchel KD. Phytoestrogens and breast cancer-commentary. Lancet. 1997;350:971–72. doi: 10.1016/S0140-6736(05)64062-7. [DOI] [PubMed] [Google Scholar]
- 53.Ingram D, Sanders K, Kolybaba M, Lopez D. Case-control study of phyto-oestrogens and breast cancer. Lancet. 1997;350:990–4. doi: 10.1016/S0140-6736(97)01339-1. [DOI] [PubMed] [Google Scholar]
- 54.Lee HP, Gourley L, Duffy SW, Estéve J, Lee J, Day NE. Dietary effects on breast-cancer risk in Singapore. Lancet. 1991;337:1197–200. doi: 10.1016/0140-6736(91)92867-2. [DOI] [PubMed] [Google Scholar]
- 55.Adlercreutz H, Mazur W. Phyto-oestrogens and Western diseases. Ann Med. 1997;29:95–120. doi: 10.3109/07853899709113696. [DOI] [PubMed] [Google Scholar]
- 56.Nikov GN, Hopkins NE, Boue S, Alworth WL. Interactions of dietary estrogens with human estrogen receptors and the effect on estrogen receptor-estrogen response element complex formation. Environ Health Perspect. 2000;108:867–72. doi: 10.1289/ehp.00108867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsieh CY, Santell RC, Haslam SZ, Helferich WG. Estrogenic effects of genistein on the growth of estrogen receptor-positive human breast cancer (MCF-7) cells in vitro and in vivo. Cancer Res. 1998;58:3833–8. [PubMed] [Google Scholar]
- 58.Maggiolini M, Bonofiglio D, Marsico S, Panno ML, Cenni B, Picard D. Estrogen receptor α mediates the proliferative but not the cytotoxic dose-dependent effects of two major phytoestrogens on human breast cancer cells. Mol Pharmacol. 2001;60:595–602. [PubMed] [Google Scholar]
- 59.Wang TT, Sathyamoorthy N, Phang JM. Molecular effects of genistein on estrogen receptor mediated pathways. Carcinogenesis. 1996;17:271–5. doi: 10.1093/carcin/17.2.271. [DOI] [PubMed] [Google Scholar]
- 60.Santell RC, Chang YC, Nair MG, Helferich WG. Dietary genistein exerts estrogenic effects upon the uterus, mammary gland and the hypothalamic/pituitary axis in rats. J Nutr. 1997;127:263–9. doi: 10.1093/jn/127.2.263. [DOI] [PubMed] [Google Scholar]
- 61.Powis G, Hill SR, Frew TJ, Sherrill KW. Inhibitors of phospholipid intracellular signaling as antiproliferative agents. Med Res Rev. 1995;15:121–38. doi: 10.1002/med.2610150204. [DOI] [PubMed] [Google Scholar]
- 62.Toi M, Mukaida H, Wada T, Hirabayashi N, Toge T, Hori T, et al. Antineoplastic effect of erbstatin on human mammary and esophageal tumors in athymic nude mice. Eur J Cancer. 1990;26:722–4. doi: 10.1016/0277-5379(90)90126-e. [DOI] [PubMed] [Google Scholar]
- 63.Constantinou A, Kiguchi K, Huberman E. Induction of differentiation and DNA strand breakage in human HL-60 and K-562 leukemia cells by genistein. Cancer Res. 1990;50:2618–24. [PubMed] [Google Scholar]
- 64.Yamashita Y, Kawada S, Nakano H. Induction of mammalian topoisomerase II dependent DNA cleavage by nonintercalative flavonoids, genistein and orobol. Biochem Pharmacol. 1990;39:737–44. doi: 10.1016/0006-2952(90)90153-c. [DOI] [PubMed] [Google Scholar]
- 65.Alhasan SA, Pietrasczkiwicz H, Alonso MD, Ensley J, Sarkar FH. Genistein-induced cell cycle arrest and apoptosis in a head and neck squamous cell carcinoma cell line. Nutr Cancer. 1999;34:12–9. doi: 10.1207/S15327914NC340102. [DOI] [PubMed] [Google Scholar]
- 66.Li Y, Upadhyay S, Bhuiyan M, Sarkar FH. Induction of apoptosis in breast cancer cells MDA-MB-231 by genistein. Oncogene. 1999;18:3166–72. doi: 10.1038/sj.onc.1202650. [DOI] [PubMed] [Google Scholar]
- 67.Pagliacci MC, Smacchia M, Migliorati G, Grignani F, Riccardi C, Nicoletti I. Growth-inhibitory effects of the natural phyto-oestrogen genistein in MCF-7 human breast cancer cells. Eur J Cancer. 1994;30A:1675–82. doi: 10.1016/0959-8049(94)00262-4. [DOI] [PubMed] [Google Scholar]
- 68.Yousefi S, Blaser K, Simon HU. Activation of signaling pathways and prevention of apoptosis by cytokines in eosinophils. Int Arch Allergy Immunol. 1997;112:9–12. doi: 10.1159/000237424. [DOI] [PubMed] [Google Scholar]
- 69.Chang KL, Kung ML, Chow NH, Su SJ. Genistein arrests hepatoma cells at G2/M phase: Involvement of ATM activation and upregulation of p21waf1/cip1 and Wee1. Biochem Pharmacol. 2004;67:717–26. doi: 10.1016/j.bcp.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 70.Ravindranath MH, Muthugounder S, Presser N, Viswanathan S. Anticancer therapeutic potential of soy isoflavone, genistein. Adv Exp Med Biol. 2004;546:121–65. doi: 10.1007/978-1-4757-4820-8_11. [DOI] [PubMed] [Google Scholar]
- 71.Chodon D, Banu SM, Padmavathi R, Sakthisekaran D. Inhibition of cell proliferation and induction of apoptosis by genistein in experimental hepatocellular carcinoma. Mol Cell Biochem. 2007;297:73–80. doi: 10.1007/s11010-006-9324-2. [DOI] [PubMed] [Google Scholar]
- 72.Huang EJ, Wu CC, Huang HP, Liu JY, Lin CS, Chang YZ, et al. Apoptotic and anti-proliferative effects of 17beta-estradiol and 17beta-estradiol-like compounds in the Hep3B cell line. Mol Cell Biochem. 2006;290:1–7. doi: 10.1007/s11010-005-9000-y. [DOI] [PubMed] [Google Scholar]
- 73.Rajah TT, Peine KJ, Du N, Serret CA, Drews NR. Physiological concentrations of genistein and 17ß-estradiol inhibit MDA-MB-231 breast cancer cell growth by increasing BAX/BCL-2 and reducing pERK1/2. Anticancer Res. 2012;32:1181–91. [PubMed] [Google Scholar]
- 74.Moutsatsou P. The spectrum of phytoestrogens in nature: Our knowledge is expanding. Hormones (Athens) 2007;6:173–93. [PubMed] [Google Scholar]
- 75.Adlercreutz H. Phyto-oestrogens and cancer. Lancet Oncol. 2002;3:364–73. doi: 10.1016/s1470-2045(02)00777-5. [DOI] [PubMed] [Google Scholar]
- 76.Barone M, Di Leo A. Estrogen receptor beta in colorectal cancer prevention: Do we have conclusive proof? J Genet Syndr Gene Ther. 2013;4:11–9. [Google Scholar]
- 77.Cotterchio M, Boucher BA, Manno M, Gallinger S, Okey A, Harper P. Dietary phytoestrogen intake is associated with reduced colorectal cancer risk. J Nutr. 2006;136:3046–53. doi: 10.1093/jn/136.12.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saxena S, Jyoti, Sharma A. Soybean seeds – An approach to treatment of breast cancer. World J Pharm Pharma Sci. 2009;3:1972–82. [Google Scholar]
- 79.Huang YT, Cha TL, Sun GH. Estrogen inhibits renal cell carcinoma cell progression through estrogen receptor-β activation. PLoS One. 2013;10:137–46. doi: 10.1371/journal.pone.0056667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luo Y, Waladali W, Li S, Zheng X, Hu L, Zheng H, et al. 17beta-estradiol affects proliferation and apoptosis of rat prostatic smooth muscle cells by modulating cell cycle transition and related proteins. Cell Biol Int. 2008;32:899–905. doi: 10.1016/j.cellbi.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 81.Kim MJ, Cho SI, Lee KO, Han HJ, Song TJ, Park SH. Effects of 17ß-estradiol and estrogen receptor antagonists on the proliferation of gastric cancer cell lines. J Gastric Cancer. 2013;13:172–8. doi: 10.5230/jgc.2013.13.3.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Messa C, Russo F, Pricci M, Di Leo A. Epidermal growth factor and 17beta-estradiol effects on proliferation of a human gastric cancer cell line (AGS) Scand J Gastroenterol. 2000;35:753–8. doi: 10.1080/003655200750023444. [DOI] [PubMed] [Google Scholar]
- 83.Messa C, Pricci M, Linsalata M, Russo F, Di Leo A. Inhibitory effect of 17beta-estradiol on growth and the polyamine metabolism of a human gastric carcinoma cell line (HGC-27) Scand J Gastroenterol. 1999;34:79–84. doi: 10.1080/00365529950172871. [DOI] [PubMed] [Google Scholar]
- 84.Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–72. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- 85.Cross HS, Kallay E, Lechner D, Gerdenitsch W, Adlercreutz H, Armbrecht HJ. Phytoestrogens and vitamin D metabolism: A new concept for the prevention and therapy of colorectal, prostate, and mammary carcinomas. Am Soc Nutr Sci. 2004;13:54–61. doi: 10.1093/jn/134.5.1207S. [DOI] [PubMed] [Google Scholar]
- 86.Miodini P, Fioravanti L, Di Fronzo G, Cappelletti V. The two phyto-oestrogens genistein and quercetin exert different effects on oestrogen receptor function. Br J Cancer. 1999;80:1150–5. doi: 10.1038/sj.bjc.6690479. [DOI] [PMC free article] [PubMed] [Google Scholar]