

Abstract
Joubert syndrome is a rare autosomal recessive disorder. It is characterized by congenital ataxia, hypotonia, developmental delay and at least one of the following features: neonatal respiratory disturbances and abnormal eye movements; including nystagmus and oculomotor apraxia. Molar tooth appearance is an essential finding for the diagnosis of Joubert syndrome. We report a five-days-old newborn with mild hypotonia, abnormal pattern of respiration, abnormal eye movements and molar tooth sign on brain CT scan. Joubert syndrome is an uncommon inherited condition and delayed diagnosis is usually related to its variable, non-specific presentation. Awareness of the characteristic clinical and radiological findings in Joubert syndrome will help in early diagnosis, appropriate counseling and proper rehabilitation.
Keywords: Joubert syndrome, Hyperpnoea, Molar tooth sign, Neonate, Oculomotor apraxia, Yemen
Introduction
Joubert syndrome (JS) is a rare genetic heterogeneously inherited disorder, named after Marie Joubert in 1969 [1–3]. It is characterized by brain stem and cerebellar malformations and multi-systemic affection involving nervous, ocular, gastrointestinal and urogenital system in varying proportions [4,5]. Molar tooth sign (MTS) and “Bat-wing” appearance on brain CT are basic findings in JS [6,7]. We report a newborn with mild hypotonia, abnormal respiration (episodic tachypnea and apnea), oculomotor abnormalities and MTS on brain CT scan. To our knowledge this is the first case of JS reported in Yemen.
Case Report
A five-days-old term, 3.4 kg, female baby was admitted to our hospital with frequent attacks, every 5–15 min, of apnea; each lasting 20–60 seconds, associated with bradycardia and alternating with hyperpnoea (up to 120 breaths/min). No history of fever, cyanosis, decreased feeding or activity, or history of trauma. There was no reported consanguinity or similar cases in the family. On examination, she was active, alert, crying normally, has low set ears, head circumference 35 cm (at 50th centile) and heart rate was 140 b/min. Systemic examination was normal apart from faint systolic murmur on cardiac auscultation. Laboratory investigations, including hemogram, blood sugar, electrolytes, C-reactive protein, liver enzymes, urea and creatinine were all normal. Blood culture was not available.
Three days later, left side upper limb seizures appeared in association with tongue protrusion and cessation of respiration but no bradycardia. An abnormal slow eye movement upon turning the head to the sides was noticed too. Chest X ray, brain and abdominal ultrasound were normal, echocardiography showed mild Epstein’s anomaly with mild pulmonary hypertension. Funduscopic examination was normal. Brain CT scan showed MTS (Figure 1). Genetic analysis was not available
Figure 1.
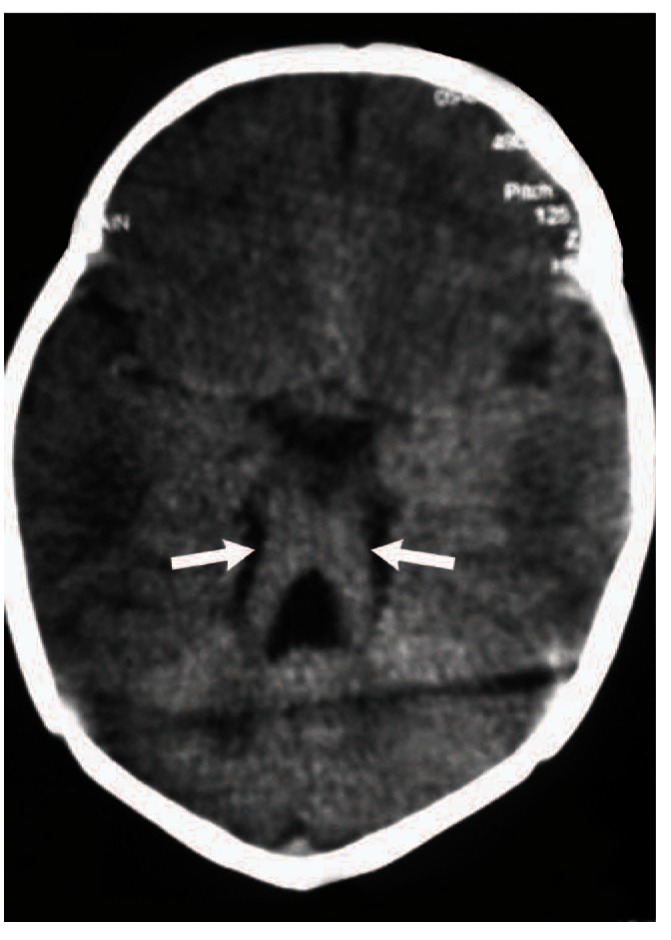
Axial brain computed tomography (CT) scan of the patient showing the characteristic molar tooth sign (arrows).
After three weeks on supportive treatment, the frequency and the duration of the apneic attacks improved, as she only had 1–2 attacks/day, each lasting < 5 seconds. The patient was discharged home on O2 therapy, as per need. On a follow up visit, at the age of three months, she had regular breathing, mild hypotonia with hand fisting and hyperreflexia. She did not follow objects on eye examination. Her head circumference was 36 cm (below the 3rd centile). Unfortunately, since that time, we have lost contact with the patient.
Discussion
Marie Joubert and associates first described JS in 1969 in four siblings and one sporadic case that exhibited episodic rapid breathing, abnormal eye movements, ataxia and mental retardation with agenesis of the cerebellar vermis [2]. By 2009, approximately 200 cases have been reported worldwide [8]. Most cases are sporadic, but some families may show a recessive pattern of inheritance [9]. The clinical manifestations usually start in the neonatal period, but the correct diagnosis is often delayed months or years after birth because of its variable phenotypes [10]. The term ‘JS and related disorders’ (JSRD) refers to a group of disorders presenting the pathognomonic features of JS in association with multiple systemic congenital abnormalities. It has an estimated incidence of 1/80000–1/100000 live birth [1,11]. These disorders have been classified as ciliopathies [12]; and were attributed to 23 genes [13]. Mutations in the AHI1and CEP290 genes are causal in 10–15% and 10% of cases, respectively. Homozygous deletion of NPHP1 gene results in 1–2% of cases [14].
Joubert syndrome is characterized by hypotonia, congenital ataxia, developmental delay and at least one of the following supportive features: neonatal respiratory abnormalities and abnormal ocular movements; including nystagmus and oculomotor apraxia [4,15]. Valente, et al, reported six clinical subgroups of JS [9], which were recently replaced with a more practical clinical – genetic classification [11,16,17]. Seizures and cardiac anomalies were rarely reported in JS [6,11].
Typical dysmorphic features (hypertelorism, small ear lobes, broad forehead, arched eyebrows, broad mouth with intermittent tongue protrusion, ptosis and anteverted nostrils) are present and hypotonia is important only if associated with other features [4,18]. Respiratory manifestations are characterized by alternating episodic attacks of hyperpnoea and central apnea or episodic hyperpnoea alone, which intensifies when the patient is stimulated. It starts typically in the neonatal period, gradually diminishes with age and disappears around the 6th month of life [2,11,19]. Ergun et al [20] reported persistence of the respiratory symptoms till the age of 17 years. The severity of respiratory irregularities can range from mild to severe and prolonged attacks of apnea [11,19].
Abnormal eye movements are characterized by nystagmus and oculomotor apraxia [15].
The similarity in clinical and radiological aspects between JS and both hypotonic cerebral palsy and rhombencephalosynapsis can lead to delayed or erroneous diagnosis [21,22].
Characteristic radiological findings in CT and MRI can help to guide and establish the diagnosis of JS [7]. The molar tooth sign is an essential part in the diagnosis of JS and JSRD [11]. It results from a combination of midline hypo-dysplasia of the cerebellar vermis, deepened interpeduncular fossa (at the level of isthmus and upper pons) and horizontal, thick, elongated superior cerebellar peduncles [1,4,12,15,23]. Batwing or umbrella sign results from the dilated or distorted fourth ventricle [2,7,12]. “Buttock sign” appears in the coronal images, resulting from the absence of the posterior vermian lobe, leaving the normal cerebellar hemispheres separated by a midline cleft [7]. The cerebrum is not affected but moderate lateral ventricular enlargement was noticed in 6%–20% of cases, with corpus callosum dysgenesis in 6%–10% [24]. Features resembling Dandy-Walker malformation were observed in 10% of cases with JS [5]. Recently, “Shepherd’s Crook sign” was reported. It appears in the sagittal views of the posterior fossa where the brain stem and the pons form the shaft of the crook and the abnormal superior cerebellar peduncles and cerebellar hemisphere complete the arc of the crook [25].
Multidisciplinary approach with supportive and symptomatic treatment is needed depending on the presence of neurological deficits and cognitive impairment, considering respiratory and feeding problems a priority [11,12,23,26]. Apnea monitoring is required for possible intensive care management and assisted ventilation, especially; peri-operatively as JS patients are very sensitive to the respiratory depressant effects of anesthetic agents [19,26,27].
In general, the prognosis of JS is poor [8]. It largely depends on the severity of different organ systems involvement [12]. Steinlin et al [28] described a variable course for developmental outcome in JS: some patient’s die early in infancy, some have severe developmental handicaps and others survive with mild developmental delay. The 5-year survival rate is 50% [8]. Death is usually due to feeding difficulties and respiratory infections [7]. Genetic counseling is an important part of management. The recurrence risk for JS is 25%. Once a diagnosis is made in a neonate, serial antenatal ultrasonography screening should be performed in subsequent pregnancies combined with fetal MRI at 20–22 weeks gestation to maximize the accuracy of prenatal diagnosis [18,27,29,30]. Annual screening for progressive JSRD complications is recommended [12,27].
Conclusion
Joubert syndrome is an uncommon inherited condition. Delayed diagnosis is usually related to its variable, nonspecific presentation. Awareness of the characteristic clinical and radiological findings in JS will help in early diagnosis, appropriate counseling and proper rehabilitation.
Acknowledgement
We are thankful for Miss Amani Saeed Barfaah for her help in revising the language and text of the manuscript.
References
- 1.Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM213300). Eur J Hum Genet. 2007; 15(5):511–521. [DOI] [PubMed] [Google Scholar]
- 2.Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the vermis: a syndrome of episodic hyperpnoea, abnormal eye movements, ataxia, and retardation. Neurology. 1969; 19(5):813–825. [DOI] [PubMed] [Google Scholar]
- 3.Boltshauser E, Isler W. Joubert syndrome: episodic hyperpnoea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie. 1977; 8:57–66. [DOI] [PubMed] [Google Scholar]
- 4.Maria BL, Boltshauser E, Palmer SC, Tran TX. Clinical Features and Revised Diagnostic Criteria in Joubert Syndrome. J Child Neurol. 1999; 14(9):583–590. [DOI] [PubMed] [Google Scholar]
- 5.Sharma NC. Jouberts Syndrome - A Case Presentation. Ind J Radiol Imag. 2006; 16(4):743–744. [Google Scholar]
- 6.Rehman IU, Bett Z, Husen Y, Akhtar AS, Khan FA. The ‘Molar Tooth Sign’ in Joubert Syndrome. Case Report. J Pak Med Assoc. 2009; 59(12)851–853. [PubMed] [Google Scholar]
- 7.Alorainy IA, Sabir S, Seidahmed MZ, Farooqu HA, Salih MA. Brain Stem and Cerebellar Findings in Joubert Syndrome. J Comput Assist Tomogr. 2006; 30:116–121. [DOI] [PubMed] [Google Scholar]
- 8.Choh SA, Choh NA, Bhat SA, Jehangir M. MRI findings in Joubert syndrome. Indian J Pediatr. 2009; 76(2):231–235. [DOI] [PubMed] [Google Scholar]
- 9.Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet. 2008; 51:1–23. [DOI] [PubMed] [Google Scholar]
- 10.Akcakus M, Gunes T, Kumandas S, Kurtoglu S, Coskun A. Joubert syndrome: Report of a neonatal case. Paediatr Child Health. 2003; 8(8):499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brancati F, Dallapiccola B, Valente EM. Joubert syndrome and related disorders. Orphanet J Rare Dis. 2010; 5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.İncecik F, Hergüner MÖ, Altunbaşak Ş, Gleeson JG. Joubert syndrome: report of 11 cases. Turk J Pediatr. 2012; 54(6): 605–611. [PMC free article] [PubMed] [Google Scholar]
- 13.Ben-Salem S, Al-Shamsi AM, Gleeson JG, Ali BR and Al-Gazali L. Mutation spectrum of Joubert syndrome and related disorders among Arabs. Human Genome Variation 2014; 1:14020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bandichhode ST, Anitha MS, Pandav A. Joubert Syndrome - A Case Report. JKIMSU 2013; 2(2):138–140. [Google Scholar]
- 15.Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, et al. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997; 12(7):423–430. [DOI] [PubMed] [Google Scholar]
- 16.Travaglini L, Brancati F, Silhavy J, Iannicelli M, Nickerson E, Elkhartoufi N, et al. Phenotypic spectrum and prevalence of INPP5E mutations in Joubert Syndrome and related disorders. Eur J Hum Genet. 2013; 21(10):1074–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alazami AM, Alshammari MJ, Salih MA, Alzahrani F, Hijazi H, Seidahmed MZ, et al. Molecular characterization of Joubert Syndrome in Saudi Arabia. Hum Mutat. 2012; 33(10):1423–1428. [DOI] [PubMed] [Google Scholar]
- 18.Aslan H, Ulker V, Gulcan EM, Numanoglu C, Gul A, Agar M, et al. Prenatal diagnosis of Joubert syndrome: A case report. Prenat Diagn. 2002; 22:13–16. [DOI] [PubMed] [Google Scholar]
- 19.Boltshauser E, Herdan M, Dumermuth G, Isler W. Joubert syndrome: Clinical and polygraphic observations in a further case. Neuropediatrics. 1981; 12(2):181–191. [DOI] [PubMed] [Google Scholar]
- 20.Ergun H, Teber S, Kafali M, Sezer T, Kendirli, T, Deda G. Case report: Joubert syndrome. Abstracts: Poster Presentations, the Seventh European Paediatric Neurology Society (EPNS) Congress. [Google Scholar]
- 21.Dekair LH, Kamel H, El-Bashir HO. Joubert syndrome labeled as hypotonic cerebral palsy. Neurosciences. 2014; 19(3):233–235. [PMC free article] [PubMed] [Google Scholar]
- 22.Cakirer S. Joubert Syndrome vs Rhombencephalosynapsis: Differentiation on the Basis of MRI Findings. Clinical Radiology Extra. 2003; 58(3):13–17. [Google Scholar]
- 23.Maria BL, Quisling RG, Rosainz LC, Yachnis AT, Gitten J, Dede D, et al. Molar tooth sign in Joubert syndrome: Clinical, radiologic, and pathologic significance. J Child Neurol. 1999; 14(6):368–376. [DOI] [PubMed] [Google Scholar]
- 24.Adamsbaum C, Moreau V, Bulteau C, Burstyn J, Lair Milan F, Kalifa G. Vermian agenesis without posterior fossa cyst. Pediatr Radiol. 1994; 24(8):543–546. [DOI] [PubMed] [Google Scholar]
- 25.Manley AT, Maertens PM. The Shepherd’s crook Sign: A new neuroimaging Pareidolia in Joubert syndrome. J Neuroimaging. 2014; 25(3):510–512. [DOI] [PubMed] [Google Scholar]
- 26.Barkovich AJ. Pediatric Neuroimaging. (3rd ed.). New York, NY: Raven; 2000. [Google Scholar]
- 27.Habre W, Sims C, D’Souza M. Anaesthetic management of children with Joubert syndrome. Paediatr Anaesth. 1997; 7(3): 251–253. [DOI] [PubMed] [Google Scholar]
- 28.Steinlin M, Schmid M, Landau K, Boltshauser E. Follow-up in children with Joubert syndrome. Neuropediatrics. 1997; 28:204–211. [DOI] [PubMed] [Google Scholar]
- 29.Doherty D, Glass IA, Siebert JR, Strouse PJ, Parisi MA, Shaw DW, et al. Prenatal diagnosis in pregnancies at risk for Joubert syndrome by ultrasound and MRI. Prenat Diagn. 2005; 25(6):442–447. [DOI] [PubMed] [Google Scholar]
- 30.Saleem SN, Zaki MS. Role of MR Imaging in Prenatal Diagnosis of Pregnancies at Risk for Joubert Syndrome and Related Cerebellar Disorders. Am J Neuroradiol. Mar 2010; 31(3):424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]