

Abstract
Importance
The “amyloid hypothesis” posits that disrupted amyloid-beta (Aβ) homeostasis initiates the pathological process resulting in Alzheimer disease (AD). Autosomal dominant Alzheimer disease (ADAD) has an early symptomatic onset and is caused by single gene mutations that result in overproduction of Aβ42. To the extent that “sporadic” late-onset Alzheimer disease (LOAD) also results from dysregulated Aβ42, the clinical phenotypes of ADAD and LOAD should be similar when controlling for the effects of age.
Objective
To use a family with late-onset ADAD caused by a presenilin 1 (PSEN1) mutation to mitigate the potential confound of age when comparing ADAD and LOAD.
Design
Case-control study of a family with late-onset ADAD and individuals with histopathologically-proven LOAD.
Setting
The Knight Alzheimer Disease Research Center, Washington University, St Louis, and other National Institutes of Aging-funded Alzheimer Disease Centers in the United States.
Participants
Ten PSEN1 A79V mutation carriers from multiple generations of a family with late-onset ADAD (median age-at-onset, 75.0 [63–77] years) and 12 noncarrier family members, followed at the Knight Alzheimer Disease Research Center (1985–2015); and 1115 individuals with neuropathologically confirmed LOAD (median age-at-onset, 74.0 [60–101] years) from the National Alzheimer Coordinating Center database (09/2005-12/14).
Main Outcome and Measure
Planned comparison of clinical characteristics between cohorts, including age at symptomatic onset, associated symptoms and signs, rates of progression, and disease duration.
Results
Seven (70%) mutation carriers developed AD dementia, while three are yet asymptomatic in their 7th and 8th decades of life. No differences were observed between mutation carriers and individuals with LOAD concerning age at symptomatic onset, presenting symptoms and duration, and rate of progression of dementia. Early emergence of comorbid hallucinations and delusions were observed in 57% of individuals with ADAD versus 19% of individuals with LOAD (p=.03). Three of twelve (25%) noncarriers from the PSEN1 A79V family are potential phenocopies as they also developed AD dementia (median age at onset=76.0 years).
Conclusions and Relevance
In this family, the amyloidogenic PSEN1 A79V mutation recapitulates the clinical attributes of LOAD. Previously reported clinical phenotypic differences between individuals with ADAD and LOAD may reflect age- or mutation-dependent effects.
Keywords: Alzheimer disease, autosomal dominant, presenilin 1, PSEN A79V, amyloid hypothesis
Introduction
A central tenet of the “amyloid hypothesis” is that accumulation and aggregation of insoluble amyloid beta peptide (Aβ42) represents a critical, causative step in the pathogenesis of Alzheimer disease (AD).1 Perhaps the strongest supporting evidence for the amyloid hypothesis is the finding that rare, dominantly-inherited pathogenic mutations in the amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes lead to clinically-symptomatic autosomal dominant Alzheimer disease (ADAD) by enhancing the conversion of APP to amyloidogenic Aβ42.1–5 Longitudinal analyses of cohorts of individuals with ADAD have informed the understanding of the clinical and paraclinical disease phenotypes resulting from disrupted cerebral Aβ42 metabolism.6,7 However, compared with sporadic late-onset AD (LOAD), the symptomatic onset of ADAD usually begins at a much earlier age (46.2 versus 72.0 years).7,8 Also, ADAD may be characterized by clinical features that are unusual in LOAD, including pyramidal signs, hypo/hyperkinetic movement disorders, ataxia, and early myoclonus and seizures.9–18 Additional differences in the pattern of cerebral Aβ42 deposition are reported in some ADAD cases, including 6,16,19,20 higher Aβ42 burden and greater densities of neurofibrillary tangles;21–23 increased Aβ42 deposition within the cerebellum24–26 and basal ganglia;19,20; and higher prevalence of cerebral amyloid angiopathy and large diffuse so-called “cotton wool” plaques.12,22,24,27 Such clinical and pathological differences may challenge whether findings from studies of early-onset ADAD, including current trials of anti-amyloid therapies,28 can be extrapolated to the understanding of the far more common LOAD.29,30 Clinical differences may also be interpreted to suggest that ADAD and LOAD are etiologically distinct.
A pathogenic PSEN1 A79V mutation was identified in members of a family followed at the Knight Alzheimer Disease Research Center (ADRC) who developed late-onset AD dementia, with symptoms beginning in the 7th and 8th decades of life, and who were initially presumed to have sporadic LOAD.31 The PSEN1 A79V mutation is known to cause early-onset ADAD in other families.11,32,33 PSEN1 knockout cells transfected with the A79V mutant PSEN1 showed significantly increased Aβ42 secretion, confirming the amyloidogenic potential of this mutation.31 Reports of individuals with late-onset ADAD are rare16,33,34 but offer the opportunity to mitigate the confound of age in comparisons with LOAD. Because dysregulation of Aβ42 metabolism appears central to both ADAD and LOAD,29,30,35 we hypothesized that the clinical characteristics of our cohort of PSEN1 A79V mutation carriers (MCs) with late-onset ADAD would overlap with those of LOAD.
Methods
Participant identification and recruitment
Twenty-two members of a family now known to carry the PSEN1 A79V mutation were research participants in longitudinal studies at the Knight ADRC across a 30 year period (1985–2015). Ten of these 22 individuals were PSEN1 A79V MCs. MCs were clinically assessed with semi-structured interviews with a knowledgeable collateral source and with the symptomatic individual, and detailed neurological examinations were performed on the individual. The remaining two MCs were not examined; history was obtained through interviews with a reliable collateral source. Dementia severity was determined at each assessment with the Clinical Dementia Rating (CDR),36 a global dementia rating scale that determines the presence or absence of dementia, rates dementia severity when present, and provides a composite measure of dysfunction across six cognitive and functional domains. Summation of scores across these domains yields the CDR Sum of Boxes score (CDR-SB), which can be used longitudinally to quantify AD progression.37 All staff involved in assessments were blinded to mutation status. All studies were approved by the Washington University School of Medicine Human Studies Committee. Informed consent was obtained from all research participants, including permission for autopsy.
Comparative data from deceased individuals with LOAD was obtained from the National Alzheimer’s Coordinating Center (NACC) Uniform Data Set (UDS)38 and Neuropathology Data Set (NPDS), including participants assessed from September 2005 to December 2014 from 34 current and past National Institute on Aging (NIA) Alzheimer Disease Centers. NACC data were selected to provide a national representation of LOAD. Individuals were included from NACC if they met the following criteria: (1) neuropathologic assessment that yielded an “intermediate” or “high” probability of causing AD dementia;39 (2) UDS data for at least one clinical visit preceding death; and, (3) a diagnosis of AD dementia at their last clinical assessment, with age-at-symptomatic onset (CDR ≥0.5) ≥60 years. AD neuropathologic change was based on a modification of the NIA-AA criteria for neuropathologic AD “ABC score”,39 reported previously.40,41
The phenotypes of PSEN1 A79V MCs were compared with those of LOAD across predetermined clinical measures. Demographic details were reported at the time of first assessment for all individuals, including age, gender and education. Dementia features were extrapolated from the UDS for NACC individuals, and from the UDS recorded within the Knight ADRC research records for PSEN1 A79V family members assessed since implementation of the UDS (i.e., 2005).38 Assessed features included dementia mode of symptom onset (i.e., gradual, subacute, abrupt, other); presenting symptom(s) of dementia (i.e., impairment of memory, orientation, language, visuospatial function, executive function, attention, other); rate of dementia progression, as operationalized by annualized change in CDR-SB); and duration from symptomatic onset to death. Similarly, the prevalence of associated symptoms, signs, and comorbidities in individuals with very mild (CDR 0.5) and mild (CDR 1.0) dementia were determined from review of UDS data for all participants assessed since 2005. The prevalence of depressive symptoms was estimated based on clinician reporting of active depression (UDS Form D1: Clinician Diagnosis) and/or the presence of depressive symptoms for two weeks or more in the past year (UDS Form B9, Clinician Judgement of Symptoms). “Psychiatric symptoms or signs” were deemed present when hallucinations (visual or auditory) or delusions were reported by assessing clinicians (UDS Form B9: Clinician Judgement of Symptoms), or when severe agitation was noted within the month preceding the clinical assessment (UDS, Neuropsychiatric Inventory Questionnaire42). Language deficits, visuospatial impairment, and parkinsonism were inferred from the UDS Form B9 (Clinician Judgement of Symptoms) or D1 (Clinician Diagnosis). A history of recent/active seizures was extracted from the UDS Form A5 (Subject Health History). In Knight ADRC participants assessed prior to 2005, clinical information was derived through direct review of research records, including detailed interviews with knowledgeable collateral sources who were directly asked about the presence of symptoms, signs and comorbidities associated with dementia, and neurological examinations performed on participants. Apolipoprotein E (APOE) ε4 allele status was reported for NACC participants, and determined within the Knight ADRC Genetics Core for PSEN1 A79V MCs.
Neuropathology
By the end of 2015, five PSEN1 A79V MCs with late-onset ADAD had died; by December 31, 2015 all had brain autopsy. Autopsies were limited to the removal of the brain. Four of these cases were examined at the Knight ADRC; the fifth case was examined at an outside site. The four Knight ADRC brains were examined macroscopically, tissue blocks dissected, and histological stains and immunohistochemistry applied following established protocols.43 Formalin-fixed, paraffin wax-embedded tissue blocks representing 16 areas were taken, including the middle frontal gyrus, superior and middle temporal gyri, inferior parietal lobe (angular gyrus), occipital lobe (to include the calcarine sulcus and peristriate cortex), anterior cingulate gyrus at the level of the genu of the corpus callosum, posterior cingulate gyrus and precuneus at the level of the splenium, amygdala and entorhinal cortex, hippocampus and parahippocampal gyrus at the level of the lateral geniculate nucleus, striatum (caudate nucleus and putamen) at the level of the anterior commissure, lentiform nuclei (globus pallidus and putamen), thalamus and subthalamic nucleus, midbrain, pons, medulla oblongata, cerebellum with dentate nucleus, and spinal cord. Sections were stained with hematoxylin and eosin and a modified Bielschowsky silver impregnation. Immunohistochemistry was performed using the following antibodies: Aβ (10D5; Eli Lilly, Indianapolis, IN, USA), phospho-tau (PHF1; a gift of Dr. P. Davies), phospho-α-synuclein (phospho-α-synuclein (Ser129); Cell Applications, Inc., San Diego, CA, USA), and phospho-TDP-43 (Cosmo Bio USA, Inc., Carlsbad, CA, USA). The National Institute on Aging-Alzheimer’s Association criteria for the neuropathologic diagnosis of AD were applied.39
Statistical Analysis
Clinical variables from PSEN1 A79V MCs and individuals with LOAD were summarized using descriptive statistics. Continuous numerical variables were compared using the non-parametric Mann-Whitney U-test. Categorical data was compared using Fisher’s exact test. The relationship between APOE ε4 carrier status and age at symptomatic onset (AAO) was considered through a Kaplan-Meier survival analysis. Overall differences in survival were assessed using the Mantel-Cox (log-rank) statistic. Statistical analyses were conducted using statistical software (IBM SPSS Statistics 23, IBM Corporation; and R Statistical Computing Software, www.r-project.org). Statistical significance was reported when p<0.05.
Results
Ten of 22 genotyped family members were PSEN1 A79V MCs (45% carrier rate). All individuals were part of a single multigenerational family of European ancestry living in the United States. All spoke English as their primary language. MCs underwent a median of 2.5 [1–9] annual research evaluations, across 0 to 11.6 years. Seven of the MCs developed progressive cognitive impairment due to AD dementia, with a median AAO of 75.0 [63–77] years. The remaining 3 MCs (all ≥60 years-old at their last assessment) still were cognitively normal (CDR 0) by December 31, 2015. Clinical information from 1115 participants with LOAD was obtained from the NACC for comparison. Participants with LOAD had a median AAO of 74 [60–101] years. Considerable overlap was observed in AAO, disease duration, and annualized rate of dementia progression between PSEN1 A79V MCs and individuals with LOAD (Figure 1).
Figure 1.
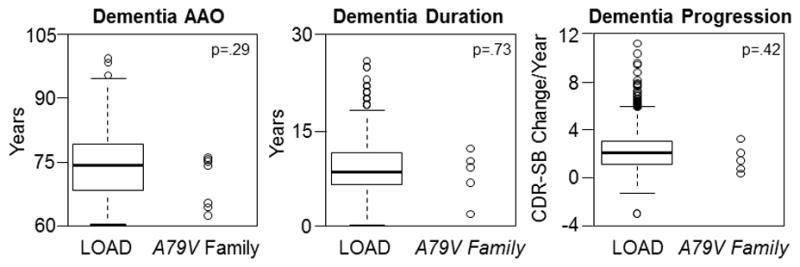
Age at symptomatic onset (AAO), duration of symptomatic dementia, and rate of dementia progression in sporadic late-onset Alzheimer disease (LOAD) and A79V family members with dementia. Control data from participants in the National Alzheimer’s Coordinating Center is shown as boxplots (N=1115 for Dementia AAO and Disease Duration; N=837 for Dementia Progression). The distribution of dementia AAO, disease duration (years), and rate of dementia progression (annualized change in CDR sum of boxes) are shown through scatterplots for individual cases (N=7 for Dementia AAO; N=5 for Dementia Duration and Progression).
Demographic characteristics; dementia features; associated symptoms, signs and comorbidities; and APOE allele status are reported for individuals with ADAD and LOAD (Table 1). Only educational attainment was lower in the ADAD cohort. Dementia most commonly presented insidiously, with a gradually progressive course, featuring memory loss as the presenting symptom in the vast majority of individuals. The early emergence of hallucinations and delusions distinguished PSEN1 A79V MCs from individuals with LOAD, occurring in 57% (4/7) of MCs with very mild or mild AD dementia, but in only 19% (137/706) of individuals with LOAD (p=.03). In 3 MCs, mild psychoses was noted concurrent with presenting symptoms of memory loss, manifesting with paranoid delusions in two individuals (suspicions that others were stealing money or food, in two MCs), and non-threatening complex auditory hallucinations of a baby crying in one MC. The remaining affected MC developed psychosis 3 years following dementia onset, manifesting with a non-threatening delusion that someone else was in the room. An additional affected MC experienced severe agitation/aggression requiring hospitalization and titration of antipsychotic medications 3 years after dementia onset. The remainder of clinical features were similarly distributed between populations, including depressive symptoms, language deficits, visuospatial impairment, parkinsonism and seizures. There were no differences in the frequency of APOE ε4 carriers between groups, and a survival analysis showed no effect of APOE ε4 carrier status and dementia AAO in PSEN1 A79V MCs.
Table 1.
Summary of clinical information for individuals with LOAD and PSEN1 A79V mutation carriers with ADAD
| Family members with dementia
|
LOAD | p value, carriers versus LOAD | ||
|---|---|---|---|---|
| PSEN1 A79V carriers | Noncarriers | |||
|
| ||||
| Demographic details | N=7 | N=3 | N=1115 | |
|
| ||||
| Median age at symptomatic onset (range), years | 75.0 (63–77) | 76.0 | 74.0 (60–101) | .29 |
|
| ||||
| Female gender (%) | 4 (57) | 2 (67) | 529 (47) | .71 |
|
| ||||
| Median education (range), years | 12 (8–15) | 12 (8–15) | 16 (2–24) | .01 |
|
| ||||
| Dementia features | ||||
|
| ||||
| Mode of onset of dementia: insidious onset, gradual progression (%) | 7 (100) | 3 (100) | 1105 (99) | >.99 |
|
| ||||
| Presenting symptom of dementia, memory loss (%) | 7 (100) | 3 (100) | 958/1063 (90) | >.99 |
|
| ||||
| Median duration of dementia (range), years | 9.9+ (2.3–12.8) | 5.1+ (<10 years) | 9.0 (1–27.0) | .73 |
|
| ||||
| Rate of dementia progression | ||||
|
| ||||
| Change in CDR sum of box scores/year (range) | 1.2 (0.1–3.3) N=5 |
1.64 (1.04–1.68) N=3 |
1.9 (−3.5–11.9) N=837 |
.42 |
|
| ||||
| Associated symptoms, signs and comorbidities early in the disease course (CDR≤1) | ||||
|
| ||||
| Depressive symptoms (low mood; %) | 3 (43) | 3 (100) | 309/706 (44) | >.99 |
|
| ||||
| Psychiatric symptoms/signs | 5 (71) | 1 (33) | 163/706 (24) | .01 |
| Hallucinations or delusions, % | 4/7 (57) | 1 (33) | 137/706 (19) | .03 |
| Severe agitation, % | 1/7 (14) | 0 (0) | 42/706 (6) | .35 |
|
| ||||
| Language deficit (%) | 3 (43) | 0 | 454/706 (58) | .26 |
|
| ||||
| Visuospatial impairment (%) | 4 (57) | 0 | 413/706 (64) | >.99 |
|
| ||||
| Parkinsonism (%) | 2 (29) | 1 (33) | 64/706 (9) | .13 |
|
| ||||
| Seizures (active; %) | 0 (0) | 0 | 4/706 (1) | >.99 |
|
| ||||
| APOE allele status | ||||
|
| ||||
| APOE ε4 carrier (%) | 5/7 (71) | 2 (67) | 568 (51) | .45 |
| - Unknown | 1/7 (14) | 0 | 167 (15) | >.99 |
Calculation limited to participants in whom complete duration was known
Histopathological analyses revealed an intermediate (n=1) or high (n=4) level of AD neuropathologic change in the 5 MCs who came to autopsy (Table 2). Aβ plaques and neurofibrillary tangles were prominent in cortical association areas including the medial temporal lobe. A lower density of neuritic pathology was noted in deep gray nuclei. Mild to moderate cerebral amyloid angiopathy was a consistent finding. Lewy bodies limited to the substantia nigra pars compacta were present in one case (Case E). Arteriolosclerosis with lacunar infarcts were present in two cases (B and E). “Cotton wool” plaques27 were absent.
Table 2.
Neuropathological findings in PSEN1 A79V carriers
| Case | Age at death (decade) | Brain weight (g) | ABC score for AD neuropathologic change (NIA-AA)39 | Other | |||
|---|---|---|---|---|---|---|---|
| A (Aβ plaques) | B (neurofibrillary tangles) | C (neuritic plaques) | Level of AD neuropathologic change | ||||
| A | 9th | 875 | 3 | 3 | 3 | High | - |
| B | 8th | 1,050 | 3 | 2 | 2 | Intermediate | CVD: lacunar infarcts in thalamus and basal ganglia |
| D | 9th | 940 | 3 | 3 | 3 | High | - |
| E* | 8th | 960 | 3 | 3 | 3 | High | CVD: lacunar infarcts in inferior parietal gyrus LBD: brainstem-predominant |
| F | 9th | 1,250 | 3 | 3 | 3 | High | - |
NIA-AA, National Institute on Aging-Alzheimer’s Association criteria for the neuropathologic assessment of AD
Information extracted from autopsy report issued by outside center; CVD, cerebrovascular disease; LBD, Lewy body disease.
Three of 12 (25%) genotyped noncarrier family members developed dementia (CDR≥0.5) in the 8th to 9th decades of life. One noncarrier was diagnosed with symptomatic AD and had neuropathologic AD confirmed at autopsy (lacunar infarcts also were present). One individual with parkinsonism developed dementia with visual and auditory hallucinations with paranoid delusions; the neuropathologic assessment met criteria for AD with cortical Lewy bodies. The third noncarrier was diagnosed with normal pressure hydrocephalus at age 70 and managed with ventriculoperitoneal shunting.
Discussion
We report a generally comparable clinical phenotype between MCs from a family with late-onset ADAD due to a PSEN1 A79V mutation, and individuals with LOAD. Similar AAO; duration of dementia; rates of progression; and associated symptoms, signs and comorbidities were found for this ADAD family and LOAD. Hallucinations and delusions were reported more frequently in PSEN1 A79V MCs from this family, as has been noted previously in individuals with ADAD.44,45 These findings demonstrate that the clinical phenotypes of late-onset ADAD and LOAD can overlap, and suggest that previously reported clinical (e.g., seizures, early myoclonus, spastic paraparesis, dysarthria, and rapid decline9–18) and neuropathological differences (e.g., altered cerebral Aβ42 and neurofibrillary tangle deposition, increased prevalence of cerebral amyloid angiopathy, and formation of “cotton wool” plaques6,12,16,19–27) may reflect age- or mutation-dependent effects.
The factors contributing to the later dementia AAO in this PSEN1 A79V family remain unknown. AAO was not influenced by APOE allele carrier status, suggesting that additional, as of yet unexplained, genetic, epigenetic or host factors must account for the late AAO, consistent with reports that family history and mutation type account for a significant proportion of the variance in AAO across a broad range of ADAD families.7 Within PSEN1 families, this variance may be influenced by Aβ-independent effects resulting from disruption of other functions of PSEN1, including its role as an endoplasmic reticulum calcium leak channel.46 These Aβ-independent effects could contribute to both the observed variability in AAO and phenotypic discordance between PSEN1 ADAD and LOAD. A79V is unusual among PSEN1 mutations as it was the only one of six screened PSEN1 mutations that did not impact PSEN1-related calcium channel function, as measured by calcium imaging experiments using presenilin-null mouse embryonic fibroblasts.46 The limited secondary effects on cellular calcium homeostasis associated with the PSEN1 A79V mutation may contribute to the later AAO reported in this cohort and others.16,33 Additionally, variability may reflect alterations in the metabolism of other known PSEN1 cleavage substrates, including Notch, a transmembrane receptor involved in neuronal development and function.47 PSEN1 A79V mutations have been shown to alter Aβ42 levels and Aβ42/40 ratio in cell culture, and MCs from the family described herein were previously found to have high CSF Aβ42 and Aβ42/40 ratios: findings that suggest gamma-secretase function may be altered in PSEN1 A79V MCs within this family.31 To our knowledge, the effect of PSEN1 A79V mutations on gamma-secretase-mediated Notch cleavage has not been previously investigated. However, the finding that many other pathogenic PSEN1 mutations which alter Aβ42 production also impact Notch cleavage,48 suggests that PSEN1 A79V mutations could have a similar effect. Future studies are required to address this issue, and to explore other molecular mechanisms through which the PSEN1 A79V mutation may impact disease phenotype.
Because of the late AAO, family members were enrolled in research studies with presumptive LOAD until the unexpected discovery of a pathogenic mutation in an affected family member.31 That the PSEN1 A79V mutation was responsible for symptoms in 7 of 10 family members who developed dementia late in life suggests that it may be necessary to reconsider the criteria used to select individuals with symptomatic AD for genetic testing. Testing for ADAD-associated mutations is considered for individuals with both early-onset AD and ≥2 affected family members (third-degree relatives or closer).49 These criteria are seldom applied to individuals with LOAD given the higher baseline prevalence of sporadic AD in this population.
Previous reports suggest that the early expression of prominent behavioral and psychiatric symptoms may represent a clinical marker for PSEN1 families.44,45 Although psychiatric symptoms and signs were observed more frequently in PSEN1 A79V MCs within this family, the specificity of this finding is expected to be low when all causes of dementia are considered across a broader population of patients. Indeed, visual hallucinations were reported in one noncarrier family-member with pathologically-proven AD and Lewy bodies. Absent a specific marker of late-onset ADAD, therefore, we support the recommendation that genetic counselling be considered in symptomatic individuals from families with 4 or more affected individuals with LOAD.50 The emergence of psychosis early in the course of dementia in such family members may further justify testing. This recommendation has potentially important implications for clinical practice, where the emphasis on genetic evaluation in individuals with early-onset AD has likely obscured prevalence estimates of late-onset ADAD, and identification of associated mutations.
The present work is subject to several limitations, including the small number of affected individuals available for study, and potential variability in reporting of clinical results across the multiple Alzheimer Disease Centers that contribute to the NACC. Lack of power may have affected our ability to quantify differences between individuals with ADAD and LOAD, and to investigate the clinical factors that may have contributed to the later-than-expected dementia AAO in PSEN1 A79V MCs. Similarly, the modest number of PSEN1 A79V cases reviewed at autopsy limited the ability to perform a quantitative comparison of neuropathological findings between this cohort and LOAD cases. Finally, we present clinical information from three PSEN1 A79V MCs who remain cognitively normal beyond the 7th decade of life. This finding may raise questions concerning whether amyloidogenic mutations invariably lead to AD, although all 3 asymptomatic MCs remain well within the expected range of onset of symptomatic AD observed within affected family members and may yet develop AD dementia. This issue will be further clarified through continued prospective follow-up.
At least for this PSEN1 A79V family an amyloidogenic mutation largely recapitulates the clinicopathological attributes of sporadic LOAD. This recapitulation indirectly supports the concept that ADAD and LOAD arise from parallel pathways, and suggests that at least some research findings involving individuals with ADAD may be extended to individuals with LOAD. The degree to which variability in the severity of amyloidogenic mutations, host genetic, epigenetic and environmental factors contribute to the AAO within and between PSEN1 A79V families should be further considered through adequately powered prospective observational studies.
Acknowledgments
The Authors are grateful for the commitment of participants, patients and their families to advancing research and clinical care. The Knight Alzheimer Disease Research Center Clinical Core provided information concerning participant evaluations; the Genetics Core provided information concerning APOE ε4 and PSEN1 A79V status; the Neuropathology Core undertook the autopsies and neuropathologic assessments of 4 of 5 cases. Ms Sarah E Monsell provided advice and assistance with obtaining NACC UDS and NPDS information, for which we are grateful. Dr. Virginia Buckles provided expert comment and helpful advice pertinent to this manuscript.
This project was supported by NIA grants to the Knight Alzheimer Disease Research Center [P50 AG05681 (PI John C. Morris, MD), P01 AG03991 (PI John C. Morris, MD), P01 AG26276 (PI John C. Morris, MD), R01 AG044546 (PI Carlos Cruchaga,, PhD)] and NACC. The NACC database is funded by NIA/National Institutes of Health (NIH) Grant U01 AG016976. NACC data are contributed by the NIA-funded Alzheimer diseases centers: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Steven Ferris, PhD), P30 AG013854 (PI M. Marsel Mesulam, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG016570 (PI Marie-Francoise Chesselet, MD, PhD), P50 AG005131 (PI Douglas Galasko, MD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P50 AG005136 (PI Thomas Montine, MD, PhD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John C. Morris, MD), and P50 AG047270 (PI Stephen Strittmatter, MD, PhD). GSD is supported by the Eugene M Johnson, Jr. Weston Brain Institute Postdoctoral Fellowship and the Neiss-Gain Family Endowment for Alzheimer Disease Research. EM is supported by NINDS grant K08NS079405.
Footnotes
Conflicts of Interest
GS Day is involved in research supported by an in-kind gift of radiopharmaceuticals from Avid Radiopharmaceuticals, and is currently participating in clinical trials of anti-dementia drugs sponsored by the A4 (The Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease) trial. He holds stocks (>$10,000) in ANI Pharmaceuticals (a generic pharmaceutical company).
ES Musiek is currently participating in clinical trials of anti-dementia drugs sponsored by the A4 (The Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease) trial.
CM Roe has nothing to declare.
J Norton has nothing to declare.
AM Goate is a member of the scientific advisory board of Denali Therapeutics and Cognition Therapeutics. She received research funding and honoraria from Genentech, Pfizer and AstraZeneca during the course of this study.
C Cruchaga has nothing to declare.
NJ Cairns has nothing to declare.
JC Morris has participated or is currently participating in clinical trials of anti-dementia drugs sponsored by the following companies: SNIFF (The Study of Nasal Insulin to Fight Forgetfulness) study, and A4 (The Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease) trial. Dr. Morris has served as a consultant for Lilly USA and Takeda Pharmaceuticals. He receives research support from Eli Lilly/Avid Radiopharmaceuticals and is funded by NIH grants # P50AG005681; P01AG003991; P01AG026276 and UF01AG032438. Neither Dr. Morris nor his family owns stock or has direct equity interest in any pharmaceutical or biotechnology company.
Role of the Funding Agencies
Design and conduct of the study = none.
Collection, management, analysis, and interpretation of the data = none.
Preparation, review, or approval of the manuscript = none.
Decision to submit the manuscript for publication = none.
Author Contributions
GS Day participated in the conception and design of the study; acquisition and interpretation of data; and drafting, revising and finalizing the manuscript. GS Day was primarily responsible for statistical analysis.
ES Musiek participated in the conception and design of the study; acquisition and interpretation of data; and revising and finalizing the manuscript.
CM Roe participated in acquisition and interpretation of data, revised the manuscript for critical content and assisted with statistical analysis.
J Norton participated in acquisition and interpretation of data, and revised the manuscript for critical content.
AM Goate participated in acquisition and interpretation of data, and revised the manuscript for critical content.
C Cruchaga participated in acquisition and interpretation of data, and revised the manuscript for critical content.
NJ Cairns participated in acquisition and interpretation of data, and revised the manuscript for critical content.
JC Morris participated in the conception and design of the study; acquisition and interpretation of data; and revision and finalization of the manuscript. Dr. Morris had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002 Jul 19;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nature Medicine. 1996 Aug;2(8):864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 3.Borchelt DR, Thinakaran G, Eckman CB, et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996 Nov;17(5):1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 4.Duff K, Eckman C, Zehr C, et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996 Oct 24;383(6602):710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 5.Citron M, Westaway D, Xia WM, et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nature Medicine. 1997 Jan;3(1):67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 6.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. The New England journal of medicine. 2012 Aug 30;367(9):795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryman DC, Acosta-Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014 Jul 15;83(3):253–260. doi: 10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waring SC, Doody RS, Pavlik MN, Massman PJ, Chan WY. Survival among patients with dementia from a large multi-ethnic population. Alz Dis Assoc Dis. 2005 Oct-Dec;19(4):178–183. doi: 10.1097/01.wad.0000189033.35579.2d. [DOI] [PubMed] [Google Scholar]
- 9.Joshi A, Ringman JM, Lee AS, Juarez KO, Mendez MF. Comparison of clinical characteristics between familial and non-familial early onset Alzheimer’s disease. Journal of neurology. 2012 Oct;259(10):2182–2188. doi: 10.1007/s00415-012-6481-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snider BJ, Norton J, Coats MA, et al. Novel presenilin 1 mutation (S170F) causing Alzheimer disease with Lewy bodies in the third decade of life. Arch Neurol. 2005 Dec;62(12):1821–1830. doi: 10.1001/archneur.62.12.1821. [DOI] [PubMed] [Google Scholar]
- 11.Finckh U, Muller-Thomsen T, Mann U, et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet. 2000 Jan;66(1):110–117. doi: 10.1086/302702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Houlden H, Baker M, McGowan E, et al. Variant Alzheimer’s disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Annals of neurology. 2000 Nov;48(5):806–808. [PubMed] [Google Scholar]
- 13.Smith MJ, Kwok JB, McLean CA, et al. Variable phenotype of Alzheimer’s disease with spastic paraparesis. Annals of neurology. 2001 Jan;49(1):125–129. doi: 10.1002/1531-8249(200101)49:1<125::aid-ana21>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 14.Kwok JB, Taddei K, Hallupp M, et al. Two novel (M233T and R278T) presenilin-1 mutations in early-onset Alzheimer’s disease pedigrees and preliminary evidence for association of presenilin-1 mutations with a novel phenotype. NeuroReport. 1997 Apr 14;8(6):1537–1542. doi: 10.1097/00001756-199704140-00043. [DOI] [PubMed] [Google Scholar]
- 15.Crook R, Verkkoniemi A, Perez-Tur J, et al. A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nature Medicine. 1998 Apr;4(4):452–455. doi: 10.1038/nm0498-452. [DOI] [PubMed] [Google Scholar]
- 16.Larner AJ, Doran M. Clinical phenotypic heterogeneity of Alzheimer’s disease associated with mutations of the presenilin-1 gene. Journal of neurology. 2006 Feb;253(2):139–158. doi: 10.1007/s00415-005-0019-5. [DOI] [PubMed] [Google Scholar]
- 17.Kennedy AM, Newman SK, Frackowiak RS, et al. Chromosome 14 linked familial Alzheimer’s disease. A clinico-pathological study of a single pedigree. Brain. 1995 Feb;118( Pt 1):185–205. doi: 10.1093/brain/118.1.185. [DOI] [PubMed] [Google Scholar]
- 18.Lopera F, Ardilla A, Martinez A, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997 Mar 12;277(10):793–799. [PubMed] [Google Scholar]
- 19.Klunk WE, Price JC, Mathis CA, et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007 Jun 6;27(23):6174–6184. doi: 10.1523/JNEUROSCI.0730-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benzinger TL, Blazey T, Jack CR, Jr, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2013 Nov 19;110(47):E4502–4509. doi: 10.1073/pnas.1317918110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomez-Isla T, Growdon WB, McNamara MJ, et al. The impact of different presenilin 1 and presenilin 2 mutations on amyloid deposition, neurofibrillary changes and neuronal loss in the familial Alzheimer’s disease brain - Evidence for other phenotype-modifying factors. Brain. 1999 Sep;122:1709–1719. doi: 10.1093/brain/122.9.1709. [DOI] [PubMed] [Google Scholar]
- 22.Mann DM, Iwatsubo T, Cairns NJ, et al. Amyloid beta protein (Abeta) deposition in chromosome 14-linked Alzheimer’s disease: predominance of Abeta42(43) Annals of neurology. 1996 Aug;40(2):149–156. doi: 10.1002/ana.410400205. [DOI] [PubMed] [Google Scholar]
- 23.Mann DM, Iwatsubo T, Ihara Y, et al. Predominant deposition of amyloid-beta 42(43) in plaques in cases of Alzheimer’s disease and hereditary cerebral hemorrhage associated with mutations in the amyloid precursor protein gene. Am J Pathol. 1996 Apr;148(4):1257–1266. [PMC free article] [PubMed] [Google Scholar]
- 24.Lemere CA, Lopera F, Kosik KS, et al. The E280A presenilin 1 Alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat Med. 1996 Oct;2(10):1146–1150. doi: 10.1038/nm1096-1146. [DOI] [PubMed] [Google Scholar]
- 25.Lippa CF, Saunders AM, Smith TW, et al. Familial and sporadic Alzheimer’s disease: neuropathology cannot exclude a final common pathway. Neurology. 1996 Feb;46(2):406–412. doi: 10.1212/wnl.46.2.406. [DOI] [PubMed] [Google Scholar]
- 26.Ringman JM, Gylys KH, Medina LD, et al. Biochemical, neuropathological, and neuroimaging characteristics of early-onset Alzheimer’s disease due to a novel PSEN1 mutation. Neuroscience letters. 2011 Jan 10;487(3):287–292. doi: 10.1016/j.neulet.2010.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norton JB, Cairns NJ, Chakraverty S, et al. Presenilin1 G217R mutation linked to Alzheimer disease with cotton wool plaques. Neurology. 2009 Aug 11;73(6):480–482. doi: 10.1212/WNL.0b013e3181b163ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mills SM, Mallmann J, Santacruz AM, et al. Preclinical trials in autosomal dominant AD: implementation of the DIAN-TU trial. Revue neurologique. 2013 Oct;169(10):737–743. doi: 10.1016/j.neurol.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herrup K. The case for rejecting the amyloid cascade hypothesis. Nature neuroscience. 2015 Jun;18(6):794–799. doi: 10.1038/nn.4017. [DOI] [PubMed] [Google Scholar]
- 30.Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nature neuroscience. 2015 Jun;18(6):800–806. doi: 10.1038/nn.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kauwe JS, Jacquart S, Chakraverty S, et al. Extreme cerebrospinal fluid amyloid beta levels identify family with late-onset Alzheimer’s disease presenilin 1 mutation. Annals of neurology. 2007 May;61(5):446–453. doi: 10.1002/ana.21099. [DOI] [PubMed] [Google Scholar]
- 32.Cruts M, van Duijn CM, Backhovens H, et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet. 1998 Jan;7(1):43–51. doi: 10.1093/hmg/7.1.43. [DOI] [PubMed] [Google Scholar]
- 33.Brickell KL, Leverenz JB, Steinbart EJ, et al. Clinicopathological concordance and discordance in three monozygotic twin pairs with familial Alzheimer’s disease. Journal of neurology, neurosurgery, and psychiatry. 2007 Oct;78(10):1050–1055. doi: 10.1136/jnnp.2006.113803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larner AJ, Ray PS, Doran M. The R269H mutation in presenilin-1 presenting as late-onset autosomal dominant Alzheimer’s disease. Journal of the neurological sciences. 2007 Jan 31;252(2):173–176. doi: 10.1016/j.jns.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 35.St George-Hyslop PH. Molecular genetics of Alzheimer’s disease. Biol Psychiatry. 2000 Feb 1;47(3):183–199. doi: 10.1016/s0006-3223(99)00301-7. [DOI] [PubMed] [Google Scholar]
- 36.Morris JC. The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology. 1993;43(11):2412–2412. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 37.Berg L, Miller JP, Baty J, Rubin EH, Morris JC, Figiel G. Mild senile dementia of the Alzheimer type. 4. Evaluation of intervention. Annals of neurology. 1992 Mar;31(3):242–249. doi: 10.1002/ana.410310303. [DOI] [PubMed] [Google Scholar]
- 38.Morris JC, Weintraub S, Chui HC, et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord. 2006 Oct-Dec;20(4):210–216. doi: 10.1097/01.wad.0000213865.09806.92. [DOI] [PubMed] [Google Scholar]
- 39.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta neuropathologica. 2012 Jan;123(1):1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monsell SE, Mock C, Hassenstab J, et al. Neuropsychological changes in asymptomatic persons with Alzheimer disease neuropathology. Neurology. 2014 Jul 29;83(5):434–440. doi: 10.1212/WNL.0000000000000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hassenstab J, Monsell SE, Mock C, et al. Neuropsychological markers of cognitive decline in persons with Alzheimer’s disease neuropathology. Journal of Neuropathology and Experimental Neurology. 2015 doi: 10.1097/NEN.0000000000000254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cummings JL. The Neuropsychiatric Inventory: assessing psychopathology in dementia patients. Neurology. 1997 May;48(5 Suppl 6):S10–16. doi: 10.1212/wnl.48.5_suppl_6.10s. [DOI] [PubMed] [Google Scholar]
- 43.Cairns NJ, Perrin RJ, Franklin EE, et al. Neuropathologic assessment of participants in two multi-center longitudinal observational studies: the Alzheimer Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN) Neuropathology: official journal of the Japanese Society of Neuropathology. 2015 Aug;35(4):390–400. doi: 10.1111/neup.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tedde A, Forleo P, Nacmias B, et al. A presenilin-1 mutation (Leu392Pro) in a familial AD kindred with psychiatric symptoms at onset. Neurology. 2000 Nov 28;55(10):1590–1591. doi: 10.1212/wnl.55.10.1590. [DOI] [PubMed] [Google Scholar]
- 45.Raux G, Gantier R, Thomas-Anterion C, et al. Dementia with prominent frontotemporal features associated with L113P presenilin 1 mutation. Neurology. 2000 Nov 28;55(10):1577–1578. doi: 10.1212/wnl.55.10.1577. [DOI] [PubMed] [Google Scholar]
- 46.Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. The Journal of clinical investigation. 2007 May;117(5):1230–1239. doi: 10.1172/JCI30447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ables JL, Breunig JJ, Eisch AJ, Rakic P. Not(ch) just development: Notch signalling in the adult brain. Nat Rev Neurosci. 2011;12(5):269–283. doi: 10.1038/nrn3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bentahir M, Nyabi O, Verhamme J, et al. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. Journal of Neurochemistry. 2006 Feb;96(3):732–742. doi: 10.1111/j.1471-4159.2005.03578.x. [DOI] [PubMed] [Google Scholar]
- 49.Goldman JS, Hahn SE, Catania JW, et al. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genetics in medicine: official journal of the American College of Medical Genetics. 2011 Jun;13(6):597–605. doi: 10.1097/GIM.0b013e31821d69b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cruchaga C, Haller G, Chakraverty S, et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLOS one. 2012;7(2):e31039. doi: 10.1371/journal.pone.0031039. [DOI] [PMC free article] [PubMed] [Google Scholar]