

Abstract
Background:
Congenital hereditary endothelial dystrophy (CHED) is an autosomal recessive disorder characterized by bilateral, symmetrical, noninflammatory corneal clouding (edema) present at birth or shortly thereafter. This study reports on an unusual delayed presentation of CHED with compound heterozygous SLC4A11 mutations.
Materials and Methods:
A 45-year-old female, presenting with bilateral decreased vision since childhood that deteriorated in the last 5 years, was evaluated to rule out trauma, viral illness, chemical injury, glaucoma, and corneal endothelial dystrophies. Tear sample was sent for herpes simplex viral (HSV) antigen testing. Genomic DNA from peripheral blood was screened for mutations in all exons of SLC4A11 by direct sequencing. Full-thickness penetrating keratoplasty was done and corneal button was sent for histopathological examination.
Results:
Slit-lamp findings revealed bilateral diffuse corneal edema and left eye spheroidal degeneration with scarring. Increased corneal thickness (762 μm and 854 μm in the right and left eyes, respectively), normal intraocular pressure (12 mmHg and 16 mmHg in the right and left eyes, respectively), inconclusive confocal scan, and specular microscopy, near normal tear film parameters, were the other clinical features. HSV-polymerase chain reaction was negative. Histopathological examination revealed markedly thickened Descemet's membrane with subepithelial spheroidal degeneration. SLC4A11 screening showed a novel variant p.Ser415Asn, reported mutation p.Cys386Arg and two polymorphisms, all in the heterozygous state and not identified in 100 controls.
Conclusions:
The study shows, for the first time, compound heterozygous SLC4A11 mutations impair protein function leading to delayed onset of the disease.
Key words: Age at onset, CHED, compound heterozygous, SLC4A11, spheroidal degeneration
Congenital hereditary endothelial dystrophy (CHED) is a rare congenital corneal condition which primarily affects corneal endothelium and usually manifests at birth or within a few years of life in the form of diffuse corneal haze, diminution of vision, with or without the presence of nystagmus and epiphora. Previously, CHED used to be classified as autosomal recessive (CHED2) and autosomal dominant (CHED1); however, recent ICD3 classification has done away with this distinction due to insufficient evidence for the existence of the autosomal dominant form.[1]
CHED gene (OMIM 217700) was identified as the bicarbonate transporter-related (BTR) SLC4A11 (solute carrier family 4, member 11; also known as BTR1, or sodium borate cotransporter 1)[2] and involvement of SLC4A11 was later confirmed in several studies.[3,4,5,6,7,8,9,10]
The disease is diagnosed on the basis of its characteristic clinical features of corneal appearance, age at onset, bilateral involvement, absence of vascularization, and inheritance pattern. The diagnosis is further supported by histopathological and genetic findings.[11,12,13]
We report here, CHED in a middle-aged female with unusually delayed presentation of the dystrophy and heterozygous SLC4A11 mutations diagnosed on the basis of extensive clinical, histopathological, and genetic evaluations.
Materials and Methods
Clinical profile
Informed written consent was taken for clinical examination, histopathological study of corneal button and genetic analysis. A detailed clinical history was taken to establish the natural history of the disease process. The patient was evaluated using slit-lamp biomicroscopy, applanation tonometry (Tono-Pen), ultrasound pachymetry; tear film breakup time and Schirmer test, specular and confocal microscopy. Ultrasound biometry was done to evaluate posterior segment as the fundus was not visible in either eye. The patient underwent full-thickness penetrating keratoplasty in the left eye and the corneal button obtained was fixed in 10% buffered formalin and cut into 4-μM thick sections using paraffin blocks. The sections were stained with hematoxylin and eosin, periodic acid-Schiff, and Congo red dyes and analyzed under a light microscope.
Genetic profile
About 5-ml peripheral blood was drawn from the patient and 100 controls in ethylenediaminetetraacetic acid vials and processed for genomic DNA isolation. The DNA samples were used for polymerase chain reaction (PCR) amplification, using primers as described previously.[6] The reaction mixture was prepared using 80-120 ng DNA, specific primers (0.5 μM each), MgCl2 (1.5 mM), deoxyribonucleotide triphosphates (dNTPs; 0.2 mM), 1 × PCR buffer (Promega, USA), and Taq polymerase (0.5 U) (Go-Taq, Promega, USA) in a thermocycler (ABI 9700, Applied Biosystems Inc., Foster City, CA, USA). The PCR products were processed for gel purification using QIAamp gel extraction kits (Qiagen, GmBH, Hilden). The amplified products were sequenced directly with BigDye Terminator Mix Version 3.1 (Applied Biosystems Inc., Foster City, CA, USA) according to the manufacturer's instructions and were then analyzed on an ABI-3100 Genetic Analyzer (Applied Biosystems Inc., Foster City, CA, USA). Nucleotide sequences for the coding regions were compared with the nucleotide sequence of the published SLC4A11 human complementary DNA (NM_032034). In silico analyses was done using sorting intolerant from tolerant (SIFT), PolyPhen-2, MutationTaster, PANTHER, and I-Mutant v2.0 tools to analyze the pathogenicity of the missense mutations identified in the patient. Human Splicing Finder (HSF), Version 3.0, was used to predict the splicing effect of the missense mutation.
Results
The patient aged 45 years presented with a complaint of diminution of vision in both eyes for the past 5 years. She had decreased vision in both eyes since childhood that deteriorated in the last 5 years. No documentation was available as the patient had presented for the first time to an ophthalmologist. There was no history of watering, recurrent pain, redness, and involuntary movements such as nystagmus. The patient was evaluated for evidence and history of ocular trauma, recurrent viral illness, chemical exposure, thermal burn, and glaucoma which were ruled out. The patient had no history of the past episodes of symptomatic viral keratitis. Tear sample PCR was done for herpes simplex viral antigen which was found to be negative. Family history was negative for similar ocular illness.
Slit-lamp evaluation revealed bilateral diffuse corneal haze extending from limbus to limbus. Yellow golden spherules located in the subepithelial region, consistent with the diagnosis of spheroidal degeneration,[14] were visible in central cornea of the left eye [Fig. 1]. The distribution of spheroidal degeneration was mostly limited to the interpalpebral area. Descemet's layer appeared thickened in both eyes on slit-lamp examination. Pachymetry showed bilateral increased central corneal thickness (762 μm and 854 μm in the right and left eyes, respectively). Intraocular pressure and tear film parameters were found to be within normal limits. Histopathological examination of the left eye and the corneal button under a light microscope revealed subepithelial and anterior stromal spheroidal degeneration lesions, stromal edema with lamellar separation, and markedly thickened Descemet's membrane with rare degenerative endothelial cells [Fig. 2a and b]. Screening of the SLC4A11 gene revealed the presence of a novel change in exon 10 at position c. 1244G>A (p.Ser415Asn) replacing serine with asparagine and a second reported change in exon 9 at position c. 1156T>C (p.Cys386Arg) replacing cysteine with arginine.[15] Apart from this, two single nucleotide polymorphisms (SNPs) rs2281575 and rs3803953 were also seen. All these changes were in the heterozygous state and were absent in 100 normal controls.
Figure 1.
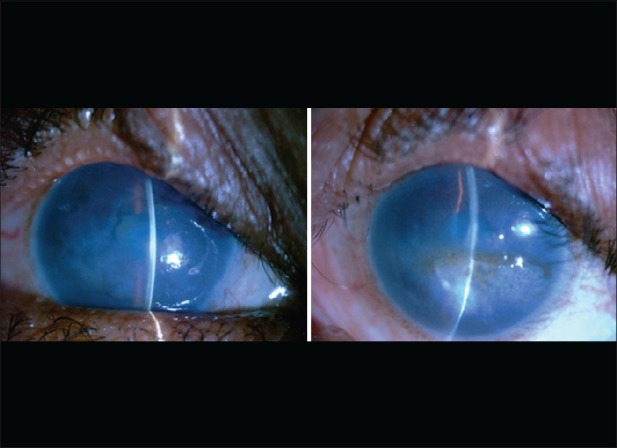
Bilateral diffuse corneal haze with left eye showing spheroidal degeneration and corneal scarring
Figure 2.
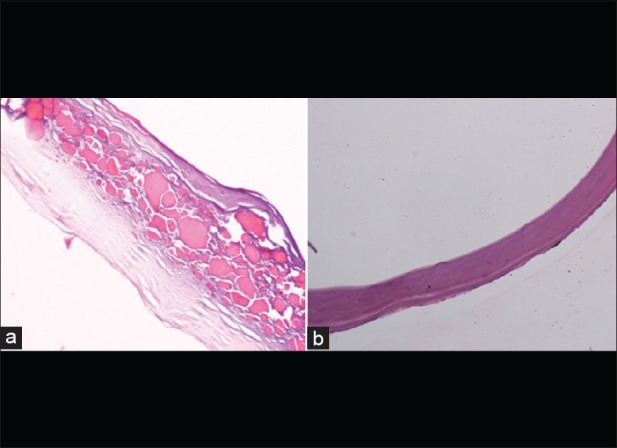
(a) Histopathological specimen showing subepithelial spheroidal degeneration (×10) in the form of multiple eosinophilic globules with stromal lamellar separation (Descemet's membrane separated from stroma during processing of the specimen). (b) Grossly thickened Descemet's membrane with degenerated endothelial cells (×20)
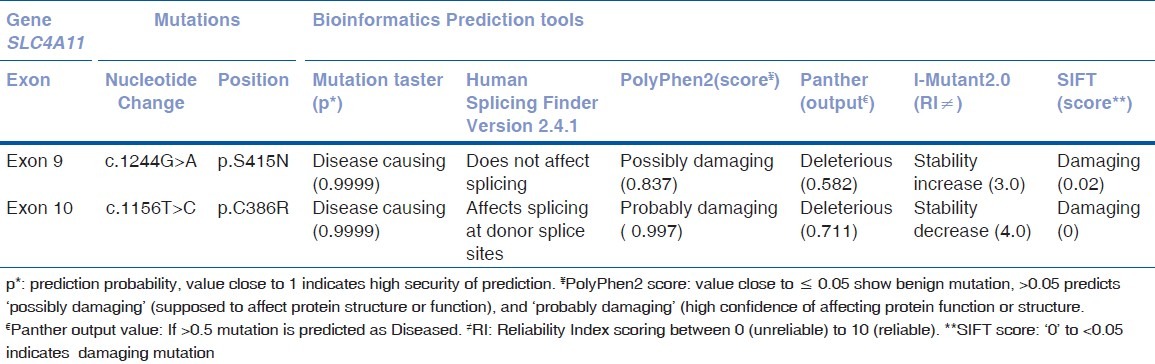
The SIFT, PolyPhen-2, MutationTaster, PANTHER, and I-Mutant v2.0 analysis showed the mutations to be damaging which affected the SLC4A11 protein function or structure and not tolerated across the species. The HSF, Version 3.0, revealed that p.Cys386Arg mutation to be affecting the splicing site [Table 1].
Table 1.
Pathogenicity prediction of the mutations using various bioinformatics tools
Discussion
CHED patients present with severe corneal clouding at birth and the condition is relatively stationary. Usually, these patients have associated nystagmus with minimal or no photophobia and watering. Besides these clinical features, CHED patients on histopathological examination present with diffuse stromal edema, lamellar separations within the stromal layers, thickened Descemet's membrane, and sparse degenerative endothelial cells irrespective of the type of CHED. Genetic studies in patients with CHED in recent years have revealed chromosome 20p13 as an important locus,[15] and SLC4A11 gene in this region has been linked to its causation.[2,3,4,5,6,7,8]
CHED which normally presents within a few years of birth is very unusual to be presented at an age of 45 years. To the best of our knowledge, this is the first case to be reported at this age. The probable explanation for this late presentation is comparatively slow progression or relatively stationary nature of the disease that might be the reason behind the good ambulatory vision of the patient till the age of 40 years. Female gender along with poor socioeconomic conditions in a developing country appears to be the additional reasons for not seeking medical attention for the extended duration.
The common differential diagnoses were bilateral hazy cornea which included bilateral physical trauma, chemical injury, glaucoma, recurrent viral keratitis, etc., before reaching the diagnosis of CHED. History of bilateral physical trauma could be easily ruled out by clinical history. There was no evidence of conjunctival cicatrization, and corneal vascularization, tear film parameters were normal; history for chemical injury was negative which ruled out bilateral chemical burns in the past. Optic disc was not visible in both eyes making it difficult to comment on evidence of long-term glaucoma, but with the negative clinical history of the recurrent painful red eye and stationary ambulatory vision for such a long time ruled out glaucoma. Similarly, it is highly unlikely for the patient to develop bilateral diffuse corneal haze without symptomatic recurrences of viral keratitis. Tear film PCR for herpes simplex was negative which rules out evidence of viral keratitis. Fuchs’ endothelial corneal dystrophy (FECD) is another differential diagnosis but is unlikely again due to poor vision since childhood and relatively stationary nature of the disease while FECD is a progressive disease of the fourth to fifth decade.[16] Further, no evidence of guttae changes was visible on slit-lamp examination to support this diagnosis.
Histopathological features suggested severe endothelial damage evident by very sparse degenerative endothelial cells with markedly thickened Descemet's membrane along with secondary changes in corneal stroma denoting long-term endothelial insult. These histopathological features were compatible with the diagnosis of CHED.[2] Further spheroidal degeneration in subepithelial as well as anterior stroma has also been described as an association with CHED.[1,17] Evidence from genetic analysis of SLC4A11 also supported CHED diagnosis.
SLC4A11 proteins facilitate the water flux across membranes.[18] Mutations in SLC4A11 are reported to result in proteins which are misfolded and retained in the endoplasmic reticulum.[18] A study has shown that heterozygous SLC4A11 mutations partially rescue cell surface trafficking of the wild type (WT) SLC4A11 proteins as it was functionally able to carry out the role of transport at the cell surface.[19] The study measured functional activity of SLC4A11 protein which differed in CHED affected and CHED carriers (heterozygous) and suggested that about 25% of the water flux activity helps in restoration of WT function and delays the disease onset, and about 60% of the WT activity prevents the disease symptoms.[20]
This study reports, for the first time, a case of CHED presented at the age of 45 years in a female patient. One novel and one reported heterozygous SLC4A11 change have been identified along with two SNPs. The changes as predicted by bioinformatics tools are seen to be damaging and deleterious. HSF-V3.0 revealed that the p.Cys386Arg mutation occurs in the exonic position and thereby may affect the exonic splicing enhancer (ESE) site. This ESE site is necessary for recognition of splice sites by other cellular molecules. Therefore, mutation on this site might alter the splicing and thus affect the protein structure. CHED is a recessive disorder, but in our case, all variants identified were in the heterozygous state and it is speculated that the disease might occur due to compound heterozygous changes.[3,7] A study on transfected human embryonic kidney cells, analyzed the effect of heterozygous mutations on SLC4A11 protein level, were analyzed in a study which revealed that two of the three heterozygous mutations r significantly lowered the expression levels of SLC4A11 protein as compared to the WT.[21] Therefore, it is envisaged that the heterozygous mutations of the present study may have a mild effect on the protein function and levels and thereby may have resulted in the late onset of the disease.
Conclusions
The diagnosis of CHED at this unusual age might be missed easily, but it becomes mandatory to suspect CHED, especially if the patient presents with a history of poor bilateral vision that is relatively stationary since childhood and when the obviously common causes are excluded.
Financial support and sponsorship
All necessary funding for the study was provided by the All India Institute of Medical Sciences, New Delhi, India.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
The authors would like to thank the patient for the participation.
References
- 1.Weiss JS, Møller HU, Aldave AJ, Seitz B, Bredrup C, Kivelä T, et al. IC3D classification of corneal dystrophies – edition 2. Cornea. 2015;34:117–59. doi: 10.1097/ICO.0000000000000307. [DOI] [PubMed] [Google Scholar]
- 2.Vithana EN, Morgan P, Sundaresan P, Ebenezer ND, Tan DT, Mohamed MD, et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2) Nat Genet. 2006;38:755–7. doi: 10.1038/ng1824. [DOI] [PubMed] [Google Scholar]
- 3.Aldave AJ, Yellore VS, Bourla N, Momi RS, Khan MA, Salem AK, et al. Autosomal recessive CHED associated with novel compound heterozygous mutations in SLC4A11. Cornea. 2007;26:896–900. doi: 10.1097/ICO.0b013e318074bb01. [DOI] [PubMed] [Google Scholar]
- 4.Desir J, Moya G, Reish O, Van Regemorter N, Deconinck H, David KL, et al. Borate transporter SLC4A11 mutations cause both Harboyan syndrome and non-syndromic corneal endothelial dystrophy. J Med Genet. 2007;44:322–6. doi: 10.1136/jmg.2006.046904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiao X, Sultana A, Garg P, Ramamurthy B, Vemuganti GK, Gangopadhyay N, et al. Autosomal recessive corneal endothelial dystrophy (CHED2) is associated with mutations in SLC4A11. J Med Genet. 2007;44:64–8. doi: 10.1136/jmg.2006.044644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar A, Bhattacharjee S, Prakash DR, Sadanand CS. Genetic analysis of two Indian families affected with congenital hereditary endothelial dystrophy: Two novel mutations in SLC4A11. Mol Vis. 2007;13:39–46. [PMC free article] [PubMed] [Google Scholar]
- 7.Ramprasad VL, Ebenezer ND, Aung T, Rajagopal R, Yong VH, Tuft SJ, et al. Novel SLC4A11 mutations in patients with recessive congenital hereditary endothelial dystrophy (CHED2). Mutation in brief #958. Online. Hum Mutat. 2007;28:522–3. doi: 10.1002/humu.9487. [DOI] [PubMed] [Google Scholar]
- 8.Sultana A, Garg P, Ramamurthy B, Vemuganti GK, Kannabiran C. Mutational spectrum of the SLC4A11 gene in autosomal recessive congenital hereditary endothelial dystrophy. Mol Vis. 2007;13:1327–32. [PubMed] [Google Scholar]
- 9.Hemadevi B, Veitia RA, Srinivasan M, Arunkumar J, Prajna NV, Lesaffre C, et al. Identification of mutations in the SLC4A11 gene in patients with recessive congenital hereditary endothelial dystrophy. Arch Ophthalmol. 2008;126:700–8. doi: 10.1001/archopht.126.5.700. [DOI] [PubMed] [Google Scholar]
- 10.Aldahmesh MA, Khan AO, Meyer BF, Alkuraya FS. Mutational spectrum of SLC4A11 in autosomal recessive CHED in Saudi Arabia. Invest Ophthalmol Vis Sci. 2009;50:4142–5. doi: 10.1167/iovs.08-3006. [DOI] [PubMed] [Google Scholar]
- 11.Maumenee AE. Congenital hereditary corneal dystrophy. Am J Ophthalmol. 1960;50:1114–24. doi: 10.1016/0002-9394(60)90998-3. [DOI] [PubMed] [Google Scholar]
- 12.Kenyon KR, Maumenee AE. The histological and ultrastructural pathology of congenital hereditary corneal dystrophy: A case report. Invest Ophthalmol. 1968;7:475–500. [PubMed] [Google Scholar]
- 13.Pearce WG, Tripathi RC, Morgan G. Congenital endothelial corneal dystrophy. Clinical, pathological, and genetic study. Br J Ophthalmol. 1969;53:577–91. doi: 10.1136/bjo.53.9.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young JD, Finlay RD. Primary spheroidal degeneration of the cornea in Labrador and northern Newfoundland. Am J Ophthalmol. 1975;79:129–34. doi: 10.1016/0002-9394(75)90470-5. [DOI] [PubMed] [Google Scholar]
- 15.Toma NM, Ebenezer ND, Inglehearn CF, Plant C, Ficker LA, Bhattacharya SS. Linkage of congenital hereditary endothelial dystrophy to chromosome 20. Hum Mol Genet. 1995;4:2395–8. doi: 10.1093/hmg/4.12.2395. [DOI] [PubMed] [Google Scholar]
- 16.Hogan MJ, Wood I, Fine M. Fuchs’ endothelial dystrophy of the cornea 29 th Sanford Gifford Memorial lecture. Am J Ophthalmol. 1974;78:363–83. doi: 10.1016/0002-9394(74)90224-4. [DOI] [PubMed] [Google Scholar]
- 17.Vemuganti GK, Sridhar MS, Edward DP, Singh S. Subepithelial amyloid deposits in congenital hereditary endothelial dystrophy: A histopathologic study of five cases. Cornea. 2002;21:524–9. doi: 10.1097/00003226-200207000-00017. [DOI] [PubMed] [Google Scholar]
- 18.Vilas GL, Loganathan SK, Liu J, Riau AK, Young JD, Mehta JS, et al. Transmembrane water-flux through SLC4A11: A route defective in genetic corneal diseases. Hum Mol Genet. 2013;22:4579–90. doi: 10.1093/hmg/ddt307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vilas GL, Loganathan SK, Quon A, Sundaresan P, Vithana EN, Casey J. Oligomerization of SLC4A11 protein and the severity of FECD and CHED2 corneal dystrophies caused by SLC4A11 mutations. Hum Mutat. 2012;33:419–28. doi: 10.1002/humu.21655. [DOI] [PubMed] [Google Scholar]
- 20.Loganathan SK, Casey JR. Corneal dystrophy-causing SLC4A11 mutants: Suitability for folding-correction therapy. Hum Mutat. 2014;35:1082–91. doi: 10.1002/humu.22601. [DOI] [PubMed] [Google Scholar]
- 21.Vithana EN, Morgan PE, Ramprasad V, Tan DT, Yong VH, Venkataraman D, et al. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum Mol Genet. 2008;17:656–66. doi: 10.1093/hmg/ddm337. [DOI] [PubMed] [Google Scholar]