

Abstract
The increasing application of soil metaproteomics is providing unprecedented, in-depth characterization of the composition and functionality of in situ microbial communities. Despite recent advances in high-resolution mass spectrometry, soil metaproteomics still suffers from a lack of effective and reproducible protein extraction protocols and standardized data analyses. This review discusses the opportunities and limitations of selected techniques in soil-, and leaf litter metaproteomics, and presents a step-by-step guideline on their application, covering sampling, sample preparation, extraction and data evaluation strategies. In addition, we present recent applications of soil metaproteomics and discuss how such approaches, linking phylogenetics and functionality, can help gain deeper insights into terrestrial microbial ecology. Finally, we strongly recommend that to maximize the insights environmental metaproteomics may provide, such methods should be employed within a holistic experimental approach considering relevant aboveground and belowground ecosystem parameters.
Keywords: environmental proteomics; protein extraction, matrix effects; bioinformatics, functional databases; meta-analysis
The presented review provides an overview of the problems that may arise during the various soil and litter metaproteomic analyses steps and summarizes our current knowledge on possible solutions strategies.
INTRODUCTION
Soil is an essential natural resource and a regulator of ecosystem provision. Biogeochemical processes occurring in soil environments such as decomposition and mineralization of organic matter (OM) significantly affect nutrient cycling, subsequently influencing the climate and the biosphere. Moreover, soil is an important habitat for soil microbes and animals, and serves as physical and cultural environment for humankind (Blum, Busing and Montanarella 2004).
Soil microbes are major drivers of biogeochemical cycles and are a considerable pool of belowground terrestrial biomass. Every gram of soil harbors thousands of bacterial, archaeal and eukaryotic taxa, and this taxonomic diversity is mirrored by the diversity of their physiologies, life styles (i.e. oligotrophy-copiotrophy) and associated functional classes of proteins (Fierer et al.2012b). Microbial diversity is highly variable in terrestrial ecosystems, depending on many factors, such as plant cover, animal activity, soil moisture, temperature, aeration, porosity, nutrient availability, pH and salinity (Kennedy et al.2004; Maron, Mougel and Ranjard 2011; Van Horn et al.2014).
When comparing a broad range of soil ecosystem types Acidobacteria and Verrucomicrobia turned out to be the most abundant taxonomic groups followed by Actinobacteria, Bacteroidetes, Planctomycetes and Archaea (Barberán et al.2012). These groups vary across different biomes e.g. Actinobacteria, Bacteroidetes and Cyanobacteria phyla dominate in desert soils (Fierer et al. 2012), while arctic permafrost peatland soils were dominated by Actinobacteria, Verrucomicrobia and Bacteroidetes (Tveit et al.2013). This phylogenetic information enables the determination of changes in ecological life styles in response to treatments, as has been shown for N gradients (Fierer et al. 2012). From the functional perspective, a variety of genes expressed for plant degradation were comparable among climatic zones, including arctic permafrost peatland soil and temperate and subtropical soils (Tveit et al.2013), displaying similar metabolic potential. However, N-fertilization resulted in increased gene abundances for DNA/RNA replication, electron transport and protein metabolism (Fierer et al. 2012), while desert microbial communities are characterized by a high abundance of genes associated with osmoregulation and dormancy, and genes associated with nutrient cycling and catabolism of plant-derived organic compounds are less abundant (Fierer et al. 2012). However, to which extent these genes are actually expressed and hence become physiologically active has yet to be determined. Notably, changes in microbial composition might be of minor relevance for soil ecosystem functions, due to functional redundancy (Souza et al.2015). Metagenome information thus represents only the ‘functional potential’ and giving no indication of the relative activity of the phyla present. Therefore, to assess function and potentially link biodiversity and ecosystem functioning, it is of upmost importance to not only measure gene abundance, but also the actual expression and activity of functional proteins (Prosser 2015; Delgado-Baquerizo et al.2016).
Reflecting the value of the insights provided the number of studies that have successfully applied metaproteomics on soil and leaf litter environments continues to grow, including e.g. metaproteome analysis of permafrost soil (Hultman et al.2015), hydrocarbon degradation in soils (Bastida et al.2016), deforestation (Bastida et al.2015a), soil restoration and ecosystem processes (Bastida et al.2015b) and a recent study that focused on the active microbial players in short-term degradation of plant-derived N (Starke et al.2016). The latter is a novel protein stable isotope probing (SIP) approach applying isotopic-N labeled plant material in a metaproteomics experiment, to track N from plants into microbes. A bacterial dominated short-term assimilation of plant-derived N was shown, and oligotrophic and copitrophic life styles of soil organisms in terms of temporal leaf litter N utilization patterns illustrate a new cutting edge approach to determine ecological attributes of soil microbes (Starke et al.2016).
Due to its large potential for providing a link between functional and phylogenetic information of soil microbial communities, as exemplified by the aforementioned studies, there has been growing interest in the application of metaproteomics in soil ecology to study microbially driven ecosystem functions (e.g. methanogensis in permafrost soils; Hultman et al.2015). However, soil metaproteomics still faces several challenges, including the heterogeneity of soil matrices, high microbial diversity, the ecosystem-specific dominance of few microbial species and limited metagenomic information and data handling (Keller and Hettich 2009; Schneider and Riedel 2010; Siggins, Gunnigle and Abram 2012; Becher et al.2013).
Protein extraction of soils is often difficult due to the presence of other organic compounds, such as complex carbohydrates, lipids and phenolic compounds (e.g. lignin), and humic substances (HS) as well as inorganic compounds from the soil matrix, such as silt and clay minerals. Coextraction of HS, which are contained in litter and soil, as well as the presence of large reactive surfaces of soil minerals (e.g. clay) not only complicate protein extraction but also interfere with the separation of peptides (Bastida et al.2009), protein identification (Arenella et al.2014) and quantification (Criquet, Farnet and Ferre 2002; Ogunseitan 2006) due to protein modifications. These limitations for extraction are due to the fact that proteins can be adsorbed, linked anchored or embedded on/to/in solid particles such as clay, clay minerals, and soil OM organo-mineral complexes (Nielsen, Calamai and Pietramellara 2006; Tomaszewski, Schwarzenbach and Sander 2011), which thereby reduce extraction efficiency (Sander, Tomaszewski and Schwarzenbach 2011).
Adsorption of proteins to clays is a rapid process, which is only partly reversible (Nielsen, Calamai and Pietramellara 2006), and is based on the large specific surface area of clay minerals (Giagnoni et al.2011). While it was shown that even whole cells can be sorbed to mineral surfaces (i.e. clays), which depend on the pH, the charge of the clay mineral and the Mg concentration (Jiang et al.2007). However, the adhesion of cells to soil particles is governed by their surface charges and global hydrophobic and hydrophilic characteristics (Doyle 2000). HS and proteins are bound reversibly by a cation exchange process, which depends on the cation exchange capacity (CEC) of the soil, the amino acid composition and the isoelectric point of the target proteins. Moreover, protein polarity may affect sorption in aqueous solution through hydrophobic interactions (Norde, Tan and Koopal 2008), though hydrophobic surfaces may reduce proteins sorption in soils (Keiblinger et al.2015). Reduced protein availability through clay-enzyme complexes has been shown for artificial soil mixtures with high CEC or clay content by lower numbers of protein spots (Giagnoni et al.2011). To this end, the choice of purification methods or the extraction buffer and additives to it depends not only on the soil type but also on the goal of the investigation. Potential strategies are discussed below. From an experimental point of view, soil metaproteomics include the following steps: (i) sample handling (including obtaining a representative sample, homogenization, pooling and storage conditions, Fig. 1A), (ii) soil protein extraction (Fig. 1B), (iii) processing of soil protein extracts (including removal of interfering substances, pre-fractionation of proteins or peptides and mass spectrometry (MS) analysis (Fig. 1C), (iv) data analysis (including spectra handling and database assembly for peptide and protein identification, Fig. 2A), (v) data evaluation and interpretation (Fig. 2B) and finally (vi) data storage and visualization. All steps are crucial for obtaining, holding and sharing high-quality soil metaproteome data, and some of these steps have recently been reviewed in detail (Keller and Hettich 2009; Schneider and Riedel 2010; Siggins, Gunnigle and Abram 2012; Becher et al.2013). Here we focus on differences in sample preparation and published protocols (Table 1) and try to synthesize knowledge to provide a ‘step-by-step’ guideline of how to best proceed in soil and leaf litter protein extraction (Fig. 1). In addition, the current work presents recent advances in data analysis and data interpretation using novel bioinformatic tools (Fig. 2). The wider objective of the present work is to (i) highlight the need for standardized methodology, which would ensure better comparability of future soil metaproteomic analyses, and to (ii) provide the basis for future meta-analysis by including additional environmental parameters and different ecosystem properties into metaproteome datasets.
Figure 1.
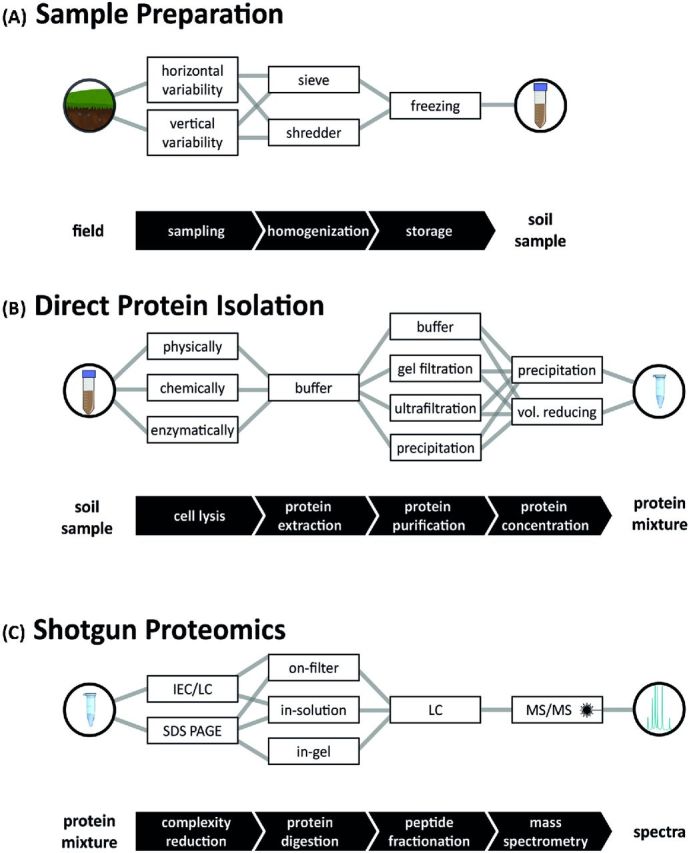
From sampling to data. Schematic representation of workflows. Researchers are confronted with various sampling methods and procedures that have to be carefully selected and combined for (A) sample preparation, including soil sampling homogenization and storage, (B) protein isolation and (C) shotgun proteomics (from top to down). Consecutive steps are connected by lines. Abbreviations are explained in the text.
Figure 2.

From data to understanding. Schematic representation of workflows discussed in this review. Researchers can select or combine various methods for (A) data analysis, and (B) data interpretation (from top to down). Consecutive steps are connected by lines (dashed lines represent workflows not suitable for high-throughput analyses). Abbreviations are explained in the text.
Table 1.
Soil Metaproteomic studies.
| Nr | Matrix | Depth (cm) | pH | Corg | N content | Texture (sand/silt/clay) | CEC | Preparation/sample storage | Extraction ratio (soil/buffer) | Extraction protocol/precipitation method | Protein yield/number of proteins | Analysis method | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Forest soils | |||||||||||||
| 1 | Populus spp., Zhengzhou, China | 15–25 | 6.2 | 2.2% | n.d. | 41/49/10 | n.d. | Sieved < 2 mm/dried soil | 1 g / 10 ml | Two sequential extractions (SEM), 0.25 M citrate (pH 8.0), and 1% SDS buffer (0.1 M Tris-HCl, 20 mM DTT, pH 6.8) | 95 μg protein; ∼250 protein spots | 2DE PAGE | Chen, Rillig and Wang (2009) |
| Phenol (pH 8.0) extraction of combined extracts (C-S-P-M); | |||||||||||||
| precipitation: 5 volumes of 0.1 M ammonium acetate in MeOH; MeOH and acetone washing steps | |||||||||||||
| 2 | Pinus halepensis Mill., sandy loam, Murcia, Spain | 0–15 | 7.6 | 3.8% | 2.7 mg g−1 | 75/9/16 | n.d. | Soil 60% WHC (fresh; enrichment incubation 200 g soil with glucose and proline 15 days 28°C) | DGC –pellet 0.5 ml EB | 0.5 M Tris-HCl (pH 8.7), 0.9 M sucrose, 50 mM EDTA, 0.1 M KCl, 2% β-mercaptoethanol + phenol EB + phenol precipitation: 5 volumes of 0.1 M ammonium acetate and 1% β-mercaptoethanol in MeOH + phenol (see also #7, Taylor and Williams 2010) | 27 peptides, 11 non-redundant proteins, 2 hyp. proteins | SDS-PAGE, LC-MS/MS QSTAR-XL | Bastida et al. (2006, 2012a) |
| 3 | Pinus halepensis Mill., sandy loam, Murcia, Spain | 0–15 | 7.6 | 38 mg g−1 | 3.9 mg g−1 | 47/37/16 | n.d. | Fresh, kept at 3°C up to 1 week, sieved < 2 mm | (a, b) 5 g soil 10 ml EB (c) 1 g soil 1 ml EB |
(a) 0.1 M NaOH purification with phenol (Benndorf et al.2007) (b) SDS buffer (5% SDS, 50 mM Tris HCl; pH 8.5; 0.15M NaCl, 0.1 mM EDTA boiling (Chourey et al.2010) (c) 50 mM Tris-HCl pH 7.58, 10% sucrose, 2 mM DTT, 4 mM EDTA, 0.1% Brij58 + protease inhibitor (Singleton et al.2003; see also #8) |
(a)–(b) 112–327 (c) 6–7 proteins |
LC-MS/MS Orbitrap Velos | Bastida, Hernandez and Garcia (2014) |
| 4 | Fagus sylvatica, silty loam, Vienna, Austria | 0–10 | 4.4 | 3.8% | 2.38 mg g−1 | -/-/19 | n.d. | –80°C sieved < 2 mm, homogenized with mortar and pestle | 1:2 (w/v) 5 g soil | (a) SDS buffer (50 mM Tris, 1% SDS pH 7.5), precipitation: 10% TCA (b) SDS-Phenol, (50 mMTris, 1% SDS pH 7.5 + phenol (pH 8.0) (c) 0.1M NaOH-Phenol (Benndorf et al. 2007), (d) W-SP as (3) with prior washing steps TCA/acetone, methanol, acetone wash (Wang et al.2006) precipitation (2)-(4) : 5 volumes of 0.1 M ammonium acetate in MeOH |
(a) 226 (b) 494 (c) 293 (d) 372 proteins |
RP LC- LC-MS/MS | (Keiblinger et al.2012b) |
| 5 | Pinus halepensis Mill. and natural shrubs Lithic Calcixeroll Murcia, Spain | 1–15 | 7.7 | 54 mg g−1 | 3.42 mg g−1 | 72/11/17 | n.d. | Air-dried homogenized, sieved < 2 mm | (a) 1:5 w/v (b) 1:3 w/v |
(a) 0.1 M Na-Pyrophosphate pH 7.1 (b) 67 mM phosphate buffer pH 6 and 0.5 M K2SO4 pH 6.6 Purification with PVPP column, filtered 0.22 μm; dialysed, concentrated by Amicon PM-10 diaflomembrane (molecular cut off 10 000) precipitation 10% (v/v) TCA | (a)–(b) 242 μg BSA g−1 soil | Enzyme activities, SDS PAGE | Masciandaro et al. (2008) |
| 6 | Quercus ilex, Laurus nobilis and natural shrubs Inceptisoil Tuscany, Itlay | 1–15 | 7.8 | 38 mg g−1 | 1.46 mg g−1 | 64/19/17 | n.d. | Air dried homogenized, sieved <2 mm | 1) (a) 1:5 w/v 2) (b) 1:3 w/v | See above (#5) | (a)–(b) 118 μg BSA g−1 soil | Enzyme activities, SDS PAGE | Masciandaro et al. (2008) |
| Agricultural soils | |||||||||||||
| 7 | Herbaceous crops Hypericum perforatum, Hyssopus officinalis L.), Thermic Aquic Paleudults, Quitman, Texas, USA | 0–10 | 5.9-6.2 | n.d. | n.d. | (Fine sandy loam) | n.d. | –10°C after defreezing kept at 4°C up to 1 month; sieved < 5 mm | 20 g soil + 50 ml 0.9% NaCl, three cycles of blending, 10 ml Nycondez centrifugation; DGC –pellet 0.5 ml EB | 0.5 M Tris-HCl (pH 8.7), 0.9 M sucrose, 50 mM EDTA, 0.1 M KCl, 2% β-mercaptoethanol homogenized with mortar and pestle + 0.5 ml phenol; EB + phenol precipitation: 5 volumes of 0.1M ammonium acetate and 1% β-mercaptoethanol in MeOH | 187 proteins; 47 identified proteins | SDS-PAGE, MALDI –TOF/TOF | Taylor and Williams (2010) |
| 8 | Soil in pots, stagnogley Newcastle England | 0–10 | 6.1 | 2.5% | n.d. | 38/31/31 | n.d. | Soil 50% WHC (fresh) | 1 g/1 ml EB + 100 μl protease inhibitor cocktail | 0.05 M Tris-HCl, 10% sucrose, 2 mM DTT,4 mM EDTA, 0.1% Brij 58 pH 7.58 adj. with ammonia solution | 250 μg protein g−1 soil | SDS-PAGE | Singleton et al. (2003) |
| 9 | A fallow, sandy loam, Murcia, Spain | 0–15 | 7.9 | 0.39% | 1.2 mg g−1 | 68/16/16 | n.d. | Soil 60% WHC (fresh; enrichment incubation 200 g soil with glucose and proline 15 days 28°C) | DGC –pellet 0.5 ml EB | 0.5 M Tris-HCl, 0.9 M sucrose, 50 mM EDTA, 0.1 M KCl, 2% β-mercaptoethanol + phenol (see also #7; Williams and Taylor 2010) | 260 peptides 61 non redundant proteins 34 hypothetical proteins | SDS-PAGE, LC-MS/MS QSTAR-XL | Bastida et al. (2006, 2012a) |
| 10 | A fallow, clay loam, Murcia, Spain | 0–15 | 7.8 | 0.27% | 1.3 mg g−1 | 58/8/34 | n.d. | Soil 60% WHC (fresh; enrichment incubation 200 g soil with glucose and proline 15 days 28°C) | DGC –pellet 0.5 ml EB | 0.5 M Tris-HCl, 0.9 M sucrose, 50 mM EDTA, 0.1 M KCl, 2% β-mercaptoethanol + phenol (see also #7; Williams and Taylor 2010) | 27 peptides 11 non-redundant proteins 2 hypothetical proteins | SDS-PAGE, LC-MS/MS QSTAR-XL | Bastida et al. (2006, 2012a) |
| 11 | Xerophytic shrubs, sandy-loam, Murcia, Spain | 0–15 | 8.5 | 11.2 mg g−1 | 1.1 mg g−1 | 70/12/18 | n.d. | Fresh, kept at 3°C up to 1 week, sieved < 2 mm | (a, b) 5 g / 10 ml EB (c) 1 g / 1 ml EB |
(a) 0.1 M NaOH purification with phenol (Benndorf et al.2007) (b) SDS buffer (5% SDS, 50 mM Tris HCl; pH 8.5; 0.15 M NaCl, 0.1 mM EDTA boiling (Chourey et al.2010) (c) 50 mM Tris-HCl pH 7.58, 10% sucrose, 2 mM DTT, 4 mM EDTA, 0.1% Brij58 + protease inhibitor (Singleton et al.2003) see also (#8) |
(a)–(b) 204–215 (c) 36–68 proteins |
LC-MS/MS Orbitrap Velos | Bastida, Hernandez and Garcia (2014) |
| 12 | A fallow, clay loam, Murcia, Spain | 0–15 | 7.8 | 2.9 mg g−1 | 0.6 mg g−1 | 50/9/41 | n.d. | See above (#11) | (a)–(b) 42–149 (c) 48–74 proteins | Bastida, Hernandez and Garcia (2014) | |||
| Other soils | |||||||||||||
| 13 | Oligotrophic dryland soil, Namib dessert, Namibia | - | 6.7 | 0.1% | n.d. | 85/11/4 | 5.2 | RNA later | 10 g / 10% w/v | 1% SDS (10 mM Tris, 5 mM MgCl2; pH 8); protease inhibitor (10 μl ml−1); second bead-beating step with subsequent benzonase treatment (250 U μl−1); phenol: chloroform : isoamylalcohol (25:24:1; pH 8); precipitation: 5 volumes of 0.1 M ammonium acetate in MeOH; MeOH & acetone washing steps | 504.4 ng μl−1 110 proteins | Q-Exactive LC-MS/MS | Gunnigle et al. (2014) |
| 14 | Organic rich, Gauteng, South Africa | – | 8.1 | 1.3% | n.d. | 57/17/26 | 22.1 | RNA later | 10 g / 10% w/v | See above (#13) | 690.2 ng μl−1 | – | Gunnigle et al. (2014) |
| 15 | Potting soil, commercial | – | n.d. | n.d. | n.d. | n.d./n.d./25 | n.d. | Fresh/–80°C | 1:3 (w/v) 5 g soil | See above (#4) | (a) 237 (b) 198 (c) 124 (d) 80 proteins | RP LC- LC-MS/MS | Keiblinger et al. (2012a) |
| 16 | Greenhouse soil, Entisol, Wachi, central Kyoto, Japan | 1–10 | 5.5-6.8 | 12-126 mg g−1 | 1.2-10.8 mg g−1 | n.d. | 12-25 | Fresh stored at 4°C, sieved <2 mm | 100 g soil/300 ml 1:3 w/v | 67 mM phosphate buffer (NaHPO4*12H2O + KH2PO4 pH 6); precipitation: 5% TCA | 5 proteins | SDS-PAGE, N-terminal sequencing | Murase et al. (2003) |
| 17 | Mixed grassland soil, ultic haploxeralf, California, USA | – | 5.2 | 12 mg g−1 | 1 mg g−1 | (sandy clay loam) | n.d. | Processed freshly or frozen in liquid N2 and stored at -80°C thawed to 4°C | 5 g / 10 ml EB direct extraction pure soil | SDS-TCA 5% SDS, 50 mM Tris-HCl pH 8.5 0.15 M NaCl, 0.1 mM EDTA; 1 mM MgCl2; 50 mM DTT precipitation with TCA re-dissolved in guanidine solution (6 mM guanidine HCl, 10 mM DTT dissolved in 50 mM Tris/10 mM CaCl2 pH 7.6 | 333 non redundant 716 redundant proteins | 2D nano LC-MS/MS LTQ XL | Chourey et al.2010 |
| 18 | Permafrost soil, Picea mariana Alaska, USA | 65–75 | 5.8 | n.d. | n.d. | n.d. | n.d. | Frozen soil | 5 g / 5 ml EB direct extraction | SDS-TCA 4% SDS, 100 mM Tris-HCl pH 8.0 boiling 5 min, sonification pulses 10 s on 10 s off for 2 min centrifugation, DTT added to supernatant (24 mM) precipitation with TCA 20% re-dissolved 8 Urea with 100 mM Tris-HCl, pH 8.0 incubating at RT for 30 min and sonicated in an ice bath | 284 identified proteins | RP 2D-LC-MS/MS, hybrid Velos/Orbitrap | Hultman et al. (2015) |
| 19 | Xerophytic shrubs, Haplic calcisol low degraded soil Murcia Spain | 0–15 | n.d. | 5.3 mg g−1 | 0.5 mg g−1 | 71.7/9.5/18.8 | n.d. | Sieved <2 mm, incubation in microcosms, | 5 g / 10 ml EB direct extraction | See above (#17) | total 2882 proteins identified; control soil 1030–1167; petroleum+soil 2 days 1433–1579; petroleum+soil 50 days 983-914 soil + compost 1189–1247 | SDS PAGE, Orbitrap Fusion LC-MS/MS | Bastida et al. (2016) |
| 20 | Mediterranean scrubs Aridic calcisol Fallow + 12 kg ha−1 compost or sewage sludge Murcia Spain | 0–15 | 7.47 | 20.7 mg g−1 | 1.6 mg g−1 | n.d | n.d. | Sieved <2 mm, 8 samples per plot were pooled | 5 g / 10 ml EB direct extraction | See above (#17) | total 10818 proteins identified 1351 protein groups | LC-MS/MS, hybrid Q-Exactive | Bastida et al. (2015) |
| 21 | Tobacco field, Typic Ariudoll Mochkrena, Saxony, Germany | 10 | 6.46 | 11.6 mg g−1 | 1.6 mg g−1 | Silty clay soil | n.d. | sieved < 2 mm, incubation in microcosms | (1) 50 g / 50 ml EB (2) 2 g / 5.4 ml EB + 0.6 ml 10% SDS buffer |
(1) EB 50 mM Tris-HCl pH 7.5, 1mM PMSF, 0.1 mg/mL chloramphenicol shaking for 2 h; centrifugation, (2) 3 freeze thaw cylces, and 2 cycles of sonication (3) Phenol purification for (1) and (2) precipitation: 5 volumes of 0.1 M ammonium acetate in MeOH | Novel 15N SIP-protein approach, with 11–26 peptides per time point to clacluate relative isotope abundances | SDS PAGE LC-MS/MS, hybrid Velos/Orbitrap | Starke et al. (2016) |
| Rhizosphere soils | |||||||||||||
| 22 | Rice, Fujian China | 0–10 | 5.5 | 6.7% | n.d. | 6.3 | Sieved < 2 mm/dried soil | 1 g / 5 ml SDS + 1 g / 5 ml citrate buffer | 0.25 M citrate (pH 8.0), or 1.25% SDS buffer (0.1 M Tris-HCl, 20 mM DTT, pH 6.8) Phenol (pH 8.0) extraction of combined extracts (C-S-P-M); precipitation: 6 volumes of 0.1 M ammonium acetate in MeOH; MeOH and acetone washing steps | 286 protein spots; 189 identified (107 plants, 72 microflora, 10 fauna | 2D PAGE; MALDI-TOF/TOF | Wang et al. (2011) | |
| 23 | Control soil, a fallow, Fujian, China | Roots uprooted, tightly connected soil | n.d. | n.d. | n.d. | n.d. | Dried at 70°C for 2 h; sieved < 2 mm | 1 g / 5 ml citrate buffer + 5 ml SDS buffer | SEM (C-S-P-M) 0.05 M citrate buffer pH 8.0; SDS buffer (1.25% w/v SDS, 0.1 M Tris-HCl, pH 6.8., 20 mM DTT) phenol purification (pH 8.0) precipitation: 6 volumes 0.1 M ammonium actetate in MeOH | 759 protein spots | 2D PAGE | Lin et al. (2013) | |
| 24 | Sugarcane after fallow, Fujian, China | See above (#19) | n.d. | n.d. | n.d. | n.d. | Dried at 70°C for 2 h; sieved <2 mm | 1 g / 5 ml citrate buffer + 5 ml SDS buffer | See above (#23) | 788 protein spots | 2D PAGE | Lin et al. (2013) | |
| 25 | Sugarcane and then ratooned, Fujian, China | See above (#19) | n.d. | n.d. | n.d. | n.d. | Dried at 70°C for 2 h; sieved < 2 mm | 1 g / 5 ml Citrate buffer + 5 ml SDS buffer | See above (#23) | 844 protein spots | 2D PAGE | Lin et al. (2013) | |
CONCEPTUALIZATION OF SOIL PROTEOMICS BY BASIC SOIL DATA
As soils of the globe are multifaceted, they are classified into groups based on their soil morphology, behavior or genesis in soil science. Due to their varying characteristics in multiple scales, a case-by-case evaluation of sample handling as well as protein extraction strategies (see also Fig. 1) are necessary for proper metaproteomics experiments, to ensure that the material extracted from the particular soil and/or site is representative for the entire soil community. Small differences in sample handling and preparation can introduce variability and may thereby dramatically alter the recovered species abundance and diversity to the measured data (Rubin et al.2013). To minimize artificially introduced variability, sample handling and preparation should involve as few steps as possible. In the following paragraphs, we will guide the reader step by step from the soil sampling to the analysis of metaproteomic data.
We believe that meta-omics studies of soil ecosystems should also provide contextual data such as soil pH, organic carbon (Corg), N-content, sampling depth, soil texture and CEC (for soils) (Table 1). As for instance these parameters might help to evaluate the potential of extracellular enzymes and, moreover, intracellular proteins that are released from the inner cells during extraction attaching to HS and mineral surfaces (for more details, see also Section ‘Sample matrix –interference of HS and physico-chemical parameters’). In addition, information on the study site including latitude and longitude, altitude, climate including mean annual temperature and precipitation, nutrient concentrations and bedrock material should be provided. Usually basic soil/environmental parameters obtained in a study are highly dependent on the hypotheses and the experimental design. However, for choosing an appropriate protein extraction, protocol knowledge on the before mentioned parameters (partly displayed in Table 1) is needed. As with any technique, metaproteomics ‘per se’ is not sufficient to provide comprehensive information on highly complex systems such soils. Hence, we need to implement additional data i.e. chemical background, soil history, microbial biomass and enzyme activities, to provide the basis to unravel the major biotic and abiotic drivers of the active abundant communities, not only for individual experiments but also for future, cross-biome meta-analysis.
Sample handling—sampling, homogenization and storage
The spatial and temporal heterogeneity of the soil matrix need to be considered by obtaining a representative sample of the natural situation for metaproteome analysis. So far, analysis of replicates in soil metaproteomic studies has been hampered by large costs and time-consuming analysis, resulting in numerous studies based on only one or few replicates (Myrold, Zeglin and Jansson 2014). As analysis costs per sample will drop, future studies should employ well-established soil sampling strategies and a larger number of biological and technical replicates. However, without giving any further details, such strategies might include sampling time, sample amount, sampling device, stratified sampling (horizontal and vertical distribution), composite samples (pooling) when appropriate (Pettitt and McBratney 1993) and/or apply a replicated sampling design (for details, see Boeddinghaus et al.2015). The individual sampling design is, however, dependent on the ecosystem type and the research objective. Soils are also strongly stratified horizontally with one or more organic horizons on top of mineral horizons, depending on soil type. These layers generally harbor the highest abundance of microbes and are also more prone to fluctuations in temperature and moisture compared to subsoil. Most metaproteomic studies thus focus on top soil horizons (0–15 cm; see Table 1).
Apart from the spatial variability, it is necessary to evaluate the seasonal impact or temporal variation, as environmental conditions such as aeration, nutrient diffusion and redox potential can vary strongly over time. While field conditions by definition include seasonal variation in a specific environment, these fluctuations can be reduced or controlled by changing only a few parameters in laboratory studies as has been demonstrated for soil (Bastida et al.2012b; Starke et al.2016) and leaf litter decomposition (Keiblinger et al.2012a).
Given the spatio-temporal variability of climatic and pedologic characteristics in the field scale that shape the active soil microbial community, we highlight the importance of measuring these covariables in metaproteomic studies as already mentioned before. Samples for soil metaproteome analysis are routinely sieved (<2 mm, see Table 1) to homogenize the sample and minimize contamination with plant and animal protein (Fig. 1A). High clay and/or moisture content, however, can inhibit effective sieving in which case removal of visible organic debris and sample homogenization has to be done manually. Homogenized soil samples are often stored until further processing. Several studies investigated the effect of storage conditions (mainly freezing and drying) on microbial parameters (Lee et al.2007; Wallenius et al.2010). Results suggest that responses to storage are strongly soil dependent (Bandick and Dick 1999) and seems to become more critical with increasing organic matter (OM) content (Lee et al.2007; Wallenius et al.2010). In previous metaproteomic, studies the chosen soil storage strategies (Fig. 1A) are summarized in Table 1, including air-drying, freeze-drying, freezing as well as deep freezing at –80°C and storage in RNA later. Unfortunately, OM content and texture of the soils processed are not always given (Table 1) hampering systematic investigations of storage conditions on soil metaproteomes. Processing fresh samples whenever possible or storage at –80°C is recommended to minimize the activity of naturally occurring proteases to avoid detrimental effects on protein abundance of environmental samples. This is supported by the findings from Hultman et al. (2015), who suggest active gene expression and translation even in permafrost soil where proteins can be preserved for long periods under subzero conditions. However, a detailed comparison of the influence of storage conditions in terms of temperature and time on the stability and activity of soil proteins is urgently needed.
Protein extraction: how to establish the optimal protocol
An optimal protein extraction protocol contains at least three important steps: (i) quantitative extraction of proteins from the environmental matrix (including steps for cell lysis, choice of buffer for solubilization and chemical reduction), (ii) protein purification (i.e. to remove lysed cellular debris, residual sample matrix, interfering chemical substances) and (iii) protein concentration (Fig. 1B).
Although a universal extraction protocol that provides good protein yields from wide range of soils would be desirable, this goal might be ‘certainly impractical’ given the heterogeneity of soil matrices (Becher et al.2013). Therefore, several protein extraction methods have been developed for specific research questions (Wang et al.2006; Benndorf et al.2007; Chourey et al.2010). As a first step towards standardization, some of these have been optimized and compared regarding their efficiency by our group (Keiblinger et al.2012b) and others (Nicora et al.2013; Bastida, Hernandez and Garcia 2014).
Direct protein extraction and cell lysis
Several studies aimed at extracting the entire protein complement of an environmental sample by employing different strategies such as (i) indirect extraction, where microbes become enriched prior to extraction (see Table 1, i.e #9, 10), (ii) separation by means of density gradient centrifugation (DGC) prior to protein extraction (to separate microorganisms from the environmental matrix, Table 1, i.e. #2, 7) and (iii) direct extraction (lysis in the environmental matrix, Table 1, i.e. #1, 3, 4). The first two options reduce or eliminate problems that derive from interfering substances such as HS or mineral surfaces (Bastida et al.2009; Giagnoni et al.2013), which can reduce extraction efficiency (Sander, Tomaszewski and Schwarzenbach 2011) but are confined by (i) focusing only on the cultivable fraction or (ii) strongly biased extractions (Bastida et al.2012).
However, direct extraction might lead to a more comprehensive protein recovery from bacteria, fungi, protozoa and multicellular organisms (Wohlbrand, Trautwein and Rabus 2013). Generally, direct extraction includes a direct cellular lysis step (Fig. 1B), which is obtained via (i) physical/mechanical lysis including heat, pressure (French press, sonication or bead milling using glass beads) (Mueller and Pan 2013), snap-freezing and grinding in liquid nitrogen with mortar and pistil; freeze-thaw cycles, (ii) chemical lysis (using detergents and stabilizing agents; Mueller and Pan 2013); or (iii) enzymatic lysis that involves lysozyme cleavage of gycosidic bondages. For the choice of cell lysis method, the target proteins and soil texture should be considered.
Physical cell rupture is usually more effective for Gram-negative bacteria, due to their thinner peptidoglycan layer compared to Gram-positive bacteria (Bakken and Frostegård 2006). Fungal lysis in soils samples can be obtained by bead beating or grinding in liquid N2 resulting in similar recoveries (van Elsas et al.2000). However, grinding is laborious; it might be also inefficient for sandy soils, as it is not possible to pulverize them with mortar and pistil. To this end, grinding seems to be most applicable for plant material, leaf litter and soils with high humic and low sand content or compost. Among physical procedures, sonication is a commonly used method for protein extraction from soils, as it favors the solubilization of stabilized proteins, and also breaks soil aggregates (Nannipieri 2006; Ogunseitan 2006).
Chemical methods use lysis buffers for cell disruption they include either ionic detergents or non-ionic detergents. Among ioinic detergents, anionic such as sodium dodecyl sulfate (SDS) or cationic such as ethylenediaminetetraacetic acid (EDTA) or zwitter ionic reagents such as CHAPS (3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate) are applied to dissolve cell membranes to release proteins. On the other hand, non-ionic detergents (i.e. Triton X-100, nonylphenoxypolyethoxyethanol (NP-40)) offer the advantage that proteins are not denatured, by still solubilizing membrane proteins. Although detergents such as EDTA also inhibit polyphenol oxidases and metalloproteases, by building complexes with metal ions, β-mercaptoethanol is often added to soil protein extraction buffers as a reducing agent, as it prevents oxidation of proteins.
Alternatively, enzymes can either be used alone or in combination with chemicals and/or physical means to lyse cells (Gianfreda and Rao 2014). A combination of mild mechanical methods (i.e. sonication) in detergents (i.e. SDS) with other additives, such as enzymes and/or protease inhibitors cocktails, is a good strategy for direct cell lysis in soil samples, depending on the target cells and soil type and further downstream processing.
Sample matrix—interference of HS and physico-chemical parameters
Basic knowledge of soil and environmental characteristics might aid the choice of an extraction procedure appropriate for the research question. Thus, it will be at least possible to evaluate which challenges during protein extraction can be expected (such as high humus content or clay-rich soils with high CEC) and to adopt existing protocols that provided promising results on similar soils, in comparable habitats. However, these parameters should not be taken individually, as clay and OM are often well related with HS because clays retard the decomposition of OM (Nannipieri 2006).
Together with the aforementioned cell lysis, the extraction buffer should often meet the requirements for the removal of HS and/or to target stabilized proteins. Specifically, salt solutions (i.e. CaCl2) of inorganic divalent cations (10–100 mM) have been used to release naturally immobilized proteins from HS by desorption (Criquet, Farnet and Ferre 2002) from HS. The extraction buffer often contains polyvinylpolypyrrolidone (PVPP) and hexadecyltrimethylammonium bromide (CTAB) because they form complexes with humic acids. Stabilized enzymes are efficiently extracted with buffers at slightly alkaline conditions (Nannipieri 2006). This illustrates the importance of the pH of the soil and the extraction buffer, as it governs sorption of proteins to minerals and removal of interfering substances, and it also influences protein structure (Bastida et al.2009). The pH of the extraction buffer has a strong influence on cell extraction, and considerably increases with pH in the range from 5 to 8 (Bakken and Frostegård 2006). Therefore, for direct extraction of soils, a pH of 7 or somewhat higher should result in sufficient amount of cells. To achieve alkaline conditions, a weak NaOH or buffers adjusted to 7.5–8.5 can be used. NaOH (Benndorf et al.2007) or alkaline pyrophosphate (Masciandaro et al.2008) supplemented extraction buffers desorb proteins bound covalently to clay particles. However, with high pH the yield of HS also increases. Alternatively, a subsequent phenolic extraction protocol has been used (Wang et al.2006; Benndorf et al.2007; Chen, Rillig and Wang 2009; Keiblinger et al.2012b) to separate proteins from HS. This phenol including extraction preferentially dissolves nucleic acids, carbohydrates and cell debris in the aqueous phase, while proteins and lipids are contained in the phenolic phase. The application to samples that contain interfering compounds resulted in more protein bands or spots on the gels and less proteolysis, and also downstream processing including bioinformatic analysis resulted better results for phenol-extracted proteins for plant tissue (Pavoković, Križnik and Krsnik-Rasol 2012). The major drawbacks of phenol-based extractions are the corrosivity and toxicity of the chemical, and the time intensive extraction with the phase separation. To ease the phase separation, the addition of sucrose pushes the phenol phase to the top and facilitates recovery (Faurobert, Pelpoir and Chaïb 2007).
The former shows already that a combination of strategies can be useful for sufficient protein yields from soils. Similarly, Nicora et al., (2013) suggested to combine the use of desorption buffers and positive polar amino acids that bind to the sorption sites of the soil prior to cell lysis. This strategy might be useful for silty and clayey soil, soils that are characterized by a high CEC.
Beside the choice of extraction buffer, the potential steps for getting rid of HS are based on physico-chemical separation principles. These strategies can be easily applied with various protein extraction buffers either before (using PVPP during grinding in liquid nitrogen; Keiblinger et al.2012b) and/or after cell lysis. Proteins and HS can be fractionated by size, using gel filtration raisins (Sepharose 4B, Sephadex or Sephacryl) or ultrafiltration with spin filters (10 KMWCO cut off), Fig. 1B. Columns packed with PVPP (Kabir et al.2003; Masciandaro et al.2008) as well as commercial ones are used to separate HS from proteins by the aid of different binding abilities to a polymeric matrix. The precipitation of HS by AlNH4(SO4)2 has to our knowledge not been used for the extraction of proteins from soil so far, but might be a potential solution (Braid, Daniels and Kitts 2003). Electrophoresis separates proteins based on their molecule size and charge density (Fig. 1C). Elimination of coextractants consequently may also reduce target proteins; to this end, recoveries of extraction should be monitored during all extractions by adding a standard protein spike to evaluate the extraction efficiency.
There are several factors that might affect protein yields during extraction (i.e. cell lysis, pH and detergents of the extraction buffer, denaturation agents and application of phenol and precipitation method, Table 1). Table 1 lists different extraction protocols for forest soils, agricultural soils and rhizosphere soils together with soil physicochemical parameters such as pH, CEC, organic C, N content, soil texture, extraction strategy applied, extracted protein concentration or number of proteins (spots) or (if applicable) assigned proteins. Owing to the complexity of the soil matrix, the reader would not be surprised that a unified extraction protocol for soils cannot be recommended at the moment. Although some suggestions are given above, based on the strong variation of conditions for sample handling and extraction, and further downstream processing as well as samples from strongly differing biomes given in Table 1, it is not even possible for agricultural and forest soils.
Extraction of the subcellular proteomes
The entire proteome of a microorganism consists of all its extracellular, cytoplasmic and membrane proteins. Many extracellular proteins have successfully been recovered from cultures grown on leaf litter (Schneider et al.2010). While studies on leaf litter (Keiblinger et al.2012a; Schneider et al.2012) aimed at capturing the entire metaproteome, these analyses include information on the extracellular fraction recovered by the extraction with SDS buffer (extraction conditions recently reviewed by Becher et al.2013). In contrast to leaf litter, the complexity of the soil matrix (Vos et al.2013) complicates the targeted extraction of extracellular proteins. Extracellular enzymes are often reached by indirect extraction or prior washing, as soil washing releases cells from the soil matrix. However, this step introduces another level of uncertainty as stabilized enzymes are not reached. In general, it should be mentioned that alkaline conditions are unfavorable for extraction of extracellular enzymes as cell lysis can occur, thereby including untargeted intracellular proteins. Extracellular proteins have been isolated from a greenhouse soil and forest soils, using extraction buffers containing phosphate (Murase et al.2003; Masciandaro et al.2008) at pH 6 (see also Table 1).
In a recent study, Bastida, Hernandez and Garcia (2014) found that Chourey's method (2010) was better suited than Singleton's (2003) to recover more extracellular proteins in metaproteomics from forest and agricultural soils.
Concentration of proteins
After extraction, it is often necessary to concentrate proteins (Fig. 1B) as amplification of low-abundant proteins is not possible (in contrast, e.g. DNA amplification via PCR). To this end, proteins can either be concentrated by reducing the sample volume (through freeze drying, heating, ultrafiltration or by vacuum centrifugation; Criquet, Farnet and Ferre 2002) by dialysis or desalting methods (Ogunseitan 2006); however, most commonly in soil metaproteomics is precipitation (Chourey et al.2010; Keiblinger et al.2012b) followed by a washing step and resolubilization (Fig. 1B). While reducing sample volume can also increase the concentration of interference compounds (i.e. humics), precipitation includes purification from undesirable substances. For soil protein extracts, most often trichloroacetic acid (TCA) or methanol–ammonium acetate precipitation (Table 1) is employed to concentrate proteins. TCA precipitation is achieved by changing the pH, and reducing the solubility of proteins in solution. In contrast, methanol–ammonium acetate precipitation in methanol combines salt-induced precipitation and organic solvents. Although adding a 4-fold amount of methanol efficiently precipitates most proteins, adding an organic base, ammonium acetate, increases yields for acidic solutions.
While TCA is known to be an efficient precipitation agent for soil proteins extracted with SDS buffers, it has several disadvantages. Among them are (i) a potential loss of large proteins (Carpentier et al.2005); (ii) the coprecipitation of interfering substances such as DNA and protein-DNA aggregates (Pavoković, Križnik and Krsnik-Rasol 2012); (iii) protein pellets need to be washed with acetone or a base to remove the remaining acid from the proteins; (iv) the risk that proteins are non-functional afterwards, which is problematic with 2DE; (v) and finally TCA precipitated proteins are difficult to re-solubilized, here preferentially small proteins are redissolved (Carpentier et al.2005). Methanol–ammonium acetate precipitation is often used in combination with phenol-based extraction procedures (Carpentier et al.2005; Benndorf et al.2007; Pavoković, Križnik and Krsnik-Rasol 2012), and might be more suitable for soils with large amounts of HS.
As mentioned above, rehydration of precipitated proteins is sometimes problematic as the protein pellets do not dissolve well. For this a variety of buffers (i.e. guanidine buffer, SDS sample buffer) can be applied; for more details, see Table 1. Rehydration buffers containing chaotropes (typically urea and thiourea) might improve protein yields (Weiss and Görg 2008).
Prior to further processing, the evaluation of the protein concentration is helpful. As most colorimetric assays such as Bradford (Whiffen, Midgley and Mcgee 2007) interfere with HS, Roberts and Jones (2008) suggested that total protein concentrations should be determined by acid hydrolysis followed by amino acid measurements. This strategy has been successfully applied recently in soil metaproteomics (Bastida, Hernandez and Garcia 2014).
Processing of soil protein extracts—complexity reduction and MS approaches
The complexity of environmental samples still outstrips the capabilities of state-of-the-art MS approaches. Thus, separation of proteins/peptides is mandatory to reduce sample complexity before MS analysis (Fig. 1C). In early (soil) metaproteomic studies, 2D gel-based protein separation methods were successfully employed (Klaassens, de Vos and Vaughan 2007; Benndorf et al.2007; Wilmes, Wexler and Bond 2008). However, this technology has major drawbacks, particularly regarding the analysis of proteins with extreme molecular weights, isoelectric points or hydrophobicity values. These restrictions were relaxed by 1D gel-based or gel-free fractionation methods. Gel-free approaches include different protein extraction procedures, followed by in-solution digestion to peptides. Peptides are further separated by reversed-phase RP-LC or a chromatographic separation in two dimensions using strong cation exchange chromatography in combination with RP-LC.
Proteins separated by gels can be enzymatically digested in-gel while gel-free approaches take advantage of in-solution or on-filter protein digestion (Fig. 1C). Identification rate of particularly low-abundant proteins after 1D gel-based fractionation can be improved by normalizing the size of fractions (gel pieces) to the contained protein amount (Yin et al.2015). Weston, Bauer and Hummon (2013) showed that filter-based digestion resulted in an 18% higher protein identification rate compared to in-solution digestion, which might be due to an additional denaturating protein solubilization step. The advantage of a gel-based fractionation is the combination of protein denaturation and separation, while it is more time consuming than gel-free fractionation that benefits from reduced processing time, and therefore has a greater high-throughput potential. One of the most frequently used strategies in such proteomic experiments is tandem MS of peptides after enzymatic protein digestion.
FROM DATA TO UNDERSTANDING
MS analysis is followed by subsequent correlation of resulting spectra with those of theoretic peptides from a given protein database (protein DB or target DB) (Eng, McCormack and Yates 1994; Yates et al.1995). Due to its high efficiency and degree of automation, this approach evolved to the preferred strategy for protein identification, quantification and detection of chemical peptide modifications in large-scale soil metaproteomic studies (Aebersold and Mann 2003). However, this approach does not allow direct protein identifications but is based on two matching steps: (i) matching the experimental spectra to theoretical spectra obtained from a given protein DB after in silico digestion and (ii) inferring the original proteins based on the resulting peptide-to-spectrum matches (PSMs). Thus, only protein sequences represented in the target DB can be identified (Fig. 2A). Alternatively, spectral libraries can be used to correlate experimental spectra directly with identified reference spectra (Fig. 2A). These reference spectra have to meet high-quality criteria and, thus, their generation is costly and not practicable in dimensions demanded by metaproteomics. However, high-quality spectra can be used as a reference even if they are identified not yet. Tools such as ScanRanker support selection of unidentified high-quality spectra by automatic routines (Ma et al.2011) whose occurrence can be then followed across different samples and ecosystems (Muth et al.2013). Promising spectra can be then submitted to de novo sequencing (Hughes, Ma and Lajoie 2010).
Data analyses
Spectra handling and database assembly
As mentioned before, correlating experimental spectra with theoretic spectra of peptides from a given protein DB is the most frequently used proteomics approach. Quality and performance of spectra correlation crucially depend on the size of the search space that is defined by both (i) the number of recorded spectra to compare and (ii) the number of theoretic or reference spectra compared to. An increased search space inevitably leads to an increase in (i) computational costs, (ii) potential of false positives (or false negatives) and (iii) frequency of PSMs matching to two or more proteins.
To reduce the number of spectra submitted to further analyses, effective filtering and clustering algorithms can be employed (Fig. 2A) (Flikka et al.2006; Ding, Shi and Wu 2009; Zou et al.2009; Lin et al.2012). Redundant spectra can be clustered into metaspectra to further reduce the number of spectra to correlate that positively affects not only false discovery rates (FDR) but also analysis speed (Flikka et al.2006; Frank et al.2008; Saeed, Hoffert and Knepper 2014). Thus, protein DB selection plays a pivotal role in metaproteomics (Tanca et al.2013). A customized protein DB is ideally assembled based on a matched full metagenome from the same sample as analyzed by metaproteomics. By this, optimal identification rates can be achieved as previously shown (Morris et al.2010). Alternatively, the taxonomic sample composition revealed by 16S and/or 18S RNA sequencing data can be used to deduce the pseudo-metagenome. Using a six-frame translation of the metagenome sequence produces more complex protein DBs, but can be helpful to increase the metaproteome coverage. Finally, unmatched metagenome data can be also successfully used for protein DB assembly as previously shown (Verberkmoes et al.2008). Here the greatest difficulty is the selection of customized subcollections from public resources since it has to be based on assumptions on the metaproteome composition. Thus, resulting protein DBs are generally large (>>106 sequences). To overcome this, an iterative DB search method that uses matches from a primary DB search to assemble a customized database of reduced size has previously been proposed (Jagtap et al.2013). This example shows a reduction in DB size to <0.1% of the original size. Tanca et al. (2013) evaluated the impact of different protein database types, one based on matched metagenome data and another one based on sequences of expected genes from TrEMBL. An interesting yet alarming result was that an overlap of only 36% of all identified peptides was found when using both protein DBs.
Peptide identification
There are various algorithms available to compare experimental and theoretic peptide fragmentation spectra, the computational basics of which are comprehensibly described elsewhere (Colinge and Bennett 2007). All have in common that they produce multiple testing effects increasing the number of wrongly accepted PSMs. The proportion of false positives can be controlled by the FDR (Benjamini and Hochberg 1995). Meanwhile, various methods for FDR assessment have been entered in metaproteomics analyses (Nesvizhskii 2010). For instance, target-decoy DBs composed of all protein sequences in forward (target) and reverse (decoy) direction have been applied as an easy and powerful method (Elias and Gygi 2007). However, this strategy leads to a doubling of the target DB size that in turn increases the search space (see above). With Percolator, a semi-supervised machine learning algorithm trained by scrambled decoy peptides and best scoring target peptides is available (Kall et al.2007; Spivak et al.2009). Combined with accurate scoring functions for PSM, the use of this approach can increase the number of peptide identifications in a variety of data sets as previously shown (Granholm et al.2014; Howbert and Noble 2014).
Protein identification and clustering
Inferring proteins (Fig. 2A) from the list of identified peptides can be surprisingly difficult. Nesvizhskii and Aebersold coined the term ‘protein inference problem’ and provided a statistical model for MS-based protein identification (Nesvizhskii et al.2003; Nesvizhskii and Aebersold 2005). Distinct protein identifications need at least one identified peptide that uniquely maps to the respective protein. The proportion of unique peptides drops with an increasing number of closely related organisms considered by the target DB, which complicates soil metaproteome data analyses. Meanwhile, there are several approaches to calculate probabilities of identified proteins (Higdon and Kolker 2007; Serang and Noble 2012; Shi and Wu 2012; Yang, He and Yu 2013). However, it should be noted that protein probabilities are experiment specific since they correlate with factors such as spectra number, protein DB size and protein abundances (Xue et al.2006). To ease protein inference, peptides can be attributed exclusively to the protein with the highest probability. This strategy is followed by the Scaffold software (Searle 2010), for instance. An alternative approach is provided by ProteomeDiscoverer (Thermo Scientific, Waltham, Massachusetts, USA) assigning peptides to all possible proteins matching the quality criteria, and a combination of DB searches and de novo sequencing is provided to maximize metaproteome coverage. However, at least peptides matching to proteins with equal probabilities cannot be uniquely attributed. Koskinen et al. (2011) introduced a hierarchical protein clustering approach by means of those shared peptide matches. Peptide-sharing proteins are grouped together and represented by a single anchor protein. However, at this time, this approach is beneficial rather for single-organism proteomics than metaproteomics where resulting clusters can be taxonomically and functionally diverse.
Data interpretation
Protein quantification
The knowledge about the abundance of proteins is essential for a systems biological perspective on microbial consortia. Various technologies have been established to assess whole protein inventories (von Bergen et al.2013; Otto, Becher and Schmidt 2014). However, only a few are applicable in a scale needed for environmental proteomics. Using 1D gel-based or gel-free approaches, protein amounts can be estimated based on spectral counts. Normalized spectral abundance factors (NSAF) account for protein length and sample-to-sample variation (Zybailov et al.2006). An improved approach (distributed normalized spectral abundance factors or dNSAF) considers shared peptides by distributing shared spectral counts based on the number of unique spectral counts (Fig. 2B) (Zhang et al.2010; McIlwain et al.2012). The application of metabolic labeling in environmental proteomics is hindered by the fact that the metabolic label has to be provided in sufficient amounts.
Functional and taxonomic assessment
In contrast to metagenomics, metaproteomics provides insights into the metabolically active species and their metabolic performance within the analyzed microbial consortium or ecosystem. However, the vast mass of data provided by metaproteome analyses complicates data interpretation (Fig. 2B). For both functional and taxonomic analyses (which should ideally be combined), quality of protein annotation is crucial and should be considered already during protein DB assembly. Several online resources provide expertly curated data sets for a high number of proteins (e.g. SWISSPROT, RefSeq) (Table 2). However, two major problems persist—(i) limited (functional) annotation standards and (ii) missing global (DB-independent) sequence identifiers—which both considerably complicate meta-physiological research. Thus, approaches to globalize sequence identifiers (e.g. SEGUID; Babnigg and Giometti 2006) or to classify functions (e.g. TIGR role categories; Haft et al.2013) are urgently needed. For metabolic pathway analyses, different repositories provide functional categories and corresponding profiles. With the Cluster of Orthologous Groups, a widely distributed classification system is available for prokaryotic (COG) and eukaryotic (KOG) proteins (Tatusov et al.2003; Koonin et al.2004). However, this system has not been updated since 2003; therefore, eggNOG as actively curated derivate can be recommended (Powell et al.2014). With TIGRFAMs and PFAMs, expertly curated Hidden Markov Models based on multiple sequence alignments of proteins fulfilling the same function are available (Haft et al.2013; Finn et al.2014). Combined with TIGR roles, an excellent classification system organized in (i) main roles (e.g. energy metabolism), (ii) subroles (e.g. glycolysis) and (iii) functions (e.g. enolase) is provided (Fig. 3). Specific metabolic functions might be underrepresented in general collections. Considering data from resources specialized to distinct protein functions can support detailed analyses on specific activities. A prominent example for such specialized resources is CAZY (http://www.cazy.org/) (Cantarel et al.2009, Lombard et al.2014) listing more than 330 families of carbohydrate-active enzymes that have been already successfully employed in several environmental studies to estimate the amount of polymer-degrading enzymes (Aylward et al.2012; López-Mondéjar et al.2016). For metabolic pathway reconstruction, different repositories such as KEGG, BiGG or BioCyc are available (Schellenberger et al.2010; Caspi et al.2014; Kanehisa et al.2014).
Table 2.
Number of annotated protein sequences provided by UniProt and NCBI (as of 28 January 2015).
| Protein sequences | |||||
|---|---|---|---|---|---|
| Resource/section | Archaea | Bacteria | Eukaryotes | Viruses | Totala |
| UniProtKBb | |||||
| TrEMBL | 888 257 | 73 062 005 | 12 775 469 | 2171 639 | 89 451 166 |
| SwissProtc | 19 312 | 331 887 | 179 679 | 16 479 | 547 357 |
| NCBId | |||||
| Protein | 2137 968 | 125 291 208 | 26 123 069 | 2760 918 | 163 229 525 |
| RefSeqe | 1094 656 | 42 822 180 | 9709 585 | 213 314 | 53 839 396 |
Including unclassified and other sequences.
The Universal Protein Resource Knowledgebase (http://www.uniprot.org).
Biologically non-redundant, expertly curated annotation.
The National Center of Biotechnology Information (http://www.ncbi.nlm.nih.gov), as of 19 February 2013.
Biologically non-redundant, annotation partially curated by experts.
Figure 3.

Voronoi Treemaps. Voronoi treemaps can visualize highly complex hierarchically organized data in a space optimized manner. Here, functional classification of TIGRFAMs (Release 15.0) is depicted based on TIGR roles main (left) and (right) subclasses.
Based on standardized taxonomic annotation, proteins in peptide sharing clusters can be reduced to the lowest common anchor (LCA) (Fig. 2B). The Unipept web application provides a robust LCA approach considering all occurrences of identified tryptic peptides in UniprotKB. Alternatively, Pipasic estimates the peptide level similarity between reference proteomes allowing differentiation on strain level. The PROPHANE web service provides a combined fully automated workflow for both (i) functional analyses using various resources (COG/KOG, TIGRFAMs and PFAMs) and (ii) LCA-based taxonomic assessment (www.prophane.de) (Schneider et al.2011). In addition, MetaProteomeAnalyzer software is a tool that features four freely available DB search algorithms (X!Tandem, OMSSA, Crux, InsPect), and is also highly suitable for comprehensive analysis and visualization of metaproteomic datasets (https://code.google.com/archive/p/meta-proteome-analyzer/) (Muth et al.2015).
Data storage and visualization
For several reasons, data storage is a major issue in metaproteomics. The generated data take valuable space and are barely standardized that both makes data handling and integration difficult (Jimenez and Vizcaino 2013). Meanwhile, several commercial (Stephan et al.2010) and open-source (Perez-Riverol et al.2014) in-house solutions exist. However, at least after publication spectral data should be made publicly available making online repositories such as PRIDE, PeptideAtlas and Tranche (for review, see Jimenez and Vizcaino 2013). Furthermore, the enormous progress of analytical tools and the tremendous increase of available protein sequences require non-traditional data storage for keeping the data in an active state. Thus, data storage should be never the end of the analysis pipeline but much more the beginning of a new improved analysis circle (see also Muth et al.2013).
The complexity of metaproteome data demands for sophisticated visualization strategies. Different approaches have been comprehensively reviewed recently (Mehlan et al.2013). Voronoi treemaps have proven to be an excellent tool to visualize hierarchical data structures in a space optimized manner (Fig. 3). Two additional dimensions (such as protein amounts or ratios) can be projected using area and/or color encoding. Stream graphs allow even one more dimension and are, thus, perfectly suited for time courses. However, there are two major drawbacks. First, biological data cannot be always reduced to a non-redundant hierarchical organization. For instance, proteins can have more than one function and, thus, have multiple places in a treemap. Second, with increasing data complexity, the human eye is overtaxed, particularly when viewing print media where space and resolution is limited.
CONCLUDING REMARKS
There are several important steps that must be carefully planned when employing soil metaproteome analysis. First, the sampling strategy must be well considered to cover the spatial and temporal heterogeneity of (i) the soil matrix and (ii) the microbial community that varies in diversity, size, generation time, functions and favored soil physical and chemical conditions. Second, an optimal sample handling procedure has to be established and should be discussed within the scientific community to generate comparable data for meta-metanalysis. Studies that compare storage conditions for soil and leaf litter from a wide variety of climates are still missing, but would be highly useful. We have reviewed the application of different extraction protocols for proteins present in soil and litter, and how soil characteristics may influence the protein extraction. However, it is important to mention that protein extraction methods need to be further explored and improved. In particular, more emphasis in the identification of extracellular proteins is required, as those are directly linked to biogeochemistry processes. So far dynamic succession of soil and leaf litter microbial populations, including their community structure and respective functions, are poorly investigated. In this regard, metaproteomics allows the untargeted assignment of proteins involved in a broad variety of biochemical processes. We thus expect that environmental metaproteomics, so far a mainly descriptive approach, will significantly contribute to hypothesis-driven research aiming at a deeper understanding of the highly complex metabolic network and multispecies interactions in terrestrial habitats. Subsequent research aiming to develop sophisticated bioinformatic tools constantly facilitates the application of metaproteomics even in such complex habitats such as soil and leaf litter and will be a central prerequisite for the hypothesis-driven evaluation of metaproteome data. The power of metaproteomics can even be further enhanced, when combined or complemented with other ‘omics’ technologies, i.e. metagenomics and metatranscriptomics and also classical soil analytics such as microbial biomass, potential enzyme activities and physico-chemical indicators. Given the environmental challenges facing society today, the need for in-depth understanding of soil functioning is critical. This review therefore concludes that the continued and increased application of soil metaproteomes within holistic ecosystem experimental frameworks constitutes a research priority.
Acknowledgments
Special thanks to Bradley Matthews for native English proof reading of the manuscript, and two anonymous reviewers for their valuable comments. The authors also thank Sonja Leitner and Stefan Forstner for comments on earlier drafts on the manuscript. The authors acknowledge the European Science Foundation (ESF) for a ClimMani exchange grant [#5306] for KMM.
FUNDING
This work was supported by the Fund FWF [] and the Program [].
Conflict of interest.None declared.
REFERENCES
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Arenella M, Giagnoni L, Masciandaro G, et al. Interactions between proteins and humic substances affect protein identification by mass spectrometry. Biol Fert Soils. 2014;50:447–54. [Google Scholar]
- Aylward FO, Burnum KE, Scott JJ, et al. Metagenomic and metaproteomic insights into bacterial communities in leaf-cutter ant fungus gardens. ISME J. 2012;6:1688–701. doi: 10.1038/ismej.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babnigg G, Giometti CS. A database of unique protein sequence identifiers for proteome studies. Proteomics. 2006;6:4514–22. doi: 10.1002/pmic.200600032. [DOI] [PubMed] [Google Scholar]
- Bakken LR, Frostegård Å. Nucleic acid extraction from soil. In: Nannipieri P, Smalla K, editors. Nucleic Acids and Proteins in Soil. Vol. 8. Berlin, Heidelberg: Springer; 2006. pp. 49–73. [Google Scholar]
- Bandick AK, Dick RP. Field management effects on soil enzyme activities. Soil Biol Biochem. 1999;31:1471–9. [Google Scholar]
- Barberán A, Bates ST, Casamayor EO, et al. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012;6:343–51. doi: 10.1038/ismej.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastida F, Algora C, Hernandez T, et al. Feasibility of a cell separation-proteomic based method for soils with different edaphic properties and microbial biomass. Soil Biol Biochem. 2012a;45:136–8. [Google Scholar]
- Bastida F, García C, von Bergen M, et al. Deforestation fosters bacterial diversity and the cyanobacterial community responsible for carbon fixation processes under semiarid climate: a metaproteomics study. Appl Soil Ecol. 2015a;93:65–7. [Google Scholar]
- Bastida F, Hernandez T, Garcia C. Metaproteomics of soils from semiarid environment: Functional and phylogenetic information obtained with different protein extraction methods. J Proteomics. 2014;101:31–42. doi: 10.1016/j.jprot.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Bastida F, Jehmlich N, Lima K, et al. The ecological and physiological responses of the microbial community from a semiarid soil to hydrocarbon contamination and its bioremediation using compost amendment. J Proteomics. 2016;135:162–9. doi: 10.1016/j.jprot.2015.07.023. [DOI] [PubMed] [Google Scholar]
- Bastida F, Jindo K, Moreno JL, et al. Effects of organic amendments on soil carbon fractions, enzyme activity and humus–enzyme complexes under semi-arid conditions. Eur J Soil Biol. 2012b;53:94–102. [Google Scholar]
- Bastida F, Moreno JL, Hernandez T, et al. Microbiological degradation index of soils in a semiarid climate. Soil Biol Biochem. 2006;38:3463–73. [Google Scholar]
- Bastida F, Moreno JL, Nicolas C, et al. Soil metaproteomics: a review of an emerging environmental science. Significance, methodology and perspectives. Eur J Soil Sci. 2009;60:845–59. [Google Scholar]
- Bastida F, Selevsek N, Torres IF, et al. Soil restoration with organic amendments: linking cellular functionality and ecosystem processes. Sci Rep. 2015b;5:15550. doi: 10.1038/srep15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher D, Bernhardt J, Fuchs S, et al. Metaproteomics to unravel major microbial players in leaf litter and soil environments: challenges and perspectives. Proteomics. 2013;13:2895–909. doi: 10.1002/pmic.201300095. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B-Met. 1995;57:289–300. [Google Scholar]
- Benndorf D, Balcke GU, Harms H, et al. Functional metaproteome analysis of protein extracts from contaminated soil and groundwater. ISME J. 2007;1:224–34. doi: 10.1038/ismej.2007.39. [DOI] [PubMed] [Google Scholar]
- Blum WEH, Busing J, Montanarella L. Research needs in support of the European thematic strategy for soil protection. Trend Anal Chem. 2004;23:680–5. [Google Scholar]
- Boeddinghaus RS, Nunan N, Berner D, et al. Do general spatial relationships for microbial biomass and soil enzyme activities exist in temperate grassland soils? Soil Biol Biochem. 2015;88:430–40. [Google Scholar]
- Braid MD, Daniels LM, Kitts CL. Removal of PCR inhibitors from soil DNA by chemical flocculation. J Microbiol Meth. 2003;52:389–93. doi: 10.1016/s0167-7012(02)00210-5. [DOI] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier SC, Witters E, Laukens K, et al. Preparation of protein extracts from recalcitrant plant tissues: an evaluation of different methods for two-dimensional gel electrophoresis analysis. Proteomics. 2005;5:2497–507. doi: 10.1002/pmic.200401222. [DOI] [PubMed] [Google Scholar]
- Caspi R, Altman T, Billington R, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014;42:D459–71. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SN, Rillig MC, Wang W. Improving soil protein extraction for metaproteome analysis and glomalin-related soil protein detection. Proteomics. 2009;9:4970–3. doi: 10.1002/pmic.200900251. [DOI] [PubMed] [Google Scholar]
- Chourey K, Jansson J, VerBerkmoes N, et al. Direct Cellular Lysis/Protein Extraction Protocol for Soil Metaproteomics. J Proteome Res. 2010;9:6615–22. doi: 10.1021/pr100787q. [DOI] [PubMed] [Google Scholar]
- Colinge J, Bennett KL. Introduction to computational proteomics. Plos Comput Biol. 2007;3:e114. doi: 10.1371/journal.pcbi.0030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criquet S, Farnet AM, Ferre E. Protein measurement in forest litter. Biol Fert Soils. 2002;35:307–13. [Google Scholar]
- Delgado-Baquerizo M, Giaramida L, Reich PB, et al. Lack of functional redundancy in the relationship between microbial diversity and ecosystem functioning. J Ecol. 2016;104:936–46. [Google Scholar]
- Ding J, Shi J, Wu FX. Model based clustering for tandem mass spectrum quality assessment. Conf Proc IEEE Eng Med Biol Soc. 2009;2009:6747–50. doi: 10.1109/IEMBS.2009.5332499. [DOI] [PubMed] [Google Scholar]
- Doyle RJ. Contribution of the hydrophobic effect to microbial infection. Microbes Infect. 2000;2:391–400. doi: 10.1016/s1286-4579(00)00328-2. [DOI] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–89. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Faurobert M, Pelpoir E, Chaïb J. Phenol extraction of proteins for proteomic studies of recalcitrant plant tissues. Methods Mol Biol. 2007;355:9–14. doi: 10.1385/1-59745-227-0:9. [DOI] [PubMed] [Google Scholar]
- Fierer N, Lauber CL, Ramirez KS, et al. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012a;6:1007–17. doi: 10.1038/ismej.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, Leff JW, Adams BJ, et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. P Natl Acad Sci USA. 2012b;109:21390–5. doi: 10.1073/pnas.1215210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Bateman A, Clements J, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flikka K, Martens L, Vandekerckhove J, et al. Improving the reliability and throughput of mass spectrometry-based proteomics by spectrum quality filtering. Proteomics. 2006;6:2086–94. doi: 10.1002/pmic.200500309. [DOI] [PubMed] [Google Scholar]
- Frank AM, Bandeira N, Shen Z, et al. Clustering millions of tandem mass spectra. J Proteome Res. 2008;7:113–22. doi: 10.1021/pr070361e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giagnoni L, Magherini F, Landi L, et al. Extraction of microbial proteome from soil: potential and limitations assessed through a model study. Eur J Soil Sci. 2011;62:74–81. [Google Scholar]
- Giagnoni L, Migliaccio A, Nannipieri P, et al. High montmorillonite content may affect soil microbial proteomic analysis. Appl Soil Ecol. 2013;72:203–6. [Google Scholar]
- Gianfreda L, Rao MA. Enzymes in Agricultural Sciences. OMICS Group International; 2014. [Google Scholar]
- Granholm V, Kim S, Navarro JC, et al. Fast and accurate database searches with MS-GF+Percolator. J Proteome Res. 2014;13:890–7. doi: 10.1021/pr400937n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnigle E, Ramond JB, Frossard A, et al. A sequential co-extraction method for DNA, RNA and protein recovery from soil for future system-based approaches. J Microbiol Meth. 2014;103:118–23. doi: 10.1016/j.mimet.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Haft DH, Selengut JD, Richter RA, et al. TIGRFAMs and genome properties in 2013. Nucleic Acids Res. 2013;41:D387–95. doi: 10.1093/nar/gks1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higdon R, Kolker E. A predictive model for identifying proteins by a single peptide match. Bioinformatics. 2007;23:277–80. doi: 10.1093/bioinformatics/btl595. [DOI] [PubMed] [Google Scholar]
- Howbert JJ, Noble WS. Computing exact p-values for a cross-correlation shotgun proteomics score function. Mol Cell Proteomics. 2014;13:2467–79. doi: 10.1074/mcp.O113.036327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes C, MA B, Lajoie GA. De novo sequencing methods in proteomics. Methods Mol Biol. 2010;604:105–21. doi: 10.1007/978-1-60761-444-9_8. [DOI] [PubMed] [Google Scholar]
- Hultman J, Waldrop MP, Mackelprang R, et al. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature. 2015;521:208–12. doi: 10.1038/nature14238. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Goslinga J, Kooren JA, et al. A two-step database search method improves sensitivity in peptide sequence matches for metaproteomics and proteogenomics studies. Proteomics. 2013;13:1352–7. doi: 10.1002/pmic.201200352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Huang Q, Cai P, et al. Adsorption of Pseudomonas putida on clay minerals and iron oxide. Colloid Surface B. 2007;54:217–21. doi: 10.1016/j.colsurfb.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Jimenez RC, Vizcaino JA. Proteomics data exchange and storage: the need for common standards and public repositories. Methods Mol Biol. 2013;1007:317–33. doi: 10.1007/978-1-62703-392-3_14. [DOI] [PubMed] [Google Scholar]
- Kabir S, Rajendran N, Amemiya T, et al. Quantitative measurement of fungal DNA extracted by three different methods using real-time polymerase chain reaction. J Biosci Bioeng. 2003;96:337–43. doi: 10.1016/S1389-1723(03)90133-2. [DOI] [PubMed] [Google Scholar]
- Kall L, Canterbury JD, Weston J, et al. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4:923–5. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiblinger KM, Liu D, Mentler A, et al. Biochar application reduces protein sorption in soil. Org Geochem. 2015;87:21–4. [Google Scholar]
- Keiblinger KM, Schneider T, Roschitzki B, et al. Effects of stoichiometry and temperature perturbations on beech leaf litter decomposition, enzyme activities and protein expression. Biogeosciences. 2012a;9:4537–51. [Google Scholar]
- Keiblinger KM, Wilhartitz IC, Schneider T, et al. Soil metaproteomics—comparative evaluation of protein extraction protocols. Soil Biol Biochem. 2012b;54:14–24. doi: 10.1016/j.soilbio.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller M, Hettich R. Environmental proteomics: a paradigm shift in characterizing microbial activities at the molecular level. Microbiol Mol Biol R. 2009;73:62–70. doi: 10.1128/MMBR.00028-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy N, Brodie E, Connolly J, et al. Impact of lime, nitrogen and plant species on bacterial community structure in grassland microcosms. Environ Microbiol. 2004;6:1070–80. doi: 10.1111/j.1462-2920.2004.00638.x. [DOI] [PubMed] [Google Scholar]
- Klaassens ES, de Vos WM, Vaughan EE. Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract. Appl Environ Microb. 2007;73:1388–92. doi: 10.1128/AEM.01921-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Fedorova ND, Jackson JD, et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004;5:R7. doi: 10.1186/gb-2004-5-2-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskinen VR, Emery PA, Creasy DM, et al. Hierarchical clustering of shotgun proteomics data. Mol Cell Proteomics. 2011;10:M110.003822. doi: 10.1074/mcp.M110.003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YB, Lorenz N, Dick LK, et al. Cold storage and pretreatment incubation effects on soil microbial properties. Soil Sci Soc Am J. 2007;71:1299–305. [Google Scholar]
- Lin W, Wang J, Zhang WJ, et al. An unsupervised machine learning method for assessing quality of tandem mass spectra. Proteome Sci. 2012;10(Suppl 1):S12. doi: 10.1186/1477-5956-10-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WX, Wu LK, Lin S, et al. Metaproteomic analysis of ratoon sugarcane rhizospheric soil. BMC Microbiol. 2013;13:135. doi: 10.1186/1471-2180-13-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V, Golaconda Ramulu H, Drula E, et al. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–5. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Mondéjar R, Zühlke D, Becher D, et al. Cellulose and hemicellulose decomposition by forest soil bacteria proceeds by the action of structurally variable enzymatic systems. Sci Rep. 2016;6:25279. doi: 10.1038/srep25279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma ZQ, Chambers MC, Ham AJ, et al. ScanRanker: quality assessment of tandem mass spectra via sequence tagging. J Proteome Res. 2011;10:2896–904. doi: 10.1021/pr200118r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain S, Mathews M, Bereman MS, et al. Estimating relative abundances of proteins from shotgun proteomics data. BMC Bioinformatics. 2012;13:308. doi: 10.1186/1471-2105-13-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron P-A, Mougel C, Ranjard L. Soil microbial diversity: methodological strategy, spatial overview and functional interest. Crit Biol. 2011;334:403–11. doi: 10.1016/j.crvi.2010.12.003. [DOI] [PubMed] [Google Scholar]
- Masciandaro G, Macci C, Doni S, et al. Comparison of extraction methods for recovery of extracellular beta-glucosidase in two different forest soils. Soil Biol Biochem. 2008;40:2156–61. [Google Scholar]
- Mehlan H, Schmidt F, Weiss S, et al. Data visualization in environmental proteomics. Proteomics. 2013;13:2805–21. doi: 10.1002/pmic.201300167. [DOI] [PubMed] [Google Scholar]
- Morris RM, Nunn BL, Frazar C, et al. Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction. ISME J. 2010;4:673–85. doi: 10.1038/ismej.2010.4. [DOI] [PubMed] [Google Scholar]
- Mueller RS, Pan C. Sample handling and mass spectrometry for microbial metaproteomic analyses. Methods Enzymol. 2013;531:289–303. doi: 10.1016/B978-0-12-407863-5.00015-0. [DOI] [PubMed] [Google Scholar]
- Murase A, Yoneda M, Ueno R, et al. Isolation of extracellular protein from greenhouse soil. Soil Biol Biochem. 2003;35:733–6. [Google Scholar]
- Muth T, Behne A, Heyer R, et al. The MetaProteomeAnalyzer: a powerful open-source software suite for metaproteomics data analysis and interpretation. J Proteome Res. 2015;14:1557–65. doi: 10.1021/pr501246w. [DOI] [PubMed] [Google Scholar]
- Muth T, Benndorf D, Reichl U, et al. Searching for a needle in a stack of needles: challenges in metaproteomics data analysis. Mol Biosyst. 2013;9:578–85. doi: 10.1039/c2mb25415h. [DOI] [PubMed] [Google Scholar]
- Myrold DD, Zeglin LH, Jansson JK. The potential of metagenomic approaches for understanding soil microbial processes. Soil Sci Soc Am J. 2014;78:3–10. [Google Scholar]
- Nannipieri P. Role of stabilised enzymes in microbial ecology and enzyme extraction from soil with potential applications in soil proteomics. In: Nannipieri P, Smalla K, editors. Nucleic Acids and Proteins in Soil. Vol. 8. Berlin, Heidelberg: Springer; 2006. pp. 75–94. [Google Scholar]
- Nesvizhskii AI. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteomics. 2010;73:2092–123. doi: 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics. 2005;4:1419–40. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, et al. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- Nicora CD, Anderson BJ, Callister SJ, et al. Amino acid treatment enhances protein recovery from sediment and soils for metaproteomic studies. Proteomics. 2013;13:2776–85. doi: 10.1002/pmic.201300003. [DOI] [PubMed] [Google Scholar]
- Nielsen KM, Calamai L, Pietramellara G. Stabilization of extracellular DNA and proteins by transient binding to various soil components. In: Nannipieri P, Smalla K, editors. Nucleic Acids and Proteins in Soil. Vol. 8. Berlin, Heidelberg: Springer; 2006. pp. 141–57. [Google Scholar]
- Norde W, Tan W, Koopal L. Protein adsorption at solid surfaces and protein complexation with humic acids. Rev Cienc Suelo Nutr. 2008;8:64–74. [Google Scholar]
- Ogunseitan O. Soil proteomics: extraction and analysis of proteins from soils. In: Nannipieri P, Smalla K, editors. Nucleic Acids and Proteins in Soil. Vol. 8. Berlin, Heidelberg: Springer; 2006. pp. 95–115. [Google Scholar]
- Otto A, Becher D, Schmidt F. Quantitative proteomics in the field of microbiology. Proteomics. 2014;14:547–65. doi: 10.1002/pmic.201300403. [DOI] [PubMed] [Google Scholar]
- Pavoković D, Križnik B, Krsnik-Rasol M. Evaluation of protein extraction methods for proteomic analysis of non-model recalcitrant plant tissues. Croat Chem Acta. 2012;85:177–83. [Google Scholar]
- Perez-Riverol Y, Wang R, Hermjakob H, et al. Open source libraries and frameworks for mass spectrometry based proteomics: a developer's perspective. Biochim Biophys Acta. 2014;1844:63–76. doi: 10.1016/j.bbapap.2013.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt AN, McBratney AB. Sampling designs for estimating spatial variance components. J Roy Stat Soc C-App. 1993;42:185–209. [Google Scholar]
- Powell S, Forslund K, Szklarczyk D, et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014;42:D231–9. doi: 10.1093/nar/gkt1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser JI. Dispersing misconceptions and identifying opportunities for the use of ‘omics’ in soil microbial ecology. Nat Rev Microbiol. 2015;13:439–46. doi: 10.1038/nrmicro3468. [DOI] [PubMed] [Google Scholar]
- Roberts P, Jones DL. Critical evaluation of methods for determining total protein in soil solution. Soil Biol Biochem. 2008;40:1485–95. [Google Scholar]
- Rubin BER, Gibbons SM, Kennedy S, et al. Investigating the impact of storage conditions on microbial community composition in soil samples. PLoS One. 2013;8:e70460. doi: 10.1371/journal.pone.0070460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed F, Hoffert JD, Knepper MA. CAMS-RS: clustering algorithm for large-scale mass spectrometry data using restricted search space and intelligent random sampling. IEEE/ACM Trans Comput Biol Bioinform. 2014;11:128–41. doi: 10.1109/TCBB.2013.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M, Tomaszewski JE, Schwarzenbach RP. Protein encapsulation by humic substances. Environ Sci Technol. 2011;45:6003–10. doi: 10.1021/es200663h. [DOI] [PubMed] [Google Scholar]
- Schellenberger J, Park JO, Conrad TM, et al. BiGG: a Biochemical Genetic and Genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinformatics. 2010;11:213. doi: 10.1186/1471-2105-11-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T, Gerrits B, Gassmann R, et al. Proteome analysis of fungal and bacterial involvement in leaf litter decomposition. Proteomics. 2010;10:1819–30. doi: 10.1002/pmic.200900691. [DOI] [PubMed] [Google Scholar]
- Schneider T, Keiblinger KM, Schmid E, et al. Who is who in litter decomposition? Metaproteomics reveals major microbial players and their biogeochemical functions. ISME J. 2012;6:1749–62. doi: 10.1038/ismej.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T, Riedel K. Environmental proteomics: analysis of structure and function of microbial communities. Proteomics. 2010;10:785–98. doi: 10.1002/pmic.200900450. [DOI] [PubMed] [Google Scholar]
- Schneider T, Schmid E, de Castro JV, Jr, et al. Structure and function of the symbiosis partners of the lung lichen (Lobaria pulmonaria L. Hoffm.) analyzed by metaproteomics. Proteomics. 2011;11:2752–6. doi: 10.1002/pmic.201000679. [DOI] [PubMed] [Google Scholar]
- Searle BC. Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics. 2010;10:1265–9. doi: 10.1002/pmic.200900437. [DOI] [PubMed] [Google Scholar]
- Serang O, Noble WS. Faster mass spectrometry-based protein inference: junction trees are more efficient than sampling and marginalization by enumeration. IEEE/ACM T Comput Bi. 2012;9:809–17. doi: 10.1109/TCBB.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wu FX. A feedback framework for protein inference with peptides identified from tandem mass spectra. Proteome Sci. 2012;10:68. doi: 10.1186/1477-5956-10-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggins A, Gunnigle E, Abram F. Exploring mixed microbial community functioning: recent advances in metaproteomics. Fems Microbiol Ecol. 2012;80:265–80. doi: 10.1111/j.1574-6941.2011.01284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton I, Merrington G, Colvan S, et al. The potential of soil protein-based methods to indicate metal contamination. Appl Soil Ecol. 2003;23:25–32. [Google Scholar]
- Souza RC, Hungria M, Cantão ME, et al. Metagenomic analysis reveals microbial functional redundancies and specificities in a soil under different tillage and crop-management regimes. Appl Soil Ecol. 2015;86:106–12. [Google Scholar]
- Spivak M, Weston J, Bottou L, et al. Improvements to the percolator algorithm for Peptide identification from shotgun proteomics data sets. J Proteome Res. 2009;8:3737–45. doi: 10.1021/pr801109k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke R, Kermer R, Ullmann-Zeunert L, et al. Bacteria dominate the short-term assimilation of plant-derived N in soil. Soil Biol Biochem. 2016;96:30–8. [Google Scholar]
- Stephan C, Kohl M, Turewicz M, et al. Using Laboratory Information Management Systems as central part of a proteomics data workflow. Proteomics. 2010;10:1230–49. doi: 10.1002/pmic.200900420. [DOI] [PubMed] [Google Scholar]
- Tanca A, Palomba A, Deligios M, et al. Evaluating the impact of different sequence databases on metaproteome analysis: insights from a lab-assembled microbial mixture. PLoS One. 2013;8:e82981. doi: 10.1371/journal.pone.0082981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor EB, Williams MA. Microbial protein in soil: influence of extraction method and C amendment on extraction and recovery. Microb Ecol. 2010;59:390–9. doi: 10.1007/s00248-009-9593-x. [DOI] [PubMed] [Google Scholar]
- Tomaszewski JE, Schwarzenbach RP, Sander M. Protein encapsulation by humic substances. Environ Sci Technol. 2011;45:6003–10. doi: 10.1021/es200663h. [DOI] [PubMed] [Google Scholar]
- Tveit A, Schwacke R, Svenning MM, et al. Organic carbon transformations in high-Arctic peat soils: key functions and microorganisms. ISME J. 2013;7:299–311. doi: 10.1038/ismej.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Elsas JD, Duarte GF, Keijzer-Wolters A, et al. Analysis of the dynamics of fungal communities in soil via fungal-specific PCR of soil DNA followed by denaturing gradient gel electrophoresis. J Microbiol Meth. 2000;43:133–51. doi: 10.1016/s0167-7012(00)00212-8. [DOI] [PubMed] [Google Scholar]
- Van Horn DJ, Okie JG, Buelow HN, et al. Soil microbial responses to increased moisture and organic resources along a salinity gradient in a polar desert. Appl Environ Microb. 2014;80:3034–43. doi: 10.1128/AEM.03414-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verberkmoes NC, Russell AL, Shah M, et al. Shotgun metaproteomics of the human distal gut microbiota. ISME J. 2008;3:179–89. doi: 10.1038/ismej.2008.108. [DOI] [PubMed] [Google Scholar]
- von Bergen M, Jehmlich N, Taubert M, et al. Insights from quantitative metaproteomics and protein-stable isotope probing into microbial ecology. ISME J. 2013;7:1877–85. doi: 10.1038/ismej.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M, Wolf AB, Jennings SJ, et al. Micro-scale determinants of bacterial diversity in soil. FEMS Microbiol Rev. 2013;37:936–54. doi: 10.1111/1574-6976.12023. [DOI] [PubMed] [Google Scholar]
- Wallenius K, Rita H, Simpanen S, et al. Sample storage for soil enzyme activity and bacterial community profiles. J Microbiol Meth. 2010;81:48–55. doi: 10.1016/j.mimet.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Wang W, Vignani R, Scali M, et al. A universal and rapid protocol for protein extraction from recalcitrant plant tissues for proteomic analysis. Electrophoresis. 2006;27:2782–6. doi: 10.1002/elps.200500722. [DOI] [PubMed] [Google Scholar]
- Wang HB, Zhang ZX, Li H, et al. Characterization of metaproteomics in crop rhizospheric soil. J Proteome Res. 2011;10:932–40. doi: 10.1021/pr100981r. [DOI] [PubMed] [Google Scholar]
- Weiss W, Görg A. Sample solublization buffers for two-dimensional electrophoresis. In: Posch A, editor. 2D PAGE: Sample Preparation and Fractionation. Totowa, NJ: Humana Press; 2008. pp. 35–42. [DOI] [PubMed] [Google Scholar]
- Weston LA, Bauer KM, Hummon AB. Comparison of bottom-up proteomic approaches for LC-MS analysis of complex proteomes. Anal Methods. 2013;5:15. doi: 10.1039/C3AY40853A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiffen LK, Midgley DJ, Mcgee PA. Polyphenolic compounds interfere with quantification of protein in soil extracts using the Bradford method. Soil Biol Biochem. 2007;39:691–4. [Google Scholar]
- Wilmes P, Wexler M, Bond PL. Metaproteomics provides functional insight into activated sludge wastewater treatment. PLoS One. 2008;3:e1778. doi: 10.1371/journal.pone.0001778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlbrand L, Trautwein K, Rabus R. Proteomic tools for environmental microbiology—a roadmap from sample preparation to protein identification and quantification. Proteomics. 2013;13:2700–30. doi: 10.1002/pmic.201300175. [DOI] [PubMed] [Google Scholar]
- Xue X, Wu S, Wang Z, et al. Protein probabilities in shotgun proteomics: evaluating different estimation methods using a semi-random sampling model. Proteomics. 2006;6:6134–45. doi: 10.1002/pmic.200600070. [DOI] [PubMed] [Google Scholar]
- Yang C, He Z, Yu W. A combinatorial perspective of the protein inference problem. IEEE/ACM T Comput Bi. 2013;10:1542–7. doi: 10.1109/TCBB.2013.110. [DOI] [PubMed] [Google Scholar]
- Yates JR, Eng JK, McCormack AL, et al. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67:1426–36. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- Yin X, Zhang Y, Liu X, et al. Systematic comparison between SDS-PAGE/RPLC and high-/low-pH RPLC coupled tandem mass spectrometry strategies in a whole proteome analysis. Analyst. 2015;140:1314–22. doi: 10.1039/c4an02119c. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wen Z, Washburn MP, et al. Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal Chem. 2010;82:2272–81. doi: 10.1021/ac9023999. [DOI] [PubMed] [Google Scholar]
- Zou AM, Wu FX, Ding JR, et al. Quality assessment of tandem mass spectra using support vector machine (SVM) BMC Bioinformatics. 2009;10(Suppl 1):S49. doi: 10.1186/1471-2105-10-S1-S49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zybailov B, Mosley AL, Sardiu ME, et al. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res. 2006;5:2339–47. doi: 10.1021/pr060161n. [DOI] [PubMed] [Google Scholar]