

Abstract
Ceramide accumulation is known to accompany acute myocardial ischemia, but its role in the pathogenesis of ischemic heart disease is unclear. In this study, we aimed to determine how ceramides accumulate in the ischemic heart and to determine if cardiac function following ischemia can be improved by reducing ceramide accumulation.
To investigate the association between ceramide accumulation and heart function, we analyzed myocardial left ventricle biopsies from subjects with chronic ischemia and found that ceramide levels were higher in biopsies from subjects with reduced heart function. Ceramides are produced by either de novo synthesis or hydrolysis of sphingomyelin catalyzed by acid and/or neutral sphingomyelinase. We used cultured HL-1 cardiomyocytes to investigate these pathways and showed that acid sphingomyelinase activity rather than neutral sphingomyelinase activity or de novo sphingolipid synthesis was important for hypoxia-induced ceramide accumulation. We also used mice with a partial deficiency in acid sphingomyelinase (Smpd1+/- mice) to investigate if limiting ceramide accumulation under ischemic conditions would have a beneficial effect on heart function and survival. Although we showed that cardiac ceramide accumulation was reduced in Smpd1+/- mice 24 h after an induced myocardial infarction, this reduction was not accompanied by an improvement in heart function or survival.
Our findings show that accumulation of cardiac ceramides in the post-ischemic heart is mediated by acid sphingomyelinase. However, targeting ceramide accumulation in the ischemic heart may not be a beneficial treatment strategy.
Keywords: Ceramide, bioactive lipids, acid sphingomyelinase, myocardial ischemia
1. Introduction
Ceramides are recognized not only as structural components of cellular membranes but also as bioactive lipids. These bioactive lipids may promote cellular stress responses such as inflammation, mitochondrial dysfunction, apoptosis and defects in insulin signaling.1 Ceramides consist of a sphingosine base linked to a fatty acid and are produced either by de novo synthesis (from palmitate via the precursor dihydroceramides) or hydrolysis of sphingomyelin.2
Abnormal ceramide accumulation has recently been implicated in acute myocardial ischemia.3, 4 Increasing evidence indicates that metabolic reprogramming in the ischemic heart contributes to pathological cardiac remodeling, leading to increased incidence of heart failure.5 However, it is not known if myocardial ceramide accumulation plays a role in this process. Here, we investigated how ceramides accumulate and what role they play in the pathogenesis of myocardial ischemia.
2. Material And Methods
Material and methods are described in the Supplemental Material.
3. Results
To investigate the association between ceramide accumulation and heart function, we analyzed myocardial left ventricle biopsies from subjects with chronic ischemia and found that ceramide levels were higher in biopsies from subjects with reduced heart function (Figure 1A). Furthermore, total triglycerides correlated with dihydroceramides (an upstream precursor to ceramides and a marker of de novo sphingolipid synthesis), indicating that increased accumulation of neutral lipids correlates with increased de novo sphingolipid synthesis. However, triglycerides did not correlate with ceramides (Figure 1B), suggesting that myocardial ceramide accumulation in the post-ischemic human heart is disassociated from accumulation of neutral lipids and of the sphingolipid de novo synthesis pathway (See Figure 1C for proposed pathway). Thus, our results suggest that cardiac ceramides accumulate independently of the do novo sphingolipid synthesis pathway and that they associate with heart function in the post-ischemic heart.
Figure 1. Ceramides correlate with heart function and hypoxia-induced ceramide accumulation in cardiomyocytes is mediated by acid sphingomyelinase.
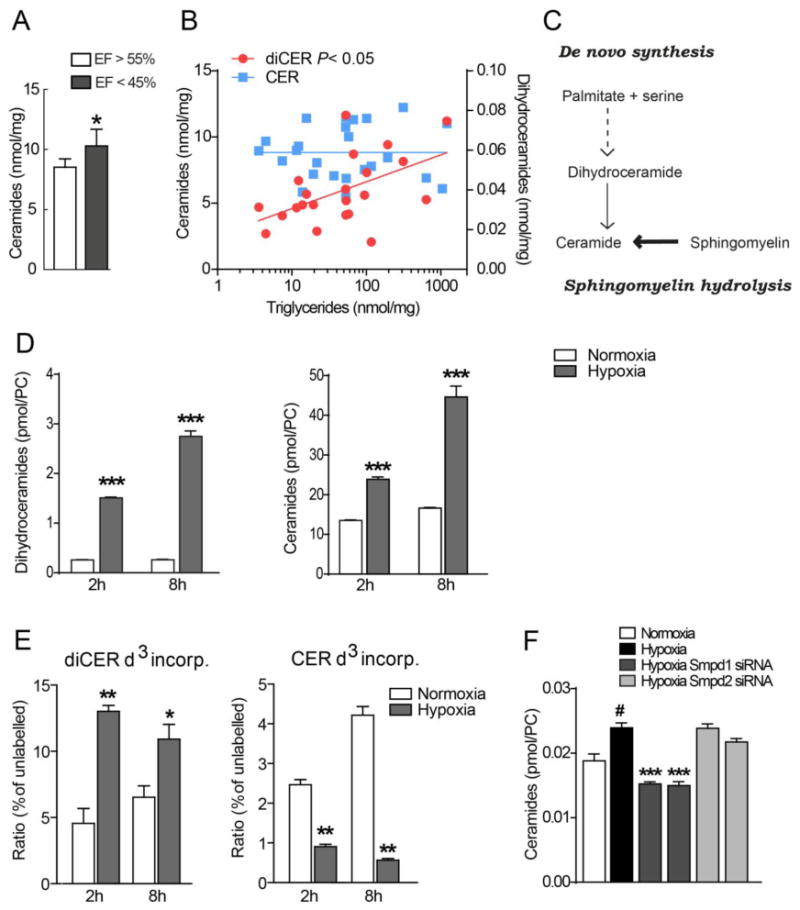
(A) Ceramide concentrations in heart biopsies from patients with chronic cardiac ischemia and ejection fraction (EF) >55% or <45% (n=30). * P<0.05, vs EF>55%. (B) Correlations between ceramides and dihydroceramides with triglycerides in heart biopsies from patients with chronic cardiac ischemia (n=30). (C) Ceramide synthesis pathway. (D) Hypoxia-induced ceramide accumulation in HL-1 cardiomyocytes. HL-1 cells were incubated in normoxia or hypoxia for 2 h or 8 h and dihydroceramides and ceramides were measured with HPLC and mass spectrometry. (n = 3). Data are shown as mean ± SEM, ***P < 0.001 vs normoxia. (E) Incorporation of deuterium-labelled serine into dihydroceramides and ceramides in HL-1 cardiomyocytes following incubation in normoxia or hypoxia for 2 h or 8 h (n=3) ***P < 0.001 vs normoxia. (F) Ceramide levels in HL-1 cardiomyocytes transfected with control siRNA, Smpd1 siRNA (ID #1 and #2) and Smpd2 siRNA (ID #1 and #2), incubated in normoxia or hypoxia for 24 h (n = 3–6). Data are mean ± SEM.
We further investigated how hypoxia induces ceramide accumulation using cultured HL-1 cardiomyocytes. Levels of ceramides and dihydroceramides were upregulated by hypoxia in HL-1 cardiomyocytes (Figure 1D). We next incubated the cells with deuterium-labelled serine (D3-serine) and found that hypoxia promoted a significant increase in the D3-serine pool in dihydroceramides but a decrease in the D3-serine pool in ceramides (Figure 1E), thus confirming that the hypoxia-induced increase in ceramides does not originate from de novo synthesis.
To test whether hypoxia-induced ceramide accumulation instead resulted from hydrolysis of sphingomyelin, we used siRNA to inhibit acid sphingomyelinase (Smpd1) and neutral sphingomyelinase (Smpd2) in HL-1 cardiomyocytes. Importantly, we showed that inhibiting Smpd1 completely blocked ceramide accumulation during hypoxia, whereas blocking Smpd2 did not have any effect (Figure 1F). Thus, our results suggest that acid sphingomyelinase mediates the ceramide accumulation induced by hypoxia in cardiomyocytes.
Next, we used mice with a heterozygous mutation in Smpd1 to test if partial inhibition of acid sphingomyelinase would reduce ceramide accumulation in the post-ischemic heart. Smpd1+/– mice had normal levels of cardiac glycerolipids and sphingolipids under baseline conditions (Figure S1). By comparing ceramide species in hearts from WT and Smpd1+/– mice before and 24 h after an induced myocardial infarction, we showed that the ischemia-induced accumulation of all ceramide species in WT hearts was significantly reduced in Smpd1+/– hearts, with the exception of the palmitate- and myristate-derived 24:0 species (Figure 2A and B). Thus, ischemia-induced cardiac ceramide accumulation can be reduced by targeting acid sphingomyelinase.
Figure 2. Inactivation of acid sphingomyelinase (Smpd1) reduces ischemia-induced cardiac ceramide accumulation but does not affect heart function.
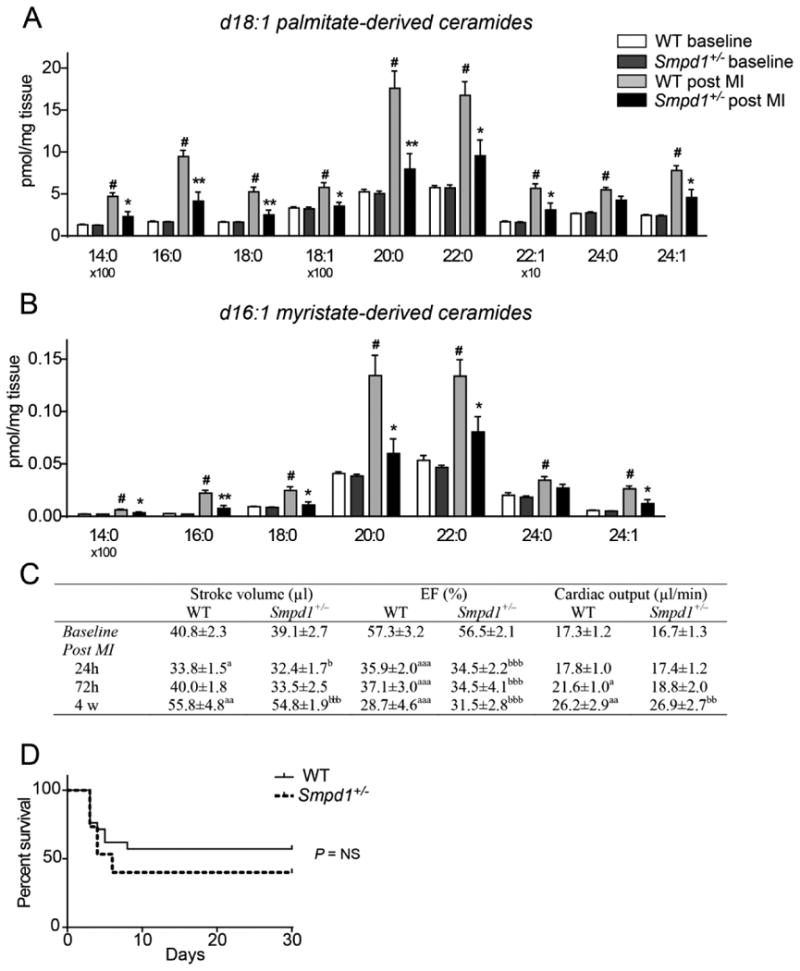
(A–B) Concentrations of d18:1 palmitate-derived (A) and d16:1 myristate-derived (B) ceramide species in WT and Smpd1+/– mice at baseline and 24 h after an induced myocardial infarction (MI) (n=5–6). Data are mean ± SEM. #P < 0.01 vs WT baseline *P < 0.05, **P < 0.01 vs WT post MI. (C) Echocardiographic analysis of WT and Smpd1+/– mice at baseline and after an induced MI (n=9 at baseline; n=15–20 at 24 h; n=7–9 at 72 h; n=6–7 at 4 weeks). aP < 0.05, aaP < 0.01 and aaaP < 0.001 vs WT baseline; bP < 0.05, bbP < 0.01 and bbbP < 0.001 vs Smpd1+/– baseline. (D) Kaplan-Meier curve showing survival of WT and Smpd1+/– mice after an induced MI (n=15–21). Data are analyzed using the log-rank test and P = 0.33.
We also assessed the cardiac function of WT and Smpd1+/– mice before and after an induced myocardial infarction. There were no differences in stroke volume, ejection fraction or cardiac output between the groups under baseline conditions nor, surprisingly, 24 h, 72 h and 4 weeks after an induced myocardial infarction (Figure 2C). In addition, we did not observe any difference in survival of these mice for up to 30 days after an induced myocardial infarction (Figure 2D). Thus, in our mouse model, reducing ischemia-induced cardiac ceramide accumulation does not result in improved heart function and survival.
4. Discussion
In this study, we showed that ceramide accumulation associates with heart function in the human post-ischemic heart and, importantly, that the increase in ceramides appears to be mediated by acid sphingomyelinase and not by de novo sphingolipid synthesis. However, we found that inhibiting acid sphingomyelinase in mice does not result in improved heart function or survival after an induced myocardial infarction despite reducing ischemia-induced ceramide accumulation.
Targeting cardiac ceramide accumulation by inhibition of de novo sphingolipid synthesis has emerged as a potential strategy to improve heart function in obesity and diabetes. Pharmacological inhibition of serine palmitoyltransferase, for example, has been shown to prevent cardiac toxicity caused by increased lipid uptake and excess ceramides in a mouse model of dilated cardiomyopathy.6 However, although de novo sphingolipid synthesis may be enhanced in the post-ischemic heart, our results from heart biopsies and cardiomyocytes indicate that ischemia/hypoxia-induced cardiac ceramide accumulation is independent of the sphingolipid de novo synthesis pathway but instead results from hydrolysis of sphingomyelin catalyzed by acid sphingomyelinase. Indeed, it is known that the enzyme catalyzing ceramide synthesis from dihydroceramides is oxygen dependent,7 and thus it is unlikely that de novo synthesis of ceramides would occur in ischemic tissue.8 Targeting acid sphingomyelinase in the heart would represent a novel approach to reduce cardiac ceramide accumulation.
We used mice with a partial deficiency in acid sphingomyelinase to test the hypothesis that limiting ceramide accumulation after a myocardial infarction would improve cardiac function and outcome. However, contrary to our expectations, cardiac function and outcome following a myocardial infarction in Smpd1+/– mice were not improved despite reduced ceramide accumulation compared with WT mice. We used Smpd1+/– mice as they have normal cardiac ceramide levels under baseline conditions. However, because they have 50% of normal acid sphingomyelinase activity,9 ischemia-induced ceramide accumulation is not entirely abolished in these mice. It is plausible that the reduction of ceramides was not robust enough to counteract the effects of this multifactorial pathological process, and we cannot rule out the possibility that elevated post-ischemic ceramide levels remain a factor in impaired cardiac function. Furthermore, although long-chained ceramides were efficiently reduced in the Smpd1+/– mice, very long-chained ceramides were reduced to a lesser extent. Very long-chained ceramides may be more harmful than long and medium-chained ceramides,4, 10 and we cannot exclude the possibility that these species preferentially mediate the harmful effects on heart function in our mouse model. An additional limitation to the study is that the Smpd1+/– mouse model is a global partial deficiency model, and any differences in heart function or outcome after a myocardial infarction could potentially be counteracted or enhanced by systemic effects.
In conclusion, our study reveals that ceramide accumulation in the post-ischemic heart is mediated by acid sphingomyelinase. However, inhibiting acid sphingomyelinase in mice does not result in improved heart function or survival after an induced myocardial infarction despite reducing ischemia-induced ceramide accumulation. Further investigations are therefore needed to determine the potential of targeting ceramide accumulation as a treatment for ischemic heart disease.
Supplementary Material
Highlights.
Ceramide accumulation in post-ischemic human heart associates with heart function.
Ceramide accumulation is mediated by acid sphingomyelinase.
Normalizing ceramide levels in the post-ischemic heart does not improve function and outcome.
Acknowledgments
We thank Azra Miljanovic for technical assistance, and Rosie Perkins for scientific editing of the manuscript.
Sources of Funding: This work was supported by the Swedish Research Council, the Swedish Heart and Lung Foundation, Novo Nordisk Foundation, Swedish Diabetes Foundation, Diabetes Research Wellness Foundation, the Sahlgrenska University Hospital ALF research grants.
Footnotes
Disclosures: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bikman BT, Summers SA. Ceramides as modulators of cellular and whole-body metabolism. J Clin Invest. 2011;121:4222–30. doi: 10.1172/JCI57144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Breslow DK, Weissman JS. Membranes in balance: mechanisms of sphingolipid homeostasis. Molecular cell. 2010;40:267–79. doi: 10.1016/j.molcel.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drevinge C, Karlsson LO, Stahlman M, Larsson T, Perman Sundelin J, Grip L, Andersson L, Boren J, Levin MC. Cholesteryl esters accumulate in the heart in a porcine model of ischemia and reperfusion. PloS one. 2013;8:e61942. doi: 10.1371/journal.pone.0061942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perman JC, Bostrom P, Lindbom M, Lidberg U, StAhlman M, Hagg D, Lindskog H, Scharin Tang M, Omerovic E, Mattsson Hulten L, Jeppsson A, Petursson P, Herlitz J, Olivecrona G, Strickland DK, Ekroos K, Olofsson SO, Boren J. The VLDL receptor promotes lipotoxicity and increases mortality in mice following an acute myocardial infarction. J Clin Invest. 2011;121:2625–40. doi: 10.1172/JCI43068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatham JC, Young ME. Metabolic remodeling in the hypertrophic heart: fuel for thought. Circulation research. 2012;111:666–8. doi: 10.1161/CIRCRESAHA.112.277392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res. 2008;49:2101–12. doi: 10.1194/jlr.M800147-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devlin CM, Lahm T, Hubbard WC, Van Demark M, Wang KC, Wu X, Bielawska A, Obeid LM, Ivan M, Petrache I. Dihydroceramide-based response to hypoxia. J Biol Chem. 2011;286:38069–78. doi: 10.1074/jbc.M111.297994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ji R, Liao X, Zhang XJH, Drosatos K, Kennel P, Castillero E, Chang J, Homma S, Goldberg I, Schulze C. De novo ceramide synthesis is upregulated by cardiac ischemia and is associated with cardiomyocyte apoptosis and mitochondrial dysfunction. Paper presented at: American Heart. 2015 [Google Scholar]
- 9.Horinouchi K, Erlich S, Perl DP, Ferlinz K, Bisgaier CL, Sandhoff K, Desnick RJ, Stewart CL, Schuchman EH. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nature genetics. 1995;10:288–93. doi: 10.1038/ng0795-288. [DOI] [PubMed] [Google Scholar]
- 10.Stiban J, Perera M. Very long chain ceramides interfere with C16-ceramide-induced channel formation: A plausible mechanism for regulating the initiation of intrinsic apoptosis. Biochim Biophys Acta. 2015;1848:561–7. doi: 10.1016/j.bbamem.2014.11.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.