

Abstract
Benzocyclobutenes (BCBs) are important entities in a multitude of areas, such as complex organic synthesis, materials and polymer chemistry, and electronics. Whereas reactions between arynes and ketene acetals have been well studied, reactions with cyclic enol ethers are unknown. A cis olefin geometry in cyclic enol ethers makes them well suited for formal [2 + 2] cycloaddition with arynes than for competing ene reactions, making them effective reactants. Reactions of 2,3-dihydrofuran, 2,3-dihydro-3H-pyran, 5-butyl-2,3-dihydrofuran, (S)-2-((benzyloxy)methyl)-2,3-dihydrofuran, and 1,4-dioxene with various arynes were successful. An advantage of the use of cyclic enol ethers is that despite the plausible intermediacy of zwitterionic intermediates, the products are limited to a cis ring junction. This can be exploited for potential access to stereochemically-defined 1,2-disubstituted BCBs. As a demonstration, ether ring cleavage with BBr3 provided trans-functionalized BCBs and displacement with azide then provided cis derivatives. DFT computations have been utilized to understand the structures of three arynes in relation to the cycloadditions and for a predictive evaluation of product ratios in two cases. A comparative evaluation of the HOMO energies of a related series of cyclic olefins has also been performed by DFT computations.
Keywords: Arynes, Cycloaddition, Fused-ring systems, Regioselectivity, Enol ethers, Density functional calculations
Introduction
From the time of its discovery over a century ago,[1] and its structural characterization about half a century later,[2] bicyclo[4.2.0]octa-1,3,5-triene (also commonly known as benzocyclobutene, BCB) and its derivatives have emerged as powerful, reactive entities. Applications of BCBs range from complex organic syntheses,[3,4] materials and polymer chemistry, and organic light-emitting diodes to electronics.[4,5]
The exceptional utility of BCBs has therefore resulted in a number of creative approaches to their syntheses. However, the most obvious route to BCBs is through a formal [2 + 2] cycloaddition between arynes and olefins. On the basis of its unusual C≡C stretch (1846 cm−1), benzyne is predicted to not have significant alkyne character,[6] and is considered a diradical.[7] This can be associated with its chemical reactivity in [2 + 2] reactions with alkenes, although zwitterionic intermediates are plausible with dipolar reactants.[8] The low-lying LUMO of benzyne[9] renders it generally electrophilic and thus capable of reactions with both charged and charge-neutral nucleophiles. On the other hand, the structures, and therefore the reactions, of substituted arynes can be influenced by the nature of the substituents. This has been the subject of reactivity and computational analyses, as well as synthetic applications.[10–12]
Formal [2 + 2] cycloaddition between arynes and alkenes has been of significant interest and has led to the development of powerful synthetic methods. With rare exceptions, reactions between simple alkenes and benzyne generally give low yields of cycloaddition products by diradial pathways, with ene reactions predominating.[13–17] Reactions between enamines and arynes[18] have led to a ring-expansion methodology involving BCB intermediates.[19] However, these are also prone to ene reactions.[20] Enolate additions to arynes have proven to be of exceptional synthetic value. These reactions proceed through the intermediacy of benzocyclobutenols and directly yield products of σ-bond insertion.[21–23]
Reaction between arynes and simple enol ethers are rare in the literature. Benzyne, produced by the diazotization of anthranilic acid, reacts with ethyl vinyl ether and vinyl acetate to yield the corresponding [2 + 2] products in 40–45% yields.[24] However, use of 1-ethoxyvinyl acetate gave benzocyclobutenone in only 4% yield, with the ethyl ester of o-acetylphenylacetic acid being the major product (25%).[24] The reaction between benzyne and trans-methoxypropene gave significant amounts of 3-methoxy-3-phenylpropene (ca. 50 % of the product mixture), whereas that between benzyne and trans-acetoxypropene gave 1-phenylallyl acetate (>80% of the product mixture), by the ene pathway.[13] Interestingly, cis-methoxypropene gave a greater proportion of the [2 + 2] products, as a cis/trans mixture of 3-methoxy-4-methylbenzocyclobutenes, albeit only in low to moderate yields.[13]
In contrast with the limited literature on the reactions between arynes and enol ethers, literature on the reactions between arynes and ketene acetals is more extensive,[25,26] and the utility of reactions between alkoxy-substituted arynes and silyl ketene acetals has been substantially documented.[26] These reagents have provided access to benzocyclobutenones and related complex architectures.[26]
A drawback to many of these methods is the need for strong bases for aryne generation. The advent of o-silyl aryltriflates[27] has had a major contribution to synthetic chemistry due to the ease of aryne generation under mild conditions.[23] Notably, reactions between arynes and enamides, leading both to indolines and to isoquinolines and also to BCBs en route to natural product scaffolds, have been achieved by this method.[28,29]
Results and Discussion
In this overall context we were intrigued by the fact that ethyl vinyl ether reacts modestly with benzyne[24] in a [2 + 2] manner as does cis-1-methoxypropene, but not the trans isomer.[13] This prompted the consideration that cyclic enol ethers fit the geometric requirements for favorable [2 + 2] cycloaddition over the undesirable ene reaction. Furthermore many cyclic enol ethers are readily available. Cycloadditions between arynes and cyclic enol ethers have not been evaluated to date, but these reactions appear to offer a variety of synthetic advantages. The relative stereochemistry at the ring junction involving the cyclobutene moiety would be cis. A similar stereochemical outcome has been reported for the significantly electron-rich, acyclic E/Z β-silyloxy ketene silyl acetals derived from α-(tert-butyldimethylsilyloxy)methyl acetate and the E/Z-ketene silyl acetals drived from ethyl propionate.[26g] However, in that study as well, the E-ketene silyl acetal underwent an ene-type process to a significant extent, and the minor erosion of stereochemistry observed at the ring junction was attributed to this side reaction (called a [4 + 2] process).[26g] All such problems are likely avoidable in reactions of cyclic enol ethers. Further, the ether rings in the cycloadducts should be cleavable to unmask classical BCBs, which could be used for further manipulations. Finally, the ability to introduce olefinic substituents by metalation alpha to the ether oxygen allows for greater synthetic utility.
With these considerations, we began our evaluation of the scope and feasibility of [2 + 2] cycloaddition between benzyne and cyclic enol ethers. As shown in Table 1, diazotization of anthranilic acid and subsequent reaction with 2,3-dihydrofuran (DHF) gave cycloadduct 1 in 48% yield (entry 1), comparable to the literature result with ethyl vinyl ether.[24] Use of 2,3-dihydro-3H-pyran (DHP) in place of DHF gave a similar yield of cycloadduct 2 (entry 2). Attempts to generate the aryne by lithium/halogen exchange processes were ineffective (entries 3 and 4) so such an approach was not pursued at this time. Generation of benzyne from 2-(trimethylsilyl)phenyl triflate, with the aid of 1:1 CsF/18-Cr-6 in MeCN as described,[30] and subsequent reaction with DHF gave product 1 in an excellent 82% isolated yield (entry 5).
Table 1.
Initial Screening Results for the [2 + 2] Cycloaddition.

| |||
|---|---|---|---|
| Entry | X and Y | Conditions | Result |
| 1 | X = NH2, Y = CO2H | tBuONO (2 equiv), MeCN, 80 °C, 4 h | 1: 48% yield[a] |
| 2 | X = NH2, Y = CO2H | tBuONO (2 equiv), MeCN, 80 °C, 4 h | 2: 46% yield[a] |
| 3 | X = Y = Br | nBuLi (1.1 equiv), THF, –78 °C, 1 h, then room temp., 16 h | 1: Trace |
| 4 | X = Br, Y = OTs | nBuLi (1.1 equiv), THF, –78 °C, 1 h, then room temp., 16 h | 1: Trace |
| 5 | X = TMS, Y = OTf | CsF (2 equiv), 18-Cr-6 (2 equiv), MeCN, room temp., 2 h | 1: 82% yield[a] |
Yield of isolated and purified product. The products appear to be volatile.
In its 1H NMR spectrum, compound 1 shows a doublet at δ = 5.56 ppm (J = 3.4 Hz). Cycloadduct 2 displays a doublet at δ = 5.16 ppm (J = 4.5 Hz). These data are comparable with those reported in the literature for similar products obtained from Fisher carbene complexes.[31,32] However, in the literature procedures, alkenyl cyclobutenyl Fisher carbenes were first obtained by [2 + 2] cycloaddition between enol ethers (alkenylethynyl)carbene complexes. Thermal CO insertion and cyclization then led to o-methoxyphenol derivatives containing the fused BCB-cyclic ether core, through a process similar to the Dötz reaction.[31] A similar approach has also been used for the synthesis of methoxy naphthalene derivatives by a metathesis pathway.[32]
The results in Table 1 clearly demonstrated the feasibility and simplicity of the approach under relatively mild aryne-generation conditions. With these initial results, we then proceeded to evaluate the broader applicability of the reactions between benzyne and other enol ethers, as well as vinylene carbonate. These data are shown in Table 2.
Table 2.
Reactions between benzyne and other enol ethers or vinylene carbonate.[a]
| Entry | Reactant | Product | Time (h), yield |
|---|---|---|---|
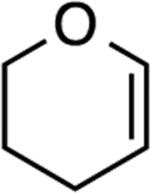
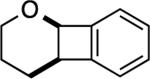
| 1 |
|
|
2: 2 h, 72% |
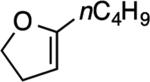
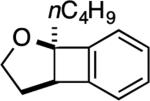
| 2 |
|
|
3: 6 h, 69% |
| 3 |
|
|
4a, 4b: 3 h, 48% 1:1 mixture |
| 4 |
|
|
5: 1 h, 50%[b] |
| 5 |
|
NA[c] | – |
| 6 |
|
NA[c] | – [d] |
| 7 |

|
|
6: 2 h, 84% |
Reactions were conducted at a 0.125 m concentration of 2-(trimethylsilyl)phenyl triflate in MeCN, in the presence of 1.2 equiv. of the enol ether (or vinylene carbonate), 2 equiv. of CsF, and 2 equiv. of 18-Cr-6 at room temperature.
Yield obtained with 3 equiv. of ethyl vinyl ether.
n.a.: not applicable.
About 86% of the glycal was recovered.
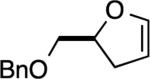
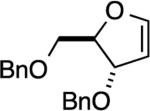
The reaction with DHP gave a good product yield (entry 1). As mentioned earlier, the ability to introduce a substituent at the vinylic position adjacent to the oxygen atom is exemplified in entry 2 (2-n-butyl-DHP was readily synthesized by lithiation/alkylation[33]). Importantly, the enol ether can be substituted at the nonvinylic position adjacent to the ether oxygen, as demonstrated by the use of (S)-2-[(benzyloxy)methyl]-2,3-dihydrofuran in entry 3. In this case a 1:1 diastereomeric pair of chromatographically separable cycloadducts 4a and 4b was obtained (see discussion below for additional details).
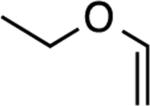
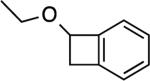
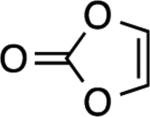
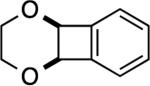
A simple enol ether also reacted (entry 4), albeit in modest yield. Not surprisingly, vinylene carbonatedid not yield a product (entry 5). What was surprising however, was the absence of product formation with the glycal derived from deoxyribose[34] (entry 6). We speculate that the allylic, inductively electron-withdrawing substituent is responsible, although other factors cannot be ruled out. Finally, the reaction with 1,4-dioxene gave an excellent yield of compound 6 (entry 7), a potential precursor to other stereochemically defined BCB derivatives.
The presence of the chiral center in the newly formed four-membered ring was clearly evident in the 1H NMR of cycloadduct 5, in which the diastereotopic OCH2 protons appear as apparent quintets at δ = 3.75 and 3.69 ppm (JAB = 15.2 Hz and Jvic = 7.3 Hz in each case). For additional confirmation of the BCB structural element, compound 5 was subjected to ring opening and cycloaddition with maleic anhydride (Scheme 1, 82% yield of compound 7). The anticipated all cis stereochemistry in the product 7 was confirmed by NOESY experiments, which showed correlations between the protons indicated with the grey ovals (Scheme 1). The OCH2 appear at higher field than might be expected (cf., for example, compound 5), likely due to the anisotropy of both the anhydride carbonyl and the aromatic moiety. The substantial differences in the chemical shifts of the CH2 resonances of the central ring are also likely due to these two functionalities. It should be noted is that these data were obtained in C6D6, in which overlap of aliphatic resonances observed in CDCl3 was eliminated.
Scheme 1.

Reaction between BCB 5 and maleic anhydride.
The next stage in the evaluation of the methodology was an assessment of the reaction scope with various arynes and enol ethers. Figure 1 shows the products synthesized, the reaction times, and yields obtained.
Figure 1.

Structures of the various BCB derivatives that were prepared, reaction times, and yields. Pairs of isomeric products formed are indicated by a and b. In the cases of 10 and 15, structures of major and minor regioisomers could be unequivocally established. Structures of major and minor regioisomers of 11 and 16 could not be determined unequivocally at this stage.
Reactions of DHF and DHP proceeded in very good to excellent yields to afford products 8–16 (73–86 %). Substitution with the n-butyl group at the vinylic position adjacent to the oxygen atom in DHF gave a slightly lower yield of 17, as in the reaction with benzyne (Table 2, entry 2). The isolated yields of 3 and 17 are both comparable and reasonable. The reaction between ethyl vinyl ether and 4,5-dimethoxybenzyne gave a 51% yield of compound 19, also comparable to that obtained with benzyne (Table 2, entry 4). A longer reaction time in this case did not provide a yield improvement. Products 20 and 21 were obtained in good yields from the reactions of 1,4-dioxene with 4,5-dimethoxybenzyne and 3-methoxybenzyne, respectively.
Structure Evaluations of the Cycloadducts
With 3-methoxybenzyne, single regioisomers of 9 and 14 were formed (Figure 1), consistently with previous results on cycloaddition between 3-alkoxyarynes and ketene acetals. Here NOESY analysis showed correlations between the OCH3 and bridgehead OCH resonances (shown in bold and by the arrows in Figure 2).
Figure 2.

Spectroscopic data supporting the structural assignments to the cycloadducts.
With 4-methoxy-, 3-methyl-, and 4-methylbenzynes, pairs of regioisomers were produced (10, 11, 12, 15, and 16). The ratios of the regioisomers in each case are indicated in Figure 1, and structure assignments of 10 and 15 were made on the basis of HMQC and HMBC data. These spectroscopic correlations are shown in Figure 2 (correlated pairs are shown in bold). At the current time, we have not been able to establish structures of the major and minor isomers in the other cases due to overlapping methyl and methylene resonances of the isomers.
The reaction between (S)-2-[(benzyloxy)methyl]-2,3-dihydrofuran and 3-methoxybenzyne gave a 1:1 ratio of diastereomeric products 18a and 18b, which were chromatographically separable. The result from this reaction was comparable to that of the reaction with benzyne (entry 3 in Table 2). In the case of the slower-eluting isomer 4b obtained from the reaction with benzyne, the ABq pattern of the benzyloxy methylene protons is better separated (ΔνAB = 74.9 Hz), and the resonances are shifted upfield relative to the corresponding protons in the faster-eluting isomer 4a (ΔνAB = 16.2 Hz, Figure 3). We propose that this is due to the all cis relative stereochemistry in the slower-eluting isomer, which places the methylene protons in the shielding region of the BCB aryl ring. Additional evidence for the proposed structure assignment of 4a was obtained from NOESY data (Figure 3). Here a furanyl methylene proton (Hβ) at δ = 1.67 ppm showed strong correlations with the BCB bridgehead proton at δ = 4.11 ppm and a methylene proton at δ = 3.58 ppm. The correlation of this furanyl methylene proton to the methine proton at δ = 3.99–3.94 ppm was very weak (shown by the dashed arrow).
Figure 3.

Key data for the structure elucidation of the cycloadducts resulting from reactions with (S)-2-((benzyloxy)methyl)-2,3-dihydrofuran.
The regiochemistry of the cycloaddition leading to isomers 18a and 18b, which was anticipated from products 9 and 14, was supported by NOESY correlations between the OCH3 and the bridgehead OCH resonances in both diastereomers (Figure 3). The 1H NMR spectra of the two isomers of compound 18 shared common features with those of compound 4. In 18b, just as in 4b, the ABq resonances for the benzylic methylene protons are shifted upfield relative to 18a and are better separated (ΔνAB = 57.7 Hz). However, in the faster-eluting 18a the central lines of the ABq are very closely spaced (ΔνAB = 5.3 Hz) relative to the case of cycloadduct 4a. On the basis of the upfield shifts of the benzylic methylene protons in 18b (slower-eluting isomer), and the very weak NOESY correlation observed between the protons on either side of the furanyl-ether linkage (Figure 3), an all cis geometry is proposed in this case. Although this NOESY correlation was not observed in the corresponding isomer 4b the other close spectral features of pairs 4a and 18a as well as 4b and 18b, combined with the NOESY data for 4a supported the assignments. Additionally, compounds 4a and 18a share similarities in the splitting patterns of protons resonating at 1.6–2.0 and 3.5–3.7 ppm. Correspondingly, there are similarities in the splitting patterns of protons resonating at 1.9–2.3, 2.7–3.1, and 4.1–4.7 ppm in compounds 4b and 18b (see the spectra in the Supporting Information).
Evaluation of the Regiochemistry of the Cycloaddition and Computational Analyses
Electronic and steric influences have been argued to explain the regioselectivity in the cycloaddition reactions of unsymmetrical arynes with substituted furans.[10] Recently, computational methods have been utilized to better understand how cycloaddition and nucleophilic addition reactions to arynes are influenced by aryne structures.[11,12] In these computations, both the bond angle and the charge at each triply bonded aryne carbon atom have been considered.
For example, in DFT calculations, the C-1 atom of 3-methoxybenzyne is flatter (θ = 135°) than the C-2 atom (θ = 119°) and nucleophilic attack is predicted at C-1.[12c] This regioselectivity is also consistent with the calculated Mulliken charges (C-1 = 0.01e and C-2 = −0.10e). The regiochemistry observed here in the reactions of DHF and DHP with 3-methoxybenzyne are fully consistent with the computational results, leading to products 9 and 14, respectively. Hence, we adopted a similar analysis for 4-methoxy-, 3-methyl-, and 4-methylbenzyne (3-methoxybenzyne was reanalyzed for comparison to the literature data[12c]).
From the DFT-computed structure, 4-methoxybenzyne is quite different from the 3-methoxy isomer (Figure 4). In the former, angles at both alkynyl carbon atoms are very similar, and the calculated Mulliken charges are −0.021e at the C-1 atom and −0.004e at the C-2. On the basis of the bond angles, nucleophilic reaction by the electron-rich β-carbon atom of the enol ether could occur at both the C-1 and C-2 atoms. The Mulliken charges indicate that the C-2 atom may be preferred over C-1 for nucleophilic reaction. Interestingly, it was reported that cycloaddition of 4-methoxybenzyne with 3-methoxyfuran gave a 1:1 mixture of regioisomers,[10a] which seems to indicate that Mulliken charge partitioning might not be a good indicator of the regioselection with this aryne. Reactions of 4-methoxybenzyne with both DHF and DHP showed a significant preference for one product, which contrasts with the results obtained with 3-methoxyfuran. In both cases, the major product resulted from reaction of the electron-rich β-carbon atom of the enol ether at the C-1 atom of the aryne. One plausible explanation for this would be an asynchronous mechanism, in which nucleophilic reaction by the enol ether at the C-1 atom of the aryne results in accumulation of electron density more proximal to the inductively electron-withdrawing methoxy group (Scheme 2). In contrast, initial reaction at the C-2 position of the aryne would cause accumulation of electron density at the para-position to the methoxy group, a location that is influenced by its mesomeric electronic-donating effect.
Figure 4.

Structures of 4-methoxybenzyne, 3-methylbenzyne, and 4-methylbenzyne obtained from the B3LYP/6-31G(d) analysis.
Scheme 2.

Formation of regioisomeric products from 4-methoxybenzyne.
Although we have not been able to ascertain structures of the major/minor products unambiguously in the reactions of 3-methylbenzyne with DHF (11a and 11b) and DHP (isomers 16a and 16b), there is clearly a preference for one product in each case. On the basis of the DFT computed structure of 3-methylbenzyne (Figure 4) and the weak inductive effect of a methyl group, no selectivity would be expected. However, a 2.9:1 ratio of products 16a and 16b was obtained in the reaction with DHP and a modest 1.3:1 ratio of compounds 11a and 11b was observed in the reaction with DHF. If steric factors had some role in the regioselection, then 11a and 16a where the aryl methyl group and the oxygen atom are proximal would be expected as the major regioisomers. On the basis of this assumption, we performed energy minimizations by DFT on the two plausible minor isomers. The Haasnoot model was used to constrain only the bridgehead protons with the observed coupling constants.[35] The ball-and-stick and corresponding space-filling models are shown in Figure 5.
Figure 5.

B3LYP/6-31G(d) analysis of two regioisomeric products arising from the reactions of 3-methylbenzyne with DHP (Panel A) and with DHF (Panel B).
In the product from the reaction with DHP (Panel A in Figure 5), the distance between the carbon atoms of the aryl methyl and proximal methylene group is 3.96 Å, and the closest H–H distance between these groups is 3.20 Å. In the corresponding product arising from DHF (Panel B in Figure 5) the same C–C and H–H distances are 4.22 Å and 3.26 Å, respectively. Thus, if steric buttressing indeed has an influence, then this could account for the higher selectivity in the reaction with DHP, as compared to that with DHF.
Finally, with 4-methylbenzyne, in which the bond angles at the alkynyl carbon atoms of the aryne are again comparable, and where steric influence of the aryne methyl group is eliminated, a 1:1 ratio of regioisomeric products (12a and 12b) was observed, as would be expected.
From the standpoint of orbital interactions, ketene silyl acetals are expected to have higher HOMO energies than simple enol ethers, and this is implicated in their higher reactivity with arynes.[26g] In order to gain insight into the plausible reactivity order of various olefins with benzyne, we performed a DFT analysis. The LUMO energy of the benzyne triple bond was calculated at the Becke3LYP/6-311G++(d,p) level. The obtained value of – 2.351 eV was comparable to that reported (−2.355 eV) using the Becke3LYP/6-311G9(d,p).[36] Next, the HOMO energies of a related series of alkenes were calculated (Figure 6).
Figure 6.

HOMO energies of the olefins calculated by DFT at the Becke3LYP/6-311++(d,p) level.
From these data, the relative HOMO-energy-based ordering of olefins that emerges is 2-(tert-butyldimethylsilyloxy)DHF > 1,4-dioxene > DHF > DHP > 1-acetyl-2,3-dihydro-1H-pyrrole (a representative enamide) > cyclopentene. As expected, the HOMO energy of DHF is about 10.8 Kcal/mol lower than that of the structurally related 2-(t-butyldimethylsilyloxy)DHF. Although enol silyl ethers have been reported to produce lower yields in [2 + 2] reactions with arynes, possibly as a consequence of their lower HOMO energies,[26g] it is notable that the reactions of cyclic enol ethers reported here are generally good to excellent. All of the enol ethers in Figure 6 have now been shown to display meaningful reactivity towards arynes, complementing the reactions with some enamides and ketene acetals described earlier.
Further Transformations of the Cycloadducts
For characterization purposes as well as to demonstrate the possibilities for conversion into other substituted BCBs, compounds 1 and 2 were subjected to ether-bond cleavage (Scheme 3) with BBr3 in CH2Cl2, to afford the corresponding bromo alcohols in 86% and 94% yield. The bromo alcohols were acetylated to produce compounds 22 and 23. Here, a two-step conversion of cycloadduct 1 into bromo acetate 22 proceeded in 80% yield without purification of the intermediate bromo alcohol. Acetylation of the purified bromo alcohol obtained from cycloadduct 2 proceeded in 85% yield. Each bromo acetate was then converted into the corresponding azido acetate in good yield (24: 70%, 25: 74%). These experiments and the following NMR analyses proved to be important because the ether ring cleavage presented an intriguing scenario.
Scheme 3.

Unmasking BCBs by ether cleavage in the cycloadducts by reaction with using BBr3.
HMBC data on compounds 23 and 25 showed correlations between the acetate carbonyl resonances (δ = 171.3 ppm in 23 and δ = 171.4 ppm in 25) and the ester OCH2 resonances (multiplet at δ = 4.18–4.12 ppm in 23 and triplet at δ = 4.16 ppm in 25), indicative of the regiochemistry of the ring-opening step. In the case of compound 24, HMBC data showed correlations between the acetate carbonyl resonance (δ = 171.3 ppm) and the OCH2 proton resonances, which appear independently (each a triplet of doublets at δ = 4.36 ppm and δ = 4.30 ppm).
From a regiochemistry standpoint, we had initially assumed that complexation of BBr3 to the ether oxygen would be followed by delivery of the bromide to the less hindered carbon atom. However, the spectroscopic evidence described above indicated that this was not the case. Initial complexation of BBr3 to the oxygen atom can lead to two possible delivery modes of bromide (Scheme 3). In fact, this delivery occurs at the secondary carbon atom, plausibly because cleavage of a benzylic bond ensues. This selectivity in the ether-ring cleavage appears to parallel observations in which the cleavage reactivity order by BBr3 in CH2Cl2 has been shown to be benzylic > propargylic > methyl.[37] This is also consistent with computational results on the BBr3-mediated ether cleavage.[38]
NOESY correlations and coupling constant data provided insight into the relative stereochemistry in products. In the case of ring-opening product 23 a very weak correlation was observed between the CHBr resonance (singlet at δ = 5.01 ppm) and the vicinal methine resonance (broad resonance at δ = 3.74 ppm). Also, in compound 23 a singlet was observed for the CHBr resonance and a 1.5 Hz coupling constant was observed in its bromo alcohol precursor. Similarly, in the bromo alcohol leading to compound 22, the CHBr resonance appeared as a singlet (δ = 5.10 ppm), whereas a 1.8 Hz coupling constant was observed after acetylation. These are consistent with the small coupling constants reported (<2 Hz) between the methine protons in BCBs containing trans substituents.[39] Either intramolecular or bimolecular delivery of bromide at the benzylic carbon[38] will result in the same outcome.
Azido acetates 24 and 25, obtained by displacement of bromide with azide ion, should then have a cis relative stereochemistry. In compound 25, a strong NOESY correlation was observed between the CHN3 resonance (doublet at δ = 5.01 ppm) and the vicinal methine multiplet (δ = 3.78–3.74 ppm). Furthermore, in compounds 24 and 25 the coupling constants between the BCB methine protons are 4.8 and 4.7 Hz, respectively. These values are consistent with those reported for BCBs containing cis substituents,[39] and are comparable with that in the less strained cycloadduct 2 (4.5 Hz). The IR spectra of compounds 24 and 25 show bands at 2101 and 2102 cm−1, consistent with the N3 functionality. Whereas the examples in Scheme 3 are representative, conversions to other BCBs can be envisioned for various purposes.
Conclusions
In summary, a previously unstudied simple and modular [2 + 2] cycloadditions of readily availed cyclic enol ethers and arynes are described. They lead to facile functionalization of the cyclobutenyl ring and aryl moieties of BCBs, although with unsymmetrical arynes the regiochemistry is dependent upon the nature of the aryne. The reactions reported here are generally superior to those of acyclic enol ethers and acetates, which are plagued by low yields and significant competing side reactions. Further, the offer stereocontrol because each cycloaddition product possesses a cis ring junction despite the potentially stepwise process,[8,14,40] likely via zwitterionic intermediates. This is an advantage for additional manipulations, as shown by the preliminary demonstration of the ether cleavage leading to bromo alcohols, and conversion into cis-functionalized compounds. Facile, high-yielding reactions of 1,4-dioxene and arynes lead to cycloadducts that can be utilized to yield other BCBs, also with a fixed relative stereochemistry. In contrast, reactions between arynes and ketene acetals generally produce cyclobutenones, whereas the reactions described here yield products with two functionalized sp3 carbon atoms. Thus, this cycloaddition methodology appears to be a convenient platform from which a range of new stereochemically defined 1,2-disubstituted BCBs can be obtained. Because of the easy access to various aryne precursors[41–43] and the various functionalized BCBs that can be obtained from them, this chemistry is likely to see utility in wide-ranging areas, from organic synthesis to materials chemistry, and electronics.
Experimental Section
General Experimental Considerations
Thin-layer chromatography was performed on 200 μm aluminum-foil-backed silica gel plates. Column chromatographic purifications were performed on 200–300 mesh silica gel. CH3CN and CH2Cl2 were distilled from CaH2 prior to use. EtOAc and hexanes were distilled from CaSO4, and commercial CH2Cl2 was redistilled for chromatography. Commercially available aryne precursors were used in this study. Other commercially available compounds and reagents were used without further purification. 1H NMR spectra were recorded at 500 MHz in the solvents indicated and are referenced to residual protonated solvent resonances. 13C NMR spectra were recorded at 125 MHz in the solvents indicated and are referenced to the solvent resonances. Chemical shifts (δ) are reported in parts per million (ppm) and coupling constants (J) are in Hertz (Hz). Standard abbreviations are used to designate resonance multiplicities. For convenience, where identified, we have designated resonances of major isomers with a subscripted M and those of minor isomers with a subscripted m. When NMR spectroscopic assignments have been made, 2D NMR techniques such as COSY, NOESY, HMQC, and HBMC have been utilized.
General Procedure for the [2 + 2] Cycloaddition of Arynes and Enol Ethers
An oven-dried screw-cap vial containing a magnetic stirring bar was charged with the enol ether (0.36 mmol, 1.2 equiv), CsF (0.6 mmol, 2.0 equiv), 18-crown-6 (0.6 mmol, 2.0 equiv), and anhydrous CH3CN (volume used was to keep the mixture 0.125 M in the o-(trimethylsilyl)aryl triflate). Then the o-(trimethylsilyl)aryl triflate (0.3 mmol, 1.0 equiv) was added and the mixture was stirred at room temperature for 2–6 h. Upon completion of the reaction, the mixture was diluted with CH2Cl2 and washed with water. The organic layer was dried with anhydrous Na2SO4 and concentrated under reduced pressure. Purification of the crude product by chromatography on silica gel (EtOAc in hexanes) afforded the desired benzocyclobutanes. For specific details, please see the individual compound descriptions that follow.
2,3,3a,7b-Tetrahydrobenzo[3,4]cyclobuta[1,2-b]furan (1)

This compound was purified by column chromatography with 5% EtOAc in hexanes and isolated as a colorless oil (36.0 mg, 82 %). Rf (SiO2/10% EtOAc in hexanes) = 0.60. 1H NMR (500 MHz, CDCl3): δ = 7.34 (t, J = 7.3 Hz, 1H, Ar-H), 7.29 (t, J = 7.3 Hz, 1H, Ar-H), 7.21 (d, J = 7.3 Hz, 1H, Ar-H), 7.16 (d, J = 7.3 Hz, 1H, Ar-H), 5.56 (d, J = 3.4 Hz, 1H), 4.14–4.10 (m, 2H), 3.58 (ddd, J = 5.2, 9.3, 11.7 Hz, 1H), 1.94 (dd, J = 5.2, 12.4 Hz, 1H), 1.90–1.82 (m, 1H). 13C NMR (125 MHz, CDCl3): δ = 145.6, 144.3, 129.8, 128.1, 123.4, 122.5, 81.4, 67.2, 49.4, 29.5. HRMS (ESI/TOF): calcd for C10H11O [M + H]+ 147.0804; found 147.0821.
3,4,4a,8b-Tetrahydro-2H-benzo[3,4]cyclobuta[1,2-b]pyran (2)

This compound was purified by column chromatography with 5% EtOAc in hexanes and isolated as a colorless oil (34.4 mg, 72 %). Rf (SiO2/10% EtOAc in hexanes) = 0.61. 1H NMR (500 MHz, CDCl3): δ = 7.35 (t, J = 7.5 Hz, 1H, Ar-H), 7.32–7.29 (m, 2H, Ar-H,), 7.18 (d, J = 7.0 Hz, 1H, Ar-H), 5.16 (d, J = 4.5 Hz, 1H), 3.86–3.76 (m, 2H), 3.71 (app q, Japp = 5.5 Hz, 1H), 2.25–2.18 (m, 1H), 1.96–1.90 (m, 1H), 1.62–1.57 (m, 1H), 1.51–1.46 (m, 1H). 13C NMR (125 MHz, CDCl3): δ = 147.9, 146.2, 129.5, 127.4, 123.7, 122.9, 73.8, 62.1, 43.1, 23.6, 20.1. HRMS (ESI/TOF): calcd for C11H13O [M + H]+ 161.0961; found 161.0984.
7b-Butyl-2,3,3a,7b-tetrahydrobenzo[3,4]cyclobuta[1,2-b]furan (3)

This compound was purified by column chromatography with 5% EtOAc in hexanes and isolated as a light yellow oil (41.7 mg, 69 %). Rf (SiO2/10% EtOAc in hexanes) = 0.59. 1H NMR (500 MHz, CDCl3): δ = 7.29 (t, J = 7.4 Hz, 1H, Ar-H), 7.25 (t, J = 7.2 Hz, 1H, Ar-H), 7.16 (d, J = 7.1 Hz, 1H, Ar-H), 7.12 (d, J = 7.0 Hz, 1H, Ar-H), 4.10–4.07 (m, 1H, OCHaHb), 3.71 (t, J = 4.0 Hz, 1H, H-3a), 3.49 (app q, J = 8.6 Hz, 1H, OCHaHb), 2.02 (dt, J = 5.8, 13.8 Hz, 1H), 1.98 (dt, J = 5.8, 13.8 Hz, 1H), 1.88–1.85 (m, 2H, H-3), 1.59–1.44 (m, 2H, H-2’), 1.38 (app sextet, J = 7.3 Hz, 2H, H-3’), 0.92 (t, J = 7.2 Hz, 3H, 4’-CH3). 13C NMR (125 MHz, CDCl3): δ = 146.8, 144.0, 129.6, 128.0, 122.7, 122.4, 92.3, 67.8, 52.7, 34.6, 30.1, 27.9, 23.4, 14.3. HRMS (ESI/TOF): calcd for C14H19O [M + H]+ 203.1430; found 203.1431.
2-[(Benzyloxy)methyl]-2,3,3a,7b-tetrahydrobenzo[3,4]cyclobuta[1,2-b]furan (faster-eluting isomer 4a)

This compound was obtained by column chromatography of the mixture with 15% EtOAc in hexanes and isolated as a colorless gummy oil (19.1 mg, 24 %). Rf (SiO2/10% EtOAc in hexanes) = 0.43. 1H NMR (500 MHz, CDCl3): δ = 7.33–7.31 (m, 4H, Ar-H), 7.29–7.24 (m, 3H, Ar-H), 7.20 (d, J = 7.0 Hz, 1H, Ar-H), 7.11 (d, J = 6.9 Hz, 1H, Ar-H), 5.59 (d, J = 3.3 Hz, 1H, H-7a), 4.59, 4.56 (ABq, J = 12.3 Hz, 2H, OCH2Ph), 4.11 (dd, J = 3.4, 8.2 Hz, 1H, H-3a), 3.99–3.94 (m, 1H, H-2), 3.64 (dd, J = 3.3, 10.2 Hz, 1H), 3.58 (dd, J = 5.8, 10.6 Hz, 1H), 1.96 (dd, J = 4.6, 12.6 Hz, 1H, H-3), 1.67 (ddd, J = 8.4, 10.8, 12.2 Hz, 1H, H-3). 13C NMR (125 MHz, CDCl3): δ = 147.8, 144.6, 138.4, 129.9, 128.6, 128.2, 128.0, 127.8, 123.8, 122.5, 95.0, 82.0, 73.6, 71.4, 49.6, 32.1. HRMS (ESI/TOF): calcd for C18H18O2Na [M + Na]+ 289.1199; found 289.1188.
2-[(Benzyloxy)methyl]-2,3,3a,7b-tetrahydrobenzo[3,4]cyclobuta[1,2-b]furan (slower-eluting isomer 4b)
This compound was obtained by column chromatography of the mixture with 15% EtOAc in hexanes and isolated as a colorless gummy oil (19.5 mg, 24 %). Rf (SiO2/10% EtOAc in hexanes) = 0.41. 1H NMR (500 MHz, CDCl3): δ = 7.30–7.22 (m, 5H, Ar-H), 7.20–7.18 (m, 2H, Ar-H), 7.14 (d, J = 7.3 Hz, 1H, Ar-H), 7.05 (d, J = 7.0 Hz, 1H, Ar-H), 5.59 (d, J = 4.0 Hz, 1H, H-7a), 4.66–4.61 (m, 1H, H-2), 4.41, 4.26 (ABq, J = 12.1 Hz, 2H, OCH2Ph), 4.19–4.16 (m, 1H), 2.93 (dd, J = 7.3, 9.9 Hz, 1H), 2.77 (dd, J = 5.9, 9.9 Hz, 1H), 2.20 (td, J = 8.8, 13.2 Hz, 1H), 1.92 (dt, J = 2.4, 13.2 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ = 147.9, 147.6, 138.3, 129.8, 128.5, 128.2, 127.9, 127.7, 123.0, 122.7, 84.4, 83.1, 73.6, 73.3, 49.6, 31.9. HRMS (ESI/TOF): calcd for C18H18O2Na [M + Na]+ 289.1199; found 289.1198.
7-Ethoxybicyclo[4.2.0]octa-1,3,5-triene (5)

This compound was purified by column chromatography with 5% EtOAc (5 %) in hexanes and isolated as a colorless oil (22.0 mg, 50 %). Rf (SiO2/10% EtOAc in hexanes) = 0.70. 1H NMR (500 MHz, CDCl3): δ = 7.33–7.28 (m, 2H, Ar-H), 7.26–7.25 (m, 1H, Ar-H), 7.17 (d, J = 6.8 Hz, 1H, Ar-H), 5.08 (dd, J = 1.0, 3.9 Hz, 1H), 3.75 (app quint, JAB = 15.2 Hz and Jvic = 7.3 Hz, 1H), 3.69 (app quint, JAB = 15.2 Hz and Jvic = 7.3 Hz, 1H), 3.50 (dd, J = 4.4, 14.1 Hz, 1H), 3.16 (d, J = 14.2 Hz, 1H), 1.32 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ = 146.4, 142.8, 129.5, 127.2, 123.7, 122.9, 76.8, 64.5, 38.9, 15.6. HRMS (ESI/TOF): calcd for C10H13O [M + H]+ 149.0961; found 149.0988.
2,3,4a,8b-Tetrahydrobenzo[3,4]cyclobuta[1,2-b][1,4]dioxine (6)

This compound was purified by column chromatography with 5% EtOAc in hexanes and isolated as a colorless oil (40.9 mg, 84 %). Rf (SiO2/10% EtOAc in hexanes) = 0.40. 1H NMR (500 MHz, CDCl3): δ = 7.39–7.38 (m, 2H, Ar-H), 7.35–7.33 (m, 2H, Ar-H), 5.36 (s, 2H), 3.78 (s, 4H). 13C NMR (125 MHz, CDCl3): δ = 146.0, 130.1, 123.9 , 74.0, 61.9. HRMS (ESI/TOF): calcd for C10H11O2 [M + H]+ 163.0754; found 163.0758.
4-Ethoxy-3a,4,9,9a-tetrahydronaphtho[2,3-c]furan-1,3-dione (7)

Benzocyclobutene 5 (29.6 mg, 0.2 mmol) and maleic anhydride (22.0 mg, 0.22 mmol) in toluene (2 mL) were placed in an oven-dried screw-cap vial also containing a magnetic stirring bar. The mixture was stirred at 120 °C for 18 h, after which time the reaction was complete. The reaction mixture was partitioned between CH2Cl2 and water. The organic layer was dried with anhydrous Na2SO4 and concentrated under reduced pressure. Purification of the crude reaction mixture by chromatography on a silica gel column with 30% EtOAc in hexanes as eluting solvent afforded compound 7 as a light yellow solid (40.4 mg, 82 %). Rf (SiO2/10% EtOAc in hexanes) = 0.11. 1H NMR (500 MHz, C6D6): δ = 6.99 (t, J = 7.3 Hz, 1H, Ar-H), 6.92 (t, J = 7.3 Hz, 1H, Ar-H), 6.78 (d, J = 7.3 Hz, 1H), 6.72 (d, J = 7.3 Hz, 1H), 4.42 (d, J = 3.4 Hz, 1H, H-4), 3.16 (dd, J = 8.8, 14.7 Hz, 1H, CHaHb), 2.86, 2.76 (q of ABq, J = 7.3, 14.0 Hz, 2H, OCHaHb), 2.69 (dd, J = 8.8, 14.7 Hz, 1H, CHaHb), 2.47–2.41 (m, 1H), 2.39 (dd, J = 3.5, 10.5 Hz, 1H), 0.69 (t, J = 6.8 Hz, 3H). 13C NMR (125 MHz, C6D6): δ = 174.5, 171.5, 136.4, 135.1, 130.0, 129.4, 129.0, 127.1, 76.4, 64.5, 48.1, 38.1, 27.7, 15.3. HRMS (ESI/TOF): calcd for C14H15O4 [M + H]+ 247.0965; found 247.0966.
5,6-Dimethoxy-2,3,3a,7b-tetrahydrobenzo[3,4]cyclobuta[1,2-b]furan (8)

This compound was purified by column chromatography with 15% EtOAc in hexanes and isolated as a white solid (50.5 mg, 82 %). Rf (SiO2/20% EtOAc in hexanes) = 0.49. 1H NMR (500 MHz, CDCl3): δ = 6.75 (s, 1H, Ar-H), 6.68 (s, 1H, Ar-H), 5.44 (d, J = 3.4 Hz, 1H), 4.06 (t, J = 8.3 Hz, 1H), 3.98 (dd, J = 3.4, 7.6 Hz, 1H), 3.86 (s, 3H, OMe), 3.84 (s, 3H, OMe), 3.55 (ddd, J = 5.0, 9.0, 11.7 Hz, 1H), 1.86 (dd, J = 4.9, 12.2 Hz, 1H), 1.79–1.72 (m, 1H). 13C NMR (125 MHz, CDCl3): δ = 151.5, 150.3, 137.0, 135.2, 106.8, 105.9, 80.8, 67.1, 56.3 (2 × C), 48.7, 29.3. HRMS (ESI/TOF): calcd for C12H15O3 [M + H]+ 207.1016; found 207.1013.
7-Methoxy-2,3,3a,7b-tetrahydrobenzo[3,4]cyclobuta[1,2-b]furan (9)

This compound was purified by column chromatography with 10% EtOAc in hexanes and isolated as a colorless gummy oil (44.5 mg, 84 %). Rf (SiO2/10% EtOAc in hexanes) = 0.53. 1H NMR (500 MHz, CDCl3): δ = 7.22 (dd, J = 7.8, 8.8 Hz, 1H, Ar-H), 6.72 (d, J = 8.8 Hz, 1H, Ar-H), 6.69 (d, J = 6.8 Hz, 1H, Ar-H), 5.64 (d, J = 3.4 Hz, 1H), 4.10 (t, J = 8.0 Hz, 1H), 4.04 (dd, J = 3.6, 8.0 Hz, 1H), 3.97 (s, 3H, OMe), 3.66 (ddd, J = 4.8, 8.7, 11.2 Hz, 1H), 1.92 (dd, J = 5.1, 12.5 Hz, 1 H), 1.88–1.80 (m, 1 H). 13C NMR (125 MHz, CDCl3): δ = 154.9, 147.3, 131.7, 126.8, 115.0, 114.5, 81.4, 67.0, 57.4, 49.2, 29.5. HRMS (ESI/TOF): calcd for C11H13O2 [M + H]+ 177.0910; found 177.0920.
5- and 6-Methoxy-2,3,3a,7b-tetrahydrobenzo[3,4]cyclobuta[1,2-b]furan (10a and 10b: ratio of isomers = 2.2:1)

This mixture of compounds was purified by column chromatography with 10% EtOAc in hexanes and isolated as a yellow gummy oil (43.1 mg, 82 %). Rf (SiO2/10% EtOAc in hexanes) = 0.53. 1H NMR (500 MHz, CDCl3): δ = 7.08 (d, J = 8.3 Hz, 1H, Ar-Hm), 7.02 (d, J = 7.8 Hz, 1H, Ar-HM), 6.86 (dd, J = 2.2, 8.0 Hz, 1H, Ar-HM), 6.80 (dd, J = 1.9, 8.3 Hz, 1H, Ar-Hm), 6.76 (d, J = 1.9 Hz, 1H, Ar-HM), 6.68 (d, J = 1.4 Hz, 1H, Ar-Hm), 5.46 (d, J = 3.4 Hz, 2HM+m), 4.07 (t, J = 8.3 Hz, 1HM), 4.06 (t, J = 8.0 Hz, 1Hm), 4.00 (dd, J = 3.2, 7.6 Hz, 2HM+m), 3.78 (s, 3H, OMem), 3.77 (s, 3H, OMeM), 3.57–3.52 (m, 2HM+m), 1.90–1.74 (m, 4HM+m). The ratio of isomers was determined by integration of the methoxy resonances at δ = 3.78 and 3.77 ppm. 13C NMR (125 MHz, CDCl3): δ = 161.4 (Cm), 160.1 (CM), 146.5 (Cm), 145.0 (CM), 137.1 (CM), 135.8 (Cm) 124.8 (Cm), 123.7 (CM), 117.1 (CM), 114.9 (Cm), 108.1 (CM), 107.6 (Cm), 80.59 (Cm), 80.57 (CM), 67.3 (CM), 67.0 (Cm), 55.6 (CM), 55.5 (Cm), 48.6 (Cm), 48.4 (CM), 29.6 (CM), 29.3 (Cm). HRMS (ESI/TOF): calcd for C11H13O2 [M + H]+ 177.0910; found 177.0927.
4- and 7-Methyl-2,3,3a,7b-tetrahydrobenzo[3,4]cyclobuta[1,2-b]furan (11a and 11b: ratio of isomers = 1.3:1)

Unambiguous determination of the major isomer was not possible.
This mixture of compounds was purified by column chromatography with 5% EtOAc in hexanes and isolated as a yellow oil (41.1 mg, 86 %). Rf (SiO2/10% EtOAc in hexanes) = 0.58. 1H NMR (500 MHz, CDCl3): δ = 7.22 (t, J = 7.6 Hz, 1H, Ar-H), 7.19 (t, J = 8.3 Hz, 1H, Ar-H), 7.10 (d, J = 7.8 Hz, 1H, Ar-H), 7.05 (d, J = 7.8 Hz, 1H, Ar-H), 7.01 (d, J = 7.3 Hz, 1H, Ar-H), 6.94 (d, J = 7.3 Hz, 1H, Ar-H), 5.55 (d, J = 3.4 Hz, 1H), 5.51 (d, J = 3.5 Hz, 1H), 4.12–4.03 (m, 4H), 3.59–3.54 (m, 2H), 2.28 (s, 3H, Me), 2.25 (s, 3H, Me), 1.94–1.88 (m, 2H), 1.86–1.78 (m, 2H). The ratio of isomers was determined by integration of the methyl resonances at δ = 2.28 and 2.25 ppm, and of OCH doublets at 5.55 and 5.51 ppm. 13C NMR (125 MHz, CDCl3): δ = 145.2, 143.82, 143.81, 142.8, 134.1, 132.8, 130.5, 130.2, 128.9, 128.6, 120.6, 119.5, 80.83, 80.80, 67.24, 67.21, 48.8, 48.3, 29.4, 28.5, 16.8. HRMS (ESI/TOF): calcd for C11H13O [M + H]+ 161.0961; found 161.0960.
5- and 6-Methyl-2,3,3a,7b-tetrahydrobenzo[3,4]cyclobuta[1,2-b]furan (12a and 12b: ratio of isomers = 1:1)
![]()
This mixture of compounds was purified by column chromatography with 5% EtOAc in hexanes and isolated as a colorless oil (37.6 mg, 78 %). Rf (SiO2/10% EtOAc in hexanes) = 0.58. 1H NMR (500 MHz, CDCl3): δ = 7.13 (d, J = 7.7 Hz, 1H, Ar-H), 7.10–7.07 (m, 2H, Ar-H), 7.02 (d, J = 6.6 Hz, 1H, Ar-H), 7.01 (s, 1H, Ar-H), 6.95 (s, 1H, Ar-H), 5.51–5.50 (m, 2H), 4.08 (t, J = 8.2 Hz, 2H), 4.05–4.03 (m, 2H), 3.58–3.53 (m, 2H), 2.36 (s, 3H, Me), 2.35 (s, 3H, Me), 1.92–1.88 (m, 2H), 1.85–1.77 (m, 2H). The ratio of isomers was determined by integration of the methyl resonances at δ = 2.36 and 2.35 ppm. This is a best possible estimate because of overlapping resonances. 13C NMR (125 MHz, CDCl3): δ = 145.6, 144.3, 142.3, 141.1, 139.8, 137.8, 130.7, 129.0, 123.9, 123.2, 123.0, 122.2, 81.1, 81.0, 67.2, 67.1, 49.0, 48.8, 29.6, 29.5, 22.4, 22.2. HRMS (ESI/TOF): calcd for C11H13O [M + H]+ 161.0961; found 161.0968.
6,7-Dimethoxy-3,4,4a,8b-tetrahydro-2H-benzo[3,4]cyclobuta[1,2-b]pyran (13)

This compound was purified by column chromatography with 15% EtOAc in hexanes and isolated as a colorless oil (51.7 mg, 78 %). Rf (SiO2/20% EtOAc in hexanes) = 0.49. 1H NMR (500 MHz, CDCl3): δ = 6.84 (s, 1H, Ar-H), 6.71 (s, 1H, Ar-H), 5.02 (d, J = 4.5 Hz, 1H), 3.86 (s, 3H, OMe), 3.85 (s, 3H, OMe), 3.81–3.76 (m, 1H), 3.74–3.68 (m, 1H), 3.57 (app q, Japp = 5.0 Hz, 1H), 2.14–2.08 (m, 1H), 1.85 (app sext, Japp = 6.5 Hz, 1H), 1.55–1.52 (m, 1H), 1.48–1.43 (m, 1H). 13C NMR (125 MHz, CDCl3): δ = 151.5, 149.9, 139.4, 137.1, 107.2, 106.4, 73.2, 61.9, 56.32, 56.31, 42.6, 23.7, 20.0. HRMS (ESI/TOF): calcd for C13H17O3 [M + H]+ 221.1172; found 221.1169.
8-Methoxy-3,4,4a,8b-tetrahydro-2H-benzo[3,4]cyclobuta[1,2-b]pyran (14)

This compound was purified by column chromatography with 10% EtOAc in hexanes and isolated as a yellow oil (48.0 mg, 84 %). Rf (SiO2/10% EtOAc in hexanes) = 0.53. 1H NMR (500 MHz, CDCl3): δ = 7.24 (dd, J = 7.3, 8.3 Hz, 1H, Ar-H), 6.74 (d, J = 8.3 Hz, 1H, Ar-H), 6.73 (d, J = 7.0 Hz, 1H, Ar-H), 5.23 (d, J = 4.9 Hz, 1H), 3.95 (s, 3H, OMe), 3.88–3.83 (m, 1H), 3.80–3.75 (m, 1H), 3.66 (app q, Japp = 5.4 Hz, 1H), 2.20–2.13 (m, 1H), 1.92 (dt, J = 6.0, 12.1 Hz, 1H), 1.63–1.55 (m, 1H), 1.52–1.44 (m, 1H). 13C NMR (125 MHz, CDCl3): δ = 155.7, 149.6, 131.5, 128.9, 114.9, 114.4, 73.6, 61.8, 57.1, 43.0, 23.5, 19.9. HRMS (ESI/TOF): calcd for C12H15O2 [M + H]+ 191.1067; found 191.1068.
6- and 7-Methoxy-3,4,4a,8b-tetrahydro-2H-benzo[3,4]cyclobuta[1,2-b]pyran (15a and 15b: ratio of isomers = 2:1)

This mixture of compounds was purified by column chromatography with 10% EtOAc in hexanes and isolated as a colorless gummy oil (41.8 mg, 73 %). Rf (SiO2/10% EtOAc in hexanes) = 0.54. 1H NMR (500 MHz, CDCl3): δ = 7.18 (d, J = 8.3 Hz, 1H, Ar-Hm), 7.05 (d, J = 7.9 Hz, 1H, Ar-HM), 6.88 (d, J = 9.0 Hz, 1H, Ar-HM), 6.87 (s, 1H, Ar-HM), 6.81 (dd, J = 1.8, 8.3 Hz, 1H, Ar-Hm), 6.72 (s, 1H, Ar-Hm), 5.06 (d, J = 5.1 Hz, 1HM), 5.05 (d, J = 4.8 Hz, 1Hm), 3.82–3.70 (m, 4H), 3.80 (s, 3H, OMem), 3.79 (s, 3H, OMeM), 3.60 (app q, Japp = 5.4 Hz, 2HM+m), 2.18–2.10 (m, 2HM+m), 1.91–1.82 (m, 2HM+m), 1.59–1.51 (m, 2HM+m), 1.50–1.41 (m, 2HM+m). The ratio of isomers was determined by integration of the methoxy resonances at δ = 3.80 and 3.79 ppm. 13C NMR (125 MHz, CDCl3): δ = 161.3 (Cm), 159.7 (CM), 148.8 (CM), 146.9 (Cm), 139.4 (CM), 137.7 (Cm), 125.0 (Cm), 124.1 (CM), 116.8 (CM), 114.4 (Cm), 108.6 (CM), 108.1 (Cm) 73.01 (CM), 72.99 (Cm), 62.2 (CM), 61.8 (Cm), 55.6 (CM), 55.5 (Cm), 42.5 (Cm), 42.1 (CM), 23.7 (CM), 23.4 (Cm), 20.0 (CM), 19.9 (Cm). HRMS (ESI/TOF): calcd for C12H15O2 [M + H]+ 191.1067; found 191.1071.
5- and 8-Methyl-3,4,4a,8b-tetrahydro-2H-benzo[3,4]cyclobuta[1,2-b]pyran (16a and 16b: ratio of isomers = 2.9:1)

Unambiguous determination of the major isomer was not possible.
This mixture of compounds was purified by column chromatography with 5% EtOAc in hexanes and isolated as a colorless gummy oil (42.9 mg, 82 %). Rf (SiO2/10% EtOAc in hexanes) = 0.58. 1H NMR (500 MHz, CDCl3): δ = 7.22 (t, J = 7.6 Hz, 1H, Ar-HM), 7.17 (t, J = 7.3 Hz, 1H, Ar-Hm), 7.09 (d, J = 7.4 Hz, 2H, Ar-Hm), 7.04 (d, J = 7.8 Hz, 1H, Ar-HM), 6.96 (d, J = 7.4 Hz, 1H, Ar-HM), 5.11 (d, J = 4.9 Hz, 1HM), 5.08 (d, J = 4.8 Hz, 1Hm), 3.81–3.71 (m, 4H), 3.66 (app q, Japp = 5.6 Hz, 1Hm), 3.63 (app q, Japp = 5.6 Hz, 1HM), 2.30 (s, 3H, MeM), 2.22 (s, 3H, Mem), 2.20–2.12 (m, 2HM+m), 1.94–1.85 (m, 2HM+m), 1.62–1.52 (m, 2HM+m), 1.48–1.39 (m, 2HM+m). The ratio of isomers was determined by integration of the methyl resonances at δ = 2.30 and 2.22 ppm. 13C NMR (125 MHz, CDCl3): δ = 147.5 (CM), 146.3 (Cm), 145.7 (Cm), 144.9 (CM), 134.6 (CM), 133.4 (Cm), 130.3 (Cm), 129.9 (CM), 128.3 (CM), 127.9 (Cm), 120.9 (Cm), 120.0 (CM), 73.5 (CM), 73.3 (Cm), 62.1 (Cm), 62.1 (CM), 42.8 (CM), 42.4 (Cm), 23.5 (CM), 22.8 (Cm), 20.2 (Cm), 20.0 (CM), 17.1 (2 × CM+m). HRMS (ESI/TOF): calcd for C12H15O [M + H]+ 175.1117; found 175.1116.
7b-Butyl-5,6-dimethoxy-2,3,3a,7b-tetrahydrobenzo[3,4]cyclobuta[1,2-b]furan (17)

This compound was purified by column chromatography with 15% EtOAc in hexanes and isolated as a yellow oil (53.0 mg, 67 %). Rf (SiO2/20% EtOAc in hexanes) = 0.37. 1H NMR (500 MHz, CDCl3): δ = 6.71 (s, 1H, Ar-H), 6.68 (s, 1H, Ar-H), 4.07–4.04 (m, 1H), 3.86 (s, 3H, OMe), 3.85 (s, 3H, OMe), 3.62 (d, J = 7.7 Hz, 1H), 3.50–3.45 (m, 1H), 1.99–1.94 (m, 2H), 1.83–1.78 (m, 2H), 1.53–1.42 (m, 2H), 1.36 (app sext, J = 7.3 Hz, 2H), 0.91 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ = 151.2, 150.1, 137.6, 135.1, 106.1, 105.8, 91.6, 67.6, 56.3, 56.2, 52.0, 34.7, 29.8, 27.9, 23.3, 14.2. HRMS (ESI/TOF): calcd for C16H23O3 [M + H]+ 263.1642; found 263.1625.
2-[(Benzyloxy)methyl]-7-methoxy-2,3,3a,7b-tetrahydrobenzo[3,4]cyclobuta[1,2-b]furan (faster-eluting isomer 18a)

This compound was obtained by column chromatography of the mixture with 20% EtOAc in hexanes and isolated as a colorless oil (22.3 mg, 25 %). Rf (SiO2/20% EtOAc in hexanes) = 0.35. 1H NMR (500 MHz, CDCl3): δ = 7.36–7.32 (m, 4H, Ar-H), 7.30–7.27 (m, 1H, Ar-H), 7.22 (dd, J = 7.3, 8.4 Hz, 1H, Ar-H), 6.71 (d, J = 8.4 Hz, 1H, Ar-H), 6.69 (d, J = 7.3 Hz, 1H, Ar-H), 5.71 (d, J = 3.3 Hz, 1H, H-7a), 4.59, 4.58 (ABq, J = 12.1 Hz, 2H, OCH2Ph), 4.12–4.09 (m, 2H), 3.99 (s, 3H, OMe), 3.70 (dd, J = 3.3, 10.6 Hz, 1H), 3.61 (dd, J = 5.5, 10.6 Hz, 1H), 1.97 (dd, J = 4.8, 12.5 Hz, 1H, H-3), 1.74 (ddd, J = 8.5, 11.0, 12.5 Hz, 1H, H-3). 13C NMR (125 MHz, CDCl3): δ = 154.9, 147.5, 138.3, 131.8, 128.5, 127.9, 127.8, 127.0, 115.2, 114.4, 81.9, 77.4, 73.6, 71.2, 57.6, 49.4, 32.1. HRMS (ESI/TOF): calcd for C19H20O3Na [M + Na]+ 319.1305; found 319.1286.
2-[(Benzyloxy)methyl]-7-methoxy-2,3,3a,7b-tetrahydrobenzo[3,4]cyclobuta[1,2-b]furan (slower-eluting isomer 18b)
This compound was obtained by column chromatography of the mixture with 20% EtOAc in hexanes and isolated as a colorless oil (24.2 mg, 27 %). Rf (SiO2/20% EtOAc in hexanes) = 0.33. 1H NMR (500 MHz, CDCl3): δ = 7.31–7.25 (m, 3H, Ar-H), 7.21 (d, J = 7.0 Hz, 2H, Ar-H), 7.16 (dd, J = 7.0, 8.2 Hz, 1H, Ar-H), 6.66 (d, J = 8.2 Hz, 1H, Ar-H), 6.64 (d, J = 7.3 Hz, 1H, Ar-H), 5.70 (d, J = 4.0 Hz, 1H, H-7a), 4.66–4.62 (m, 1H, H-2), 4.41, 4.29 (ABq, J = 12.1 Hz, 2H, OCH2Ph), 4.14 (dd, J = 3.2, 8.6 Hz, 1H), 4.00 (s, 3H, OMe), 3.04 (dd, J = 7.5, 9.7 Hz, 1H), 2.87 (dd, J = 6.2, 9.5 Hz, 1H), 2.21 (td, J = 9.1, 13.2 Hz, 1H), 1.94 (td, J = 2.2, 13.2 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ = 154.1, 149.5, 138.3, 131.7, 130.1, 128.5, 127.9, 127.7, 115.4, 114.6, 84.1, 83.0, 73.6, 73.4, 57.7, 49.4, 32.1. HRMS (ESI/TOF): calcd for C19H20O3Na [M + Na]+ 319.1305; found 319.1301.
7-Ethoxy-3,4-dimethoxybicyclo[4.2.0]octa-1,3,5-triene (19)

This compound was purified by column chromatography with 15% EtOAc in hexanes and isolated as a colorless oil (31.9 mg, 51 %). Rf (SiO2/20% EtOAc in hexanes) = 0.39. 1H NMR (500 MHz, CDCl3): δ = 6.83 (s, 1H, Ar-H), 6.71 (s, 1H, Ar-H), 4.96 (dd, J = 1.3, 4.4 Hz, 1H), 3.85 (s, 3H, OMe), 3.84 (s, 3H, OMe), 3.70–3.61 (m, 2H), 3.36 (dd, J = 4.0, 13.6 Hz, 1H), 3.02 (d, J = 13.6 Hz, 1H), 1.28 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ = 151.4, 149.7, 137.2, 134.3, 107.3, 106.8, 76.1, 64.2, 56.39, 53.35, 38.3, 15.6. HRMS (ESI/TOF): calcd for C12H17O3 [M + H]+ 209.1172; found 209.1159.
6,7-Dimethoxy-2,3,4a,8b-tetrahydrobenzo[3,4]cyclobuta[1,2-b][1,4]dioxine (20)

This compound was purified by column chromatography with 15% EtOAc in hexanes and isolated as a light yellow solid (52.2 mg, 78 %). Rf (SiO2/20% EtOAc in hexanes) = 0.37. 1H NMR (500 MHz, CDCl3): δ = 6.87 (s, 2H, Ar-H), 5.27 (s, 2H), 3.88 (s, 6H, OMe), 3.76 (d, J = 2.2 Hz, 4H). 13C NMR (125 MHz, CDCl3): δ = 152.0, 137.6, 106.5, 73.6, 61.5, 56.3. HRMS (ESI/TOF): calcd for C12H15O4 [M + H]+ 223.0965; found 223.0945.
5-Methoxy-2,3,4a,8b-tetrahydrobenzo[3,4]cyclobuta[1,2-b][1,4]dioxine (21)

This compound was purified by column chromatography with 10% EtOAc in hexanes and isolated as a colorless oil (43.7 mg, 76 %). Rf (SiO2/20% EtOAc in hexanes) = 0.43. 1H NMR (500 MHz, CDCl3): δ = 7.30 (t, J = 7.7 Hz, 1H, Ar-H), 6.92 (d, J = 7.0 Hz, 1H, Ar-H), 6.83 (d, J = 8.5 Hz, 1H, Ar-H), 5.45 (d, J = 4.0 Hz, 1H), 5.31 (d, J = 4.0 Hz, 1H), 3.94 (s, 3H, OMe), 3.84–3.78 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 155.9, 147.6, 132.0, 129.1, 116.5, 115.8, 73.8, 73.7, 61.6, 61.5, 57.2. HRMS (ESI/TOF): calcd for C11H13O3 [M + H]+ 193.0859; found 193.0852.
2-[8-Azidobicyclo[4.2.0]octa-1(6),2,4-trien-7-yl]ethyl acetate (24)
Step 1: synthesis of 2-[8-bromobicyclo[4.2.0]octa-1(6),2,4-trien-7-yl]ethyl acetate (22).

Benzocyclobutene 1 (51.2 mg, 0.35 mmol) and BBr3 in CH2Cl2 (0.1 m, 3.5 mL) were placed in an oven-dried 10 mL round-bottomed flask, also containing a magnetic stirring bar, at 0 °C. The mixture was stirred at 0 °C for 30 min. Upon completion, the reaction was quenched with water, and the layers were separated. The aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried with anhydrous Na2SO4, and concentrated under reduced pressure. The crude material was dissolved in CH2Cl2 (3.5 mL) at 0 °C. Ac2O (40 µL, 0.42 mmol) and DMAP (4.3 mg, 35 µmol) were added, and the mixture was stirred at 0 °C for 1 h. Upon completion, the reaction was quenched with water, and the layers were separated. The aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried with anhydrous Na2SO4, and concentrated under reduced pressure. The crude material was chromatographed on a silica gel column with 5–10 % EtOAc in hexanes to yield compound 22 as a colorless liquid (75.1 mg, 80 %). Rf (SiO2/10% EtOAc in hexanes) = 0.39. 1H NMR (500 MHz, CDCl3): δ = 7.33–7.31 (m, 2H, Ar-H), 7.21–7.20 (m, 1H, Ar-H), 7.14–7.13 (m, 1H, Ar-H), 5.08 (d, J = 1.8 Hz, 1H), 4.35–4.30 (m, 1H), 4.26 (dt, J = 5.9, 11.7 Hz, 1H), 3.79 (t, J = 7.1 Hz, 1H), 2.19–2.06 (m, 2H), 2.10 (s, 3H). 13C NMR (125 MHz, CDCl3): δ = 171.2, 144.7, 143.6, 130.3, 129.0, 123.0, 122.8, 62.8, 55.2, 46.9, 31.6, 21.2. HRMS (ESI/TOF): calcd. for C12H13BrO2Na [M + Na]+ 290.9991; found 290.9999.
Step 2: synthesis of 2-[8-azidobicyclo[4.2.0]octa-1(6),2,4-trien-7-yl]ethyl acetate (24).

Compound 22 (26.9 mg, 0.1 mmol) and NaN3 (13.0 mg, 0.2 mmol) in DMF (1.0 mL) were placed in an oven-dried screw-cap vial also containing a magnetic stirring bar. The mixture was stirred at 80 °C for 1 h. Upon completion of the reaction, the mixture was diluted with EtOAc and washed with aq. NH4Cl. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water followed by brine, dried with anhydrous Na2SO4, and concentrated under reduced pressure. The crude material was chromatographed on a silica gel column with 5% EtOAc in hexanes to yield compound 24 as a pale yellow liquid (16.1 mg, 70 %). Rf (SiO2/10% EtOAc in hexanes) = 0.40. IR (neat): 3068, 2957, 2101, 1736, 1366, 1239, 1036, 747 cm−1. 1H NMR (500 MHz, CDCl3): δ = 7.40–7.26 (m, 3H, Ar-H), 7.21 (d, J = 7.3 Hz, 1H, Ar-H), 5.07 (d, J = 4.8 Hz, 1H), 4.36 (td, J = 6.6, 11.0 Hz, 1H), 4.30 (td, J = 6.6, 11.0 Hz, 1H), 3.89–3.85 (m, 1H), 2.20–2.10 (m, 1H), 2.13 (s, 3H), 2.00–1.95 (m, 1H). 13C NMR (125 MHz, CDCl3): δ = 171.3, 146.8, 141.6, 130.2, 128.4, 123.5, 123.3, 63.0, 62.1, 47.2, 29.5, 21.2. HRMS (ESI/TOF): calcd for C12H13N3O2Na [M + Na]+ 254.0900; found 254.0898.
Synthesis of an intermediate: 2-[8-bromobicyclo[4.2.0]octa-1(6),2,4-trien-7-yl]ethan-1-ol.

Benzocyclobutene 1 (36.5 mg, 0.25 mmol) and BBr3 in CH2Cl2 (0.1 m, 2.5 mL) were placed in an oven-dried 10 mL round-bottomed flask, also containing a magnetic stirring bar, at 0 °C. The mixture was stirred at 0 °C for 30 min. Upon completion, the reaction mixture was quenched with water and extracted with EtOAc. The organic layer was separated, dried with anhydrous Na2SO4, and concentrated under reduced pressure. The crude material was chromatographed on a silica gel column with 30% EtOAc in hexanes to yield the bromo alcohol as a yellow oil (48.9 mg, 86 %). Rf (SiO2/20% EtOAc in hexanes) = 0.20. 1H NMR (500 MHz, CDCl3): δ = 7.33–7.30 (m, 2H, Ar-H), 7.21 (m, 1H, Ar-H), 7.12 (m, 1H, Ar-H), 5.10 (s, 1H), 3.88 (t, J = 6.4 Hz, 2H), 3.84 (t, J = 7.6 Hz, 1H), 2.15 (s, 1H, OH), 2.06 (app q, Japp = 6.9 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ = 145.3, 143.7, 130.3, 128.9, 123.1, 122.9, 61.3, 55.4, 47.2, 35.6.
3-[8-Azidobicyclo[4.2.0]octa-1(6),2,4-trien-7-yl]propyl acetate (25)
Step 1: synthesis of intermediate 3-[8-bromobicyclo[4.2.0]octa-1(6),2,4-trien-7-yl]propan-1-ol.

Benzocyclobutene 2 (40.0 mg, 0.25 mmol) and BBr3 in CH2Cl2 (0.1 m, 2.5 mL) were placed in an oven-dried 10 mL round-bottomed flask, also containing a magnetic stirring bar, at 0 °C. The mixture was stirred at 0 °C for 30 min. Upon completion, the reaction was quenched with water, and the layers were separated. The aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried with Na2SO4, and concentrated under reduced pressure. The crude material, which was obtained in quantitative yield, can be chromatographed on a silica gel column with 30% EtOAc in hexanes to yield the bromo alcohol as a yellow oil (56.9 mg, 94 %). Rf (SiO2/10% EtOAc in hexanes) = 0.17. 1H NMR (500 MHz, CDCl3): δ = 7.32–7.30 (m, 2H, Ar-H), 7.21–7.18 (m, 1H, Ar-H), 7.12–7.11 (m, 1H, Ar-H), 5.01 (d, J = 1.5 Hz, 1H), 3.74 (app t, J = 6.4 Hz, 3H), 1.88 (app quint, Japp = 6.9 Hz, 2H), 1.82–1.76 (m, 2H). 13C NMR (125 MHz, CDCl3): δ = 145.7, 143.7, 130.2, 128.8, 123.1, 122.8, 62.8, 57.8, 47.0, 30.7, 29.2.
Step 2: synthesis of 3-[8-bromobicyclo[4.2.0]octa-1(6),2,4-trien-7-yl]propyl Acetate (23).

3-[8-Bromobicyclo[4.2.0]octa-1(6),2,4-trien-7-yl]propan-1-ol (62.0 mg, 0.25 mmol) in CH2Cl2 (1.5 mL) was placed in an oven-dried screw-cap vial, also containing a magnetic stirring bar, at 0 °C. Ac2O (28.4 µL, 0.3 mmol) and DMAP (3.1 mg, 25 µmol) were added, and the mixture was stirred at 0 °C for 1 h. Upon completion, the reaction was quenched with water, and the layers were separated. The aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried with anhydrous Na2SO4, and concentrated under reduced pressure. The crude material was chromatographed on a silica gel column with 5–10 % EtOAc in hexanes to yield compound 23 as a pale yellow liquid (60.0 mg, 85 %). Rf (SiO2/10% EtOAc in hexanes) = 0.36. 1H NMR (500 MHz, CDCl3): δ = 7.32–7.30 (m, 2H, Ar-H), 7.22–7.19 (m, 1H, Ar-H), 7.13–7.10 (m, 1H, Ar-H), 5.01 (s, 1H), 4.18–4.12 (m, 2H), 3.74 (br, 1H), 2.06 (s, 3H, Me), 1.90–1.85 (m, 4H). 13C NMR (125 MHz, CDCl3): δ = 171.3, 145.4, 143.7, 130.3, 128.9, 123.1, 122.8, 64.3, 57.6, 46.8, 29.5, 26.8, 21.2. HRMS (ESI/TOF): calcd for C13H15BrO2Na [M + Na]+ 305.0148; found 305.0177.
Step 3: synthesis of 3-[8-azidobicyclo[4.2.0]octa-1(6),2,4-trien-7-yl]propyl Acetate (25).

Compound 23 (28.3 mg, 0.1 mmol) and NaN3 (13.0 mg, 0.2 mmol) in DMF (1.0 mL) were placed in an oven-dried screw-cap vial also containing a magnetic stirring bar. The mixture was stirred at 80 °C for 1 h. Upon completion of the reaction, the mixture was diluted with EtOAc and washed with aq. NH4Cl. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with water followed by brine, dried with anhydrous Na2SO4, and concentrated under reduced pressure. The crude material was chromatographed on a silica gel column with 5% EtOAc in hexanes to yield compound 25 as a pale yellow liquid (18.1 mg, 74 %). Rf (SiO2/10% EtOAc (10 %) in hexanes) = 0.38. IR (neat): 3055, 2958, 2102, 1734, 1367, 1265, 1045, 738 cm−1. 1H NMR (500 MHz, CDCl3): δ = 7.36–7.28 (m, 3H, Ar-H), 7.17 (d, J = 7.0 Hz, 1H, Ar-H), 5.01 (d, J = 4.7 Hz, 1H), 4.16 (t, J = 6.2 Hz, 2H), 3.78–3.74 (m, 1H), 2.06 (s, 3H, Me), 1.89–1.83 (m, 3H), 1.76–1.72 (m, 1H). 13C NMR (125 MHz, CDCl3): δ = 171.4, 147.5, 141.7, 130.2, 128.3, 123.6, 123.1, 64.5, 62.3, 50.2, 27.1, 27.0, 21.2. HRMS (ESI/TOF): calcd for C13H16N3O2 [M + H]+ 246.1237; found 246.1209.
Supplementary Material
Acknowledgments
Infrastructural support at CCNY was provided by National Institutes of Health (NIH) Grant G12MD007603 from the National Institute on Minority Health and Health Disparities. The computational analysis was supported in part by National Science Foundation (NSF) Grants CNS-0958379 and CNS-0855217, and by The City University of New York High Performance Computing Center at the College of Staten Island. We thank Dr. Satish Sakilam, Dr. Prasanna Vuram and Mr. Hari Akula for assistance.
Footnotes
Supporting Information (see footnote on the first page of this article): Synthetic procedures, characterization details, copies of 1H NMR spectra with expanded spectra and 13C NMR spectra of all products, gCOSY, NOESY, gHMQC, and HMBC spectra of selected products for structure elucidation purposes, and IR spectra of compounds 24 and 25.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/ejoc.XXXXXXXXX.
References
- 1.Finkelstein H. Doctoral Dissertation. University of Strasbourg; 1909. [Google Scholar]
- 2.Cava MP, Napier DR. J. Am. Chem. Soc. 1956;78:500. [Google Scholar]
- 3.For reviews, see: Sadana AK, Saini RK, Billups WE. Chem. Rev. 2003;103:1539–1602. doi: 10.1021/cr010022j.Pellisier H, Santelli M. Tetrahedron. 2003;59:701–730.Mehta G, Kotha S. Tetrahedron. 2001;57:625–659.Michellys P-Y, Pellissier H, Santelli M. Org. Prep. Proc. Int. 1996;28:545–608.Kametani T, Fukumoto K. Acc. Chem. Res. 1976;9:319–325.Klundt IL. Chem. Rev. 1970;70:471–487.
- 4.Segura JL, Martín N. Chem. Rev. 1999;99:3199–3246. doi: 10.1021/cr990011e. [DOI] [PubMed] [Google Scholar]
- 5.See for examples [Google Scholar]; a Ribas-Arino J, Marx D. Chem. Rev. 2012;112:5412–5847. doi: 10.1021/cr200399q. [DOI] [PubMed] [Google Scholar]; b Yang J, Huang Y, Cao K. Recent Progress in Benzocyclobutene Related Polymers. In: De Souza Gomes A, editor. Polymerization. Chapter 9. InTech –Open Access Company; 2012. pp. 201–222. [Google Scholar]; c Zuniga CA, Barlow S, Marder SR. Chem. Mater. 2011;23:658–681. [Google Scholar]; d So H-Y, Garrou PE, Im J-H, Ohba K. Benzocyclobutene-Based Polymers for Microelectronic Applications. polymers for Microelectronics and Nanoelectronics [Google Scholar]; Lin Q, Pearson RA, Hedrick JC, editors. ACS Symposium Series Vol. Vol. 874. American Chemical Society; Washington, DC: 2004. pp. 279–293. Chapter 21. [Google Scholar]
- 6.Wenk HH, Winkler M, Sander W. Angew. Chem. Int. Ed. 2003;42:502–528. doi: 10.1002/anie.200390151. [DOI] [PubMed] [Google Scholar]
- 7.Radziszewski JG, Hess BA, Jr., Zahradnik R. J. Am. Chem. Soc. 1992;114:52–57. [Google Scholar]
- 8.Maier WF, Lau GC, McEwen AB. J. Am. Chem. Soc. 1985;107:4724–4727. [Google Scholar]
- 9.Rondan NG, Domelsmith LN, Houk KN, Bowne AT, Levin RH. Tetrahedron Lett. 1979;20:3237–3240. [Google Scholar]
- 10.a Baker RW, Baker (née Nicoletti) TM, Birkbeck AM, Giles RGF, Sargent MV, Skelton BW, White AH. J. Chem. Soc., Perkin Trans. 1991;1:1589–1600. [Google Scholar]; b Giles RGF, Hughes AB, Sargent MV. J. Chem. Soc., Perkin Trans. 1991;1:1581–1587. [Google Scholar]; c Giles RGF, Sargent MV, Sianipar H. J. Chem. Soc., Perkin Trans. 1991;1:1571–1579. [Google Scholar]; d Gribble GW, Keavy DJ, Branz SE, Kelly WJ, Pals MA. Tetrahedron Lett. 1988;29:6227–6230. [Google Scholar]; e Newman MS, Kannan R. J. Org. Chem. 1976;41:3356–3359. [Google Scholar]
- 11.a Nerurkar A, Chandrasoma N, Maina L, Brassfield A, Luo D, Brown N, Buszek KR. Synthesis. 2013;45:1843–1852. [Google Scholar]; b Garr AN, Luo D, Brown N, Cramer CJ, Buszek KR, VanderVelde D. Org. Lett. 2010;12:96–99. doi: 10.1021/ol902415s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Goetz AE, Bronner SM, Cisneros JD, Melamed JM, Paton RS, Houk KN, Garg NK. Angew. Chem. Int. Ed. 2012;124:2812–2816. doi: 10.1002/anie.201108863. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Im G-YJ, Bronner SM, Goetz AE, Paton RS, Cheong P-HY, Houk KN, Garg NK. J. Am. Chem. Soc. 2010;132:17933–17944. doi: 10.1021/ja1086485. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cheong PH-Y, Paton RS, Bronner SM, Im G-YJ, Garg NK, Houk KN. J. Am. Chem. Soc. 2010;132:1267–1269. doi: 10.1021/ja9098643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedman L, Osiewicz RJ, Rabideau PW. Tetrahedron Lett. 1968;9:5735–5737. [Google Scholar]
- 14.Gassman PG, Benecke HP. Tetrahedron Lett. 1969;10:1089–1092. [Google Scholar]
- 15.Ahlgren G, Åkermark B. Tetrahedron Lett. 1970;11:3047–3048. [Google Scholar]
- 16.Bowne AT, Christopher TA, Levin RH. Tetrahedron Lett. 1976;17:4111–4114. [Google Scholar]
- 17.Crews P, Beard J. J. Am. Chem. Soc. 1973;38:522–528. [Google Scholar]
- 18.Kuehne ME. J. Am. Chem. Soc. 1962;84:837–847. [Google Scholar]
- 19.Keyton DJ, Griffin GW, Kuehne ME, Bayha CE. Tetrahedron Lett. 1969;10:4163–4168. [Google Scholar]
- 20.Gingrich HL, Huang Q, Morales AL, Jones M., Jr. J. Org. Chem. 1992;57:3803–3806. [Google Scholar]
- 21.a Caubère P, Guillaumet G, Mourad MS. Tetrahedron Lett. 1971;12:4673–4676. [Google Scholar]; b Carre M-C, Gregoire B, Caubere P. J. Org. Chem. 1984;49:2050–2052. [Google Scholar]
- 22.See for examples [Google Scholar]; a Kita Y, Higuchi K, Yoshida Y, Iio K, Kitagaki S, Ueda K, Akai S, Fujioka H. J. Am. Chem. Soc. 2001;123:3214–3222. doi: 10.1021/ja0035699. [DOI] [PubMed] [Google Scholar]; b Shair MD, Yoon T-Y, Mosny KK, Chou TC, Danishefsky SJ. J. Am. Chem. Soc. 1996;118:9509–9525. [Google Scholar]; c Danheiser RL, Helgason AL. J. Am. Chem. Soc. 1994;116:9471–9479. [Google Scholar]; d Guyot M, Molho D. Tetrahedron Lett. 1973;14:3433–3436. [Google Scholar]
- 23.Reviewed in: Tadross PM, Stoltz BM. Chem. Rev. 2012;112:3550–3577. doi: 10.1021/cr200478h.
- 24.Wasserman HH, Solodar J. J. Am. Chem. Soc. 1965;87:4002–4003. [Google Scholar]
- 25.1,1-Dimethoxyethylene: Stevens RV, Bisacchi GS. J. Org. Chem. 1982;47:2393–2396.; 2-methylene-1,3-dioxepane: Maurin P, Ibrahim-Ouali M, Santelli M. Eur. J. Org. Chem. 2002:151–156.Mariet N, Ibrahim-Ouali M, Santelli M. Tetrahedron Lett. 2002;43:5789–5791.Maurin P, Ibrahim-Ouali M, Santelli M. Tetrahedron Lett. 2001;42:8147–8149.
- 26.a Hamura T, Ibusuki Y, Uekusa H, Matsumoto T, Siegel JS, Baldridge KK, Suzuki K. J. Am. Chem. Soc. 2006;128:10032–10033. doi: 10.1021/ja064063e. [DOI] [PubMed] [Google Scholar]; b Hamura T, Arisawa T, Matsumoto T, Suzuki K. Angew. Chem. Int. Ed. 2006;45:6842–6844. doi: 10.1002/anie.200602539. [DOI] [PubMed] [Google Scholar]; c Hamura T, Ibusuki Y, Uekusa H, Matsumoto T, Suzuki K. J. Am. Chem. Soc. 2006;128:3534–3535. doi: 10.1021/ja0602647. [DOI] [PubMed] [Google Scholar]; d Hamura T, Ibusuki Y, Sato K, Matsumoto T, Osamura Y, Suzuki K. Org. Lett. 2003;5:3551–3554. doi: 10.1021/ol034877p. [DOI] [PubMed] [Google Scholar]; e Hamura T, Hosoya T, Yamaguchi H, Kuriyama Y, Tanabe M, Miyamoto M, Yasui Y, Matsumoto T, Suzuki K. Helv. Chim. Acta. 2002;85:3589–3604. [Google Scholar]; f Hosoya T, Hamura T, Kuriyama Y, Miyamoto M, Matsumoto K, Suzuki K. Synlett. 2002:520–522. [Google Scholar]; g Hosoya T, Hasegawa T, Kuriyama Y, Suzuki K. Tetrahedron Lett. 1995;36:3377–3380. [Google Scholar]; h Hosoya T, Hasegawa T, Kuriyama Y, Matsumoto T, Suzuki K. Synlett. 1995:177–179. [Google Scholar]
- 27.Himeshima Y, Sonoda T, Kobayashi H. Chem. Lett. 1983:1211–1214. [Google Scholar]
- 28.a Allan KM, Stoltz BM. J. Am. Chem. Soc. 2008;130:17270–17271. doi: 10.1021/ja808112y. [DOI] [PubMed] [Google Scholar]; b Gilmore CD, Allan KM, Stoltz BM. J. Am. Chem. Soc. 2008;130:1558–1559. doi: 10.1021/ja0780582. [DOI] [PubMed] [Google Scholar]
- 29.a Ma Z-X, Feltenberger JB, Hsung RP. Org. Lett. 2012;14:2742–2745. doi: 10.1021/ol300967a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Feltenberger JB, Hayashi R, Tang Y, Babiash ESC, Hsung RP. Org. Lett. 2009;11:3666–3669. doi: 10.1021/ol901434g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campbell-Verduyn L, Elsinga PH, Mirfeizi L, Dierckx RA, Feringa BL. Org. Biomol. Chem. 2008;6:3461–3463. doi: 10.1039/b812403e. [DOI] [PubMed] [Google Scholar]
- 31.a Barluenga J, Aznar F, Palomero MA, Barluenga S. J. Org. Chem. 2002;68:537–544. doi: 10.1021/jo0264574. [DOI] [PubMed] [Google Scholar]; b Barluenga J, Aznar F, Palomero MA, Barluenga S. Org. Lett. 1999;1:541–543. [Google Scholar]
- 32.Barluenga J, Andina F, Aznar F, Valdés C. Org. Lett. 2007;9:4143–4146. doi: 10.1021/ol701604g. [DOI] [PubMed] [Google Scholar]
- 33.Kessler SN, Neuberger M, Wegner HA. Eur. J. Org. Chem. 2011:3238–3245. [Google Scholar]
- 34.a Bravo F, Kassou M, Díaz Y, Castillión S. Carbohydr. Res. 2001;336:83–97. doi: 10.1016/s0008-6215(01)00256-7. [DOI] [PubMed] [Google Scholar]; b Marcotte S, Gérard B, Pannecoucke X, Feasson C, Quirion J-C. Synthesis. 2001:929–933. [Google Scholar]
- 35.Haasnoot CAG, de Leeuw FAAM, Altona C. Tetrahedron. 1980;36:2783–2792. [Google Scholar]
- 36.Maurin P, Ibrahim-Ouali M, Parrain J-L, Santelli M. J. Mol. Struct. (Theochem) 2003;637:91–100. [Google Scholar]
- 37.Punna S, Meunier S, Finn MG. Org. Lett. 2004;6:2777–2779. doi: 10.1021/ol0489898. [DOI] [PubMed] [Google Scholar]
- 38.Sousa C, Silva PJ. Eur. J. Org. Chem. 2013:5195–5199. [Google Scholar]
- 39.a Coll G, Costa A, Deyá PM, Flexas F, Rotger C, Saá JM. J. Org. Chem. 1992;57:6222–6231. [Google Scholar]; b Jung ME, Lam PY-S, Mansuri MM, Speltz LM. J. Org. Chem. 1985;50:1087–1105. [Google Scholar]; c Dhawan KL, Gowland BD, Durst T. J. Org. Chem. 1980;45:922–924. [Google Scholar]; d Tabushi I, Oda R, Okazaki K. Tetrahedron Lett. 1968;9:3743–3747. [Google Scholar]
- 40.Jones M, Jr., Levin RH. J. Am. Chem. Soc. 1969;91:6411–6415. [Google Scholar]
- 41.a Tadross PM, Gilmore CD, Bugga P, Virgil SC, Stoltz BM. Org. Lett. 2010;12:1224–1227. doi: 10.1021/ol1000796. [DOI] [PubMed] [Google Scholar]; b Atkinson DJ, Sperry J, Brimble MA. Synthesis. 2010:911–913. [Google Scholar]; c Bronner SM, Garg NK. J. Org. Chem. 2009;74:8842–8843. doi: 10.1021/jo9020166. [DOI] [PubMed] [Google Scholar]; d Peña D, Pérez D, Guitián E, Castedo L. J. Org. Chem. 2000;65:6944–6950. doi: 10.1021/jo000535a. [DOI] [PubMed] [Google Scholar]
- 42.Peña D, Cobas A, Pérez D, Guitián E. Synthesis. 2002:1454–1458. [Google Scholar]
- 43.Kirkham JD, Delaney PM, Ellames GJ, Row EC, Harrity JPA. Chem. Comm. 2010:5154–5156. doi: 10.1039/c0cc01345e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.