

Abstract
Photoremovable protecting groups are important for a wide range of applications in peptide chemistry. Using Fmoc-Cys(Bhc-MOM)-OH, peptides containing a Bhc-protected cysteine residue can be easily prepared. However, such protected thiols can undergo isomerization to a dead-end product (a 4-methylcoumarin-3-yl thioether) upon photolysis. To circumvent that photoisomerization problem, we explored the use of nitrodibenzofuran (NDBF) for thiol protection by preparing cysteine-containing peptides where the thiol is masked with an NDBF group. This was accomplished by synthesizing Fmoc-Cys(NDBF)-OH and incorporating that residue into peptides by standard solid-phase peptide synthesis procedures. Irradiation with 365 nm light or two-photon excitation with 800 nm light resulted in efficient deprotection. To probe biological utility, thiol group uncaging was carried out using a peptide derived from the protein K-Ras4B to yield a sequence that is a known substrate for protein farnesyltransferase; irradiation of the NDBF-caged peptide in the presence of the enzyme resulted in the formation of the farnesylated product. Additionally, incubation of human ovarian carcinoma (SKOV3) cells with an NDBF-caged version of a farnesylated peptide followed by UV irradiation resulted in migration of the peptide from the cytosol/Golgi to the plasma membrane due to enzymatic palmitoylation. Overall, the high cleavage efficiency devoid of side reactions and significant two-photon cross-section of NDBF render it superior to Bhc for thiol group caging. This protecting group should be useful for a plethora of applications ranging from the development of light-activatable cysteine-containing peptides to the development of light-sensitive biomaterials.
Introduction
The ability of light to traverse various chemical and biological barriers and be modulated by time and amplitude makes light-regulated molecules unique tools for a plethora of applications in the areas of chemistry and biology.1−4 Photoremovable protecting groups, also known as caging groups, are one of the most important light-regulated tools, which can be utilized to mask specific functional groups in molecules such that they can be cleaved on demand upon irradiation.5,6 In biological applications, this typically involves masking a biomolecule with a caging group to produce a compound whose biological activity is either increased or decreased upon uncaging.7−9 The recent development of two-photon-sensitive protecting groups, which allow uncaging using near-infrared (near-IR) irradiation, has resulted in significant improvements in the spatiotemporal resolution of uncaging as well as increased penetration with lower phototoxicity;10−14 the latter attribute is of particular importance for the use of caged molecules in tissue samples or intact organisms that are essentially opaque to UV light. Due to inherent differences in the chemical reactivity of various functional groups, there is no single photocleavable protecting group that works efficiently for caging all functionalities. Hence, protecting group selection must be performed on a case by case basis.15,16
Thiol-containing compounds play vital roles in many aspects of biology (e.g., controlling cellular redox state),17 protein chemistry (e.g., protein and peptide folding, native chemical ligation18), and enzymology.19 Hence, significant efforts have gone into the preparation of proteins and ligands/substrates containing caged thiols that can be triggered with light to reveal bioactive species;20−24 for that purpose, several protecting groups have been explored.25−29 The most widely used approach for thiol protection involves caging with o-nitrobenzyl (ONB) groups. Among the advantages of ONB groups are high one-photon quantum efficiency and high yield of free compound produced upon photolysis.30 However, they are poor chromophores with low two-photon sensitivities which limit their biological applications. To address this issue, coumarin-based protecting groups have been recently employed, which absorb light at longer wavelengths and possess significantly higher one- and two-photon absorptivity.31 In one important study, Hagen and co-workers harnessed the chromatic orthogonality of ONB- and coumarin-based protecting groups to introduce a wavelength-selective thiol caging system.32 Using a combination of those protecting groups, they were able to selectively uncage different thiols in a peptide sequence using different wavelengths for UV irradiation; however, no two-photon photochemistry was explored. In another novel study, Shoichet and co-workers incorporated brominated hydroxycoumarin (Bhc)-protected thiols into hydrogels that allowed them to perform light-induced protein patterning within those materials with high spatial control.33,34 The high two-photon sensitivity of Bhc allowed them to create 3D protein patterns inside these polymeric scaffolds for tissue engineering applications.
In recent work, we demonstrated that Bhc could also be used for thiol protection of a peptidomimetic enzyme inhibitor.35 The high one- and two-photon sensitivity of Bhc allowed efficient uncaging of the inhibitor inside cells for biological applications. Inspired by these results, we reasoned that Bhc could also be used for thiol protection of cysteine-containing peptides. In the work reported here, we first explored the use of Bhc-protected cysteine in peptides. While they are straightforward to prepare using Fmoc-Cys(Bhc)-OH as a building block in solid-phase peptide synthesis (SPPS), irradiation of such caged peptides was complicated by the generation of an unwanted photoisomer instead of the free thiol. Using NMR analysis, we were able to confirm the structure of the photoisomer to be a 4-methylcoumarin-3-yl thioether, in agreement with a previous prediction by Hagen and co-workers.32 Further analysis of several different peptide sequences revealed that the photocleavage efficiency of Bhc-protected thiols is context dependent and typically leads to formation of a photoisomer as the major product. To circumvent this problem, we next explored using nitrodibenzofuran (NDBF)36 as a thiol caging group since it manifests a two-photon cross-section comparable to that of Bhc. Thus, cysteine-containing peptides were prepared where the thiol was protected with an NDBF group. This was accomplished by preparing Fmoc-Cys(NDBF)-OH and incorporating it into peptides via standard SPPS. In contrast to that of Bhc-caged thiols, irradiation of NDBF-protected thiols at 365 nm resulted in clean conversion to the free mercaptan. Deprotection was also obtained via irradiation at 800 nm, where the two-photon action cross-section was measured to be comparable to that of Bhc-protected acetate (Bhc-OAc). To probe the utility of this protecting group for biological experiments, thiol group uncaging was carried out using a K-Ras-derived peptide containing an NDBF-protected cysteine. Irradiation of that molecule in the presence of protein farnesyltransferase (PFTase) and farnesyl diphosphate (FPP) resulted in the formation of the free thiol form and subsequent enzymatic conversion to a prenylated species. Those experiments indicate that one- and two-photon deprotection can be performed under mild conditions that allow enzymatic activity to be retained. In order to illustrate the utility of this strategy for the development of caged peptides that can be activated via irradiation inside live cells, the thiol of a cell-penetrating peptide known to be a substrate for palmitoyl acyltransferase was protected as a NDBF thioether. Irradiation of human ovarian carcinoma (SKOV3) cells, preincubated with the probe, resulted in migration of the peptide from the cytosol/Golgi to the plasma membrane (visualized via confocal microscopy) due to enzymatic palmitoylation. These data suggest that the NDBF group should be useful for caging thiols in peptides and potentially larger proteins assembled via native chemical ligation18 for biological applications. The high uncaging yield of NDBF-caged thiols upon one- and two-photon irradiation, together with the facile incorporation of caged cysteine via standard SPPS into peptides containing multiple cysteines, make this a highly versatile strategy for studying cysteine-containing peptides and proteins.
Experimental Section
General Details
All reagents needed for SPPS were purchased from Peptide International (Louisville, KY). All other solvents and reagents used for synthesis and other experiments were purchased from Sigma-Aldrich (St. Louis, MO). High-performance liquid chromatography (HPLC) analysis (analytical and preparative) was performed using a Beckman model 125/166 instrument, equipped with a UV detector and C18 columns (Varian Microsorb-MV, 5 μm, 4.6 × 250 mm and Phenomenex Luna, 10 μm, 10 × 250 mm, respectively). 1H NMR data of synthetic compounds were recorded at 500 MHz on a Varian Instrument at 25 °C. 1H NMR data for the products of photolysis reactions were recorded using a Bruker Advance III 700 MHz spectrometer with 1.7 mm TCI cryoprobe.
General Procedure for Solid-Phase Peptide Synthesis (SPPS)
Peptides were synthesized using an automated solid-phase peptide synthesizer (PS3, Protein Technologies Inc., Memphis, TN) employing Fmoc/HCTU-based chemistry. Fmoc-Met-Wang resin (0.25 mmol) was transferred into a reaction vessel, and peptide chain elongation was performed using HCTU and N-methylmorpholine. Standard amino acid coupling was performed by incubation of 4 equiv of both HCTU and Fmoc-protected amino acid with the resin for 30 min. Coupling of Fmoc-Cys(Bhc)-OH or Fmoc-Cys(NDBF)-OH was performed by incubation of 1.5 equiv of both the amino acid and HCTU with the resin for 6 h. Peptide chain elongation was completed by N-terminus deprotection using 10% piperidine in DMF (v/v). 5-Fam coupling was conducted by incubation of 1.2 equiv of 5-Fam, 1.2 equiv of HCTU, and 2 equiv of N,N-diisopropylethylamine (DIEA) with the resin for 8 h. Resin was then transferred into a syringe filter, washed three times with DCM, and dried in vacuo. Global deprotection and resin cleavage were accomplished via treatment with Reagent K. Peptides were then precipitated with Et2O and pelleted by centrifugation, and the residue was rinsed twice with Et2O. The resulting crude peptide was dissolved in MeOH and purified by preparative reversed-phase (RP)-HPLC.
General Procedure for Synthesis of Peptides Containing C-Terminal Methyl Esters
Trityl chloride resin (1 equiv) was transferred into a fitted syringe and washed three times with DMF. In a separate flask, Fmoc-Cys-OMe (3 equiv) was treated with DIEA (6 equiv) in DCM, transferred into the resin-containing fitted syringe, and then mixed overnight using a rotisserie. Resins were treated with MeOH to cap any unreacted trityl moiety, followed by washing three times with DMF. The prepared Fmoc-Cys-OMe-loaded resins were used to synthesize peptides containing C-terminal methyl esters via traditional Fmoc/HCTU-based chemistry as described in “General Procedure for Solid-Phase Peptide Synthesis”.
5-Fam-KKKSKTKC(Bhc)VIM (5): ESI-MS calcd for [C87H126BrN16O23S2 + 3H]3+ 635.2597, found 635.2568.
5-Fam-KKKSKTKC(NDBF)VIM (17b): ESI-MS calcd for [C91H130N17O23S2 + 3H]3+ 630.9650, found 630.9658.
C(Bhc)VLS: ESI-MS calcd for [C27H37BrN4O9S + H]+ 673.1537, found 673.1575.
Dansyl-GC(Bhc)VLS: ESI-MS calcd for [C41H51BrN6O12S2 + H]+ 963.2363, found 963.2302.
Dansyl-GC(NDBF)VLS: ESI-MS calcd for [C45H55N7O12S2 + Na]+ 972.3248, found 972.3280.
KKKSKC(Bhc)CVIM (5): ESI-MS calcd for [C63H109BrN15O17S3 + 3H]3+ 507.5485, found 507.5497.
KKKSKTCC(Bhc)IM: ESI-MS calcd for [C64H112BrN16O17S3 + 3H]3+ 517.2240, found 517.2240.
KKKSKTKC(NDBF)VIM (17a): ESI-MS calcd for [C70H117N17O17S2 + 3H]3+ 511.6158, found 511.6238.
NBD-Hex-C(NDBF)LC-OMe (20): ESI-MS calcd for [C54H70N8O11S2 + H]+ 1071.4684, found 1071.4284.
Fmoc-Cys(MOM-Bhc)-OCH3 (3)
Chloride 2 (93 mg, 0.28 mmol) and Fmoc-Cys-OCH3 (200 mg, 0.56 mmol) were dissolved in 3 mL of a solution of 2:1:1 DMF/CH3CN/H2O/0.1% TFA (v/v). Zn(OAc)2 was then added (154 mg, 0.70 mmol) and the reaction monitored by thin-layer chromatography (TLC) (1:1 Hex/EtOAc). After 2 days, the solvent was removed and the reaction purified via column chromatography (1:1 Hex/EtOAc) to give 149 mg of 3 as a yellow powder (81% yield): 1H NMR (CDCl3) δ 7.83 (1H, s) 7.76 (2H, d, J = 7.5), 7.6 (2H, d, J = 7.5 Hz), 7.38 (2H, m), 7.29 (2H, m), 7.13 (1H, s), 6.36 (1H, s), 5.74 (2H, s), 4.68 (1H, m), 4.38–4.48 (2H, m), 4.20 (1H, t), 3.74 (3H, s), 3.50 (3H, s); HR-MS (ESI) m/z calcd for (C31H28BrNO8S + Na)+ 676.0611 (79Br) and 678.0596 (81Br), found 676.0639 (79Br) and 678.0636 (81Br).
Fmoc-Cys(MOM-Bhc)-OH (4)
Ester 3 (100 mg, 0.15 mmol) and Me3SnOH (69 mg, 0.38 mmol) were dissolved in CH2Cl2 (5 mL) and brought to reflux. After 7 h the reaction was judged complete by TLC (1:1 Hex/EtOAc). The solvent was removed in vacuo and the resulting oil redissolved in EtOAc (20 mL). The organic layer was washed with 5% HCl (3 × 10 mL) and brine (3 × 10 mL), dried with Na2SO4, and evaporated to give 92 mg of 4 as a yellow powder (90% yield): 1H NMR (d6-acetone) δ 8.12 (1H, s), 7.86 (2H, d, J = 7.5), 7.73 (2H, t, J = 7), 7.41 (2H, t, J = 7.5), 7.33 (2H, m), 7.16 (1H, s), 6.42 (1H, s), 5.64 (1H, s), 5.42 (2H, s), 4.51 (1H, b), 4.37–4.41 (2H, m), 4.32 (1H, t), 4.25 (1H, t), 4.07 (2H, d), 3.49 (3H, s); HR-MS (ESI) m/z calcd for [C30H26BrNO8S + Na]+ 662.0455 (79Br) and 664.0439 (81Br), found 662.0472 (79Br) and 664.0428 (81Br).
Fmoc-Cys(NDBF)-OCH3 (15)
NDBF-Br (1.00 g, 3.12 mmol) and Fmoc-Cys-OCH3 (2.2 g, 6.25 mmol) were dissolved in 60 mL of a solution of 2:1:1 DMF/ACN/0.1% TFA in H2O (v/v/v). A 0.5 M aqueous solution of Zn(OAc)2 was prepared in 0.1% TFA (v/v), and 25 μL of that solution was added to the reaction mixture. The reaction was monitored by TLC (1:1 Hex/Et2O) and stopped after 36 h of stirring at room temperature. Solvent was evaporated in vacuo, and the final product was purified via column chromatography (1:1 Hex/Et2O) to give 0.90 g of a diastereomeric mixture of 15 as a yellow oil (48% yield): 1H NMR (CDCl3) δ 8.38–8.40 (1H), 8.01–8.06 (2H, m), 7.77 (2H, m), 7.54–7.63 (4H, m), 7.30–7.45 (5H, m), 5.58–5.59 (1H, m), 4.84–4.88 (1H, m), 4.53–4.59 (1H, m), 4.14–4.40 (3H, m), 3.72–3.78 (3H), 2.84–3.03 (2H, m), 1.72–1.74 (3H, m); HR-MS (ESI) m/z calcd for [C33H28N2O7S + Na]+ 619.1515, found 619.1537.
Fmoc-Cys(NV)-OMe
This compound was synthesized following the same procedure described above for synthesis of 15, except NDBF-Br was replaced with 1-(bromomethyl)-4,5-dimethoxy-2-nitrobenzene (NV-Br, 80% yield): 1H NMR (CDCl3) δ 7.78 (2H, d, J = 7.5), 7.68 (1H, s), 7.62 (2H, t, J = 8.0 Hz), 7.42 (2H, t, J = 7.5 Hz), 7.33 (2H, t, J = 8 Hz), 6.89 (1H, s), 5.66 (H, d, J = 8.0 Hz), 4.64 (1H, q, J = 7.5 Hz), 4.42 (2H, d, J = 7.0 Hz), 4.25 (1H, t, J = 7.5 Hz), 4.00 (3H, s), 3.94 (3H, s), 3.81 (3H, s), 3.00 (2H, m); HR-MS (ESI) m/z calcd for [C28H28N2O8S + Na]+ 575.1464, found 575.1493.
Fmoc-Cys(NDBF)-OH (16)
Ester 15 (900 mg, 1.50 mmol) was dissolved in 25 mL of CH2Cl2, and Me3SnOH (678 mg, 3.75 mmol) was added. The reaction was refluxed for 7 h and monitored by TLC (1:1 Hex/EtOAc), at which point the solvent was removed in vacuo and the resulting oil dissolved in EtOAc (30 mL). The organic layer was washed with 5% HCl (3 × 10 mL) and brine (3 × 10 mL), dried with Na2SO4, and evaporated to give 786 mg of 16 as a yellow powder (90% yield) as a diastereomeric mixture: 1H NMR (CDCl3) δ 8.36–8.39 (1H), 7.97–8.03 (2H, m), 7.75–7.77 (2H, m), 7.53–7.62 (4H, m), 7.30–7.42 (5H, m), 5.58–5.62 (1H, m), 4.88–4.91 (1H, m), 4.54–4.66 (1H, m), 4.16–4.40 (3H, m), 2.88–3.04 (2H, m), 1.71–1.74 (3H, m); 13C NMR (CDCl3) δ 174.42, 158.28, 155.88, 153.66, 147.66, 143.75, 141.31, 133.37, 129.45, 128.95 127.73, 127.11, 125.21, 123.77, 122.41, 121.87, 120.99, 119.97, 112.18, 108.25, 67.49, 53.48, 47.02, 39.60, 33.70, 23.66; HR-MS (ESI) m/z calcd for [C32H26N2O7S + Na]+ 605.1358, found 619.1335.
General Procedure for UV Photolysis of Caged Molecules
The caged compound was dissolved in photolysis buffer (50 mM phosphate buffer (PB), pH 7.4 containing 1 mM DTT) at a final concentration of 25–250 μM. The solutions were transferred into a quartz cuvette (10 × 50 mm) and irradiated with 365 nm UV light using a Rayonet reactor (2 × 14 W RPR-3500 bulbs). After each reaction the samples were analyzed by RP-HPLC or liquid chromatography–mass spectrometry (LC-MS).
General Procedure for LC-MS Analysis
Aliquots (100 μL) containing 5–20 μM caged compound in photolysis buffer were irradiated in a Rayonet UV photoreactor or using an 800 nm laser (see below for description). Each irradiated sample was then analyzed by LC-MS. The general gradient for LC-MS analysis was 0–100% H2O (0.1% HCO2H) to CH3CN (0.1% HCO2H) in 25 min.
Photolysis Study of Bhc-Protected Boc-Cysteamine (11) and NMR Analysis of the Photoisomerization Reaction
Aliquots (200 μL) containing compound 11 (200 μM in photolysis buffer) were irradiated at 365 nm for 80 and 400 s. After each illumination, samples were analyzed via RP-HPLC by monitoring the absorbance at 220 nm. To obtain sufficient photoisomer for NMR analysis, 10 mL of a 300 μM solution of 11 was irradiated for 6 min and purified via preparative RP-HPLC. The collected eluate was lyophilized to yield ∼1 mg of the desired compound, which was then dissolved in 500 μL of d6-acetone and subjected to 1H NMR analysis.
Photolysis Rate and Quantum Efficiency of 17b Using UV Excitation
Aliquots (100 μL) containing 17b (200 μM in photolysis buffer) were irradiated at 365 nm in a Rayonet UV reactor. The duration of irradiation ranged from 5 to 90 s. After each irradiation interval, 80 μL of the sample was analyzed by RP-HPLC. The reaction samples were eluted with a gradient of 0.1% TFA in H2O (Solvent A) and 0.1% TFA in CH3CN (Solvent B) (gradient of a 3% increase in Solvent B/min, flow rate 1 mL/min) and monitored by fluorescence (λex = 492 nm, λem = 518 nm). Reaction progress data were plotted in KaleidaGraph 3.0 and fitted via nonlinear regressional analysis to a first-order process. The quantum efficiency (Qu) was calculated using the formula Qu = (Iσt90%)−1, where I is the irradiation intensity in einstein cm–2 s–1, σ is the decadic extinction coefficient (103 × ε, molar extinction coefficient) in cm2 mol–1, and t90% is the irradiation time in seconds for 90% conversion to the product.31 The UV intensity of the lamps (I) in the photoreactor was measured using potassium ferrioxalate actinometry.37
Laser Apparatus for Two-Photon Irradiations
The light source that was utilized for two-photon irradiation is a home-built, regeneratively amplified Ti:sapphire laser system. This laser operates at 1 kHz with 210 mW pulses centered at a wavelength of 800 nm. The laser pulses have a Gaussian full width at half-maximum of 80 fs. Samples were irradiated in a 15 μL quartz cuvettes (Starna Cells Corp.).
Two-Photon Uncaging Cross-Section (δu) of 17a at 800 nm
The two-photon action cross-section for 17a was measured by comparing the photolysis rate of 17a with that of Bhc-OAc as a reference (δu = 0.45 at 800 nm). Aliquots (100 μL) containing 17a (300 μM in photolysis buffer) were irradiated with the 800 nm laser system for varying amounts of time, ranging from 2.5 to 30 min. Each sample was analyzed by HPLC using the method described above. Similar photolysis experiments were conducted using 100 μL aliquots of Bhc-OAc (100 μM in 50 mM PB, pH 7.2). Photolyzed Bhc-OAc solutions were also analyzed by RP-HPLC. The compounds were eluted with a gradient of Solvent A and Solvent B (gradient of a 1% increase in Solvent B/min, flow rate 1 mL/min) and monitored by absorbance at 220 nm. Reaction progress data were analyzed as described above, and the first-order decay constants for the two compounds were used in the formula δuΦu(17a) = δuΦu(reference) × Kobs(17a)/Kobs(reference) to calculate the value of δu for 17a, where δuΦu(reference) = 0.45 GM.
UV- and Two-Photon-Triggered Farnesylation of 17
A 7.5 μM solution of compound 17 was prepared in prenylation buffer (15 mM DTT, 10 mM MgCl2, 50 μM ZnCl2, 20 mM KCl, and 22 μM FPP) and divided into three 100 μL aliquots. Yeast PFTase was added to the first aliquot to give a final concentration of 30 nM, but the resulting sample was not subjected to photolysis. The second aliquot was irradiated in the absence of yeast PFTase, while the third sample was supplemented with yeast PFTase (50 nM) and then photolyzed with either UV or laser light. UV photolysis was conducted for 1 min at 365 nm, while two-photon irradiation was performed for 2.5 min at 800 nm. Each sample was incubated for 30 min at room temperature and then analyzed by LC-MS as described above.
Cell Culture and Microscopy
SKOV3 cells were grown in Mcoy’s 5a medium containing 10% FBS at 37 °C under CO2 (5.0%). For microscopy experiments, cells were seeded into 35 mm glass-bottomed dishes at a density of 8 × 103 cells/cm2. To monitor the trafficking of 20 before and after UV irradiation inside cells, SKOV3 cells were incubated with 5 μM 20 for 3 h. The medium was then replaced with RPMI (10% FBS) medium without phenol red. Half of the plates were irradiated at 330 nm for 5 min using a transilluminator (Fotodyne Inc.), and then all of the plates were incubated for 120 min at 37 °C under CO2 (5.0%). Cells were then incubated with Hoecht 33342 (2 μg/mL) and AF488-WGA (15 μg/mL) in Mcoy’s 5a (10% FBS) medium for 10 min. The medium was removed, and cells were washed three times with warm phosphate-buffered saline (PBS), followed by RPMI medium (10% FBS, no phenol red). Cells were directly imaged using an Olympus FluowView IX2 inverted confocal microscope with a 60× objective. Colocalization of the peptide with the plasma membrane, in the presence and absence of UV exposure, was statistically quantified using Pearson correlation coefficient analysis calculated using FIJI software.
Cell Viability Assay
SKOV3 cells were grown and irradiated for 0, 5, and 50 min following the same procedure described above. In each sample, medium was replaced with 1 mL of Mcoy’s 5a medium (10% FBS) containing 0.5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and incubated for 1 h at 37 °C under CO2 (5.0%). Medium was removed from each plate, and cells were washed once with warm PBS. Next, 1.5 mL of DMSO was added to each plate to lyse the cells. The cells were placed on an orbital shaker for 15 min until they were completely dissolved. Absorbance was obtained at 570 nm using a UV spectrometer. Data were normalized such that cells that were not exposed to irradiation had a cell viability of 100%.
Results and Discussion
1. Synthesis and Studies of the Photolysis of Bhc-Protected Cysteine-Containing Peptides
1.1. Synthesis of Bhc-Protected Fmoc-Cysteine and Incorporation into Peptides
Previous studies in our laboratory have shown that Bhc can be used for photolabile thiol protection of a peptidomimetic enzyme inhibitor. The caged molecule manifested efficient cleavage to yield a free thiol upon one- and two-photon irradiation, allowing it to be used for biological applications in cell culture. Therefore, in order to develop photolabile S-protected cysteine-containing peptides, we initially used Bhc as a caging group. Our strategy was to prepare Bhc-protected Fmoc-cysteine and incorporate that into a peptide of interest through SPPS; the synthesis of a form of cysteine suitable for SPPS is depicted in Scheme 1. The phenolic hydroxyl group of Bhc-chloride (1) was protected using chloromethyl methyl ether and triethylamine to give MOM-protected Bhc-Cl (2) in 89% yield. This chloride was subsequently used to alkylate Fmoc-cysteine methyl ester under mild acidic conditions, using Zn(OAc)2 as a catalyst,38 to produce 3 in 80% yield. Saponification of the methyl ester using trimethyl tin hydroxide39 under reflux generated the desired caged Fmoc-cysteine derivative (4) in 81% yield.
Scheme 1. Synthesis of Bhc-Protected Fmoc-Cys and Incorporation into a Peptide via SPPS.
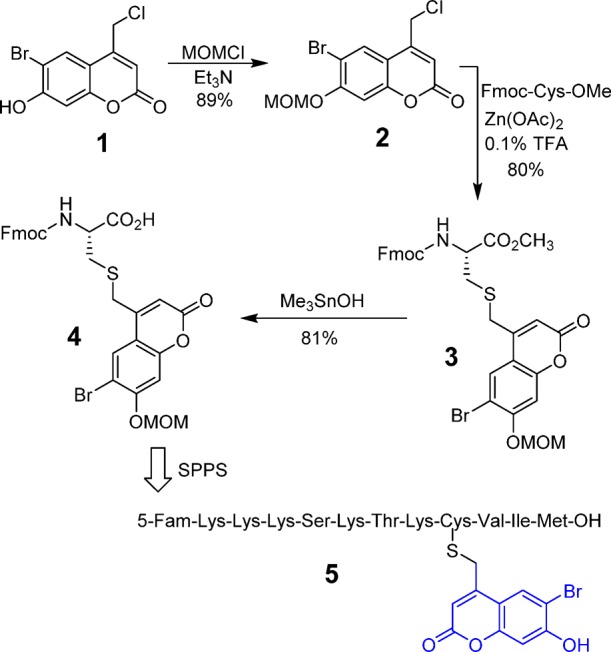
The general route for synthesis of caged peptides employed standard SPPS conditions, in which Fmoc-protected residues were added sequentially to a peptide anchored on Wang resin. The only exception involved the incorporation of the caged Fmoc-Cys residue, where the coupling time was increased to 6 h to ensure quantitative incorporation of the nonstandard residue. Final treatment of the resin-bound peptide with acid (standard conditions using Reagent K) removed all side-chain protecting groups, including the MOM group present on the Bhc moiety, and cleaved the polypeptide from the resin to generate the desired caged molecule. This strategy was successfully employed to synthesize a caged form of K-Ras peptide, 5, that includes an N-terminal fluorescein group. While the uncaged form of that peptide is a known substrate for the enzyme, PFTase, the Bhc-protected form is not. The goal was to use light to uncage the peptide and restore its ability to be recognized by the enzyme and undergo farnesylation.
1.2. Photolysis of Bhc-Protected Cysteine-Containing Peptides
Once the fluorescein-labeled caged peptide was successfully synthesized and purified, the next step was to verify its uncaging efficiency upon photolysis. Hence, solutions of caged peptide in photolysis buffer (1 mM DTT in 50 mM PB at pH 7.2) were irradiated using 365 nm light in a Rayonet photoreactor for varying amounts of time (Figure 1A). Each sample was analyzed by RP-HPLC and monitored by fluorescence. Inspection of the HPLC traces (Figure S1) revealed that the starting peptide peak disappeared over time, with concomitant formation of a new peak with a later retention time. Surprisingly, further analysis of the reaction mixture via ESI-MS revealed that the photolytic product and non-irradiated starting peptide had identical masses, indicating that irradiation causes isomerization instead of uncaging.
Figure 1.

(A) Uncaging reaction of Bhc-protected cysteine-containing peptide 5 upon UV irradiation. LC-MS analysis of photolysis of peptide 5: (B) EIC chromatogram (m/z = 635.20–635.30) of a sample of 5 in photolysis buffer, (C) EIC chromatogram (m/z = 635.20–635.30) of sample of 5 after 120 s photolysis showing the formation of a photoisomer, and (D) EIC chromatogram (m/z = 551.20–551.30) of a standard sample of free peptide 6b showing that no uncaged product was detected upon photolysis of 5.
Extracted ion current (EIC) chromatograms obtained by LC-MS analysis (Figure 1B,C) clearly revealed the disappearance of the starting peptide (tR = 8.45 min, m/z = 635.26) upon photolysis and concomitant formation of a new peak with identical mass (tR = 8.70 min, m/z = 635.26) that corresponds to the photoisomerized product. To study whether any free (uncaged) peptide 6 was produced upon irradiation, 6 was synthesized by an independent route and subjected to LC-MS. Analysis of the LC-MS traces of the irradiated peptide showed no evidence for the presence of free peptide ions that match with the authentic standard (Figure 1C,D).
We initially hypothesized that the observed isomerization might be due to photoinduced migration of the Bhc group to a side-chain amine group of a neighboring lysine residue. This hypothesis was tested by MS/MS fragmentation analysis, since migration of Bhc to other residues would change the fragmentation of the photoisomer relative to the starting peptide. Interestingly, MS/MS analysis revealed that the two isomers have the same backbone fragmentation pattern (Table S1). Of particular importance, two of the main fragments present in both the photoisomer and the caged peptide are the doubly charged X4 and B7 ions (Figure S2), which clearly indicate that the Bhc group remains connected to cysteine even after irradiation. This observation is not consistent with the hypothesis of phototriggered Bhc migration to lysine residues and indicates that a rearrangement occurs directly on the caged cysteine residue.
The high extent of photoisomerization observed upon photolysis of compound 5 was somewhat contrary to our previous results with Bhc-protected inhibitors, where photolysis led to efficient (>85%) uncaging. Thus, we considered the possibility that the photocleavage of Bhc-caged thiols may be context dependent. To test this hypothesis, two additional peptides were synthesized in which the location of caged cysteine was moved by one residue along the peptide sequence. Each peptide was photolyzed separately, and the resulting products were analyzed by LC-MS. Based on the EIC data (Figures S3 and S4), photolysis of KKKSKTCC(Bhc)IM produced only the photoisomer and no detectable uncaged peptide. In contrast, photolysis of KKKSKTC(Bhc)CVIM generated a mixture of both the photoisomer and free peptide. This hypothesis was also tested with shorter peptides where C(Bhc)VLS showed formation both uncaged and isomerized product (Figure S5), whereas photolysis of dansyl-GC(Bhc)VLS did not produce any uncaged peptide (Figure S6). These data confirm that the efficiency of Bhc photocleavage is highly dependent on its surrounding chemical environment. It is worth noting that photoisomerization was also observed upon two photon excitation of C(Bhc)VLS (Figure S7).
Previously reported mechanistic studies have demonstrated that the photocleavage of coumarin-based protected carboxylic acids results in scission of the C–O bond to produce a reactive carbocation which is rapidly quenched by water when the latter is used as the solvent.40 However, an alternative pathway could involve reaction of the cationic intermediate with an internal nucleophile via an intramolecular process; such a mechanism would give rise to an isomeric product consistent with the observations reported here. To examine this possibility, photolysis reactions were performed in the presence of high concentrations of thiols in order to increase the rate of trapping. Thus, aqueous solutions of 5 were irradiated in the presence of excess DTT (up to 200 mM), and analyzed by RP-HPLC. Interestingly, none of those experiments revealed any measurable change in the extent of photoisomerization. These results suggest that the photo-rearrangement may proceed through a concerted intramolecular mechanism; however, additional experiments are needed to thoroughly address this question.
1.3. NMR Analysis of Bhc Photo-rearrangement
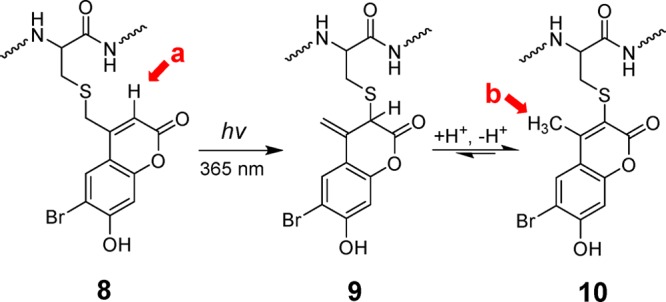
The possibility of photo-rearrangement of related amino-coumarin-protected cysteines has been previously suggested by Hagen and co-workers; however, in their publication, no analysis was performed to conclusively identify the structure of photoisomer generated.32 Therefore, after first observing the photo-rearrangement of Bhc-caged thiols by mass-spectroscopy, we decided to determine the structure of the isomeric product using NMR methods. In order to obtain sufficient material for NMR analysis, a solution of caged peptide 5 was irradiated and the photoisomer was isolated via preparative RP-HPLC purification. 1H NMR spectra were obtained using a sensitive cryoprobe instrument. As shown in Figure S8A,B for 5 and the photoisomer, respectively, both compounds have very similar spectra, with the exception of a distinctive peak at 6.2 ppm present in the spectrum of 5 that is missing in the spectrum of the photoisomer; concomitantly, a new signal appears at 2.4 ppm in the spectrum of the isomer that is not present in the spectrum of 5. Comparison of those spectra with the Bhc 1H NMR spectrum31 indicates that the signal at 6.2 ppm corresponds to the aryl proton at the 3 position of Bhc (Scheme 2). The disappearance of that peak and the appearance of the new signal at 2.4 ppm are consistent with a photoinduced 1,3 shift of the sulfur atom from the exocylic position to the 3 position to give intermediate 9 followed by tautomerization to yield a 4-methylbromohydroxycoumarin-3-yl thioether (10) as previously suggested by Hagen and co-workers.32 In such a case, the resonance at 2.4 ppm could be attributed to the presence a methyl group in the final photoproduct 10.
Scheme 2. Hypothesized Mechanism of Photoisomerization of Bhc-Caged Cysteine.
Due to the complicated 1H NMR spectra of the peptides, we decided to validate the proposed hypothesis using a simpler model system. Hence, Bhc-protected cysteamine (11, Figure 2), which has a simple structure and a straightforward synthetic route, was chosen as the model system. Additionally, this specific molecule has been previously used by Shoichet and co-workers for phototriggered uncaging of thiol functionality inside hydrogels.28 Therefore, knowing all the complexities due to context dependence of Bhc photocleavage, we were also interested to see how efficient this compound could undergo uncaging.
Figure 2.
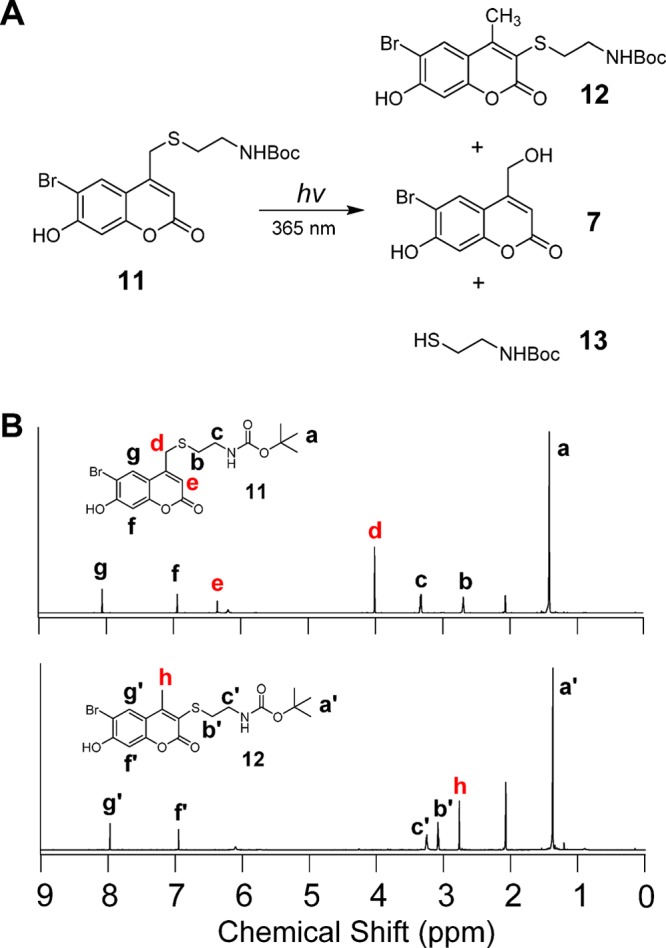
(A) Photolysis of Bhc-protected Boc-cysteamine and the resulting photolytic products. (B) 1H NMR spectrum of Bhc-protected Boc-cysteamine (top) and the corresponding photoisomer (bottom).
Compound 11 was synthesized following a previously reported procedure.33 Solutions of 11 were irradiated using 365 nm light in a Rayonet photoreactor for varying amounts of time followed by analysis via RP-HPLC with UV detection. That allowed the disappearance of 11 as well as the formation of the isomeric rearrangement product 12 and Bhc-OH (7) (formed from the desired uncaging reaction) to be monitored. Inspection of the HPLC data (Figure S9) indicates that the major product of photolysis of compound 11 under these conditions is the photoisomer 12, the product with a higher retention time) with a smaller amount of the desired uncaged product formed, as indicated by the low intensity peak corresponding to 7.
Reactions containing Bhc-cysteamine (11) and its corresponding photoisomer 12 were separated by RP-HPLC and the purified compounds analyzed by NMR spectroscopy. Comparison of the 1H NMR spectrum of 11 with that of compound 12 revealed characteristic changes in the proton signals corresponding to those observed in the peptide experiment (Figure 2). Methylene (Hd) and aryl (He) protons present in the starting material are absent in the spectrum of the photoisomer. In addition, a new signal at 2.72 ppm (Hh) corresponds to the new methyl group that is generated. Also of note, the triplet signal (H′b), corresponding to the methylene protons of cysteamine, shifts downfield relative to that of the starting compound (Hb) as a result of thiol conjugation with double bonds which renders the thiol a stronger electron-withdrawing group; alternatively, this shift may be due to a ring current effect. These observations convincingly support the suggested mechanism for photo-rearrangement and the proposed structure of the photoisomer. A similar photolysis experiment was performed in D2O. LC-MS analysis clearly indicates formation of a monodeuterated photoisomer (Figure S10). This data demonstrates that there is a solvent-derived proton incorporated in the product, consistent with the mechanism proposed for photo-rearrangement described in Scheme 2.
To obtain an accurate ratio of the extent of uncaging versus photo-rearrangement, a sample of compound 11 that had been subjected to irradiation, and thus contained both uncaged and photoisomerized product, was analyzed by 1H NMR spectroscopy (Figure S11). Using integration values obtained from characteristic protons from each product, a ratio for uncaging versus photoisomerization of 1:10 was calculated. Thus, while Shoichet and co-workers have used 11 (under different conditions) to successfully uncage a thiol upon photolysis, the experiments reported here revealed that the major product of this reaction is an unwanted photoisomer. Overall, the variability obtained using Bhc suggests that it is not generally applicable as a caging group for thiols and that there is a real need for an alternative caging group for general usage.
2. Use of Nitrodibenzofuran for Development of Caged Cysteine-Containing Peptides
2.1. Alternative Strategy Using NDBF
The initial goal of our work was to identify a protecting group that could be used to cage the thiol group of cysteine when present within a peptide that would be efficiently deprotected through UV irradiation as well as near-IR light via a two-photon process. Although Bhc has shown reasonable one- and two-photon uncaging efficiencies for protection of various functionalities including carboxylates, phosphates and carbamates, the experiments described above revealed its photocleavage efficiency is unpredictable when used with thiols; moreover, the main product formed upon irradiation is often an unwanted rearrangement byproduct in lieu of the desired free thiol. To address these limitations, we elected to examine another type of caging group that undergoes uncaging via a process significantly different from coumarin-based compounds.
o-Nitrobenzyl (ONB)-based caging groups have been extensively used for thiol photocaging. Despite, their relatively slower uncaging rate (compared with coumarins), they undergo photolysis with minimal byproduct formation. However, ONB-based compounds suffer from low one-photon and especially low two-photon absorptivity which limits their applications in cellular media and live tissue. In 2006, Momotake et al. introduced NDBF, a more extensively conjugated form of ONB, as a new caging group with high one- and two-photon sensitivity.36 This compound has previously been used for protection of hydroxyl functionalities and showed rapid and efficient uncaging upon one- and two-photon irradiation.41 Due to its advantages over traditional ONB-based molecules, we decided to explore its applicability for photocaging of thiols in order to prepare peptides containing caged cysteine residues.
2.2. Synthesis of NDBF-Protected Fmoc-Cys-OH and Incorporation via SPPS
Similar to the synthesis of Bhc-caged peptides, the strategy pursued here was to first synthesize Fmoc-cysteine containing an NDBF-protected thiol [Fmoc-Cys(NDBF)-OH] and then incorporate that into a peptide through standard SPPS. Starting from dibenzofuran, NDBF-Br (13) was synthesized in four steps (Scheme S1). Next, as described in Scheme 3, Fmoc-cysteine methyl ester was first reacted with NDBF-Br under acidic conditions, to produce compound 15 in 70% yield. The resulting methyl ester was then hydrolyzed using (CH3)3SnOH to yield Fmoc-Cys(NDBF)-OH (16) in 75% yield.
Scheme 3. Synthesis of NDBF-Protected Fmoc-Cys-OH and Incorporation into Peptide Sequence via SPPS.
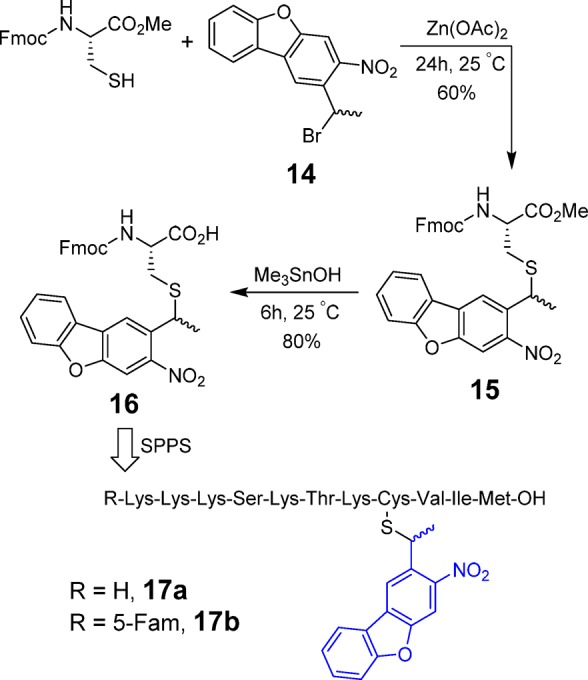
The resulting protected cysteine residue was successfully incorporated into several K-Ras-derived peptides (17a,b) via standard SPPS as described for the related Bhc-protected peptides noted above; the final products were characterized by ESI-MS-MS to confirm the presence of the NDBF group after the global deprotection step (Table S2). Since NBDF protection of cysteine involves a thioether bond, there was no evidence of any S-to-N shift or deprotection occurring during synthesis, a problem that does occur when thiocarbamate-based protection strategies are used.32
2.3. One- and Two-Photon Photolysis of NDBF-Caged Cysteine Peptides
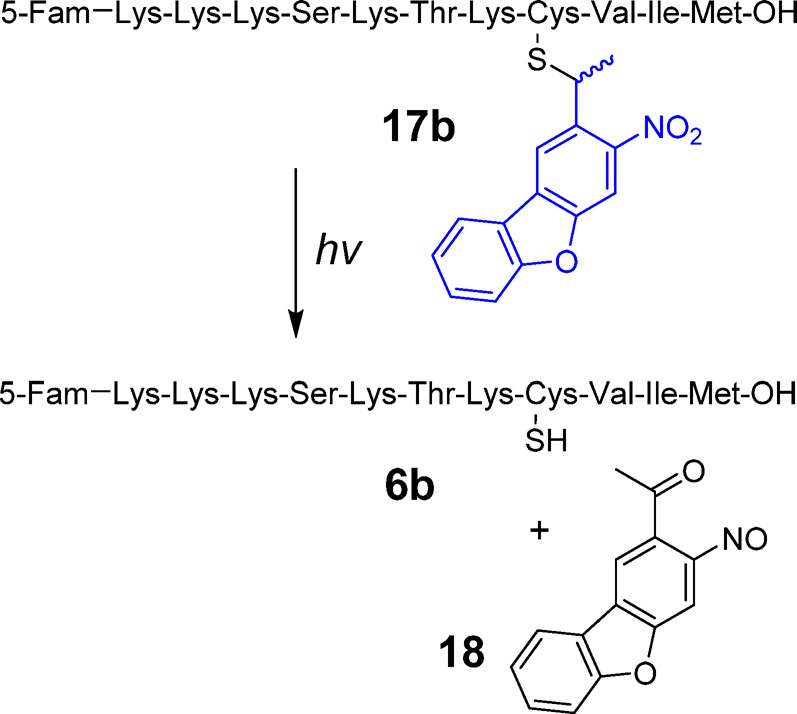
After completion of the synthesis of the fluorescently labeled caged peptide 17b, photolysis experiments were conducted to probe for the formation of the uncaged peptide containing a free thiol upon photolysis (Scheme 4). In this experiment, a solution of 17b was irradiated at 365 nm for 45 s and subjected to RP-HPLC. As can be seen from the chromatograms shown in Figure S12, photolysis resulted in the disappearance of the peak corresponding to the starting peptide 17b and concomitant appearance of a new peak tentatively assigned as 6b. ESI-MS/MS analysis confirmed that the newly formed peak corresponds to the expected free peptide (Figure S12, Table S3). The absence of any unwanted photoproducts based on an HPLC trace devoid of any other significant products, suggests that that photolysis of NDBF-caged peptides undergo conversion to free peptide upon UV irradiation with high efficiency. In order to further evaluate the general applicability of this strategy, a second peptide, dansyl-GC(NDBF)VLS was also synthesized and studied. Analysis of a photolysis reaction containing that peptide showed complete conversion to the free peptide upon irradiation (Figure S13), unlike its Bhc-protected counterpart (compare with Figure S6). These data suggest that NDBF lacks the limitations and undesired reactivity manifested by Bhc for thiol caging.
Scheme 4. Light-Triggered Uncaging of NDBF-Protected K-Ras Peptide (17b).
One-photon uncaging kinetics of compound 17b were evaluated by irradiating solutions of 17b for varying periods of time followed by analysis via RP-HPLC (Figure 3A). Based on those data, the uncaging quantum yield (εΦ) of peptide 17b was measured to be 0.2. The quantum yield measured in this experiment is somewhat lower than the value reported for NDBF used for caging hydroxyl functionality (0.7),36 which may be due to the light absorption of fluorescein attached to the peptide. However, due to the high molar absorptivity of NDBF, which results in a high εΦ value, the uncaging t1/2 was quite short (25 s) under the photolysis conditions (standard Rayonet reactor).
Figure 3.

(A) HPLC quantification of disappearance of the starting peptide (17b) and formation of the uncaged peptide (6b) as a function of irradiation time at 365 nm. (B) HPLC quantification of uncaging of 17a as a function of two-photon irradiation time (800 nm, pulsed Ti:Saphire laser, 210 mw, 80 fs pulse width). Photolysis reactions were performed in 200 and 300 μM solutions of 17b and 17a respectively, containing 1 mM DTT in 50 mM PB, pH 7.5.
Since the NDBF-caged peptide showed useful uncaging properties upon one-photon irradiation, further experiments were performed to evaluate its two-photon uncaging efficiency. Thus, solutions of 17a were irradiated at 800 nm using a Ti:Saphire laser and the photolysis products were again analyzed by RP-HPLC and confirmed by LC-MS (Figure S14). The two-photon action cross-section of 17a was measured using 6-bromo-7-hydroxycoumarin-4-ylmethyl acetate (Bhc-OAc) as a reference.31 Even though the extinction coefficient of the NDBF chromophore at 400 nm is less than 10% of that at λmax (325 nm), a value of 0.13 GM for 17a uncaging at 800 nm was calculated (Figure 3B). It should be noted that this value would be greater if the two-photon irradiation was performed at lower wavelengths where the extinction coefficient is closer to the maximum value although tissue penetration would be less. Overall, these results demonstrate that NDBF is an efficient thiol caging group that undergoes clean photocleavage to generate a free thiol upon one- or two-photon irradiation.
2.4. One- and Two-Photon-Triggered in Vitro Farnesylation of a Caged K-Ras Peptide
Since the NDBF-caged peptide demonstrated good uncaging efficiency, we next sought to examine its utility in a more biologically relevant context. Protein prenylation is a critical process that affects key signaling mechanisms within cells involved in a plethora of functions from growth to differentiation.42 Prenyl groups are appended to proteins via thioether linkages formed by alkylation of specific cysteine residues catalyzed by prenyltransferases including PFTase, which transfers a farnesyl (C15) group.43 Thus, a K-Ras-derived peptide incorporating a caged cysteine residue at the site of prenylation should not be a substrate for PFTase; however, upon irradiation, the resulting peptide manifesting a free thiol produced by photocleavage of the protecting group should be an efficient substrate and become farnesylated (Figure 4A). In order to test this, a series of experiments was conducted in which a caged peptide was treated with FPP in prenylation buffer (15 mM DTT, 10 mM MgCl2, 50 μM ZnCl2, 20 mM KCl), with or without enzyme addition and UV light exposure.
Figure 4.
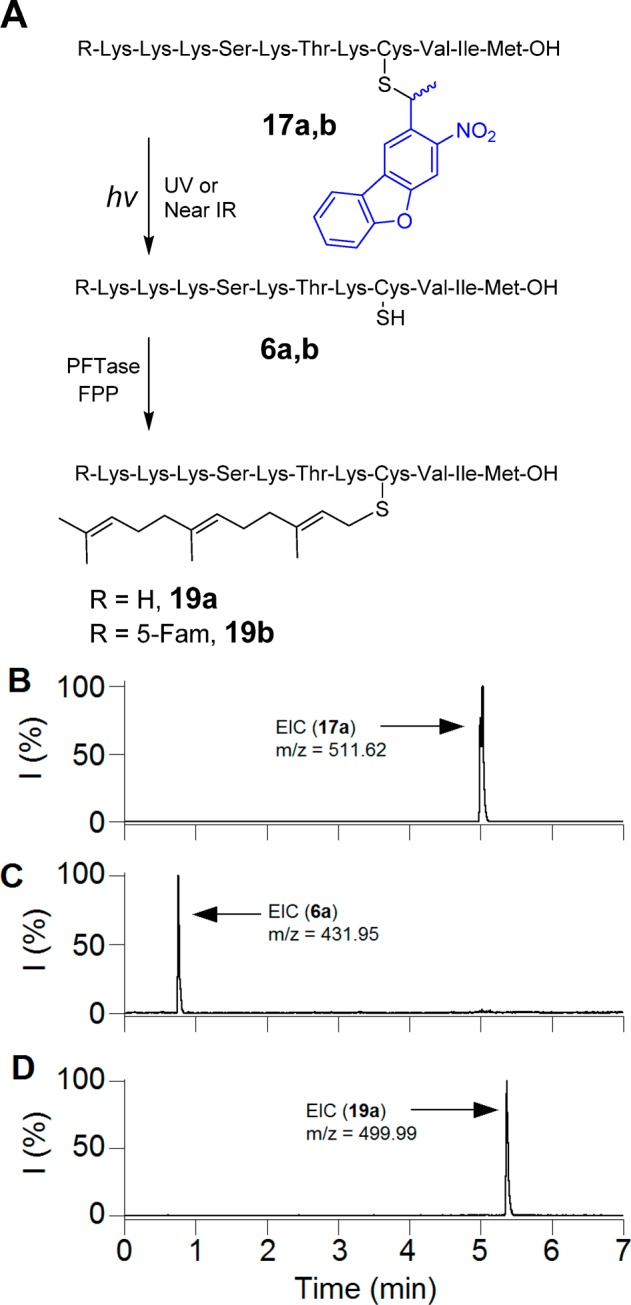
(A) Photo-uncaging of 17 and its subsequent farnesylation by enzyme. (B) EIC chromatogram (m/z = 511.62) of a 7.5 μM solution of 17a in prenylation buffer containing PFTase without irradiation. (C) EIC chromatogram (m/z = 431.95) of a solution of 17a after 2.5 min irradiation at 800 nm (Ti:sapphire laser, 170 mW, 90 fs) in prenylation buffer without PFTase. (D) EIC chromatogram (m/z = 499.99) of 17a after 2.5 min irradiation at 800 nm (Ti:sapphire laser, 210 mW, 90 fs) in the presence of PFTase, showing the formation of farnesylated peptide 19a.
As predicted, the caged peptide 17b was not farnesylated when treated with yeast PFTase. LC-MS analysis of reaction mixture indicates only the presence of the caged peptide (m/z = 630.87, Figure 4A). Photolysis of 17b for 60 s at 365 nm, in the absence of enzyme, generated the uncaged peptide, as confirmed by the formation of a new peak with the expected m/z value (m/z = 551.21, Figure S15). However, photolysis of 17b in the presence of PFTase led to the generation of a farnesylated peptide (19b). The new peak with the retention time of 10.55 min has a mass to charge ratio of 619.26 which is in good agreement with the calculated value (C92H145N16O20S23+, 619.34) for the farnesylated peptide. This observation clearly illustrates that the peptide undergoes UV-dependent farnesylation which could make it a useful probe for studying prenylation reactions in a spatiotemporally controllable manner.
Since, it would be useful to employ such caged peptides for studies in tissue or whole organisms where UV light cannot efficiently penetrate, the ability of 17a to undergo farnesylation by irradiation at longer wavelengths via two-photon excitation was examined. Accordingly, in vitro farnesylation reactions, similar to those described above for UV irradiation, were conducted using an 800 nm laser light source.
As was noted before, treatment of the caged peptide with enzyme in the presence of FPP without irradiation did not alter the starting peptide (Figure 4). Irradiation of 17a (m/z = 511.62) at 800 nm for 2.5 min in the absence of enzyme, generated the free peptide 6a, as confirmed from the EIC chromatogram (m/z = 431.95, Figure 4C). Treatment of 17a with the enzyme along with 2.5 min two-photon irradiation generated the farnesylated peptide 19a, as shown by the appearance of an ion of m/z = 499.99 (Figure 4D). In summary, these data demonstrate that an NDBF-caged K-Ras peptide (17a) is capable of undergoing farnesylation triggered by 800 nm light via two-photon excitation (also see Figure S16).
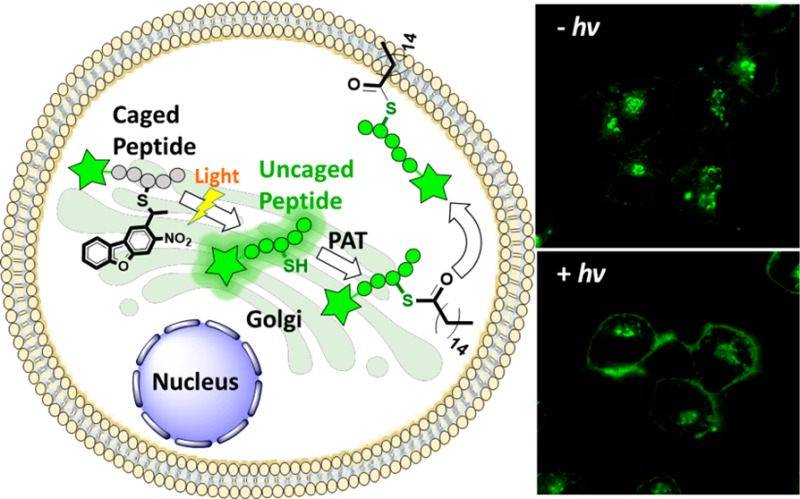
3. Light Activation of a Caged Peptide inside Live Cells
One of the important goals for photocaging of bioactive molecules, including peptides, is to modulate their activity by irradiation inside cells in order to study biological processes. Since farnesylation of NDBF-caged K-Ras peptide was efficiently and rapidly triggered by UV and IR irradiation in vitro, we decided to explore the same strategy to develop peptides that can be efficiently activated upon irradiation inside live cells.
Protein palmitoylation is a post-translational modification that plays critical roles in subcellular protein localization. In this process, palmitoyl acyltransferases (PATs) covalently attach a C16 palmitate group via thioesterification to one or more specific cysteine residues present in protein targets.44 This modification causes proteins to be more hydrophobic and migrate to the plasma membrane where they are active;45 prenylated proteins including H-Ras and N-Ras are commonly palmitoylated. Draper et al. have developed several fluorescently labeled cell-penetrating peptides including NBD-Hex-CLC(S-farnesyl)-OMe (21, Scheme 5), which they have used to study palmitoylation inside cells.46 When the free cysteine in the peptide is not modified, it localizes mainly in the cytosol and the Golgi; however, palmitoylation of the free cysteine by PATs inside cells results in the migration of the peptide to the plasma membrane. Therefore, a caged version of Hex-CLC(S-farnesyl)-OMe (20), cannot be a substrate for PAT and would thus localize in the cytosol/Golgi; however, irradiation should uncage the peptide, revealing a free thiol that would become palmitoylated and hence gradually migrate to plasma membrane. While peptide 21 has previously been shown to traffic to the plasma membrane, it was impossible to temporally control that process since cellular uptake and trafficking could not be uncoupled. However, the availability of a caged form makes this possible.
Scheme 5. Schematic Representation of NBD-Hex-C(NDBF)LC-OMe Uncaging and Subsequent Palmitoylation.
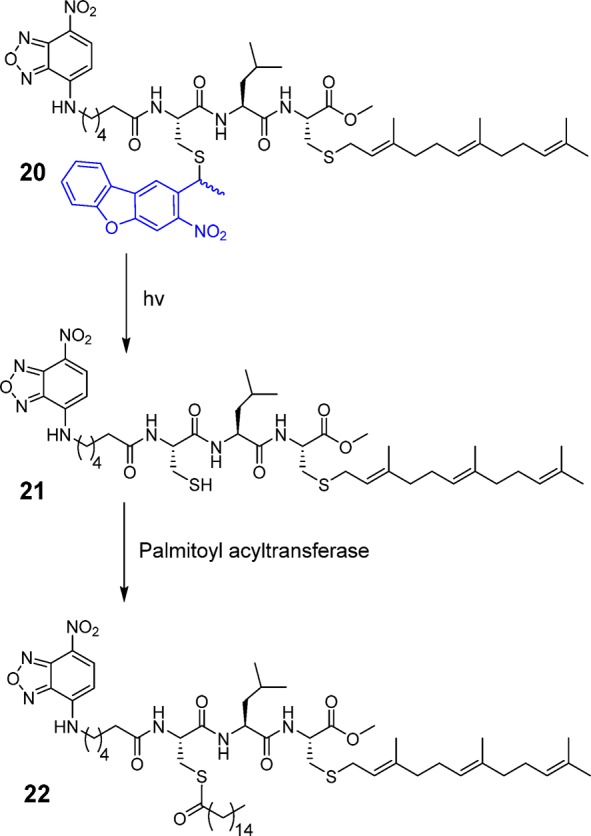
Peptide 20 was prepared using a cysteine anchoring method developed by our group for the synthesis of C-terminal methyl esters (Figure S17).47 Trityl chloride resin was first treated with Fmoc-Cys-OMe and DIEA in CH2Cl2 overnight to afford Fmoc-cysteine-loaded resin. The peptide was extended on the resin through standard SPPS employing Fmoc-based chemistry. Reagent K treatment cleaved the peptide from the resin which was then farnesylated via treatment with farnesyl bromide and Zn(OAc)2 under acidic conditions. The final caged peptide was purified by preparative RP-HPLC. Despite the presence of two cysteines in the sequence, there was no evidence of NDBF scrambling between the two thiols during the synthesis, consistent with the stability afforded by the NDBF thioether linkage. Those results are in contrast to those obtained when thiocarbonate strategies are used for thiol protection in peptides containing multiple cysteines where migration via acyl transfer readily occurs. Moreover, studies have shown that carbonates and thiocarbonates are prone to hydrolysis via esterases, thus limiting their applicability in living systems.48 In general, the efficient assembly of caged peptide 20 highlights the utility of NDBF-protected cysteine and how it can be employed for the synthesis of a variety of caged peptides including those containing multiple cysteines, with no risk of caging group migration.
Next, light-triggered intracellular palmitoylation of caged peptide 20 was studied using human ovarian carcinoma SKOV3 cells. Cells were incubated with 20 in for 3 h at 37 °C to allow cellular uptake. The cells were then divided into two groups and one was subjected to 5 min of UV irradiation at 330 nm. Both irradiated and non-irradiated cells were incubated for an additional 120 min at 37 °C, stained with nuclear and membrane markers and imaged by confocal microscopy. As observed in the fluorescence microscopy images (Figure 5A), before irradiation, the peptides reside primarily in the cytosol and Golgi. However, after UV exposure, the peptides traffic to the plasma membrane where they colocalize with the membrane dye; this change in colocalization occurs due to enzymatic palmitoylation. The degree of colocalization of the peptide and plasma membrane dye was quantified by calculating Pearson’s coefficients for both the non-irradiated (0.26) and irradiated cells (0.76) which clearly indicates a significant increase in membrane localization of the peptide after irradiation (Figure 5B). It is important to note that while all of the peptide did not localize to the membrane upon photolysis, this is unlikely to be due to incomplete uncaging since UV-mediated uncaging is quite fast and efficient (see Figure 3). In their original work, Draper et al.46 reported incomplete localization even when starting with the fully deprotected form of the peptide used here.46 Similar results (only partial localization in pulse-chase labeling experiments) have been reported in work with fluorescent proteins that are prenylated and palmitoylated and have been attributed to competing pathways involving degradation versus membrane targeting.49
Figure 5.

Live-cell experiments showing temporal control of enzymatic palmitoylation via NDBF-thiol caging. (A) Images obtained by fluorescence confocal microscopy illustrating intracellular localization of fluorescently labeled peptide 20 in SKOV3 cells, before (top) and after (bottom) UV exposure. (B) Quantification of colocalization of peptide and membrane dye via Pearson’s coefficient analysis, indicating a significant increase in plasma membrane localization of peptide upon irradiation.
To study the localization process in more detail, samples of the caged peptide were allowed to internalize for 3 h and then uncaged by UV exposure. Analysis of the cellular distribution of the peptide in those samples at different times ranging from 30 to 120 min showed that the membrane colocalization reached a peak after 30 min followed by a slow decrease (Figure S18). Such behavior is consistent with observations made with fluorescent proteins that have been shown to undergo dynamic cycling involving palmitoylation and concomitant membrane localization followed by depalmitoylation and internalization.50,51 Overall, these live cell experiments illustrate how NDBF caging of cysteine allows an enzyme substrate for palmitoylation to be temporally activated, thus permitting the processes of cellular entry and subsequent enzymatic modification to be deconvoluted.
Finally, in order to highlight the improved uncaging efficiency of NDBF and its utility in live cell experiments, a comparison was made between the uncaging efficiency of NDBF versus the 6-nitroveratryl (NV) group which is one of the most frequently used caging groups.15 We elected to study this experimentally since a range of values for the quantum yield of NV have been reported.15 Thiol-protected forms of Fmoc-Cys-OMe were prepared using the two caging groups which were then subjected to UV irradiation (365 nm) and the extent of deprotection was monitored. Uncaging data obtained by HPLC analysis shown in Figure S19, shows that the NDBF uncaging efficiency is greater than 10-fold higher than that of NV. This difference reflects the high molar absorptivity (ε = 15 300 M–1 cm–1) and high quantum yield (Φ = 0.2) of NDBF leading to a high εΦ (∼3060) value versus that of NV (εΦ ≈ 6000 × 0.01 = 60).52 Thus, the lower uncaging efficiency manifested by NV requires longer irradiation times to obtain comparable levels of uncaging. When SKOV3 cells were subjected to 50 min of irradiation (10-fold longer than was used to uncage 20), a significant decrease in cell viability was observed. Figure S20 shows a 5% loss of viability after 5 min of irradiation and a 57% decrease after 50 min. Clearly this excessive loss of viability precludes the use of NV protection in this experiments and serves to underscore the increased efficiency and utility of NDBF. However, it should be noted that the two-photon action cross-section of NDBF thiol uncaging is comparable to those manifested by Bhc-carboxylates31 and NDBF-alcohols.36 To date, two-photon activation of such caged molecules has been restricted to experiments where only a small fraction of uncaging is required to obtain a biological response. At present, it is unclear whether a large fraction of an NDBF-caged bioactive thiol can be released via two-photon excitation in cells since longer irradiation times may result in IR heating or phototoxicity; cell-based experiments to answer these questions are currently in progress. Nevertheless, the results reported here highlight the utility of NDBF caging for a variety of different biological applications. Coupled with its utility for the synthesis of peptides containing multiple cysteines, the data described here make it clear why NDBF is a superior choice for thiol caging.
Conclusion
In this work, we analyzed the photolysis of several Bhc-protected thiol-containing peptides and small molecules. Those experiments revealed that Bhc-caged thiols exhibit variable uncaging yields and that their photolysis frequently leads to the formation of an unwanted rearrangement product. Using NMR analysis, the structure of the photochemically produced isomer was established to be a 4-methylcoumarin-3-yl thioether.
The poor uncaging yield of Bhc-caged thiols led us to search for a more efficient thiol caging group that would be useful for biological applications; accordingly, NDBF caging, which has previously been shown to be effective for hydroxyl group protection, was explored. NDBF-protected Fmoc-cysteine was synthesized and successfully incorporated into a K-Ras-derived peptide via standard solid-phase synthesis. The resulting caged peptide was photolyzed and completely converted to free peptide with a photolysis quantum yield of 0.2. The two-photon action cross-section of the caged peptide was measured to be 0.13 GM at 800 nm comparable to that of Bhc-OAc. The one- and two-photon photolysis of a caged K-Ras peptide in the presence of PFTase revealed that the free peptide formed upon irradiation is efficiently converted by the enzyme to its biologically relevant prenylated form. The NDBF-protected cysteine was also used to develop a light-activatable, cell-penetrating peptide containing a caged and a farnesylated cysteine. Confocal microscopy analysis showed that the caged peptide could be activated inside cells upon light exposure which resulted in intracellular migration due to enzymatic palmitoylation. Taken together, this work for the first time reports an efficient, robust, and broadly applicable strategy for the synthesis of a variety of peptides and related small molecules containing caged thiols that can be activated by both one- and two-photon processes in live cells. These results set the stage for a variety of studies where spatiotemporal control of thiol reactivity is required, including a diverse span of applications ranging from chemical biology to material science.
Acknowledgments
We thank Dr. Joseph Dalluge for helpful discussion regarding mass spectrometry. We thank Dr. Guillermo Marquez and John Oja at University of Minnesota imaging center for helpful discussion regarding confocal microscopy experiments. We also thank Prof. Emil Lou, Dr. Venugopal Thayanithy, and Snider Desir for providing us with SKOV-3 cells and helpful discussion regarding the cell culture. Financial support for these studies through National Institutes of Health Grant GM084152, the University of Minnesota Supercomputer Institute, the University of Minnesota Imaging Center, a Nikon Center of Excellence for confocal microscopy, and the University of Minnesota Graduate School is gratefully acknowledged. This work is dedicated to Prof. Peter B. Dervan on the occasion of his 70th birthday.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b11759.
Synthesis procedures, HPLC and LC-MS data, and MS/MS fragmentation data, including Scheme S1, Tables S1–S3, and Figures S1–S20 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Brieke C.; Rohrbach F.; Gottschalk A.; Mayer G.; Heckel A. Angew. Chem., Int. Ed. 2012, 51, 8446. 10.1002/anie.201202134. [DOI] [PubMed] [Google Scholar]
- Adamantidis A. R.; Zhang F.; Aravanis A. M.; Deisseroth K.; de Lecea L. Nature 2007, 450, 420. 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A. S.; Deiters A. ACS Chem. Biol. 2014, 9, 1398. 10.1021/cb500176x. [DOI] [PubMed] [Google Scholar]
- Gautier A.; Gauron C.; Volovitch M.; Bensimon D.; Jullien L.; Vriz S. Nat. Chem. Biol. 2014, 10, 533. 10.1038/nchembio.1534. [DOI] [PubMed] [Google Scholar]
- Ellis-Davies G. C. R. Nat. Methods 2007, 4, 619. 10.1038/nmeth1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.-M.; Larson D. R.; Lawrence D. S. ACS Chem. Biol. 2009, 4, 409. 10.1021/cb900036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea A. C.; Vandenberg L. N.; Ball R. E.; Snouffer A. A.; Hudson A. G.; Zhu Y.; McLain D. E.; Johnston L. L.; Lauderdale J. D.; Levin M.; Dore T. M. Chem. Biol. 2013, 20, 1536. 10.1016/j.chembiol.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer F.; Wagner J.; Knau A.; Dimmeler S.; Heckel A. Angew. Chem., Int. Ed. 2013, 52, 13558. 10.1002/anie.201307502. [DOI] [PubMed] [Google Scholar]
- Walsh S.; Gardner L.; Deiters A.; Williams G. J. ChemBioChem 2014, 15, 1346. 10.1002/cbic.201400024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bort G.; Gallavardin T.; Ogden D.; Dalko P. I. Angew. Chem., Int. Ed. 2013, 52, 4526. 10.1002/anie.201204203. [DOI] [PubMed] [Google Scholar]
- Donato L.; Mourot A.; Davenport C. M.; Herbivo C.; Warther D.; Léonard J.; Bolze F.; Nicoud J.-F.; Kramer R. H.; Goeldner M.; Specht A. Angew. Chem. 2012, 124, 1876. 10.1002/ange.201106559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X.; Shestopalov I. A.; Sinha S.; Zheng G.; Pitt C. L. W.; Li W.-H.; Olson A. J.; Chen J. K. J. Am. Chem. Soc. 2009, 131, 13255. 10.1021/ja809933h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson J. P.; Kwon H.-B.; Takasaki K. T.; Chiu C. Q.; Higley M. J.; Sabatini B. L.; Ellis-Davies G. C. R. J. Am. Chem. Soc. 2013, 135, 5954. 10.1021/ja4019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatrudo J. M.; Olson J. P.; Lur G.; Chiu C. Q.; Higley M. J.; Ellis-Davies G. C. R. ACS Chem. Neurosci. 2014, 5, 64. 10.1021/cn400185r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klán P.; Šolomek T.; Bochet C. G.; Blanc A.; Givens R.; Rubina M.; Popik V.; Kostikov A.; Wirz J. Chem. Rev. 2013, 113, 119. 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelliccioli A. P.; Wirz J. Photochem. Photobiol. Sci. 2002, 1, 441. 10.1039/b200777k. [DOI] [PubMed] [Google Scholar]
- Schafer F. Q.; Buettner G. R. Free Radical Biol. Med. 2001, 30, 1191. 10.1016/S0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Dawson P.; Muir T.; Clark-Lewis I.; Kent S. Science 1994, 266, 776. 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- Barron E. S. G. In Advances in Enzymology and Related Areas of Molecular Biology; Nord F. F., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2006; pp 201–266. [Google Scholar]
- Philipson K. D.; Gallivan J. P.; Brandt G. S.; Dougherty D. A.; Lester H. A. Am. J. Physiol. Cell Physiol. 2001, 281, C195. [DOI] [PubMed] [Google Scholar]
- Li H.; Hah J.-M.; Lawrence D. S. J. Am. Chem. Soc. 2008, 130, 10474. 10.1021/ja803395d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uprety R.; Luo J.; Liu J.; Naro Y.; Samanta S.; Deiters A. ChemBioChem 2014, 15, 1793. 10.1002/cbic.201400073. [DOI] [PubMed] [Google Scholar]
- Shao Q.; Xing B. Chem. Soc. Rev. 2010, 39, 2835. 10.1039/b915574k. [DOI] [PubMed] [Google Scholar]
- Cysteine: biosynthesis, chemical structure and toxicity; Nova Science Publishers, Inc.: Hauppauge, NY, 2012. [Google Scholar]
- Pan P.; Bayley H. FEBS Lett. 1997, 405, 81. 10.1016/S0014-5793(97)00165-8. [DOI] [PubMed] [Google Scholar]
- Arabaci G.; Guo X.-C.; Beebe K. D.; Coggeshall K. M.; Pei D. J. Am. Chem. Soc. 1999, 121, 5085. 10.1021/ja9906756. [DOI] [Google Scholar]
- Jones P. B.; Pollastri M. P.; Porter N. A. J. Org. Chem. 1996, 61, 9455. 10.1021/jo961638p. [DOI] [Google Scholar]
- Wosnick J. H.; Shoichet M. S. Chem. Mater. 2008, 20, 55. 10.1021/cm071158m. [DOI] [Google Scholar]
- DeGraw A. J.; Hast M. A.; Xu J.; Mullen D.; Beese L. S.; Barany G.; Distefano M. D. Chem. Biol. Drug Des. 2008, 72, 171. 10.1111/j.1747-0285.2008.00698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieboldt R.; Gee K. R.; Niu L.; Ramesh D.; Carpenter B. K.; Hess G. P. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 8752. 10.1073/pnas.91.19.8752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta T.; Wang S. S.-H.; Dantzker J. L.; Dore T. M.; Bybee W. J.; Callaway E. M.; Denk W.; Tsien R. Y. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 1193. 10.1073/pnas.96.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzur N.; Briand B.; Beyermann M.; Hagen V. J. Am. Chem. Soc. 2009, 131, 16927. 10.1021/ja907287n. [DOI] [PubMed] [Google Scholar]
- Wylie R. G.; Shoichet M. S. Biomacromolecules 2011, 12, 3789. 10.1021/bm201037j. [DOI] [PubMed] [Google Scholar]
- Wylie R. G.; Ahsan S.; Aizawa Y.; Maxwell K. L.; Morshead C. M.; Shoichet M. S. Nat. Mater. 2011, 10, 799. 10.1038/nmat3101. [DOI] [PubMed] [Google Scholar]
- Abate-Pella D.; Zeliadt N. A.; Ochocki J. D.; Warmka J. K.; Dore T. M.; Blank D. A.; Wattenberg E. V.; Distefano M. D. ChemBioChem 2012, 13, 1009. 10.1002/cbic.201200063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momotake A.; Lindegger N.; Niggli E.; Barsotti R. J.; Ellis-Davies G. C. R. Nat. Methods 2006, 3, 35. 10.1038/nmeth821. [DOI] [PubMed] [Google Scholar]
- Hatchard C. G.; Parker C. A. Proc. R. Soc. London, Ser. A 1956, 235, 518. 10.1098/rspa.1956.0102. [DOI] [Google Scholar]
- Xue C.-B.; Becker J. M.; Naider F. Tetrahedron Lett. 1992, 33, 1435. 10.1016/S0040-4039(00)91640-X. [DOI] [Google Scholar]
- Nicolaou K. C.; Estrada A. A.; Zak M.; Lee S. H.; Safina B. S. Angew. Chem., Int. Ed. 2005, 44, 1378. 10.1002/anie.200462207. [DOI] [PubMed] [Google Scholar]
- Schmidt R.; Geissler D.; Hagen V.; Bendig J. J. Phys. Chem. A 2007, 111, 5768. 10.1021/jp071521c. [DOI] [PubMed] [Google Scholar]
- Chen X.; Tang S.; Zheng J.-S.; Zhao R.; Wang Z.-P.; Shao W.; Chang H.-N.; Cheng J.-Y.; Zhao H.; Liu L.; Qi H. Nat. Commun. 2015, 6, 7220. 10.1038/ncomms8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F. L.; Casey P. J. Annu. Rev. Biochem. 1996, 65, 241. 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- Palsuledesai C. C.; Distefano M. D. ACS Chem. Biol. 2015, 10, 51. 10.1021/cb500791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resh M. D. Cell. Signalling 1996, 8, 403. 10.1016/S0898-6568(96)00088-5. [DOI] [PubMed] [Google Scholar]
- Dudler T.; Gelb M. H. J. Biol. Chem. 1996, 271, 11541. 10.1074/jbc.271.19.11541. [DOI] [PubMed] [Google Scholar]
- Draper J. M.; Xia Z.; Smith C. D. J. Lipid Res. 2007, 48, 1873. 10.1194/jlr.M700179-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Rodriguez V.; Mullen D. G.; Ganusova E.; Becker J. M.; Distefano M. D. Org. Lett. 2012, 14, 5648. 10.1021/ol302592v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T. L.; Székács A.; Uematsu T.; Kuwano E.; Parkinson A.; Hammock B. D. Pharm. Res. 1993, 10, 639. 10.1023/A:1018987111362. [DOI] [PubMed] [Google Scholar]
- Choy E.; Chiu V. K.; Silletti J.; Feoktistov M.; Morimoto T.; Michaelson D.; Ivanov I. E.; Philips M. R. Cell 1999, 98, 69. 10.1016/S0092-8674(00)80607-8. [DOI] [PubMed] [Google Scholar]
- Rocks O.; Peyker A.; Kahms M.; Verveer P. J.; Koerner C.; Lumbierres M.; Kuhlmann J.; Waldmann H.; Wittinghofer A.; Bastiaens P. I. H. Science 2005, 307, 1746. 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- Rocks O.; Gerauer M.; Vartak N.; Koch S.; Huang Z.-P.; Pechlivanis M.; Kuhlmann J.; Brunsveld L.; Chandra A.; Ellinger B.; Waldmann H.; Bastiaens P. I. H. Cell 2010, 141, 458. 10.1016/j.cell.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules; Goeldner M., Givens R. S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, FRG, 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.