

Abstract
Hydrogenase enzymes efficiently process H2 and protons at organometallic FeFe, NiFe, or Fe active sites. Synthetic modeling of the many H2ase states has provided insight into H2ase structure and mechanism, as well as afforded catalysts for the H2 energy vector. Particularly important are hydride-bearing states, with synthetic hydride analogues now known for each hydrogenase class. These hydrides are typically prepared by protonation of low-valent cores. Examples of FeFe and NiFe hydrides derived from H2 have also been prepared. Such chemistry is more developed than mimicry of the redox-inactive monoFe enzyme, although functional models of the latter are now emerging. Advances in physical and theoretical characterization of H2ase enzymes and synthetic models have proven key to the study of hydrides in particular, and will guide modeling efforts toward more robust and active species optimized for practical applications.
Graphical abstract
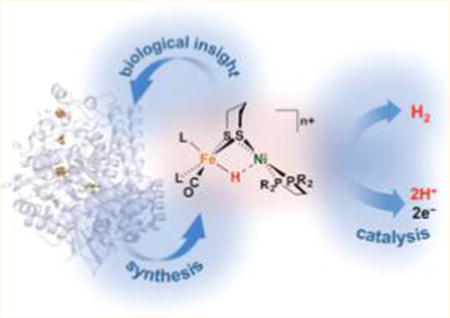
1. INTRODUCTION
Certain prokaryotes live off energy liberated from the metabolism of simple oxidants and dihydrogen. While this was known even in 1892,1 it would be another 40 years before bacterial dehydrogenase enzymes were implicated in such processes. In what must have been a startling observation, Bacterium aliphaticum liquefaciens, grown under H2, O2, and CO2, was found to reduce methylene blue (E = 11 mV vs NHE, the normal hydrogen electrode) under a stream of H2.2 This early work was followed by a landmark paper by Stephenson and Stickland, who described bacterial cultures from river mud that reduced NO3−, O2, and fumarate.3,4 They wrote that “hydrogen is in some way activated, and this activation can be conveniently expressed H2 ⇌ 2H without implying anything about the nature of the reaction”, the catalyst for which was termed “hydrogenase”. These H2-processing metalloenzymes are the basis for many anaerobic bacteria, protozoa, fungi, and algae5 collectively producing and consuming an estimated 0.3 Gt of H2 each year.6
Three major classes of hydrogenase (H2ase) have been identified, members of each featuring either an FeFe, a NiFe, or an Fe core.7 The [FeFe]- and [NiFe]-H2ases have been shown to mediate the interconversion of dihydrogen (H2) with protons (H+) and electrons (e−) at high rates8 and at potentials very close to those bounded by thermodynamics (eq 1).
| (1) |
In contrast to these two redox enzymes, the [Fe]-H2ases (also known as H2-forming methylenetetrahydromethanopterin dehydrogenases, Hmd) cleave H2 to then deliver H− to the organic substrate methenyltetrahydromethanopterin, with H+ passing to the bulk solvent (eq 2).9
| (2) |
For each reaction, as was hypothesized in 1931, the catalysts are often bidirectional3 and deactivated by CO. Unbeknownst to Stephenson and Stickland, the H2ases each catalyze the isotope exchange between H2O and D2 (eq 3).10, 11
| (3) |
A further hallmark of H2ase behavior is their interconversion of ortho- and para-H2 (eq 4),12 the two nuclear spin isomers whose differing thermal properties have implications for H2 storage.13
| (4) |
Interest in renewable, carbon-neutral energy technologies has motivated considerable biological research into the expression (including biosynthesis),14 structure, and function of H2ases. Each of the three enzyme cores and catalytic cycles is described in individual sections below (sections 3, 4, and 5). Before considering details, we must emphasize that the H2ase families share several mechanistic themes—principal among them H2 heterolysis—that are in no small part borne of the structural similarities revealed by even a cursory glance at their active sites (Figure 1).
Figure 1.
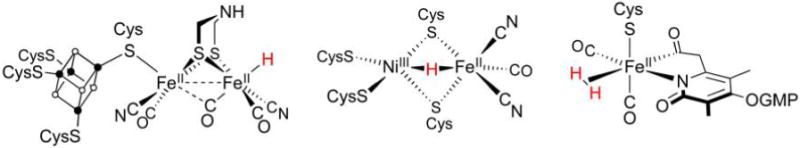
Schematic representations of the active sites in [FeFe]-H2ase (left), [NiFe]-H2ase (center), and [Fe]-H2ase (right). The presence of H− and H2 ligands in [FeFe]-H2ase and [Fe]-H2ase, respectively, has yet to be confirmed.
Central to the H2ases is Fe(II),15 whose ligation to thiolate and CO sees it adopt low-spin configurations most suited to the binding of H− and H2. Despite the three enzyme classes being phylogenically unrelated,16 “convergent evolution” causes expression of these common features that are apparently expedient for H2 processing.14 Inorganic chemists are motivated to prepare structural, spectroscopic, and/or functional mimics to elucidate the mechanisms of the very enzymes that inspired their synthesis.17 Given that H2 processing is championed by the Pt group metals, more cost-effective alternatives would involve the use of purely organic catalysts18 or those based on earth-abundant metals, like the H2ases.19–27 Better yet, their synthetic models may be more robust and mass-producible, with smaller spatial footprints that allow densely decorated, highly active electrodes to be prepared and incorporated into devices such as fuel cells.28–30
Having identified the importance of Fe–CO–thiolato ensembles in H2-processing enzymes, one can also reasonably expect complexes of H− and H2 to be somehow involved in Nature’s plans. These substrate-bearing intermediates, and the synthetic models proposed to mimic them most closely, are the focus of this review. Particular emphasis is placed on work in the past decade, which has seen great progress not only in biochemical and chemical synthesis, but also in the development of experimental and computational methods for the characterization of metal dihydrogen and hydride complexes. Before turning to these themes, some background into the chemistry relevant to these natural and synthetic catalysts is presented.
2. FORMATION AND CHARACTERIZATION OF METAL HYDRIDES
2.1. Formation of Metal Hydrides from H+ or H2
Nature’s handling of H+ and H2 with Fe and Ni follows patterns familiar in synthetic chemistry and catalysis.31 For example, electron-rich, low-valent metal sites are susceptible to protonation (oxidative addition of H+, Figure 2). In the case of a single metal center, the resulting hydride may vary greatly in its polarity and acid–base properties. When two metals are involved, consideration must also be paid to regiochemistry: will the product feature a terminal hydrido (t-H−) or a bridging hydrido (μ-H−) ligand?
Figure 2.
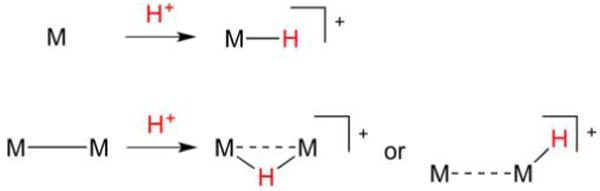
Formation of mono- and dinuclear metal hydrides from low valent metals and H+. In the latter case, both bridging and terminal hydrido products are possible.
When low- or mid-valent metal centers are exposed to H2 instead of H+, σ-complexes of the type M(η2-H2) may form, in which intact H2 binds side-on to M (Figure 3).32 This bonding motif has been described by analogy with the Dewar–Chatt–Duncanson model for metal complexes of olefins.33 Since η2-H2 acts as a σ-base and π-acid, the bond between H atoms (bond dissocation energy (BDE) = 436 kJ mol−1 for free H2)34 is weakened upon coordination, and in the case of electron-rich metals, can be entirely broken to afford M(H)2 dihydrides through a formal oxidative addition (homolysis).35
Figure 3.

Interactions of low-valent metal sites with H2. The η2-H2 ligand can cleave through oxidative addition to afford dihydride complexes.
The homolytic route for hydride formation, while common for group 10 metals and previously proposed based on computations of the [NiFe]-H2ases,36 is no longer thought to be relevant to the H2ases. Indeed, the kinetic barriers associated with homolytic H2ase pathways are prohibitive, and the enzymes instead operate through heterolysis (Figure 4). Heterolysis typically involves higher-valent metals, suitably unsaturated examples of which can ligate H2. The resulting M(η2-H2) complexes, when in the presence of a Brønsted base, may experience H2 fission, but not in the homolytic sense. Rather, the base deprotonates acidic η2-H2 to afford a metal hydride and a conjugate acid. Such a tug-of-war over the H2 substrate, which does not result in any change in metal oxidation state,37 is typical of that played out between frustrated Lewis acid and base pairs (FLPs).38,39 Cleavage is enabled by the remarkable influence electrophilic metal centers have on H2, whose acidity when bound is highly variable, as is evident from the contrasting behaviors of trans-[Fe(dppe)2(η2-H2)H]+ (pKa(THF) = 12; THF = tetrahydrofuran; dppe =1,2-bis(diphenylphosphino)ethane)40,41 and trans-[Fe(dppe)2(η2-H2)CO]2+ (pKa(THF) ≈ pKa (CH2Cl2) = −5).42,43 Such pKa values depart greatly from that of the free gas (pKa(THF) ≈ 50,44 pKa(MeCN) ≈ 50),40 underscoring Nature’s need for metals to activate this otherwise very strong bond.
Figure 4.
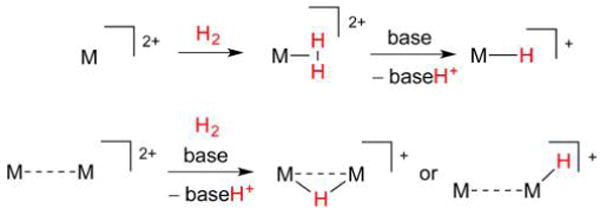
Interactions of high-valent metal sites with H2. The η2-H2 ligand in high-valent complexes can be acidic and is cleaved through deprotonation (heterolysis).
Pentacoordinate, low-spin 16e− centers represent archetypal metal fragments for H2 binding, and the H2 adducts of three such motifs are presented in Figure 5. The Fe(II) electrophiles [Fe(dppe)2CN]+ ([1]+ and [Fe(dppe)2CO]2+ ([2]2+) both form stable η2-H2 complexes, despite the contrasting donor/acceptor properties of CN− and CO. This is further astonishing in that it is the ligand trans to the H2 binding site that has the most influence on whether or not a η2-H2 complex can persist. It turns out that η2-H2 complexes are stabilized by CN− and CO ligands, although for different reasons in each case. According to Pauling’s principle of electroneutrality, the strong σ-basicity of CN− reduces the electron density Fe requires from the H–H σ-bond, such that the latter remains largely intact.45 It may be somewhat counterintuitive that a strong donor such as CN− does not, in this case, promote H2 oxidative addition. But since σ-bonding between Fe and H2 is weak, the Fe(dπ)–H2(σ*) π-backdonation is insufficient to induce splitting. When instead a strongly π-acidic CO ligand35 is ligated to Fe, the system once more attracts the H–H σ-bond. Yet since CO competes for backdonation, the η2-H2 moiety persists as Fe(dπ)–H2(σ*) interactions are weak.33
Figure 5.
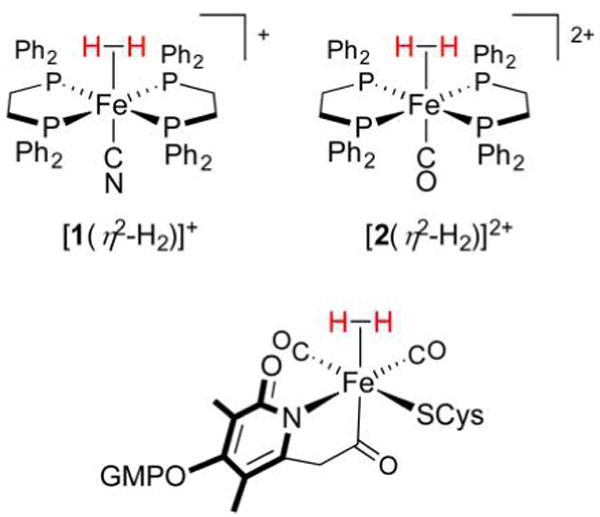
Fe(η2-H2) complexes in the laboratory (top left and right) and in Nature (bottom). Note the presence of five strong-field ligands that serve to support the low-spin Fe(II) electrophile.
A continuum exists between labile M(η2-H2) and bona fide M(H)2 complexes, between which are metal complexes where H2 is activated to the point that heterolysis, but not homolysis, can occur. Residing in the middle of the first row of transition metals, Fe seems to be in such a “sweet spot”. With the judiciously chosen coligands CO and CN−, Fe(η2-H2) species not only form readily, but, as demonstrated by the three H2ase families, often do so in preference to binding ubiquitous substrates such as H2O or N2. Once formed, η2-H2 complexes may undergo heterolysis to afford hydrido species, general aspects of which are discussed in section 2.2.
2.2. Properties of Hydride Complexes
The strong donor properties of H− are very familiar to inorganic chemists. In general, the donicity of a given ligand L can be quantified by several parameters, including the so-called PL value.46 Defined in terms of ligand effects on the Cr+/0 couple (PL = E1/2[Cr(I/0)(CO)5L] – E1/2[Cr(I/0)(CO)6]), PL ≡ 0 V for CO, with NO+ (1.4 V) and OH− (−1.55 V) having the expected opposite effects on redox. The value for H− (−1.22 V) is less negative than the latter extreme, but more negative than the value for CN− (−1.00 V), consistent with the strong donicity and anionic nature of H−. Closely related to the PL value is the Lever parameter (EL), which describes redox effects of ligands on Ru(III/II) couples.47 The EL parameter estimated for H− is −0.5 V, in the −0.63 to 0 V range characteristic of strongly donating ligands and π-bases including OH− and RS−. While the two scales are certainly influenced by ligand charge, they do provide useful measures of donicity, although they do not account for any steric interactions and synergistic effects between ligands.
Other ligands can be quantitatively compared to H−, but how can different complexes of H− be compared? Nominally an anionic ligand, hydride can exist in anything from “hydridic” M–Hδ− moieties to “protic” M–Hδ+ groups. A key parameter in describing hydrides is pKa, which, for an arbitrary species MH+ (eq 5), is found by treating it with a comparable amount of a suitable base (denoted B, eq 6) with a known pKa (BH+) in the solvent being used. If the Brønsted acidities of MH+ and BH+ differ greatly, then only an upper or lower bound for pKa(MH+) is measurable in practice. If the acidities are comparable, then Keq (eq 7) and pKa(MH+) may be easily determined.
| (5) |
| (6) |
| (7) |
Matters are more complicated when a base forms an H-bonded adduct with its conjugate acid (eq 8), a process referred to as homoconjugation or self-association.48
| (8) |
For certain weak acid/base pairs in organic solvents (e.g., RCO2−/RCO2H in MeCN), such effects must be taken into account when the concentration of added base exceeds 1/Kf.49 Homoconjugation constants Kf for several acids are known,50 and pKa(MH+) can still be computed in a straightforward manner (see the Appendix).48 Homoconjugation can be largely avoided by employing aniliniums/anilines, which are expected to exhibit low values of Kf.51 Values for pKa(MH+) span a wide range,44 and rough predictions based on metal and ligand parametrization are possible.41 The values are typically lower for electron-poor metal complexes and higher for their electron-rich counterparts, with a M–Hδ− description perhaps being more relevant in the latter case (eq 9).
| (9) |
The reaction in eq 9 is clearly related to that in eq 5, with the metal hydride now releasing H− rather than H+. The Gibbs free energy change associated with eq 9, known as the hydricity (ΔG°(H−)),52 is readily computed if MH+ can transfer H− to acceptors of known hydricity, such a competitive binding method being analogous to determining pKa by adding a base. Alternatively, if pKa(MH+) and the half-wave potentials (E1/2) of the 1e− couples [M]+/0 and [M]2+/+ (or the 2e− couple [M]2+/0) are known, hydricity (expressed in J mol−1) can also be calculated according to eq 10 or 11, derivations of which are in the Appendix.52,53
| (10) |
| (11) |
where F = 96 485 C mol−1, R = 8.314 J K−1 mol−1 and T is reported in kelvin. It is noted that very few H2ase models have two consecutive reversible 1e− couples (or, alternatively, a reversible 2e− couple) necessary for this calculation.
The hydricity of a compound is a measure of its propensity to donate H−, with the lowest values corresponding to the most hydridic species. As with the pKa’s of metal complexes, hydricities are also strongly influenced by solvation,54 with a narrower range of values expected for more polar solvents. For example, H2, {Ni[(1,2-bis(dihydroxylmethylphosphino)ethane]2H}+, and HCO2− have vastly different hydricities in MeCN (76.6, 57.4, and 44.0 kcal mol−1, respectively), the solvent most commonly used for such determinations. The values measured in the stronger dielectric H2O are much closer (34.2, 30.0, and 24.1 kcal mol−1).54 Hydricity is intuitively also related to the electron density at the metal in question, with ΔG°(H−) for the relatively electron-rich and -poor complexes [Ni(dmpe)2H]+ (dmpe =1,2-bis(dimethylphosphino)ethane) and [Ni(dppe)2H]+ determined as 48.9 and 62.7 kcal mol−1, respectively.53 Lastly, hydricity is also sensitive to coordination geometry, an aspect that has been explored with [M(diphosphine)2H]+ complexes, and one that is not surprising given that bite angles of chelating ligands can favor certain coordination numbers.55,56 In particular, ligands that span large angles will stabilize a hydride-free complex, leading to lower hydricity values. Overall, the many factors contributing to hydricity mean that it is certainly possible for a species to have a high pKa and modest hydricity, as the influence of redox may swing the result either way.
The interrelation between hydride redox, pKa, and hydricity is summarized in Figure 6. Simple thermodynamic cycles enable determination of free energies, such as hydricity ΔG°(H−, II), from experimentally determined pKa(II), E°(II/I), and E°(I/0) (or E°(II/0)) values as in eq 10. In this context, pKa(n), ΔG°(H•,n), and ΔG°(H−,n) are respectively associated with equilibria for H+, H•, and H− donation from [MnH](n−1)+.
Figure 6.
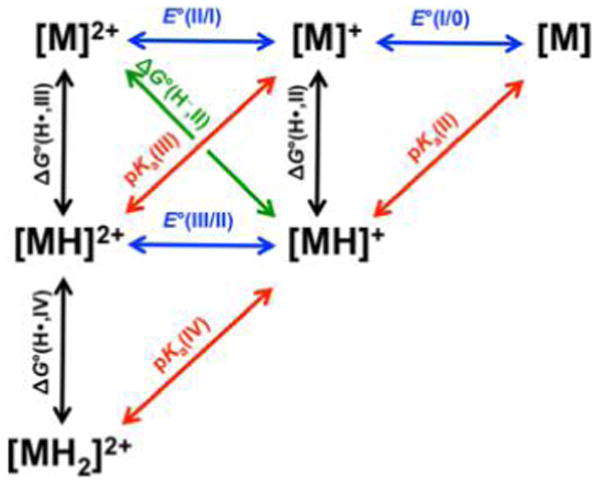
Thermodynamic parameters for redox and acid–base processes for a metal complex M. Colored arrows denote transfer of an electron (blue), proton (red), H• (black), and H− (green). Adapted from ref 57. Copyright 2012 American Chemical Society.
Until now, metal hydrides have been viewed here as potential sources of H+ or H−. Yet another important scenario is homolysis of an M–H bond,58,59 which in practice does not involve free H• but rather its delivery to a substrate.60 The facility of the homolysis reaction can be calculated analogously to hydricity: if pKa(II) and E°(I/0) (written in eq 13 as pKa and E1/2([M]+/0), respectively) are known, one can compute the H• donor strength (ΔG°(H•), eq 12, 13). Once more, details of the cycles used to derive eqs 12 and 13 are presented in the Appendix.
| (12) |
| (13) |
Our discussion now returns to considering pKa’s of metal hydrides and the significant and intuitive perturbations redox has on these.61,62 Indeed, the oxidation of metal hydrides causes large increases in acidity, as the reductive elimination of H+ is certainly more favorable when the metal site is more electron poor. One family of species for which the thermodynamics are well studied are the bis(diphosphine) complexes of Ni.57 Consider tetrahedral [Ni(dppe)2], which readily protonates to give the respective 5-coordinate divalent hydride [Ni(dppe)2H]+ (eq 14, pKa(II) = 16.7). The same cannot be said for oxidized [Ni(dppe)2]+, whose protonation product [Ni(dppe)2H]2+ is a strong acid in MeCN (eq 15, pKa(III) = −8.6).57
| (14) |
| (15) |
The marked drop in pKa upon oxidation of [Ni(dppe)2H]+ is a typical consequence of redox on a mononuclear metal hydride, with this acidity increase often leading to hydride-related redox events being irreversible. One reason the [FeFe]- and [NiFe]-H2ases employ bimetallic active sites may be to disperse the strong effects of redox over two metal sites.63 This dampens the impact felt by the H− ligand, enhances the reversibility of redox couples, and perhaps lowers activation barriers to catalysis. The [Fe]-H2ase active site, which is a Lewis acid not required to perform redox, features only a single Fe center, with Nature apparently dispensing with the need for a second metal. A detailed understanding of redox and acid–base properties of H2ases and functional models requires a number of experimental techniques. Some key methods are discussed in section 2.3, particularly in the context of characterizing hydride-containing species.
2.3. Physical Characterization of Hydrogenase Enzymes and Models
2.3.1. Electrochemistry
In addition to the two hydrogenic substrates H+ and H2, the [FeFe]- and [NiFe]-H2ases also handle electrons to drive eq 1 in a forward or reverse sense. According to the Nernst equation, at pH 7, p(H2) = 1 bar, and T = 298 K, the 2H+/H2 couple is at −414 mV vs NHE. Yet hydrogenases only have access to H2 partial pressures of 1–10 Pa (10−5–10−4 bar), such that they must operate between −266 and −296 mV, the narrow range mandated by thermodynamics.6 In vivo, electrons travel via Fe–S clusters positioned close enough together (<14 Å is optimal)64 to allow tunneling to and from an electron transport protein as the source/sink. In place of the transport protein, in vitro studies of H2ases instead often employ suitable small molecule electron donors (e.g., methylviologen radical, E1/2 = −446 mV vs NHE) and acceptors (e.g., benzylviologen, E1/2 = −358 mV), whose potentials lie on either side of the 2H+/H2 couple at p(H2) = 1 bar.65 Such redox reagents are also used when studying model compounds, which, with notable exceptions,66,67 almost always lack redox-active Fe–S clusters or other electron shuttling moieties. Organic solvents may be necessary, as many models feature lipophilic ligands (e.g., phosphines) to stabilize their low-valent states. These studies have benefited from a growing body of work on electrochemistry in polar aprotic solvents,68 and although pKa scales are still not uniform, certain reference couples have become standard.69 While redox potentials of the enzymes in aqueous solutions are typically reported relative to NHE, potentials of model complexes are most often reported against Fc+/0 (Fc = FeCp2; Cp− = cyclopentadienide), a couple which lies at 0.4, 0.63, and 0.69 V relative to NHE in H2O,68 MeCN,69 and CH2Cl2, respectively.68 In terms of electrolyte salts, model studies make extensive use of noncoordinating anions,70 with the borates BF4−, B(C6F5)4−, and BArF4−(3,5-(CF3)2C6H3)4B−),71 as well as PF6−, being typical counteranions for both voltammetry72 and synthesis. Such anions possess diffuse charge and allow for coordinatively unsaturated cations to be studied “in isolation” and to be given every possible chance to interact with weak ligands like H2.
An important alternative to sacrificial electron donors and acceptors is the use of an electrode as the electron source/sink, with voltammetric methods73 (as well as associated spectroelectrochemical techniques)74 proving extremely useful for characterizing analytes when immobilized or in solution,75 and for determining catalytic rates76,77 and overpotentials (vide infra).49,78,79 Cyclic voltammetry (CV) has become popular in the study of synthetic hydrides, and many hydride-bearing mimics of [FeFe]- and [NiFe]-H2ase exhibit electrochemistry that has informed their HER mechanisms, including uncovering the existence of paramagnetic (S = 1/2) hydrides. In contrast to conventional metal hydride chemistry, focused for example on hydrogenation reactions, H2ases characteristically operate by one-electron (1e−) pathways. As described for [Ni-(dppe)2H]2+/+, redox imparts large changes in the acidity of hydride species, leading to the reductions and oxidations often being rather irreversible. This reflects the high reactivity of certain hydrides and may hint at desirable catalytic properties.
Redox waves of hydrides are often perturbed when H2 or H+ substrate is present. Consider the latter case, for which CV is a typical assay for the hydrogen evolution reaction (HER, the reverse of eq 1) catalytic activity. Assume, for instance, a metal hydride gives rise to a 1e− reduction mechanistically relevant to the HER. The potential of this couple in the absence of H+, denoted here Eredox, will certainly depend on the relative electron density at the metal(s) and thus the donor set. When an acid substrate HA is titrated into the hydride/electrolyte mixture, the growth of a wave implicates a situation in which the hydride complex is continually regenerated, and thus participates in a catalytic reaction involving protons. The resultant “catalytic wave” is often described by its potentials at full height (Ecat) or half-height (Ecat/2),80 values that may differ from Eredox. The current associated with the catalytic wave, ic, can be several times ip, the current at Eredox in the absence of HA. The value ic will be clear for an ideal (sigmoidal) wave, such that the potentials Ecat and Ecat/2 are precisely determined.79 More complicated examples can introduce variance in ic and, correspondingly, the potentials Ecat and Ecat/2. The latter value is least affected by nonidealities, and is thus the more reliable. If the quotient ic/ip is proportional to [HA], then the process is second order in [H+], consistent with the HER. At a certain point, one reaches the so-called “acid-independent regime”, in which the addition of HA no longer leads to an increase in ic, and pseudo-first-order conditions with respect to the catalyst are reached. The maximum obtainable ic/ip is proportional to the square root of the turnover frequency (TOF) for H2 evolution. The latter is an important metric, yet some have questioned its general applicability and instead advocate use of other values including catalytic efficiency (CE = ic[catalyst]/ip[HA]).81
Catalytic currents and/or TOF values are key descriptors of reactivity, yet these kinetic parameters should not be quoted without their associated potentials, on which they are dependent. The potentials Ecat and Ecat/2 ideally will be more positive than the HA reduction in the absence of hydride catalyst, a wave associated with direct reaction at the electrode (e.g., glassy carbon).82 Such a process may compete with catalyst-mediated HER at strongly reducing potentials, and one must be aware that currents arising from direct reductions have often been erroneously ascribed to catalysis from model complexes. In any case, Ecat/2 will be more negative than E(H2/HA), the thermodynamic potential for the H2/HA couple, although Ecat/2 for an ideal catalyst will be very close to this upper bound. When comparing catalysts, the values Ecat/2, Ecat, and Eredox are to be used in preference to the oft-quoted but rather subjective term “onset potential” (Eonset), the voltage near the base of the catalytic wave at the “onset” of catalysis.49,73
An important electrochemical figure of merit is overpotential (η = E(H2/HA) – E), defined as the additional voltage, beyond that required thermodynamically, used to drive a reaction at a specific rate.83 The reaction is driven at a potential E, which may be taken as Ecat/2, Ecat, or another potential. Whatever potential (and thus overpotential) one selects, it is essential that this thermodynamic value is reported if and only if a kinetic value derived from the current is reported.80,84 Given that η is proportional to the excess electrical energy required, its minimization, while maintaining an acceptable rate of reaction, is highly desirable. Although electron-rich metal catalysts give rise to more negative Ecat/2 values than their electron-poor counterparts, the former can still have reasonably small overpotentials if they can catalyze H2 evolution from weaker acids, which have more negative thermodynamic potentials E(H2/HA). In general, one aims to find the weakest acid that will quantitatively protonate the reduced form of the catalyst, whence η and TOF will be optimized. In addition to these catalytic data, stoichiometric conversions can allow one to develop a picture of the HER mechanism(s) at play. Two photons and two electrons must be transferred during the catalytic cycle; these reactions are often denoted as C (chemical) and E (electrochemical) steps. As will become clear, the sequence of these steps (e.g., ECEC vs CEEC) varies from catalyst to catalyst.
Cyclic voltammograms associated with catalytic reactions may be complicated by certain processes including acid homoconjugation, whence H2 evolves not only following reduction of HA, but also from other sources such as the homoconjugated species H+[AHA]−.49 Digital simulation of voltammograms and supplementary data from chronoamperometry are often beneficial in elucidating mechanisms.85 Overall, information from CV contributes in no small part to a detailed picture of processes involved in catalytic H2 evolution or oxidation. Some case studies involving CV in catalysis are presented in the [FeFe]- and [NiFe]-H2ase sections 3 and 4. Readers unfamiliar with electrocatalysis and its figures of merit are referred to some useful introductions.73,80,86,87
2.3.2. Crystallography
Possessing only a single electron, H atoms are often challenging to locate by X-ray crystallography, as any bond they form automatically means a significant amount of electron density is in this bond rather than about the H nucleus. Furthermore, metal hydrides can be difficult to characterize in that Fourier truncation ripples surrounding heavy atoms can overwhelm smaller electron density peaks. These two effects result in H atoms being difficult to locate and their M–H bond lengths typically being underestimated.88 Despite these problems, laboratory X-ray analysis of high-quality small molecule metal hydride crystals routinely allows identification and refinement of H− ligand positions. In contrast, synchrotron X-ray diffraction of protein crystals can only afford H atom positions under very favorable circumstances. The sample must diffract to <1 Å (high resolution by protein standards), and only then can careful modeling and refinement reveal hydrides and other H nuclei.89,90 The resulting crystallographic models are best compared to those from neutron diffraction,91,92 a technique that exploits the larger scattering cross section of D vs H, and is suitable to the study of macromolecules.93 For example, one might characterize a H2ase or its hydride-bearing model by X-ray diffraction and compare the metrics to neutron data of the relevant small molecule deuteride (crystals of suitable size for neutron diffraction are realistically more accessible for small molecules than for H2ases). Such deuterides are typically simple to prepare owing to the H/D exchange properties of many metal hydrides.
2.3.3. Magnetic Resonance Spectroscopy
When considering metal hydrides in particular, certain spectroscopic methods are more informative than others. Among the most useful is nuclear magnetic resonance (NMR) spectroscopy, with spin–orbit coupling causing even diamagnetic Fe–1H moieties to resonate over a wide range, although typically in the region upfield of Si(CH3)4 in which other ligand signals are rarely observed. The chemical shift, as will become clear, is not only dependent on spin–orbit interactions94 and the electron-rich or -poor character of the metal(s) to which H− is bound, but also on the terminal or bridging nature of its coordination. NMR has not been successfully applied to the [FeFe]- and [NiFe]-H2ase enzymes themselves, even in cases where H− is ligated at an S = 0 core. In cases where H− is bound to open-shell metal centers (S > 0), electron paramagnetic resonance (EPR) spectroscopy, as well as its pulsed variants electron nuclear double resonance (ENDOR) and electron spin echo envelope modulation (ESEEM),95 have proven extremely powerful. Indeed, the first direct evidence for a metal hydride in Nature was obtained by ENDOR spectroscopic measurement of a [NiFe]-H2ase (vide infra).96 Overall, the latter two techniques are particularly useful for resolution of anisotropic coupling between unpaired spins and 1H nuclei diagnostic of H− ligation.
2.3.4. Vibrational Spectroscopy
Vibrational spectroscopy provided early breakthroughs in H2ase structure elucidation, particularly the identification of the CO and CN− cofactors present at the active sites. These cofactors represent key spectroscopic handles because νCO and νCN bands appear in spectral regions devoid of protein bands. The number and intensities of νCO and νCN bands not only report on the number of such moieties but also the C–M–C angles between them. In turn, the vibrational frequencies report on metal electron density and the extent to which π-backbonding occurs. But what of M–H and M–H–M chromophores—how easily detected and assigned are their vibrations? Although their intensities are often rather low, νMH modes can be identified using infrared (IR) and Raman spectroscopies,97 in the latter case taking due consideration of the photolability of many CO- and H−-containing complexes. Terminal hydride νMH bands often appear in IR spectral regions populated by νCO stretches (particularly those of bridging CO ligands).98 Bridging hydrides give rise to weaker, lower energy vibrations best observed with resonance Raman, although vibrational mixing with other bands can convolute matters. The challenges discussed here can partly be overcome by studying M–D and M–13CO isotopologues, in which case the bands are shifted to lower energy (νMH ≈ 21/2νMD and ν12CO ≈ (91/87)1/2ν13CO, assuming uncoupled harmonic vibrations and mM ≫ mD).97 Nevertheless, unambiguous assignment of hydride bands is not always possible, and such data are unknown for many hydrides.
The difficulties in identifying metal hydride bands are well met by using a technique known as either nuclear resonance vibrational spectroscopy (NRVS) or nuclear inelastic scattering (NIS),99–101 which now represents a valuable complement to the two more established vibrational methods. While Raman provides vibrational information following electronic excitation, NRVS does the same with nuclear excitation, requiring inelastic scattering of highly monochromatic (meV-resolved) synchrotron γ- or X-rays102 by Mössbauer-active nuclides (e.g., 57Fe, 61Ni). The resulting spectra feature bands corresponding to only (and all) vibrations in which the metal nuclide moves. The unparalleled selectivity for probing the inner coordination sphere (even when surrounded by many kilodaltons of proteic mass) has already proven invaluable, as examples in this review will highlight. The intensities of NRVS bands are related to the displacement of the Mössbauer-active nuclide along the beam path. Many NRVS studies focus on low energy vibrations, such as bending and wagging modes, as they often involve large amplitude motion of metals and thus have relatively large NRVS intensities. The disadvantage in using these modes is that their coupled nature necessitates detailed density functional theory (DFT, vide infra) analyses for confident assignments. On the other hand, high energy vibrations, such as M–H/D stretches, see the metal move only very slightly, as the mass of M is large relative to that of H or D. Pure νMH modes are thus difficult—although not impossible—to observe.103
The inherently high frequency and low intensity of M–H/D modes leads to such NRVS measurements being far from routine, but this will inevitably change given the ever-increasing flux and resolution of synchrotron light sources. At the time of this review, NRVS-equipped facilities include beamlines at the Advanced Photon Source (Chicago, IL, USA), European Synchrotron Radiation Facility (Grenoble, France), and SPring-8 (Hyogo, Japan). Alternatively, seeded free-electron lasers can also provide the necessary monochromatic radiation, although such facilities are currently of limited accessibility and their applications to NRVS are yet to be well developed.104
When a complex exists for only a short time, its characterization calls for stopped-flow techniques and transient (time-resolved) spectroscopies. For example, the pulsed nature of free-electron lasers, while resulting in lower average fluxes, would enable NRVS measurements on the femtosecond scale. The most widespread transient methods use more traditional laser sources to obtain vibrational105 and optical spectra.106 These have been applied to H+ transfer dynamics in H2ases107 as well as the detection of synthetic mixed-valent hydride species,108 and are well complemented by computational investigations into such fleeting species. Described in section 2.4 are some of the theoretical approaches used to understand H2ases and their models, as well as the information that is afforded by such work.
2.4. Theoretical Characterization of Hydrogenase Enzymes and Models
2.4.1. Approaches
The physical characterization of synthetic H2ase models is typically much simpler than analogous studies on the proteins themselves. The same can be said for computational investigations, and thus while many synthetic species are amenable to in silico characterization, the enzyme structures are typically truncated to contain only the active site and perhaps the second coordination sphere for fully quantum mechanical calculations. Alternatively, mixed quantum mechanical/molecular mechanical (QM/MM) methods, in which only the active site is treated quantum mechanically while the remainder of the enzyme and solvent are treated with a MM force field,109–113 have been used to study the larger H2ase enzyme systems.114–116 The computationally expensive nature of metallic cores, as well as the intractability associated with high levels of theory such as coupled cluster methods, have led to DFT117,118 being widely used in the study of H2ases and synthetic models.119–121 As used with other molecular transition metal electrocatalysts,122–124 popular functionals include B3LYP,125,126 BP86,127,128 B3P86,126,127 PBE0,129–131 TPSSh,132 M06-L,133 and ωB97XD.134 These functionals are typically used with double- or triple-ζ basis sets, as well as polarization for specific atoms including moving H+ or H− groups,135 and diffuse basis functions for anionic systems.136 Additionally, effective core potentials and their corresponding basis sets, such as those of Hay and Wadt (LANL)137 or Stuttgart–Dresden (SDD),138 may be used for the transition metals to decrease computational cost.139 The application of DFT to such complexes has been reviewed in detail.140
2.4.2. Predicting Observables
Molecular geometries of H2ase models optimized using the above functionals have been shown to agree quite well with X-ray crystal structures. Geometries can be optimized either in the gas phase, subsequently including solvation free energies using a Born–Haber thermodynamic cycle, or in the solution phase directly.141 Solvation effects are typically incorporated with a polarizable continuum model, in which the solvent is represented as a homogeneous dielectric. These models, such as PCM,142–144 C-PCM,145 SMD,146 and COSMO,147 are advantageous for treating solute–solvent interactions without requiring conformational sampling, although they do so by inherently neglecting specific solute–solvent interactions such as hydrogen bonding.
With optimized structures in hand, DFT calculations are invaluable for the prediction of further observables such as redox potentials and pKa values. Accurate predictions of these parameters are necessary given their importance in the H+ reduction reaction. The accuracy of calculated reduction potentials and pKa values has been assessed for many molecular electrocatalysts and provides validation for the computational methods employed.57,141 Several methods can be implemented to account for the reference electrode in the calculations to allow comparison with experimental measurements. One option is to subtract an experimental or theoretical value for the absolute reduction potential of the reference electrode.148–152 An alternative is to subtract the value of the reduction potential of the reference electrode calculated using the same level of theory and basis set.153–156 Yet another option is to calculate the reduction potential of a related half-cell reaction, which has been experimentally measured under similar experimental conditions and with respect to the same reference electrode.52,57,141,157,158 Application of a thermodynamic cycle, also called an isodesmic reaction, results in the reference electrode potentials canceling, as has been demonstrated for the calculation of quantitatively accurate redox potentials for synthetic H2ase models.159–161 This approach may also be applied to calculate pKa values, although experimental pKa measurements are not performed as often. One particular advantage of this approach is the cancellation of systematic errors associated with the level of theory and specified functional and basis set. Furthermore, application of a thermodynamic cycle also avoids the calculation of the free energies of H+ and e−, as well as the potential of the reference electrode. The application of this thermodynamic cycle is equivalent to the calculation of reduction potentials and pKa’s relative to related systems with experimentally measured values. Such an approach appears to be the most reliable at the current time.
Along with redox and acid–base behavior, spectroscopic observables are also available through DFT calculations. In particular, the diagnostic IR signatures of the CN− and CO ligands are readily observed and can also be predicted by DFT calculations. However, since the harmonic approximation is often invoked to compute vibrational frequencies, calculated absolute frequencies are generally shifted systematically. For this reason, empirical scaling factors, dependent on the functional and basis set used, are often employed.162,163 While the absolute vibrational frequencies calculated with DFT are often not quantitatively accurate, shifts in frequencies due to protonations and/or reductions are generally reliable. Nevertheless, absolute vibrational frequencies calculated using the BP86 functional tend to agree remarkably well with the experimental data without the use of scaling factors, which has been attributed to a fortuitous cancellation of errors.164
Although EPR spectroscopy is widely used to characterize paramagnetic states of the enzyme and models,95 the accurate calculation of magnetic spectroscopic parameters is typically a challenging computational task. When calculated within the framework of DFT, the accuracy of parameters such as g-values, hyperfine couplings, zero-field splittings, and quadrupole couplings are very sensitive to the particular system being studied, especially the metal center(s). For example, while DFT calculated values are in good agreement with measurements for organic radicals and biradicals,165–167 the calculated values for transition metal complexes typically deviate from those observed. In particular, g-shifts are notably underestimated by many functionals for transition metal complexes.168,169 Hyperfine couplings for transition metal complexes present an additional challenge due to the significant contributions from spin–orbit coupling and spin polarization,170 with the latter commonly underestimated by DFT.171,172 However, predicted magnetic spectroscopic parameters are not always unreliable,173 with calculated Mössbauer isomer shifts for 57Fe being especially promising.174–176 It has also been suggested that hybrid functionals, which contain a portion of Hartree–Fock exchange, tend to provide better agreement with experiment,169,177 although at the risk of spin contamination.178 Nonetheless, the application of these approaches to H2ase models has seen successes over the years.179–185
The value of theoretical calculations is not limited to rationalizing experimental observations. The rapid nature of many H+ and e− transfers ensures that many species may evade conclusive identification even by transient spectroscopies. Indeed, calculations come to the fore when practical experimental solutions are not to be found. Computational studies are not only useful for the characterization of known H2ase models, but also have guided the design of new synthetic targets.186–189 Experimental and theoretical studies in the field are thus driven by one another to a deeper understanding of H2ases and their models. Our general discussion on characterization ends here, and we now present the state-of-the-art in the characterization and synthetic modeling of the [FeFe]-H2ases (section 3), [NiFe]-H2ases (section 4), and [Fe]-H2ases (section 5).
3. [FeFe]-H2ASES
3.1. Enzyme Structure and Function
Expressed in bacteria and lower eukaryotes,190 the [FeFe]-H2ases are arguably the fastest and most evolved of the H2ases. While the [FeFe]-H2ases vary in size (45–130 kDa), each features an active site ensemble known as the H-cluster. With six Fe centers in total, the H-cluster comprises a [4Fe–4S] metallocubane linked to an apical Cys-S− ligand in a [2Fe] moiety of formula {(Cys-S)(NC)(OC)Fe[(SCH2)2NH]Fe-(CO)2(CN)}2−/3− (Figure 7).
Figure 7.
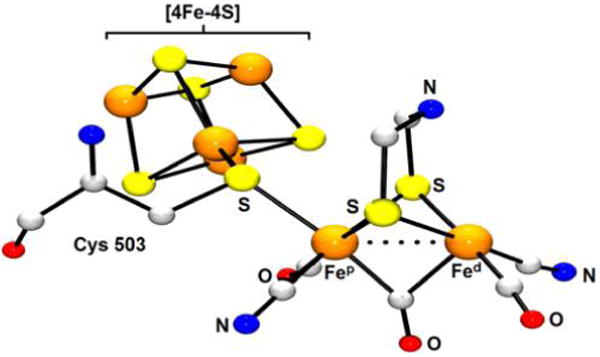
X-ray structure of the [FeFe]-H2ase active site from Clostridium pasteurianum (PDB code 3C8Y).191 H atoms are omitted for clarity.
Perhaps the more striking feature of the H-cluster structure is the [4Fe–4S] cluster, a common redox cofactor positioned in an uncommon location—covalently bound at the active site—that emphasizes the importance of e− transfer in catalysis. The Fe centers in the [2Fe] unit are labeled Fep (“proximal” to 4Fe–4S) and Fed (“distal” to 4Fe–4S), with the latter featuring a vacant site for substrate binding. The low-spin Fep and Fed centers are linked through S atoms of the azadithiolate (adt2− = −SCH2N-(H)CH2S−) cofactor. While the 4Fe–4S unit and three or four “auxiliary” Fe–S clusters define an e− transport chain,192 azadithiolate plays a key role in shuttling H+ to or from the active site. Poised over the apical site of Fed, the secondary amine influences interconversion of Fe, Fe–H, and Fe(η2-H2) species by virtue of its acid–base properties. Beyond this, the amine further participates in N⋯H–S-Cys hydrogen-bonding interactions, beginning a pathway for H+ transfer between the active site and the protein surface. The arrangement of the amine and Fed is constrained such that no bond can form between the two, with this Lewis base and acid constituting a FLP.39 However, this system is unlike most synthetic FLPs in that the [2Fe] site is also redox-active, and is thus well-suited to perform electrocatalysis.
Completing the H-cluster are five “organometallic ligands” (3 × CO, 2 × CN−), whose presence at the active site initially came as a surprise, although foreshadowed by the crystallographic analysis of [NiFe]-H2ases. These chromophores absorb strongly in an uncluttered IR region,191,193 serving as crucial reporters on the purity of protein samples, which is greatly decreased by any exposure of [FeFe]-H2ases to O2. The diatomic ligands are key spectroscopic handles, and while νCN modes are less sensitive to H-cluster redox (CN− is a weak π-acceptor), the νCO energies can vary over a 200 cm−1 range.7 Consequently, IR analyses of [FeFe]-H2ases are particularly informative, although interpretation can be complicated by the highly coupled nature of these modes.
After accounting for all the ligands, one can now consider the geometry of the [2Fe] unit. If the μ-CO ligand were trans to its current location, at a terminal Fed site, one might consider the [2Fe] unit as a pseudosymmetric union of two pyramidal Fe units, much like the vast majority of synthetic [L3Fe(SR)2FeL3] complexes (see section 3.2.1). But the presence of the CO bridge stabilizes an unsymmetrical “rotated” structure, wherein a coordination site on the Fed trans to this π-acid is free for substrate binding. The “rotated” structure is further rigidified by a strong FedCN⋯+H3N-Lys358 interaction involving a conserved residue.194 In addition to the structural role played by the CN− ligands, cyanides also enhance the basicity and lower the redox potentials of the [2Fe] site and, along with the CO ligands, enforce low-spin configurations ideal for H2 binding.35
On the other hand, have all the ligands been accounted for? Structural analyses suffer from a major complication: protein X-ray crystallography is typically unable to resolve the presence—much less the location—of hydrogenic substrates. Consequently, the protonation state of the amine is not clarified by crystallography. The presence of substrate at the Fed binding site is also an open question, and even if an Fed–H terminal hydride were a long-lived intermediate, the ligand would typically not be crystallographically locatable. Furthermore, Fe–H and Fe(η2-H2) moieties are very weak IR and Raman chromophores (see section 2.3.4), and are usually EPR-inactive, such that their identification based on hyperfine interactions is unlikely. Despite the lack of direct evidence for hydride ligation,195 Fed–H intermediates are assumed to exist at least transiently in the catalytic cycle.24,196 It is altogether conceivable that hydrides might not exist as stabile entities on a nearly flat potential energy landscape associated with an efficient catalytic cycle. While often slow,197 the protonation and deprotonation of metal centers might be accelerated by proton-coupled electron transfer (PCET), which would exploit the juxtaposition of [4Fe–4S] and adt2− cofactors to keep Fed hydride-free. Information on Fe–H species, albeit indirect, has been obtained by studying a mutated [FeFe]-H2ase from Chlamydomonas reinhardtii. Such work builds on the hypothesis that Fe–H intermediates are destabilized by the PCET machinery. Indeed, introduction of Ser in place of Cys in the H+ transfer pathway results in a significant drop in activity, and IR and EPR analyses of the reduced mutant indicated a new form with a H-cluster best described as [4Fe–4S]+/Fe(II)Fe(II). Such an assignment is consistent with an Fe–H species, and the presence of the hydride is further indicated by the shift in the bridging CO band upon changing the solvent from H2O to D2O.198
Taking in the active site as a whole, the H-cluster is conformationally rigid, and its overall structure persists throughout the catalytic cycle. Despite this, there is some flexibility within certain components. First, substrate turnover necessitates interaction at the apical site on Fed, a metal that must switch between octahedral and square-pyramidal geometries. Second, to function as a H+ relay, the secondary amine in the adt2− cofactor is required to undergo rapid inversion (this aspect is illustrated in Appendix A.1, Figure 58). Lastly, the CO bridging Fep and Fed serves as a “shock absorber”, with its linearity dependent on redox and the chemistry occurring at Fed. This reactive site is the location for H−/H2 binding, and its coordination to exogenous CO leads to deactivation of the enzyme. In contrast, H2O does not poison the active site, consistent with a low-spin π-donor description for Fed.
Figure 58.

Proton transport to and from the [2Fe] site in [FeFe]-H2ase (formal charges omitted). This involves H+ movement, denoted by red arrows, between Fed, the adt2− cofactor, and the protein (via Cys299), and likely requires pyramidal inversion of the amine.
Particularly important structure–function relationships have been elucidated by reconstitution of the semi-apoenzyme derived from algal H2ases. The semi-apoenzyme, which contains the Fe4S4 cofactor but not the [2Fe] subunit, is readily reconstituted to a fully active enzyme upon treatment with the synthetic diiron complex [NC(OC)2Fe(adt)Fe(CO)2CN]2−([3]2−, Figure 8). The reconstitution is accompanied by loss of CO, attachment of the [4Fe–4S] cluster, repositioning of CN− ligands to transoid, basal positions, and adoption of a “rotated” structure.199,200
Figure 8.
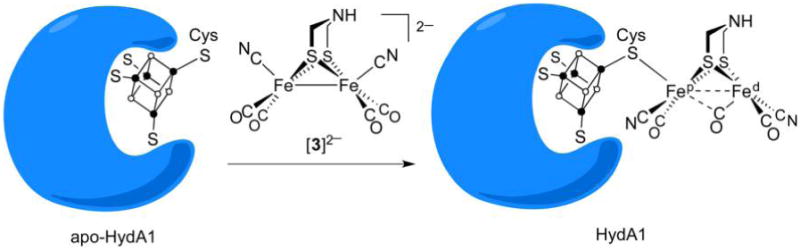
Reconstitution of apo-[FeFe]-H2ase from Chlamydomonas reinhardtii with [NC(CO)2Fe(adt)Fe(CO)2CN]2−.199,200
The artificial maturations have also been performed using isostructurual, although subtly different, diiron dithiolates including {Fe2[(SCH2)2–X](CN)x(CO)6−x}−/2−, where X = CH2, O, S, NMe and x = 1, 2.201,202 While these abiological analogues are readily accepted by the protein, catalytic activity assays indicated that the adt2− cofactor is crucial for H+ transfer, and the only semisynthetic [FeFe]-H2ase that exhibits significant activity is that obtained by reconstitution with [(OC)3Fe(adt)-Fe(CO)2CN]−. With the functional importance of each H-cluster component now clear, our discussion moves to the various enzyme states and their roles in the catalytic cycle.
The understanding of enzyme mechanisms rests on the identification of characterized states,203 which number three in the case of [FeFe]-H2ase. Several crystallographic and spectroscopic studies confirm that the structure of the H-cluster is virtually invariant in all redox forms. In considering the proposed catalytic cycle (Figure 9), focus is consequently placed on the oxidation and protonation states of the Fe centers and adt2− cofactor. Spectroscopic parameters for the three known [FeFe]-H2ase states are presented in Table 1, and the properties of each are now individually discussed.
Figure 9.
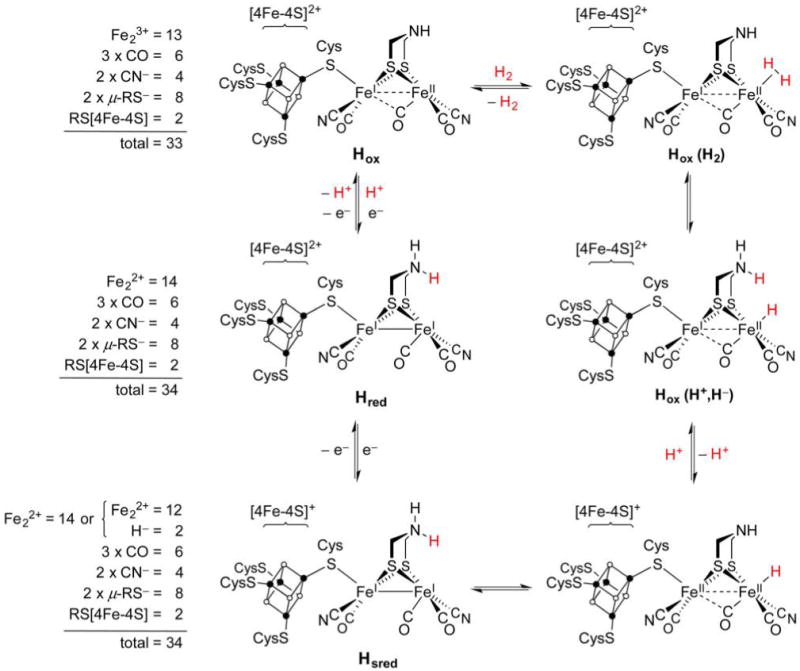
Catalytic cycle proposed for [FeFe]-H2ase (right)203 and electron counting for the [2Fe] unit in the three characterized states (left). The totals include Fe 3d electrons and bonding electron pairs from donor atoms. The substrate H atoms are in red for emphasis, although they cannot be distinguished from the amine H atoms.
Table 1.
Oxidation States and Spectroscopic Parameters for [FeFe]-H2ase HydA1 from Chlamydomonas reinhardtiia
| state | [Fe4S4]n+ | [2Fe] | νCO/cm−1 | νCN/cm−1 | g |
|---|---|---|---|---|---|
| Hox | [Fe4S4]2+ | Fep(II)Fed(I) or Fep(I)Fed(II) | 1964, 1940, 1800 | 2088, 2072 | 2.10, 2.04, 2.00 |
| Hred | [Fe4S4]2+ | Fep(I)Fed(I) | 1935, 1891, 1793 | 2088, 2072 | – |
| Hsred | [Fe4S4]+ | Fep(I)Fed(I) | 1954, 1919, 1882 | 2070, 2026 | 2.08, 1.94, 1.87 |
The most common state for biophysical studies is Hox, so denoted as it is “oxidized”, although not oxygenated (an important distinction for such O2-sensitive organometallic enzymes). Early Mössbauer studies on Hox indicated strong coupling between the proximal [4Fe–4S]2+ cofactor and the [2Fe] center, with the latter initially described as a low-spin (S = 1/2) Fe(II)Fe(III) tandem.204 Yet, the inadequacy of Mössbauer spectroscopy in distinguishing oxidation states of low-spin Fe led some astray, particularly when low oxidation states such as Fe(I) (“subferrous”) were not considered to be biologically relevant.
A suite of advanced EPR techniques has greatly informed the current picture of Hox. While its EPR signals at g = 2.10, 2.04, and 2.00 shift slightly depending on the organism, these invariably are consistent with an Fe(II)Fe(I) description for the [2Fe] core. Although biophysical reports indicate Fep to be monovalent, analyses of synthetic models consistently point to the reverse assignment, supported on general grounds by considering the favorability of a +I oxidation state for pentacoordinate sites such as Fed in Hox. In any case, the electronic coupling of the [4Fe–4S]2+ cofactor and [2Fe] core allows mixing of excited states and enhanced hyperfine coupling (isotropic A(57Fe) ≈11–12 MHz for each moiety). The value of paramagnetic centers as spectroscopic handles also comes to the fore in the identification of 14N hyperfine, which represented the first evidence for the identity of the adt2− cofactor.205
Poised to bind H2, the Hox state also strongly binds CO at the apical Fed site adjacent to the secondary amine.206 The binding of 13CO is stereospecific,207 which adds weight to our picture of the relatively rigid active site. When exogenous CO is taken up by Hox, its spin becomes more delocalized across the H-cluster, although EPR analysis indicates that the cubane remains in a [4Fe–4S]2+ state. An Fe(1.5)Fe(1.5) description for [2Fe] is thus logical, given that both Fep and Fed are now octahedrally coordinated with similar ligand sets. The deactivated, CO-bound state features strong coupling between the [4Fe–4S] and [2Fe] units, which is significant mechanistically in that binding of H2 (and especially of H−) might also enhance interaction between these two subsites and thus be involved in PCET.208
When [FeFe]-H2ase exists at potentials more negative than −395 mV, it is not Hox, but rather its product Hred, that predominates. The latter state is EPR-silent and is thought to feature a [4Fe–4S]2+ cluster (also EPR-silent) and an S = 0 [2Fe] site. It was formerly assumed that Hred features a H− ligand as part of an Fe(II)Fe(II), H− core. However, X-ray spectroscopy does not support the presence of an Fe–H moiety in Hred, which is instead considered to be an isoelectronic Fe(I)Fe(I) form. This description is consistent with IR data indicating that the Hred core is more electron-rich than that of Hox, with the terminal νCO bands undergoing, on average, a bathochromic shift of 25 cm−1 on forming Hred. The two states also differ in the geometry of the semibridging CO ligand. In Hox, the CO bridges to Fep, with the Fed–C–O unit being highly bent,209 perhaps in order to enable some spin delocalization. In the diamagnetic Hred state, the Fed–C–O is almost linear.
A third active state has been uncovered through studies on an [FeFe]-H2ase that, unlike most such proteins, lacks Fe–S clusters outside of the H-cluster. This recently identified Hsred (“super reduced”) form in Chlamydomonas reinhardtii is observed at or below −540 mV, about 150 mV more negative than the Hox/red couple. Both the [4Fe–4S]+ cubane and the Fe(I)Fe(I) [2Fe] site adopt their lowest oxidation states, a situation that would not be long-lived were auxiliary Fe–S clusters present to accept electrons. Nevertheless, Hsred is assumed to be a transient intermediate in all [FeFe]-H2ases. Its IR spectrum features νCO bands more similar to Hox than Hred, but one νCN band shifts by 45 cm−1 to lower energy, perhaps indicating a structural change as such stretches are insensitive to metal electron density.210 Described now are current proposals regarding the enzyme catalytic cycle and mechanism, subjects that are still topics of debate.
In considering the stoichiometry of the catalytic cycle, one notes that the two redox equivalents demanded by the H2/2H+ couple cannot be provided by the Hox/red couple alone, a problem conveniently addressed by invoking Hsred, which is 2e− more reduced than Hox (Figure 9). Consistent with present biophysical information, it is suggested that oxidation of H2 involves its binding at the Fed site in Hox to give Hox (H2).203 The Fe(η2-H2) fragment in the latter species is deprotonated by the amine of the adt2− cofactor, resulting in the ammonium hydride heterolysis product Hox(H+, H−). A PCET step is now proposed, in which deprotonation is associated with e− transfer from [2Fe] to the [4Fe–4S]2+ cluster. The resulting [4Fe–4S]+,Fe(II)Fe(II),H− species may tautomerize to Hsred, whose rapid oxidation (e− transfer) affords Hred and then Hox, thereby completing the cycle. The reverse processes apply to H+ reduction, which involves H+ transfer from adt2− to a reduced Fed center in concert with e− transfer from [4Fe–4S]+ to give the same intermediate invoked for H2 oxidation.
The catalytic mechanism can be further appreciated from the perspective of e− counting. Summing 3d electrons and electron pairs for each Fe–ligand bond in the [2Fe] subsite, one notes that Hox is a 33e− dimer, and Hred is a 34e− dimer (Figure 9), with 34e− being consistent with a bimetallic complex featuring a metal–metal bond. In order to maintain this electron count, binding of exogenous CO to Fed in Hred is accompanied by dissociation of the Cys-S–Fep bond.211 If the [2Fe] site carried a H− ligand, as seems likely in at least a transient form, the 18e− rule is still obeyed, since the H− complex would remain 34e− regardless of its description as Fe(II)Fe(II),H− or Fe(I)Fe(I),H+. To reiterate, as the Hox/Hred couple differs by only one electron, it is clear that [4Fe–4S] must supply the extra electron or hole required for the H2 ⇌ 2H+ + 2e− reaction.
While characterization data for each catalytic intermediate are not available, what we do know is that the [FeFe]-H2ases are some of the fastest enzymes.14 Rates vary by 2 orders of magnitude for H2 evolution but only by a factor of 2 or 3 for H2 oxidation. Compared to the rates for H2 evolution, H2 oxidation is always faster, sometimes by as much as 3 orders of magnitude.212 Perhaps the best characterized in terms of structure and rates is the H2ase I (note: organisms often have two or more different H2ases) from Clostridium pasteurianum. In this case, the turnover frequencies (TOFs) at 30 °C for H2 oxidation and evolution are 25 000 and 5700 s−1, respectively.212
The remarkably high catalytic rates of [FeFe]-H2ases are competitive with those exhibited by Pt metal.213 Yet, as stated in the Introduction, one motivation for the study of [FeFe]-H2ases is the preparation of base metal catalysts with the hope that reproducing the native structure will afford the native function. While long-lived hydrides are unlikely to be involved in the [FeFe]-H2ase mechanism, it is assumed that complexes of the form Fe(II/I)Fe(II),H− do have a transient presence (Figure 9). Presented in section 3.2 are synthetic hydrides relevant to the FeFe active site. The high fidelity hydride models are typically terminal, but, for historical purposes, some bridging species will be described in discussion of early work. Moreover, catalytically important bridging hydrides will also be considered.
3.2. [FeFe]-H2ase Synthetic Modeling
3.2.1. Early Diiron Dithiolates and Their Hydrido Complexes
The crystallographic analysis of [FeFe]-H2ase attracted particular interest from organometallic chemists, for whom the active site immediately brought to mind “classic” diiron dithiolates that predated the determination of the enzyme structure by 70 years.214 Renewed attention was paid to Fe(I)Fe(I) carbonyls such as [(OC)3Fe(SEt)2Fe(CO)3]23 and, later, [(OC)3Fe(pdt)Fe(CO)3] ([4], pdt2− = 1,3-propanedithiolate; Figure 10),215 owing to the presence of Fe, RS−, and CO in these complexes as well as the diiron (and, in fact, all) H2ase active sites.
Figure 10.
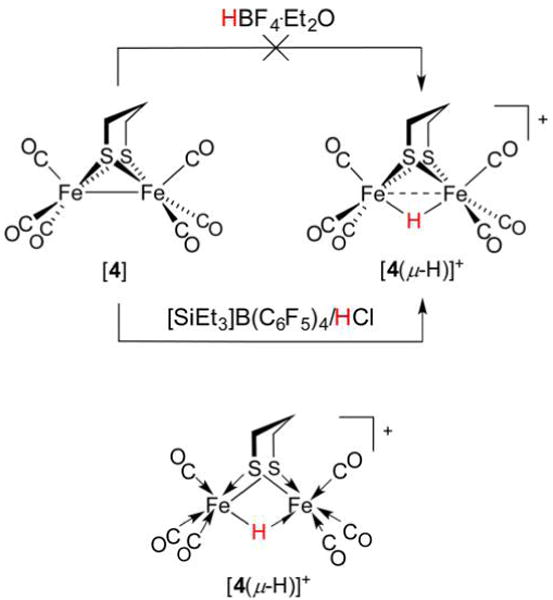
Archetypal diiron thiolato hexacarbonyl [4] can be converted to its hydride [4(μ-H)]+ only with very strong acids.44 An alternative structural representation of [4(μ-H)]+, using full arrow and half-arrow notation, is provided below.
There exists an uncanny and promising resemblance between [FeFe]-H2ase active sites and the archetypal synthetic low-valent hexacarbonyls, with their 34e−, stable Fe–Fe bonded motif.216 Yet, in reality the latter are too stable, to the point that their oxidations are strongly anodic (e.g., E1/2([4]+/0) = 0.65 V vs Fc+/0)217 and their Brønsted basicities are low. As discussed above, a requirement of the [FeFe]- and [NiFe]-H2ases is their participation in both acid–base and redox at mild pH and potentials. Although [4] is inert to HBF4·Et2O, it is protonated by HOTf (trifluoromethanesulfonic acid) such that lower and upper bounds for the acidity of [4(μ-H)]+ in C6D5F are known: −9 <pKa([4(μ-H)]+) < 0.44 However, protonation with HOTf does not proceed smoothly, and isolation of an Fe(II)(μ-H)Fe(II) derivative of [4] was found to necessitate action of the superacid generated from [SiEt3]B(C6F5)4 and HCl.218 In the preparation of [4(μ-H)]+ from [4], while the 2e− Fe–Fe bond in the latter formally reduces H+ to afford a H− ligand, the internuclear separation in the resulting hydride is still typically comparable to twice the covalent radius219 of low-spin Fe (2 × 1.32 Å = 2.64 Å). Thus, although the 3d6 Fe(II) sites are not expected to interact strongly with each other, a dashed bond between metals is nevertheless an oft-adopted notation. While not used in this review, a more rigorous notation exists for the representation of Fe(μ-H)Fe systems and three-center two-electron bonds in general.220 This convention avoids depicting direct Fe–Fe bonds (Figure 10, bottom), and employs the typical arrows for L-type (charge-neutral, 2e−) donors, and a half-arrow for the bridging hydride. Such a method is useful when counting electrons, with the half-arrow making clear that Fe(μ-H)Fe units involve three-center two-electron rather than three-center four-electron bonds, as the simple line drawing might imply.
The acid–base and redox reactivity of the diiron(I) dithiolato hexacarbonyls is greatly enhanced upon replacement of π-acidic CO ligands with stronger donors—a strategy that has proven very generalizable. For example, the installation of one or two CN− ligands occurs under mild conditions to afford, in the latter case, a complex bearing all the diatomic ligands present in the [FeFe]-H2ase active site.221 While acids only protonate the N atom in [(OC)3Fe(pdt)Fe(CO)2CN]− (the Fe–Fe bond being insufficiently basic), the dicyanide [3]2− protonates to give hydrides.222–224 In situ analysis revealed an N-protonated intermediate prior to the formation of [NC(OC)2Fe(pdt)(μ-H)Fe(CO)2CN]− ([3(μ-H)]−).
The asymmetric derivative [Me3P(OC)2Fe(pdt)Fe-(CO)2CN]−([5]−) could be converted to its stable conjugate acid [Me3P(OC)2Fe(pdt)(μ-H)Fe(CO)2CN] ([5(μ-H)], Figure 11),23 which exhibits a 1H NMR resonance at −17 ppm typical of a μ-H− ligand. The charge-neutral hydride [5(μ-H)] undergoes electrochemical reduction at −1.57 V vs Fc+/0, the pseudoreversibility of which indicates that a mixed-valent complex is accessible (vide infra). Further protonation of [5(μ-H)] affords [Me3P(CO)2Fe(pdt)(μ-H)Fe(CO)2(CNH)]+ ([5(μ-H)H]+), whose irreversible reduction at Epc = −1.47 V is accompanied by H2 evolution. In what is a common indirect indicator of catalytic activity, the reductive current increases upon titration with strong acids, including HCl, H2SO4, and toluenesulfonic acid (HOTs), with a TOF of 0.0067 s−1 at −1.2 V vs Ag/AgCl in the latter case.225 While modest, the activity of species such as [5(μ-H)] contrasts the inactivity of the related bis(phosphine) hydride [Me3P(CO)2Fe(SMe)2(μ-H)Fe-(CO)2PMe3]+ (δ(1H) −15.6 ppm),226 which lacks an effective H+ relay. This role is apparently well-served by CN−, despite its arrangement perhaps not being ideal for H+ transfer to the metal sites. Overall, these findings highlight an important design feature for functional [FeFe]-H2ase (and [NiFe]-H2ase) models: the basic/oxidizable metal(s) must be proximal to a basic moiety.
Figure 11.

Catalytic cycle for electrocatalytic proton reduction mediated by [5(μ-H)].23
The ambidentate nature of CN− was recognized as a complicating factor that saw it largely replaced in models by simpler ligands of (ideally) comparable σ-donicity. While donors such as carbenes,227–231 isonitriles,232–235 and even nitrosyls236,237 have been used, tertiary phosphines have proven the most useful.23
3.2.2. Mixed-Valent Hydrides
The 34e− Fe(II)(μ-H)Fe-(II) species discussed above represent models for a putative [FeFe]-H2ase form tautomeric to the Hsred state, an unobserved species which likely bears a terminal hydride (Figure 9, bottom right). The catalytic cycle for H2 evolution necessitates the reduction of diferrous hydrides to mixed-valent derivatives, both for synthetic catalysts and in the native catalytic cycle (the state Hox (H+, H−)). While there is reason to believe that synthetic versions of these hydrides may be unstable (e.g., toward bimolecular decomposition and H2 evolution), the pseudoreversibility of Fe(II)(μ-H)Fe(II/I) couples in certain complexes suggested that a mixed-valent species might be persistent. Such odd-electron dinuclear species are often classified according to the system of Robin and Day.238,239 A Robin–Day class I complex features structural asymmetry associated with localization of the singly occupied orbital on one site. A class III complex features structurally indistinguishable metal sites, each with a “genuinely nonintegral valence”.240 Between these two extremes lie the class II complexes, whose metal sites are distinguishable, but not very different.
At this point it is important to acknowledge a large, but quite distinct body of work concerning hydrogen evolution reaction (HER) electrocatalysis mediated by hexacarbonyls [(OC)3Fe-(dithiolate)Fe(CO)3] and pentacarbonyls [(OC)3Fe-(dithiolate)Fe(CO)2L]. Such ligand sets, in contrast to that of the CN−-containing active site and models, result in higher oxidation potentials but make accessible very reduced (but abiological) states such as Fe(0)Fe(I),241 particularly when electron-poor ligands, such as 3,6-dichloro-1,2-benzeneditholate,242 are used. The protonation of these (often monoanionic) Fe(0)Fe(I) complexes would afford mixed-valent Fe(II)Fe(I)/Fe(1.5)Fe(1.5) hydrides, species that have been invoked during the HER as mediated by the [FeFe]-H2ases. However, the necessary formation of Fe(0)Fe(I) species leads to hexa- and pentacarbonyls displaying high overpotentials,67 and these parent compounds can be considered to have less fidelity to [FeFe]-H2ases than their more substituted analogues, with which this subsection is now concerned.
An early study on mixed-valent diiron hydrides reported the reduction of [Me3P(OC)2Fe(pdt)(μ-H)Fe(CO)2PMe3]+ ([6(μ-H)]+) with acenaphthylene anion radical to generate the neutral complex [Me3P(OC)2Fe(pdt)(μ-H)Fe(CO)2PMe3] ([6(μ-H)], Figure 12).108 The product features an EPR resonance at g = 2.0066 (near that of the free electron ge = 2.0023) split by 1H (Aiso = −41.7 MHz) and two equivalent 31P nuclei (Aiso = −75.8 MHz). A Robin–Day class III delocalized Fe(1.5)(μ-H)Fe(1.5) description for the complex was supported by DFT studies, which suggested that 70% of the unpaired spin density resides on the two Fe centers, with approximately 35% on each site.108
Figure 12.
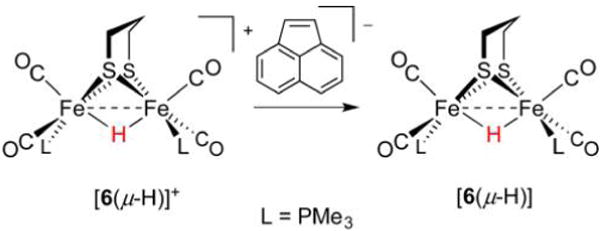
The 1e− reduction of a diamagnetic Fe(II)(μ-H)Fe(II) hydride affords a Fe(1.5)(μ-H)Fe(1.5) mixed-valent hydride.108
The radical hydride [6(μ-H)] is rather labile, as evidenced by its poorly reversible oxidation and an estimated t1/2 ~ 1 s at 25°C. Relatives of the present system include Fe(edt)(μ-H)Fe (edt2− = 1,2-ethanedithiolate),108 Fe(bdt)(μ-H)Fe (bdt2− = 1,2-benzenedithiolato),243 and Fe(SH)2(μ-H)Fe derivaties,244 all of which are similarly fragile. However, protonation of [dppv-(OC)Fe(pdt)Fe(CO)dppv] ([7], dppv =1,2-bis-(diphenylphosphino)ethene) affords a more sterically encumbered diferrous hydride [dppv(OC)Fe(pdt)(μ-H)Fe(CO)-dppv]+ ([7(μ-H)]+, Figure 13), which sustains reduction to afford the isolable neutral hydride [dppv(OC)Fe(pdt)(μ-H)Fe(CO)dppv] ([7(μ-H)]).245 X-ray crystallography revealed the product to feature an asymmetric Fe(II)(μ-H)Fe(I) core. The structural and EPR data are consistent with DFT calculations indicating that the Fe(I), which bears two-thirds of the spin density, is more distant from the hydride than is Fe(II) (1.82 vs 1.61 Å), on which most of the remaining spin resides. In solution, a small fraction of this asymmetric Robin–Day class II species converts to a C2-symmetric complex with the expected Fe(1.5)(μ-H)Fe(1.5) class III description. The latter product features dppv ligands with so-called “apical–basal” stereochemistry, in which they each bind an apical site that is trans to μ-H, as well as a basal site trans to pdt2−.
Figure 13.
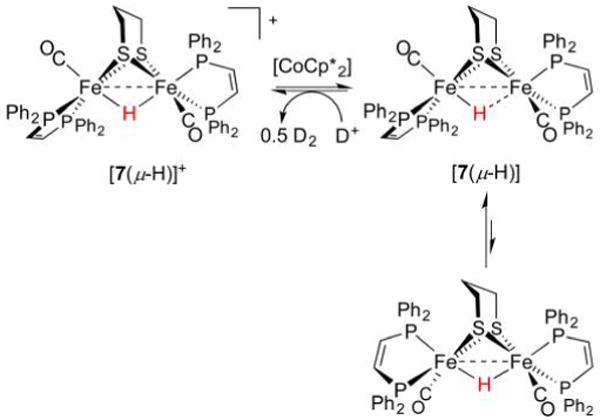
Formation and isomerism of the mixed-valent hydride [7(μ-H)]. The μ-H− ligand is unaffected by acid.245
Treatment of mixed-valent bis(dppv) hydride [7(μ-H)] with D+ yields the diferrous hydride and 1/2D2 rather than [dppv(OC)Fe(pdt)Fe(CO)dppv]+ ([7]+) and HD. This surprising observation implicates a spectator role for μ-H− in this complex during HER, probably reflecting the influence of the two bulky diphosphines and the robustness of Fe(II)(μ-H)Fe(II) motifs in general.
The relatively high-valent paramagnetic hydride [(Cp*)Fe-(bdt)(μ-H)Fe(Cp*)] (Cp*− = pentamethylcyclopentadienide) is generated by reduction of the unusual diferric species [(Cp*)Fe(bdt)(μ-H)Fe(Cp*)]+ (E1/2 = −0.91 V vs Fc+/0). Protonation of the Fe(II)(μ-H)Fe(III) complex returns the Fe(III)(μ-H)Fe(III) complex and induces evolution of H2 (0.5 equiv).246 These bridging hydride ligands, even on reduced diiron cores, can often be so inert they resist protonation even with strong acids. Indeed, the HER reactions described here can be considered to proceed by an outer-sphere mechanism, throughout which the coordination sphere of the catalyst is unchanged. If instead HER occurs with protons contacting the metal site(s), then it corresponds to an inner sphere mechanism, involving the intermediacy of a mixed-valent dihydride complex. Diiron dihydrides are described in section 3.2.3.
3.2.3. Dihydrides
The privileged bis(dppv) motif is well-suited to stabilizing many Fe oxidation and protonation states, among which are FeFe complexes in which two hydride ligands are present. Displacement of CO from [7(μ-H)]+ in MeCN affords the activated species [dppv(OC)Fe(pdt)(μ-H)Fe-(MeCN)dppv]+ ([8(μ-H)(MeCN)]+), and subsequent treatment with BH4− cleanly gives [dppv(OC)Fe(pdt)(μ-H)Fe(t-H)dppv] ([8(μ-H)(t-H)], Figure 14).247 The displacement of a labile MeCN ligand by a hydride source was inspired by the related synthesis of [(Me3P)2(CO)Fe(edt)Fe(CO)(PMe3)2(t-H)]+ discussed below. The product features not only a bridging hydride but also a terminal hydride (denoted t-H−); these cis ligands give rise to 1H NMR resonances at δ −18.9 and −12.2 ppm, respectively (intramolecular exchange rate ≈1 s−1 at −25°C).
Figure 14.
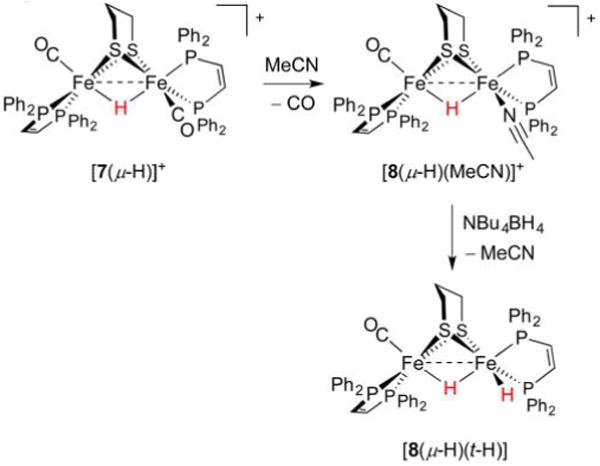
Preparation of the dihydride [8(μ-H)(t-H)] from sources of H+ and H−.247
While sequential addition of H+ and H− to an Fe(I)Fe(I) precursor represents one route to dihydrides, perhaps a more obvious (and generally applied) method is oxidative addition of H2. In the case of [dppv(OC)Fe(edt)Fe(CO)3] ([9], Figure 15), photolytic decarbonylation affords a transient 32e− species that binds and cleaves H2 to afford [dppv(OC)Fe(edt)(μ-H)Fe(t-H)(CO)2] ([10(μ-H)(t-H)], δ −12.8 (t-H), −14.9 ppm (μ-H)).248 DFT calculations indicate a product of Cs symmetry, with the trans nature of the hydride ligands contrasting the cis arrangement in [8(μ-H)(t-H)].248
Figure 15.
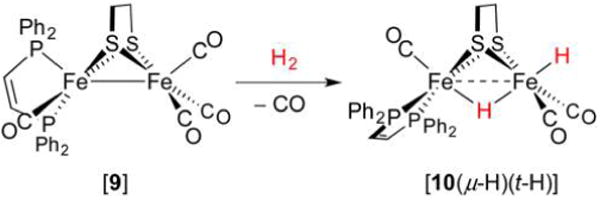
Preparation of a dihydride through oxidative addition.248
A closely related process of CO dissociation and H2 activation occurs for diruthenium analogues. For example, irradiation of [Cy3P(OC)2Ru(pdt)Ru(CO)2PCy3] under an H2 atmosphere affords the trans dihydride [Cy3P(OC)2Ru(pdt)(μ-H)Ru(t-H)(CO)PCy3],249 which yields [Cy3P(CO)2Ru(pdt)(μ-H)Ru-(η2-H2)(CO)PCy3]+ on treatment with [H(OEt2)2]BArF4 in CH2Cl2. When [D(OEt2)2]BArF4 is instead used, D incorporation is only observed as η2-HD and not as a D− ligand, a result that highlights the inherent stability of μ-H− over t-H− ligands.
The discussion on synthetic models has, until now, focused on bridging hydride complexes. Yet, both [8(μ-H)(t-H)] and [10(μ-H)(t-H)] feature t-H− ligands that are a key motif in the [FeFe]-H2ase mechanism. The preparation of terminal hydride complexes has been the subject of intense research activity, much of which is summarized in section 3.2.4.
3.2.4. Terminal Hydrides
In considering the structure of dihydride [8(μ-H)(t-H)], one might say that the presence of the strongly donating μ-H− ligand directs the second H− to adopt the important terminal position. But what of the situation when only a single H− ligand is present? Can a terminal monohydride, similar to that proposed in the enzyme mechanism, be observed in a model complex?
The large number of bridging hydrides reported is in part due to such species typically being thermodynamic products of [L3Fe(dithiolate)FeL3] protonation. Yet it just so happens that terminal hydrides, of varying kinetic stability, feature commonly (but not always)250 as intermediates in this reaction. Their presence was initially inferred from electrochemical studies on the HER activity of the related diphosphide [(OC)3Fe(μ-PPh2)2Fe(CO)3].251 The first direct observation of terminal hydrides arising from protonation was found by studying the action of HBF4·Et2O on [OC(dppe)Fe(pdt)Fe(CO)3].252 At 298 K in CH2Cl2 solution, the sole product was bridging hydride [(dppe)(CO)Fe(pdt)(μ-H)Fe(CO)3]+, in which a dibasal phosphine (i.e., Cs symmetry) was indicated by a characteristic high-field 1H NMR triplet at −14.1 ppm (2JPH = 21 Hz). When protonation was instead monitored at 203 K, a singlet at −4.33 ppm could be observed, such a shift being significantly downfield of resonances expected for bridging hydrides. The lack of coupling to any 31P nuclei indicated that initial protonation, perhaps counterintuitively, occurred at the relatively electron-poor Fe(CO)3 fragment to afford [(dppe)(OC)Fe(pdt)Fe-(CO)3(t-H)]+. This terminal hydride, upon warming to 243 K, converts to a mixture of apical–basal and dibasal forms of the bridging hydride, with the latter being the only FeFe species at 298 K. As many further examples will demonstrate, low-temperature protonation studies of this type are a powerful tool in studying the formation of terminal hydrides for a variety of FeFe systems.
Despite efforts in monitoring protonations of Fe(I)Fe(I) species, the first diiron terminal hydride complex to be fully characterized arose from a rather different route involving hydride transfer to an Fe(II)Fe(II) precursor. Treatment of [(Me3P)2(CO)Fe(edt)Fe(CO)(PMe3)2MeCN]2+([11(MeCN)]2+) with either AlH4− or BH4− at −25°C induced the formation of [(Me3P)2(CO)Fe(edt)Fe(CO)(PMe3)2(t-H)]+ ([11(t-H)]+) by displacement of MeCN (Figure 16). Isolated as a strikingly green species,253 [11(t-H)]+ exhibits a 1H NMR resonance at −4.6 ppm coupled to just two 31P nuclei. The product features an IR-active νFeH band at 1844 cm−1, consistent with the terminal nature of the hydride ligand. While M–H vibrations are often not easily assigned,254 the frequency is comparable to that calculated using DFT (1908 cm−1), with two lower frequency bands (1352 and 1151 cm−1) predicted for the analogous bridging isomer.253
Figure 16.
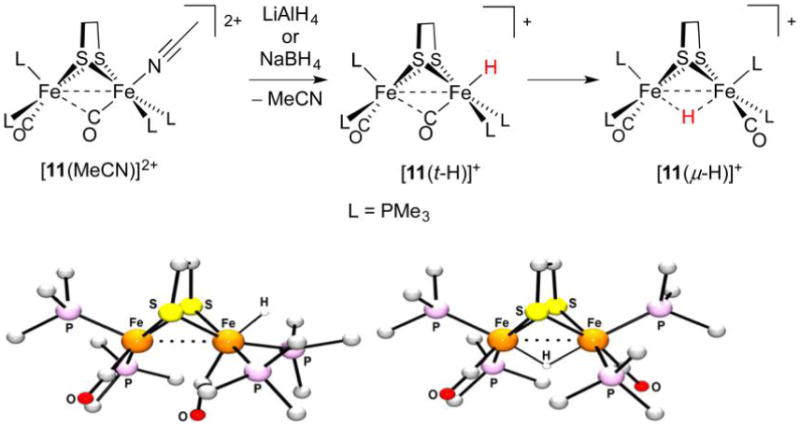
Preparation (top) and X-ray structures of [11(t-H)]+ and ([11(μ-H)]+ (bottom). Non-hydride H atoms are omitted for clarity.253
Although terminal hydride [11(t-H)]+ could be crystallized (rFeFe = 2.565 Å, rFeFe= = 1.498 Å), it is of limited thermal stability; in solution at room temperature it isomerizes to the red, C2-symmetric bridging hydride ([11(μ-H)]+ (−20.6 ppm, rFeFe = 2.610 Å, rFeFe= = 1.656, 1.602 Å) by a first-order process (k = 2 × 10−4 s−1 at 294 K). Both t-H and μ-H isomers are unreactive toward H2O, but the terminal form does liberate H2 upon treatment with the strong acids HOTf or [H(OEt2)2]BArF4 in the presence of MeCN to give back [11(MeCN)]2+.255 This finding highlights the considerably more hydridic nature of t-H− ligands.249 Such work further emphasizes the need to suppress the isomerization of terminal to bridging species. This process is typically irreversible, although theoretical work has suggested that excitation of Fe(pdt)(μ-H)Fe species to a low-lying triplet state can afford the terminal isomer.256
The t-H → μ-H isomerization has been examined using DFT calculations on [(dppv)(CO)Fe(edt)Fe(CO)3(t-H)]+ ([12(t-H)]+), [(dppv)(CO)Fe(edt)Fe(PMe3)(CO)2(t-H)]+, and [(Me3P)2(CO)Fe(edt)Fe(PMe3)2CO(t-H)]+ ([11(t-H)]+). These isomerizations may conceivably occur through Bailar (trigonal) or Ray–Dutt (rhombic) twists, with the pathway dependent on the ligand set.257 For these complexes, the free energy barriers to Ray–Dutt twisting are lower by 4.4–7.7 kcal/mol. When bulky ligands are present, certain isomerizations are unlikely to occur by Ray–Dutt twists since these would involve high-energy intermediates such as bridging phosphine complexes (Figure 17). Such motifs need not be invoked if isomerization proceeds through Bailar twists. For example, [12(t-H)]+ was predicted to convert to its bridging tautomer through a multistep pathway involving only Ray–Dutt twists, while [11(t-H)]+ was calculated to isomerize by both Ray–Dutt and Bailar twists.
Figure 17.
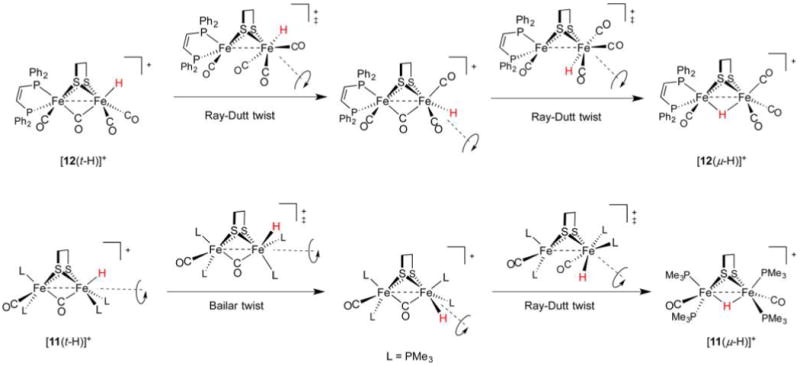
Mechanisms for the isomerization of terminal to bridging hydride complexes.
Since the first preparation of a diiron terminal hydride, several more such complexes of varying stability have been prepared. Their syntheses typically follow a biomimetic route involving (low-temperature) protonation of Fe(I)Fe(I) species. In the case of the bis(dppv) complex [dppv(OC)Fe(pdt)Fe(CO)dppv] ([7]) protonation with strong acids such as HBF4·Et2O (weak acids will not suffice) at −25 °C affords the terminal hydride [dppv(OC)Fe(pdt)Fe(CO)(t-H)dppv]+ ([7(t-H)]+, δ(1H) −3.5 ppm) as an isolable kinetic product. Solutions of this species are stable for minutes at 0 °C,258 an effect of the bulky, strongly electron-donating ligands that hinder isomerization.259 At room temperature, the hydride converts to asymmetric and symmetric isomers of the bridging species [dppv(OC)Fe(pdt)-(μ-H)Fe(CO)dppv]+ ([7(μ-H)]+, δ(1H) −14.5 and −15.6 ppm, respectively) described above. While stereodynamics and HER mechanisms are discussed in section 3.2.6, both the t- and μ-H propanedithiolates catalyze the HER, although with modest TOFs of 5 and 3 s−1, respectively.
Protonation at a terminal site in preference to the Fe–Fe bond is puzzling260 because most synthetic Fe(I)Fe(I) species do not have an empty apical coordination site. Due consideration must also be paid to the thiolate S atoms, whose nonbonding electron pairs allow for thiol intermediates in the protonation reaction. The S atoms in synthetic [FeFe]-H2ase models have indeed been shown to be competent Brønsted bases. For example, protonation of [(OC)2(Me3P)Fe(edt)(μ-H)Fe(CO)2(PMe3)] affords, according to IR spectroscopy, the expected hydride [(OC)2(Me3P)Fe(edt)(μ-H)Fe(CO)2(PMe3)]+, as well as the thiol [(OC)2(Me3P)Fe(Hedt)Fe(CO)2(PMe3)]+.261 Similarly, treatment of [(OC)3Fe(3,4-dichlorobenzene-1,2-dithiolate)Fe-(dppp)CO] with HBF4·OEt2 (1 equiv) results in the protonation of one S atom.262 The involvement of heteroatom-protonated species can greatly influence the regiochemistry of hydride formation because heteroatom proton relays are known to play key roles in reducing barriers in what would otherwise be intermolecular protonations of metal sites. Indeed, heteroatom bases of comparable thermodynamic basicity to low-valent metal centers have much greater kinetic basicities.263 These effects are borne out in the contrasting protonations of dithiolate [dppv(OC)Fe(pdt)Fe(CO)3]264 ([13], Figure 18) and a related diphosphide [dppv(OC)Fe(pdpp)Fe(CO)3] ([14], pdpp2− = 1,3-propanedi(phenylphosphide)).265 Treatment of [13] with [H(OEt2)2]BArF4 at −90 °C rapidly affords [dppv(OC)Fe-(pdt)Fe(t-H)(CO)3]+ ([13(t-H)]+), which undergoes slow isomerization to the bridging hydride [13(μ-H)]+. Under identical conditions, the diphosphide transforms slowly and exclusively to the bridging species [dppv(OC)Fe(pdpp)(μ-H)Fe(CO)3]+ ([14(μ-H)]+) with no intermediates observed. With lone electron pairs available on ligated pdt2− but not pdpp2−, it is apparent that heteroatom participation not only accelerates metal protonation, but also directs it to terminal sites.
Figure 18.
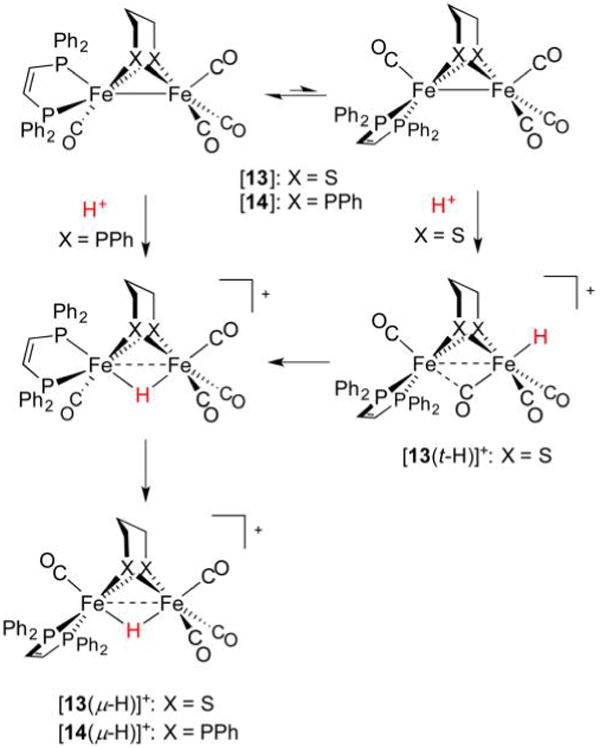
Bridging secondary phosphido ligands do not represent sites for protonation, and only bridging hydrides are observed.265 In contrast, bridging thiolato ligands can be protonated, and serve as relays to afford terminal and bridging hydride complexes.264
Assuming the dppv(CO)FeI fragment is more basic than FeI(CO)3, it may seem strange that the t-H− ligands in complexes such as [12(t-H)]+ are located at the tricarbonyl sites. This stereochemistry is rationalized in terms of the steric bulk of the diphosphine, as well as by considering a “rotated” form of the neutral complex in which a bridging CO ligand is trans to a vacant terminal Fe site (Figure 19). In the extreme case, one CO ligand moves to a bridging position to afford [dppv(CO)Fe(edt)(μ-CO)Fe(CO)2]. This isomer may be considered a 2e− mixed-valent species in which the pseudooctahedral site is Fe(II), with the square-pyramidal basic site being Fe(0). According to DFT calculations, the two isomers of this “rotated” species are both transition states, of which an asymmetric form, in which dppv occupies an apical and basal site (apical–basal), is only slightly more stable.265
Figure 19.
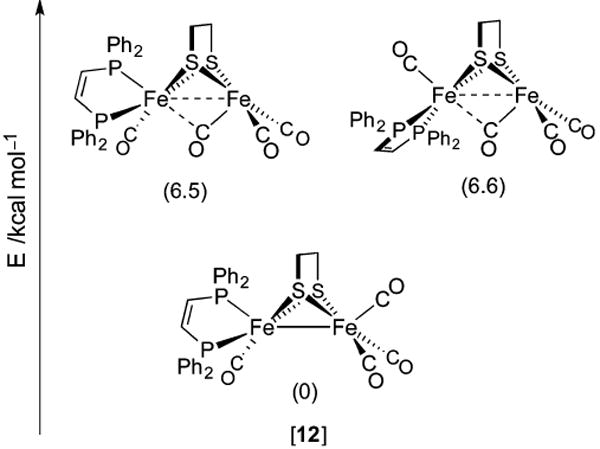
Rotated and unrotated isomers of [12], an Fe(I)Fe(I) model for Hred.265
The role of S atoms in hydride formation was also implicated in the protonation and isomerization of the electron-rich tetraphosphine [(Me3P)2(OC)Fe(pdt)Fe(CO)(PMe3)2] ([15], Figure 20).250 Its treatment with [H(OEt2)2]BArF4 at −90 °C gave a 2:1 mixture of symmetrical bridging hydride and a terminal hydride (δ(1H) −18.8 and −2.2 ppm, respectively). Also formed was a nonhydride (putatively thiol) species that converts to both bridging and terminal hydrides at −60 °C, with the t-H → μ-H conversion being, encouragingly, rather slow (t1/2 = 2.5 h at 20 °C) in the absence of acid catalyst.
Figure 20.
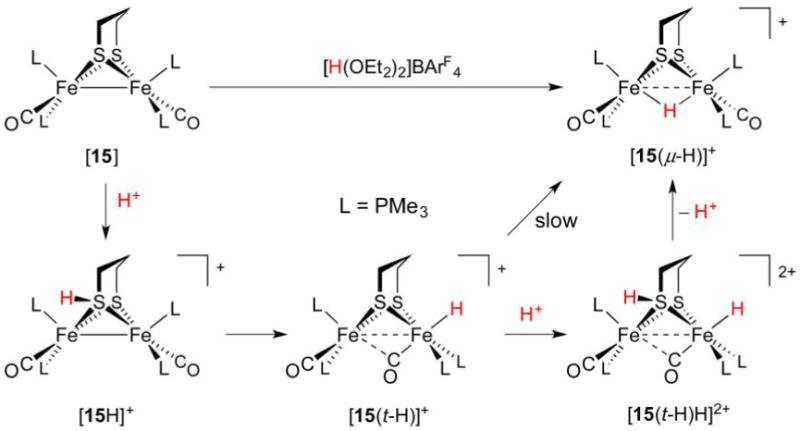
Protonation of [15] affords a thiol intermediate en route to a terminal hydride. Formation of the bridging hydride thermodynamic product is acid-catalyzed.250
While models discussed in this section share much in common with the [FeFe]-H2ase active site, the synthetic terminal hydrides lack the conformational rigidity of the enzyme H-cluster. In the latter case, the distal Fe adopts a reactive “rotated” state in which a terminal site is available for binding H− or H2 substrate.266 This rotated structure is perhaps stabilized by hydrogen bonding to the protein, but electronic factors may also be important. In model systems, this geometry can only be reproduced in unsymmetrically substituted complexes, including the Fe(I)Fe-(I) compounds [dppv(OC)Fe(2,2-diethyl-1,3-propanedithiolate)(μ-CO)Fe(CO)2]266 and [dmpe(OC)Fe-(adtBn)Fe(CO)3].267
3.2.5. Comparison of Bridging and Terminal Hydrides
While the thermal stability of bridging hydrides was discussed above, these isomers are also typically more resistant to electrochemical reduction than their terminal counterparts. In view of the strong σ-donicity of the H− ligand,47 1e− reduction of Fe(II)Fe(II)(t-H) occurs at the ferrous center not bound to the hydride (analogous to Fep in [FeFe]-H2ase). This situation is not possible in the case of Fe(II)(μ-H)Fe(II), as H− exerts its cathodic effect on the couples of both metals.259 A corollary is that H− binds Fe(II) considerably more tightly than Fe(I), as is illustrated by the disparate Fe–H distances in the mixed-valent complex [dppv(OC)Fe(pdt)(μ-H)Fe(CO)dppv] ([7(μ-H)]). Theoretical calculations also indicate that reductions of terminal hydrides occur at more positive potentials than those of bridging hydrides,186 although some special cases have been identified in which this trend is reversed. These situations arise when bulky ligands are present, with reduction of the μ-H complex alleviating strain by allowing the metals to distance themselves from one another.186
Discussion of μ-H isomers can be illustrative and guide the synthesis of more stable terminal hydrides, but one must keep in mind that μ-H species do not come into consideration in the [FeFe]-H2ase mechanism. For the enzyme catalytic cycle (Figure 9), DFT studies indicate that t-H to μ-H isomerization comes with a prohibitively large electronic energy barrier (29 kcal mol−1),268 in no small part due to the H-bonding of CN− ligands to nearby residues preventing turnstile rotation. Moreover, direct protonation of the Fe–Fe bond—bypassing any t-H species—is even more unlikely, and the endothermic H+ transfers from Lys358 to Hox and Hred are accompanied by kinetic barriers of 52 and 39 kcal mol−1, respectively. These large barriers arise because direct H+ transfer would require movement of Lys358 and cleavage of its H-bond to Glu361. In contrast to such processes, terminal hydride formation is exothermic and requires an electronic activation energy of only 6.9 kcal mol−1, with the barrier to H2 formation being 4.1 kcal mol−1.268 The redox properties of diiron dithiolates allows one to estimate rates of protonation of the Fe–Fe bond (the highest occupied molecular orbital (HOMO)).217 In the case of an [FeFe]-H2ase protein operating at −414 mV vs NHE at pH 7, the estimated rate is an order of magnitude lower than the observed rates for the enzyme. This once more underscores that the enzyme operates through terminal hydrides, and it does so because of distinct kinetic advantages. Of course, the key component facilitating terminal protonation is the azadithiolate cofactor. Synthetic models featuring adt2− (or its derivatives) are discussed in section 3.2.6, along with their mechanisms for H2 evolution.
3.2.6. Azadithiolato Hydrides
The synthesis of the archetypal [(OC)3Fe(pdt)Fe(CO)3] simply involves treatment of an Fe(0) carbonyl with the dithiol H2pdt. In contrast, the instability of free H2adt or H2adtR ((adtR)2− = −SCH2N(R)-CH2S−); R = alkyl, etc.) species or simple salts thereof269 necessitates indirect synthetic routes for [(OC)3Fe(adt)Fe-(CO)3] derivatives. Among the methods used, the Mannich-type condensation of CH2O, amines, and [(OC)3Fe(SH)2Fe(CO)3] has proven useful in affording [(OC)3Fe(adt)Fe(CO)3],270 N-substituted species [(OC)3Fe(adtR)Fe(CO)3], as well as oxadithiolate [(OC)3Fe(odt)Fe(CO)3] (odt2− = (−SCH2)2O).271 Conveniently, the ligand substitution chemistry of these heteroatom-bridged hexacarbonyls mirrors that of the pdt2− parent, and several tertiary phosphine- and CN−-substituted derivatives are known. These models thus feature an ideal arrangement with an Fe(I)Fe(I) core of high thermodynamic basicity proximal to an amine proton relay with high kinetic basicity.
A major contributor to the kinetic favorability of terminal hydrides is the azadithiolate cofactor, which plays a key role in the catalytic activity of models and the enzyme. The azadithiolate in the enzyme was confirmed by an elegant series of experiments in which apo-HydA (the [FeFe]-H2ase from Chlamydomonas reinhardtii lacking the [2Fe] subsite) was reconstituted (“maturated”) with the synthetic diiron dicyanides [NC-(CO)2Fe(adt)Fe(CO)2CN]2− ([3]2−), [NC(CO)2Fe(odt)Fe-(CO)2CN]2−, and [NC(CO)2Fe(pdt)Fe(CO)2CN]2−.199,200,272 Each maturation product exhibited νCO and νCN bands matching those of native HydA, indicating that the diiron complexes had been accepted by the apoprotein. Yet only the apo-HydA + [NC(CO)2Fe(adt)Fe(CO)2CN]2− product (believed to be the holoenzyme) could reproduce the activity of authentic HydA. Because the activity of the enzyme is so high, the protein maturated with the flawed cofactor pdt2− is still catalytically competent, albeit with <1% activity of HydA.201 Such work confirms not only that the dithiolate cofactor contains an amine, but further that this H+ relay is essential to the efficiency of [FeFe]-H2ases.
The divergent reactivity of propanedithiolates and azadithiolates, apparent also in purely synthetic species, is of great importance. While [(Me3P)2(OC)Fe(pdt)Fe(CO)(PMe3)2] ([15]) converts primarily to the bridging hydride upon treatment with [H(OEt2)2]BArF4 (vide supra), azadithiolate [(Me3P)2(OC)Fe(adt)Fe(CO)(PMe3)2] ([16]) converts rapidly and exclusively to the terminal hydride [(Me3P)2(OC)Fe-(adt)Fe(t-H)(CO)(PMe3)2]+ ([16(t-H)]+, δ(1H) −2.29 ppm).250 Considering the similar electronic properties of the low-valent species (νCO = 1860, 1839 and 1857, 1836 cm−1 for pdt2− and adt2− complexes, respectively), this regiochemical selectivity is ascribed to the influence of the secondary amine, which provides a kinetic protonation pathway via an ammonium intermediate. The effect is so marked that even weak acids (e.g., NH4PF6) can induce rapid t-H formation in the azadithiolate complex, with no reactivity observed for the propanedithiolate complex. While S-protonation was invoked when describing the reactivity of [15], such thiol intermediates are no longer relevant when a basic amine is present, especially when it is poised above the terminal site. Such an arrangement has been crystallographically confirmed in the diiron azadithiolato terminal hydrides [(Me3P)2(OC)Fe(adt)Fe(t-H)(CO)-(PMe3)2]+([16(t-H)]+) and [dppv(CO)Fe(adtH)Fe(t-H)-(dppv)(CO)]2+ ([17(t-H)H]2+, Figure 21).250,258
Figure 21.

X-ray structures of [16(t-H)]+ and [17(t-H)H]2+. Nonionizable H atoms are omitted for clarity.250,258
The structure of [16(t-H)]+ features not only the synergistic trans arrangement of the t-H− donor and μ-CO acceptor, but also a positioning of the amine such that N–H⋯H–Fe dihydrogen bonding (2.042 Å) is possible, as also indicated by one-dimensional nuclear Overhauser effect NMR experiments. It does not take much imagination to see that the Fe–H (1.487 Å) moiety could arise from tautomerization of an ammonium intermediate, followed by inversion at N such that this interaction (as well as the anomeric-type interactions featuring the lone pair) can be established. The related tetraphosphine [17(t-H)H]2+, perhaps owing to the lower donicity of dppv versus bis(PMe3) ligation, has a slightly shorter Fe–H bond (1.438 Å). Given that the dppv complex was crystallized with the dithiolate in the ammonium state, the difference could also be ascribed to the stronger dihydrogen bonding (1.879 Å) of this now more acidic moiety to the H− ligand. DFT studies have shown that the N–H⋯H–Fe interaction is strongly affected by the presence of H-bond acceptors such as BF4−, and that in the absence of the counteranions the H⋯H distance contracts to 1.40 Å.161 The participation of an ammonium hydride state for the enzyme has been proposed, with such a species (Figure 9, middle right) arising from the protonation of Hsred.
The thermodynamic influence of dihydrogen bonding has been the subject of many experimental and theoretical studies. In particular, vibrational frequency shifts induced by such bonding, as well as the final H⋯H distances, have been used in combination with DFT calculations to correlate observables with the strengths of these interactions.273 In the present case of [16(t-H)]+ and [17(t-H)H]2+, the stabilization afforded by dihydrogen bonding amounts to approximately 2 and 4 kcal mol−1, respectively, based on the H⋯H distances. In all, such interactions play a small but not insignificant role in reactivity, and they have also been implicated in the [Fe]-H2ase catalytic cycle (section 5.1).274
Although the amine group plays a key role in second coordination sphere interactions and directing terminal protonation, it is perhaps unfortunate that the t-H → μ-H isomerization in synthetic models occurs regardless of which dithiolate is present.264 Thus, both complexes in Figure 21 irreversibly convert to their respective bridging hydride thermodynamic products. In the dppv-containing system, the conformational dynamics have been extensively studied both in situ and in silico (Figure 22). DFT calculations indicate the kinetic protonation product of [17] to be the ammonium [17H]+.161 The proton is rapidly relayed to a terminal coordination site at the nearest Fe center, which is 1.3 pKa units (on an MeCN scale) more basic than the amine. Experimentally, low-temperature protonation of [17] readily affords terminal hydride [17(t-H)]+ (δ(1H) −4.2 ppm), which eventually isomerizes to bridging hydrides of pseudo C2 and then C1 symmetry. The final hydride complex [17(μ-H)]+ (−14.3 ppm; Figure 22, center left) is much less acidic (pKa > 18.6) than [17(t-H)]+ (pKa = 16), reflecting the higher thermodynamic basicity of the Fe–Fe bond relative to the terminal sites in Fe(I)Fe(I) species. Consistent with NMR data, the pKa values suggest the conversion should be almost quantitative, as [[17(μ-H)]+]/[[17(t-H)]+] > 102.6.258 A similar t-H → μ-H isomerization process is at play for the doubly protonated derivatives. The ammonium terminal hydride [17(t-H)H]2+ is 9.5 pKa units more acidic than [17(t-H)]+, in line with the former only being relevant when strong acids are employed. It turns out that [17(t-H)H]2+ plays an important role in catalysis. Having addressed the stoichiometric protonations of diiron species, the discussion now moves to their catalytic reactions. As will become clear, such complexes—most notable of which is the dication [17(t-H)H]2+—can exhibit a broad range of HER activity that can be readily appreciated by considering their structures.
Figure 22.
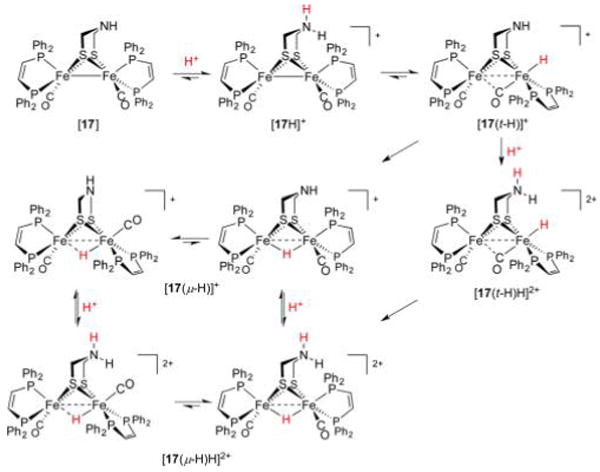
Conformational dynamics accompanying the protonation of [17] (inversion of ammonium/amine centers not depicted).161
3.2.7. Proton Reduction Catalysis
Understanding the protonation of [FeFe]-H2ase models is a prerequisite for the study of HER electrocatalysis, in which acid–base chemistry plays a central role. Hydrides of the form FeFe(t-H) or Fe(μ-H) Fe are important intermediates, but few HER investigations on diiron models include conclusive characterization of hydride-bearing states. Several surveys on the HER mediated by FeFe species are available,81,275–277 and presented here is rather a description of the salient components of diiron dithiolate catalysts, such that one can identify the often subtle structural and electronic differences between systems and rationalize their influence on catalytic H+ reduction. For instance, the donicity of ligands bound to the FeFe cores will govern which oxidation states are accessible during the catalytic cycle. The electron density at the metal further influences which protonation states predominate, an aspect that is also affected by protic groups in the second coordination sphere and the nature of the acid employed. With these aspects in mind, the design challenges associated with [FeFe]-H2ase modeling, as well as some potential solutions, will become more apparent. In terms of evaluating electrocatalysis, it is once more emphasized that a description should include both thermodynamic information (e.g., potential) and a corresponding kinetic parameter (e.g., TOF).73 In all, catalysts acting at low overpotentials and high turnover frequencies are the most desirable.
Before turning to specific examples, possible pathways for inner-sphere HER mediated by an arbitrary reduced metal complex LnM are presented in Figure 23. With respect to FeFe complexes, catalysis typically involves conversion of an Fe(I)-Fe(I) species (an Hred model denoted LnM) to the doubly protonated and doubly reduced state LnMH2. Poised to release H2, this latter state may take the form of a dihydrogen complex LnM(η-H2), a dihydride LnM(H)2, or another tautomer. The exact nature of the relevant hydride-bearing intermediates for a given catalyst depends on the mechanism at play, which is influenced by metal coordination environment, Brønsted acid strength, and catalytic conditions. As described in section 3.2.2, a scarce few diiron hydrides have been characterized aside from the 34e− diferrous systems.196 Furthermore, it is rare for even the latter well-studied compounds to be directly observed in catalysis.
Figure 23.
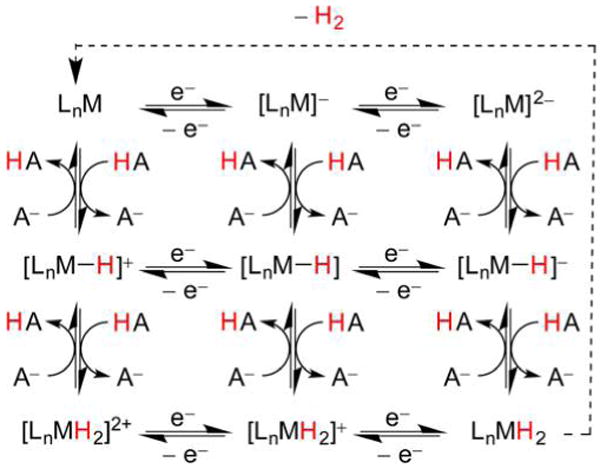
Many proton and electron transfers that can be involved in the HER.
Discussed now are examples of [(OC)3Fe(dithiolate)(CO)3] catalysts, the “first-generation” [FeFe]-H2ase models on which the majority of HER studies have focused,276 in part due to the straightforward preparation and robust nature of such hexacarbonyl compounds. With respect to the archetypal [(OC)3Fe(pdt)Fe(CO)3] ([4]), the use of electrochemical and spectroelectrochemical measurements, digital simulations,278,279 and DFT calculations has revealed much about its HER mechanism (Figure 24).280 The electron-poor Fe(I)Fe(I) core in [4] is not readily protonated (section 3.2.1), and catalytic reduction of HOTs instead begins with reduction of [4] (E1/2 = −1.55 V vs Fc+/0). The monoanion is now sufficiently basic to undergo protonation by HOTs, and the resulting μ-H species is more easily reduced (E > −1.55 V) than [4]− such that an additional reduction occurs. The formally Fe(I)Fe(I) anion [(OC)3Fe(pdt)(μ-H)Fe(CO)3]− ([4(μ-H)]−) can now undergo protonation at either of its Fe or S atoms. Protonation at a metal site affords an Fe(II)Fe(II) dihydride poised to release H2 and regenerate [4] (albeit slowly) in an overall ECEC mechanistic cycle. Another possible form of the doubly-protonated species is a Fe(I)Fe(I) hydride in which one thiol binds only one Fe site. The latter species readily undergoes reduction (−1.75 V) and quickly releases H2 as part of a CECE cycle. A common reduced hydride intermediate can thus perform catalysis by two separate mechanisms occurring at distinct potentials and rates.
Figure 24.
HER mechanism associated with [4].280
Electrocatalytic proton reduction by the 1,2-benzenedithiolate congener [(OC)3Fe(bdt)Fe(CO)3] ([18]) proceeds by a mechanism different from those described above. While the exact pathway is dependent on the acid pKa, experimental and DFT studies indicate that this very electron-poor complex must receive 2e− (−1.31, −1.33 V vs Fc+/0 in MeCN) to give the formally Fe(0)Fe(0) dianion [18]2−, protonation of which yields the Fe(I)(μ-H)Fe(I) species [18(μ-H)]− (Figure 25).85 This hydride product is susceptible to further reaction with acids of pKa < 23 (e.g., HOTs), resulting in slow release of H2 that regenerates [18] and completes the EECC catalytic cycle. Additional reduction of [18(μ-H)]− is necessary when weaker acids (pKa > 23, e.g. HOAc) are used. This is followed by protonation and rapid H2 release to complete an overall ECEC mechanism.
Figure 25.

HER mechanism associated with [18].85
The reader may recall that it is an Fe(I)Fe(I) form of [FeFe]-H2ase, namely Hsred, that is proposed to undergo tautomerization/protonation to afford a terminal hydride (section 3.1). Contrasting the HER mechanism of the enzyme with those of its hexacarbonyl models allows one to tease out the influences of ligand sets and acid–base cofactors: the first and second coordination spheres. With respect to ligand effects, electron-poor models (i.e., those with six CO ligands) require reduction below an Fe(I)Fe(I) state before their protonation can occur. Even then, it is a μ-H rather than t-H species that invariably forms. In terms of the second coordination sphere, the presence of Brønsted basic sites within a model complex can have a strong influence on the electrocatalytic mechanism and potential at which catalysis occurs.281 Basic residues are protonated in acidic media, resulting in the catalyst bearing less negative charge and giving rise to milder reductions relative to those of more anionic species. For example, the mechanism proposed for HOTs reduction282 catalyzed by [(OC)3Fe(adtR)Fe(CO)3] ([19], R = iPr, CH2CH2OCH3) begins with protonation of the amino group to form [19H]+ (Figure 26). The resulting Fe(I)Fe(I) cation can be reduced at −1.2 V vs Fc+/0, nearly 450 mV more positive than the wave for the neutral complex [(OC)3Fe(adtR)Fe(CO)3]. The reduced ammonium [19H] is then protonated to generate a mixed-valence ammonium hydride [19(μ-H)H]+, which immediately reduces at potentials below −1.2 V and slowly releases H2. While the structure of the [19(H)2] product is not obvious, what is clear is that an acid–base cofactor enables a mechanism involving mixed-valence hydrides in place of abiological, highly reduced Fe(I)(μ-H)Fe(I) intermediates. In the presence of the strong acid HBF4·Et2O, [19(H)2] can not only undergo slow H2 loss, but instead may also be protonated to afford [19(H)3]+, whose reduction (−1.4 V) causes fast release of H2. Of the two CECE mechanisms, as was illustrated in the previous two examples, the reduction occurring at the more negative potential results in faster H2 production.
Figure 26.

HER mechanism associated with [19].282
The low basicity of hexacarbonyl derivatives means that their conversion to hydride-bearing species necessitates either the use of superacids or electrochemical reduction prior to protonation. The second process requires highly negative potentials and, thus, can incur significant overpotentials, even when an azadithiolate (adtR)2− is present. At no point does ammonium [19H]+ convert to an amine hydride form, reflecting a poorly basic metal core. The observation that another Fe(I)Fe(I) species, namely Hred, readily protonates to form a hydride indicates deficiencies in the inner coordination sphere of these “first-generation” models. As described earlier in this section, these are addressed by substitution of CO ligands for strong σ-donors such as CN−, phosphines, and N-heterocyclic carbenes. But how many substitutions must be made? Replacing two carbonyls is apparently insufficient, as exemplified in the reduction of HOTs catalyzed by [(dppe)(OC)Fe(pdt)Fe(CO)3]. In this case, HER requires reduction of either a bridging hydride [(dppe)(OC)Fe(pdt)(μ-H)Fe(CO)3]+ (−1.3V vs Fc+/0) or the parent complex (−1.5 V).283 Of course, Nature eschews bridging hydrides and does not operate via Fe(I)Fe(0) intermediates, allowing for much milder HER potentials. The first model to exhibit H+ reduction electrocatalysis through a terminal hydride was [(OC)(dppv)Fe(pdt)Fe(dppv)(CO)] ([7]), whose treatment with HBF4·Et2O affords the Fe(II)Fe(II)(t-H) complex [7(t-H)]+, a species that persists for minutes at 20 °C. The hydride reduces at −1.64 V vs Fc+/0, and catalytic reduction of HBF4·Et2O at this potential proceeds with a TOF of 5 s−1. In contrast, the Fe(II)(μ-H)Fe(II) isomer [7(μ-H)]+ reduces at −1.84 V with an associated TOF of 3 s−1 (Table 2).
Table 2.
Redox and Catalytic Properties of FeFe Dithiolato Hydrides
| hydride complex | abbreviation | δ(1H)/ppm | E1/2a | Ecata | acid used | TOF/s−1 | η/V |
|---|---|---|---|---|---|---|---|
| [(OC)2(Me3P)Fe(pdt)(μ-H)Fe(CO)2(CN)] | [5(μ-H)] | −17.08 | −1.57 | −1.47 | HOTs | – | 0.75 |
| [(OC)2(PMe3)Fe(pdt)(μ-H)Fe(CO)2(PMe3)]+ | [6(μ-H)]+ | −15.3 | −1.39b | −1.4 | HOTs | – | 0.75 |
| [dppv(OC)Fe(adt)Fe(t-H)(dppv)(CO)]+ | [17(t-H)]+ | −4.2 | −1.64b | −1.49 | ClCH2CO2H | 5000 | 0.71 |
| [dppv(OC)Fe(adtH)Fe(t-H)(dppv)(CO)]2+ | [17(t-H)H]2+ | −4.95 | −1.4b | −1.11 | CF3CO2H | 58000 | 0.51 |
| [dppv(OC)Fe(adt)(μ-H)Fe (dppv)(CO)]+ | [17(μ-H)]+ | −14.8,c −13.7d | −1.86 | −1.72 | ClCH2CO2H | 20 | 0.90 |
| [dppv(OC)Fe(adtH)(μ-H)Fe(dppv)(CO)]2+ | [17(μ-H)H]2+ | −15.5,c −14.3d | −1.77 | ||||
| [dppv(OC)Fe(pdt)Fe(t-H)(dppv)(CO)]+ | [7(t-H)] + | −3.5 | −1.67e | −1.49e | HBF4·Et2O | 5 | 1.32 |
| [dppv(OC)Fe(pdt)(μ-H)Fe(dppv)(CO)]+ | [7(μ-H)] + | −15.6,c −3.5d | −1.8e | −1.78e | ClCH2CO2H | 3 | 0.95 |
Relative to Fc+/0 in CH2Cl2/NBu4BArF4.
Irreversible.
Symmetric isomer.
Unsymmetric isomer.
In CH2Cl2/NBu4PF6.
The above example illustrates that, as described in section 3.2.5, terminal hydrides are privileged in that their reductions are both mild (due to the availability of a hydride-free Fe(II) site as an oxidant) and can give rise to higher TOFs (due to t-H− ligands being more reactive than μ-H− ligands). The 1e− reduction of [7(t-H)]+ is likely followed by direct protonation of its t-H− ligand to evolve H2, a process in which no doubly protonated and singly reduced derivative of [7] (analogous to Hox (H+, H−)) is formed. A much faster HER would result if a heteroatom base, proximal to H−, could serve as a “landing site” for H+. The important combination of an electron-rich bimetallic core and a pendant base is present in [dppv(OC)Fe(adt)Fe(dppv)(CO)] ([17], Figure 27), whose terminal hydride derivative [dppv-(OC)Fe(adt)Fe(t-H)(dppv)(CO)]+ ([17(t-H)]+) is relatively long-lived and catalyzes the mild and rapid HER (−1.49 V vs Fc+/0, TOF = 5000 s−1) from ClCH2CO2H in CH2Cl2. The bridging hydride tautomer [17(μ-H)]+ is an inferior catalyst: Ecat = −1.72 V, TOF = 20 s−1. Furthermore, a remarkable effect is observed if [17(t-H)]+ acts on the stronger acid CF3CO2H, whence catalysis occurs at −1.11 V with a TOF of 58 000 s−1. The improvement in both catalytic potential and TOF was attributed to the high activity of the oxidizing, doubly protonated dication [dppv(OC)Fe(adtH)Fe(t-H)(dppv)(CO)]2+ ([17(t-H)H]2+).
Figure 27.

Highly active HER catalyst [17] operates by different mechanisms that depend on acid strength.258
The attractive catalytic properties of [17] have motivated computational investigations into its activity.160 Unlike other synthetic models described in this review so far, the two protonations of [17] precede any reduction events, a situation that parallels [FeFe]-H2ase. Reduction of [17(t-H)H]2+ induces a contraction of the N–H⋯H–Fe interaction, such that a spin-delocalized intermediate Fe(1.5)Fe(1.5)(η2-H2) forms.95,208 Release of H2 from this labile species affords a substrate-free Fe(II)Fe(I) species, whose rapid reduction affords the starting Fe(I)Fe(I) complex. This study underscores once more the synergistic roles that terminal hydrides and proximal protic moieties play in efficient HER catalysis.
The FeFe HER catalysts discussed here make use of an electrode to mimic the native [4Fe–4S] electron relay cluster. An electron relay can be incorporated into synthetic models in the form of a redox-active moiety that may serve as an electron source/sink. Pentacarbonyl [20] features such a ligand—a phosphole—and the mechanism of its HER has been studied in CH2Cl2 solution.284 Precatalyst [20] enters the HER cycle upon double protonation and 2e− reduction (−1.44 V vs Fc+/0, Figure 28). An ECCE mechanism affords the active species [20(μ-H)H], which takes the form of a mixed-valence bridging hydride featuring a singly reduced, protonated ligand. Catalytic reduction of [Et3NH]BF4 occurs at −2.0 V, at which [20(μ-H)H] undergoes a second 2e− reduction and protonation in an ECE process, resulting in an Fe(I)(μ-H)Fe(I) species of formula [20(μ-H)(H)2]−. This species is indicated by spectroelectrochemical and DFT studies to adopt a structure in which an Fe–S bond is cleaved and a doubly protonated, doubly reduced ligand is present. Intermediate [20(μ-H)(H)2]− is the catalyst resting state; its protonation and release of H2 is rate-determining (kcat = 105 M−1 s−1). Overall, the phosphole ligand serves as a reservoir of reducing equivalents, enhancing PCET in a system that is both O2-tolerant and very fast. This system also catalyzes reduction of H2SO4 at −0.66 V at TOF = 70 000 s−1 (kcat = 3.5 × 104 M−1 s−1 in acid-independent regime). The value of the redox-active ligand is emphasized in that catalysis by [20(μ-H)(H)2]− is so fast even though its mechanism is “handicapped” by operation via bridging hydrides.
Figure 28.
HER catalytic cycle proposed for [20], which bears a redox-active phosphole ligand.284
3.2.8. Hydrides from H2
Mimicking the catalytic activity of [FeFe]-H2ases requires a synthetic FeFe complex to mediate not only the evolution of H2, but also its oxidation. Such a conversion is of interest for many reasons, a principle one being the need for cost-effective alternatives to Pt electrodes in fuel cells. As noted in section 2.1, the heterolysis of H2 is challenging in that free H2 is only weakly acidic (pKa(THF)44 ≈ pKa(MeCN) ≈ 50),40 a hurdle that can be overcome by its binding to a cationic metal site such as Fe(II). While successes in catalytic H2 oxidation are less numerous than those in the HER, several synthetic Fe catalysts can cleave H2. Some of these bear little structural resemblance to the [2Fe] subsite; these “bioinspired” models have been reviewed elsewhere.24,26,281 The focus here is on “biomimetic” systems—diiron thiolates in particular—that either catalytically oxidize H2 or convert to a hydride form under H2. The ideal biomimetic system would mimic Hox, the Fe(I)Fe(II) enzyme state that binds and cleaves H2, using it as a source of H−.
Early work on H2 activation by [FeFe]-H2ase models did not result in oxidation of H2 or its conversion to hydrides. Rather, it was shown that [(Me3P)(CO)2 Fe(pdt)(μ-H)Fe-(CO)2(PMe3)]+ ([6(μ-H)]+), a Fe(II)(μ-H)Fe(II) complex, mediates H/D scrambling under D2 and ambient light (Figure 29).285 The process is rather slow, with exchange of the hydride in CH2Cl2 solution under 7–8 bar of D2 requiring 4 weeks. In a related experiment, a H2/D2 mixture, exposed to direct sunlight and [6(μ-H)]+ in CH2Cl2, afforded HD. H/D exchange was markedly slower in the absence of light, and did not occur at all when the coordinating solvent CH3CN was used. It was thus proposed that light opens a coordination site by inducing either reversible decarbonylation or isomerization of the bridging hydride to a terminal hydride (the latter possibility was addressed in section 3.2.4).256 Further studies of H/D exchange with [6(μ-H)]+ or [(Me3P)(CO)2Fe(pdt)(μ-SMe)2Fe(CO)2(PMe3)]+ indicated that both complexes catalyzed D2/H2O exchange, a characteristic reaction of H2ases. The basic requirements for H/D exchange catalysis could thus be formulated: one needs a pentacoordinate Fe(II) Lewis acid to bind (H/D)2 and an accessible Brønsted base to deprotonate coordinated (H/D)2. These roles are possibly fulfilled by the μ-H complex and water, respectively.
Figure 29.
H/D scrambling mediated by [6(μ-H)]+. This complex may undergo either decarbonylation or μ-H → t-H isomerization such that a vacant site for D2 binding is accessible.285
While the [2Fe] subsite extracts a H− ligand from the H2 substrate, early models could not replicate this activity. Photolytic decarbonylation of [(dppv)(CO)Fe(edt)Fe(CO)3] ([9]) affords an all-important free Fe site, yet the unsaturated intermediate, instead of heterolyzing H2, oxidatively adds it through a very slow process.248 As mentioned above, oxidative addition of H2 is not observed for H2ases, which operate exclusively by heterolysis. It is also conceivable that metals extract H+ (rather than H−) from H2, as in the complete conversion of [6] to its hydride [6(μ-H)]+ under H2 (1 atm) in the presence of the H− acceptor B(C6F5)3. This FLP can be viewed as the reverse of the enzymatic system, in which the amine accepts H+ and Fed receives H−.
Initial attempts at activating and oxidizing H2 with synthetic [FeFe]-H2ase models utilized compounds in their Fe(I)Fe(I) or Fe(II)Fe(II) forms, neither of which is relevant to the biological mechanism, which proceeds through an Fe(I)Fe(II) state. As discussed in section 3.1, the mixed-valent core of Hox exhibits a rotated Fed coordination geometry exposing a site on an electrophilic Fe center to bind H2 (or the inhibitor CO). More recent synthetic studies have thus involved the preparation and reactivity studies of Hox models. While the first such complexes, obtained by oxidation of Fe(I)Fe(I) Hred models, were only observed transiently in spectroelectrochemical and stopped-flow studies,286 a series of stable Hox models appeared following reports of [(CO)2(PMe3)Fe(pdt)Fe(CO)2(IMes)]+(IMes =1,3-bis(2-mesityl)imidazolylidine) and [Me3P(CO)2Fe(edt)-Fe(CO)(dppv)]+.230,287 The products, readily prepared by oxidation of Fe(I)Fe(I) parent complexes with Fc+, reproduced many features of Hox. For example, the [(CO)2(Me3P)Fe] fragment in [(CO)2(Me3P)Fe(pdt)Fe(CO)2(IMes)]+ adopts a rotated structure with a semibridging CO ligand. Meanwhile, [Me3P(CO)2Fe(edt)Fe(CO)(dppv)]+ mimics Hox behavior in that it binds CO.
A more complete Hox model also contains an azadithiolate ligand, [Me3P(CO)2Fe(adtBn)Fe(CO)(dppv)]+ ([21]+). In considering the structure of [21]+, one notices that it features not only the mixed-valent core but also a H+ relay poised for H2 heterolysis.200,288,289 This prototype was the first Hox model to activate H2 and afford a hydride product, albeit in a bridging form and at very high H2 pressure (Figure 30).290 The reaction mechanism is not obvious a priori, but could be proposed based on the redox potentials of the relevant species. A likely scenario involves [21]+ binding and heterolyzing H2 to afford the mixed-valence ammonium hydride [21(t-H)H]+. An additional [21]+ complex may then oxidize and deprotonate (or abstract H• from) [21(t-H)H]+. The slow nature of the H2 activation is likely due to a combination of weak H2 binding, as well as the necessity for two complexes to act on a single H2 molecule, one as an H2 binder and the other as an oxidant.
Figure 30.
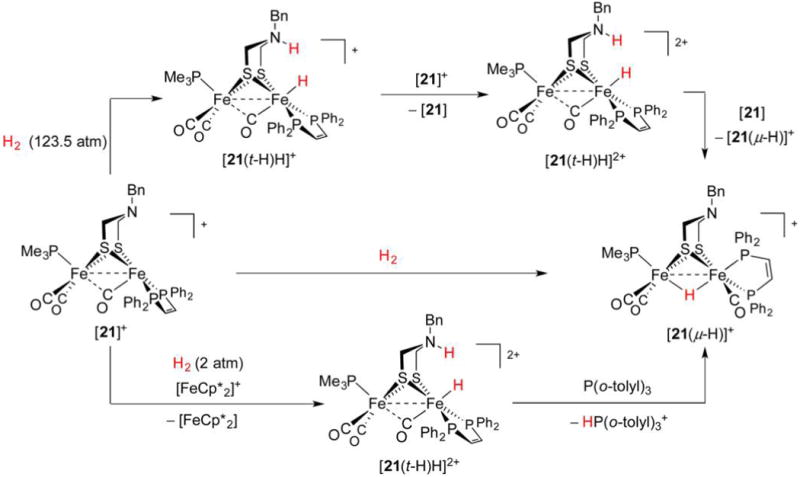
Stoichometric activation of H2 with [21]+ in the presence291 and absence290 of an external oxidant.
The above result prompted investigation of a potentially catalytic and biomimetic route, in which external base and oxidant were employed.291 In fact, [21]+ effects H2 heterolysis in the presence of the mild oxidant [FeCp*2]+ (Fc*+) as well as the (optional) weak base P(o-tolyl)3. The reaction proceeds over the course of hours under only 2 atm H2 at 0 °C to quantitatively afford [21(μ-H)]+ and [HP(o-tolyl)3]+ (kobs = 2.2 × 10−5 s−1 at 0°C). Given that the rate-determining step is likely H2 binding, it was thought that the stronger electrophile [(OC)3Fe(adtBn)Fe-(CO)(dppn)]+ ([22]+, dppn = 1,8-bis(diphenylphosphino)-naphthalene) might more effectively cleave H2. Under identical conditions (although using the slightly stronger oxidant Fc+) [22]+ heterolyzes H2 20 times faster (kobs = 4.8 × 10−4 s−1 at 0 °C) than [21]+ and ~104 times faster than [21]+ in the absence of oxidant at room temperature. The kinetic isotope effect measured for H2/D2 heterolysis by [22]+ and Fc+ was inverse (KIE = 0.8).
The above results bring to light the three requirements for H2 activation. Not only does the system require a Lewis acidic Fe site and a Brønsted basic amine, but it also requires an electron sink. In Nature, these roles are served by Fed, adt2−, and the proximal [4Fe–4S] cluster, respectively. The first synthetic model addressing these three design principles was an analogue of [21]+ in which the PMe3 ligand was replaced by the redox-active phosphine Cp*Fe(C5Me4CH2PEt2) (abbreviated FcP*).66 In this way, the complex [(FcP*)(CO)2Fe(adtBn)Fe(CO)(dppv)] ([23], Figure 31) preserves the electronic properties of [21], as the redox auxiliary is insulated by a CH2 spacer group. The triiron complex reversibly oxidizes at its Fe(I)Fe(I) core (−700 mV vs Fc+/0) and at the FcP* metalloligand (−393 mV) to afford the unique Hox model [23]2+ featuring Fe(I), Fe(II), and Fe(III) centers. Encouragingly, [23]2+ exhibited increased affinity toward CO, such that at low temperature the diamagnetic triferrous complex [23(CO)]2+ forms quantitatively. While the inclusion of redox cofactor Fc*P resulted in only a 2-fold increase in H2 activation rate relative to the [21]+/Fc*+ system, [23]2+ catalytically oxidizes H2 to protons and electrons. Exposure of [23]2+ to H2 in the presence of excess Fc+ and P(o-tolyl)3 resulted in complete conversion to Fc and [HP(o-tolyl)3]+, although at an extremely slow rate (TOF = 0.4 h−1). However, the triiron system is bidirectional in that the Hred model [23] is a catalyst for the HER,98 such that its combination with [H(OEt2)2]BArF4 (10 equiv) and Fc* (5 equiv) resulted in three turnovers during a 3 h experiment. The functional [FeFe]-H2ase model [23] represented a significant step forward, although its activity does not approach that of the native system. The markedly lower activity of [23]2+ is thought to result from its low affinity for H2. DFT calculations on [23]2+ indicate that H2 binding requires the relatively bulky benzyl group to shift to an energetically unfavorable conformation to allow H2 access to an open Fe site.188
Figure 31.
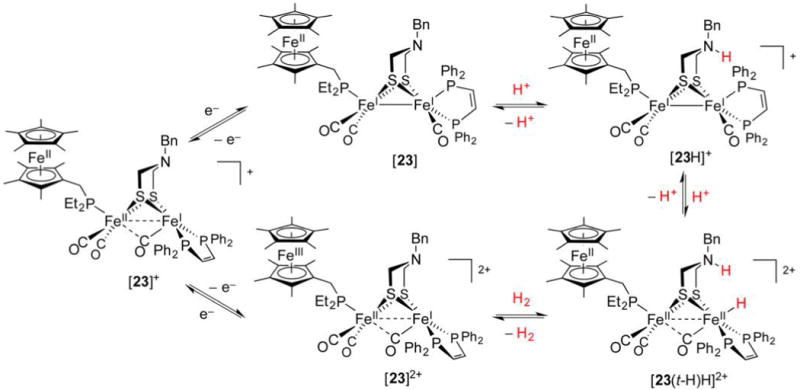
Catalytic cycle proposed for [23]-mediated H2 evolution and oxidation (clockwise and counterclockwise, respectively).66,292
Catalytic H2 oxidation and formation mediated by the triiron system are plagued by the formation of μ-H complexes, which are more inert than their active t-H tautomers. The bridging species [23(μ-H)]+ is not a HER catalyst, although it maintains some H2 oxidation activity. This deactivation is apparent as rates for catalytic reactions are much lower than those for stoichiometric reactions, implying sluggish deprotonation of the μ-hydride might restrict catalytic turnover.
The oxidation of H2 by Hox models does not require the acid–base cofactor to take the form of an azadithiolate, nor does it require the redox cofactor to be attached to the FeFe core. This is demonstrated with [(OC)3Fe(pdt)(μ-dppf)Fe(CO)] (dppf =1,1′-bis(diphenylphosphino)ferrocene), which catalyzes H2 oxidation at the surface of an electrode in the presence of pyridine as the base.293 A separate body of work on azadiphosphines294 culminated in the preparation of [(OC)3Fe-(pdt)Fe(PNP)(CO)] ([24], Figure 32), whose H+ relay Ph2PCH2N(nPr)CH2PPh2 (PNP)295 causes bridging hydride [24(μ-H)]+ to undergo rapid H/D exchange with CH3CO2D. Importantly, these hydrides are almost instantaneously deprotonated by relatively weak bases such as aniline and water, suggesting μ-H complexes might undergo turnover during H2 oxidation.
Figure 32.

Catalytic cycle proposed for H2 oxidation mediated by PNP complex [24(μ-H)]+.295
It was found that [24]+ catalytically oxidizes H2 in the presence of excess Fc+ and P(o-tolyl)3, illustrating that the redox auxiliary need not be directly appended to the FeFe core to achieve catalytic turnover.296 However, the proximity of the H+ relay is absolutely necessary, as replacement of PNP’s nPrN group with a nonbasic CH2 fragment affords a catalytically inactive species. Oxidation of H2 by the [24]+/Fc+ system (TOF = 0.6 h−1 at 25 °C) is only marginally greater than the triiron system (0.4 h−1), suggesting that the extremely low rates of catalytic H2 oxidation by model complexes are primarily a result of their inherently poor affinity for H2.
3.3. Concluding Remarks and Future Challenges
Considerable efforts have gone into synthetic modeling of the [FeFe]-H2ases, no doubt targeted at reproducing the activity of these remarkably efficient enzymes. The ease with which “first generation” models (i.e., hexacarbonyls) can be prepared has led the majority of synthetic and catalytic studies to focus on these systems. Yet their structural and functional resemblance to the active site is inferior to that of more substituted, electron-rich systems. The latter can operate biomimetically through terminal hydride intermediates and at oxidation states matching those proposed for [FeFe]-H2ase. Synthetic studies concerned with fine-tuning the first and second coordination spheres by CO substitution and acid–base incorporation, respectively, have been rewarded with HER activities comparable to the enzymatic catalysts, albeit at overpotentials greater than 0.5 V. The quest for fully functional bidirectional models has led to the development of sophisticated models featuring not only a pendant base proximal to an electron-rich FeFe core, but also a redox cofactor.297 These complete models for the H-cluster mediate H2 oxidation, yet they do so at rates several orders of magnitude less than the native system.
Given the successes and shortcomings of functional models, what avenues seem promising from the vantage? Certainly the [FeFe]-H2ase active site is subject to an array of weak interactions that are difficult to replicate in small molecule models. Further, the highly evolved mechanisms allowing for H2 and H+ transport to and from the Fed site cannot be found in purely synthetic systems. Whereas chemists are jubilant in replicating the role of the first and even the second coordination spheres, mutation studies make it clear that the third coordination sphere is also crucial to high catalytic rates.298
Despite the challenges inherent to synthetically approximating the protein environment around the active site, there are some specific shortcomings in current models that restrict their activities. In the case of Hox models, these are poised to activate H2 yet bind it rather weakly. This relative unreactivity of Hox models is also reflected in their weak binding of CO, a strong inhibitor of [FeFe]-H2ase. Until synthetic models show higher affinity for CO, they are unlikely to exhibit high rates for H2 oxidation.297 Remarkably little emphasis has been placed on a rather important design aspect—matching redox potentials of synthetic models to those of [FeFe]-H2ases, which must operate between −266 and −296 mV vs NHE. Most desirable are diiron dithiolates with reversible electrochemistry around −900 mV vs Fc+/0 mimicking the Fe(I)Fe(II/I) Hox/Hred couple. This criterion is satisfied by only a handful of models, including bis(dppv) complexes. While H2ases operate at low H2 pressures, the most sought after synthetic catalysts are those that can handle higher pressures by exhibiting more cathodic redox waves. Such reducing models are yet to be reported, and the cationic nature of most systems contrasts the tetra- and trianionic Hred and Hox states, respectively (assuming adt2− is not doubly N-protonated). Finally, one obvious problem with models is that reduced states almost invariably adopt nonbiomimetic structures, not unlike the age-old [(OC)3Fe(SR)2Fe(CO)3] species. In the enzyme, the [2Fe] subsite adopts a preorganized rotated structure necessary for catalysis. Reproducing this is naturally a difficult task as CO and CN− are free to move and are not functionalizable. But by employing bulky dithiolates and unsymmetrical substitution, the rotated structure can be stabilized in select cases.266,267,299 It will be necessary to target more conformationally rigid models to enforce this geometry and prevent bridging hydride formation.
It is anticipated that efforts in further tuning biomimetic FeFe models will bring us closer to the enzyme activity. This goal may also be approached from an entirely different angle, and much success has been reported in research concerning bioinspired systems, whose structures are considerably different from that of the native catalyst. Among these is a remarkably active monoiron PNP (azadiphosphine) complex,300 which merges the pendant base motif of [24] into an otherwise rather different structure that nevertheless serves as an efficient bidirectional catalyst. The discussion concerning [FeFe]-H2ases and their models ends here, and we now consider the [NiFe]-H2ases in section 4. As will become apparent, despite being biologically unrelated, these two classes of H2ase share similar mechanistic themes that make their study even more instructive.
4. [NiFe]-H2ASES
4.1. Enzyme Structure and Function
The [NiFe]-H2ases are older301 and more abundant and O2-tolerant302 than the [FeFe]-H2ases. The former, more robust family of catalysts is expressed in bacteria, but unlike [FeFe]-H2ases is also found in archaea and cyanobacteria (though not in lower eukaryotes).190 The [NiFe]-H2ases are heterodimeric (63 + 29 kDa),303–305 with the larger subunit housing an active site featuring a redox-active Ni(S-Cys)4 center, of which two −S-Cys ligands bridge to an organometallic, redox-inactive Fe-(CN)2(CO) fragment (Figure 33).
Figure 33.
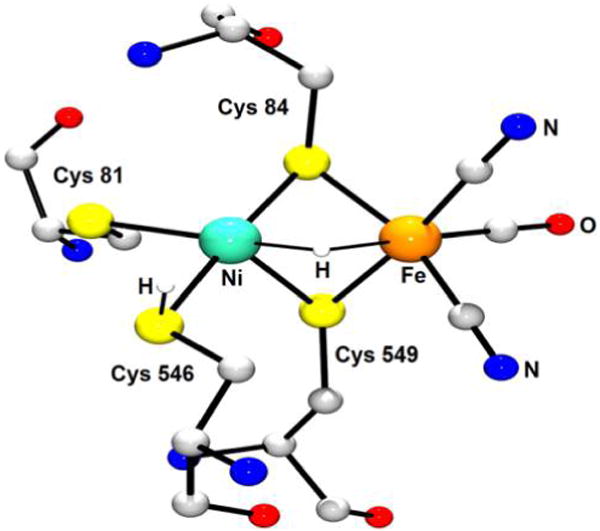
X-ray structure of the [NiFe]-H2ase active site from Desulfovibrio vulgaris Miyazaki F (PDB code 4U9H).89 Nonionizable H atoms are omitted for clarity.
A striking aspect of the active site is the unusual arrangement of S donor atoms around the Ni center. While (Cys546-S)Ni(S-Cys84) defines an almost straight line (176.3°), the (Cys549-S)Ni(S-Cys81) angle is considerably smaller (107.9°) such that Ni adopts a seesaw geometry approaching that of SF4. As for the (Cys-S)2Fe(CN)2(CO) center, it is best described as a distorted square pyramid in which CO occupies the apical site. The structural parameter τ, which ranges from 0 to 1 for the square-pyramidal and trigonal bipyramidal extremes, is in this case equal to (173.7° − 162.7°)/60° = 0.18.306 The metals are separated by a distance (2.57 Å) comparable to the sum of the Ni and Fe covalent radii (1.24 Å + 1.32 Å = 2.56 Å).219 The (Cys-S)2Ni(μ-S-Cys)2Fe(CN)2(CO) core is common to all enzyme states, and while the metrics described here are for the Ni-R form (vide infra), they are very similar to those determined experimentally or computationally for other states. One aspect that does change between redox states is the nature of a third coordination site between the metals. When occupied, this site completes the distorted square-pyramidal and octahedral ligand arrangements about Ni and Fe, respectively. The reduced, active forms of [NiFe]-H2ase have the site either vacant or home to a bridging hydrido ligand. These catalytically relevant states are presented in Figure 34.
Figure 34.
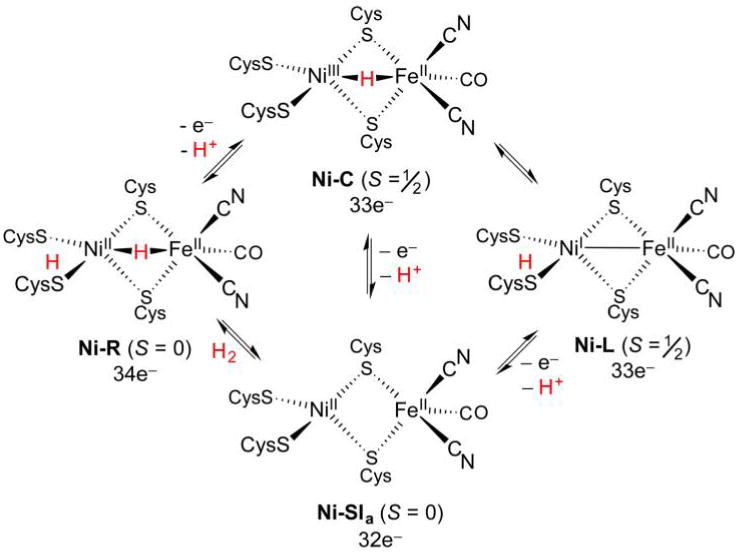
Catalytic cycles proposed for [NiFe]-H2ase. The electronic spin (S) and electron count of the NiFe cores (the sum of 3d electrons and electrons in metal–ligand bonds) are presented below each structure.307 A H2O ligand may be weakly bound to Ni in Ni-SIa.
The name “Ni-R” is given to the most reduced enzyme state. In a biophysical context, the “most reduced” species has the greatest number of electrons and prevails at low redox potentials (“reduced” does not necessarily refer to low metal oxidation states, as might be understood by synthetic chemists). Although (t-H)Ni(II)(μ-H)Fe(II) dihydrides181,308 and Ni(II)Fe(II)(η-H2) dihydrogen-bound formulations have been proposed based on computations,309 the current picture for Ni-R is that of a 34e− Ni(II)(μ-H)Fe(II) core. This is further believed to be low-spin,103 despite some earlier computational predictions to the contrary.310,311 While the identities of the active site heavy atoms have been agreed upon for some time, only recently has X-ray crystallographic data been of sufficient resolution to allow unambiguous location of the μ-H− ligand.89 The hydrido binds Ni (1.58 Å) somewhat tighter than Fe (1.78 Å), and its presence has since been verified by 57Fe NRVS, with the observation of a characteristic δNiHFe bending vibration at 675 cm−1.103,312
IR spectroscopy indicates the existence of three isoelectronic Ni-R subspecies, whose pH-dependent speciation indicates that they differ in their protonation states. More basic than bridging thiolates, the terminal thiolates are the most obvious H+ acceptors. The highest-resolution X-ray diffraction data for Ni-R have been modeled with an active site featuring protonated Cys546 (Figure 33).89 However, as the difference map has electron density on either side of the Cys546 S atom, an argument can be made for S to be unprotonated and undergo anisotropic motion perpendicular to the Ni–S bond. Indeed, aside from the terminal thiolates, an Arg509 residue proximal to Ni represents another viable H+ relay (vide infra).313 In any case, the situation is much clearer when it comes to electron transport, with a chain of 4Fe–4S, 3Fe–4S, and 4Fe–4S clusters clearly defining a tunneling path to and from the active site.
The stepwise oxidation and deprotonation107 (−436 mV vs NHE, pH 7.4)314 of EPR-silent Ni-R affords the 33e− Ni-C state (Ni(III)(μ-H)Fe(II), “catalytic intermediate”), perhaps the most well-characterized of the active forms. This EPR-active (g = 2.21, 2.15, 2.01) low-spin (S = 1/2) species has been the subject of several X-ray structural determinations, although none with sufficient resolution to unambiguously locate the μ-H− ligand. Rather, this moiety has been confirmed by the pulsed EPR methods HYSCORE and ENDOR, with signals for the paramagnetic Ni(III) site featuring anisotropic spin coupling to a 1H nucleus.96,315 The five anionic ligands surrounding the Ni center undoubtedly play a role in taming the otherwise oxidizing +III state, whose polarizing nature results in terminal cysteinato ligands almost certainly being deprotonated,316 in contrast to the situation with Ni-R.
Oxidation and deprotonation (−375 mV vs NHE, pH 7.4)314 of Ni-C gives Ni-SIa (“silent active”), a low-spin hydride-free 32e− Ni(II)Fe(II) state. This electron count does not take into account a weakly Ni-bound H2O ligand,316 upon release of which an electrophilic core is exposed to heterolyze H2. Yet before this transformation is described, the potentially important 33e− Ni-L (Ni(I)Fe(II), “light”) form, intermediary between Ni-C and Ni-SIa, should also be acknowledged. Originally observed by low-temperature irradiation of Ni-C, the isoelectronic Ni-L is a tautomer in which reductive elimination of the proton allows formation of a Ni–Fe bond,179 although with little change in the internuclear separation.85 ENDOR and HYSCORE point to the proton residing on a terminal Cys546,317 with prolonged irradiation leading to H+ departure from the active site.318 Should interconversion between Ni-C and Ni-SIa involve concerted PCET, then Ni-L need not be invoked in mechanisms. Computational studies had proposed involvement of Ni-L in catalysis,36 but this was contraindicated by the apparent need for light irradiation that would not necessarily be available in vivo. However, Ni-L formation has been demonstrated to occur in the dark319 as well as during catalysis, although its presence is fleeting.107,307,320 The divergent observations regarding the stability of Ni-L have been attributed to the redox state of the proximal [4Fe–4S]2+/+ cluster.321 When this cluster is oxidized, Ni-C converts to Ni-SIa, either directly through PCET, or by tautomerization to Ni-L and donation of an e− to the cluster. If instead the cluster is in its reduced state, the intermediate Ni-L may persist. Analogous to these redox effects, pH also has an influence on the Ni-C to Ni-SIa conversion. At low pH, this reaction is slowed, presumably due to the protonation of the residue that must accept a proton from the Ni(III)HFe(II) core of Ni-C.322 While the participation of Ni-L in turnover is still a point of contention, selected data for this state, as well as the three confirmed catalytic states Ni-R, Ni-C, and Ni-SIa, are given in Table 3.
Table 3.
Structural and Spectroscopic Parameters of Active [NiFe]-H2ase States
With the active states described, one can now complete a catalytic cycle by activation of H2 with Ni-SIa. Upon dissociation of any aquo ligand present, Ni-SIa features seesaw Ni(II) and square-pyramidal Fe(II), both of which are electrophilic 16e− metal centers that may conceivably bind and cleave H2. On general grounds, one might consider this to be more likely to occur at Fe(II), a middle transition metal with just enough nuclear charge to impart electrophilicity while still retaining π-basicity for synergistic H2 binding. Yet the consensus is that while both metals may participate, Ni does the bulk of the work, not only because it is optimally positioned near the end of a hydrophobic H2 transport channel,324 but also because its unusual geometry is particularly reactive, as indicated by DFT calculations.310,325 Recent work has suggested that the orientation of Cys ligands has a strong influence on spin densities at Ni and S.326,327 Furthermore, several computational studies on truncated active sites indicate that rotation of terminal Cys modulates the relative stability of low- and high-spin Ni,310,328,329 although the protein environment may also have nontrivial effects on Ni electronic structure.325,330 In the case of a Ni(II)Fe(II) core such as that in Ni-SIa, one would expect square-planar and tetrahedral Ni to give rise to S = 0 and 1 states, respectively. The seesaw Ni(S-Cys)4 coordination present in all states is rigidly poised between these two typical geometries, and while Ni-SIa has no EPR signal, its singlet–triplet gap is not large.329
The conversion of Ni-SIa to Ni-R involves the reactive Ni(II)Fe(II) core being “tamed” upon receiving the μ-H− ligand, with the fate of the other heterolysis product, a proton, still unclear. A substantial body of computational work331 as well as the high-resolution Ni-R X-ray structure89 implicates Cys546, although questions have arisen as to whether the terminal thiolate is sufficiently basic24 to deprotonate the as-yet-unobserved Ni(II)(η2-H2)Fe(II) intermediate. In an alternative mechanism, a more basic Arg509 serves as the H+ acceptor, much as the azadithiolate cofactor does for [FeFe]-H2ase (Figure 35). Notably, Arg509 is highly conserved, and its basic guanidine group (pKa ≈ 13.8)332 is only 4.5 Å away from Ni.313
Figure 35.
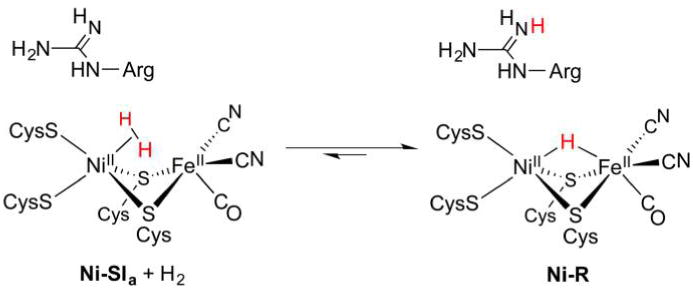
Alternative mechanism for H2 activation by [NiFe]-H2ase, in which a proximal Arg residue (rather than a terminal Cys ligand) relays H+ to and from the active site.313
While there is debate as to exactly how [NiFe]-H2ases process H2, there is little doubt that they do it incredibly quickly. A graphite-supported film of [NiFe]-H2ase isolated from Allochromatium vinosum exhibited diffusion-limited TOF values for H2 oxidation (>6000 s−1, pH 7, 45 °C, 0.2 V vs SHE). The corresponding current density was similar to that of a Pt electrode, although for the latter case this could be achieved at lower potentials of −0.4 V.333 Given the large footprint of [NiFe]-H2ases, their activity must be superior to that of the individual Pt sites, although the accessibility of the active site to the substrate is cited as a source of overpotential.334 Alternatively, the H2 oxidation can be driven using benzylviologen as a sacrificial electron acceptor, as demonstrated with [NiFe]-H2ase from Desulfovibrio gigas (2200 s−1, 30 °C, −358 mV).335 Many examples of [NiFe]-H2ase are bidirectional, and the same Desulfovibrio gigas enzyme also catalyzes H+ reduction (640 s−1, 30 °C, methylviologen at −446 mV).335
The activities of [NiFe]-H2ases can vary greatly. These differences may arise from several factors, an important one being the strength with which the protein binds H2. Stronger H2 binding is exhibited by enzymes expressed for H2 uptake, such as the Ralstonia [NiFe]-H2ases. Their H2 production rates are modest (70 s−1, pH 5.5, 40 °C, −0.45 V vs SHE), yet they can operate in the presence of O2. Indeed, the strong binding of H2 makes it an even more potent inhibitor than O2 and CO.336 Notably, O2 and CO irreversibly deactivate [FeFe]-H2ase and Pt catalysts, respectively.
Inhibition of the HER by H2 can be quantified by considering the Michaelis constant KM,337,338 which is related to how much the activity is attenuated by a given H2 concentration.339 In the case of [NiFe]-H2ases, KM values are typically ~10−7−10−4 M, whereas those for [FeFe]-H2ase are greater and fall in the range ~10−4−10−3 M.339 Thus, while trace H2 can inhibit the HER in many [NiFe]-H2ases, the [FeFe]-H2ases bind H2 more weakly and are less prone to product inhibition. Such measurements are consistent with the typical biasing of [FeFe]-H2ases toward H2 formation, with [NiFe]-H2ases usually being expressed to oxidize H2. Interestingly, under high pressures of H2, certain [FeFe]-H2ases can still mediate H2 oxidation faster than [NiFe]-H2ases.
As has been described above, hydrides are central to the catalytic activity of [NiFe]-H2ases, the only H2ases for which hydride-bearing species have been confirmed. Further comparison of the H2ases points to the contrasting bridging hydride cores of Ni-R and Ni-C with respect to the terminal hydride proposed for [FeFe]-H2ase. Lacking azadithiolate, the [NiFe]-H2ases shuttle protons around entirely with amino acid residues (and a H-bonded H2O network) and feature a dinuclear core more strongly anchored to the protein scaffold. Relative to the diiron enzymes, one might say that the [NiFe]-H2ases make up for in robustness what they lose in activity. The heteronuclear active sites represent attractive targets for synthetic modeling studies, which, through reproducing [NiFe]-H2ase structure and spectroscopy (Table 3), aim to reproduce enzyme function. Several reports have described NiFe complexes strongly resembling the [NiFe]-H2ase active site. These efforts are summarized in section 4.2, with particular emphasis placed on complexes bearing hydride ligands.
4.2. [NiFe]-H2ase Synthetic Modeling
4.2.1. Hydrides from H+
Synthetic modeling of the [NiFe]-H2ases, relative to that of the [FeFe]-H2ases, is a much younger area of study.340,341 The low-spin Fe(CN)2(CO) fragment in the former is reminiscent of the distal Fe(CN)(CO)2 site in the latter, and has similarly caught the eye of synthetic chemists. Certain difficulties associated with preparing heterobimetallics led early [NiFe]-H2ase modeling to be directed toward mononuclear Ni or Fe models of their respective subsites. With respect to metal hydrides, particular attention was paid to Fe–CN–CO systems, including [Fe(CN)2(CO)3]2−. This dianion undergoes protonation, not at the N atoms, but rather at Fe(0) to give three isomers of the octahedral Fe(II) product [HFe(CN)2(CO)3]−, one of which is shown in Figure 36.342 With four of the six ligands in the Ni-R state of the enzyme, this hydride remains an attractive synthetic module, and its potential aqueous solubility may lend itself to reconstitution studies akin to those of [FeFe]-H2ases.
Figure 36.

Initial work on hydride-containing [NiFe]-H2ase models focused on the Fe subsite, whose Fe(CO)(CN)2H fragment is replicated by [25]−.342
Two CO ligands in [HFe(CN)2(CO)3]− are liberated during its conversion to [Fe(dppv)(CO)(CN)2H]− ([25]−), a product that replicates the enzyme structure well, with the diphosphine dppv representing a prosthesis for the bridging Cys thiolates. IR spectroscopy (νCO = 1936 cm−1; νCN = 2087, 2080 cm−1) indicates the monocarbonyl product to be electronically similar to the enzyme subsite, although the νCN values are lower than those for Ni-R, whose CN− ligands participate in H-bonding.
Hydrides containing both Fe and Ni are rare, and at one point examples of these were limited to clusters such as the μ3-H−-containing [NiFe3H(CO)12]− metallotetrahedron343 or the μ-H−-bridged [NiFe5HN(CO)14]2− and [Ni2Fe4HN(CO)14]2− octahedra bearing interstitial N3−.344 Of much greater relevance to the [NiFe]-H2ase active site was the report of the first NiFe thiolato hydride, [(dppe)Ni(pdt)(μ-H)Fe(CO)3]+ ([26(μ-H)]+, Figure 37).345
Figure 37.
Synthesis of a Ni(I)Fe(I) thiolate [26],63 which can accept H+ either from acid or from H2 heterolysis to afford the hydride derivative [26(μ-H)]+.345
Yet before discussing hydride [26(μ-H)]+ and its derivatives, one must consider the low-valent conjugate bases from which they are prepared. The Ni(I)Fe(I) species [(dppe)Ni(pdt)Fe-(CO)3] ([26]),346 diamagnetic on account of its metal–metal bond (2.467 Å),347 is a 34e− complex similar to the homobimetallic [dppe(OC)Fe(pdt)Fe(CO)3]. Comproportionation of the Ni(II) complex [(dppe)Ni(pdt)] with an Fe(0) source such as [Fe(benzylideneacetone)(CO)3] or [Fe2(CO)9] affords [26], which was initially purported to be unstable,346 but is in fact rather robust. A more generalizable synthesis for [26] was later developed in which [(diphosphine)Ni(dithiolate)] and cis-[FeI2(CO)4] are combined, with 2e− reduction of the resulting iodide [(diphosphine)Ni(dithiolate)(μ-I)Fe(CO)3]+ affording the neutral species [(diphosphine)Ni(dithiolate)Fe-(CO)3].348 Such complexes reproduce the Ni(SR)2Fe “butterfly” and apical CO ligand at the [NiFe]-H2ase active site, although differences in the coordination spheres of the synthetic and native systems are not insignificant. Much like the modeling of [FeFe]-H2ases, in which phosphines are extensively used as prostheses for CN− groups and the Cys-S–[4Fe–4S] metalloligand, the diphosphine in [(diphosphine)Ni(dithiolate)Fe-(CO)3] is a substitute for terminal Cys-S− ligands. Another significant difference can be found in the set of diatomic ligands, with the three CO ligands in these models contrasting Nature’s choice of one CO and two CN− groups. The stereoelectronic consequences of these changes will be discussed in the context of the first generation of synthetic NiFe hydrides.
The finding that the archetypal Ni(I)Fe(I) complex [26] sustains protonation to afford a well-defined hydride product represented a significant advance in the field of [NiFe]-H2ase modeling. This protonation reaction was found to proceed via a 2e− mixed-valent conformer, with dppe rotation resulting in a square-planar environment around Ni (see section 4.2.3).160 Oxidized (divalent) Ni is now strongly stabilized, with concomitant cleavage of the NiFe bond resulting in Fe being in a reduced state. Overall, this redox isomerization closely parallels the “rotation” of Fe(I)Fe(I) to afford a reactive Fe(II)Fe(0) species. In the present case, DFT calculations indicate that the latent Ni(II)Fe(0) isomer is ~108 times more basic than the Ni(I)Fe(I) form and is geometrically preorganized to form hydride [26(μ-H)]+.160 With its Ni(II)(μ-H)Fe(II) core, the robust, air-stable product [26(μ-H)]+ represented the first model for the Ni-R enzyme state. However, Ni-R does not form by protonation of a low-valent species—indeed no [NiFe]-H2ase states feature a Ni(I)Fe(I) or Ni(II)Fe(0) core. The 26 to [26(μ-H)]+ reaction is rather a low-valent analogue of the Ni-L to Ni-C conversion, in which oxidative addition of a H+ transforms Ni(I)Fe(II) to Ni(III)(μ-H)Fe(II). Yet [26(μ-H)]+ is nevertheless an important model with instructive characterization. Curiously, the H− ligand gives rise to a high-field 1H NMR resonance (−3.53 ppm) that appears in a chemical shift region typical of FeFe(t-H) terminal hydrides rather than Fe(μ-H)Fe species. The ligand was found by X-ray crystallography (Figure 38) to be somewhat closer to Fe (1.460 Å) than to Ni (1.637 Å).345 Relative to [26], the metals in [26(μ-H)]+ are further apart (2.613 Å) such that they are out of bonding distance, although a Ni⋯Fe interaction is sometimes drawn. The H− ligand has a rigidifying influence on the structure, resulting in a single sharp 31P NMR resonance for the dppe ligand and two 13C resonances for the CO ligands.
Figure 38.
X-ray structure of [26(μ-H)]+. Non-hydride H atoms are omitted for clarity.345
The structure of [26(μ-H)]+ is supported by vibrational spectroscopy in the form of Raman349 and 57Fe NRVS measurements.103 The latter method, applied to 57Fe-labeled isotopologue [(dppe)Ni(pdt)(μ-H)57Fe(CO)3]+ ([26′(μ-H)]+), indicated that H− does indeed bond more strongly to Fe (νHFe = 1479 cm−1) than to Ni (νNiH = 1022 cm−1). The vibrational frequencies of the corresponding deuteride agree with the νMH ≈ 21/2νMD approximation for M–H/D harmonic oscillators in which M is much heavier than H/D. Relative to stretches, bending modes associated with μ-H− ligands are much lower in energy, with the frequency of the δNiHFe bending vibration for [(dppe)Ni(pdt)(μ-H)57Fe(CO)3]+ (758 cm−1) approaching that for the enzyme (675 cm−1). The asymmetric hydride binding in [26(μ-H)]+ has also been probed by valence-to-core X-ray emission spectroscopy.350 In this case, the Fe spectra exhibit strong Fe–H signatures, while the Ni spectrum shows no evidence for its interaction with a light bridging atom. The method further allows estimation of M–H bond lengths, and although these come with moderately high uncertainties (~0.15 Å), such information is a useful complement to other analyses.
The tightness with which hydride binds Fe is rationalized in terms of the high electrophilicity of the Fe(CO)3 center in [26(μ-H)]+, whose weighted average νCO frequency (2043 cm−1) is considerably higher than that of the Fe(CO)(CN)2 fragment in Ni-R (1944 cm−1). The electronic influences described here result in [26(μ-H)]+ being somewhat acidic (pKa = 10.7, Table 4), with the μ-H group being rather protic (e.g., it readily exchanges with D2O) and insensitive to excess acid. The latter fact simplifies the synthesis of this and related NiFe hydrides, and is in contrast to the preparation of FeFe hydrides, which often involve strictly stoichiometric quantities of acid to avoid H2 formation.
Table 4.
Spectroscopic and Structural Parameters for NiFe Thiolato Hydrides
| hydride complexa | δ(1H)/ppm | νCO/cm−1 | pKad | rNi–Fe/Å | rNi–H/Å | rFe–H/Å | |
|---|---|---|---|---|---|---|---|
| [(dppe)Ni(pdt)(μ-H)Fe(CO)3]+ | [26(μ-H)]+ | −3.53 | 2082, 2024 | 10.7 | 2.613 | 1.637 | 1.460 |
| [(dcpe)Ni(pdt)(μ-H)Fe(CO)3]+ | [27(μ-H)]+ | −3.00 | 2078, 2017 | 13.6 | 2.684 | 1.905 | 1.535 |
| [(dppe)Ni(edt)(μ-H)Fe(CO)3]+ | [28(μ-H)]+ | −5.7 | 2084, 2025 | 11.3 | 2.596 | 1.843 | 1.578 |
| [(dppe)Ni(pdt)(μ-H)Fe(CO)2PPh3]+ | [29(μ-H)]+ | −3.08b | 2016, 1964 | 14.9 | 2.643 | 1.890b | 1.487b |
| [(dppe)Ni(pdt)(μ-H)Fe(CO)2PPh2(2-py)]+ | [30(μ-H)]+ | −3.19b | 2022, 1971 | ||||
| [(dppe)Ni(pdt)(μ-H)Fe(CO)2P(OPh)3]+ | [31(μ-H)]+ | −3.45b | 2031, 1981 | ||||
| [(dcpe)Ni(edt)(μ-H)Fe(CO)3]+ | [32(μ-H)]+ | −5.3 | 2080, 2019 | 13.6 | |||
| [(dppe)Ni(edt)(μ-H)Fe(CO)2PPh3]+ | [33(μ-H)]+ | −8.15,b −5.13c | 2068, 1912 | 14.0 | |||
| [(dppe)Ni(pdt)(μ-H)Fe(dppe)CO]+ | [34(μ-H)]+ | −6.34,b −3.01c | 1954, 1938 | 2.656c | 1.847c | 1.555c | |
| [(dppe)Ni(pdt)(μ-H)Fe(dppbz)CO]+ | [35(μ-H)]+ | −6.01,b −3.14c | 1949, 1935 | 2.671a | 1.788a | 1.798a | |
| [(dppv)Ni(pdt)(μ-H)Fe(dppv)CO]+ | [36(μ-H)]+ | −5.79b | 1962, 1952 | 16.6 | 2.646b | 1.79b | 1.56b |
Isolated as BF4− salts.
Asymmetric isomer.
Symmetric isomer.
pKa is in PhCN.
Several derivatives of [26(μ-H)]+ have been prepared in order to bring the structure and spectroscopy of models more in line with Ni-R. One component in such compounds that is not often varied is the dithiolate, with pdt2− and edt2− giving rise to the most stable species owing to their high basicity and favorable chelate ring sizes. Indeed, while FeFe complexes of many mono-and di-, alkyl-, and arylthiolates are known, the NiFe systems are less modular. Although the variety of diphosphines at Ni is not large (dppe, dcpe, and dppv have been reported), the Fe(CO)3 center allows more substitutional scope. The hydrides [(diphosphine)Ni(pdt)(μ-H)Fe(CO)3]+ undergo reaction with mono-63 and diphosphines351 to afford [(diphosphine)Ni-(pdt)(μ-H)Fe(PR3)(CO)2]+ and [(diphosphine)Ni(pdt)(μ-H)Fe(diphosphine)(CO)]+ complexes, respectively (Figure 39). One limitation of this method is its potential incompatibility with highly basic phosphines such as PCy3, which deprotonate [26(μ-H)]+ rather than participate in ligand substitution at Fe.
Figure 39.
Synthesis of phosphine-substituted Ni(pdt)(μ-H)Fe derivatives is best performed from hydrido tricarbonyl [26(μ-H)]+.63
Complexes of the type [(diphosphine)Ni(pdt)(μ-H)Fe(PR3)-(CO)2]+ feature basal PR3 ligands and are thus of lower symmetry than their Cs-symmetric tricarbonyl parents. When edt2− is present, appreciable quantities of Cs-symmetric apical PR3 isomers coexist with the C1-symmetric species. In either case, substitution of one CO ligand for PPh3 causes negligible change in ΔG°(H−), although the weighted average νCO decreases by ~50 cm−1 and the pKa increases by 3–4 units.63 Even greater effects are observed when diphosphines are employed, and the first such complex [(dppe)Ni(pdt)(μ-H)Fe(dppe)(CO)]+ ([34(μ-H)]+) was initially prepared by decarbonylation of [(dppe)Ni(pdt)Fe(dppe)(CO)2]2+ with BH4−. A cleaner and more general route involves photolysis of a solution containing tricarbonyl hydride [(diphosphine)Ni(pdt)(μ-H)Fe(CO)3]+ and diphosphine. The complexes [(diphosphine)Ni(pdt)(μ-H)Fe(diphosphine)(CO)]+ typically exist as two isomers: one with the Fe-ligated diphosphine being apical–basal and another in which it is dibasal. The structure of the latter isomer corresponds well to that of Ni-R, whose two basal CN− ligands are mimicked by the diphosphine in the present species. Such bis(diphosphine) species also approach Ni-R in terms of their electron density at Fe, with the two isomers of [26(μ-H)]+ exhibiting low νCO frequencies (1954 and 1938 cm−1). The effect of substitution on the weighted average νCO is approximately additive, with these values being ~100 cm−1 lower than that for the tricarbonyl. As expected, changes in Fe ligation have large effects on νCO and pKa, and the latter is consistent with the μ-H− being closely associated with the Fe site. Changes at Ni (e.g., substitution of dppe with dcpe) are less influential on these parameters but do affect redox (section 4.2.3).
The present complexes feature Ni(μ-H)Fe cores whose geometries certainly vary with the identity of the coligand(s). While the bond length difference rNi–H − rFe–H is 0.177 Å in the case of [26(μ-H)]+, this doubles to 0.370 Å for the Ni(dcpe) analogue [27(μ-H)]+.348 This latter complex features the most electron-rich Ni site and the most electron-poor Fe site, and one might argue that an increase in donicity of the Ni ligand set means that it need not bind H− so tightly. However, a conclusive trend in H− binding is difficult to tease out as ligand substitution often has steric consequences. Indeed, interactions between the phosphine substituents in the PPh3 complex [(dppe)Ni(pdt)(μ-H)Fe(PPh3)(CO)2]+ ([29(μ-H)]+)63 lead to a bond length difference of 0.403 Å that is greater than that in [26(μ-H)]+, a counterintuitive result if just considering electronic effects. Such steric interactions are also at play in the bis(diphosphine) complexes, and while there is structural variation, it is significant that the rNi–H − rFe–H value for [(dppe)Ni(pdt)(μ-H)Fe-(dppbz)(CO)]+ ([35 (μ-H)]+, dppbz = 1,2-bis-(diphenylphosphino)benzene)351 of −0.010 Å indicates that increasing electron density at Fe can shift H− toward Ni such that the structures approach that of [NiFe]-H2ase.
4.2.2. Related Dithiolato Hydrides
As might be expected, the properties of heterobimetallic dithiolato hydride complexes are sensitive to not only variations in the ligand sets, but also changes in the metals used. The ease of preparation and high stability of the Ni(pdt)(μ-H)Fe models above have inspired the synthesis of congeners in which Ni or Fe is replaced by other metals, typically heavier elements from their respective groups. While such work departs somewhat from replicating the [NiFe]-H2ase active site, it may provide information about the factors governing reactivity of NiFe systems. It is also possible that these “less biomimetic” systems perform even better catalytically. The ligand set chosen for Fe (or its replacement) often varies, with that of the group 10 metal typically bearing two neutral donors in addition to two thiolates. For example, square-planar building blocks of the form [ML2(SR)2] feature prominently, and although the following examples have phosphines as the neutral ligand (e.g., L2 = dppe), the formation of bimetallics can in principle occur with a range of building blocks.
The most closely related complexes to [26(μ-H)]+ are its analogues [(dppe)Pd(pdt)(μ-H)Fe(CO)3]+ ([37(μ-H)]+) and [(dppe)Pt(pdt)(μ-H)Fe(CO)3]+ ([38(μ-H)]+). These are spectroscopically almost identical to their NiFe relative [26(μ-H)]+ and are also isostructurual, including the asymmetry of the μ-H− bridge toward Fe. Additionally, each is prepared in high yield by treatment of a [(dppe)M(pdt)Fe(CO)3] precursor with HBF4·Et2O. A distinguishing feature of the PtFe example is that the low-valent species [(dppe)Pt(pdt)Fe(CO)3] ([38]) is a bona fide M(II)Fe(0) complex. No Pt–Fe bond exists in this species, which is “frozen” in a reactive conformation rather than in a tetrahedral arrangement observed for the NiFe system.
More often than is the case with the Ni site, the Fe site can also be altered to feature other elements. The most common substitute is Ru, which has rich organometallic chemistry and forms strong, stable bonds to H−, H2, and C-donor ligands, as evidenced by the formation of [Cy3P(CO)2Ru(pdt)(μ-H)Ru(t-H)(CO)PCy3] from H2 (section 3.2.3).249 Indeed, the synthesis of NiRu complexes [(dxpe)Ni(pdt)Ru(cymene)] ([39], dxpe = dppe; [40], dxpe = dcpe; Figure 40)352 suggests that 14e− FeL3 and Ru(cymene) fragments are to some extent interchangeable. The tetrahedral Ni coordination might suggest a Ni(I)Ru(I) description for these complexes, but the geometry is also consistent with a Ni(0)Ru(II) assignment, to which DFT calculations point. Combination of either [39] or [40] with [H(OEt2)2]BArF4 affords the corresponding hydrides [39(μ-H)]+ and [40(μ-H)]+. The zerovalent Ni center is thought to be the initial site of protonation, although the X-ray structure of [39(μ-H)]+ reveals a familiar motif in which the H− ends up closer to Ru (1.54 Å) than to Ni (1.65 Å).
Figure 40.
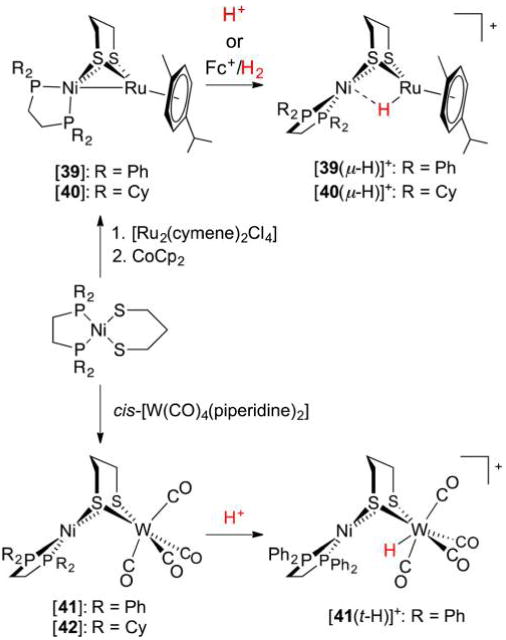
Synthesis of NiRu352 and NiW dithiolates353 and their respective hydrides.
There is no particular reason why the metal to replace Fe must be chosen from group 8. However, it is thought that 14e− fragments are most suitable, and indeed Mo(CO)4 and W(CO)4 fragments readily bind the Ni chelates [Ni(R2PCH2CH2S)2] and [(R2PCH2CH2PR2)Ni(pdt)] to form Ni(II)M(0) bimetallics.354 While the products do not feature metal–metal bonds and M is octahedrally bound, the propensity of group 6 metals to form 7-coordinate complexes can be exploited to prepare the Ni(II)(μ-H)W(II) species ([41(t-H)]+).353 The high acidity of this hydride (which is deprotonated by excess Et2O) and low catalytic activity result from the electron-poor nature of the W(II) center (average νCO = 2006 cm−1), and perhaps also from its coordination geometry not allowing H− to interact with Ni.
An ever-increasing number of hydrides have been prepared that reproduce many aspects of the [NiFe]-H2ase active site. In terms of structure and spectroscopy, some are certainly closer than others to Nature’s blueprint, while some do not even include Ni and/or Fe. The key question is, how do the differences between the enzyme and its models affect catalysis? This question is addressed in section 4.2.3 with respect to some complexes already described here, as well as some further examples.
4.2.3. Electrochemistry and HER Catalysis
With active sites that exhibit both acid–base and redox behavior, the [NiFe]-H2ases are suitably equipped to mediate proton reduction (HER). The acid–base and redox criteria are met by several synthetic [NiFe]-H2ase models, and their electrochemistry, studied using methods described in section 3.2.7, is now described. Considered first are the family of complexes [(diphosphine)Ni(dithiolate)(μ-H)Fe(CO)3–nLn]+, not only because they were the first HER-active models to be reported, but also because variation in the ligand sets allows development of structure–function relationships. All but the most electron-rich of these Ni(μ-H)Fe species are not oxidizable. Surprisingly, the oxidation of the bis(dppe) complex [34(μ-H)]+ occurs at a mild potential (0.15 V vs Fc+/0), although with low reversibility. When treated with a chemical oxidant (acetylferrocenium) at −25 °C, the νCO bands are shifted by large amounts (68 cm−1), indicative of a Ni(II)(μ-H)Fe(III) product whose oxidation states are reversed relative to the Ni(III)(μ-H)Fe(II) description for Ni-C. In contrast to the oxidations, the reductions of Ni(μ-H)Fe are more well-defined and more relevant to the HER mechanisms of these synthetic catalysts. Some key electrochemical measurements are presented in Table 5.
Table 5.
Redox and Catalytic Properties of NiFe Dithiolato Hydrides
| hydride complexa | E(NiHFe+/0)b | Ecat/2b,c | acid used | TOF/s−1 | η/V | |
|---|---|---|---|---|---|---|
| [(dppe)Ni(pdt)(μ-H)Fe(CO)3]+ | [26(μ-H)]+ | −1.34 | −1.20 | CF3CO2H | 20 | 0.50 |
| [(dcpe)Ni(pdt)(μ-H)Fe(CO)3]+ | [27(μ-H)]+ | −1.56 | −1.46 | ClCH2CO2H | 50 | 0.59 |
| [(dppe)Ni(edt)(μ-H)Fe(CO)3]+ | [28(μ-H)]+ | −1.33 | −1.23 | CF3CO2H | 240–310 | 0.49 |
| [(dppe)Ni(pdt)(μ-H)Fe(CO)2PPh3]+ | [29(μ-H)]+ | −1.49 | −1.30 | CF3CO2H | 50 | 0.60 |
| [(dppe)Ni(pdt)(μ-H)Fe(CO)2PPh2(2-py)]+d | [30(μ-H)]+ | −1.49 | −1.30 | CF3CO2H | 50 | 0.60 |
| [(dppe)Ni(pdt)(μ-H)Fe(CO)2P(OPh)3]+ | [31(μ-H)]+ | −1.44 | −1.32 | CF3CO2H | 50 | 0.62 |
| [(dcpe)Ni(edt)(μ-H)Fe(CO)3]+ | [32(μ-H)]+ | −1.47 | −1.45 | ClCH2CO2H | 20 | 0.59 |
| [(dppe)Ni(edt)(μ-H)Fe(CO)2PPh3]+ | [33(μ-H)]+ | −1.47 | −1.45 | ClCH2CO2H | 60–120 | 0.54 |
| [(dppe)Ni(pdt)(μ-H)Fe(dppe)CO]+ | [34(μ-H)]+ | N/D | −1.61, −1.65 | ClCH2CO2H | 23 | 0.85 |
| [(dppe)Ni(pdt)(μ-H)Fe(dppbz)CO]+ | [35(μ-H)]+ | N/D | −1.61, −1.70 | ClCH2CO2H | 30 | 0.85 |
| [(dppv)Ni(pdt)(μ-H)Fe(dppv)CO]+ | [36(μ-H)]+ | N/D | −1.54 | ClCH2CO2H | 29 | 0.75 |
Isolated as BF4− salts.
In volts, determined in CH2Cl2, relative to Fc+/0.
Potential at icat/2.
2-py =2-pyridyl.
Potentials for the [(diphosphine)Ni(dithiolate)(μ-H)-FeL3]+/0 couple are strongly related to the nature and number of phosphine donors at both the Ni and Fe sites, being most negative when strongly donating ligand sets are involved. As is the case with the reduction of Fe(II)(μ-H)Fe(II) species, the initial 1e− reduction product of Ni(II)(μ-H)Fe(II) may take multiple forms, viz., Ni(I)(μ-H)Fe(II), Ni(II)(μ-H)Fe(I), and Ni(1.5)(μ-H)Fe(1.5). The reversibility of this couple varies greatly and correlates with the stability of the radical hydride complex, the formation of which is a key step in catalysis (vide infra). The reduction is irreversible in the case of tri- and dicarbonyls with the exception of [31(μ-H)]+/0, where the [31(μ-H)]0 radical is thought to be stabilized by spin delocalization onto the P(OPh)3 ligand in a manner similar to that observed for [31]+.180 When two diphosphines are incorporated into Ni(II)(μ-H)Fe(II) species, the reversibility of the 1e− reductions approaches unity (e.g., for [36(μ-H)]+, Figure 41, |ipa/ipc| = 0.94 at 0.1 V s−1, room temperature). Electron transfer can also be induced chemically, and treatment of [(dppv)Ni(pdt)(μ-H)Fe(dppv)CO]+ with CoCp*2 allows isolation of the stable charge-neutral hydride. Reduction causes only a modest red shift, with ΔνCO = −37 cm−1, consistent with Ni-centered redox and thus a Ni(I)(μ-H)Fe(II) description. This is further corroborated by EPR and DFT studies, the latter indicating that significant spin density resides on Ni.351
Figure 41.
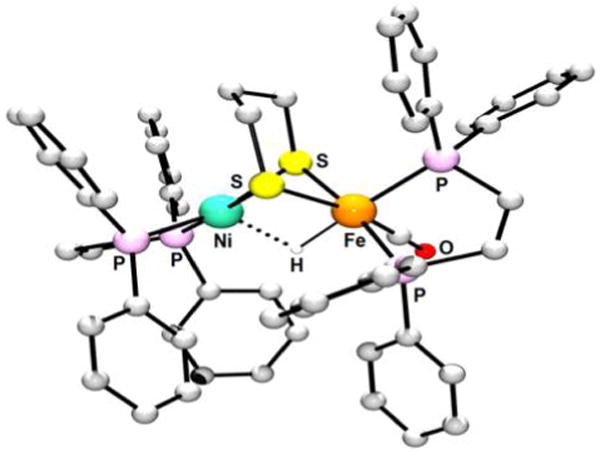
X-ray structure of [36(μ-H)]+, one of the most active synthetic NiFe HER catalysts. Non-hydride H atoms are omitted for clarity.351
The electron-rich di- and monocarbonyl catalysts operate at high rates, although their Ecat/2 values are, unsurprisingly, rather negative. Such electron-rich catalysts give rise to more negative Ecat/2 values, yet can still have reasonably small overpotentials if they can mediate H2 evolution from weaker acids. As stated in section 2.3.1, one should find the weakest acid that will protonate the reduced form of the catalyst, which is the Ni(I)Fe(I) species in the present case. As is clear from the data in Table 5, the more cathodic Ecat/2 values have yet to be offset by employing weaker acids for the NiFe systems. Overpotentials for the current catalysts remain >0.5 V, and reducing this key metric while maintaining high TOFs is a fundamental challenge in HER catalysis.
The mechanism by which [(diphosphine)Ni(dithiolate)(μ-H)FeL3]+ catalysts operate involves sequential e− and H+ transfers. Hydrides of the form Ni(II)(μ-H)Fe(II) (Figure 42, top right) are typically unaffected by acid, and the catalytic cycle thus involves their 1e− reduction to a mixed-valent complex. Examples of these reduced species have been observed to react instantaneously with acid,351 perhaps with S atoms serving to relay protons to the metal sites. Following protonation, loss of H2 affords mixed-valent species Ni(II)Fe(I) complexes (for tri- and dicarbonyls) or Ni(I)Fe(II) complexes (for monocarbonyls), the latter case matching the assignment for the Ni-L state of [NiFe]-H2ase. Reduction of the S = 1/2 intermediates gives the neutral, hydride-free species. The reduced hydride-free species can exist in a reactive Ni(II)Fe(0) form in which Ni is planar, as well as a Ni(I)Fe(I) resting state with tetrahedral Ni.160 The tetrahedral isomer can readily convert to the reactive form (with a DFT-calculated free energy barrier of ΔG‡ ≈ 6.7 kcal mol−1 and an experimentally estimated barrier of 9.5 kcal mol−1 for [26]160), whose high basicity allows for protonation and completion of the catalytic cycle.
Figure 42.
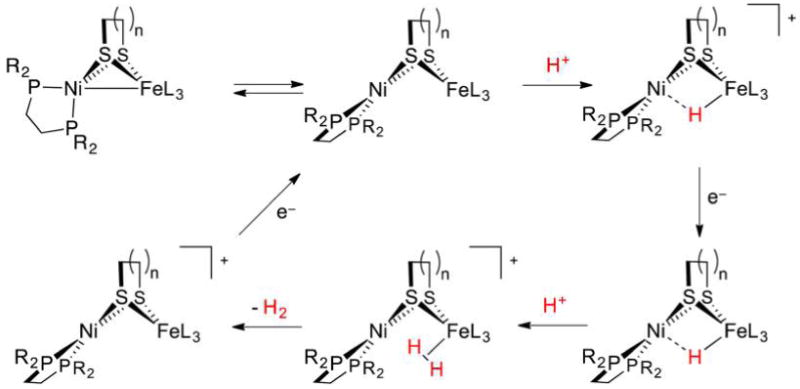
HER catalytic cycle proposed for the present NiFe dthiolato hydrides.160
Given that HER catalysts featuring a mono- or diphosphine at Fe operate at the highest rates, it is likely that the basicity conferred by these ligands is important for catalysis. Indeed, one or both of the H+ transfers may be rate-determining, and the lack of an effective H+ relay apart from the bridging pdt2− may represent a shortcoming of these models, despite the negligible effect replacing PPh3 with P(2-pyridyl)Ph2 has on the TOF. This relay aspect has been investigated with the unusual Ni(I)Fe(I) complex Ni(xbsms)Fe(CO)3 ([43], Figure 43), in which only one of the thiolates is bridging.355 The xbsms2− ligand and its relatives are popular ligands in [NiFe]-H2ase synthetic modeling, with flexible polydentate donors of this type often being referred to as podands. Treatment with HBF4·Et2O affords conjugate acid [43H]+, whose average νCO is only modestly (43 cm−1) shifted relative to [43] (cf. 66 cm−1 for [26] → [26(μ-H)]+). Crystallographic analysis confirms the product as a Ni(I)Fe(I) thiol, although DFT calculations suggest that the Ni(II)(μ-H)Fe(II) hydride tautomer is only slightly higher in free energy (~5 kcal mol−1). Cyclic voltammetry studies indicate [43] mediates electrocatalytic reduction of CF3CO2H (Ecat/2 = −1.43 V vs Fc+/0, η = 540 mV), although bulk electrolysis suggests that the TOF is low in this case (0.0014 s−1 at −1.6 V). The poor basicity of [43], which is not completely protonated by CF3CO2H, may well be responsible for this sluggishness. The use of stronger acids and/or substitution of CO ligands for more basic donors is likely to increase the activity of the present system.
Figure 43.
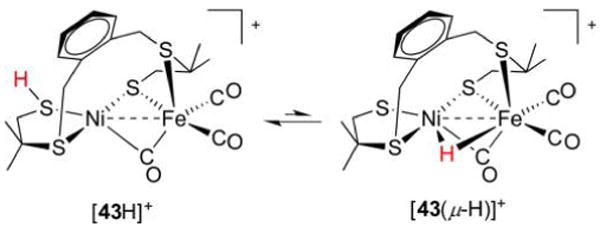
Interconversion between thiol and hydride tautomers of [43H]+.355 There is evidence for both thiol and hydride moieties in the structure for Ni-R.
Given the scope of this review, the focus here has largely been on bimetallic HER catalysts that are isolable in one or more hydride-bearing forms. However, these represent a subset of catalytically active [NiFe]-H2ase models, and many others have not been characterized as their hydride derivatives, although hydrides are assumed as HER intermediates.356,357 Consider the Ni(I)Fe(II) radical [CpNi(pdt)Fe(dppe)CO] ([44], νCO = 1901 cm−1),159 a very electron-rich model for the Ni-L state. The conjugate acid [CpNi(pdt)(μ-H)Fe(dppe)CO]+ ([44(μ-H)]+, Figure 44), also a radical, is predicted by DFT studies to have spin density localized mainly on the Ni coordination sphere, with almost twice as much density at Ni than Fe. Such a finding is significant, as the Ni(III)(μ-H)Fe(II) core implicated would match that of Ni-C. The hydride [44(μ-H)]+ has so far eluded characterization, as while it is generated by addition of acid (1 equiv) to [44], the hydride is instantly reduced to [44(μ-H)] by surrounding [44]. After reaction of [44(μ-H)] with remaining acid, the overall stoichiometry is [44] + H+ → [44]+ + 1/2H2, a reaction that also proceeds electrocatalytically using CF3CO2H as the H+ source (Ecat/2 = −1.16 V, TOF = 4 s−1). The DFT calculations suggest that the H+ reduction mechanism can proceed through one of two pathways, depending on whether the dppe ligand is dibasal or apical–basal. Overall, this example demonstrates well that the challenges in [NiFe]-H2ase modeling cannot solely be met by preparing more electron-rich NiFe dithiolates. The DFT structure calculated for [44(μ-H)]+ features asymmetrically bound μ-H−, with the Ni–H bond (1.80 Å) being longer than the Fe–H bond (1.60 Å).159 Despite the presence of the Cp− ligand, the Ni(III) site remains a strong oxidant, as it does not bind H− as tightly as Fe. The oxidizing power and reducing power of [44(μ-H)]+ and [44], respectively, illustrate the large effects protonation can have on redox (and vice versa). Such NiFe models rely on the geometric persistence of the NiCp subunit, and the square planar ⇌ tetrahedral isomerization accompanying the reduction of 4-coordinate NiII does not come into play here.
Figure 44.
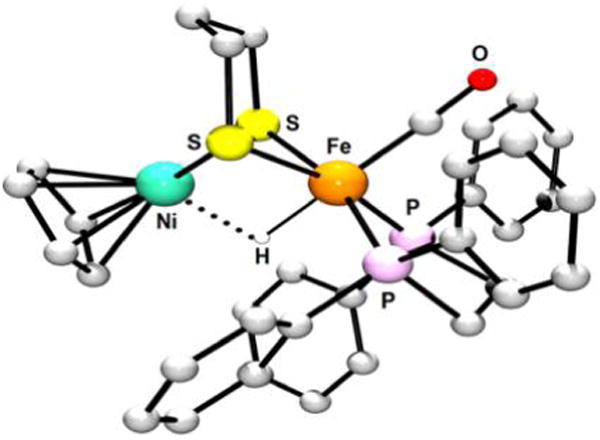
DFT-calculated structure for the putative species [44(μ-H)]+, whose Ni(III)(μ-H)Fe(II) core mimics that in Ni-C. Non-hydride H atoms omitted for clarity.159
This review has so far highlighted examples from the growing list of thiolate- and hydride-containing heterobimetallic complexes, many of which exhibit electrocatalytic behavior.356,358 Even rather electron-poor bimetallic species can often be converted to hydrides because the proton chemical potential is readily increased by changing acid concentration and/or employing a superacid. The preparation of Ni(μ-H)Fe species from H2 is an arguably more challenging matter and is addressed in section 4.2.4.
4.2.4. Hydrides from H2
As is the case for the modeling of [FeFe]-H2ases, very few [NiFe]-H2ase models exist in which a H− ligand is derived from H2 heterolysis.26 Before these examples are described, two less-biomimetic situations are addressed, in which model complexes instead accept either a H+ or a H• from H2.
The first case is exemplified by the conversion of the Ni(I)Fe(I) species [(dppe)Ni(pdt)Fe(CO)3] ([26]) to the Ni(II)Fe(II) hydride [(dppe)Ni(pdt)(μ-H)Fe(CO)3]+ ([26(μ-H)]+) not by using the typical acid HBF4·Et2O, but rather by using a combination of H2 (1 atm) and B(C6F5)3.63 This presumably heterolytic pathway makes use of the [26]/B(C6F5)3 FLP, although the roles are reversed relative to the enzyme. Indeed, the Ni-SIa + H2 ⇌ Ni-R conversion involves a Lewis acidic metal site and a basic Cys (or Arg) residue. The present situation is perhaps more relevant to the [Fe]-H2ase mechanism (vide infra), in which the Lewis acidic substrate methenyl-H4MPT+, whose role here is played by B(C6F5)3, accepts H− in a metal-mediated process.
Metal complexes can also use H2 as a source of H•, although this does not necessarily implicate a homolytic activation pathway. Consider the low-valent Ni(0)Ru(II) species [39], which is readily oxidized by Fc+ to afford the Ni(I)Ru(II) cation [39]+ (Figure 40). Under H2, the latter complex converts to the diamagnetic Ni(II)(μ-H)Ru(II) product ([39(μ-H)]+), whose alternative synthesis is from [39] and [H(OEt2)2]BArF4. The reaction with H2 is slow (~12 h) and poorly understood,359 and it is as yet unclear whether the process is first or second order in [39]+. The latter case would be less relevant to the H2ases, whose active sites are deeply nested in their protein shells and cannot work in concert with a second H2ase.
Biomimetic [NiFe]-H2ase models that utilize H2 as a H− source are best designed by mimicking the structure of Ni-SIa, whose electrophilic Ni(II)Fe(II) core is amenable to the binding and cleavage of H2. A popular class of building blocks exists in Ni complexes of N2S2- (2 × amine + 2 × thiolate) or S4-donor (2 × thioether + 2 × thiolate) podands.360 As with (diphosphine)-Ni(dithiolate) species, low-spin Ni(II) podands have been incorporated into many bimetallic systems, one example of which is [Ni(podandMe)Ru(OH2)(C6Me6)]2+ ([45(OH2)]2+, Figure 45, top left).361
Figure 45.
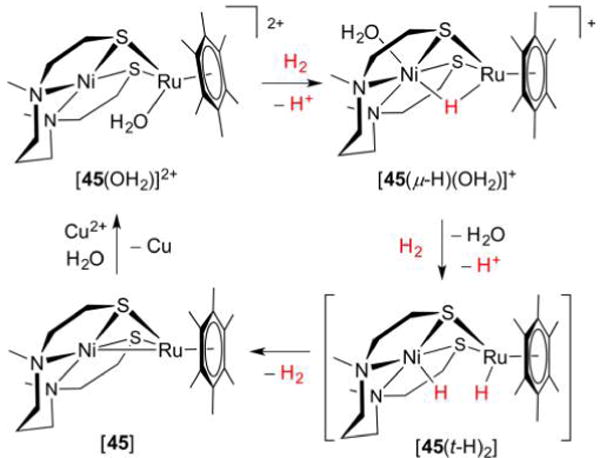
NiRu dithiolate [45(OH2)]2+ activates H2 to afford hydride [45(μ-H)(OH2)]+. Catalytic H2 oxidation, using Cu2+ as the electron acceptor, is proposed to involve a dihydride intermediate.361
As proposed for Ni-SIa, the Ni(II)Ru(II) complex [45(OH2)]2+ incorporates a weakly bound OH2 ligand into its structure, with a 14e− Ru(C6Me6) fragment serving as a Fe(CN)(CO)2 surrogate. Under H2 (1 atm) at room temperature, diamagnetic [45(OH2)]2+ converts to the paramagnetic hydride [45(μ-H)(OH2)]+ with concomitant release of H+ to the (buffered) solution. Octahedral on account of an apical aquo ligand, the Ni(II) site in the product (S = 1) contrasts that in Ni-R (S = 0), wherein no H2O is bound. Precise atomic coordinates of the synthetic model were obtained by X-ray and neutron diffraction, the latter utilizing deuteride [45(μ-D)(OH2)]+ (prepared from [45(OH2)]2+ and D2) and affording accurate Ni–D (1.859 Å) and Ru–D (1.676 Å) distances. The two proposed mechanisms for H2 activation are (i) arene slippage to η4-hapticity, with subsequent H2 ligation and cleavage at Ru, and (ii) H2 ligation and cleavage at Ni. In both cases it was suggested that the Ru-bound aquo deprotonates η2-H2, but an alternative route that appears at least as likely involves a free H2O molecule serving as the base.
Hydride [45(μ-H)(OH2)]+ is a catalyst for H2 oxidation, mediating 2e− reduction of excess Cu2+ under H2, with the concomitant release of 2H+. The proposed mechanism is unusual in that each cycle requires activation of two molecules of H2 and involves a putative dihydride intermediate.362 The latter species is invoked based on H/D-exchange experiments, as well the observation that [45(μ-H)(OH2)]+, despite its protic nature, requires H2 to convert to the low-valent Ni(I)Ru(I) complex. The hydride [(H2O)Ni(podandMe)(μ-H)Ru(C6Me6)]+ was further demonstrated to mediate hydrogenation of aldehydes to their corresponding alcohols,363 as well as to serve as an anodic catalyst in a fuel cell28,29 in which the cathodic reaction was mediated by [Ni(podandMe)(η2-O2)Ru(Cp*)]+.364 The processing of H2 is accompanied here also by O2 activation, with the more electron-rich Cp*− derivative allowing access to Ni(II)-Ru(IV) peroxos. More recently, the Ni(II)Fe(IV) peroxo [Ni(podandMe)(η2-O2)Fe(Cp*)]+ could also be prepared from O2,365 a reaction mimicking the oxidase behavior exhibited by certain O2-tolerant [NiFe]-H2ases.366
The structure of hydride [45(μ-H)(OH2)]+ certainly differs from that of Ni-R, not least in the use of Ru(II) for Fe(II). The promising reactivity and aqueous solubility of the synthetic model362 inspired further research into related podand complexes, including those with NiFe cores. A close relative of [45(μ-H)(OH2)]+ is the 34e− complex {Ni(podandEt)Fe-(MeCN)[P(OEt)3]3}2+ ([46(MeCN)]2+, Figure 46), which preserves many important features while also having the targeted NiFe composition. The organometallic Fe(CN)2(CO) site at [NiFe]-H2ase is mimicked here by a Fe[P(OEt)3]3 fragment, with the phosphite ligands chosen for their σ-donating and π-accepting properties.367
Figure 46.
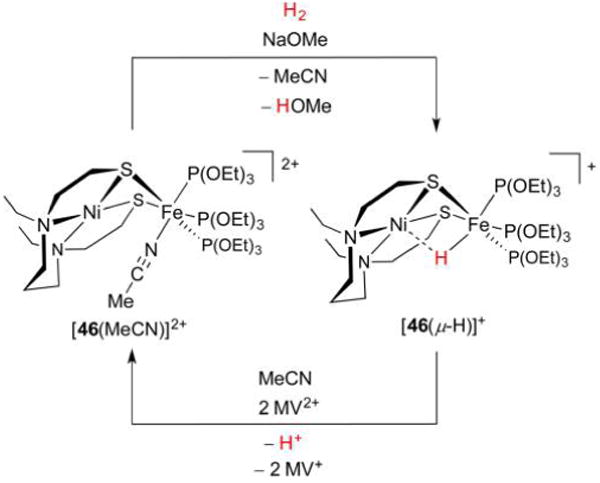
Activation of H2 by Ni(II)Fe(II) species [46(MeCN)]2+ affords hydride [46(μ-H)]+ (top). X-ray structure of [46(μ-H)]+ (bottom; non-hydride H atoms omitted for clarity).367
Demonstrating reactivity akin to its NiRu analogue, [46(MeCN)]2+ transforms under H2 to the hydride [46(μ-H)]+, although not without the use of a strong base to accept the H+ byproduct. The product here is diamagnetic, allowing for assignment of the H− resonance (δ(1H) −3.57 ppm), whose shift is on par with values for [(diphosphine)Ni(dithiolate)(μ-H)Fe(CO)3–nLn]+. The use of D2 in place of H2 confirms both the resultant hydride structure and the origin of the new bridging ligand identified according to IR (νNiHFe = 1687 cm−1, νNiDFe = 1218 cm−1) and ESI-MS data. X-ray crystallography of the hydride afforded bond lengths for Ni–H (2.16 Å) and Fe–H (1.57 Å) slightly smaller than those obtained by neutron diffraction using the deuteride (Ni–D 2.18 Å, Fe–H 1.577 Å). The latter measurements provide a better picture of the true nuclear positions, although in both cases the Fe-biased binding of H− is clear. Nevertheless, the complex [46(μ-H)]+ is interesting in that it exhibits hydridic properties, liberating H2 upon treatment with HBF4·Et2O. Similar to Ni-R, it also undergoes oxidation, although only irreversibly and at a rather high potential (0.45 V). A reversible wave at −0.42 V may arise from a [46(μ-H)]+/0 couple, which indicates that the mixed-valent hydride [46(μ-H)] is potentially isolable.
The report that hydride [46(μ-H)]+ could be prepared from H2 was followed almost immediately by one describing synthetic hydrides of even higher fidelity to Ni-R, both in structure and in function. One model in question was prepared from [(dppe)-Ni(pdt)Fe(CN)2(CO)2], one of many complexes that mimic the Ni-SCO state (Ni-SIa + CO ⇌ Ni-SCO), an inactive enzyme form in which Fe(II) is coordinatively saturated due to its 2CO + 2CN− ligands. The bis(triarylborane) adduct ([47(CO)]) undergoes decarbonylation with Me3NO to presumably afford a coordinatively unsaturated electrophile that, despite its lack of a positive charge, extracts H− from H2 in the presence of the Me3N decarbonylation coproduct (Figure 47).368
Figure 47.
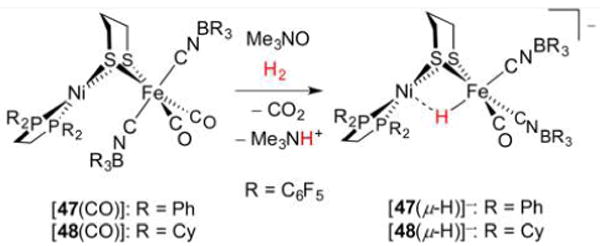
Activation of H2 by in situ generated coordinatively unsaturated Ni(II)Fe(II) species affords hydride models for Ni-R (top). X-ray crystal structure of anion [47(μ-H)]− (bottom; F and non-hydridic H atoms omitted for clarity).368
The product [47(μ-H)]− and its relatives are the first [NiFe]-H2ase models containing both CN− and H− groups. Aside from its CO occupying a basal site, the Fe coordination sphere in [47(μ-H)]− completely reproduces that in Ni-R, with the BR3 groups simulating the H-bonding exhibited by the CN− ligands in Ni-R. Nevertheless, the H− ligand (δ(1H) −7.05 ppm) is still closer to Fe (1.516 Å) than to Ni (1.710 Å), although the asymmetry is less pronounced than in [46(μ-H)]−. A subtlety differentiating [47(μ-H)]− from the many Ni(II)(pdt)(μ-H)Fe(II) species is the distortion in the Ni coordination, with the lesser of the P–Ni–S angles (147.7°) departing from linearity, although not to the extent of the respective S–Ni–S angle in Ni-R (107.9°). The distorted Ni geometry in these systems is unlikely to be reproduced by podand-containing complexes such as those described above, for which square-planar Ni sites are apparently favored.
The hydridic behavior of [47(μ-H)]− is evidenced in its dihydrogen bonding interactions with weak acids (e.g., Fe– H−⋯+HNMe3) and its liberation of H2 when treated with the stronger acid PhNH3+. These interactions are no doubt promoted by the anionic nature of [47(μ-H)]−, an aspect that also affects redox. Hydride [47(μ-H)]− and its dcpe analogue [48(μ-H)]− oxidize at −0.08 and −0.10 V vs Fc+/0, respectively, with the similarity of these values suggesting that oxidation is Fe-centered and generates a Ni(II)(μ-H)Fe(III) species (cf. Ni-C Ni(III)(μ-H)Fe(II)). Significantly, [47(μ-H)]− is an electrocatalyst for H2 oxidation (Ecat/2 ≈ −0.08 V, TOF = 0.98 s−1) in the presence of 1,8-diazabicycloundec-7-ene (DBU, pKa = 24.3 in MeCN) as a base. The observation that DBU does not deprotonate [47(μ-H)]− mandates that the anion first be oxidized in the catalytic cycle, with the implicated Ni(II)(μ-H)Fe(III) species serving as a rare example of a synthetic high-valent NiFe hydride. Work aimed at a more accurate synthetic model for Ni-C will certainly address the −CNBR3 ligands, whose high νCN frequencies (2162, 2137 cm−1) and low donicity (2 × −CNBR3 is less donating than dppe) contrast the −CN ligands in the enzyme. Concomitantly, the installation of stronger donors at Ni will be required to ensure that Ni(II) is more easily oxidized than Fe(II) in complexes of the present type.
4.2.5. Bioinspired Systems
The mechanism by which [NiFe]-H2ase operates involves redox at Ni, with the Fe center serving as a Lewis acid permanently in the +II oxidation state. This observation has led many chemists to investigate mononuclear Ni catalysts with the requisite redox and acid–base properties. Such species are often termed “bioinspired” as they have less in common with H2ases than the “biomimetic” examples described here thus far. Although several Ni complexes, particularly those featuring dithiolenes,369 are known to mediate the HER, discussed here are the more privileged examples that exhibit H2 activation. There are certainly fewer stable examples of Ni(η2-H2) complexes than of Fe(η2-H2) analogues, in part a reflection of η2-H2 complexes almost always being octahedral. This rarity had initially led to doubts about Ni performing H2 cleavage in [NiFe]-H2ase, although this is now thought to be the case. Early evidence for Ni interaction with H2 could be found from [49H], which converts to its D isotopologue [49D] under D2 (Figure 48).370 The formation of a short-lived D2 complex is thought to be followed by heterolysis of D2, with Ni receiving D− and thiolate accepting D+.
Figure 48.

H/D scrambling implies a Ni(η2-H2) intermediate (top).370 Such Ni(η2-H2) species can exhibit heterolysis with371 and without an external base372 (middle and bottom, respectively).
One isolable dihydrogen complex is [50(η2-H2)]+, whose 1H NMR resonance at −3.21 ppm is characteristic of a η2-H2 ligand. Illustrating the H2 heterolysis reaction, this cation reacts with NEt3 to afford the corresponding hydride [50(H)].371 A similar example exists in [51(η2-H2)]+, although this H2 adduct exhibits H2 fission without the need for an external base.372 Indeed, the amido group is well-placed to accept a H+ while remaining bonded to Ni, resulting in the low thermal stability of [51(η2-H2)]+. In general, it is important to note that observation of H2 heterolysis does not rule out a homolytic pathway. Indeed, an oxidative addition route involving a Ni(IV) dihydride is proposed in the case of [51(η2-H2)]+. Lastly, the most stable Ni(II)(η2-H2) complex to date is {[(2-Ph2PC6H4)3Si]Ni(η2-H2)}+, which features a tetradentate, anionic SiP3-donor tripod.373 In a sense, this complex mirrors the 5-coordinate Ni(II) center in the (Cys-S)2Ni(η2-H2)(Cys-S)2Fe(CN)2(CO) species proposed as an enzyme intermediate. While the complexes [50(η2-H2)]+ and [51(η2-H2)]+ illustrate biorelevant facets of nickel hydrides, their redox chemistry has not been developed.
Molecular Ni complexes with favorable acid–base and redox properties allow mediation of both H2 oxidation and evolution. Such bidirectional catalysts remain key synthetic targets. One prominent class of these blends the well-understood protonation behavior of bis(diphosphine)nickel complexes55 with the H+ relay motif in [FeFe]-H2ases. These complexes feature heterocycles with phosphine donor groups and amine relays (denoted “P2N2”, Figure 49).86
Figure 49.
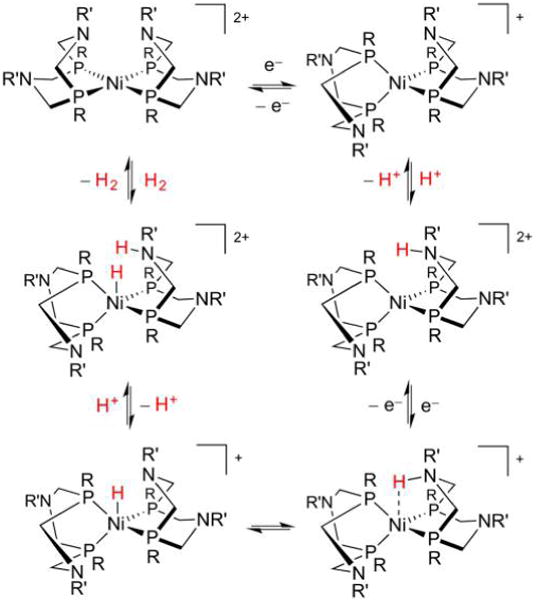
Examples of the [Ni(P2N2)2] family of electrocatalysts mediate both H2 oxidation (counterclockwise) and evolution (clockwise).86 This cycle is a simplification that neglects certain side pathways, and the order of the steps depends on the identities of substituents R and R′.
Variation of R and R′ substituents in [Ni(P2N2)2] allows tuning of steric and electronic properties such that certain reactions are favored. For example, when R = Cy and R′ = tBu, the Ni derivative is a fast H2 oxidation electrocatalyst, at least by synthetic standards (TOF = 51 s−1 under H2 (1 atm) at −0.77 V vs Fc+/0).27,374 When instead R = Ph and R′ = CH2CH2OMe, a bidirectional catalyst is afforded, albeit one that effects H2 oxidation and production very slowly. Bidirectional catalysts typically operate at low overpotentials, in this case possibly facilitated by concerted PCET.375,376 It was later found that the HER activity of the Ni complexes is greatly enhanced by using a “P2N” ligand, a seven-membered analogue of “P2N2” ligands bearing only one NR′ substituent. When R = R′ = Ph, [Ni(P2N)2] mediates the HER from protonated N,N-dimethylformamide (pKa = 6.1) extremely rapidly (TOF > 100 000 s−1 at −1.13 vs Fc+/0), albeit at a high overpotential (η = 625 mV).377 Whether performing H2 oxidation or evolution, it is clear that H+ relays play a large role in facilitating substrate transfer. The present class of compounds combines Ni with amines reminiscent of adt2−, the latter being positioned far enough away such that Ni–N bonding is unfavorable, yet close enough to rapidly relay H+ to and from the metal.281
4.3. Concluding Remarks and Future Challenges
The synthetic modeling of [NiFe]-H2ases has afforded an ever-growing collection of heterobimetallic complexes, the structures and reactivities of which are incrementally approaching those of Nature’s blueprint. The high efficiency of H2ases requires that each catalytically active state be of similar free energy375 and exist in an almost flat free energy landscape in which kinetic barriers to H2 oxidation or evolution are minimized.141 Reproducing these rates in synthetic systems will necessitate more rigid complexes, and no doubt motivate the synthesis of new polydentate ligands. Indeed, Ni(diphosphine)2-based catalysts oscillate between tetrahedral Ni(0) and planar Ni(II) during catalysis in contrast to the rigid NiS4 coordination sphere of the enzyme. This molecular reorganization may well be a contributing factor to the high overpotentials several conformationally flexible models exhibit. While rigidity appears important, not just any ligand will suffice, and ideal designs include those that constrain the complex, particularly the Ni center, in the unique geometry between square planar and tetrahedral. The seesaw coordination, indicated by DFT studies to be important in H2 scission,325 has yet to be replicated. Synthetic chemists will need to look beyond classic ligands, such as podands, which strongly bias square-planar coordination. This biasing of one redox state over another may be a reason why no biomimetic catalyst has been characterized in all of the states proposed for its catalytic cycle. On the other hand, ligand flexibility has been shown to be important for PCET in the [Ni(P2N2)2] and related catalysts with proton relays. In such cases, thermal motions enable the pendant amine to move closer to the Ni center with a relatively low energy penalty in order to facilitate proton transfer.376 Although the geometry of the Fe site in models is more easily managed, holding the CO ligand trans to the hydride binding site poses a challenge.378
A key shortcoming with models prepared thus far is that no system can exist in a Ni(III)(μ-H)Fe(II) form analogous to Ni-C. The continuing quest to prepare more electron-rich complexes can be readily tracked by considering redox potentials and νCO frequencies. This work will involve shifting the μ-H− ligand toward Ni to stabilize a +III oxidation state.379,380 Additionally, this state would be favored by the inclusion of stronger anionic ligands including thiolates or possibly even phosphides or amides. Such ligands may also serve as H+ relays, although their propensity to bridge metals must be suppressed. The innovations necessary to tackle these and other challenges in the modeling of [NiFe]-H2ases will not only bring chemists closer to useful synthetic analogues, but also deepen our understanding of the unusual chemistry in Nature’s toolkit.
5. [Fe]-H2ASES
5.1. Enzyme Structure and Function
Of all the H2 produced in Nature, approximately half is used in the conversion of CO2 to CH4, a process that underpins the metabolism of methanogens. To this end, these archaea express four [NiFe]-H2ases, as well as, when starved for Ni, one [Fe]-H2ase381 (the “third hydrogenase”, 43 kDa).382 Discovered after383 the more O2-sensitive FeFe and NiFe proteins,384 [Fe]-H2ase is not a redox enzyme and thus does not feature Fe–S clusters. In fact, it was initially purported to be completely free of Fe,385 although it is now known as a metalloenzyme after all,386 whose activity centers around a Cys-ligated Fe-guanylylpyridinol cofactor (Fe-GP). This structure was shown by IR (νCO = 2011, 1944 cm−1)387 and NRVS388 analyses to contain a Fe(CO)2(S-Cys) fragment bound to a unique guanylylpyridinol/pyridonate (GP) through its acyl (νCO = 1697 cm−1) and pyridyl donors. The GP ligand can be isolated, although in a form with the acyl group oxidized to the corresponding carboxylate.389 Initial holoenzyme structural studies included this additional O atom, although subsequent mutagenesis and crystallography390 confirmed that the acyl form is indeed at the active site (Figure 50).391
Figure 50.
X-ray structure of the [Fe]-H2ase active site from Methanocaldococcus jannaschii (PDB code 3F4Z).390 H atoms are omitted for clarity.
The Fe(CO)2(S-Cys)(guanylylpyridinol/pyridonate) unit is square pyramidal (τ = (175.1° – 173.7°)/60° = 0.02),306 with the two CO ligands being cis: one trans to the basic thiolate and the other trans to the pyridine/pyridonate. The acyl ligand exerts its trans effect on a sixth site, which when not passivated by a H2O ligand, is available to bind H2 (as well as inhibitors such as CO). When the enzyme adopts an “open” state, H2O is displaced by methenyl-H4MPT+ (Figure 51). After folding to the closed state, there remains a 4 Å-wide hydrophobic channel accessible to solvent and H2. Diffusion of the latter to the active site and release of a proton from the pyridinol moiety to a neighboring His residue affords a Fe(η2-H2)(CO)2(S-Cys)-(guanylylpyridonate) complex proximal to methenyl-H4MPT+. The H2 substrate is now primed to undergo heterolysis, with the basic O atom of the pyridonate392 and methenyl-H4MPT+ serving as the H+ and H− acceptors, respectively. Release of the product and ligation of H2O completes the cycle.393 Overall, the consumption of H2 is close to isoergic (ΔG = −5.5 kJmol−1), as one would expect given the comparable potentials for the H+/H2 (−414 mV) and methenyl-H4MPT+/methylene-H4MPT (−390 mV) couples at pH 7, as well as the rates of H2 splitting (TOF = 215 s−1, pH 7.5) and formation (TOF = 555 s−1, pH 6.5) mediated by [Fe]-H2ase.383
Figure 51.
Catalytic cycle proposed for [Fe]-H2ase.9
Inasmuch as pyridonate is similar to CN− in terms of donor/acceptor properties,394 the active site of [Fe]-H2ase shares certain features with that of [FeFe]-H2ase.395 Each has a basic cofactor (secondary amine or pyridonate O atom) adjacent to the transiently vacant Fe coordination site. Yet [Fe]-H2ase does not perform redox and is not highly O2-sensitive,394 and its Fe site is invariably found to exist in a low-spin divalent state, certainly a good fit for H2 binding and heterolysis.
The catalytic cycle for [Fe]-H2ase, like that for [FeFe]- or [NiFe]-H2ase, is proposed to involve hydride species. One mechanism, based on DFT calculations of a truncated active site model, invokes an elongated dihydrogen bond of the form Fe(II)–H⋯H–Opyridinol.274 However, calculations in which all amino acids are considered indicate that Fe hydrides need not be considered if H2 scission is concerted. In such a case, the Fe(η2-H2)(guanylylpyridonate) group, in which the Fe(η2-H2) moiety has an estimated pKa of 9,396 converts to Fe(guanylylpyridinol) without the intermediacy of a hydride, as H+ abstraction by pyridonate and H− migration to the substrate occur simultaneously.397 The methylene-H4MPT product then leaves the active site, but not before the protein undergoes large-scale movement to open its binding pocket. This study employs molecular dynamics (MD) simulations to probe the opening and closing of the binding pocket, and quantum mechanical/molecular mechanical (QM/MM) calculations to investigate H2 splitting on select MD snapshots.397 In contrast to [Fe]-H2ase, no large displacements are required to shuttle small substrates to the cores of [FeFe]-H2ase and [NiFe]-H2ase.
While not an electrocatalyst (and thus not relevant to fuel cell applications), [Fe]-H2ase does deliver H− stereoselectively to its substrate. The enzyme could thus serve as a basis for catalysts in organic synthesis, although perhaps due to the many specific supramolecular interactions involved in binding substrate, no molecules other than methenyl-H4MPT+ induce H2 cleavage.398 Time will tell whether the methenyl-H4MPT+/H2/Fe-GP ternary complex is absolutely necessary or whether the H− delivery can be generalized. Motivated by this, as well as the prospect of learning more about the catalytic cycle, synthetic chemists have taken to reproducing the [Fe]-H2ase active site.399
5.2. [Fe]-H2ase Synthetic Modeling
5.2.1. Models of CO-Inhibited [Fe]-H2ase
H2ases are inhibited by CO, which often (but not always, see section 3.1) outcompetes H2 for metal binding sites, thereby filling their coordination spheres. In the case of [Fe]-H2ase, the inhibited enzyme features a low-spin facial Fe(II)(CO)3 fragment giving rise to νCO bands at 2074, 2020, and 1981 cm−1 (Table 6). Synthetic modeling of [Fe]-H2ase began with mimics of the inhibited enzyme, which are described here before highlighting efforts to mimic the coordinatively unsaturated Fe site in the active states.
Table 6.
CO-Stretching Bands for Active and CO-Inhibited Forms of [Fe]-H2ase, as well as for Selected Synthetic Models
| species | νCO/cm−1 |
|---|---|
| [Fe]-H2ase | 2011, 1944 |
| [Fe]-H2ase-CO | 2074, 2020, 1981 |
| [52] | 2075, 2020, 1981 |
| [53] | 2032, 1987 |
| [54] | 2026, 1961 |
| [55] | 2027, 1964 |
| [56] | 2022, 1958 |
| [57] | 2068, 2050, 1996 |
| [58] | 2084, 2018, 2003 |
| [59] | 2013, 1950 |
| [60] | 2022, 1958 |
| [61HBr] | 2030, 1974 |
| [62] | 2003, 1940 |
| [63I] | 1945 |
| [RuCp(CO)2]− | 1887, 1802 |
The finding in 2009 that existing X-ray data for [Fe]-H2ase indicated an Fe-bound acyl ligand inspired a flurry of synthetic activity to reproduce this common organometallic fragment. Despite the multitude of octahedral ferrous complexes, the enzyme has a unique ligand set, and work in the area has focused on reproducing the structure (inner coordination sphere) and spectroscopy (IR bands). For example, the first acyl thiolato monoiron complex [Fe(CO)3(SPh)(2-diphenylphosphinobenzoyl)] ([52], Figure 52) features five of the six donors present at the [Fe]-H2ase active site and gives rise to νCO bands closely matching those of the enzyme.400,401 Moreover, the synthesis of [52] by oxidative addition of a thioester to Fe(0) is biomimetic in that the guanylylpyridinol/pyridonate (GP) cofactor is also derived from a thioester.402 Much like the CO-inhibited enzyme, [52] readily loses CO, although the 5-coordinate product converts to a dinuclear species.
Figure 52.
Early models for CO-inhibited [Fe]-H2ase feature some of the donor groups present in the enzyme.400,401,403
Complexes of Fe featuring 2-pyridonate moieties have also been reported, one example of which is [Fe(CO)2(PPh3)I(6-methyl-2-pyridonate)] ([53]).403 A dicarbonyl, this complex further contrasts the active site in that the pyridonate group in [53] binds through both N and O atoms.404 A key feature of [Fe]-H2ase is the positioning of the basic O atom near but not bonded to the Fe site. Early efforts toward octahedral analogues of Nature’s 5-ccordinate FLP are now described.
Synthetic investigations into 2-pyridylcarbamoyl405,406 and pyridylacyl complexes have afforded models closely resembling the CO-inhibited [Fe]-H2ase active site. Indeed, this motif has been incorporated into complexes of the form [Fe(CO)2(2-pyridinethiolate)(2-pyridylacetyl)] ([54–56], Figure 53), which feature donors resembling the native ligands, with an additional N-donor.407–409 While [54–56] feature protected acylpyridinols, Fe complexes of the unmasked ligand have also been prepared, the coordination sphere in tricarbonyl [57] being similar to that in the CO-inhibited enzyme with I− in place of Cys-S−. These pyridylacyl complexes are not assembled through a biomimetic thioester oxidative addition, but instead form by more traditional organometallic routes. In the case of [54]–[56], this involves nucleophilic attack of [Fe(CO)4]2− onto a 2-picolyl tosylate, with subsequent insertion of CO into the Fe–alkyl bond.408–410 In the synthesis of [58], Fe instead serves as an electrophile, with [Fe(CO)5] undergoing addition of 2-picolide anion such that a CO ligand is converted to an acyl.407
Figure 53.
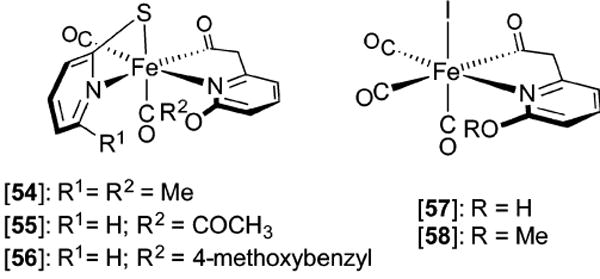
Acyl- and carbamoylpyridine models for CO-inhibited [Fe]-H2ase.
The tricarbonyls [57] and [58] were employed in an [Fe]-H2ase reconstitution study analogous to that described for [FeFe]-H2ase (vide supra). Incubation of [57] or [58] with apo-[Fe]-H2ase, which lacks the Fe-GP cofactor,411 afforded products whose IR spectra indicated ligation of Fe(CO)2(acylpyridinol/acylmethylpyridinol) to Cys176. Although the methylated derivative was unreactive toward H2/methenyl-H4+MPT+ or H+/methylene-H4MPT, the acylpyridinol-containing protein catalyzed these reactions. The semisynthetic system was 2 orders of magnitude slower than the native one, a result attributed to subtle electronic differences between GP and the acylpyridinol ligand. Nevertheless, this work highlights the importance of pyridinol/pyridonate as a H+ relay, and refutes some older DFT studies that, by analogy with [NiFe]-H2ases, implicated Cys-S− as a relay.114,274
Given that pentacoordinate Fe is crucial to the activity of H2ases, section 5.2.2 highlights 16e− Fe complexes that most closely mimic the [Fe]-H2ase active site in both structure and reactivity. This will serve as a prelude to studies into H2 activation with [Fe]-H2ase models.
5.2.2. Models of Active [Fe]-H2ase States
A significant breakthrough in modeling the [Fe]-H2ases came with the finding that a combination of thiolate, pyridylacyl, and other π-bonding ligands can stabilize unsaturated Fe(II) sites. Addition of thiolate to [58] replaces not only I− but also CO, such that square-pyramidal complex [59] forms (Figure 54).412 However, this species does not bind H2O or H2 (or O2), a result attributed to its electron richness. Although the similarity of its νCO frequencies to those of the enzyme suggests that other factors may also play a role, DFT calculations indicate that H2 binding is thermodynamically unfavorable (ΔG = 10.2 kcal mol−1).413
Figure 54.
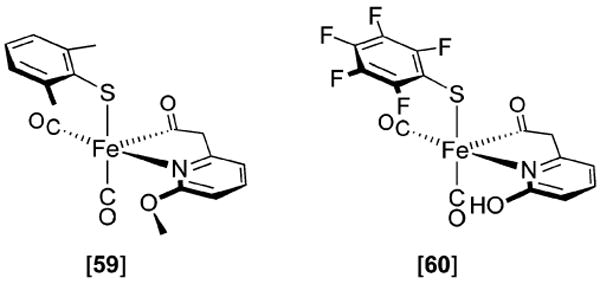
Pentacoordinate acylpyridine models [59] and [60] replicate inner Fe coordination sphere of the active [Fe]-H2ase states, yet do not bind H2.412,414
Building on the chemistry of [59], the higher-fidelity structural model [60] features the hydroxyl group characteristic of [Fe]-H2ase.414 The species was identified according to NMR and IR spectroscopies, but was found to be rather labile (although less so than its 2,6-dimethylphenylthiolato and 1-propanethiolato congeners). Although spectroscopically and structurally similar to the active site, it is unreactive toward H2. This may indicate that secondary interactions involving the [Fe]-H2ase pocket, the inclusion of methenyl-H4MPT+, and the presence of a more substituted pyridyl ligand are important factors in the activation of H2.26
Another example of a five-coordinate [Fe]-H2ase model could be prepared from dehydrobromination of octahedral Fe(II) carbamoyl [61HBr] (Figure 55).415 Similar in structure to [60], the proposed kinetic product could not be characterized, as it rapidly converted to the cyclometalated species [62]. The latter product can also be prepared from hydride [61(H)H] (δ(1H) −5.1 ppm), which, in turn, is generated from [61HBr] and HBEt3−. While displacement of Br− with H− is not a biomimetic transformation, the donor atoms around [61(H)H] match those of the active site in a transient hydride-bearing intermediate. Such an enzyme state has yet to be experimentally confirmed, although some theoretical studies invoke it as a short-lived species, very similar to [61(H)H], which is unstable above −40 °C. While the generation of [61(H)H] indicates that the present type of carbamoyl species can support a hydride ligand, the second coordination sphere of [61(H)H] does not feature a basic moiety to engage in H2 heterolysis.
Figure 55.
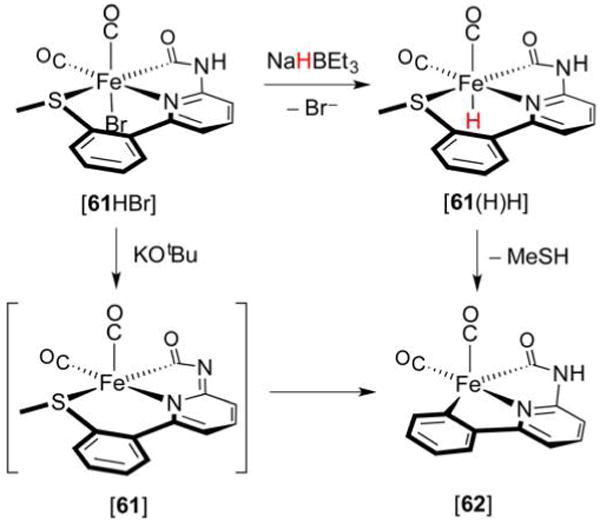
Preparation and reactivity of the Fe hydrido carbamoyl [61(H)H].415
The close resemblance that the Fe coordination spheres of acylpyridine [61] and related species have to that of [Fe]-H2ase indicates that differences in reactivity may arise from secondary interactions. In search of H2 heterolysis, such structural models have been modified to incorporate more basic fragments into their architecture, even though this may decrease their resemblance to the active site. For example, combination of tricarbonyl [58] with the bidentate ligand Et2PCH2N(Me)-CH2PEt2 affords monocarbonyl [63I], from which I− dissociation can occur (Figure 56).416 The putative 5-coordinate species is well set up to cleave H2, although no direct evidence of the ammonium hydride has been reported, even under high H2 pressures. The activation of H2 by [63]+ was evidenced in the scrambling of H2/D2 mixtures, with the additional scrambling of H2 and CH3CO2D suggesting a heterolytic mechanism is at play. Bearing some similarity to bioinspired Ni(PNP) and Fe(PNP) electrocatalysts, the present species represents the first functional model for [Fe]-H2ase and underscores the importance of a proximal base for FLP activity. Even though no hydride complex could be detected, such work importantly demonstrates that an Fe-based [Fe]-H2ase model can activate H2 outside of the enzyme in the absence of the native methenyl-H4MPT+ substrate.
Figure 56.
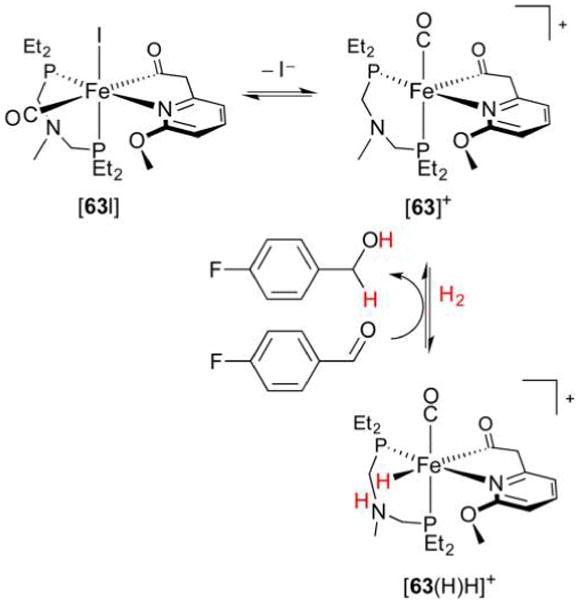
Polar hydrogenation of an aldehyde catalyzed by [63]+.416
The inertness toward H2 shown by many 5-coordinate Fe complexes described above comes as a disappointment, but is unsurprising given that ferrous dihydrogen complexes are characteristically cationic.35 Furthermore, when observed, either directly with [61(H)H] or indirectly in the case of [63]+, the hydrides are unstable. This has led efforts in functional mimicry to also consider structures departing from the Fe(pyridylacyl) paradigm. For example, triarylimidazolium salts of [RuCp-(CO)2]− represent FLPs that parallel the biological combination of methenyl-H4MPT+ and the Fe-GP cofactor (Figure 57).417 Indeed, the electrophilic C centers of the triarylimidazolium (δ(13C) 169.6 ppm) and methenyl-H4MPT+ (165.34 ppm)418 would appear to be comparably reactive. When treated with H2, the synthetic Lewis pair heterolyzes H2 with the cation receiving H− and the Ru0 center accepting a H+ by oxidative addition.
Figure 57.
Complex [RuCp(CO)2]− exhibits [Fe]-H2ase behavior, effecting H2 heterolysis and delivery of H− to an imidazolium substrate resembling methenyl-H4MPT+.417
The heterolysis reaction proceeds fastest when a polymeric Ru–CO–Cp species is used in place of [RuCp(CO)2]−. A slightly more biomimetic choice would of course be [FeCp-(CO)2]−, but it directly reduces the imidazolium cation. Overall, H2 heterolysis is similar to the H2 fission mediated by the [(dppe)Ni(pdt)Fe(CO)3]/B(C6F5)3 FLP (section 4.2.3).63 Since the H− and H+ heterolysis products are delivered respectively to the organic substrate and metal center, the process is not biomimetic.
5.3. Concluding Remarks and Future Challenges
Synthetic modeling of [Fe]-H2ases, particularly since the clarification of the active site structure in 2009, has afforded several high-fidelity structural mimics. This work began with the preparation of octahedral Fe(II) complexes featuring some or all of the thiolate, carbonyl, acyl, and pyridonate/pyridinol ligands present in the native protein. More relevant to the active enzyme are 5-coordinate species, in which a site is free for H2 or H− binding. Such 16e− species have been reported, but typically do not exhibit [Fe]-H2ase reactivity despite closely mimicking the structure and spectroscopy. This points to the need for tuning the second coordination sphere, the importance of which is demonstrated by artificial reconstitution studies of [Fe]- and [FeFe]-H2ase. Indeed, the high H–H bond strength means that H2 heterolysis can only be facile when both Lewis acidic and basic components are present and well arranged. To this end, incorporation of an artificial amine H+ relay in [63]+ afforded a catalyst that activates and transfers H2. No η2 –H2 or H− complex intermediates have been identified in this case, yet the same is true for [Fe]-H2ases, with the possible role of Fe–H moieties still a subject of debate. Future efforts in this area may address these questions and also aim to synthetically replicate the cooperative binding of methenyl-H4MPT+ and H2 to the enzyme active site.
6. CONCLUSIONS
The study of H2ases reminds us that organometallic chemistry is literally alive and well, providing the machinery that allows Nature to process the most abundant element in the universe. It is worth mentioning here that examples of metal hydrides in biology are not limited to H2ases. For example, ENDOR studies on nitrogenase (N2ase) suggest the presence of two hydrides attached to its Fe7MoS9C cofactor during H+ reduction.419 Each of the chemically equivalent hydrides is thought to bridge two Fe centers, a situation rather reminiscent of Fe(pdt)(μ-H)Fe model complexes. It is hardly surprising that hydrides play a role in N2ases, as these exhibit H2ase behavior in that they can process protons and electrons independently of N2 reduction to NH3.420 An inner sphere mechanism for this HER would necessitate metal hydride participation. Such chemistry is also at play in the related enzyme nickel–iron carbon monoxide dehydrogenase ([NiFe]-CODHase), which utilizes protons and electrons in the interconversion of CO2 with CO and H2O.421 In this case, the Fe4NiS4 cofactor can feature a Ni–H or Ni–H–Fe moiety, into which CO2 inserts during the catalytic cycle.422,423 The similarities between the enzymes discussed here indicate that acid–base and redox reactions are tasks carried out well by FeS-containing enzymes, especially when they are low-spin.
Returning to the H2ases, each of their unique organoiron sites15 facilitates rapid H2 cleavage and/or formation, motivating chemists to unravel the mechanisms by which they operate. Characterizing the many H2ase intermediates, including key hydride-bearing states, necessitates the use of several complementary experimental and theoretical techniques. These techniques are also applicable to synthetic hydrides inspired by the H2ase active sites, with small molecules often being more tractable and easily studied. Many catalytically competent examples are known, and the prospect of their use in a future hydrogen economy is attractive.
One reality of H2ase modeling is that there will always be differences between enzymes and their small molecule analogues. The [FeFe]- and [NiFe]-H2ases tightly encapsulate their active sites, ruling out bimolecular pathways and permitting only minor solvation changes and ligand reorganization during catalysis.424 This is not the case for model complexes, which are conformationally less restricted, as exemplified by the t-H → μ-H conversion at play in [dppv(OC)Fe(adt)Fe(H)(dppv)-(CO)]+ ([17(H)]+),258 which does not occur for the [FeFe]-H2ases, whose cores are held in place by intricate H-bonding networks. Similarly, Ni isomerization in [(dppe)Ni(pdt)Fe-(CO)3] ([26]) is necessary to accommodate protonation or oxidation,160 contrasting the [NiFe]-H2ases, in which Ni is rigidly held in a seesaw coordination. Indeed, preorganized, rigid metal sites and second coordination spheres play key roles in minimizing both overpotential and the possibility of side reactions. On the other hand, some degree of conformational flexibility is required for the systems to find the optimal distances and angles for intramolecular proton or hydride transfer reactions.376
Incorporated into H2ase protein scaffolds are hydrophobic channels for H2 propagation to and from their active sites. This is advantageous in selectively introducing substrate to the catalytic metals, but in the case of [NiFe]-H2ase, its channels may be prone to blockage when H2 partial pressures are high.337 The more exposed nature of model catalysts relative to the enzyme active sites ensures that the former will not suffer from this blockage, and access to H2, protons, and electrons (as well as inhibitors) is enhanced. With compact profiles, model complexes may also bind electrode surfaces closely and densely such that electron transfer might be rapid and catalytic current densities might be high.425 Ideally, synthetic catalysts would operate at high rates and low overpotentials, as well as exhibit a robustness that enables operation over wide temperature and pH ranges that may be necessary for implementation in fuel cells.426
The amino acids surrounding the H2ase active sites serve to shield mixed-valent or coordinatively unsaturated states from participating in any reactions other than those of the catalytic cycle. Many mimics of coordinatively saturated, inactive enzyme states have been prepared, but certainly the reactive forms have proven more elusive. For example, terminal Fe(II)Fe(II)(t-H) models for [FeFe]-H2ase have been isolated, yet their 1e− reduced mixed-valent forms have not. Absent from [NiFe]-H2ase modeling are satisfactory 33e− Ni(I)Fe(II) models for Ni-L, an isolable 32e− Ni(II)Fe(II) model for Ni-SIa (without its coordinated H2O), as well as any Ni(III)(μ-H)Fe(II) complex that would resemble the key hydride-bearing Ni-C state. Efforts toward the latter two will involve incorporation of strongly basic ligands to stabilize coordinatively unsaturated and/or high-valent metal sites.
In general, it is debatable whether characterizing catalysts in their many states is necessary or even possible if the species in question is highly active. But it is undeniable that the native H2ases are significantly faster and more energy-efficient than model complexes. While many models show reactivity toward protons, the discrepancy is most apparent when considering H2 activation.427 Despite considerable progress over the past decade, the few H2ase models that heterolyze H2 do so very slowly. Thus, bidirectional synthetic catalysts are scarce, and those that exist exhibit slow rates for electrocatalytic H2 oxidation and/or evolution arising from significant overpotentials associated with their mechanisms. The bidirectionality of many [FeFe]- and [NiFe]-H2ases has evolved not only from a biological need to both consume and dispense reducing equivalents, but also to operate in very narrow potential ranges and at low concentrations of H2 and H+.6 Furthermore, for a H2-processing catalyst to be bidirectional, it follows that interconversion of H2 and hydrides must be rapid and close to isoergic.428 Such a transformation has been identified as a major challenge in organometallic chemistry,429 and the viability of H2 as an energy vector is reliant on both its efficient production and consumption. Electrocatalysis is certainly not the only means of H2 processing, but it is notable that the reaction of H2 and O2430 in fuel cells is potentially more efficient than traditional combustion, in which energy losses from the Carnot cycle are unavoidable.
Regardless of the motivation for modeling the H2ases—be it fundamentally learning more about enzymes or perhaps developing base-metal hydrogen-processing catalysts—this work is invariably instructive, and intricacies associated with apparently simple reactions are continually uncovered. At the heart of H2ases and their functional models are metal hydrides,431 which are more pervasive and of broader relevance than many would have guessed. A deeper understanding of these hydrides, obtained through a confluence of biological and organometallic chemistry research, will place us closer to a sustainable future.
Acknowledgments
D.S. is supported by the Institute for Basic Science (IBS-R019-D1). T.B.R. is supported by the National Institutes of Health (GM061153, GM-65440). S.H.-S. acknowledges support from the National Science Foundation (CHE-13-61293). M.T.H. acknowledges support from the National Science Foundation Graduate Research Fellowship Program under Grant DGE-1144245.
ABBREVIATIONS
- Arg
arginine residue
- adt2−
azadithiolate (−SCH2N(H)CH2S−)
- (adtR)2−
substituted azadithiolate (−SCH2N(R)-CH2S−)
- BArF4−
(3,5-(CF3)2C6H3)4B−
- bdt2−
1,2-benzenedithiolate
- Bn
benzyl
- Bu
butyl
- CODHase
carbon monoxide dehydrogenase
- Cp−
cyclopentadienide
- Cp*−
pentamethylcyclopentadienide
- CV
cyclic voltammetry
- Cy
cyclohexyl
- Cys
cysteine residue
- dcpe
1,2-bis(dicylcohexylphosphino)ethane
- DBU
1,8-diazabicycloundec-7-ene
- DFT
density functional theory
- dmpe
1,2-bis(dimethylphosphino)ethane
- dppbz
1,2-bis(diphenylphosphino)benzene
- dppe
1,2-bis(diphenylphosphino)ethane
- dppn
1,8-bis(diphenylphosphino)naphthalene
- dppv
1,2-bis(diphenylphosphino)ethane
- edt2−
1,2-ethanedithiolate (−S(CH2)2S−)
- ENDOR
electron nuclear double resonance
- EPR
electron paramagnetic resonance
- Et
ethyl
- Fc
ferrocene
- Fc*
decamethylferrocene
- Fe-GP
Fe-guanylylpyridinol cofactor
- FLP
frustrated Lewis pair
- IR
infrared
- H2ase
hydrogenase
- HER
hydrogen evolution reaction
- Hmd
H2-forming methylenetetrahydromethanopterin dehydrogenase
- HYSCORE
hyperfine sublevel correlation
- Lys
lysine residue
- Me
methyl
- methenyl-H4MPT+
methenyltetrahydromethanopterin
- methylene-H4MPT
methylenetetrahydromethanopterin
- N2ase
nitrogenase
- NHE
normal hydrogen electrode
- NMR
nuclear magnetic resonance
- nPr
n-propyl
- NRVS
nuclear resonance vibrational spectroscopy
- OTf−
triflate (CF3SO3−)
- PCET
proton-coupled electron transfer
- pdpp2−
1,3-propanedi(phenylphosphide) (−PhP-(CH2)3PPh−)
- pdt2−
1,3-propanedithiolate (−S(CH2)3S−)
- Ph
phenyl
- ppm
parts per million
- Ser
serine residue
- Tf
trifluoromethanesulfonyl
- THF
tetrahydrofuran
- TOF
turnover frequency
- Ts
4-toluenesulfonyl
Biographies
David Schilter was educated at The University of Sydney, from which he received B.Sc. (Hons.) and Ph.D. degrees in 2005 and 2009, respectively. These studies were followed by postdoctoral research at the University of Illinois at Urbana–Champaign and at the IBS Center for Multidimensional Carbon Materials at UNIST, where he has served as a research scientist since 2014. His interests include coordination, organometallic, and materials chemistry.
James M. Camara earned his Ph.D. in chemistry from Columbia University in 2009, where he studied the polymerization and transmetalation chemistry of zirconium and aluminum complexes. Following the completion of his Ph.D., he was a postdoctoral fellow at the University of Illinois at Urbana–Champaign where he conducted synthetic and mechanistic studies on models of the [FeFe]-H2ase active site. He is currently an assistant professor of chemistry at Yeshiva University in New York City. His research interests are centered around various aspects of mechanistic organometallic chemistry including the use of redox-active ligands in catalysis, development of new transmetalation reactions, and small molecule activation by metal centers.
Mioy T. Huynh received his B.S. in chemistry from the University of California, Los Angeles, in 2012. He is currently a graduate student at the University of Illinois at Urbana–Champaign in Sharon Hammes-Schiffer’s research group. His research interests include the investigation of proton-coupled electron transfer reactions in catalytic systems.
Sharon Hammes-Schiffer received her B.A. in chemistry from Princeton University in 1988 and her Ph.D. in chemistry from Stanford University in 1993, followed by two years at AT&T Bell Laboratories as a postdoctoral research scientist. She was the Clare Boothe Luce Assistant Professor of Chemistry and Biochemistry at the University of Notre Dame from 1995 to 2000 and spent the next 12 years at The Pennsylvania State University, initially as the Shaffer Associate Professor of Chemistry and later as the Eberly Professor of Biotechnology. In 2012 she became the Swanlund Chair and professor of chemistry at the University of Illinois Urbana–Champaign. Dr. Hammes-Schiffer’s research centers on the investigation of electron, proton, and proton-coupled electron transfer reactions in chemical, biological, and interfacial processes. Her work encompasses the development of analytical theories and computational methods, as well as applications to a wide range of experimentally relevant systems.
Thomas B. Rauchfuss was born in 1949 in Baltimore, MD. He received his undergraduate degree from the University of Puget Sound (1971) and his Ph.D. from Washington State University (1976). After a postdoc with David Buckingham at the Australian National University, he started his independent career at the University of Illinois at Urbana–Champaign in 1978, where he has remained. He has also studied at the following institutions: the University of Auckland, University of Louis Pasteur, and the Technical University of Karlsruhe. His research focuses on synthetic inorganic and organometallic chemistry, with an emphasis on environmentally motivated themes. For the past decade, his group has conducted research on synthetic modeling of the active sites of the hydrogenase enzymes.
APPENDIX
A.1. Proposed Pyramidal Inversion of Azadithiolate During Catalysis
Figure 58 shows proton transport to and from the [2Fe] site in [FeFe]-H2ase.
A.2. Determining the pKa of a Metal Hydride
If titrating a metal hydride MH+ with a base B50,51
A known amount of base B (=[B]total) is added to a known amount of MH+. The values [MH+] and [M] in solution are determined, typically using IR or NMR spectroscopy with an internal standard.
- [BH+] is determined according to
- [BHB+] is determined using mass balance on M:
- [B] is determined using mass balance on B:
-
All the values [B], [BH+], [M], and [MH+] are now known such that Keq and thus pKa(MH+) = pKeq + pKa(BH+) can be computed.
If titrating a metal complex M with a weak acid HA:
A known amount of acid HA (=[A−]total) is added to a known amount of M. The values [MH+] and [M] in solution are determined, typically using IR or NMR spectroscopy with an internal standard present.
- [A−] is determined according to
-
[AHA−] is determined using mass balance on A−:One does not directly know [HA], but the H+ from reacted HA formed either [MH+] or [AHA−] (mass balance on H+):
andSubstituting this expression for [HA] into the mass balance above: - [HA] is then computed from the mass balance:
All the values [A−], [HA], [M], and [MH+] are now known such that Keq and thus pKa(MH+) = pKa(HA) – pKeq can be computed.
A.3. Expressions for Hydricity and H Atom Donation
The hydricity of a metal hydride MH+ can be determined by thermodynamic cycles.53,58,59 If the potentials E1/2[M]+/0 and E1/2[M]2+/+ are known, one can sum the four reactions
This affords
| (A9) |
Summing the free energy changes for the four reactions gives the change for eq A9:
| (A10) |
where F = 96 485 C mol−1, R = 8.314 J K−1 mol−1, e = 2.718… and T is reported in kelvin.
Note that the last term accounts for the hydricity of H2 itself. When data are collected in MeCN at T = 298 K, and E1/2 values are each relative to Fc+/0, eq A10 becomes eq A10′, in which the free energy change, in kcal mol−1, is
| (A10′) |
When data are instead collected in H2O vs NHE, eq A10″ is appropriate:
| (A10″) |
Alternatively, if the potential for the 2e− couple E1/2[M]2+/0 is known, then the following equations are summed:
This once more gives eq A9, the free energy change of which is now equated in terms of
| (A11) |
Again, when data are collected in MeCN at T = 298 K, and E1/2 values are each relative to Fc+/0, eq A11 becomes eq A11′, in which the free energy change, in kcal mol−1, is
| (A11′) |
When data are instead collected in H2O vs NHE, eq A11″ is appropriate:
| (A11″) |
One can also consider MH+ as a source of H• instead of H+. In this case, summing the reactions
affords the net conversion:
| (A12) |
The free energy change of this reaction is
| (A13) |
When data are collected in PhCN/MeCN at T = 298 K, and E1/2 values are each relative to Fc+/0, eq A13 becomes eq A13′, in which the free energy change, in kcal mol−1, is
| (A13′) |
When data are instead collected in H2O vs NHE, eq A13″ is appropriate:
| (A13″) |
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Immerdorf H. Contributions to the solution of the “nitrogen question”. Landwirtsch Jahrb. 1892;21:281. [Google Scholar]
- 2.Tausz J, Donath H. On the oxidation of hydrogen and hydrocarbons by bacteria. Hoppe-Seyler’s Z Physiol Chem. 1930;190:141. [Google Scholar]
- 3.Stephenson M, Stickland LH. XXVII. Hydrogenase: a bacterial enzyme activating molecular hydrogen. I. The properties of the enzyme. ochem J. 1931;25:205–214. doi: 10.1042/bj0250205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elsden SR. Hydrogenase 1931–1981. Trends Biochem Sci. 1981;6:251–253. [Google Scholar]
- 5.Stripp ST, Happe T. How algae produce hydrogen—news from the photosynthetic hydrogenase. Dalton Trans. 2009:9960–9969. doi: 10.1039/b916246a. [DOI] [PubMed] [Google Scholar]
- 6.Thauer RK. Hydrogenases and the Global H2 Cycle. Eur J Inorg Chem. 2011;2011:919–921. [Google Scholar]
- 7.Lubitz W, Ogata H, Rüdiger O, Reijerse E. Hydrogenases. Chem Rev. 2014;114:4081–4148. doi: 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- 8.Frey M. Hydrogenases: Hydrogen-Activating Enzymes. Chem-BioChem. 2002;3:153–160. doi: 10.1002/1439-7633(20020301)3:2/3<153::AID-CBIC153>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 9.Shima S, Ermler U. Structure and Function of [Fe]-Hydrogenase and its Iron–Guanylylpyridinol (FeGP) Cofactor. Eur J Inorg Chem. 2011;2011:963–972. [Google Scholar]
- 10.Farkas A, Farkas L, Yudkin J. The Decomposition of Sodium Formate by Bacterium coli in the Presence of Heavy Water. Proc R Soc London, Ser B. 1934;115:373–379. [Google Scholar]
- 11.Krasna AI. Hydrogenase: properties and applications. Enzyme Microb Technol. 1979;1:165–172. [Google Scholar]
- 12.Yagi T, Tsuda M, Inokuchi H. Kinetic studies on hydrogenase parahydrogen-orthohydrogen conversion and hydrogen-deuterium exchange reactions. J Biochem. 1973;73:1069–1081. doi: 10.1093/oxfordjournals.jbchem.a130161. [DOI] [PubMed] [Google Scholar]
- 13.Pagliaro M, Konstandopoulos AG. Solar Hydrogen: Fuel of the Future. RSC Publishing; 2012. [Google Scholar]
- 14.Peters JW, Schut GJ, Boyd ES, Mulder DW, Shepard EM, Broderick JB, King PW, Adams MWW. [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim Biophys Acta, Mol Cell Res. 2015;1853:1350–1369. doi: 10.1016/j.bbamcr.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 15.Adams MWW, Stiefel EI. Organometallic iron: the key to biological hydrogen metabolism. Curr Opin Chem Biol. 2000;4:214–220. doi: 10.1016/s1367-5931(99)00077-0. [DOI] [PubMed] [Google Scholar]
- 16.Vignais PM, Billoud B. Occurrence, classification, and biological function of hydrogenases: an overview. Chem Rev. 2007;107:4206–4272. doi: 10.1021/cr050196r. [DOI] [PubMed] [Google Scholar]
- 17.Bethel RD, Darensbourg MY. In: Bioorganometallic Chemistry: Applications in Drug Discovery, Biocatalysis, and Imaging. Jaouen G, Salmain M, editors. Wiley-VCH Verlag GmbH & Co. KGaA; 2015. [Google Scholar]
- 18.Lawrence EJ, Clark ER, Curless LD, Courtney JM, Blagg RJ, Ingleson MJ, Wildgoose GG. Metal-free electrocatalytic hydrogen oxidation using frustrated Lewis pairs and carbon-based Lewis acids. Chem Sci. 2016;7:2537–2543. doi: 10.1039/c5sc04564a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Artero V, Berggren G, Atta M, Caserta G, Roy S, Pecqueur L, Fontecave M. From enzyme maturation to synthetic chemistry: the case of hydrogenases. Acc Chem Res. 2015;48:2380–2387. doi: 10.1021/acs.accounts.5b00157. [DOI] [PubMed] [Google Scholar]
- 20.Darensbourg MY. Hydrogenase active sites: a new paradigm for natural product-inspired synthesis based on organometallic chemistry. Comments Inorg Chem. 2010;31:144–152. [Google Scholar]
- 21.DuBois DL. Development of molecular electrocatalysts for energy storage. Inorg Chem. 2014;53:3935–3960. doi: 10.1021/ic4026969. [DOI] [PubMed] [Google Scholar]
- 22.Gloaguen F, Rauchfuss TB. Small molecule mimics of hydrogenases: hydrides and redox. Chem Soc Rev. 2009;38:100–108. doi: 10.1039/b801796b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heinekey DM. Hydrogenase enzymes: recent structural studies and active site models. J Organomet Chem. 2009;694:2671–2680. [Google Scholar]
- 24.Simmons TR, Berggren G, Bacchi M, Fontecave M, Artero V. Mimicking hydrogenases: from biomimetics to artificial enzymes. Coord Chem Rev. 2014;270–271:127–150. [Google Scholar]
- 25.Tard C, Pickett CJ. Structural and functional analogues of the active sites of the [Fe]-, [NiFe]-, and [FeFe]-hydrogenases. Chem Rev. 2009;109:2245–2274. doi: 10.1021/cr800542q. [DOI] [PubMed] [Google Scholar]
- 26.Xu T, Chen D, Hu X. Hydrogen-activating models of hydrogenases. Coord Chem Rev. 2015;303:32–41. [Google Scholar]
- 27.Yang JY, Bullock RM, Rakowski DuBois M, DuBois DL. Fast and efficient molecular electrocatalysts for H2 production: using hydrogenase enzymes as guides. MRS Bull. 2011;36:39–47. [Google Scholar]
- 28.Matsumoto T, Kim K, Nakai H, Hibino T, Ogo S. Organometallic catalysts for use in a fuel cell. ChemCatChem. 2013;5:1368–1373. [Google Scholar]
- 29.Matsumoto T, Kim K, Ogo S. Molecular catalysis in a fuel cell. Angew Chem, Int Ed. 2011;50:11202–11205. doi: 10.1002/anie.201104498. [DOI] [PubMed] [Google Scholar]
- 30.Tran PD, Morozan A, Archambault S, Heidkamp J, Chenevier P, Dau H, Fontecave M, Martinent A, Jousselme B, Artero V. A noble metal-free proton-exchange membrane fuel cell based on bio-inspired molecular catalysts. Chem Sci. 2015;6:2050–2053. doi: 10.1039/c4sc03774j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakazawa H, Itazaki M. Fe–H complexes in catalysis. Top Organomet Chem. 2011;33:27–81. [Google Scholar]
- 32.Crabtree RH. Dihydrogen complexation. Chem Rev. 2016 doi: 10.1021/acs.chemrev.6b00037. [DOI] [PubMed] [Google Scholar]
- 33.Kubas GJ. Metal–dihydrogen and σ-bond coordination: the consummate extension of the Dewar–Chatt–Duncanson model for metal–olefin π bonding. J Organomet Chem. 2001;635:37–68. [Google Scholar]
- 34.Gordon JC, Kubas GJ. Perspectives on how nature employs the principles of organometallic chemistry in dihydrogen activation in hydrogenases. Organometallics. 2010;29:4682–4701. [Google Scholar]
- 35.Kubas GJ. Fundamentals of H2 binding and reactivity on transition metals underlying hydrogenase function and H2 production and storage. Chem Rev. 2007;107:4152–4205. doi: 10.1021/cr050197j. [DOI] [PubMed] [Google Scholar]
- 36.Lill SON, Siegbahn PEM. An autocatalytic mechanism for NiFe-hydrogenase: reduction to Ni(I) followed by oxidative addition. Biochemistry. 2009;48:1056–1066. doi: 10.1021/bi801218n. [DOI] [PubMed] [Google Scholar]
- 37.Kubas GJ. Catalytic processes involving dihydrogen complexes and other sigma-bond complexes. Catal Lett. 2005;104:79–101. [Google Scholar]
- 38.Rokob TA, Bakó I, Stirling A, Hamza A, Pápai I. Reactivity models of hydrogen activation by frustrated Lewis pairs: synergistic electron transfers or polarization by electric field? J Am Chem Soc. 2013;135:4425–4437. doi: 10.1021/ja312387q. [DOI] [PubMed] [Google Scholar]
- 39.Stephan DW, Erker G. Frustrated Lewis pair chemistry: development and perspectives. Angew Chem, Int Ed. 2015;54:6400–6441. doi: 10.1002/anie.201409800. [DOI] [PubMed] [Google Scholar]
- 40.Abdur-Rashid K, Fong TP, Greaves B, Gusev DG, Hinman JG, Landau SE, Lough AJ, Morris RH. An acidity scale for phosphorus-containing compounds including metal hydrides and dihydrogen complexes in THF: toward the unification of acidity scales. J Am Chem Soc. 2000;122:9155–9171. [Google Scholar]
- 41.Morris RH. Estimating the acidity of transition metal hydride and dihydrogen complexes by adding ligand acidity constants. J Am Chem Soc. 2014;136:1948–1959. doi: 10.1021/ja410718r. [DOI] [PubMed] [Google Scholar]
- 42.Landau SE, Morris RH, Lough AJ. Acidic dicationic iron(II) dihydrogen complexes and compounds related by H2 substitution. Inorg Chem. 1999;38:6060–6068. doi: 10.1021/ic990876a. [DOI] [PubMed] [Google Scholar]
- 43.Forde CE, Landau SE, Morris RH. Dicationic iron(II) complexes with dihydrogen trans to π-acid ligands: trans-[Fe(H2)(L)-(dppe)2]2+ (L = CO or CNH). Is there Fe H2 π-back bonding? J Chem Soc, Dalton Trans. 1997:1663–1664. [Google Scholar]
- 44.Morris RH. Brønsted-Lowry acid strength of metal hydride and dihydrogen Complexes. Chem Rev. 2016 doi: 10.1021/acs.chemrev.5b00695. [DOI] [PubMed] [Google Scholar]
- 45.Fong TP, Forde CE, Lough AJ, Morris RH, Rigo P, Rocchini E, Stephan T. Synthesis of the acidic dihydrogen complexes trans-[M(H2)(CN)L2]+ and trans-[M(H2)(CNH)L2]2+ where M = Fe, Ru, Os and L = dppm, dppe, dppp, depe, and dihydrogen substitution by the trifluoromethanesulfonate anion to give trans-[Ru(OTf)(CN)L2] or trans-[Ru(OTf)(CNH)L2]OTf. J Chem Soc, Dalton Trans. 1999:4475–4486. [Google Scholar]
- 46.Chatt J, Kan CT, Leigh GJ, Pickett CJ, Stanley DR. Transition-metal binding sites and ligand parameters. J Chem Soc, Dalton Trans. 1980:2032. [Google Scholar]
- 47.Lever ABP. Electrochemical parametrization of metal complex redox potentials, using the ruthenium(III)/ruthenium(II) couple to generate a ligand electrochemical series. Inorg Chem. 1990;29:1271–1285. [Google Scholar]
- 48.Moore EJ, Sullivan JM, Norton JR. Kinetic and thermodynamic acidity of hydrido transition-metal complexes. 3. Thermodynamic acidity of common mononuclear carbonyl hydrides. J Am Chem Soc. 1986;108:2257–2263. doi: 10.1021/ja00269a022. [DOI] [PubMed] [Google Scholar]
- 49.Fourmond V, Jacques P-A, Fontecave M, Artero V. H2 evolution and molecular electrocatalysts: determination of overpotentials and effect of homoconjugation. Inorg Chem. 2010;49:10338–10347. doi: 10.1021/ic101187v. [DOI] [PubMed] [Google Scholar]
- 50.Izutsu K. Acid-Base Dissociation Constants in Dipolar Aprotic Solvents. Blackwell Scientific Publications; Oxford, U.K: 1990. [Google Scholar]
- 51.Kaljurand I, Kütt A, Sooväli L, Rodima T, Mäemets V, Leito I, Koppel IA. Extension of the self-consistent spectrophotometric basicity scale in acetonitrile to a full span of 28 pKa units: unification of different basicity scales. J Org Chem. 2005;70:1019–1028. doi: 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
- 52.Qi X-J, Fu Y, Liu L, Guo Q-X. Ab initio calculations of thermodynamic hydricities of transition-metal hydrides in acetonitrile. Organometallics. 2007;26:4197–4203. [Google Scholar]
- 53.Berning DE, Noll BC, DuBois DL. Relative hydride, proton, and hydrogen atom transfer abilities of [HM(diphosphine)2]PF6 complexes (M = Pt, Ni) J Am Chem Soc. 1999;121:11432–11447. [Google Scholar]
- 54.Tsay C, Livesay BN, Ruelas S, Yang JY. Solvation effects on transition metal hydricity. J Am Chem Soc. 2015;137:14114–14121. doi: 10.1021/jacs.5b07777. [DOI] [PubMed] [Google Scholar]
- 55.Nimlos MR, Chang CH, Curtis CJ, Miedaner A, Pilath HM, DuBois DL. Calculated hydride donor abilities of five-coordinate transition metal hydrides [HM(diphosphine)2]+ (M = Ni, Pd, Pt) as a function of the bite angle and twist angle of diphosphine ligands. Organometallics. 2008;27:2715–2722. [Google Scholar]
- 56.Raebiger JW, Miedaner A, Curtis CJ, Miller SM, Anderson OP, DuBois DL. Using ligand bite angles to control the hydricity of palladium diphosphine complexes. J Am Chem Soc. 2004;126:5502–5514. doi: 10.1021/ja0395240. [DOI] [PubMed] [Google Scholar]
- 57.Chen S, Rousseau R, Raugei S, Dupuis M, DuBois DL, Bullock RM. Comprehensive thermodynamics of nickel hydride bis(diphosphine) complexes: a predictive model through computations. Organometallics. 2011;30:6108–6118. [Google Scholar]
- 58.Tilset M, Parker VD. Solution homolytic bond dissociation energies of organotransition-metal hydrides. J Am Chem Soc. 1989;111:6711–6717. [Google Scholar]
- 59.Tilset M, Parker VD. Solution homolytic bond dissociation energies of organotransition-metal hydrides [J. Am. Chem. Soc. 1989, 111, 6711] J Am Chem Soc. 1990;112:2843. [Google Scholar]
- 60.Choi J, Pulling ME, Smith DM, Norton JR. Unusually weak metal–hydrogen bonds in HV(CO)4(P–P) and their effectiveness as H• donors. J Am Chem Soc. 2008;130:4250–4252. doi: 10.1021/ja710455c. [DOI] [PubMed] [Google Scholar]
- 61.Tilset M, Fjeldahl I, Hamon J-R, Hamon P, Toupet L, Saillard J-Y, Costuas K, Haynes A. Theoretical, thermodynamic, spectroscopic, and structural studies of the consequences of one-electron oxidation on the Fe-X bonds in 17- and 18-electron Cp*Fe(dppe)X complexes (X = F, Cl, Br, I, H, CH3. J Am Chem Soc. 2001;123:9984–10000. doi: 10.1021/ja0106927. [DOI] [PubMed] [Google Scholar]
- 62.Tilset M. In: Electron Transfer in Chemistry. Balzani V, editor. Wiley; 2001. [Google Scholar]
- 63.Barton BE, Rauchfuss TB. Hydride-containing models for the active site of the nickel-iron hydrogenases. J Am Chem Soc. 2010;132:14877–14885. doi: 10.1021/ja105312p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 65.Cammack R, Fernandez VM, Hatchikian EC. [5] Nickel-iron Hydrogenases. Methods Enzymol. 1994;243:43–68. [Google Scholar]
- 66.Camara JM, Rauchfuss TB. Combining acid–base, redox and substrate binding functionalities to give a complete model for the [FeFe]-hydrogenase. Nat Chem. 2011;4:26–30. doi: 10.1038/nchem.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tard C, Liu X, Ibrahim SK, Bruschi M, De Gioia L, Davies SC, Yang X, Wang L-S, Sawers G, Pickett CJ. Synthesis of the H-cluster framework of iron-only hydrogenase. Nature. 2005;433:610–613. doi: 10.1038/nature03298. [DOI] [PubMed] [Google Scholar]
- 68.Connelly NG, Geiger WE. Chemical redox agents for organometallic chemistry. Chem Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 69.Pavlishchuk VV, Addison AW. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorg Chim Acta. 2000;298:97–102. [Google Scholar]
- 70.Krossing I, Raabe I. Noncoordinating anions—fact or fiction? A survey of likely candidates. Angew Chem, Int Ed. 2004;43:2066–2090. doi: 10.1002/anie.200300620. [DOI] [PubMed] [Google Scholar]
- 71.Nishida H, Takada N, Yoshimura M, Sonoda T, Kobayashi H. Tetrakis[3,5-bis(trifluoromethyl)phenyl]borate. Highly lipophilic stable anionic agent for solvent-extraction of cations. Bull Chem Soc Jpn. 1984;57:2600–2604. [Google Scholar]
- 72.Geiger WE, Barrière F. Organometallic electrochemistry based on electrolytes containing weakly-coordinating fluoroarylborate anions. Acc Chem Res. 2010;43:1030–1039. doi: 10.1021/ar1000023. [DOI] [PubMed] [Google Scholar]
- 73.Rountree ES, McCarthy BD, Eisenhart TT, Dempsey JL. Evaluation of homogeneous electrocatalysts by cyclic voltammetry. Inorg Chem. 2014;53:9983–10002. doi: 10.1021/ic500658x. [DOI] [PubMed] [Google Scholar]
- 74.Best SP. Spectroelectrochemistry of hydrogenase enzymes and related compounds. Coord Chem Rev. 2005;249:1536–1554. [Google Scholar]
- 75.Armstrong FA, Belsey NA, Cracknell JA, Goldet G, Parkin A, Reisner E, Vincent KA, Wait AF. Dynamic electrochemical investigations of hydrogen oxidation and production by enzymes and implications for future technology. Chem Soc Rev. 2009;38:36–51. doi: 10.1039/b801144n. [DOI] [PubMed] [Google Scholar]
- 76.Nicholson RS, Shain I. Theory of stationary electrode polarography: single scan and cyclic methods applied to reversible, irreversible, and kinetic systems. Anal Chem. 1964;36:706–723. [Google Scholar]
- 77.Savéant J-M, Vianello E. Potential-sweep chronoamperometry: kinetic currents for first-order chemical reaction parallel to electron-transfer process (catalytic currents) Electrochim Acta. 1965;10:905–920. [Google Scholar]
- 78.Felton GAN, Glass RS, Lichtenberger DL, Evans DH. Iron-only hydrogenase mimics. Thermodynamic aspects of the use of electrochemistry to evaluate catalytic efficiency for hydrogen generation. Inorg Chem. 2006;45:9181–9184. doi: 10.1021/ic060984e. [DOI] [PubMed] [Google Scholar]
- 79.Roberts JAS, Bullock RM. Direct determination of equilibrium potentials for hydrogen oxidation/production by open circuit potential measurements in acetonitrile. Inorg Chem. 2013;52:3823–3835. doi: 10.1021/ic302461q. [DOI] [PubMed] [Google Scholar]
- 80.Appel AM, Helm ML. Determining the overpotential for a molecular electrocatalyst. ACS Catal. 2014;4:630–633. [Google Scholar]
- 81.Felton GAN, Mebi CA, Petro BJ, Vannucci AK, Evans DH, Glass RS, Lichtenberger DL. Review of electrochemical studies of complexes containing the Fe2S2 core characteristic of [FeFe]-hydrogenases including catalysis by these complexes of the reduction of acids to form dihydrogen. J Organomet Chem. 2009;694:2681–2699. [Google Scholar]
- 82.McCarthy BD, Martin DJ, Rountree ES, Ullman AC, Dempsey JL. Electrochemical reduction of Brønsted acids by glassy carbon in acetonitrile—implications for electrocatalytic hydrogen evolution. Inorg Chem. 2014;53:8350–8361. doi: 10.1021/ic500770k. [DOI] [PubMed] [Google Scholar]
- 83.Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. 2nd. John Wiley & Sons, Inc; Hoboken, NJ: 2001. [Google Scholar]
- 84.Costentin C, Drouet S, Robert M, Savéant J-M. Turnover numbers, turnover frequencies, and overpotential in molecular catalysis of electrochemical reactions. cyclic voltammetry and preparative-scale electrolysis. J Am Chem Soc. 2012;134:11235–11242. doi: 10.1021/ja303560c. [DOI] [PubMed] [Google Scholar]
- 85.Felton GAN, Vannucci AK, Chen J, Lockett LT, Okumura N, Petro BJ, Zakai UI, Evans DH, Glass RS, Lichtenberger DL. Hydrogen generation from weak acids: electrochemical and computational studies of a diiron hydrogenase mimic. J Am Chem Soc. 2007;129:12521–12530. doi: 10.1021/ja073886g. [DOI] [PubMed] [Google Scholar]
- 86.Bullock RM, Appel AM, Helm ML. Production of hydrogen by electrocatalysis: making the H–H bond by combining protons and hydrides. Chem Commun. 2014;50:3125–3143. doi: 10.1039/c3cc46135a. [DOI] [PubMed] [Google Scholar]
- 87.Costentin C, Passard G, Savéant J-M. Benchmarking of homogeneous electrocatalysts: overpotential, turnover frequency, limiting turnover number. J Am Chem Soc. 2015;137:5461–5467. doi: 10.1021/jacs.5b00914. [DOI] [PubMed] [Google Scholar]
- 88.Woińska M, Grabowsky S, Dominiak PM, WoŸniak K, Jayatilaka D. Hydrogen atoms can be located accurately and precisely by x-ray crystallography. Sci Adv. 2016;2:e1600192. doi: 10.1126/sciadv.1600192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ogata H, Nishikawa K, Lubitz W. Hydrogens detected by subatomic resolution protein crystallography in a [NiFe] hydrogenase. Nature. 2015;520:571–574. doi: 10.1038/nature14110. [DOI] [PubMed] [Google Scholar]
- 90.Elias M, Liebschner D, Koepke J, Lecomte C, Guillot B, Jelsch C, Chabriere E. Hydrogen atoms in protein structures: high-resolution X-ray diffraction structure of the DFPase. BMC Res Notes. 2013;6:308. doi: 10.1186/1756-0500-6-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Piccoli PMB, Koetzle TF, Schultz AJ. Single crystal neturon diffraction for the inorganic chemist - a practical guide. Comments Inorg Chem. 2007;28:3–38. [Google Scholar]
- 92.Bau R, Drabnis MH. Structures of transition metal hydrides determined by neutron diffraction. Inorg Chim Acta. 1997;259:27–50. [Google Scholar]
- 93.Gruene T, Hahn HW, Luebben AV, Meilleur F, Sheldrick GM. Refinement of macromolecular structures against neutron data with SHELXL2013. J Appl Crystallogr. 2014;47:462–466. doi: 10.1107/S1600576713027659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hrobarik P, Hrobáriková V, Meier F, Repiský M, Komorovský S, Kaupp M. Relativistic four-component DFT calculations of 1H NMR chemical shifts in transition-metal hydride complexes: unusual high-field shifts beyond the Buckingham–Stephens model. J Phys Chem A. 2011;115:5654–5659. doi: 10.1021/jp202327z. [DOI] [PubMed] [Google Scholar]
- 95.Lubitz W, Reijerse E, van Gastel M. [NiFe] and [FeFe] Hydrogenases studied by advanced magnetic resonance techniques. Chem Rev. 2007;107:4331–4365. doi: 10.1021/cr050186q. [DOI] [PubMed] [Google Scholar]
- 96.Whitehead JP, Gurbiel RJ, Bagyinka C, Hoffman BM, Maroney MJ. The Hydrogen Binding Site in Hydrogenase: 35-GHz ENDOR and XAS studies of the Ni-C active form and the Ni-L photoproduct. J Am Chem Soc. 1993;115:5629–5635. [Google Scholar]
- 97.Socrates G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts. 3rd. Wiley; 2004. [Google Scholar]
- 98.Nakamoto K. Infrared and Raman Spectra of Inorganic and Coordination Compounds. 4th. Wiley; 1986. [Google Scholar]
- 99.Sturhahn W. Nuclear resonant spectroscopy. J Phys Condens Matter. 2004;16:S497–S530. [Google Scholar]
- 100.Sturhahn W, Toellner TS, Alp EE, Zhang X, Ando M, Yoda Y, Kikuta S, Seto M, Kimball CW, Dabrowski B. Phonon density of states measured by inelastic nuclear resonant scattering. Phys Rev Lett. 1995;74:3832–3835. doi: 10.1103/PhysRevLett.74.3832. [DOI] [PubMed] [Google Scholar]
- 101.Seto M, Yoda Y, Kikuta S, Zhang XW, Ando M. Observation of nuclear resonant scattering accompanied by phonon excitation using synchrotron radiation. Phys Rev Lett. 1995;74:3828–3831. doi: 10.1103/PhysRevLett.74.3828. [DOI] [PubMed] [Google Scholar]
- 102.Yoda Y, Yabashi M, Izumi K, Zhang XW, Kishimoto S, Kitao S, Seto M, Mitsui T, Harami T, Imai Y, Kikuta S. Nuclear resonant scattering beamline at SPring-8. Nucl Instrum Methods Phys Res, Sect A. 2001;467–478:715–718. [Google Scholar]
- 103.Ogata H, Krämer T, Wang H, Schilter D, Pelmenschikov V, van Gastel M, Neese F, Rauchfuss TB, Gee LB, Scott AD, et al. Hydride bridge in [NiFe]-hydrogenase observed by nuclear resonance vibrational spectroscopy. Nat Commun. 2015;6:7890–7897. doi: 10.1038/ncomms8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kamali S, Wang H, Mitra D, Ogata H, Lubitz W, Manor BC, Rauchfuss TB, Byrne D, Bonnefoy V, Jenny FE, Jr, et al. Observation of the Fe–CN and Fe–CO vibrations in the active site of [NiFe] hydrogenase by nuclear resonance vibrational spectroscopy. Angew Chem, Int Ed. 2013;52:724–728. doi: 10.1002/anie.201204616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nibbering ETJ, Fidder H, Pines E. Ultrafast chemistry: using time-resolved vibrational spectroscopy for interrogation of structural dynamics. Annu Rev Phys Chem. 2005;56:337–367. doi: 10.1146/annurev.physchem.56.092503.141314. [DOI] [PubMed] [Google Scholar]
- 106.Hunt NT, Wright JA, Pickett C. Detection of transient intermediates generated from subsite analogues of [FeFe] hydrogenases. Inorg Chem. 2016;55:399–410. doi: 10.1021/acs.inorgchem.5b02477. [DOI] [PubMed] [Google Scholar]
- 107.Greene BL, Wu C-H, McTernan PM, Adams MWW, Dyer RB. Proton-coupled electron transfer dynamics in the catalytic mechanism of a [NiFe]-hydrogenase. J Am Chem Soc. 2015;137:4558–4566. doi: 10.1021/jacs.5b01791. [DOI] [PubMed] [Google Scholar]
- 108.Jablonskytë A, Wright JA, Fairhurst SA, Peck JNT, Ibrahim SK, Oganesyan VS, Pickett CJ. Paramagnetic bridging hydrides of relevance to catalytic hydrogen evolution at metallosulfur centers. J Am Chem Soc. 2011;133:18606–18609. doi: 10.1021/ja2087536. [DOI] [PubMed] [Google Scholar]
- 109.Vidossich P, Magistrato A. QM/MM molecular dynamics studies of metal binding proteins. Biomolecules. 2014;4:616–645. doi: 10.3390/biom4030616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dal Peraro M, Ruggerone P, Raugei S, Gervasio FL, Carloni P. Investigating biological systems using first principles Car–Parrinello molecular dynamics simulations. Curr Opin Struct Biol. 2007;17:149–156. doi: 10.1016/j.sbi.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 111.Lin H, Truhlar DG. QM/MM: what have we learned, where are we, and where do we go from here? Theor Chem Acc. 2007;117:185–199. [Google Scholar]
- 112.van der Kamp MW, Mulholland AJ. Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry. 2013;52:2708–2728. doi: 10.1021/bi400215w. [DOI] [PubMed] [Google Scholar]
- 113.Saunders MG, Voth GA. Coarse-graining methods for computational biology. Annu Rev Biophys. 2013;42:73–93. doi: 10.1146/annurev-biophys-083012-130348. [DOI] [PubMed] [Google Scholar]
- 114.Finkelmann AR, Stiebritz MT, Reiher M. Kinetic modeling of hydrogen conversion at [Fe] hydrogenase active-site models. J Phys Chem B. 2013;117:4806–4817. doi: 10.1021/jp312662y. [DOI] [PubMed] [Google Scholar]
- 115.Ginovska-Pangovska B, Ho M-H, Linehan JC, Cheng Y, Dupuis M, Raugei S, Shaw WJ. Molecular dynamics study of the proposed proton transport pathways in [FeFe]-hydrogenase. Biochim Biophys Acta, Bioenerg. 2014;1837:131–138. doi: 10.1016/j.bbabio.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 116.Sumner I, Voth GA. Proton transport pathways in [NiFe]-Hydrogenase. J Phys Chem B. 2012;116:2917–2926. doi: 10.1021/jp208512y. [DOI] [PubMed] [Google Scholar]
- 117.Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects. Phys Rev. 1965;140:A1133–A1138. [Google Scholar]
- 118.Parr RG, Yang W. Density-Functional Theory of Atoms and Molecules. Oxford University Press; New York: 1989. [Google Scholar]
- 119.Bruschi M, Zampella G, Fantucci P, De Gioia L. DFT investigations of models related to the active site of [NiFe] and [Fe] hydrogenases. Coord Chem Rev. 2005;249:1620–1640. [Google Scholar]
- 120.Siegbahn PEM, Tye JW, Hall MB. Computational studies of [NiFe] and [FeFe] hydrogenases. Chem Rev. 2007;107:4414–4435. doi: 10.1021/cr050185y. [DOI] [PubMed] [Google Scholar]
- 121.Greco C, De Gioia L. Bioinspired Catalysis. Wiley-VCH Verlag GmbH & Co. KGaA; 2014. [Google Scholar]
- 122.Jensen KP, Roos BO, Ryde U. Performance of density functionals for first row transition metal systems. J Chem Phys. 2007;126:014103. doi: 10.1063/1.2406071. [DOI] [PubMed] [Google Scholar]
- 123.Bühl M, Reimann C, Pantazis DA, Bredow T, Neese F. Geometries of third-row transition-metal complexes from density-functional theory. J Chem Theory Comput. 2008;4:1449–1459. doi: 10.1021/ct800172j. [DOI] [PubMed] [Google Scholar]
- 124.Narendrapurapu BS, Richardson NA, Copan AV, Estep ML, Yang Z, Schaefer HF., III Investigating the effects of basis set on metal–metal and metal–ligand bond distances in stable transition metal carbonyls: performance of correlation consistent basis sets with 35 density functionals. J Chem Theory Comput. 2013;9:2930–2938. doi: 10.1021/ct4002398. [DOI] [PubMed] [Google Scholar]
- 125.Lee C, Yang W, Parr RG. Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B: Condens Matter Mater Phys. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 126.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 127.Perdew JP. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B: Condens Matter Mater Phys. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 128.Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A: At, Mol, Opt Phys. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 129.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 130.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple [Phys. Rev. Lett. 77, 3865 (1996)] Phys Rev Lett. 1997;78:1396. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 131.Adamo C, Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J Chem Phys. 1999;110:6158. [Google Scholar]
- 132.Tao J, Perdew JP, Staroverov VN, Scuseria GE. Climbing the density functional ladder: nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys Rev Lett. 2003;91:146401. doi: 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- 133.Zhao Y, Truhlar DG. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J Chem Phys. 2006;125:194101. doi: 10.1063/1.2370993. [DOI] [PubMed] [Google Scholar]
- 134.Chai J-D, Head-Gordon M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys. 2008;10:6615–6620. doi: 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- 135.Hariharan PC, Pople JA. Influence of polarization functions on MO hydrogenation energies. Theoret Chim Acta. 1973;28:213–222. [Google Scholar]
- 136.Clark T, Chandrasekhar J, Spitznagel GW, Schleyer PVR. Efficient diffuse function-augmented basis sets for anion calculations. III.† The 3-21+G basis set for first-row elements, Li–F. J Comput Chem. 1983;4:294–301. [Google Scholar]
- 137.Hay PJ, Wadt WR. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J Chem Phys. 1985;82:299–310. [Google Scholar]
- 138.Dolg M, Wedig U, Stoll H, Preuss H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J Chem Phys. 1987;86:866–872. [Google Scholar]
- 139.Frenking G, Antes I, Böhme M, Dapprich S, Ehlers AW, Jonas V, Neuhaus A, Otto M, Stegmann R, Veldkamp A, Vyboishchikov SF. Pseudopotential calculations of transition metal compounds: scope and limitations. Rev Comput Chem. 1996;8:63–144. [Google Scholar]
- 140.Cramer CJ, Truhlar DG. Density functional theory for transition metals and transition metal chemistry. Phys Chem Chem Phys. 2009;11:10757–10816. doi: 10.1039/b907148b. [DOI] [PubMed] [Google Scholar]
- 141.Solis BH, Hammes-Schiffer S. Proton-coupled electron transfer in molecular electrocatalysis: theoretical methods and design principles. Inorg Chem. 2014;53:6427–6443. doi: 10.1021/ic5002896. [DOI] [PubMed] [Google Scholar]
- 142.Miertuš S, Scrocco E, Tomasi J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of ab initio molecular potentials for the prevision of solvent effects. Chem Phys. 1981;55:117–129. [Google Scholar]
- 143.Miertuš S, Tomasi J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem Phys. 1982;65:239–245. [Google Scholar]
- 144.Barone V, Cossi M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J Phys Chem A. 1998;102:1995. [Google Scholar]
- 145.Cossi M, Rega N, Scalmani G, Barone V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J Comput Chem. 2003;24:669–681. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- 146.Marenich AV, Cramer CJ, Truhlar DG. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 147.Klamt A, Schüürmann G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc, Perkin Trans. 1993;2:799–805. [Google Scholar]
- 148.Baik M-H, Friesner RA. Computing redox potentials in solution: density functional theory as a tool for rational design of redox agents. J Phys Chem A. 2002;106:7407–7412. [Google Scholar]
- 149.Tsai M-K, Rochford J, Polyansky DE, Wada T, Tanaka K, Fujita E, Muckerman JT. Characterization of redox states of Ru(OH2)(Q)(tpy)2+ (Q = 3,5-di-tert-butyl-1,2-benzoquinone, tpy = 2,2′:6′,2″-terpyridine) and related species through experimental and theoretical studies. Inorg Chem. 2009;48:4372–4383. doi: 10.1021/ic900057y. [DOI] [PubMed] [Google Scholar]
- 150.Wang T, Brudvig G, Batista VS. Characterization of proton coupled electron transfer in a biomimetic oxomanganese complex: evaluation of the DFT B3LYP level of theory. J Chem Theory Comput. 2010;6:755–760. doi: 10.1021/ct900615b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sundstrom EJ, Yang X, Thoi VS, Karunadasa HI, Chang CJ, Long JR, Head-Gordon M. Computational and experimental study of the mechanism of hydrogen generation from water by a molecular molybdenum-oxo electrocatalyst. J Am Chem Soc. 2012;134:5233–5242. doi: 10.1021/ja210949r. [DOI] [PubMed] [Google Scholar]
- 152.Keith JA, Grice KA, Kubiak CP, Carter EA. Elucidation of the selectivity of proton-dependent electrocatalytic CO2 reduction by fac-Re(bpy)(CO)3Cl. J Am Chem Soc. 2013;135:15823–15829. doi: 10.1021/ja406456g. [DOI] [PubMed] [Google Scholar]
- 153.Roy LE, Batista ER, Hay PJ. Theoretical studies on the redox potentials of Fe dinuclear complexes as models for hydrogenase. Inorg Chem. 2008;47:9228–9237. doi: 10.1021/ic800541w. [DOI] [PubMed] [Google Scholar]
- 154.Roy LE, Jakubikova E, Guthrie MG, Batista ER. Calculation of one-electron redox potentials revisited. Is it possible to calculate accurate potentials with density functional methods? J Phys Chem A. 2009;113:6745–6750. doi: 10.1021/jp811388w. [DOI] [PubMed] [Google Scholar]
- 155.Muckerman JT, Fujita E. Theoretical studies of the mechanism of catalytic hydrogen production by a cobaloxime. Chem Commun. 2011;47:12456–12458. doi: 10.1039/c1cc15330g. [DOI] [PubMed] [Google Scholar]
- 156.Konezny SJ, Doherty MD, Luca OR, Crabtree RH, Soloveichik GL, Batista VS. Reduction of systematic uncertainty in DFT redox potentials of transition-metal complexes. J Phys Chem C. 2012;116:6349–6356. [Google Scholar]
- 157.Fernandez LE, Horvath S, Hammes-Schiffer S. Theoretical analysis of the sequential proton-coupled electron transfer mechanisms for H2 oxidation and production pathways catalyzed by nickel molecular electrocatalysts. J Phys Chem C. 2012;116:3171–3180. [Google Scholar]
- 158.Solis BH, Hammes-Schiffer S. Theoretical analysis of mechanistic pathways for hydrogen evolution catalyzed by cobaloximes. Inorg Chem. 2011;50:11252–11262. doi: 10.1021/ic201842v. [DOI] [PubMed] [Google Scholar]
- 159.Chambers GM, Huynh MT, Li Y, Hammes-Schiffer S, Rauchfuss TB, Reijerse E, Lubitz W. Models of the Ni-L and Ni-SIa states of the [NiFe]-hydrogenase active site. Inorg Chem. 2016;55:419–431. doi: 10.1021/acs.inorgchem.5b01662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Huynh MT, Schilter D, Hammes-Schiffer S, Rauchfuss TB. Protonation of nickel–iron hydrogenase models proceeds after isomerization at nickel. J Am Chem Soc. 2014;136:12385–12395. doi: 10.1021/ja505783z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Huynh MT, Wang W, Rauchfuss TB, Hammes-Schiffer S. Characterization of singly and doubly protonated intermediates and mechanistic insights. Inorg Chem. 2014;53:10301–10311. doi: 10.1021/ic5013523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Scott AP, Radom L. Harmonic vibrational frequencies: an evaluation of Hartree-Fock, Møller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J Phys Chem. 1996;100:16502–16513. [Google Scholar]
- 163.Alecu IM, Zheng J, Zhao Y, Truhlar DG. Computational thermochemistry: scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries. J Chem Theory Comput. 2010;6:2872–2887. doi: 10.1021/ct100326h. [DOI] [PubMed] [Google Scholar]
- 164.Neugebauer J, Hess BA. Fundamental vibrational frequencies of small polyatomic molecules from density-functional calculations and vibrational perturbation theory. J Chem Phys. 2003;118:7215–7225. [Google Scholar]
- 165.Schreckenbach G, Ziegler T. Calculation of the g-tensor of electron paramagnetic resonance spectroscopy using gauge-including atomic orbitals and density functional theory. J Phys Chem A. 1997;101:3388–3399. [Google Scholar]
- 166.Kaupp M. In: EPR of Free Radicals in Solids I. Lund A, Shiotani M, editors. Springer; New York: 2003. [Google Scholar]
- 167.Kaupp M, Remenyi C, Vaara J, Malkina OL, Malkin VG. Density functional calculations of electronic g-tensors for semiquinone radical anions. The role of hydrogen bonding and substituent effects. J Am Chem Soc. 2002;124:2709–2722. doi: 10.1021/ja0162764. [DOI] [PubMed] [Google Scholar]
- 168.Neese F. Prediction of electron paramagnetic resonance g values using coupled perturbed Hartree–Fock and Kohn–Sham theory. J Chem Phys. 2001;115:11080. [Google Scholar]
- 169.Kaupp M, Reviakine R, Malkina OL, Arbuznikov A, Schimmelpfennig B, Malkin VG. Calculation of electronic g tensors for transition metal complexes using hybrid density functionals and atomic meanfield spin-orbit operators. J Comput Chem. 2002;23:794–803. doi: 10.1002/jcc.10049. [DOI] [PubMed] [Google Scholar]
- 170.Neese F. Efficient and accurate approximations to the molecular spin-orbit coupling operator and their use in molecular g-tensor calculations. J Chem Phys. 2005;122:034107. doi: 10.1063/1.1829047. [DOI] [PubMed] [Google Scholar]
- 171.Munzarová M, Kaupp M. A critical validation of density functional and coupled-cluster approaches for the calculation of EPR hyperfine coupling constants in transition metal complexes. J Phys Chem A. 1999;103:9966–9983. [Google Scholar]
- 172.Munzarová M, Kubáček P, Kaupp M. Mechanisms of EPR hyperfine coupling in transition metal complexes. J Am Chem Soc. 2000;122:11900–11913. [Google Scholar]
- 173.Neese F. Prediction of molecular properties and molecular spectroscopy with density functional theory: from fundamental theory to exchange-coupling. Coord Chem Rev. 2009;253:526–563. [Google Scholar]
- 174.Zhang Y, Mao J, Oldfield E. 57Fe Mössbauer isomer shifts of heme protein model systems: electronic structure calculations. J Am Chem Soc. 2002;124:7829–7839. doi: 10.1021/ja011583v. [DOI] [PubMed] [Google Scholar]
- 175.Liu T, Lovell T, Han W-G, Noodleman L. DFT calculations of isomer shifts and quadrupole splitting parameters in synthetic iron-oxo complexes: applications to methane monooxygenase and ribonucleotide reductase. Inorg Chem. 2003;42:5244–5251. doi: 10.1021/ic020640y. [DOI] [PubMed] [Google Scholar]
- 176.Sinnecker S, Slep LD, Bill E, Neese F. Performance of nonrelativistic and quasi-relativistic hybrid DFT for the prediction of electric and magnetic hyperfine parameters in 57Fe Mössbauer spectra. Inorg Chem. 2005;44:2245–2254. doi: 10.1021/ic048609e. [DOI] [PubMed] [Google Scholar]
- 177.Neese F. Metal and ligand hyperfine couplings in transition metal complexes: the effect of spin–orbit coupling as studied by coupled perturbed Kohn–Sham theory. J Chem Phys. 2003;118:3939–3948. [Google Scholar]
- 178.Sonnenberg JL, Schlegel HB, Hratchian HP. Encyclopedia of Inorganic and Bioinorganic Chemistry. John Wiley & Sons, Inc; 2011. [Google Scholar]
- 179.Kampa M, Pandelia M-E, Lubitz W, van Gastel M, Neese F. A Metal–metal bond in the light-induced state of [NiFe] hydrogenases with relevance to hydrogen evolution. J Am Chem Soc. 2013;135:3915–3925. doi: 10.1021/ja3115899. [DOI] [PubMed] [Google Scholar]
- 180.Schilter D, Nilges MJ, Chakrabarti M, Lindahl PA, Rauchfuss TB, Stein M. Mixed-valence nickel–iron dithiolate models of the [NiFe]-hydrogenase active site. Inorg Chem. 2012;51:2338–2348. doi: 10.1021/ic202329y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Stein M, Lubitz W. Relativistic DFT calculation of the reaction cycle intermediates of [NiFe] hydrogenase: a contribution to understanding the enzymatic mechanism. J Inorg Biochem. 2004;98:862–877. doi: 10.1016/j.jinorgbio.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 182.Stein M, van Lenthe E, Baerends EJ, Lubitz W. Relativistic DFT calculations of the paramagnetic intermediates of [NiFe] hydrogenase. implications for the enzymatic mechanism. J Am Chem Soc. 2001;123:5839–5840. doi: 10.1021/ja005808y. [DOI] [PubMed] [Google Scholar]
- 183.Stadler C, de Lacey AL, Hernández B, Fernández VM, Conesa JC. Density functional calculations for modeling the oxidized states of the active site of nickel–iron hydrogenases. 1. Verification of the method with paramagnetic Ni and Co Complexes. Inorg Chem. 2002;41:4417–4423. doi: 10.1021/ic020015t. [DOI] [PubMed] [Google Scholar]
- 184.Hsieh C-H, Erdem OF, Harman SD, Singleton ML, Reijerse E, Lubitz W, Popescu CV, Reibenspies JH, Brothers SM, Hall MB, Darensbourg MY. Structural and spectroscopic features of mixed valent FeIIFeI complexes and factors related to the rotated configuration of diiron hydrogenase. J Am Chem Soc. 2012;134:13089–13102. doi: 10.1021/ja304866r. [DOI] [PubMed] [Google Scholar]
- 185.Silakov A, Olsen MT, Sproules S, Reijerse EJ, Rauchfuss TB, Lubitz W. EPR/ENDOR, Mössbauer, and quantum-chemical investigations of diiron complexes mimicking the active oxidized state of [FeFe] hydrogenase. Inorg Chem. 2012;51:8617–8628. doi: 10.1021/ic3013766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Filippi G, Arrigoni F, Bertini L, De Gioia L, Zampella G. DFT Dissection of the Reduction Step in H2 catalytic production by [FeFe]-hydrogenase inspired models: can the bridging hydride become more reactive than the terminal isomer? Inorg Chem. 2015;54:9529–9542. doi: 10.1021/acs.inorgchem.5b01495. [DOI] [PubMed] [Google Scholar]
- 187.Greco C. Towards [NiFe]-hydrogenase biomimetic models that couple H2 binding with functionally relevant intramolecular electron transfers: a quantum chemical study. Dalton Trans. 2013;42:13845–13854. doi: 10.1039/c3dt50836f. [DOI] [PubMed] [Google Scholar]
- 188.Greco C. H2 binding and splitting on a new-generation [FeFe]-hydrogenase model featuring a redox-active decamethylferrocenyl phosphine ligand: a theoretical investigation. Inorg Chem. 2013;52:1901–1908. doi: 10.1021/ic302118h. [DOI] [PubMed] [Google Scholar]
- 189.Greco C, De Gioia L. A theoretical study on the enhancement of functionally relevant electron transfers in biomimetic models of [FeFe]-hydrogenases. Inorg Chem. 2011;50:6987–6995. doi: 10.1021/ic200297d. [DOI] [PubMed] [Google Scholar]
- 190.McGlynn SE, Mulder DW, Shepard EM, Broderick JB, Peters JW. Hydrogenase cluster biosynthesis: organometallic chemistry nature’s way. Dalton Trans. 2009:4274–4285. doi: 10.1039/b821432h. [DOI] [PubMed] [Google Scholar]
- 191.Pandey AS, Harris TV, Giles LJ, Peters JW, Szilagyi RK. Dithiomethylether as a ligand in the hydrogenase H-cluster. J Am Chem Soc. 2008;130:4533–4540. doi: 10.1021/ja711187e. [DOI] [PubMed] [Google Scholar]
- 192.Adams MWW. The Mechanisms of H2 activation and CO binding by hydrogenase I and hydrogenase II of Clostridium pasteurianum. J Biol Chem. 1987;262:15054–15061. [PubMed] [Google Scholar]
- 193.Pierik AJ, Roseboom W, Happe RP, Bagley KA, Albracht SPJ. Carbon monoxide and cyanide as intrinsic ligands to iron in the active site of [NiFe]-hydrogenases. J Biol Chem. 1999;274:3331–3337. doi: 10.1074/jbc.274.6.3331. [DOI] [PubMed] [Google Scholar]
- 194.Knörzer P, Silakov A, Foster CE, Armstrong FA, Lubitz W, Happe T. Importance of the protein framework for catalytic activity of [FeFe]-hydrogenases. J Biol Chem. 2012;287:1489–1499. doi: 10.1074/jbc.M111.305797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chernev P, Lambertz C, Brünje A, Leidel N, Sigfridsson KGV, Kositzki R, Hsieh C-H, Yao S, Schiwon R, Driess M, Limberg C, Happe T, Haumann M. Hydride binding to the active site of [FeFe]-hydrogenase. Inorg Chem. 2014;53:12164–12177. doi: 10.1021/ic502047q. [DOI] [PubMed] [Google Scholar]
- 196.Tschierlei S, Ott S, Lomoth R. Spectroscopically characterized intermediates of catalytic H2 formation by [FeFe] hydrogenase models. Energy Environ Sci. 2011;4:2340–2352. [Google Scholar]
- 197.Kramarz KW, Norton JR. Slow Proton-Transfer Reactions in Organometallic and Bioinorganic Chemistry. Prog Inorg Chem. 1994;42:1–65. [Google Scholar]
- 198.Mulder DW, Ratzloff MW, Bruschi M, Greco C, Koonce E, Peters JW, King PW. Investigations on the Role of Proton-Coupled Electron Transfer in Hydrogen Activation by [FeFe]-Hydrogenase. J Am Chem Soc. 2014;136:15394–15402. doi: 10.1021/ja508629m. [DOI] [PubMed] [Google Scholar]
- 199.Berggren G, Adamska A, Lambertz C, Simmons TR, Esselborn J, Atta M, Gambarelli S, Mouesca J-M, Reijerse E, Lubitz W, Happe T, Artero V, Fontecave M. Biomimetic assembly and activation of [FeFe]-hydrogenases. Nature. 2013;499:66–70. doi: 10.1038/nature12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Esselborn J, Lambertz C, Adamska-Venkatesh A, Simmons T, Berggren G, Noth J, Siebel J, Hemschemeier A, Artero V, Reijerse E, Fontecave M, Lubitz W, Happe T. Spontaneous activation of [FeFe]-hydrogenases by an inorganic [2Fe] active site mimic. Nat Chem Biol. 2013;9:607–610. doi: 10.1038/nchembio.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Siebel JF, Adamska-Venkatesh A, Weber K, Rumpel S, Reijerse E, Lubitz W. Hybrid [FeFe]-hydrogenases with modified active sites show remarkable residual enzymatic activity. Biochemistry. 2015;54:1474–1483. doi: 10.1021/bi501391d. [DOI] [PubMed] [Google Scholar]
- 202.Adamska-Venkatesh A, Krawietz D, Siebel J, Weber K, Happe T, Reijerse E, Lubitz W. New redox states observed in [FeFe] hydrogenases reveal redox coupling within the H-cluster. J Am Chem Soc. 2014;136:11339–11346. doi: 10.1021/ja503390c. [DOI] [PubMed] [Google Scholar]
- 203.Adamska A, Silakov A, Lambertz C, Rüdiger O, Happe T, Reijerse E, Lubitz W. Identification and characterization of the “super-reduced” state of the H-cluster in [FeFe] hydrogenase: a new building block for the catalytic cycle? Angew Chem, Int Ed. 2012;51:11458–11462. doi: 10.1002/anie.201204800. [DOI] [PubMed] [Google Scholar]
- 204.Popescu CV, Münck E. Electronic structure of the H cluster in [Fe]-hydrogenases. J Am Chem Soc. 1999;121:7877–7884. [Google Scholar]
- 205.Silakov A, Wenk B, Reijerse E, Lubitz W. 14N HYSCORE investigation of the H-cluster of [FeFe] hydrogenase: evidence for a nitrogen in the dithiol bridge. Phys Chem Chem Phys. 2009;11:6592–6599. doi: 10.1039/b905841a. [DOI] [PubMed] [Google Scholar]
- 206.Lemon BJ, Peters JW. Binding of exogenously added carbon monoxide at the active site of the iron-only hydrogenase (CpI) from Clostridium pasteurianum. Biochemistry. 1999;38:12969–12973. doi: 10.1021/bi9913193. [DOI] [PubMed] [Google Scholar]
- 207.de Lacey AL, Stadler C, Cavazza C, Hatchikian EC, Fernandez VM. FTIR characterization of the active site of the Fe-hydrogenase from Desulfovibrio desulfuricans. J Am Chem Soc. 2000;122:11232–11233. [Google Scholar]
- 208.Silakov A, Reijerse EJ, Albracht SPJ, Hatchikian EC, Lubitz W. The electronic structure of the H-cluster in the [FeFe]-hydrogenase from Desulfovibrio desulfuricans: a Q-band 57Fe-ENDOR and HYSCORE study. J Am Chem Soc. 2007;129:11447–11458. doi: 10.1021/ja072592s. [DOI] [PubMed] [Google Scholar]
- 209.Nicolet Y, de Lacey AL, Vernède X, Fernandez VM, Hatchikian EC, Fontecilla-Camps JC. Crystallographic and FTIR spectroscopic evidence of changes in Fe coordination upon reduction of the active site of the Fe-only hydrogenase from Desulfovibrio desulfuricans. J Am Chem Soc. 2001;123:1596–1601. doi: 10.1021/ja0020963. [DOI] [PubMed] [Google Scholar]
- 210.Silakov A, Kamp C, Reijerse E, Happe T, Lubitz W. Spectroelectrochemical characterization of the active site of the [FeFe] hydrogenase HydA1 from Chlamydomonas reinhardtii. Biochemistry. 2009;48:7780–7786. doi: 10.1021/bi9009105. [DOI] [PubMed] [Google Scholar]
- 211.Baffert C, Bertini L, Lautier T, Greco C, Sybirna K, Ezanno P, Etienne E, Soucaille P, Bertrand P, Bottin H, Meynial-Salles I, De Gioia L, Léger C. CO disrupts the reduced H-cluster of FeFe hydrogenase. A combined DFT and protein film voltammetry study. J Am Chem Soc. 2011;133:2096–2099. doi: 10.1021/ja110627b. [DOI] [PubMed] [Google Scholar]
- 212.Adams MWW. The structure and mechanism of iron-hydrogenases. Biochim Biophys Acta, Bioenerg. 1990;1020:115–145. doi: 10.1016/0005-2728(90)90044-5. [DOI] [PubMed] [Google Scholar]
- 213.Hambourger M, Gervaldo M, Svedruzic D, King PW, Gust D, Ghirardi M, Moore AL, Moore TA. [FeFe]-hydrogenase-catalyzed H2 production in a photoelectrochemical biofuel Cell. J Am Chem Soc. 2008;130:2015–2022. doi: 10.1021/ja077691k. [DOI] [PubMed] [Google Scholar]
- 214.Reihlen H, Gruhl A, von Hessling G. Photochemical and oxidative degradation of carbonyls. Justus Liebigs Ann Chem. 1929;472:268–287. [Google Scholar]
- 215.Winter A, Zsolnai L, Huttner G. Dinuclear and trinuclear carbonyliron complexes wth 1,2- and 1,3-dithiolato bridging ligands. Z Naturforsch, B: J Chem Sci. 1982;37:1430–1436. [Google Scholar]
- 216.Li Y, Rauchfuss TB. Synthesis of diiron(I) dithiolato carbonyl complexes. Chem Rev. 2016 doi: 10.1021/acs.chemrev.5b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Jablonskytë A, Webster LR, Simmons TR, Wright JA, Pickett CJ. Electronic control of the protonation rates of Fe–Fe bonds. J Am Chem Soc. 2014;136:13038–13044. doi: 10.1021/ja506693m. [DOI] [PubMed] [Google Scholar]
- 218.Matthews SL, Heinekey DM. A carbonyl-rich bridging hydride complex relevant to the Fe-Fe hydrogenase active site. Inorg Chem. 2010;49:9746–9748. doi: 10.1021/ic1017328. [DOI] [PubMed] [Google Scholar]
- 219.Cordero B, Gómez V, Platero-Prats AE, Revés M, Echeverría J, Cremades E, Barragán F, Alvarez S. Covalent radii revisited. Dalton Trans. 2008:2832–2838. doi: 10.1039/b801115j. [DOI] [PubMed] [Google Scholar]
- 220.Green JC, Green MLH, Parkin G. The occurrence and representation of three-centre two-electron bonds in covalent inorganic compounds. Chem Commun. 2012;48:11481–11503. doi: 10.1039/c2cc35304k. [DOI] [PubMed] [Google Scholar]
- 221.Mack AE, Rauchfuss TB. (1,3-Propanedithiolato)-hexacarbonyldiiron and Cyanide Derivatives. Inorg Synth. 2010;35:142–147. [Google Scholar]
- 222.Cloirec AL, Davies SC, Evans DJ, Hughes DL, Pickett CJ, Best SP, Borg S. A di-iron dithiolate possessing structural elements of the carbonyl/cyanide sub-site of the H-centre of Fe-only hydrogenase. Chem Commun. 1999:2285–2286. [Google Scholar]
- 223.Lyon EJ, Georgakaki IP, Reibenspies JH, Darensbourg MY. Carbon monoxide and cyanide ligands in a classical organometallic complex model for Fe-only hydrogenase. Angew Chem, Int Ed. 1999;38:3178–3180. [PubMed] [Google Scholar]
- 224.Schmidt M, Contakes SM, Rauchfuss TB. First generation analogues of the binuclear site in the Fe-only hydrogenases: Fe2(μ-SR)2(CO)4(CN)22−. J Am Chem Soc. 1999;121:9736–9737. [Google Scholar]
- 225.Gloaguen F, Lawrence JD, Rauchfuss TB. Biomimetic hydrogen evolution catalyzed by an iron carbonyl thiolate. J Am Chem Soc. 2001;123:9476–9477. doi: 10.1021/ja016516f. [DOI] [PubMed] [Google Scholar]
- 226.Fauvel K, Mathieu R, Poilblanc R. Protonation of the metal-metal bond in [μ (SCH3)Fe(CO)2L]2 complexes (L = P-(CH3)3−x(C6H5)x). Experimental evidence of the variation of nucleophilicity of the metal-metal bond with donor properties of phosphorus ligands. Inorg Chem. 1976;15:976–978. [Google Scholar]
- 227.Capon J-F, El Hassnaoui S, Gloaguen F, Schollhammer P, Talarmin J. N-heterocyclic carbene ligands as cyanide mimics in diiron models of the all-iron hydrogenase active site. Organometallics. 2005;24:2020–2022. [Google Scholar]
- 228.Morvan D, Capon J-F, Gloaguen F, Le Goff A, Marchivie M, Michaud F, Schollhammer P, Talarmin J, Yaouanc J-J, Pichon R, Kervarec N. N-heterocyclic carbene ligands in nonsymmetric diiron models of hydrogenase active sites. Organometallics. 2007;26:2042–2052. [Google Scholar]
- 229.Tye JW, Lee J, Wang H-W, Mejia-Rodriguez R, Reibenspies JH, Hall MB, Darensbourg MY. Dual electron uptake by simultaneous iron and ligand reduction in an N-heterocyclic carbene substituted [FeFe] hydrogenase model compound. Inorg Chem. 2005;44:5550–5552. doi: 10.1021/ic050402d. [DOI] [PubMed] [Google Scholar]
- 230.Liu T, Darensbourg MY. A mixed-valent, Fe(II)Fe(I), diiron complex reproduces the unique rotated state of the [FeFe]hydrogenase active site. J Am Chem Soc. 2007;129:7008–7009. doi: 10.1021/ja071851a. [DOI] [PubMed] [Google Scholar]
- 231.Singleton ML, Jenkins RM, Klemashevich CL, Darensbourg MY. The effect of bridgehead steric bulk on the ground state and intramolecular exchange processes of (μ-SCH2CR2CH2S)-[Fe(CO)3][Fe(CO)2L] complexes. C R Chim. 2008;11:861–874. [Google Scholar]
- 232.Boyke CA, Rauchfuss TB, Wilson SR, Rohmer M-M, Bénard M. [Fe2(SR)2(μ-CO)(CNMe)6]2+ and analogues: a new class of diiron dithiolates as structural models for the Hoxair state of the Fe-only hydrogenase. J Am Chem Soc. 2004;126:15151–15160. doi: 10.1021/ja049050k. [DOI] [PubMed] [Google Scholar]
- 233.Hou J, Peng X, Liu J, Gao Y, Zhao X, Gao S, Han K. A binuclear isocyanide azadithiolatoiron complex relevant to the active site of Fe-only hydrogenases: synthesis, structure and electrochemical properties. Eur J Inorg Chem. 2006;2006:4679–4686. [Google Scholar]
- 234.Lawrence JD, Rauchfuss TB, Wilson SR. New class of diiron dithiolates related to the Fe-only hydrogenase active site: synthesis and characterization of [Fe2(SR)2(CNMe)7]2+ Inorg Chem. 2002;41:6193–6195. doi: 10.1021/ic025919t. [DOI] [PubMed] [Google Scholar]
- 235.Nehring JL, Heinekey DM. Dinuclear iron isonitrile complexes: models for the iron hydrogenase active site. Inorg Chem. 2003;42:4288–4292. doi: 10.1021/ic034334b. [DOI] [PubMed] [Google Scholar]
- 236.Hsieh CH, Ding S, Erdem OF, Crouthers DJ, Liu T, McCrory CCL, Lubitz W, Popescu CV, Reibenspies JH, Hall MB, Darensbourg MY. Redox active iron nitrosyl units in proton reduction electrocatalysis. Nat Commun. 2014 doi: 10.1038/ncomms4684. [DOI] [PubMed] [Google Scholar]
- 237.Olsen MT, Bruschi M, De Gioia L, Rauchfuss TB, Wilson SR. Nitrosyl derivatives of diiron(I) dithiolates mimic the structure and Lewis acidity of the [FeFe]-hydrogenase active site. J Am Chem Soc. 2008;130:12021–12030. doi: 10.1021/ja802268p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Allen GC, Hush NS. Intervalence-transfer absorption. Part 1. Qualitative evidence for intervalence-transfer absorption in inorganic systems in solution and in the solid state. Prog Inorg Chem. 1967;8:357–390. [Google Scholar]
- 239.Robin MB, Day P. Mixed-valence chemistry: a survey and classification. Adv Inorg Chem Radiochem. 1968;10:247–422. [Google Scholar]
- 240.Day P, Hush NS, Clark RJH. Mixed valence: origins and developments. Philos Trans R Soc, A. 2008;366:5–14. doi: 10.1098/rsta.2007.2135. [DOI] [PubMed] [Google Scholar]
- 241.Singh PS, Rudbeck HC, Huang P, Ezzaher S, Eriksson L, Stein M, Ott S, Lomoth R. (I,0) Mixed-valence state of a diiron complex with pertinence to the [FeFe]-hydrogenase active site: an IR, EPR, and computational study. Inorg Chem. 2009;48:10883–10885. doi: 10.1021/ic9016454. [DOI] [PubMed] [Google Scholar]
- 242.Streich D, Astuti Y, Orlandi M, Schwartz L, Lomoth R, Hammarström L, Ott S. High-turnover photochemical hydrogen production catalyzed by a model complex of the [FeFe]-hydrogenase active site. Chem - Eur J. 2010;16:60–63. doi: 10.1002/chem.200902489. [DOI] [PubMed] [Google Scholar]
- 243.Mirmohades M, Pullen S, Stein M, Maji S, Ott S, Hammarström L, Lomoth R. Direct observation of key catalytic intermediates in a photoinduced proton reduction cycle with a diiron carbonyl complex. J Am Chem Soc. 2014;136:17366–17369. doi: 10.1021/ja5085817. [DOI] [PubMed] [Google Scholar]
- 244.Keizer PN, Krusic PJ, Morton JR, Preston KF. Thiolato- and selenato-bridged dinuclear iron carbonyl radicals. J Am Chem Soc. 1991;113:5454–5456. [Google Scholar]
- 245.Wang W, Nilges MJ, Rauchfuss TB, Stein M. Isolation of a mixed valence diiron hydride: evidence for a spectator hydride in hydrogen evolution catalysis. J Am Chem Soc. 2013;135:3633–3639. doi: 10.1021/ja312458f. [DOI] [PubMed] [Google Scholar]
- 246.Yang D, Li Y, Wang B, Zhao X, Su L, Chen S, Tong P, Luo Y, Qu J. Synthesis and electrocatalytic property of diiron hydride complexes derived from a thiolate-bridged diiron complex. Inorg Chem. 2015;54:10243–10249. doi: 10.1021/acs.inorgchem.5b01508. [DOI] [PubMed] [Google Scholar]
- 247.Wang W, Rauchfuss TB, Zhu L, Zampella G. New reactions of terminal hydrides on a diiron dithiolate. J Am Chem Soc. 2014;136:5773–5782. doi: 10.1021/ja501366j. [DOI] [PubMed] [Google Scholar]
- 248.Heiden ZM, Zampella G, De Gioia L, Rauchfuss TB. [FeFe]-hydrogenase models and hydrogen: oxidative addition of dihydrogen and silanes. Angew Chem, Int Ed. 2008;47:9756–9759. doi: 10.1002/anie.200804400. [DOI] [PubMed] [Google Scholar]
- 249.Justice AK, Linck RC, Rauchfuss TB, Wilson SR. Dihydrogen activation by a diruthenium analogue of the Fe-only hydrogenase active site. J Am Chem Soc. 2004;126:13214–13215. doi: 10.1021/ja0455594. [DOI] [PubMed] [Google Scholar]
- 250.Zaffaroni R, Rauchfuss TB, Gray DL, De Gioia L, Zampella G. Terminal vs bridging hydrides of diiron dithiolates: protonation of Fe2(dithiolate)(CO)2(PMe3)4. J Am Chem Soc. 2012;134:19260–19269. doi: 10.1021/ja3094394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Cheah MH, Borg SJ, Bondin MI, Best SP. Electrocatalytic proton reduction by phosphido-bridged diiron carbonyl compounds: distant relations to the H-cluster? Inorg Chem. 2004;43:5635–5644. doi: 10.1021/ic049746e. [DOI] [PubMed] [Google Scholar]
- 252.Ezzaher S, Capon J-F, Gloaguen F, Pétillon FY, Schollhammer P, Talarmin J, Pichon R, Kervarec N. Evidence for the formation of terminal hydrides by protonation of an asymmetric iron hydrogenase active site mimic. Inorg Chem. 2007;46:3426–3428. doi: 10.1021/ic0703124. [DOI] [PubMed] [Google Scholar]
- 253.van der Vlugt JI, Rauchfuss TB, Whaley CM, Wilson SR. Characterization of a diferrous terminal hydride mechanistically relevant to the Fe-only hydrogenases. J Am Chem Soc. 2005;127:16012–16013. doi: 10.1021/ja055475a. [DOI] [PubMed] [Google Scholar]
- 254.Pelmenschikov V, Guo Y, Wang H, Cramer SP, Case DA. Fe–H/D stretching and bending modes in nuclear resonant vibrational, Raman and infrared spectroscopies: Comparisons of density functional theory and experiment. Faraday Discuss. 2011;148:409–420. doi: 10.1039/c004367m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Brookhart M, Grant B, Volpe AF., Jr [(3,5-(CF3)2C6H3)4B]−[H(OEt2)2]+: a convenient reagent for generation and stabilization of cationic, highly electrophilic organometallic complexes. Organometallics. 1992;11:3920–3922. [Google Scholar]
- 256.Bertini L, Fantucci P, De Gioia L, Zampella G. Excited state properties of diiron dithiolate hydrides: implications in the unsensitized photocatalysis of H2 evolution. Inorg Chem. 2013;52:9826–9841. doi: 10.1021/ic400818t. [DOI] [PubMed] [Google Scholar]
- 257.Zampella G, Fantucci P, De Gioia L. DFT characterization of the reaction pathways for terminal- to μ-hydride isomerisation in synthetic models of the [FeFe]-hydrogenase active site. Chem Commun. 2009;46:8824–8826. doi: 10.1039/c0cc02821e. [DOI] [PubMed] [Google Scholar]
- 258.Carroll ME, Barton BE, Rauchfuss TB, Carroll PJ. Synthetic models for the active site of the [FeFe]-hydrogenase: catalytic proton reduction and the structure of the doubly protonated intermediate. J Am Chem Soc. 2012;134:18843–18852. doi: 10.1021/ja309216v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Barton BE, Rauchfuss TB. Terminal hydride in [FeFe]-hydrogenase model has lower potential for H2 production than the isomeric bridging hydride. Inorg Chem. 2008;47:2261–2263. doi: 10.1021/ic800030y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Zampella G, Fantucci P, De Gioia L. Unveiling how stereoelectronic factors affect kinetics and thermodynamics of protonation regiochemistry in [FeFe] hydrogenase synthetic models: a DFT investigation. J Am Chem Soc. 2009;131:10909–10917. doi: 10.1021/ja902727z. [DOI] [PubMed] [Google Scholar]
- 261.Dong W, Wang M, Liu X, Jin K, Li G, Wang F, Sun L. An insight into the protonation property of a diiron azadithiolate complex pertinent to the active site of Fe-only hydrogenases. Chem Commun. 2006:305–307. doi: 10.1039/b513270c. [DOI] [PubMed] [Google Scholar]
- 262.Ezzaher S, Gogoll A, Bruhn C, Ott S. Directing protonation in [FeFe] hydrogenase active site models by modifications in their second coordination sphere. Chem Commun. 2010;46:5775–5777. doi: 10.1039/c0cc00724b. [DOI] [PubMed] [Google Scholar]
- 263.Jordan RF, Norton JR. Kinetic and Thermodynamic acidity of hydrido transition-metal complexes. 1. Periodic trends in group 6 complexes and substituent effects in osmium complexes. J Am Chem Soc. 1982;104:1255–1263. [Google Scholar]
- 264.Barton BE, Zampella G, Justice AK, De Gioia L, Rauchfuss TB, Wilson SR. Isomerization of the hydride complexes [HFe2(SR)2(PR3)x(CO)6−x]+ (x = 2, 3, 4) relevant to the active site models for the [FeFe]-hydrogenases. Dalton Trans. 2010;39:3011–3019. doi: 10.1039/b910147k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Zaffaroni R, Rauchfuss TB, Fuller A, De Gioia L, Zampella G. Contrasting protonation behavior of diphosphido vs dithiolato diiron(I) carbonyl complexes. Organometallics. 2013;32:232–238. [Google Scholar]
- 266.Wang W, Rauchfuss TB, Moore CE, Rheingold AL, De Gioia L, Zampella G. Crystallographic characterization of a fully rotated, basic diiron dithiolate: model for the Hred state? Chem - Eur J. 2013;19:15476–15479. doi: 10.1002/chem.201303351. [DOI] [PubMed] [Google Scholar]
- 267.Munery S, Capon J-F, De Gioia L, Elleouet C, Greco C, Pétillon FY, Schollhammer P, Talarmin J, Zampella G. New FeI–FeI complex featuring a rotated conformation related to the [2Fe]H subsite of [Fe–Fe] hydrogenase. Chem - Eur J. 2013;19:15458–15461. doi: 10.1002/chem.201303316. [DOI] [PubMed] [Google Scholar]
- 268.Finkelmann AR, Stiebritz MT, Reiher M. Inaccessibility of the μ-hydride species in [FeFe] hydrogenases. Chem Sci. 2014;5:215–221. [Google Scholar]
- 269.Angamuthu R, Chen C-S, Cochrane TR, Gray DL, Schilter D, Ulloa OA, Rauchfuss TB. N-Substituted derivatives of the azadithiolate cofactor from the [FeFe] hydrogenases: stability and complexation. Inorg Chem. 2015;54:5717–5724. doi: 10.1021/acs.inorgchem.5b00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Li H, Rauchfuss TB. Iron carbonyl sulfides, formaldehyde, and amines condense to give the proposed azadithiolate cofactor of the Fe-only hydrogenases. J Am Chem Soc. 2002;124:726–727. doi: 10.1021/ja016964n. [DOI] [PubMed] [Google Scholar]
- 271.Lawrence JD, Li H, Rauchfuss TB, Benard M, Rohmer M-M. Diiron azadithiolates as models for the iron-only hydrogenase active site: synthesis, structure, and stereoelectronics. Angew Chem, Int Ed. 2001;40:1768–1771. doi: 10.1002/1521-3773(20010504)40:9<1768::aid-anie17680>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 272.Gilbert-Wilson R, Siebel JF, Adamska-Venkatesh A, Pham CC, Reijerse E, Wang H, Cramer SP, Lubitz W, Rauchfuss TB. Spectroscopic investigations of [FeFe] hydrogenase maturated with [57Fe2(adt)(CN)2(CO)4]2−. J Am Chem Soc. 2015;137:8998–9005. doi: 10.1021/jacs.5b03270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Belkova NV, Epstein LM, Filippov OA, Shubina ES. Hydrogen and dihydrogen bonds in the reactions of metal hydrides. Chem Rev. 2016 doi: 10.1021/acs.chemrev.6b00091. [DOI] [PubMed] [Google Scholar]
- 274.Yang X, Hall MB. Monoiron hydrogenase catalysis: hydrogen activation with the formation of a dihydrogen, Fe–Hδ−···Hδ+–O, bond and methenyl-H4MPT+ triggered hydride transfer. J Am Chem Soc. 2009;131:10901–10908. doi: 10.1021/ja902689n. [DOI] [PubMed] [Google Scholar]
- 275.Gloaguen F. Electrochemistry of simple organometallic models of iron–iron hydrogenases in organic solvent and water. Inorg Chem. 2016;55:390–398. doi: 10.1021/acs.inorgchem.5b02245. [DOI] [PubMed] [Google Scholar]
- 276.Capon J-F, Gloaguen F, Pétillon FY, Schollhammer P, Talarmin J. Electron and proton transfers at diiron dithiolate sites relevant to the catalysis of proton reduction by the [FeFe]-hydrogenases. Coord Chem Rev. 2009;253:1476–1494. [Google Scholar]
- 277.Pandey IK, Natarajan M, Kaur-Ghumaan S. Hydrogen generation: aromatic dithiolate-bridged metal carbonyl complexes as hydrogenase catalytic site models. J Inorg Biochem. 2015;143:88–110. doi: 10.1016/j.jinorgbio.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 278.Borg SJ, Behrsing T, Best SP, Razavet M, Liu X, Pickett CJ. Electron transfer at a dithiolate-bridged diiron assembly: electrocatalytic hydrogen evolution. J Am Chem Soc. 2004;126:16988–16999. doi: 10.1021/ja045281f. [DOI] [PubMed] [Google Scholar]
- 279.Borg SJ, Bondin MI, Best SP, Razavet M, Liu X, Pickett CJ. Electrocatalytic proton reduction by dithiolate- bridged diiron carbonyl complexes: a connection to the H-cluster? Biochem Soc Trans. 2005;33:3–6. doi: 10.1042/BST0330003. [DOI] [PubMed] [Google Scholar]
- 280.Greco C, Zampella G, Bertini L, Bruschi M, Fantucci P, De Gioia L. Insights into the mechanism of electrocatalytic hydrogen evolution mediated by Fe2(S2C3H6)(CO)6: the simplest functional model of the Fe-hydrogenase active site. Inorg Chem. 2007;46:108–116. doi: 10.1021/ic061168+. [DOI] [PubMed] [Google Scholar]
- 281.Bullock RM, Helm ML. Molecular electrocatalysts for oxidation of hydrogen using earth-abundant metals: shoving protons around with proton relays. Acc Chem Res. 2015;48:2017–2026. doi: 10.1021/acs.accounts.5b00069. [DOI] [PubMed] [Google Scholar]
- 282.Capon J-F, Ezzaher S, Gloaguen F, Pétillon FY, Schollhammer P, Talarmin J. Electrochemical insights into the mechanisms of proton reduction by [Fe2(CO)6{μ-SCH2N(R)CH2S}] complexes related to the [2Fe]H subsite of [FeFe]hydrogenase. Chem -Eur J. 2008;14:1954–1964. doi: 10.1002/chem.200701454. [DOI] [PubMed] [Google Scholar]
- 283.Ezzaher S, Capon J-F, Dumontet N, Gloaguen F, Pétillon FY, Schollhammer P, Talarmin J. Electrochemical study of the role of a H-bridged, unsymmetrically disubstituted diiron complex in proton reduction catalysis. J Electroanal Chem. 2009;626:161–170. [Google Scholar]
- 284.Becker R, Amirjalayer S, Li P, Woutersen S, Reek JNH. An iron-iron hydrogenase mimic with appended electron reservoir for efficient proton reduction in aqueous media. Sci Adv. 2016;2:e1501014. doi: 10.1126/sciadv.1501014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Zhao X, Georgakaki IP, Miller ML, Mejia-Rodriguez R, Chiang C-Y, Darensbourg MY. Catalysis of H2/D2 scrambling and other H/D exchange processes by [Fe]-hydrogenase model complexes. Inorg Chem. 2002;41:3917–3928. doi: 10.1021/ic020237r. [DOI] [PubMed] [Google Scholar]
- 286.Razavet M, Borg SJ, George SJ, Best SP, Fairhurst SA, Pickett CJ. Transient FTIR spectroelectrochemical and stopped-flow detection of a mixed valence {Fe(I)–Fe(II)} bridging carbonyl intermediate with structural elements and spectroscopic characteristics of the di-iron sub-site of all-iron hydrogenase. Chem Commun. 2002:700–701. doi: 10.1039/b111613b. [DOI] [PubMed] [Google Scholar]
- 287.Justice AK, Rauchfuss TB, Wilson SR. Unsaturated, mixed-valence Diiron dithiolate model for the Hox state of the [FeFe]-hydrogenase. Angew Chem, Int Ed. 2007;46:6152–6154. doi: 10.1002/anie.200702224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Barton BE, Olsen MT, Rauchfuss TB. Aza- and oxadithiolates are probable proton relays in functional models for the [FeFe]-hydrogenases. J Am Chem Soc. 2008;130:16834–16835. doi: 10.1021/ja8057666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Esselborn J, Muraki N, Klein K, Engelbrecht V, Metzler-Nolte N, Apfel U-P, Hofmann E, Kurisu G, Happe T. A structural view of synthetic cofactor integration into [FeFe]-hydrogenases. Chem Sci. 2016;7:959–968. doi: 10.1039/c5sc03397g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Olsen MT, Barton BE, Rauchfuss TB. Hydrogen activation by biomimetic diiron dithiolates. Inorg Chem. 2009;48:7507–7509. doi: 10.1021/ic900850u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Camara JM, Rauchfuss TB. Mild redox complementation enables H2 activation by [FeFe]-hydrogenase models. J Am Chem Soc. 2011;133:8098–8101. doi: 10.1021/ja201731q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Lansing JC, Camara JM, Gray DL, Rauchfuss TB. Hydrogen production catalyzed by bidirectional, biomimetic models of the [FeFe]-hydrogenase active site. Organometallics. 2014;33:5897–5906. doi: 10.1021/om5004013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Ghosh S, Hogarth G, Hollingsworth N, Holt KB, Kabir SE, Sanchez BE. Hydrogenase biomimetics: Fe2(CO)4(μ-dppf)(μ-pdt)(dppf = 1,1′-bis(diphenylphosphino)ferrocene) both a proton-reduction and hydrogen oxidation catalyst. Chem Commun. 2014;50:945–947. doi: 10.1039/c3cc46456c. [DOI] [PubMed] [Google Scholar]
- 294.Ezzaher S, Capon J-F, Gloaguen F, Pétillon FY, Schollhammer P, Talarmin J, Kervarec N. Influence of a pendant amine in the second coordination sphere on proton transfer at a dissymmetrically disubstituted diiron system related to the [2Fe]H subsite of [FeFe]H2ase. Inorg Chem. 2009;48:2–4. doi: 10.1021/ic801369u. [DOI] [PubMed] [Google Scholar]
- 295.Wang N, Wang M, Zhang T, Li P, Liu J, Sun L. A proton–hydride diiron complex with a base-containing diphosphine ligand relevant to the [FeFe]-hydrogenase active site. Chem Commun. 2008:5800–5802. doi: 10.1039/b811352a. [DOI] [PubMed] [Google Scholar]
- 296.Wang N, Wang M, Wang Y, Zheng D, Han H, Ahlquist MSG, Sun L. Catalytic activation of H2 under mild conditions by an [FeFe]-hydrogenase model via an active μ-hydride species. J Am Chem Soc. 2013;135:13688–13691. doi: 10.1021/ja408376t. [DOI] [PubMed] [Google Scholar]
- 297.Rauchfuss TB. Diiron azadithiolates as models for the [FeFe]-hydrogenase active site and paradigm for the role of the second coordination sphere. Acc Chem Res. 2015;48:2107–2116. doi: 10.1021/acs.accounts.5b00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Leroux F, Dementin S, Burlat B, Cournac L, Volbeda A, Champ S, Martin L, Guigliarelli B, Bertrand P, Fontecilla-Camps J, Rousset M, Léger C. Experimental approaches to kinetics of gas diffusion in hydrogenase. Proc Natl Acad Sci U S A. 2008;105:11188–11193. doi: 10.1073/pnas.0803689105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Goy R, Bertini L, Elleouet C, Görls H, Zampella G, Talarmin J, De Gioia L, Schollhammer P, Apfel U-P, Weigand W. A sterically stabilized FeI–FeI semi-rotated conformation of [FeFe] hydrogenase subsite model. Dalton Trans. 2015;44:1690–1699. doi: 10.1039/c4dt03223c. [DOI] [PubMed] [Google Scholar]
- 300.Liu T, DuBois DL, Bullock RM. An iron complex with pendent amines as a molecular electrocatalyst for oxidation of hydrogen. Nat Chem. 2013;5:228–233. doi: 10.1038/nchem.1571. [DOI] [PubMed] [Google Scholar]
- 301.Vignais PM, Billoud B, Meyer J. Classification and phylogeny of hydrogenases. FEMS Microbiol Rev. 2001;25:455–501. doi: 10.1111/j.1574-6976.2001.tb00587.x. [DOI] [PubMed] [Google Scholar]
- 302.Shafaat HS, Rüdiger O, Ogata H, Lubitz W. [NiFe] hydrogenases: a common active site for hydrogen metabolism under diverse conditions. Biochim Biophys Acta, Bioenerg. 2013;1827:986–1002. doi: 10.1016/j.bbabio.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 303.Volbeda A, Charon M-H, Piras C, Hatchikian EC, Frey M, Fontecilla-Camps JC. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature. 1995;373:580–587. doi: 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]
- 304.Higuchi Y, Yagi T, Yasuoka N. Unusual ligand structure in Ni–Fe active center and an additional Mg site in hydrogenase revealed by high resolution X-ray structure analysis. Structure. 1997;5:1671–1680. doi: 10.1016/s0969-2126(97)00313-4. [DOI] [PubMed] [Google Scholar]
- 305.Volbeda A, Darnault C, Parkin A, Sargent F, Armstrong FA, Fontecilla-Camps JC. Crystal structure of the O2-tolerant membrane-bound hydrogenase 1 from Escherichia coli in complex with its cognate cytochrome b. Structure. 2013;21:184–190. doi: 10.1016/j.str.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 306.Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J Chem Soc, Dalton Trans. 1984:1349–1356. [Google Scholar]
- 307.Murphy BJ, Hidalgo R, Roessler MM, Evans RM, Ash PA, Myers WK, Vincent KA, Armstrong FA. Discovery of dark pH-dependent H+ migration in a [NiFe]-hydrogenase and its mechanistic relevance: mobilizing the hydrido ligand of the Ni-C intermediate. J Am Chem Soc. 2015;137:8484–8489. doi: 10.1021/jacs.5b03182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Amara P, Volbeda A, Fontecilla-Camps JC, Field MJ. A hybrid density functional theory/molecular mechanics study of nickel-iron hydrogenase: investigation of the active site redox states. J Am Chem Soc. 1999;121:4468–4477. [Google Scholar]
- 309.Niu S, Thomson LM, Hall MB. Theoretical characterization of the reaction intermediates in a model of the nickel-iron hydrogenase of Desulfovibrio gigas. J Am Chem Soc. 1999;121:4000–4007. [Google Scholar]
- 310.Yson RL, Gilgor JL, Guberman BA, Varganov SA. Protein induced singlet–triplet quasidegeneracy in the active site of [NiFe]-hydrogenase. Chem Phys Lett. 2013;577:138–141. [Google Scholar]
- 311.Fan H-J, Hall MB. High-spin Ni(II), a surprisingly good structural model for [NiFe] hydrogenase. J Am Chem Soc. 2002;124:394–395. doi: 10.1021/ja0171310. [DOI] [PubMed] [Google Scholar]
- 312.Wang H, Yoda Y, Ogata H, Tanaka Y, Lubitz W. A strenuous experimental journey searching for spectroscopic evidence of a bridging nickel–iron–hydride in [NiFe] hydrogenase. J Synchrotron Radiat. 2015;22:1334–1344. doi: 10.1107/S1600577515017816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Evans RM, Brooke EJ, Wehlin SAM, Nomerotskaia E, Sargent F, Carr SB, Phillips SEV, Armstrong FA. Mechanism of hydrogen activation by [NiFe] hydrogenases. Nat Chem Biol. 2015;12:46–50. doi: 10.1038/nchembio.1976. [DOI] [PubMed] [Google Scholar]
- 314.Fichtner C, Laurich C, Bothe E, Lubitz W. Spectroelectrochemical characterization of the [NiFe] hydrogenase of Desulfovibrio Vulgaris Miyazaki F. Biochemistry. 2006;45:9706–9716. doi: 10.1021/bi0602462. [DOI] [PubMed] [Google Scholar]
- 315.Brecht M, van Gastel M, Buhrke T, Friedrich B, Lubitz W. Direct detection of a hydrogen ligand in the [NiFe] center of the regulatory H2-sensing hydrogenase from Ralstonia eutropha in its reduced state by HYSCORE and ENDOR spectroscopy. J Am Chem Soc. 2003;125:13075–13083. doi: 10.1021/ja036624x. [DOI] [PubMed] [Google Scholar]
- 316.Krämer T, Kampa M, Lubitz W, van Gastel M, Neese F. Theoretical spectroscopy of the NiII intermediate states in the catalytic cycle and the activation of [NiFe] hydrogenases. ChemBioChem. 2013;14:1898–1905. doi: 10.1002/cbic.201300104. [DOI] [PubMed] [Google Scholar]
- 317.Foerster S, Stein M, Brecht M, Ogata H, Higuchi Y, Lubitz W. Single crystal EPR studies of the reduced active site of [NiFe] hydrogenase from Desulfovibrio vulgaris Miyazaki F. J Am Chem Soc. 2003;125:83–93. doi: 10.1021/ja027522u. [DOI] [PubMed] [Google Scholar]
- 318.Tai H, Nishikawa K, Inoue S, Higuchi Y, Hirota S. FT-IR Characterization of the light-induced Ni-L2 and Ni-L3 states of [NiFe] hydrogenase from Desulfovibrio vulgaris Miyazaki F. J Phys Chem B. 2015;119:13668–13674. doi: 10.1021/acs.jpcb.5b03075. [DOI] [PubMed] [Google Scholar]
- 319.Roessler MM, Evans RM, Davies RA, Harmer J, Armstrong FA. EPR spectroscopic studies of the Fe–S clusters in the O2-tolerant [NiFe]-hydrogenase Hyd-1 from Escherichia coli and characterization of the unique [4Fe–3S] cluster by HYSCORE. J Am Chem Soc. 2012;134:15581–15594. doi: 10.1021/ja307117y. [DOI] [PubMed] [Google Scholar]
- 320.Hidalgo R, Ash PA, Healy AJ, Vincent KA. Infrared spectroscopy during electrocatalytic turnover reveals the Ni-L active site state during H2 oxidation by a NiFe hydrogenase. Angew Chem, Int Ed. 2015;54:7110–7113. doi: 10.1002/anie.201502338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Tai H, Nishikawa K, Suzuki M, Higuchi Y, Hirota S. Control of the transition between Ni-C and Ni-SIa States by the Redox State of the Proximal Fe-S Cluster in the Catalytic Cycle of [NiFe]Hydrogenase. Angew Chem, Int Ed. 2014;53:13817–13820. doi: 10.1002/anie.201408552. [DOI] [PubMed] [Google Scholar]
- 322.Greene BL, Wu C-H, Vansuch GE, Adams MWW, Dyer RB. Proton inventory and dynamics in the Nia-S to Nia-C transition of a [NiFe] hydrogenase. Biochemistry. 2016;55:1813–1825. doi: 10.1021/acs.biochem.5b01348. [DOI] [PubMed] [Google Scholar]
- 323.Bleijlevens B, Faber BW, Albracht SPJ. The [NiFe] hydrogenase from Allochromatium vinosum studied in EPR-detectable states: H/D exchange experiments that yield new information about the structure of the active site. JBIC, J Biol Inorg Chem. 2001;6:763–769. doi: 10.1007/s007750100252. [DOI] [PubMed] [Google Scholar]
- 324.Montet Y, Amara P, Volbeda A, Vernede X, Hatchikian EC, Field MJ, Frey M, Fontecilla-Camps JC. Gas access to the active site of Ni-Fe hydrogenases probed by X-ray crystallography and molecular dynamics. Nat Struct Biol. 1997;4:523–527. doi: 10.1038/nsb0797-523. [DOI] [PubMed] [Google Scholar]
- 325.Bruschi M, Tiberti M, Guerra A, De Gioia L. Disclosure of key stereoelectronic factors for efficient H2 binding and cleavage in the active site of [NiFe]-hydrogenases. J Am Chem Soc. 2014;136:1803–1814. doi: 10.1021/ja408511y. [DOI] [PubMed] [Google Scholar]
- 326.Smith DMA, Raugei S, Squier TC. Modulation of active site electronic structure by the protein matrix to control [NiFe] hydrogenase reactivity. Phys Chem Chem Phys. 2014;16:24026–24033. doi: 10.1039/c4cp03518f. [DOI] [PubMed] [Google Scholar]
- 327.Kampa M, Lubitz W, van Gastel M, Neese F. Computational study of the electronic structure and magnetic properties of the Ni–C state in [NiFe] hydrogenases including the second coordination sphere. JBIC. J Biol Inorg Chem. 2012;17:1269–1281. doi: 10.1007/s00775-012-0941-9. [DOI] [PubMed] [Google Scholar]
- 328.Kaliakin DS, Zaari RR, Varganov SA. Effect of H2 binding on the nonadiabatic transition probability between singlet and triplet states of the [NiFe]-hydrogenase active site. J Phys Chem A. 2015;119:1066–1073. doi: 10.1021/jp510522z. [DOI] [PubMed] [Google Scholar]
- 329.Delcey MG, Pierloot K, Phung QM, Vancoillie S, Lindh R, Ryde U. Accurate calculations of geometries and singlet–triplet energy differences for active-site models of [NiFe] hydrogenase. Phys Chem Chem Phys. 2014;16:7927–7938. doi: 10.1039/c4cp00253a. [DOI] [PubMed] [Google Scholar]
- 330.Wu H, Hall MB. Density functional theory on the larger active site models for [NiFe] hydrogenases: Two-state reactivity? C R Chim. 2008;11:790–804. [Google Scholar]
- 331.Dong G, Ryde U. Protonation states of intermediates in the reaction mechanism of [NiFe] hydrogenase studied by computational methods. JBIC. J Biol Inorg Chem. 2016;21:383–394. doi: 10.1007/s00775-016-1348-9. [DOI] [PubMed] [Google Scholar]
- 332.Fitch CA, Platzer G, Okon M, Garcia-Moreno E B, McIntosh LP. Arginine: its pKa value revisited. Protein Sci. 2015;24:752–761. doi: 10.1002/pro.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Jones AK, Sillery E, Albracht SPJ, Armstrong FA. Direct comparison of the electrocatalytic oxidation of hydrogen by an enzyme and a platinum catalyst. Chem Commun. 2002:866–867. doi: 10.1039/b201337a. [DOI] [PubMed] [Google Scholar]
- 334.Pershad HR, Duff JLC, Heering HA, Duin EC, Albracht SPJ, Armstrong FA. Catalytic electron transport in Chromatium Vinosum [NiFe]-hydrogenase: application of voltammetry in detecting redox-active centers and establishing that hydrogen oxidation is very fast even at potentials close to the reversible H+/H2 Value. Biochemistry. 1999;38:8992–8999. doi: 10.1021/bi990108v. [DOI] [PubMed] [Google Scholar]
- 335.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases. Chem Rev. 2007;107:4273–4303. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- 336.Goldet G, Wait AF, Cracknell JA, Vincent KA, Ludwig M, Lenz O, Friedrich B, Armstrong FA. Hydrogen production under aerobic conditions by membrane-bound hydrogenases from Ralstonia Species. J Am Chem Soc. 2008;130:11106–11113. doi: 10.1021/ja8027668. [DOI] [PubMed] [Google Scholar]
- 337.Fourmond V, Baffert C, Sybirna K, Dementin S, Abou-Hamdan A, Meynial-Salles I, Soucaille P, Bottin H, Léger C. The mechanism of inhibition by H2 of H2-evolution by hydrogenases. Chem Commun. 2013;49:6840–6842. doi: 10.1039/c3cc43297a. [DOI] [PubMed] [Google Scholar]
- 338.Léger C, Dementin S, Bertrand P, Rousset M, Guigliarelli B. Inhibition and aerobic inactivation kinetics of Desulfovibrio fructosovorans NiFe hydrogenase studied by protein film voltammetry. J Am Chem Soc. 2004;126:12162–12172. doi: 10.1021/ja046548d. [DOI] [PubMed] [Google Scholar]
- 339.van Haaster DJ, Hagedoorn P-L, Jongejan JA, Hagen WR. On the relationship between affinity for molecular hydrogen and the physiological directionality of hydrogenases. Biochem Soc Trans. 2005;33:12–14. doi: 10.1042/BST0330012. [DOI] [PubMed] [Google Scholar]
- 340.Kaur-Ghumaan S, Stein M. [NiFe] hydrogenases: how close do structural and functional mimics approach the active site? Dalton Trans. 2014;43:9392–9405. doi: 10.1039/c4dt00539b. [DOI] [PubMed] [Google Scholar]
- 341.Ohki Y, Tatsumi K. Thiolate-bridged iron–nickel models for the active site of [NiFe] hydrogenase. Eur J Inorg Chem. 2011;2011:973–985. [Google Scholar]
- 342.Whaley CM, Rauchfuss TB, Wilson SR. Coordination chemistry of [HFe(CN)2(CO)3]− and its derivatives: toward a model for the iron subsite of the [NiFe]-hydrogenases. Inorg Chem. 2009;48:4462–4469. doi: 10.1021/ic900200s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Ceriotti A, Chini P, Fumagalli A, Koetzle TF, Longoni G, Takusagawa F. Synthesis of Bimetallic Fe-Ni Carbonyl Clusters: Crystal Structure of [N(CH3)3CH2Ph][Fe3Ni(CO)8(μ-CO)4(μ3-H)] Inorg Chem. 1984;23:1363–1368. [Google Scholar]
- 344.Della Pergola R, Fumagalli A, Garlaschelli L, Manassero C, Manassero M, Sansoni M, Sironi A. Iron–nickel mixed metal nitrido clusters: Synthesis and solid state structure of the anions [HFe5NiN-(CO)14]2− and [HFe4Ni2N(CO)13]2−. Inorg Chim Acta. 2008;361:1763–1769. [Google Scholar]
- 345.Barton BE, Whaley CM, Rauchfuss TB, Gray DL. Nickel-iron dithiolato hydrides relevant to the [NiFe]-hydrogenase active site. J Am Chem Soc. 2009;131:6942–6943. doi: 10.1021/ja902570u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Zhu W, Marr AC, Wang Q, Neese F, Spencer DJE, Blake AJ, Cooke PA, Wilson C, Schröder M. Modulation of the electronic structure and the Ni–Fe distance in heterobimetallic models for the active site in [NiFe]hydrogenase. Proc Natl Acad Sci U S A. 2005;102:18280–18285. doi: 10.1073/pnas.0505779102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Schilter D, Pelmenschikov V, Wang H, Meier F, Gee LB, Yoda Y, Kaupp M, Rauchfuss TB, Cramer SP. Synthesis and vibrational spectroscopy of 57Fe-labeled models of [NiFe] hydrogenase: first direct observation of a nickel–iron interaction. Chem Commun. 2014;50:13469–13472. doi: 10.1039/c4cc04572f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Carroll ME, Barton BE, Gray DL, Mack AE, Rauchfuss TB. Active-site models for the nickel-iron hydrogenases: effects of ligands on reactivity and catalytic properties. Inorg Chem. 2011;50:9554–9563. doi: 10.1021/ic2012759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Shafaat HS, Weber K, Petrenko T, Neese F, Lubitz W. Key hydride vibrational modes in [NiFe] hydrogenase model compounds studied by resonance Raman spectroscopy and density functional calculations. Inorg Chem. 2012;51:11787–11797. doi: 10.1021/ic3017276. [DOI] [PubMed] [Google Scholar]
- 350.Hugenbruch S, Shafaat HS, Krämer T, Delgado-Jaime MU, Weber K, Neese F, Lubitz W, DeBeer S. In search of metal hydrides: an X-ray absorption and emission study of [NiFe] hydrogenase model complexes. Phys Chem Chem Phys. 2016;18:10688–10699. doi: 10.1039/c5cp07293j. [DOI] [PubMed] [Google Scholar]
- 351.Ulloa OA, Huynh MT, Richers CP, Bertke JA, Nilges MJ, Hammes-Schiffer S, Rauchfuss TB. Mechanism of H2 Production by Models for the [NiFe]-Hydrogenases: Role of Reduced Hydrides. J Am Chem Soc. 2016 doi: 10.1021/jacs.6b04579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Chambers GM, Angamuthu R, Gray DL, Rauchfuss TB. Organo ruthenium–nickel dithiolates with redox-responsive nickel sites. Organometallics. 2013;32:6324–6329. [Google Scholar]
- 353.Schilter D, Fuller AL, Gray DL. Nickel-molybdenum and nickel-tungsten dithiolates: hybrid models for hydrogenases and hydrodesulfurization. Eur J Inorg Chem. 2015;2015:4638–4642. [Google Scholar]
- 354.Chojnacki SS, Hsiao Y-M, Darensbourg MY, Reibenspies JH. X-ray structure and solution characterization of Ni-(SCH2CH2PPh2)2Mo(CO)4. Inorg Chem. 1993;32:3573–3576. [Google Scholar]
- 355.Weber K, Krämer T, Shafaat HS, Weyhermüller T, Bill E, van Gastel M, Neese F, Lubitz W. A functional [NiFe]-hydrogenase model compound that undergoes biologically relevant reversible thiolate protonation. J Am Chem Soc. 2012;134:20745–20755. doi: 10.1021/ja309563p. [DOI] [PubMed] [Google Scholar]
- 356.Canaguier S, Vaccaro L, Artero V, Ostermann R, Pécaut J, Field MJ, Fontecave M. Cyclopentadienyl ruthenium–nickel catalysts for biomimetic hydrogen evolution: electrocatalytic properties and mechanistic DFT studies. Chem - Eur J. 2009;15:9350–9364. doi: 10.1002/chem.200900854. [DOI] [PubMed] [Google Scholar]
- 357.Fourmond V, Canaguier S, Golly B, Field MJ, Fontecave M, Artero V. A nickel–manganese catalyst as a biomimic of the active site of NiFe hydrogenases: a combined electrocatalytical and DFT mechanistic study. Energy Environ Sci. 2011;4:2417–2427. [Google Scholar]
- 358.Song L-C, Li J-P, Xie Z-J, Song H-B. Synthesis, structural characterization, and electrochemical properties of dinuclear Ni/Mn model complexes for the active site of [NiFe]-hydrogenases. Inorg Chem. 2013;52:11618–11626. doi: 10.1021/ic401978h. [DOI] [PubMed] [Google Scholar]
- 359.Chambers GM, Mitra J, Rauchfuss TB, Stein M. NiI/RuII model for the Ni–L state of the [NiFe]hydrogenases: synthesis, spectroscopy, and reactivity. Inorg Chem. 2014;53:4243–4249. doi: 10.1021/ic500389p. [DOI] [PubMed] [Google Scholar]
- 360.Denny JA, Darensbourg MY. Metallodithiolates as ligands in coordination, bioinorganic, and organometallic chemistry. Chem Rev. 2015;115:5248–5273. doi: 10.1021/cr500659u. [DOI] [PubMed] [Google Scholar]
- 361.Ogo S, Kabe R, Uehara K, Kure B, Nishimura T, Menon SC, Harada R, Fukuzumi S, Higuchi Y, Ohhara T, Tamada T, Kuroki R. A dinuclear Ni(μ-H)Ru complex derived from H2. Science. 2007;316:585–587. doi: 10.1126/science.1138751. [DOI] [PubMed] [Google Scholar]
- 362.Ogo S. Electrons from hydrogen. Chem Commun. 2009:3317–3325. doi: 10.1039/b900297a. [DOI] [PubMed] [Google Scholar]
- 363.Kure B, Matsumoto T, Ichikawa K, Fukuzumi S, Higuchi Y, Yagi T, Ogo S. pH-Dependent isotope exchange and hydrogenation catalysed by water-soluble NiRu complexes as functional models for [NiFe]hydrogenases. Dalton Trans. 2008:4747–4755. doi: 10.1039/b807555g. [DOI] [PubMed] [Google Scholar]
- 364.Kim K, Kishima T, Matsumoto T, Nakai H, Ogo S. Selective redox activation of H2 or O2 in a [NiRu] complex by aromatic ligand effects. Organometallics. 2013;32:79–87. [Google Scholar]
- 365.Kishima T, Matsumoto T, Nakai H, Hayami S, Ohta T, Ogo S. A high-valent iron(IV) peroxo core derived from O2. Angew Chem, Int Ed. 2016;55:724–727. doi: 10.1002/anie.201507022. [DOI] [PubMed] [Google Scholar]
- 366.Wulff P, Day CC, Sargent F, Armstrong FA. How oxygen reacts with oxygen-tolerant respiratory [NiFe]-hydrogenases. Proc Natl Acad Sci U S A. 2014;111:6606–6611. doi: 10.1073/pnas.1322393111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Ogo S, Ichikawa K, Kishima T, Matsumoto T, Nakai H, Kusaka K, Ohhara T. A functional [NiFe]hydrogenase mimic that catalyzes electron and hydride transfer from H2. Science. 2013;339:682–684. doi: 10.1126/science.1231345. [DOI] [PubMed] [Google Scholar]
- 368.Manor BC, Rauchfuss TB. Hydrogen activation by biomimetic [NiFe]-hydrogenase model containing protected cyanide cofactors. J Am Chem Soc. 2013;135:11895–11900. doi: 10.1021/ja404580r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Begum A, Moula G, Sarkar S. A nickel(II) –sulfur-based radical-ligand complex as a functional model of hydrogenase. Chem - Eur J. 2010;16:12324–12327. doi: 10.1002/chem.201001812. [DOI] [PubMed] [Google Scholar]
- 370.Sellmann D, Geipel F, Moll M. [Ni(NHPnPr3)(‘S3’)], the first nickel thiolate complex modeling the nickel cysteinate site and reactivity of [NiFe] hydrogenase. Angew Chem, Int Ed. 2000;39:561–563. [PubMed] [Google Scholar]
- 371.Connelly SJ, Zimmerman AC, Kaminsky W, Heinekey DM. Synthesis, structure, and reactivity of a nickel dihydrogen complex. Chem - Eur J. 2012;18:15932–15934. doi: 10.1002/chem.201203675. [DOI] [PubMed] [Google Scholar]
- 372.He T, Tsvetkov NP, Andino JG, Gao X, Fullmer BC, Caulton KG. Mechanism of heterolysis of H2 by an unsaturated d8 nickel center: via tetravalent nickel? J Am Chem Soc. 2010;132:910–911. doi: 10.1021/ja908674x. [DOI] [PubMed] [Google Scholar]
- 373.Tsay C, Peters JC. Thermally stable N2 and H2 adducts of cationic nickel(II) Chem Sci. 2012;3:1313–1318. [Google Scholar]
- 374.Yang JY, Chen S, Dougherty WG, Kassel WS, Bullock RM, DuBois DL, Raugei S, Rousseau R, Dupuis M, Rakowski DuBois M. Hydrogen oxidation catalysis by a nickel diphosphine complex with pendant tert-butyl amines. Chem Commun. 2010;46:8618–8620. doi: 10.1039/c0cc03246h. [DOI] [PubMed] [Google Scholar]
- 375.Raugei S, Helm ML, Hammes-Schiffer S, Appel AM, O’Hagan M, Wiedner ES, Bullock RM. Experimental and computational mechanistic studies guiding the rational design of molecular electrocatalysts for production and oxidation of hydrogen. Inorg Chem. 2016;55:445–460. doi: 10.1021/acs.inorgchem.5b02262. [DOI] [PubMed] [Google Scholar]
- 376.Horvath S, Fernandez LE, Soudackov AV, Hammes-Schiffer S. Insights into proton-coupled electron transfer mechanisms of electrocatalytic H2 oxidation and production. Proc Natl Acad Sci U S A. 2012;109:15663–15668. doi: 10.1073/pnas.1118333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Helm ML, Stewart MP, Bullock RM, Rakowski DuBois M, DuBois DL. A synthetic nickel electrocatalyst with a turnover frequency above 100,000 s−1 for H2 production. Science. 2011;333:863–866. doi: 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- 378.Schilter D, Rauchfuss TB, Stein M. Connecting [NiFe]- and [FeFe]-hydrogenases: mixed-valence nickel–iron dithiolates with rotated structures. Inorg Chem. 2012;51:8931–8941. doi: 10.1021/ic300910r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Schilter D. Nickel–iron hydrogenases: high-resolution crystallography resolves the hydride, but not the debate. ChemBioChem. 2015;16:1712–1714. doi: 10.1002/cbic.201500270. [DOI] [PubMed] [Google Scholar]
- 380.Behnke SL, Shafaat HS. Heterobimetallic models of the [NiFe] hydrogenases: a structural and spectroscopic comparison. Comments Inorg Chem. 2016;36:123–140. [Google Scholar]
- 381.Thauer RK, Kaster A-K, Goenrich M, Schick M, Hiromoto T, Shima S. Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annu Rev Biochem. 2010;79:507–536. doi: 10.1146/annurev.biochem.030508.152103. [DOI] [PubMed] [Google Scholar]
- 382.Shima S, Thauer RK. A third type of hydrogenase catalyzing H2 activation. Chem Rec. 2007;7:37–46. doi: 10.1002/tcr.20111. [DOI] [PubMed] [Google Scholar]
- 383.Zirngibl C, Hedderich R, Thauer RK. N5,N10-Methylenetetrahydromethanopterin dehydrogenase from Methanobacterium thermoautotrophicum has hydrogenase activity. FEBS Lett. 1990;261:112–116. [Google Scholar]
- 384.Stiebritz MT, Reiher M. Hydrogenases and oxygen. Chem Sci. 2012;3:1739–1751. [Google Scholar]
- 385.Berkessel A. Activation of dihydrogen without transition metals. Curr Opin Chem Biol. 2001;5:486–490. doi: 10.1016/s1367-5931(00)00245-3. [DOI] [PubMed] [Google Scholar]
- 386.Lyon EJ, Shima S, Buurman G, Chowdhuri S, Batschauer A, Steinbach K, Thauer RK. UV-A/blue-light inactivation of the ‘metal-free’ hydrogenase (Hmd) from methanogenic archaea The enzyme contains functional iron after all. Eur J Biochem. 2004;271:195–204. doi: 10.1046/j.1432-1033.2003.03920.x. [DOI] [PubMed] [Google Scholar]
- 387.Lyon EJ, Shima S, Boecher R, Thauer RK, Grevels F-W, Bill E, Roseboom W, Albracht SPJ. Carbon monoxide as an intrinsic ligand to iron in the active site of the iron-sulfur-cluster-free hydrogenase H2-forming methylenetetrahydromethanopterin dehydrogenase as revealed by infrared spectroscopy. J Am Chem Soc. 2004;126:14239–14248. doi: 10.1021/ja046818s. [DOI] [PubMed] [Google Scholar]
- 388.Guo Y, Wang H, Xiao Y, Vogt S, Thauer RK, Shima S, Volkers PI, Rauchfuss TB, Pelmenschikov V, Case DA, Alp EE, Sturhahn W, Yoda Y, Cramer SP. Characterization of the Fe Site in iron-sulfur cluster-free hydrogenase (Hmd) and of a model compound via nuclear resonance vibrational spectroscopy (NRVS) Inorg Chem. 2008;47:3969–3977. doi: 10.1021/ic701251j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Shima S, Lyon EJ, Sordel-Klippert M, Kauß M, Kahnt J, Thauer RK, Steinbach K, Xie X, Verdier L, Griesinger C. The cofactor of the iron–sulfur cluster free hydrogenase Hmd: structure of the light inactivation product. Angew Chem, Int Ed. 2004;43:2547–2551. doi: 10.1002/anie.200353763. [DOI] [PubMed] [Google Scholar]
- 390.Hiromoto T, Ataka K, Pilak O, Vogt S, Stagni MS, Meyer-Klaucke W, Warkentin E, Thauer RK, Shima S, Ermler U. The crystal structure of C176A mutated [Fe]-hydrogenase suggests an acyl-iron ligation in the active site iron complex. FEBS Lett. 2009;583:585–590. doi: 10.1016/j.febslet.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 391.Hiromoto T, Warkentin E, Moll J, Ermler U, Shima S. The crystal structure of an [Fe]-hydrogenase–substrate complex reveals the framework for H2 activation. Angew Chem, Int Ed. 2009;48:6457–6460. doi: 10.1002/anie.200902695. [DOI] [PubMed] [Google Scholar]
- 392.Tamura H, Salomone-Stagni M, Fujishiro T, Warkentin E, Meyer-Klaucke W, Ermler U, Shima S. Crystal structures of [Fe]-hydrogenase in complex with inhibitory isocyanides: implications for the H2-activation site. Angew Chem, Int Ed. 2013;52:9656–9659. doi: 10.1002/anie.201305089. [DOI] [PubMed] [Google Scholar]
- 393.Bartoschek S, Buurman G, Thauer RK, Geierstanger BH, Weyrauch JP, Griesinger C, Nilges M, Hutter MC, Helms V. Reface stereospecificity of methylenetetrahydromethanopterin and methylenetetrahydrofolate dehydrogenases is predetermined by intrinsic properties of the substrate. ChemBioChem. 2001;2:530–541. doi: 10.1002/1439-7633(20010803)2:7/8<530::AID-CBIC530>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 394.Shima S, Pilak O, Vogt S, Schick M, Stagni MS, Meyer-Klaucke W, Warkentin E, Thauer RK, Ermler U. The crystal structure of [Fe]-hydrogenase reveals the geometry of the active site. Science. 2008;321:572–575. doi: 10.1126/science.1158978. [DOI] [PubMed] [Google Scholar]
- 395.Stiebritz MT, Reiher M. A unifying structural and electronic concept for Hmd and [FeFe] hydrogenase active sites. Inorg Chem. 2010;49:5818–5823. doi: 10.1021/ic902529c. [DOI] [PubMed] [Google Scholar]
- 396.Dey A. Density functional theory calculations on the mononuclear non-heme iron active site of Hmd hydrogenase: role of the internal ligands in tuning external ligand binding and driving H2 heterolysis. J Am Chem Soc. 2010;132:13892–13901. doi: 10.1021/ja1041918. [DOI] [PubMed] [Google Scholar]
- 397.Finkelmann AR, Senn HM, Reiher M. Hydrogen-activation mechanism of [Fe] hydrogenase revealed by multi-scale modeling. Chem Sci. 2014;5:4474–4482. doi: 10.1039/d1sc90165f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Vogt S, Lyon EJ, Shima S, Thauer RK. The exchange activities of [Fe] hydrogenase (iron–sulfur-cluster-free hydrogenase) from methanogenic archaea in comparison with the exchange activities of [FeFe] and [NiFe] hydrogenases. JBIC. J Biol Inorg Chem. 2008;13:97–106. doi: 10.1007/s00775-007-0302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Wright JA, Turrell PJ, Pickett CJ. The third hydrogenase: more natural organometallics. Organometallics. 2010;29:6146–6156. [Google Scholar]
- 400.Royer AM, Rauchfuss TB, Gray DL. Oxidative addition of thioesters to iron(0): active-site models for Hmd, Nature’s third hydrogenase. Organometallics. 2009;28:3618–3620. [Google Scholar]
- 401.Royer AM, Salomone-Stagni M, Rauchfuss TB, Meyer-Klaucke W. Iron acyl thiolato carbonyls: structural models for the active site of the [Fe]-hydrogenase (Hmd) J Am Chem Soc. 2010;132:16997–17003. doi: 10.1021/ja1072228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Fujishiro T, Kahnt J, Ermler U, Shima S. Protein-pyridinol thioester precursor for biosynthesis of the organometallic acyl-iron ligand in [Fe]-hydrogenase cofactor. Nat Commun. 2015;6:6895. doi: 10.1038/ncomms7895. [DOI] [PubMed] [Google Scholar]
- 403.Obrist BV, Chen D, Ahrens A, Schünemann V, Scopelliti R, Hu X. An iron carbonyl pyridonate complex related to the active site of the [Fe]-hydrogenase (Hmd) Inorg Chem. 2009;48:3514–3516. doi: 10.1021/ic900281g. [DOI] [PubMed] [Google Scholar]
- 404.Song L-C, Xie Z-J, Wang M-M, Zhao G-Y, Song H-B. Biomimetic models for the active site of [Fe]hydrogenase featuring an acylmethyl(hydroxymethyl)pyridine ligand. Inorg Chem. 2012;51:7466–7468. doi: 10.1021/ic301146u. [DOI] [PubMed] [Google Scholar]
- 405.Turrell PJ, Hill AD, Ibrahim SK, Wright JA, Pickett CJ. Ferracyclic carbamoyl complexes related to the active site of [Fe]-hydrogenase. Dalton Trans. 2013;42:8140–8146. doi: 10.1039/c3dt50642h. [DOI] [PubMed] [Google Scholar]
- 406.Turrell PJ, Wright JA, Peck JNT, Oganesyan VS, Pickett CJ. The third hydrogenase: a ferracyclic carbamoyl with close structural analogy to the active site of Hmd. Angew Chem, Int Ed. 2010;49:7508–7511. doi: 10.1002/anie.201004189. [DOI] [PubMed] [Google Scholar]
- 407.Chen D, Scopelliti R, Hu X. [Fe]-hydrogenase models featuring acylmethylpyridinyl ligands. Angew Chem, Int Ed. 2010;49:7512–7515. doi: 10.1002/anie.201004579. [DOI] [PubMed] [Google Scholar]
- 408.Song L-C, Hu F-Q, Wang M-M, Xie Z-J, Xu K-K, Song H-B. Synthesis, structural characterization, and some properties of 2-acylmethyl-6-ester group difunctionalized pyridine-containing iron complexes related to the active site of [Fe]-hydrogenase. Dalton Trans. 2014;43:8062–8071. doi: 10.1039/c4dt00335g. [DOI] [PubMed] [Google Scholar]
- 409.Song L-C, Zhao G-Y, Xie Z-J, Zhang J-W. A novel acylmethylpyridinol ligand containing dinuclear iron complex closely related to [Fe]-hydrogenase. Organometallics. 2013;32:2509–2512. [Google Scholar]
- 410.Song L-C, Xu K-K, Han X-F, Zhang J-W. Synthetic and structural studies of 2-acylmethyl-6-R-difunctionalized pyridine ligand-containing iron complexes related to [Fe]-hydrogenase. Inorg Chem. 2016;55:1258–1269. doi: 10.1021/acs.inorgchem.5b02490. [DOI] [PubMed] [Google Scholar]
- 411.Shima S, Chen D, Xu T, Wodrich MD, Fujishiro T, Schultz KM, Kahnt J, Ataka K, Hu X. Reconstitution of [Fe]-hydrogenase using model complexes. Nat Chem. 2015;7:995–1002. doi: 10.1038/nchem.2382. [DOI] [PubMed] [Google Scholar]
- 412.Chen D, Scopelliti R, Hu X. A five-coordinate iron center in the active site of [Fe]-hydrogenase: hints from a model study. Angew Chem, Int Ed. 2011;50:5671–5673. doi: 10.1002/anie.201100201. [DOI] [PubMed] [Google Scholar]
- 413.Wodrich MD, Hu X. Electronic elements governing the binding of small molecules to a [Fe]-hydrogenase mimic. Eur J Inorg Chem. 2013;2013:3993–3999. [Google Scholar]
- 414.Hu B, Chen D, Hu X. Synthesis and reactivity of mononuclear iron models of [Fe]-hydrogenase that contain an acylmethylpyridinol ligand. Chem - Eur J. 2014;20:1677–1682. doi: 10.1002/chem.201304290. [DOI] [PubMed] [Google Scholar]
- 415.Durgaprasad G, Xie Z-L, Rose MJ. Iron hydride detection and intramolecular hydride transfer in a synthetic model of mono-iron hydrogenase with a CNS chelate. Inorg Chem. 2016;55:386–389. doi: 10.1021/acs.inorgchem.5b01733. [DOI] [PubMed] [Google Scholar]
- 416.Xu T, Yin C-JM, Wodrich MD, Mazza S, Schultz KM, Scopelliti R, Hu X. A functional model of [Fe]-hydrogenase. J Am Chem Soc. 2016;138:3270–3273. doi: 10.1021/jacs.5b12095. [DOI] [PubMed] [Google Scholar]
- 417.Kalz KF, Brinkmeier A, Dechert S, Mata RA, Meyer F. Functional model for the [Fe] hydrogenase inspired by the frustrated Lewis pair concept. J Am Chem Soc. 2014;136:16626–16634. doi: 10.1021/ja509186d. [DOI] [PubMed] [Google Scholar]
- 418.Escalante-Semerena JC, Kenneth L, Rinehart J, Wolfe RS. Tetrahydromethanopterin, a carbon carrier in methanogenesis. J Biol Chem. 1984;259:9447–9455. [PubMed] [Google Scholar]
- 419.Igarashi RY, Laryukhin M, Dos Santos PC, Lee H-I, Dean DR, Seefeldt LC, Hoffman BM. Trapping H− bound to the nitrogenase FeMo-cofactor active site during H2 evolution: characterization by ENDOR spectroscopy. J Am Chem Soc. 2005;127:6231–6241. doi: 10.1021/ja043596p. [DOI] [PubMed] [Google Scholar]
- 420.Hoffman BM, Lukoyanov D, Yang Z-Y, Dean DR, Seefeldt LC. Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem Rev. 2014;114:4041–4062. doi: 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Jeoung J-H, Dobbek H. Carbon dioxide activation at the Ni,Fe-cluster of anaerobic carbon monoxide dehydrogenase. Science. 2007;318:1461–1464. doi: 10.1126/science.1148481. [DOI] [PubMed] [Google Scholar]
- 422.Amara P, Mouesca J-M, Volbeda A, Fontecilla-Camps JC. Carbon monoxide dehydrogenase reaction mechanism: a likely case of abnormal CO2 insertion to a Ni-H− Bond. Inorg Chem. 2011;50:1868–1878. doi: 10.1021/ic102304m. [DOI] [PubMed] [Google Scholar]
- 423.Fontecilla-Camps JC, Amara P, Cavazza C, Nicolet Y, Volbeda A. Structure–function relationships of anaerobic gas-processing metalloenzymes. Nature. 2009;460:814–822. doi: 10.1038/nature08299. [DOI] [PubMed] [Google Scholar]
- 424.Armstrong FA, Hirst J. Reversibility and efficiency in electrocatalytic energy conversion and lessons from enzymes. Proc Natl Acad Sci U S A. 2011;108:14049–14054. doi: 10.1073/pnas.1103697108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Chavarot-Kerlidou M, Chenevier P, Artero V. In: Bioorganometallic Chemistry: Applications in Drug Discovery, Biocatalysis, and Imaging. Jaouen G, Salmain M, editors. Wiley-VCH Verlag GmbH & Co. KGaA; 2015. [Google Scholar]
- 426.Cabot P-L, Alcaide F, Brillas E. Hydrogen reaction at open circuit in alkaline media on Pt in a gas-diffusion electrode. J Electroanal Chem. 2009;626:183–191. [Google Scholar]
- 427.Barton BE, Olsen MT, Rauchfuss TB. Artificial hydrogenases. Curr Opin Biotechnol. 2010;21:292–297. doi: 10.1016/j.copbio.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Kiss G, Zhang K, Mukerjee SL, Hoff CD, Roper GC. Heat of reaction of the Cr(CO)3(C5Me5) radical with H2 and related reactions. Relative and absolute bond strengths in the complexes H–Cr(CO)2(L)(C5R5) J Am Chem Soc. 1990;112:5657–5658. [Google Scholar]
- 429.Gladysz JA, Bedford RB, Fujita M, Gabbaï FP, Goldberg KI, Holland PL, Kiplinger JL, Krische MJ, Louie J, Lu CC, Norton JR, Petrukhina MA, Ren T, Stahl SS, Tilley TD, Webster CE, White MC, Whiteker GT. Organometallics roundtable 2013–2014. Organometallics. 2014;33:1505–1527. [Google Scholar]
- 430.Gewirth AA, Thorum MS. Electroreduction of dioxygen for fuel-cell applications: materials and challenges. Inorg Chem. 2010;49:3557–3566. doi: 10.1021/ic9022486. [DOI] [PubMed] [Google Scholar]
- 431.Mealli C, Rauchfuss TB. Models for the hydrogenases put the focus where it should be—hydrogen. Angew Chem, Int Ed. 2007;46:8942–8944. doi: 10.1002/anie.200703413. [DOI] [PMC free article] [PubMed] [Google Scholar]