

Multidisciplinary study reveals the mechanism for second messenger–mediated regulation of a central bacterial cell cycle kinase.
Keywords: Signaling, signal transduction, histidine kinase, cyclic di-GMP, second messenger, crystallography, biochemistry
Abstract
Histidine kinases are key components of regulatory networks in bacteria. Although many of these enzymes are bifunctional, mediating both phosphorylation and dephosphorylation of downstream targets, the molecular details of this central regulatory switch are unclear. We showed recently that the universal second messenger cyclic di–guanosine monophosphate (c-di-GMP) drives Caulobacter crescentus cell cycle progression by forcing the cell cycle kinase CckA from its default kinase into phosphatase mode. We use a combination of structure determination, modeling, and functional analysis to demonstrate that c-di-GMP reciprocally regulates the two antagonistic CckA activities through noncovalent cross-linking of the catalytic domain with the dimerization histidine phosphotransfer (DHp) domain. We demonstrate that both c-di-GMP and ADP (adenosine diphosphate) promote phosphatase activity and propose that c-di-GMP stabilizes the ADP-bound quaternary structure, which allows the receiver domain to access the dimeric DHp stem for dephosphorylation. In silico analyses predict that c-di-GMP control is widespread among bacterial histidine kinases, arguing that it can replace or modulate canonical transmembrane signaling.
INTRODUCTION
Regulatory kinases represent central cellular switches in all kingdoms of life. Accurate control of their activity in time and space is vital for the execution of specific cellular programs. In bacteria, signal transduction is mediated by histidine kinases (HKs), which, in response to specific stimuli, initiate phosphotransfer to downstream response regulators (1). Many HKs are bifunctional and, by switching to phosphatase action, can effectively reverse the flux within phosphorylation cascades. The cell cycle kinase CckA controls replication initiation in Caulobacter crescentus by changing from kinase to phosphatase activity when cells enter the S phase (2–4). This reversal of activity is controlled by the second messenger cyclic di–guanosine monophosphate (c-di-GMP) (5), the concentration of which oscillates during the C. crescentus cell cycle (4). However, it has remained unclear how this works mechanistically and whether c-di-GMP control of HKs is more widely used in bacteria.
The action of HKs hinges on domain rearrangements. The core of HKs comprises the dimerization histidine phosphotransfer (DHp) domain with the conserved histidine phosphoryl acceptor and the catalytically active (CA) domain with the substrate-binding pocket (6). Both domains can form a tight complex to catalyze the transfer of the γ-phosphate of adenosine triphosphate (ATP) onto the histidine. How autophosphorylation is controlled by external signals has been the subject of several studies, and rearrangements in the dimeric DHp helix bundle in response to conformational changes in the periplasmic sensory domain appear to be central (7, 8) [for a recent review, see Bhate et al. (6)]. Next, for phosphoryl transfer, the CA domain has to disengage from the DHp stem to allow the receiver domain (Rec) of the cognate response regulator to accept the phosphoryl group from P~His (9). Many HKs are bifunctional in that they also catalyze the reverse reaction, that is, dephosphorylation of P~Rec and, as a consequence, inactivation of the response regulator. This bifunctionality not only permits HKs to act as efficient regulatory toggles to rapidly reverse cellular processes but also confers “absolute concentration robustness” to the signal output (10–12). Mechanistically, dephosphorylation entails rebinding of the P~Rec domain to the DHp stem and subsequent phosphoryl transfer to a trapped water molecule (13, 14).
Recently, we have demonstrated that c-di-GMP adopts a cyclin-like function to drive the C. crescentus cell cycle by acting as a potent allosteric effector switching the HK CckA from its default kinase to the phosphatase mode (4). Whereas c-di-GMP levels are low in the G1 phase, they rapidly rise during G1-S transition to peak upon entry into the S phase (15, 16). Binding of c-di-GMP shifts CckA into phosphatase mode to allow chromosome replication (4). Here, we exploit the allosteric effect of c-di-GMP on the structure and function of the bifunctional CckA (4) to unravel the molecular mechanism of the c-di-GMP–mediated switch.
RESULTS AND DISCUSSION
Structure of the CA domain of CckA in complex with c-di-GMP
CckA is composed of two transmembrane helices (with no intervening periplasmic domain) followed by two PAS (Per-Arnt-Sim) domains, the DHp-CA core, and a C-terminal Rec domain. Various constructs with different domain compositions were generated for structural and functional analysis (Fig. 1A). Figure 1B shows the high-resolution crystal structure of the CA domain in complex with c-di-GMP and AMPPNP/Mg2+ (table S1 and fig. S1A). The structure adopts the canonical CA fold but with an unusual insertion folded as a helix/loop “tower” firmly attached to the outer face of the β sheet via hydrophobic interactions. A monomeric c-di-GMP molecule is bound to the edge of the β sheet, where one of its guanine bases forms two (base-specific) H-bonds with the main-chain atoms of residue 524 and stacks with Trp523 and, at an angle, with Tyr514 (Fig. 1C). In addition, guanine N7 is H-bonded to Lys518. The binding site is fully consistent with previous analyses that showed lack of c-di-GMP binding for CckA_ΔTM mutants Y514D, W523A, and F539A (4) in ultraviolet (UV)–cross-linking experiments. All residues are interacting with the ligand, with the exception of F539 that is buried below the tower. C-di-GMP–induced nuclear magnetic resonance chemical shift perturbations have been observed for main-chain amides 465, 515, 518, 519, 526, 537, and 539 (4), again consistent with the crystallographically observed binding mode. The remainder of the ligand forms artifactual crystal contacts with the His-tail of an adjacent molecule. Thus, the crystal structure defines a specific c-di-GMP binding subsite (primary binding site) on the CA domain and suggests that the distal part of the ligand may be available for binding to another domain in full-length CckA.
Fig. 1. CckA crystal structures of the c-di-GMP–complexed CA domain.
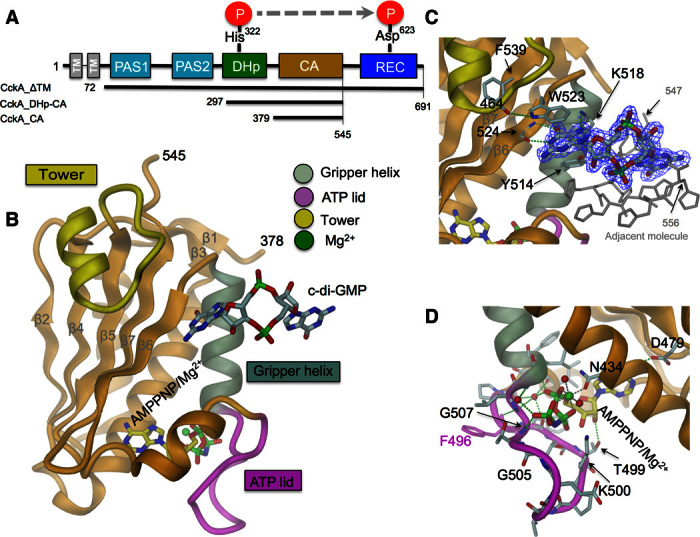
(A) CckA domain arrangement and constructs used. (B to D) Crystal structure of CckA_CA with bound c-di-GMP and AMPPNP/Mg2+. The ATP lid partly covering the mononucleotide site (magenta), the gripper helix (gray, residues 508 to 520), and the CckA-specific insertion (tower, yellow, residues 456 to 471) are distinguished by color. Detailed views of the c-di-GMP and the mononucleotide-protein interactions are shown in (C) and (D), respectively. H-bonds and Mg2+ coordination are shown with green and brown broken lines, respectively.
Structure of dimeric DHp-CA
The structure of the dimeric DHp-CA HK core in complex with adenosine diphosphate (ADP) (Fig. 2 and figs. S1B and S2A) shows a symmetric open domain constellation with the DHp helix bundle apparently accessible for Rec docking. Most of the DHp-CA interactions are mediated by the “gripper” helix and the “thumb” (Phe496) of the CA domain (6). Similar constellations have previously been found for HK853/RR468 (13) and CpxA/ADP (17) and have been discussed as being phosphatase-competent. A structure-based sequence alignment is given in fig. S3. The two CA domains of the DHp-CA dimer are swapped as compared to the above kinases (see “Structural model of the c-di-GMP–mediated DHp-CA cross-link” section), possibly, but not necessarily, as a consequence of distinct crystal contacts. The connection of the α1 and α2 helices of DHp is right-handed, as defined in the study by Ashenberg et al. (18). The ATP lid (with bound ADP) is ordered and shows a similar conformation as in the isolated CA domain in complex with the ATP analog AMPPNP (Fig. 1D). Unfortunately, we were unable to obtain crystals of the DHp-CA construct in the presence of c-di-GMP.
Fig. 2. Crystal structure of CckA_DHp-CA dimer.
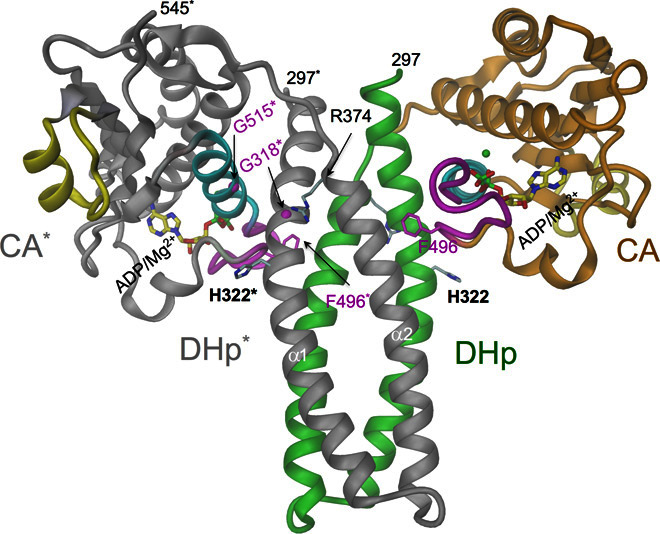
Cartoon representation with one of the subunits colored in green (DHp domain)/orange (CA domain), the other in gray (with asterisked labels); other colors are the same as in Fig. 1. Some important residues and ADP/Mg2+ are shown in full. The Cα atoms of Gly318 and Gly515 are shown as magenta spheres. The domain arrangement is similar as in CpxA/ADP/Mg2+ (17) (fig. S6) but with domains swapped in the dimer. The Phe496 thumb of CA is inserted between DHp helices, and the gripper helix (cyan) mediates most of the interdomain contact.
CckA ligand affinity
To obtain first functional insight into c-di-GMP regulation of CckA, we performed extensive ligand-binding studies (Fig. 3, fig. S4, and table S2). AMPPNP binds with high affinity [Kd (dissociation constant) = ~10 μM] to both the CA domain and the full-length cytosolic CckA_ΔTM construct, whereas ADP affinity is considerably lower (Fig. 3, A and B). C-di-GMP binding turned out to be more complex. The CA domain binds the second messenger with a Kd of about 20 μM (Fig. 3C), that is, clearly above the physiological submicromolar c-di-GMP concentration but in line with the crystal structure that shows only half of the c-di-GMP ligand bound to the domain (Fig. 1). Strikingly, the full-length protein (CckA_ΔTM) binds c-di-GMP with considerably higher affinity (1.4 μM), but only in the presence of ADP (Fig. 3C). No binding is observed in the presence of AMPPNP. These observations strongly suggest that additional domain(s), apart from CA, contribute to c-di-GMP binding and that only the ADP-stabilized domain arrangement is competent for c-di-GMP binding. Pertinently, HK structures obtained in the presence of ADP and AMPPNP are grossly distinct (compare Fig. 4, A and B) (13, 17, 19) and have been correlated with phosphatase and kinase activity, respectively. Thus, c-di-GMP and ADP appear to synergistically stabilize the CckA phosphatase constellation. Because the DHp-CA construct binds c-di-GMP less strongly than the full cytosolic construct (Fig. 3C), additional domains may contribute to the interaction, or, alternatively, the structure of the dimeric DHp bundle is perturbed. The medium-binding affinity of DHp-CA would thus reflect binding to the primary site only. Homology modeling based on the structure of a full-length canonical HK chimera (8) [Protein Data Bank (PDB) code 4gcz] suggests that PAS2-DHp of CckA is organized in the same way (fig. S5, A and B), with the N-terminal PAS2 domains associated to a dimer (fig. S5C) and followed by a parallel coiled coil, which is formed by the C-terminal PAS helix (Jα) that is uninterruptedly merged with the DHp α1 helix. Indeed, in the DHp-CA construct, these stabilizing interactions would be absent, and the N-terminal ends of the DHp bundle may thus be frayed out or unwound.
Fig. 3. Ligand binding to various CckA constructs.
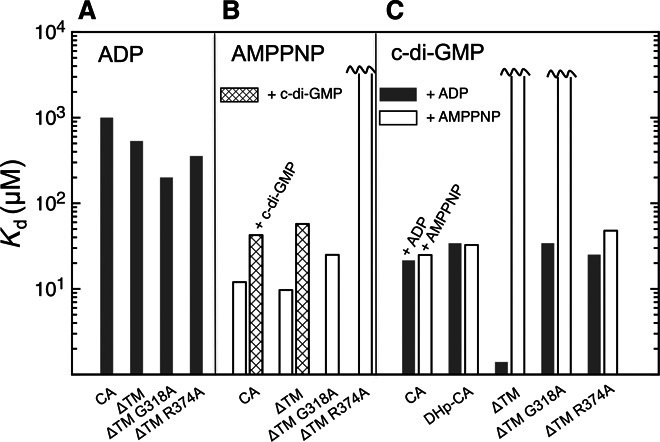
Kd determination was measured by isothermal titration calorimetry (ITC). (A) ADP. (B) AMPPNP in the absence or presence of 100 μM c-di-GMP. (C) c-di-GMP in the presence of 5 mM ADP or AMPPNP. Further information is given in table S2 and fig. S4.
Fig. 4. Modeled DHp-CA domain constellations of CckA.
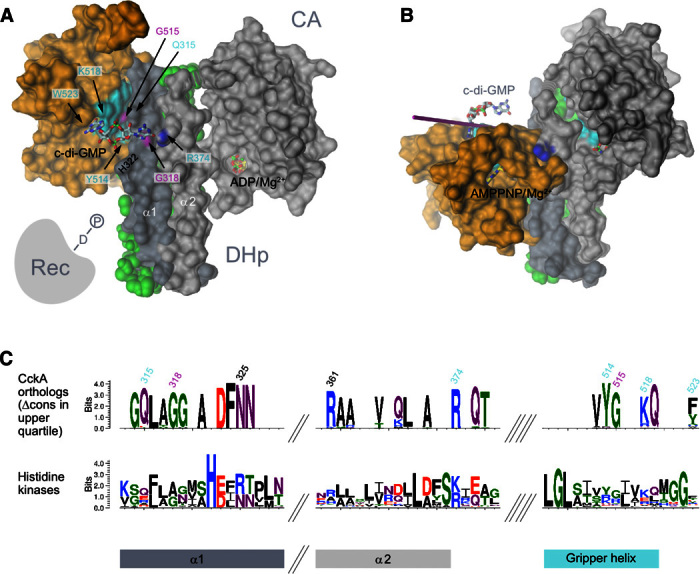
The crystal structures of the individual DHp helices and the CA domains of CckA (molecular surface representation) are shown as obtained upon superposition onto CpxA structures (17). (A) Phosphatase constellation as in the CpxA/ADP complex (PDB code 4bix). C-di-GMP is cross-linking the primary site (CA domain; Y514, K518, and W523) with the secondary site (DHp domain; G318 shown in magenta, R374 with nitrogen atoms in blue). G515 (magenta) is part of the interface. The full model is shown in fig. S6. This constellation is predicted to be competent for dephosphorylation of cognate P~Rec (represented symbolically) (13). (B) Kinase constellation as in the CpxA/AMPPNP complex (PDB code 4biw). Note that relative to the constellation in (A), the CA domain has turned around by about 70° to bring the AMPPNP substrate analog close to the phospho-acceptor His322 (not shown, buried within the interface). (C) Sequence logo excerpts for CckA orthologs (top) and HKs in general (CckA orthologs plus paralogs; bottom). For the upper logo, only residues with a conservation contrast score (Δcons) in the upper quartile are shown (see also fig. S8A). Residues related to c-di-GMP binding are labeled according to CckA numbering.
Structural model of the c-di-GMP–mediated DHp-CA cross-link
To determine possible binding sites for the distal (not CA-bound) part of c-di-GMP, we superimposed the structure of CckA_CA/c-di-GMP with CckA_DHp-CA (fig. S2B) or a CckA model based on the CpxA/ADP structure (17) (Fig. 4A), that is, open constellation structures believed to be phosphatase-competent. In both cases, the distal half of the ligand points toward Arg374 of the DHp α2 helix. With only minor manual conformational adjustments, both moieties can be brought into close juxtaposition to form a canonical O6, N7–guanidinium interaction (20) (fig. S6A). In addition, Gln315 of DHp α1 seems well poised to interact with the ligand. This arrangement is only possible when no side chain is present at position 318 (such as Gly318 of CckA) because this would cause steric interference with c-di-GMP. Through the presence of such a “secondary binding site” on the DHp domain, c-di-GMP could act as a noncovalent cross-linker to stabilize the “open” domain constellation, a critical requirement for access of the phosphorylated C-terminal Rec domain to the DHp stem and phosphatase action. Because, in CpxA, ADP stabilizes the same domain constellation (17), the mechanistic model would also explain the requirement of ADP for high-affinity c-di-GMP binding (Fig. 3C). Why c-di-GMP binding to the ATP-bound conformation of CckA_ΔTM (Fig. 4B) is abolished, although the primary binding site should be accessible, remains to be investigated. The elevated Kd for AMPPNP in the presence of (nonsaturating) c-di-GMP (Fig. 3B) may be evidence for negative cooperativity between these two ligands.
Mechanistic model probed by functional analysis
To test the regulatory model, we assayed kinase and phosphatase activities of CckA_ΔTM by incubation with [32-γP]-ATP. The time behavior in Fig. 5 (A and B) shows nonlinear accumulation of the phosphorylated species, indicating both kinase and phosphatase activity in the absence of c-di-GMP. When c-di-GMP is added, the protein is efficiently and rapidly dephosphorylated as has been seen before (4). Furthermore, phosphorylation is effectively blocked when c-di-GMP is present throughout the experiment (Fig. 5C, first two lanes). Because c-di-GMP binds to CckA only in the presence of ADP, we reasoned that ADP on its own might have a similar, phosphatase-stimulating effect. Indeed, an ADP dose-dependent boost in dephosphorylation is observed (Fig. 5, A and B), which reiterates the c-di-GMP effect, though at a higher ligand concentration. Enhanced dephosphorylation upon addition of ADP has been observed before for EnvZ/OmpR (21) and PhoQ/PhoP (22) and is the cornerstone for a feedback model (23) explaining the transient surge of phospho-PhoP upon activation (24).
Fig. 5. Net phosphorylation of CckA variants in response to c-di-GMP and ADP.
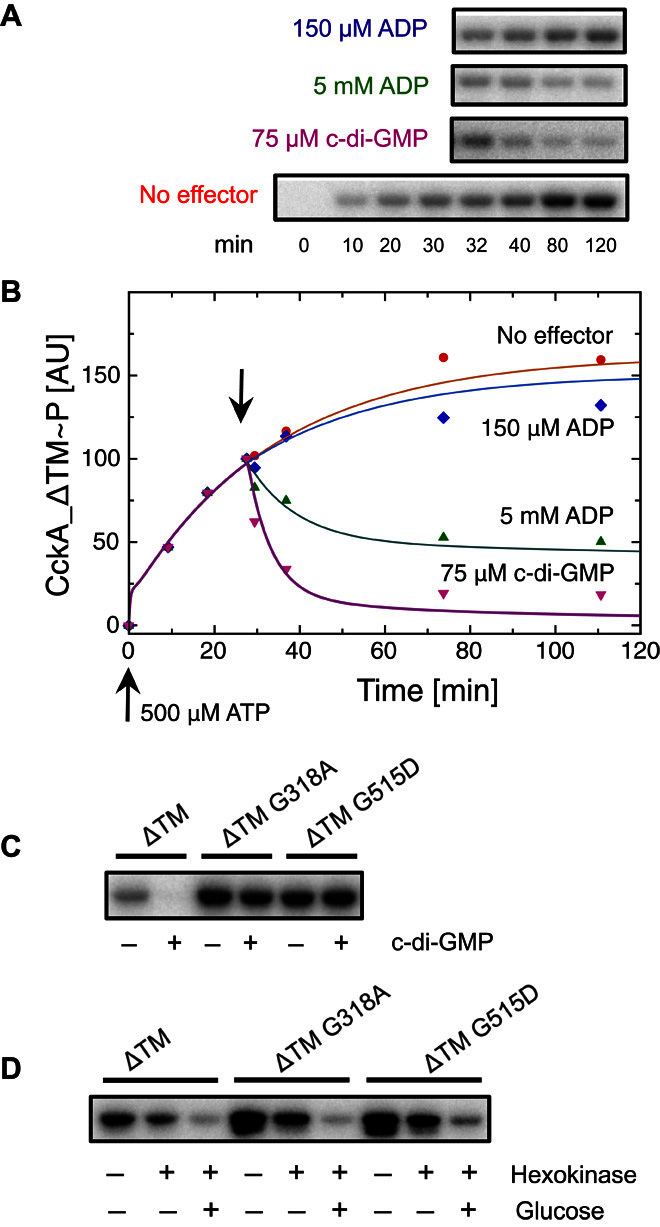
(A) Time course of net CckA_ΔTM~P formation as measured by autoradiography (bottom row). ADP or c-di-GMP was added to aliquots after 30 min of incubation (top three rows). (B) Quantification of the data in (A). Continuous lines have been calculated on the basis of the kinetic model shown in Fig. 7, with parameters (table S3) obtained from the global fit to the data. (C) Net CckA phosphorylation for the indicated variants after 15 min of ATP incubation, in the absence and presence of 75 μM c-di-GMP. (D) Net CckA phosphorylation for the same variants as in (C) but with addition of hexokinase/glucose at t = 15 min and total incubation time of 65 min.
Mechanistic model probed by mutagenesis
Next, we tested the effect of point mutations on the putative secondary binding site. Consistent with the cross-linking model, mutants G318A and R374A exhibited a significant decrease in c-di-GMP affinity (Fig. 3C). In the phosphorylation assay, G318A showed virtually no response to c-di-GMP (Fig. 5C), although the mutant protein was fully competent for mononucleotide binding (Fig. 3, A and B), autophosphorylation (Fig. 5C), and ADP-induced dephosphorylation (Fig. 5D). The mutant R374A failed to bind AMPPNP (Fig. 3A) and was, therefore, not amenable to in vitro phosphorylation assays. However, as discussed below, this mutant showed an informative in vivo phenotype. In addition, the role of residue Gly515 was tested. This residue is a part of the domain interface (Fig. 2 and fig. S2A) and interacts with Gln315, which, in turn, appears well poised to interact with c-di-GMP (fig. S6A). A side chain at position 515 would disturb the domain arrangement and, thus, may interfere with c-di-GMP–mediated domain cross-linking. Indeed, the in vitro phenotype of G515D was identical to that of G318A (Fig. 5, C and D), further strengthening the cross-linking model.
To test our model, we investigated the respective cckA mutant alleles in vivo by analyzing chromosome replication (Fig. 6). As previously reported (4), overexpression of CckA_ΔTM resulted in chromosome overreplication, indicating dominant CckA phosphatase activity. This effect depended on c-di-GMP because overreplication was strongly mitigated in a strain lacking c-di-GMP (16). Expression from a low–copy number plasmid showed virtually no change in phenotype. The (kinase-deficient) R374A caused similar overreplication, but in this case, the phenotype was independent of the c-di-GMP background and, thus, was likely due to basal (ADP-activated) phosphatase activity superseding endogenous CckA kinase activity. Mutants Y514D and G515D showed severe replication deficiency, indicating dominant kinase activity, which is explained by the loss of c-di-GMP–mediated dephosphorylation. In a strain lacking c-di-GMP, expression of these variants was lethal, possibly because of the aggravation of the mutant effect by the absence of c-di-GMP–mediated CtrA proteolytic turnover (25, 26). Finally, the G318A and Y514D mutants showed a strong replication defect even when expressed from the low–copy number plasmid. In contrast, Q315A was indistinguishable with wild-type (WT) overexpression, indicating a minor contribution (if any) of Gln315 to c-di-GMP binding. In summary, these in vivo data are fully consistent with the proposed regulatory c-di-GMP mechanism.
Fig. 6. DNA replication activity of cells expressing various CckA variants.
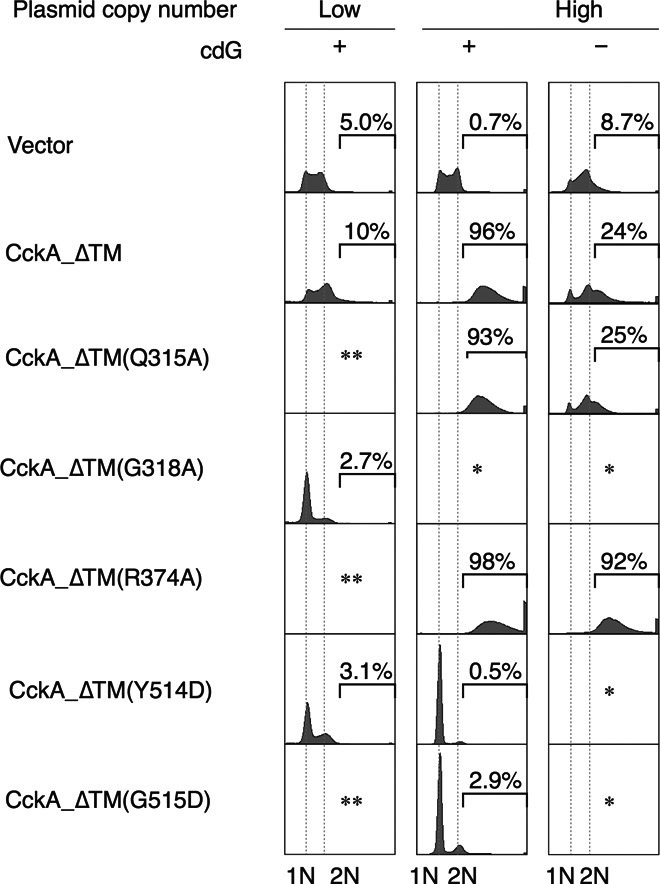
The indicated cckA(ΔTM) variants were expressed from a low– or high–copy number plasmid in the WT (+) or cdG0 (−) strain, followed by analysis of DNA content using flow cytometry. Representative DNA profiles of the two biological replicates are shown. The fraction of cells bearing more than two chromosomes is indicated and shown as percentage. *Transformed cells did not grow. **Not tested.
Evolutionary sequence analysis
Having identified residues crucial for c-di-GMP–mediated CckA control, we set out to study their conservation in other bacterial HKs. Two HK sequence families that broadly cover α-Proteobacteria were generated on the basis of CckA and a distant CckA paralog, respectively (fig. S7, A and B). Although both groups carry the canonical residues that qualify them as HKs (fig. S8A), the orthologous group shows, in addition, well-defined specificity-determining positions [SDPs (27); see also Fig. 4C]. Figure S7D demonstrates that most of the SDPs of the orthogroup (for the distribution, see fig. S7C) cluster at the CckA surface around K518. Strikingly, all of the identified c-di-GMP–related positions belong to this cluster and are highly or strictly conserved among CckA orthologs (fig. S8A). This strongly supports our regulatory model and indicates that c-di-GMP–controlled HKs are widely distributed. Indeed, a broader search for CckA orthologs with a respective hidden Markov model (HMM) profile yielded approximately 2000 unique protein sequences, which contain the characteristic CckA SDPs necessary for c-di-GMP regulation. The sequences span not only the α, β, δ, and ζ classes of Proteobacteria but also representatives from distantly related phyla, including Actinobacteria and Spirochaetes. This finding suggests that c-di-GMP control of HKs was acquired at least 2.5 to 3.0 billion years ago (28).
The CckA PAS domain preceding the DHp-CA core (PAS2) has recently been implicated in c-di-GMP–mediated regulation (29) on the basis of the observation that constructs lacking PAS2 no longer show c-di-GMP–induced inhibition of CckA phosphorylation. This is in line with our observation that the CckA_DHp_CA construct reduced c-di-GMP affinity compared to CckA_ΔTM (Fig. 3C). However, as argued above, this is likely due to a perturbed DHp bundle geometry caused by the absence of the dimeric PAS2 module (see also fig. S5). As a consequence, c-di-GMP would no longer be able to reach the secondary binding site and exert the cross-linking effect necessary for phosphatase induction. The same interpretation may hold for the data reported by Mann et al., in particular because convincing evidence for direct binding of c-di-GMP to PAS2 is still missing. Extending the sequence comparison of CckA orthologs to the region N-terminal to DHp (fig. S8B) revealed that, for most of the proteins, a proximal PAS domain can be predicted confidently. Apart from a few residues conserved for structural reasons, the conservation of the C-terminal DITE motif followed by the J linker helix [see also fig. S5 and the study by Diensthuber et al. (8)] is conspicuous, but there is no hint for a potential c-di-GMP binding site on the PAS2 domain.
Kinetic model of CckA regulation
The various structural and functional aspects of CckA regulation by c-di-GMP and ADP have been consolidated in the kinetic model shown in Fig. 7. Under physiological conditions and in the absence of the c-di-GMP effector, the enzyme would be predominantly complexed with ATP, considering its tighter affinity and larger cellular concentration compared to ADP. Not considering other input signals, the enzyme would autophosphorylate and come to a halt as an ATP complex after nucleotide exchange. The effect of c-di-GMP (Fig. 7B) is proposed to rest on the (thermodynamic and probably also kinetic) stabilization of the ADP complex, which would be the only state competent for dephosphorylation. At the same time, kinase action would be inhibited by the ADP product.
Fig. 7. Kinetic regulatory model of CckA.
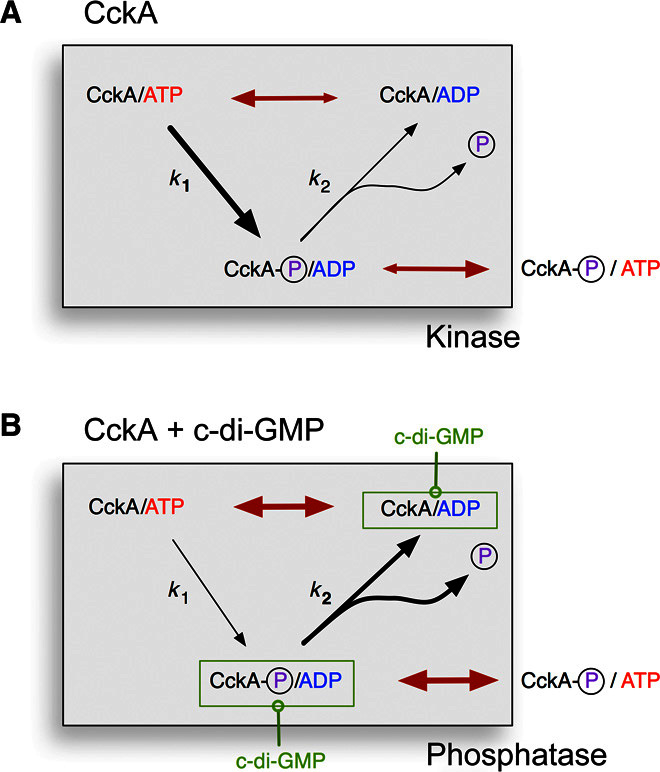
Autophosphorylation and autodephosphorylation proceed with first-order rates k1 and k2, respectively. For simplicity, no distinction is made between phosphorylation of His322 (DHp), Asp623 (Rec), or both. Red double arrows indicate ADP ↔ ATP equilibration. (A) After autophosphorylation, the ADP product gets efficiently replaced by ATP. The dephosphorylation branch is not effective, and the enzyme can catalyze phosphotransfer to the cognate downstream. (B) C-di-GMP allosterically stabilizes the ADP complex, which is competent for autodephosphorylation. Note that rephosphorylation is impeded because of ADP product inhibition. Phosphoryl groups will flow back along the phosphorelay.
This simple kinetic model fits well the progress curves of CckA phosphorylation acquired in the absence and presence of the allosteric effectors (Fig. 5B), considering the small number of free parameters (table S3). A drastically enhanced ADP affinity (modeled by a respective increase in the on-rate) in the presence of a saturating amount of c-di-GMP was obtained from the fit. Furthermore, the equivalent effects induced by c-di-GMP and ADP are reproduced, although the ADP effect at low concentration is somewhat underestimated. Simulations show that the ADP effect is very sensitive to the rate of debinding (kr), and the data can only be fit with a rather slow rate (10−2 to 10−3 s−1, similar to reaction rates k1 and k2; table S3). A similar observation has been made for PhoQ/PhoP by Yeo et al. (23). Further experiments are needed to obtain direct kinetic information.
CONCLUSION
In summary, we have shown that c-di-GMP, in synergy with ADP, reciprocally regulates kinase/phosphatase activity of an HK by stabilizing the open, phosphatase-competent constellation. In this way, the enzyme is rapidly converted to a sink for the CckA phosphorelay, resulting in the inactivation of the replication initiation inhibitor CtrA (3). Ultrasensitive effector response has been predicted theoretically for these bifunctional enzymes with antagonistic activities (30). For CckA, this may allow precise mode switching in response to the oscillating c-di-GMP level during the Caulobacter cell cycle and, thus, well-coordinated execution of the CtrA-mediated cellular processes.
MATERIALS AND METHODS
Strains
Strains used in this study are listed in database S1.
Plasmids
For structural work and binding studies, the coding sequences for various CckA constructs were cloned into pET21b vectors between Nde I and Not I restriction sites, which yielded C-terminally His6 tag variants. Primers 8688 and 9610 were used to generate CckA G72-691. Primers 7243 and 7244 were used to generate CckA G77-A545. Primers 7244 and 7247 were used to generate Q297A-A545, and primers 7244 and 7249 were used to generate Q379-A545. Internal primers were used to introduce point mutations. Plasmids and oligonucleotides are listed in databases S2 and S3, respectively.
For in vitro activity assays, the previously described CckA_ΔTM (S72-A691) construct was cloned into the pET28a-His-MBP vector between Bam HI and Sal I restriction sites using primers 5276 and 5277 (4), and the derived point mutants were used. R374A was introduced by mutagenic primers 6483 and 6484. G515D was introduced using mutagenic primers 7901 and 7902. G318A was amplified from SH339 genomic DNA using primers 6276 and 5277.
For in vivo studies, pBXMCS-cckAQ315A, pBXMCS-cckAR374A, and pBXMCS-cckAG515D were generated by splice overlap extension–polymerase chain reaction (PCR) using pBXMCS-2-cckAWT as a template DNA, 7200 and 7369 as outside primers, and internal mutagenic primers. The mutagenic primers were as follows: 8543 and 8544 for Q315A, 6483 and 6484 for R374A, and 7901 and 7902 for G515D. After fusion PCR, the products were digested with Eco RI and Nde I, followed by ligation to the Eco RI–Nde I fragment of pBXMCS-2. To construct pBXMCS-cckAG318A, PCR was performed using a genomic DNA with the cckA G318A allele and primers 7200 and 7369. The products were digested with Eco RI and Nde I, followed by ligation to the Eco RI–Nde I fragment of pBXMCS-2. To construct a low–copy number plasmid expressing cckAΔTM_WT from the xylose-dependent promoter, pBXMCScckAWT was digested with Eco RI and Hind III. The resulting 2.3-kb DNA fragment was ligated to the Eco RI–Hind III fragment of pMR10, yielding pMR10-Pxyl::cckAΔTM(WT). pMR10-Pxyl::cckAΔTM(G318A) and pMR10-Pxyl::cckAΔTM(Y514D) were constructed similarly using pBXMCScckA G318A and pBXMCScckA Y514D instead of pBXMCScckAWT.
Expression and purification
Proteins were expressed in Rosetta (DE3) cells. LB media were inoculated with 1% preculture at 37°C. Bacterial cultures were induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside at an optical density at 600 nm (0.6 to 0.8), and the incubation temperature was shifted to 22°C. The cells were finally harvested overnight after induction by spinning the cultures at 5000 rpm for 10 min (at 4°C using F8-6 rotor, Thermo Scientific). For purification, the cell pellet was resuspended in buffer A [30 mM tris-HCl (pH 7.5), 500 mM NaCl, 20 mM imidazole, and 5mM MgCl2] with protease inhibitor cocktail supplied by Roche. Cells were lysed using a microfluidizer, and the lysate was centrifuged at 16,000 rpm for 30 min (at 4°C using an SS-34 rotor, Sorvall) to remove cell debris and any suspended particles.
Clear supernatant was applied on a Ni–nitrilotriacetic acid column (5-ml prepack column, GE Healthcare) preequilibrated with buffer A. The column was preequilibrated with buffer A until the baseline was reached. Bound protein was eluted with a 0 to 100% linear gradient of buffer B [30 mM tris-HCl (pH 7.5), 500 mM NaCl, 500 mM imidazole, and 5 mM MgCl2]. Fractions containing the desired protein were pooled and concentrated to a final volume of 2 to 4 ml. The concentrated protein was then loaded on a Superdex S200-26/60 gel filtration column preequilibrated with a buffer containing 30 mM tris-HCl (pH 7.5), 100 mM NaCl, and 5 mM MgCl2. Elution peak was collected and the protein concentration was quantified by UV absorption (NanoDrop, Thermo Scientific). For in vitro activity assays, maltose-binding protein (MBP) tagged CckA_ΔTM and the derived point mutants were purified as described (4).
Crystallization, data collection, and structure solution
Crystallization was performed using the sitting-drop vapor diffusion method. The CckA_CA construct was crystallized at a concentration of 15 mg/ml in the presence of AMPPNP and c-di-GMP (1:5:3 molar ratio) in 30 mM tris-HCl (pH 7.5), 200 mM NaCl, and 5 mM MgCl2. Initial plate-like crystals appeared after 10 days in 0.15 M ammonium sulfate, 25% polyethylene glycol (PEG) 4000, and 0.1 M MES (pH 5.5) and were used for seed stock preparation. Upon seeding, well-diffracting plate-like crystals were obtained in 0.1 M ammonium sulfate, 28.4% PEG 4000, and 0.1 M MES (pH 5.5). Crystals were frozen with cryo-oil (Hampton) as a cryoprotectant.
After extensive crystallization trials for the CckA_DHp-CA construct, a single hit was obtained at a concentration of 15 mg/ml in the presence of ADP (1:5 molar ratio) in 30 mM tris-HCl (pH 7.5), 300 mM NaCl, and 5 mM MgCl2. The protein crystallized in 30% PEG 550 MME (monomethylester), 30% PEG 2000 MME, Morpheus (Molecular Dimensions) buffer 1 (pH 6.5), and Morpheus carboxylic acids after 3 to 4 weeks. The crystals were frozen without any cryoprotectant.
X-ray diffraction data sets were collected at the Swiss Light Source, Villigen, Switzerland at 100 K. Data sets were processed either with MOSFLM (31) or XDS (32), and resulting intensities were scaled using SCALA from the CCP4 suite (33). Both structures were solved by molecular replacement. For determination of the CckA_CA structure, the CA domain of DivL [PDB code 4q20 (34)] was used as a search model. For determination of the CckA_DHp-CA structure, the CckA_CA was used as a search model, and the DHp domain was obtained by iterative model building and refinement.
All structures were refined using REFMAC5 (CCP4). Model building was performed using COOT (35) and O (36). Ligand molecules and metal ions were modeled in Fo-Fc electron density maps. Finally, water molecules were added where the difference density in the Fo-Fc map exceeded 3σ and potential hydrogen bonds could be formed. Data processing, structure refinement, and validation statistics are given in table S1.
Ligand binding
Thermodynamic parameters for ligand binding to CckA variants were measured by ITC using a VP-ITC (MicroCal) instrument. All calorimetric titrations were carried out at a temperature of 25° or 12°C, a syringe stirring speed of 300 rpm, a preinjection delay of 200 s, and a recording interval of 250 s. Equilibrium association constant, binding stoichiometry (N), entropy (ΔS), and enthalpy (ΔH) of the binding reaction were derived by fitting the data to a single binding-site model using ORIGIN7 (OriginLab). Because all N values indicated (within 25%) 1:1 stoichiometry, the data were refitted with a fixed N = 1 value.
Protein phosphorylation assay
CckA phosphorylation was assayed by autoradiography following the protocol given in the study by Lori et al. (4). Reactions were run in the presence of 500 μM ATP and 5 μCi of γ32PATP (3000 Ci mmol−1, Hartmann Analytic) at room temperature. Additional nucleotides were added at indicated time points. Reactions were stopped with SDS sample buffer and subsequently loaded (or stored on ice) on 10% SDS gels. Wet gels were exposed to phosphor screen (0.5 to 3 hours) before being scanned using a Typhoon FLA 7000 imaging system (GE Healthcare). Where applicable, ATP was converted to ADP by the addition of 1.5 U of hexokinase (Roche) and 5 mM d-glucose.
Flow cytometry
This assay was performed as previously described (4). Briefly, pBXMCS-2 or its derivatives carrying a cckA(ΔTM) variant were transformed into NA1000 (WT) or UJ5065 (cdG0). After incubation on peptone yeast extract (PYE) agar supplemented with kanamycin (20 μg/ml) and 0.1% glucose at 30°C for 2 days, the transformants were grown overnight at 30°C in PYE medium supplemented with kanamycin (5 μg/ml) and 0.1% glucose. A portion (0.2 ml) was diluted in 4 ml of PYE medium supplemented with kanamycin (5 μg/ml) and 0.03% xylose and incubated for 4 hours, followed by cell fixation in 70% ethanol. After ribonuclease treatment, DNA was stained with YO-PRO-1 iodide (Invitrogen), and the fluorescence intensity was analyzed using FACSCanto II (BD Biosciences).
Fitting of kinetic data and simulations
Enzymatic reactions were fitted to standard first-order and second-order reaction kinetics according to the model shown in Fig. 4. The system of coupled ordinary differential equations was set up using the Complex Pathway Simulator (COPASI) software (37). Global fitting and simulations were performed using ProFit 6.2.14 (QuantumSoft).
Bioinformatic sequence analyses
We identified CckA orthologs within the α-Proteobacteria lineage, and γ-Proteobacteria as an outgroup, satisfying the criteria of reciprocally highest similarity [BLASTp (38, 39)] and a sequence distance tree topology that matches the topology of speciation (40). There is no clear conservation of flanking synteny for these orthologs. All of these orthologs have the same domain organization—an N-terminal region containing a variable number of transmembrane and PAS (cl21578) domains, followed by a single copy of the HisKA/DHp (cl00080), HATPase c/CA (cl00075), and REC (cl19078) domains. The only exception is the Escherichia coli CckA ortholog (gi 553959296), which has a HAMP (cl01054) domain instead of a PAS domain.
The C. crescentus CB15 genome contains at least 23 protein-coding genes with sufficiently high sequence similarity with CckA, suggesting multiple duplication events within and/or preceding the α-Proteobacteria lineage. A paralog with the same domain topology as CckA (Uniprot ID:Q9AAL1, 22/37% core identity/similarity) was used as a query to identify reciprocally highest similarity sequences among the same Proteobacteria species used to identify the group of CckA orthologs.
We derived a score for identifying SDPs by contrasting sequence conservation between the CckA orthologs and the closely related paralogs. A ClustalW2-initiated (41) and manually curated multiple sequence alignment (MSA) of the CckA orthologs and paralogs spanning the contiguous DHp and CA domains was used as input. For each column in the alignment, a conservation score was calculated using normalized 21-type Shannon’s entropy (42) and a window of size 7 (43) for the whole alignment of orthologs and paralogs (EOP), and only for the CckA orthologs (EO) and paralogs (EP) from the same MSA. In the general case, Δcons was calculated as per Eq. 1
| (1) |
For sites in the MSA with high conservation within both orthologs and paralogs but not between these groups [formalized by these two criteria: (EO − EOP) > EOP and EP > EOP], Δcons was calculated as per Eq. 2
| (2) |
A CckA ortholog–derived HMM was built using the HMMer package (44) with the contiguous MSA of the DHp and CA domains as input.
Supplementary Material
Acknowledgments
We thank F. Hamburger for her professional work in plasmid constructions, the beamline staff at the Swiss Light Source in Villigen for expert help in data acquisition, and T. Sharpe from the Biophysics facility at the Biozentrum for expert biophysical support and comments on the manuscript. Bioinformatic analyses were performed at the sciCORE (http://scicore.unibas.ch/) scientific computing core facility at the University of Basel, with support from the SIB Swiss Institute of Bioinformatics. We thank A. Eberhardt for technical help. Funding: This work was supported by grants from the Swiss National Science Foundation (31003A-138414 to T.S. and 310030B_147090 to U.J.) and by a European Research Council Advanced Research grant to U.J. Author contributions: B.N.D., U.J., and T.S. initiated the project. All authors designed the experiments. B.N.D. crystallized the CckA variants and determined the structures. B.N.D. and T.S. performed the analyses on the resulting models. B.N.D., I.P.-M., and C.L. carried out biochemical experiments. S.O. carried out genetic experiments. G.F. performed bioinformatic analyses. T.S., U.J., S.O., G.F., and B.N.D. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Coordinates and structure factors for CckA_CA/AMPPNP/c-di-GMP and CckA_DH-CA /ADP have been deposited in the PDB under accession codes 5IDM and 5IDJ, respectively.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/9/e1600823/DC1
table S1. Data collection and refinement statics.
table S2. Thermodynamic parameters of CckA-ligand interactions as measured by ITC.
table S3. Kinetic parameters of HK/phosphatase CckA_ΔTM.
fig. S1. CckA crystal packings.
fig. S2. Detailed views of domain interface of CckA_DHp-CA and comparison with c-di-GMP–bound CckA_CA.
fig. S3. Sequence alignment between CckA orthologs and paralogs.
fig. S4. ITC ligand-binding profiles for several CckA.
fig. S5. Structure prediction for PAS2 domain of CckA.
fig. S6. Proposed model of noncovalent c-di-GMP–mediated cross-linking of the CA with the DHp domain of CckA.
fig. S7. Consensus tree analysis for CckA orthologs and paralogs.
fig. S8. Residue conservation in CckA orthologs and paralogs.
database S1. Bacterial strains used in this study.
database S2. Plasmids used in this study.
database S3. Oligonucleotides used in this study.
REFERENCES AND NOTES
- 1.Gao R., Stock A. M., Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 63, 133–154 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobs C., Domian I. J., Maddock J. R., Shapiro L., Cell cycle–dependent polar localization of an essential bacterial histidine kinase that controls DNA replication and cell division. Cell 97, 111–120 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Biondi E. G., Reisinger S. J., Skerker J. M., Arif M., Perchuk B. S., Ryan K. R., Laub M. T., Regulation of the bacterial cell cycle by an integrated genetic circuit. Nature 444, 899–904 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Lori C., Ozaki S., Steiner S., Böhm R., Abel S., Dubey B. N., Schirmer T., Hiller S., Jenal U., Cyclic di-GMP acts as a cell cycle oscillator to drive chromosome replication. Nature 523, 236–239 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Hengge R., Galperin M. Y., Ghigo J.-M., Gomelsky M., Green J., Hughes K. T., Jenal U., Landini P., Systematic nomenclature for GGDEF and EAL domain-containing cyclic di-GMP turnover proteins of Escherichia coli. J. Bacteriol. 198, 7–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhate M. P., Molnar K. S., Goulian M., DeGrado W. F., Signal transduction in histidine kinases: Insights from new structures. Structure 23, 981–994 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Möglich A., Ayers R. A., Moffat K., Design and signaling mechanism of light-regulated histidine kinases. J. Mol. Biol. 385, 1433–1444 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diensthuber R. P., Bommer M., Gleichmann T., Möglich A., Full-length structure of a sensor histidine kinase pinpoints coaxial coiled coils as signal transducers and modulators. Structure 21, 1127–1136 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Casino P., Miguel-Romero L., Marina A., Visualizing autophosphorylation in histidine kinases. Nat. Commun. 5, 3258 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Batchelor E., Goulian M., Robustness and the cycle of phosphorylation and dephosphorylation in a two-component regulatory system. Proc. Natl. Acad. Sci. U.S.A. 100, 691–696 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hart Y., Alon U., The utility of paradoxical components in biological circuits. Mol. Cell 49, 213–221 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Gao R., Stock A. M., Probing kinase and phosphatase activities of two-component systems in vivo with concentration-dependent phosphorylation profiling. Proc. Natl. Acad. Sci. U.S.A. 110, 672–677 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casino P., Rubio V., Marina A., Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 139, 325–336 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Casino P., Rubio V., Marina A., The mechanism of signal transduction by two-component systems. Curr. Opin. Struct. Biol. 20, 763–771 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Christen M., Kulasekara H. D., Christen B., Kulasekara B. R., Hoffman L. R., Miller S. I., Asymmetrical distribution of the second messenger c-di-GMP upon bacterial cell division. Science 328, 1295–1297 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abel S., Bucher T., Nicollier M., Hug I., Kaever V., Abel zur Wiesch P., Jenal U., Bi-modal distribution of the second messenger c-di-GMP controls cell fate and asymmetry during the caulobacter cell cycle. PLOS Genet. 9, e1003744 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mechaly A. E., Sassoon N., Betton J.-M., Alzari P. M., Segmental helical motions and dynamical asymmetry modulate histidine kinase autophosphorylation. PLOS Biol. 12, e1001776 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashenberg O., Keating A. E., Laub M. T., Helix bundle loops determine whether histidine kinases autophosphorylate in cis or in trans. J. Mol. Biol. 425, 1198–1209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang C., Sang J., Wang J., Su M., Downey J. S., Wu Q., Wang S., Cai Y., Xu X., Wu J., Senadheera D. B., Cvitkovitch D. G., Chen L., Goodman S. D., Han A., Mechanistic insights revealed by the crystal structure of a histidine kinase with signal transducer and sensor domains. PLOS Biol. 11, e1001493 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou S.-H.,Galperin M. Y., Diversity of cyclic di-GMP-binding proteins and mechanisms. J. Bacteriol. 198, 32–46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Y., Qin L., Yoshida T., Inouye M., Phosphatase activity of histidine kinase EnvZ without kinase catalytic domain. Proc. Natl. Acad. Sci. U.S.A. 97, 7808–7813 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanowar S., Le Moual H., Functional reconstitution of the Salmonella typhimurium PhoQ histidine kinase sensor in proteoliposomes. Biochem. J. 390, 769–776 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yeo W.-S., Zwir I., Huang H. V., Shin D., Kato A., Groisman E. A., Intrinsic negative feedback governs activation surge in two-component regulatory systems. Mol. Cell 45, 409–421 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shin D., Lee E.-J., Huang H., Groisman E. A., A positive feedback loop promotes transcription surge that jump-starts Salmonella virulence circuit. Science 314, 1607–1609 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Duerig A., Abel S., Folcher M., Nicollier M., Schwede T., Amiot N., Giese B., Jenal U., Second messenger-mediated spatiotemporal control of protein degradation regulates bacterial cell cycle progression. Genes Dev. 23, 93–104 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozaki S., Schalch-Moser A., Zumthor L., Manfredi P., Ebbensgaard A., Schirmer T., Jenal U., Activation and polar sequestration of PopA, a c-di-GMP effector protein involved in Caulobacter crescentus cell cycle control. Mol. Microbiol. 94, 580–594 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Chakrabarti S., Bryant S. H., Panchenko A. R., Functional specificity lies within the properties and evolutionary changes of amino acids. J. Mol. Biol. 373, 801–810 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Battistuzzi F. U., Feijao A., Hedges S. B., A genomic timescale of prokaryote evolution: Insights into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evol. Biol. 4, 44 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mann T. H., Seth Childers W., Blair J. A., Eckart M. R., Shapiro L., A cell cycle kinase with tandem sensory PAS domains integrates cell fate cues. Nat. Commun. 7, 11454 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Straube R., Reciprocal regulation as a source of ultrasensitivity in two-component systems with a bifunctional sensor kinase. PLOS Comput. Biol. 10, e1003614 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leslie A. G. W., The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. D62, 48–57 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. D66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Collaborative Computational Project, Number 4 , The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. D50, 760–763 (1994). [DOI] [PubMed] [Google Scholar]
- 34.Childers W. S., Xu Q., Mann T. H., Mathews I. I., Blair J. A., Deacon A. M., Shapiro L., Cell fate regulation governed by a repurposed bacterial histidine kinase. PLOS Biol. 12, e1001979 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. D66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.T. A. Jones, M. Kjeldgaard, O Version 5.9 (The Manual, Uppsala University, Uppsala, Sweden, 1993).
- 37.Hoops S., Sahle S., Gauges R., Lee C., Pahle J., Simus N., Singhal M., Xu L., Mendes P., Kummer U., COPASI—A COmplex PAthway SImulator. Bioinformatics 22, 3067–3074 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 39.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T. L., BLAST+: Architecture and applications. BMC Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams K. P., Sobral B. W., Dickerman A. W., A robust species tree for the Alphaproteobacteria. J. Bacteriol. 189, 4578–4586 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G., Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Valdar W. S. J., Scoring residue conservation. Proteins 48, 227–241 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Capra J. A., Singh M., Predicting functionally important residues from sequence conservation. Bioinformatics 23, 1875–1882 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Eddy S. R., Profile hidden Markov models. Bioinformatics 14, 755–763 (1998). [DOI] [PubMed] [Google Scholar]
- 45.Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L., Schwede T., SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sundriyal A., Massa C., Samoray D., Zehender F., Sharpe T., Jenal U., Schirmer T., Inherent regulation of EAL domain-catalyzed hydrolysis of second messenger cyclic di-GMP. J. Biol. Chem. 289, 6978–6990 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saitou N., Nei M., The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 (1987). [DOI] [PubMed] [Google Scholar]
- 48.Jones D. T., Taylor W. R., Thornton J. M., The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282 (1992). [DOI] [PubMed] [Google Scholar]
- 49.Felsenstein J., Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39, 783–791 (1985). [DOI] [PubMed] [Google Scholar]
- 50.Tamura K., Stecher G., Peterson D., Filipski A., Kumar S., MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crooks G. E., Hon G., Chandonia J.-M., Brenner S. E., WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/9/e1600823/DC1
table S1. Data collection and refinement statics.
table S2. Thermodynamic parameters of CckA-ligand interactions as measured by ITC.
table S3. Kinetic parameters of HK/phosphatase CckA_ΔTM.
fig. S1. CckA crystal packings.
fig. S2. Detailed views of domain interface of CckA_DHp-CA and comparison with c-di-GMP–bound CckA_CA.
fig. S3. Sequence alignment between CckA orthologs and paralogs.
fig. S4. ITC ligand-binding profiles for several CckA.
fig. S5. Structure prediction for PAS2 domain of CckA.
fig. S6. Proposed model of noncovalent c-di-GMP–mediated cross-linking of the CA with the DHp domain of CckA.
fig. S7. Consensus tree analysis for CckA orthologs and paralogs.
fig. S8. Residue conservation in CckA orthologs and paralogs.
database S1. Bacterial strains used in this study.
database S2. Plasmids used in this study.
database S3. Oligonucleotides used in this study.