

Abstract
Sympathetic nervous system activation and catecholamine release are important events following injury and infection. The nature and timing of different pathophysiologic insults have significant effects on adrenergic pathways, inflammatory mediators, and the host response. Beta adrenergic receptor blockers (β-blockers) are commonly used for treatment of cardiovascular disease but recent data suggests that the metabolic and immunomodulatory effects of β-blockers can expand their use. β-blocker therapy can reduce sympathetic activation and hypermetabolism as well as modify glucose homeostasis and cytokine expression. It is the purpose of this review to examine either the biologic basis for proposed mechanisms or to describe current available clinical evidence for the use of β-blockers in traumatic brain injury (TBI), spinal cord injury (SCI), hemorrhagic shock, acute traumatic coagulopathy, erythropoietic dysfunction, metabolic dysfunction, pulmonary dysfunction, burns, immunomodulation, and sepsis.
Keywords: Beta blockade, beta blocker, propranolol, metoprolol, trauma, injury, catecholamines, immunosuppression
Introduction
The physiologic concept that β adrenergic receptor blocker (β-blocker) therapy can decrease tissue oxygen consumption and hypermetabolism has led several investigators to research the role of β-blockers following traumatic injury. Increased sympathetic activity and exaggerated catecholamine release may be triggered by traumatic brain injury (TBI), autonomic dysfunction associated with spinal cord injury (SCI), hemorrhagic shock, burns, infection, tissue hypoperfusion, hypoxia, and direct tissue injury. The intensity and duration of catecholamines released as part of the host response to injury can influence the activation of the acute inflammatory response.(1) Catecholamines are an integral part of the neuro-endocrine-immune/inflammatory network.(1) Not only do stress and inflammation have key roles following injury, but these responses also change over the course of critical illness in those that survive their initial insult. Therefore, it is a complicated process to consider the potential contributions of β-blockers in modulating the stress response, the inflammatory response, hypermetabolism, and host protective immunity. This review provides an overview of the β adrenergic receptor and the pharmacology of β-blockers. Subsequently, it discusses both animal and existing clinical studies regarding β-blocker use in TBI, SCI, hemorrhagic shock, acute traumatic coagulopathy, erythropoietic dysfunction, metabolic dysfunction, pulmonary dysfunction, burns, immunomodulation, and sepsis.
β adrenergic receptors and their pharmacology
β adrenergic receptors are G-protein coupled receptors of three types (β-1, β-2, and β-3), all of which activate adenylate cyclase which then increases cyclic adenosine monophosphate, activating protein kinase A.(2) The β adrenergic receptor itself can be phosphorylated by protein kinase A, which leads to its internalization and downregulation.(3) Each receptor is activated by epinephrine (EPI) and norepinephrine (NE).(2) Plasma EPI levels primarily reflect adrenal medullary release, whereas circulating NE represents spillover from sympathetic neuron terminals, and both can function as neurotransmitters.(4) While EPI and NE have nearly equal potency for β-1 receptors, NE has up to 100-fold selectivity for β-2 receptors and EPI has greater potency for β-3 receptors.(2) β-1 receptors are concentrated in cardiomyocytes, β-2 receptors predominate in blood vessels and airway smooth muscle tissues, and each receptor is found in every major organ system.(3) β-1 stimulation increases chronotropy and inotropy in cardiomyocytes, whereas β-2 stimulation causes smooth muscle dilation in blood vessels and airways.(2) β-3 receptor functions include lipolysis in white adipose tissue, thermogenesis in brown adipose tissue, and modulation of hematopoietic progenitor cell (HPC) mobilization.(2–5)
β-blockers are characterized by their variable selectivity among receptor subtypes (Table 1).(6) Some β-blockers also inhibit α-1 adrenergic receptors, and therefore potentiate vasodilation.(6) Commonly prescribed β-blockers include: (1) nonselective β-blockers that antagonize both β-1 and β-2 receptors, (2) β-1 selective antagonists, and (3) α-1 and nonselective β-blockers. β-2 and β-3 blockade may be accomplished with experimental selective antagonists or with non-selective β-blockers.(5) Experimental β-3 antagonists include L-748337, which has been shown to inhibit nitric-oxide mediated cellular proliferation,(7) and SR 59230A hydrochloride, which has been used to investigate brown adipose tissue thermogenesis and colonic motility in rodent models.(8)
Table 1.
Properties of β-blockers. Time to peak and half-life are based on clinical pharmacokinetics. Use of L-748,337 and SR 59230A have been reported only in experimental studies.
| Medication | Selectivity | Time to peak | Half-life | Other effects |
|---|---|---|---|---|
| Propranolol | nonselective | 1–1.5 hr | 3–5 hr | membrane stabilizer, ↓ portal pressure |
| Nadolol | nonselective | 3–4 hr | 20–24 hr | ↓ portal pressure |
| Pindolol | nonselective | 1 hr | 3–4 hr | ↓ atrioventricular nodal conduction |
| Timolol | nonselective | 1–2 hr | 3–4 hr | ↓ sympathetic outflow |
| Metoprolol | β1 antagonist | 0.3–2 hr | 3–4 hr | |
| Esmolol | β1 antagonist | 2–10 min | 10 min | |
| Bisoprolol | β1 antagonist | 2–4 hr | 9–12 hr | |
| Atenolol | β1 antagonist | 2–4 hr | 6–7 hr | |
| Acebutolol | β1 antagonist | 2–4 hr | 4–13 hr | membrane stabilizer, sympathomimetic |
| Nebivolol | β1 antagonist | 1.5–4 hr | 10–19 hr | potentiates nitric oxide |
| Carvedilol | alpha-1, β1, β2 antagonist | 1–5 hr | 6–10 hr | alpha-1 blockade, membrane stabilization |
| Labetalol | alpha-1, β1, β2 antagonist | 1–2 hr | 6–8 hr | |
| L-748,337 | β3 antagonist | NA | NA | ↓ nitric oxide mediated cell proliferation |
| SR 59230A | β3 antagonist | NA | NA |
There are several potential adverse effects of β-blockers. β-blockade may compromise cardiovascular function and the compensatory response by decreasing blood pressure and heart rate.(9) These effects are particularly deleterious in the context of hemorrhagic shock,(10) limiting the clinical application of β-blockers for patients with severe bleeding. In addition, because β-2 stimulation potentiates smooth muscle dilation in the airways,(2) blockade of these receptors may result in bronchoconstriction. This phenomenon may unmask or exacerbate the effects of asthma.(11) Patients with diabetes and those at increased risk for developing diabetes may be adversely affected by long term β-blocker use due to the propensity of these medications to blunt insulin sensitivity. (12) Finally, chronic β-blockade has been associated with increased incidence of sexual dysfunction, though the causal mechanism remains unclear.(13)
Traumatic brain injury
Plasma NE and urine catecholamines are significantly elevated following TBI and correlate with the severity of neurologic deficit.(14, 15) This catecholamine surge affects brain inflammatory markers and increases both cardiac and cerebral oxygen demands. β-blockade’s mechanism of action in TBI is not yet fully understood, but there are preclinical studies demonstrating that it affects both inflammatory and oxygenation pathways. In microglial cells, β adrenergic stimulation attenuates lipopolysaccharide (LPS)-induced inflammatory cytokine production, and nonselective β-blockers reverse this effect.(16, 17) In stroke patients, propranolol decreases cerebral oxygen consumption, carbon dioxide production, and glucose consumption.(18, 19) Similarly, mice that receive propranolol have improved neurologic recovery one hour after TBI, better grip test scoring, and significantly less brain edema on histological analysis.(20) Propranolol has also been shown to improve cerebral oxidative phosphorylation and lipid synthesis.(19, 21) In animal studies, β-blockers have been shown to improve cerebral perfusion by 152% and decrease cerebral hypoxia by 24%.(18) Propranolol given after TBI has been shown to improve oxygen delivery to the brain via increased cerebral perfusion on both immunohistochemical analysis and by micro-positron emission tomography analysis compared to TBI in mice not receiving β-blockade.(20)
β-blockers have displayed promising results in animal studies, case series, retrospective reviews, cohort studies, one randomized trial, and a meta-analysis.(22–30) β-blocker administration following acute TBI is associated with lower in-hospital mortality (Table 2). Safety data in one study revealed no increase in adverse events.(25) The beneficial effects of β-blockers for severe TBI may be due to improved cerebral autoregulation. The Lund protocol, which includes the use of metoprolol, is thought to decrease vasogenic edema.(31) Cotton et al.(24) excluded early deaths but demonstrated a survival advantage for β-blockers in TBI. Those receiving β-blockers had a lower mortality than the non-β-blocker group (5.1% vs 10.8%). Schroeppel et al.(30) demonstrated a smaller survival advantage after excluding deaths within the first 24 hours. β-blockers do not appear to significantly decrease heart rate variability or blood pressure among patients with TBI, though overall effects on cardiac output have not been directly measured in this patient population.(25, 27, 28) Currently, there is evidence to suggest a benefit of β-blockers following TBI but there is a lack of well-designed controlled trials to address important functional outcomes, quality of life, and long-term mortality in TBI patients. In addition, there is a lack of uniformity on which β-blocker should be given. Future prospective studies should detail appropriate dosing, ideal agent, and the population of TBI patients most likely to benefit.
Table 2.
β-blocker use in TBI patients
| Study | Design | n | Sample population | β-blocker | Outcome |
|---|---|---|---|---|---|
| Arbabi(23) | Retrospective | 524 | subgroup: admission GCS < 14 | not specified | scheduled BB associated with ↓mortality |
| Cotton(24) | Retrospective | 420 | hAIS ≥ 3, no major neck injury, HLOS 4–30d | β1, non-selective | BB for ≥ 2d associated with ↓mortality |
| Cruickshank(25) | Prospective | 114 | Primary diagnosis acute TBI | β1 | BB x 7days associated with ↓SVT, ST/T EKG changes |
| Glass(26) | Retrospective | 419 | hAIS > 3, HLOS > 3d | β1, non-selective | BB for ≥ 2d associated with delay in ↓ Hb |
| Inaba(27) | Retrospective | 1156 | hAIS < 6, no major associated injury | not specified | BB associated with ↓mortality, especially for age ≥55 |
| Murry(28) | Prospective | 38 | hAIS 4–5, no non-survivable injuries | non-selective | BB for ≥ 2d associated with ↓ICU/HLOS, no ↓HR, SBP |
| Riordan(29) | Retrospective | 446 | hAIS ≥ 5, no major neck injury | β1, non-selective | BB exposure associated with ↓mortality |
| Schroeppel(30) | Retrospective | 2601 | all blunt TBI, mean ISS 26 | β1, non-selective | ≥ 2 doses BB associated with ↓mortality |
GCS: Glasgow Coma Scale, BB: β-blocker, hAIS: head Abbreviated Injury Score, HLOS: hospital length of stay, Hb: hemoglobin, ICU: intensive care unit, HR: heart rate, SBP: systolic blood pressure, TBI: traumatic brain injury, ISS: Injury Severity Score.
Spinal cord injury
Autonomic dysfunction often accompanies spinal cord injury.(32) Following T5–T6 blunt spinal cord injury (SCI), rats have been shown to exhibit neurogenic shock for three minutes after injury with mean arterial pressure (MAP) decrease by 78% and mean heart rate decrease by 63%.(33) Both heart rate and MAP returned to basal values within 20 minutes, demonstrating the transient nature of autonomic dysfunction following SCI in this model.(33) Rats receiving propranolol before injury did not experience the dramatic decrease in heart rate that was observed among injured rats that did not receive propranolol.(33) The propranolol group also had fewer increases and decreases in blood pressure.(33)
In addition, β-blockers may prevent or reduce the severity of secondary insult following SCI. Rodents receiving metoprolol immediately after T7–T10 blunt SCI have been shown to have lower spinal cord myeloperoxidase levels, indicating decreased neutrophil activity.(34) In a rabbit model of spinal cord ischemia and reperfusion, selective β-1 blockade with nebivolol beginning two days prior to injury improved motor deficit scores compared to animals not receiving β-blockers.(35) Nebivolol may be protective in spinal cord ischemia via free radical scavenging and antioxidant activity, independent of β-blockade effects.(35) Propranolol has similarly been shown to restore axonal function after SCI by suppressing glial scar formation and astrocyte hypertrophy.(36) β-blockers may have several potential clinical applications in spinal cord injury: blunting early autonomic imbalances, decreasing post-injury inflammation, and minimizing ischemia-reperfusion injury. All of these findings must be interpreted in the context that β-blockers were administered prior to traumatic injury in these preclinical studies. So additional animal studies examining the use of β-blockers after spinal cord injury are warranted.
Hemorrhagic shock
Hypotension decreases aortic arch and carotid sinus baroreceptor stimulation, triggering increased sympathetic tone and hypercatecholaminemia.(1) β-blockers suppress the hyperdynamic cardiovascular response to hypotension, limiting clinical application in hemorrhagic shock. Taniguchi et al.(10) demonstrated that oral administration of the alpha-1, β-1, and β-2 antagonist carvedilol prior to hemorrhagic shock increased mortality and exacerbated the inflammatory response in a rodent model. The authors noted that a dose-response relationship analysis was not performed.(10) Despite these findings, β-blockers have been shown to be useful for investigating hemorrhagic shock pathophysiology. The observation that blood lactate levels rise in the absence of a hypoxic insult triggered investigation of pertinent metabolic pathways, leading to several important findings.(37) Propranolol and phenoxybenzamine administration prior to hemorrhagic shock does not affect skeletal muscle perfusion, but lowers plasma lactate levels during hemorrhage and after resuscitation.(38, 39) Propranolol alone blocks EPI-stimulated muscle lactate production, but increases plasma EPI and lactate levels.(38) Plasma EPI stimulates aerobic glycolysis in skeletal muscle, resulting in lacticemia despite adequate tissue perfusion.(38) Muscle lactate, muscle glucose-6-phosphate, and Na+-K+-ATPase pump activity each follow the same trend, supporting the hypothesis that EPI-stimulated aerobic glycolysis contributes to skeletal muscle glycolysis in shock, increasing plasma lactate levels.(39) β-2 receptor regulation is important in this metabolic pathway.(40) Although β-blockers may be clinically detrimental in hemorrhagic shock, they have been invaluable in accurately describing lactate production during shock.
Acute traumatic coagulopathy
Severe traumatic injury often leads to acute traumatic coagulopathy (ATC) that is characterized by hypocoagulability and hyperfibrinolysis.(41) Principle processes responsible for ATC include tissue hypoperfusion, post-traumatic inflammation, and activation of the neurohumoral system.(42) ATC may be induced by the circulating catecholamine surge that is associated with severe trauma, hemorrhage, and endothelial damage.(43) ATC is an independent risk factor for increased transfusion requirements, multiple organ failure, and mortality.(44)
Rodents subjected to laparotomy and hemorrhagic shock were compared to another group pretreated with chemical sympathectomy to evaluate autonomic function in ATC and its influence on endothelial and coagulation activation.(45) Chemical sympathectomy suppressed tissue type plasminogen activator, plasmin-antiplasmin complex, soluble thrombomodulin, and syndecan-1.(45) Reduction of sympathetic activation with chemical sympathectomy yielded antifibrinolytic and endothelial protective effects in rats with ATC.(45) Similarly, in a rodent laparotomy, hemorrhagic shock, and femur fracture model, rodents pre-treated with propranolol had decreased sympathetic tone, decreased fibrinolytic marker levels, and decreased tumor necrosis factor (TNF)-α and interleukin (IL)-6 without significant effects on fibrinogen or mortality.(42) β-blockers had an endothelial protective effect by reducing soluble thrombomodulin and syndecan-1 levels.(42) β-blockers were shown to be protective against inflammation, glycocalyx shedding, hyperfibrinolysis, and endothelial damage.(42)
Of note, ATC patients may present with hemorrhagic shock,(46) which is a contraindication to β-blocker therapy as discussed in the previous section. In addition, β-blockers decrease platelet aggregation, potentially limiting their clinical application for patients with ATC.(47) Finally, fibrinolytic pathways appeared to shut down in over half of all severely injured patients in one study.(46) For these patients, anti-fibrinolytic therapy may be harmful.(48) The net effect of β-blocker therapy on coagulation is not yet completely understood. Despite limitations in clinical pharmacologic management of ATC, experimental studies have shown that rodent models may effectively emulate human ATC pathophysiology, and have established a foundation on which further research may fully elucidate mechanisms and best practices for management of ATC.
Erythropoietic Dysfunction
Anemia affects 95% of patients who remain critically ill for three or more days, and is associated with longer lengths of stay, increased incidence of organ failure, and increased mortality.(49, 50) Patients who require blood transfusion within 24 hours of severe traumatic injury are at increased risk for persistent anemia.(51) Management typically involves red blood cell transfusion which is associated with immune suppression, infectious complications, and increased mortality.(51, 52) Studies examining treatment alternatives to blood transfusion have not been successful. In a large multicenter prospective randomized trial, IV iron supplementation in trauma patients was not found to significantly affect hemoglobin (Hb) concentration or transfusion rates.(53) Similarly, in critically ill patients, erythropoietin- α administration increased Hb concentration at 29 days (1.6 vs. 1.2 g/dl), but did not decrease transfusion rates, and was associated with increased incidence of thrombotic events.(54)
There are both animal and human data to suggest that post-injury anemia is related to erythropoietic dysfunction at the level of the bone marrow.(55–58) NE has dose dependent effects on bone marrow progenitor cell growth.(57) Adding NE at supraphysiologic concentrations to normal human bone marrow in vitro decreases bone marrow hematopoietic progenitor cell (HPC) growth by more than 95%.(59) Similarly, supraphysiologic NE infusions suppress erythroid blast forming unit (BFU-E) colony growth and erythroid colony forming unit (CFU-E) growth in vivo in a dose dependent fashion.(60) EPI also suppresses bone marrow CFU-E growth, but with less potency than NE.(57) High NE levels promote HPC egress from the bone marrow via G-CSF-induced CXCL12 (SDF-1) downregulation.(61, 62) Mesenchymal stem cells (MSCs) express β adrenergic receptors and are critical for maintaining hematopoietic stem cell pluripotency and facilitating orderly differentiation.(63, 64) Although NE has been shown to inhibit MSC chondrogenesis,(65) the effects of β-blockers on MSC modulation of erythropoiesis have not yet been reported. In addition, post-injury inflammation has been shown to increase hepcidin, thereby decreasing intestinal iron absorption and reducing the amount of substrate available for erythropoiesis.(66) Therefore, catecholamine effects on the bone marrow, modulation of MSCs, and systemic inflammation may each play important roles in the pathophysiology of persistent injury-associated anemia.
Critically ill trauma patients have significantly elevated urine NE levels for nearly two weeks after traumatic injury.(57) This hypercatecholaminemia is associated with a persistent injury-associated anemia.(56) Persistent injury-associated anemia lasts for more than one week after traumatic injury and occurs out of proportion to blood loss. This condition is characterized by low reticulocyte counts despite adequate iron stores and erythropoietin levels, implicating pathophysiologic bone marrow dysfunction.(58, 67) A rat model of combined lung contusion, hemorrhagic shock, and chronic stress-induced anemia (Hb 11.1 vs. 13.3 g/dl in controls) seven days after initial injury and was associated with excessive HPC egress and decreased bone marrow cellularity (Figure 1).(68)
Figure 1.

Pathophysiology of persistent injury-associated anemia after severe trauma Lung contusion immediately followed by hemorrhagic shock with a mean arterial pressure of 30–35 mm Hg for 45 minutes along with chronic restraint stress for two hours daily until sacrifice on day seven creates a persistent hypercatecholamine state. Nonselective β-blocker, propranolol, was administered 10 minutes after HS and daily after CS. (LC: lung contusion, HS: hemorrhagic shock, CS: chronic stress, HPC: hematopoietic progenitor cell, BM: bone marrow)
Since it was determined that NE played a substantial role in bone marrow dysfunction, subsequent studies have concentrated on the effects of β-blockade on bone marrow function. Using the aforementioned lung injury rodent model, the use of propranolol before lung injury reversed bone marrow HPC growth suppression (Figure 1). To determine which β adrenergic receptors were involved in this bone marrow protection, additional studies with selective β-1, β-2, and β-3 blockers given prior to lung injury demonstrated that bone marrow protection was mediated through β-2 and β-3 receptors.(5) Propranolol therapy was then tested in the hemorrhagic shock rodent model. Results mirrored what was found in the lung injury rodent model. Propranolol protected bone marrow HPC growth whether it was administered prior to or immediately after hemorrhagic shock.(55) In a combined lung contusion and hemorrhagic shock model, propranolol was administered immediately after resuscitation and was given daily for seven days. Daily propranolol use was associated with a significant increase in bone marrow HPC growth, restoration of Hb, and reduced HPC mobilization into the peripheral blood.(69) Propranolol administration in these studies was not associated with increased mortality or significant changes in blood pressure, but effective propranolol dosing did correlate with a 20% decrease in heart rate.(55) Dose studies demonstrated that the safe range of intraperitoneal propranolol injections in rats was between 0.5 mg/kg to 20 mg/kg. Daily treatments of 5 and 10 mg/kg of propranolol provided significant bone marrow protection by preserving bone marrow cellularity and also preventing prolonged bone marrow HPC growth suppression.(55) Lower doses of propranolol did not sufficiently provide bone marrow protection.(55)
A clinical translational study has verified some of the rodent findings.(56) In a pilot trial, severely injured patients were randomized to receive propranolol after resuscitation was complete (serum lactate ≤ 4 mg/dl) or to a no β-blocker therapy as a control group. (56) Those randomized to receive propranolol had propranolol doses titrated to decrease initial HR by 10–20%.(56) The β-blocker treatment group had reduced HPC mobilization, increased reticulocyte counts, and a non-significant trend toward increased Hb levels.(56) There is abundant preclinical data suggesting the beneficial effects of β-blocker therapy for erythropoietic dysfunction after traumatic injury but only one clinical trial. Thus, a large, prospective, randomized trial investigating the role of β-blockers in post-injury bone marrow protection is warranted.
Metabolic dysfunction
Increased protein catabolism and hyperglycemia is partly mediated by β-2 adrenergic signaling.(70) EPI induces insulin resistance and enhances hepatic glucose production.(71) Propranolol has been shown to reduce plasma glucose concentrations by decreasing endogenous glucose production.(72) Propranolol also improves nitrogen balance, suggesting reduced muscle proteolysis.(73) Herndon et al. have shown that propranolol treatment in children attenuates resting energy expenditure and reverses muscle catabolism. In contrast, these effects are not seen with selective β-1 blockade and have not been shown in adults.(74)
In severely injured patients, obesity (body mass index ≥ 30 kg/m2) is associated with increased incidence of sepsis, Acute Respiratory Distress Syndrome (ARDS), renal failure, and multiple organ failure as well as increased hospital length of stay, longer duration of mechanical ventilation, and increased mortality.(75, 76) Serum glucose ≥ 200 mg/dl within 24 hours of injury is a risk factor for multiple organ failure and mortality in trauma patients with hemorrhagic shock, and is associated with worse neurologic outcomes in head injury patients.(77, 78) These scenarios were combined in an experiment involving obese Zucker rats with insulin resistance subjected to extremity fracture.(79, 80) Although basal glucose levels were not significantly different between obese and lean Zucker rats, obese rats had increased glucose levels within ten minutes of injury and persisting throughout the six hour experiment.(80) Post-injury hyperglycemia in obese rats was suppressed with selective β-2 blockade.(80) Post-injury hyperglycemia was caused by impaired glucose uptake and alleviated by hepatic β-2 receptor blockade.(80) These results are consistent with propranolol’s capacity to decrease free fatty acid oxidation and lipolysis, redirecting fuel utilization toward glucose oxidation.(72) Animal studies have demonstrated a benefit for propranolol use in reducing post-injury hyperglycemia but further study examining the potential long term benefits, reduced infection, and improved metabolism have not been studied. The design of a future randomized controlled trial may consider investigating the metabolic changes as well.
Pulmonary Dysfunction
The inflammatory state after trauma and hemorrhagic shock leads to gut and pulmonary dysfunction.(81) Pulmonary dysfunction is manifested by increased neutrophil infiltration, histologic changes, and changes in pulmonary function.(82) In a rodent model of lung contusion and hemorrhagic shock with and without propranolol administration, lung injury scores were significantly attenuated three hours after injury in the propranolol treatment group.(83) Baranski et al. hypothesized that the improvement in lung injury scores was related to either the systemic effects of propranolol or a reduction in gut permeability.(83)
ARDS is characterized by acute onset, hypoxia with PaO2/FiO2 ratio <300 mm Hg, diffuse infiltrates on chest X-ray, and the absence of cardiac failure.(84) Trauma, hemorrhage, burns, and sepsis often precipitate this condition. Mortality for ARDS remains between 27–45%.(85) Pathophysiologic mechanisms include immunoregulatory cytokine dysfunction and intrapulmonary leukocyte activation preceding systemic inflammation.(86, 87) Pulmonary denervation studies in dogs have established the role of the autonomic nervous system in ARDS models.(88) The effects of β-blockers in ARDS depend upon the etiology of pulmonary insult. A murine model delineated essential differences between alpha and β adrenergic stimulation.(86) Animals subjected to hemorrhagic shock activated nuclear factor kappa (NF-κB) and cyclic adenosine monophosphate response element binding protein, thereby promoting inflammatory cytokine gene expression including IL-1 and TNF-alpha.(86) Mice that received β-blockers prior to hemorrhage had significantly elevated mRNA levels of lung mononuclear IL-1 and TNF-alpha as compared to controls.(86) Conversely, mice receiving α adrenergic blockade prior to hemorrhage had decreased lung levels of IL-1 and TNF-alpha.(86) These findings are consistent with overall effects of alpha and β stimulation on inflammatory cytokine profiles summarized in Figure 2.
Figure 2.
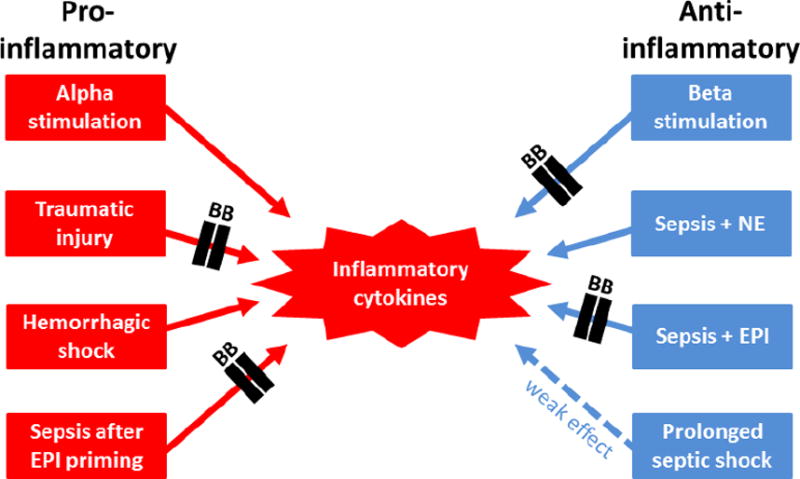
The predominant effects of pathologic insults and β-blockers on inflammatory cytokines (EPI: epinephrine, NE: norepinephrine).
When pulmonary injury was induced by a septic insult, selective β-1 blockade protected against lung injury and decreased lung tissue expression of the inflammatory protein high-mobility group box 1.(89) More recently, increased PaO2/FiO2 ratios were observed three hours after administration of esmolol in a pig model of endotoxic shock.(90) This result was consistent with overall findings of immunomodulatory β-blocker effects in numerous experimental sepsis models.(91) Differential effects of β-blockade depending on ARDS etiology may be due to variations in the control of different inflammatory cytokine gene expression.(92) Experimental data show that high pulmonary vascular flow is important in the development of ARDS, so logically selective β-1 blockade would reduce lung damage but randomized controlled trials are needed before the use of selective β-1 blockade in ARDS can be routinely advocated.(91)
Burns and wound healing
Burn injuries induce significant hypercatecholaminemia in children and adults.(93, 94) There have been several studies in burn patients demonstrating that propranolol improves wound healing, decreases cardiac work, and reduces metabolic demands without causing significant arterial hypotension or metabolic dysregulation.(95–97) Rodent models have provided the pathophysiologic basis for these clinical advances. Stress response pathways, including p38 MAPK, JNK, and NF-κB, are activated by scald burn injury and have been effectively blocked with propranolol.(93) In mice subjected to full thickness scald injury and wound sepsis with Pseudomonas aeruginosa inoculation, adrenergic stimulation modulated CD59+ monocyte progenitor maturation, decreased β adrenergic receptor cell surface density, and increased receptor affinity in a process which was reversed by selective β-2 receptor blockade.(98)
In a full thickness burn rat model, daily propranolol administration corrected stress-induced insulin receptor dysregulation.(99) In addition, β-blocker treatment resulted in enhanced wound healing at the macroscopic level, and at the microscopic level there was improved epithelialization, collagen deposition, and angiogenesis, as well as decreased protease activity.(100) β-blocker treatment also conferred wound healing advantages in the absence of comorbid metabolic dysfunction. In a partial thickness excisional wound model, rabbits were allocated to receive no treatment or propranolol infusion during a seven hour period of infusion with isotope tracers D-[U-13C6]glucose, L-[ring-13C6]phenylalanine, L-[1-13C]leucine, and L-[1,2-13C2]leucine.(101) Tracer analysis demonstrated that wound DNA fractional synthetic rates were similar, but protein fractional synthetic rates were greater in the propranolol group.(101) Local effects of β-blockade may also be due in part to the presence of β adrenergic receptors on MSCs and endothelial cells.(102) β-2 receptor blockade increases migration and proliferation of keratinocytes during wound epithelialization, accelerating epidermal barrier repair. (102) NE-depleted mice have increased wound angiogenesis,(103) and rats treated with propranolol demonstrate increased wound blood vessel density.(104) Likewise, among human burn patients, propranolol use resulted in faster donor site healing times.(95)
Immunomodulation after trauma
Following traumatic injury there is a complex interaction between the neuroendocrine and the immune systems. The β adrenergic system is a well-known modulator of the immune system, and circulating catecholamines have immunomodulatory properties.(105) β adrenergic stimulation has diverse effects on natural killer (NK) cells, CD14+ monocytes, T lymphocytes, and B lymphocytes.(105, 106) Lymphoid tissues containing these cells are innervated by the sympathetic nervous system, where NE functions as a neurotransmitter.(3) Catecholamines have also been shown to down-regulate the synthesis of pro-inflammatory cytokines such as TNF-alpha, IL-6 and IL-1 and upregulate synthesis of anti-inflammatory cytokines.(3) The pattern of cytokine production and the polarization of T-cell populations in sepsis may depend on the T-helper type 1 (Th1) and Th2 balance. Th1 cells are presumed to promote cellular immunity while Th2 cells promote the humoral response. β-2 adrenergic stimulation suppresses Th1 cell polarization, shifting the immune response away from cellular immunity in favor of humoral immunity.(3) Although the precise mechanisms have not been elucidated, the apparent lack of β-2 adrenergic receptors on Th2 cells may partially explain this phenomenon.(107)
Circulating catecholamines after trauma and hemorrhagic shock have been shown to reduce hepatic β adrenergic receptor binding capacity.(108) In a hemorrhagic shock model, mice were pretreated with either propranolol or metoprolol, and both agents significantly reduced the number of circulating natural killer cells and CD8+ lymphocytes.(105) These effects may be due in part to catecholamine-induced changes in adhesion molecule and chemoattractant receptor expression.(109, 110) The clinical effects of immunosuppression must be considered in the context of several factors relating to the nature of the physiologic insult, as discussed in the sepsis section.
In a retrospective review of 663 critically ill trauma patients, those receiving β-blockers within thirty days of ICU admission had significantly lower in-hospital mortality compared to patients with similar ISS scores not receiving β-blockers (11% vs. 19%).(111) In a large retrospective review of 4,117 trauma patients who received β-blockers during their hospital stay (60% received β-blockers for hypertension, 20% for heart rate control, and 45% were on chronic β-blockers), the β-blocker cohort all-cause mortality odds ratio was 0.3 after adjusting for age, Glasgow Coma Scale, and Injury Severity Score.(22) These improved outcomes with β-blockers could be due to decreased myocardial oxygen demand,(10) improved myocardial oxygen utilization,(112) and/or immunomodulation of hypercatecholaminemia.(105) A prospective controlled trial enrolled 42 critically ill trauma patients and allocated β-blocker naïve subjects to initiate β-blocker therapy 24 hours after resuscitation and hemodynamic normalization.(113) Metoprolol was given unless resuscitation requirements were ≥ 2 liters, in which case short-acting esmolol was initiated and then converted to longer-acting metoprolol after 24 hours of hemodynamic stability. Mean daily heart rate was lower in the β-blocker group and mean daily systolic blood pressure was not significantly affected. Baseline and hospital day one plasma IL-6 levels did not differ between groups. On hospital day four, serum IL-6 levels were lower in the β-blocker group. In addition to relating post-injury β-blocker use to IL-6 levels, this study also demonstrated that β-blockade was safe for a small cohort of critically ill trauma patients.(113) To summarize, β-1 and β-2 adrenergic receptors seem to exert opposite actions on the immune system, but more investigation is required to understand if β-blockers of selective β-blockade can offer advantages in immune function.
Sepsis
Interactions between adrenergic pathways and host protective immunity are complex. Catecholamines and β-adrenergic stimulation have exhibited net anti-inflammatory effects in animal and human sepsis models, primarily by reducing pro-inflammatory cytokine mRNA levels.(1, 114–119) Diminished immunosuppressive effects in prolonged septic shock may indicate that catecholamine-induced immunosuppression depends upon the duration of catecholamine exposure.(120) Immune cell apoptosis has also been proposed as a mechanism for immunosuppression in sepsis.(121) Septic humans exhibit significant lymphocyte apoptosis within one day of septic insult, and lymphocyte recovery appears to correlate with improved clinical outcomes.(122) Although hypercatecholaminemia has been shown to contribute to pro-apoptotic molecular up-regulation and β-blockade may prevent post-hemorrhage splenocyte apoptosis; the role of hypercatecholaminemia in sepsis-induced leukocyte apoptosis remains unclear.(105, 123) Finally, NE has been shown to decrease T cell production of the chemokine CCL3, which regulates leukocyte recruitment to sites of infection.(124) This effect was abrogated by propranolol in a murine thermal injury model.(124)
In contrast to hypercatecholaminemia-mediated immunosuppression, the septic insult itself causes a pro-inflammatory mediator surge.(125, 126) Reports of different cytokine profile responses to catecholamine challenge and septic insult may be due to variable catecholamine concentrations, timing of interventions, and differential α and β adrenergic stimulation. Exogenous alpha adrenergic stimulation can have either immunoenhancing or immunosuppressive effects on IgM production in vitro, depending on drug concentration.(127) In one experiment, an 18 hour delay between EPI administration and LPS challenge was chosen to facilitate a pro-inflammatory environment prior to septic challenge, because simultaneous EPI and LPS administration has been shown to have an anti-inflammatory effect.(125) In addition, alpha adrenergic stimulation increases TNF-alpha expression in murine peritoneal macrophages in a process that is reversed with alpha adrenergic blockade but augmented by β adrenergic blockade.(128, 129) Conversely, β adrenergic stimulation decreases TNF-alpha expression in a process that is reversed by β blockers but not alpha adrenergic blockade.(128, 129) Similarly, β adrenergic stimulation inhibits TNF-α expression in human monocytes, an effect that is prevented by β-blockers but not alpha adrenergic blockade.(130) Individual studies investigating interactions among catecholamines, adrenergic receptors, and cytokines are listed in Table 3.
Table 3.
Effects of adrenergic stimulation and septic insult on pro- and anti-inflammatory cytokines
| Study | Design | Findings |
|---|---|---|
| Bergmann(120) | Human in vitro | EPI + LPS→ ↓ plasma TNF-α and IL-6, ↑ IL-10, early effects > late effects |
| Deng(114) | Mouse in vitro | EPI + LPS→ ↓ splenic macrophage IL-6 and TNF-α, ↑ IL-10 |
| Rough(125) | Mouse in vitro | EPI + LPS→ ↑ RAW 264.7 TNF-α expression, reversed with β-2 blockade |
| Mouse in vivo | T/H + LPS→ ↑ splenic macrophage IL-6 and TNF-α, reversed with β-2 blockade | |
| Sekut(117) | Human in vitro | β stimulation + LPS→ ↓ THP-1 TNF-α, reversed with β-2 blockade |
| Mouse in vivo | β stimulation + LPS→ ↓ serum TNF-α, reversed with non-selective β blockade | |
| Severn(130) | Human in vitro | β stimulation + LPS→ ↓ serum TNF-α, reversed with non-selective β blockade |
| Human in vitro | β stimulation + LPS→ ↓ THP-1 TNF-α, reversed with non-selective β blockade | |
| Spengler(128) | Mouse in vitro | alpha stimulation + LPS→ ↑ peritoneal macrophage TNF-α, reversed with alpha-2 blockade |
| Spengler(129) | Mouse in vitro | β stimulation + LPS→ ↓ peritoneal macrophage TNF-α, augmented with alpha-2 blockade, reversed with non-selective β blockade |
| van der Poll(118) | Human in vivo | EPI + LPS (Escherichia coli 0113)→ ↓ plasma TNF-α |
| Human in vitro | EPI + LPS (Escherichia coli 0127)→ ↓ plasma TNF-α, ↑ IL-10 | |
| Wilson(126) | Rat in vivo | CLP→ ↑ lung /plasma TNF-α expression, ↓ with non-selective β blockade |
EPI: epinephrine, LPS: lipopolysaccharide, TNF: tumor necrosis factor, IL: interleukin, RAW 264.7: murine leukemic cell line, T/H: trauma and hemorrhage, THP-1: human leukemic monocyte cell line, CLP: cecal ligation and puncture.
The predominant effects of pathologic insults and β-blocker treatment on inflammation are summarized in Figure 2. Effects of catecholamine concentrations, timing of pathophysiologic events, and differential alpha and β adrenergic stimulation must be considered within context. Too much or too little inflammation may be detrimental, and adrenergic receptor blockade can facilitate either condition. In addition, the effects of β-blockade on infectious outcomes following the systemic inflammatory response syndrome (SIRS)(131) and the compensatory anti-inflammatory response syndrome (CARS)(132) are unknown. Although it remains plausible that restitution of T cell CCL3 production with β-blockers may augment adaptive immunity and thereby abrogate CARS, this hypothesis has not been tested in the clinical setting. However, several beneficial effects of β-blockers in sepsis have been described, including restoration of normal cellular metabolism, improved glucose regulation, and improved cardiac function.(91) Notably, epidemiologic data suggested that critically ill septic patients with chronic β-blocker prescriptions had lower 28 day mortality than sensitivity and pair-matched controls (17.7% vs. 22.1%).(133) The first randomized controlled trial was performed by Morelli et al.(134) Continuous esmolol infusion in septic shock patients requiring NE was associated with a significant reduction in NE and fluid requirements and a decreased 28 day mortality.(134) Large clinical trials evaluating the potential benefits of β-blockers in sepsis are warranted. More data is needed to fully describe the complex relationships among sepsis, the inflammatory response, catecholamines, and adrenergic receptors.
Future Directions
Initial sympathetic activation after injury is beneficial but persistent severe overactivation is likely detrimental. Therefore, β-blocker therapy and tight regulation of β adrenergic receptors may provide unique advantages in managing several injuries and physiologic insults (Table 4). However, more preclinical research is needed to continue to elucidate mechanisms of action, cellular targets, safety, and clinical efficacy of β-blockers. In addition, more research is needed to determine which patients would benefit most from this therapy. In all prospective studies, the timing, dosing, and which β-blocker therapy have yet to be answered. Once these objectives are met, level I evidence regarding β-blockade after TBI and models in which β-blockers are administered after SCI would be beneficial. Valuable knowledge will be gained by additional preclinical studies examining mechanisms by which β-blockers could confer protection against ATC and studies clarifying therapeutic strategies and clinical endpoints for post-injury hypermetabolism and glucose dysregulation among patients with obesity and pre-existing metabolic dysfunction. A large, prospective, randomized trial investigating the role of β-blockers in post-injury bone marrow protection is warranted. Research should continue to further elucidate the complex relationships among trauma, the inflammatory response, sepsis, catecholamines, and β adrenergic receptors with attention to catecholamine concentrations, timing of pathophysiologic events, and differential β adrenergic receptor effects.
Table 4.
Proposed mechanisms for beneficial effects of β-blockers.
| Injury/Insult | B blockade | Proposed Mechanism | Result |
|---|---|---|---|
| Traumatic brain injury | non-selective | ↓NE-induced cytokine production | ↓brain edema, ↑oxygenation |
| Spinal cord injury | non-selective | blunting autonomic dysfunction | hemodynamic stability |
| β-1 blockade | ↓PMN activation and oxidative stress | ↑motor function | |
| Acute traumatic coagulopathy | non-selective | blunting hyperadrenergic response | ↓fibrinolysis, inflammation |
| Post-injury hyperglycemia | non-selective | ↓FA oxidation and lipolysis | ↑glucose oxidation |
| β-2 blockade | hepatic β-2 receptor blockade | normoglycemia | |
| Post-injury anemia | β-2,3 blockade | ↓NE-induced bone marrow dysfunction | ↓HPC egress, ↑HPC growth, Hb |
| Burns/wounds | non-selective | block p38 MAPK/JNK/NF-κB pathway | ↑wound healing |
| Infection/sepsis | non-selective | ↓ NE-induced immune cell apoptosis | ↓immunosuppression |
| β-2 blockade | ↓EPI-induced splenic macrophage priming | ↓inflammation |
NE: norepinephrine, PMN: neutrophil, FA: fatty acid, HPC: hematopoietic progenitor cell, Hb: hemoglobin, MAPK: mitogen-activated protein kinase, JNK: c-Jun N-terminal kinase, NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells, EPI: epinephrine
Acknowledgments
This research was supported by the National Institutes of Health. AMM was supported by NIH NIGMS grant R01 GM105893-01A1. TJL was supported by a training grant in burn and trauma research T32 GM-08431. This work was also supported by grants R01 GM40586-24 and R01 GM-081923-06 awarded by the NIGMS. PAE was supported by P30 AG028740 from the National Institute on Aging and by the NIH NIGMS grant R01 GM113945-01. Finally, AMM LLM and PAE were all supported by P50 GM111152-01 (NIGMS).
References
- 1.Molina PE. Neurobiology of the stress response: contribution of the sympathetic nervous system to the neuroimmune axis in traumatic injury. Shock. 2005;24(1):3–10. doi: 10.1097/01.shk.0000167112.18871.5c. [DOI] [PubMed] [Google Scholar]
- 2.Bylund DB, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP, Molinoff PB, Ruffolo RR, Jr, Trendelenburg U. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev. 1994;46(2):121–36. [PubMed] [Google Scholar]
- 3.de Montmollin E, Aboab J, Mansart A, Annane D. Bench-to-bedside review: β-adrenergic modulation in sepsis. Critical care. 2009;17:230. doi: 10.1186/cc8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCarty R. Age-related alterations in sympathetic-adrenal medullary responses to stress. Gerontology. 1986;32(3):172–83. doi: 10.1159/000212785. [DOI] [PubMed] [Google Scholar]
- 5.Beiermeister KA, Keck BM, Sifri ZC, ElHassan IO, Hannoush EJ, Alzate WD, Rameshwar P, Livingston DH, Mohr AM. Hematopoietic progenitor cell mobilization is mediated through β-2 and β-3 receptors after injury. J Trauma. 2010;69(2):338–43. doi: 10.1097/TA.0b013e3181e5d35e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaugg M, Schaub MC, Pasch T, Spahn DR. Modulation of β-adrenergic receptor subtype activities in perioperative medicine: mechanisms and sites of action. Br J Anaesth. 2002;88(1):101–23. doi: 10.1093/bja/88.1.101. [DOI] [PubMed] [Google Scholar]
- 7.Dal Monte M, Fornaciari I, Nicchia GP, Svelto M, Casini G, Bagnoli P. β3-adrenergic receptor activity modulates melanoma cell proliferation and survival through nitric oxide signaling. Naunyn Schmiedebergs Arch Pharmacol. 2014;387(6):533–43. doi: 10.1007/s00210-014-0969-1. [DOI] [PubMed] [Google Scholar]
- 8.Manara L, Badone D, Baroni M, Boccardi G, Cecchi R, Croci T, Giudice A, Guzzi U, Landi M, Le Fur G. Functional identification of rat atypical β-adrenoceptors by the first β 3-selective antagonists, aryloxypropanolaminotetralins. Br J Pharmacol. 1996;117(3):435–442. doi: 10.1111/j.1476-5381.1996.tb15209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Bortel LM, van Baak MA. Exercise tolerance with nebivolol and atenolol. Cardiovasc Drugs Ther. 1992;6(3):239–47. doi: 10.1007/BF00051145. [DOI] [PubMed] [Google Scholar]
- 10.Taniguchi T, Kurita A, Yamamoto K, Inaba H. Effects of carvediol on mortality and inflammatory responses to severe hemorrhagic shock in rats. Shock. 2009;32:272–275. doi: 10.1097/SHK.0b013e3181a24cb3. [DOI] [PubMed] [Google Scholar]
- 11.Morales DR, Jackson C, Lipworth BJ, Donnan PT, Guthrie B. Adverse respiratory effect of acute beta-blocker exposure in asthma: a systematic review and meta-analysis of randomized controlled trials. Chest. 2014;145(4):779–86. doi: 10.1378/chest.13-1235. [DOI] [PubMed] [Google Scholar]
- 12.Pollare T, Lithell H, Selinus I, Berne C. Sensitivity to insulin during treatment with atenolol and metoprolol: a randomised, double blind study of effects on carbohydrate and lipoprotein metabolism in hypertensive patients. BMJ. 1989;298(6681):1152–7. doi: 10.1136/bmj.298.6681.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manolis A, Doumas M. Antihypertensive treatment and sexual dysfunction. Curr Hypertens Rep. 2012;14(4):285–92. doi: 10.1007/s11906-012-0276-5. [DOI] [PubMed] [Google Scholar]
- 14.Heffernan DS, Inaba K, Arbabi S, Cotton BA. Sympathetic hyperactivity after traumatic brain injury and the role of β-blocker therapy. J Trauma. 2010;69:1602–1609. doi: 10.1097/TA.0b013e3181f2d3e8. [DOI] [PubMed] [Google Scholar]
- 15.McLeod AA, Neil-Dwyer G, Meyer CH, Richardson PL, Cruickshank J, Bartlett J. Cardiac sequelae of acute head injury. Br Heart J. 1982;47(3):221–6. doi: 10.1136/hrt.47.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hetier E, Ayala J, Bousseau A, Prochiantz A. Modulation of interleukin-1 and tumor necrosis factor expression by β-adrenergic agonists in mouse ameboid microglial cells. Exp Brain Res. 1991;86(2):407–13. doi: 10.1007/BF00228965. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Li J, Sheng X, Zhao H, Cao XD, Wang YQ, Wu GC. β-adrenoceptor mediated surgery-induced production of pro-inflammatory cytokines in rat microglia cells. J Neuroimmunol. 2010;223(1–2):77–83. doi: 10.1016/j.jneuroim.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 18.Ley EJ, Scehnet J, Park R, Schroff S, Dagliyan G, Conti PS, Margulies DR, Salim A. The in vivo effect of propranolol on cerebral perfusion and hypoxia after traumatic brain injury. J Trauma. 2009;66:154–9. doi: 10.1097/TA.0b013e31819388be. [DOI] [PubMed] [Google Scholar]
- 19.Meyer JS, Okamoto S, Shimazu K, Koto A, Ouchi T, Sari A, Ericsson AD. Cerebral metabolic changes during treatment of subacute cerebral infarction by α and β adrenergic blockade with phenoxybenzamine and propranolol. Stroke. 1974;5(2):180–95. doi: 10.1161/01.str.5.2.180. [DOI] [PubMed] [Google Scholar]
- 20.Liu M. Protective effects of propranolol on experimentally headinjured mouse brains. J Formos Med Assoc. 1995;94:386–90. [PubMed] [Google Scholar]
- 21.Standefer M, Little JR. Improved neurologic outcome in experimental focal cerebral ischemia treated with propranolol. Neurosurgery. 1986;18:136–40. doi: 10.1227/00006123-198602000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Alali AS, McCreadle VA, Golan E, Shah PS, Nathens AB. β blockers for acute traumatic brain injury: a systematic review and meta-analysis. Neurocrit Care. 2014;20:215–523. doi: 10.1007/s12028-013-9903-5. [DOI] [PubMed] [Google Scholar]
- 23.Arbabi S, Campion EM, Hemmila MR, Barker M, Dimo M, Ahrns KS, Niederbichler AD, Ipaktchi K, Wahl WL. β-blocker use is associated with improved outcomes in adult trauma patients. J Trauma. 2007;62:56–61. doi: 10.1097/TA.0b013e31802d972b. [DOI] [PubMed] [Google Scholar]
- 24.Cotton BA, Snodgrass KB, Fleming SB, Carpenter RO, Kemp CD, Arbogast PG, Morris JA., Jr β-blocker exposure is associated with improved survival after severe traumatic brain injury. J Trauma. 2007;62:26–33. doi: 10.1097/TA.0b013e31802d02d0. [DOI] [PubMed] [Google Scholar]
- 25.Cruickshank JM, Neil-Dwyer G, Degaute JP, Hayes Y, Kuurne T, Kytta J, Vincent JL, Carruthers ME, Patel S. Reduction of stress/catecholamine-induced cardiac necrosis by β 1-selective blockade. Lancet. 1987;2(8559):585–9. doi: 10.1016/s0140-6736(87)92984-9. [DOI] [PubMed] [Google Scholar]
- 26.Glass NE, Kaltenbach LA, Fleming SB, Arbogast PG, Cotton BA. The impact of β-blocker therapy on anemia after traumatic brain injury. Transfusion. 2012;52(10):2155–60. doi: 10.1111/j.1537-2995.2012.03609.x. [DOI] [PubMed] [Google Scholar]
- 27.Inaba K, Teixeira PG, David JS, Chan LS, Salim A, Brown C, Browder T, Beale E, Rhee P, Demetriades D. β-blockers in isolated blunt head injury. J Am Coll Surg. 2008;206(3):432–8. doi: 10.1016/j.jamcollsurg.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 28.Murry JS, Hoang DM, Barmparas G, Harada MY, Bukur M, Bloom MB, Inaba K, Margulies DR, Salim A, Ley EJ. Prospective evaluation of early propranolol after traumatic brain injury. J Surg Res. 2015 doi: 10.1016/j.jss.2015.06.045. pii: S0022-4804(15)00723. [DOI] [PubMed] [Google Scholar]
- 29.Riordan WP, Jr, Cotton BA, Norris PR, Waitman LR, Jenkins JM, Morris JA., Jr β-blocker exposure in patients with severe traumatic brain injury (TBI) and cardiac uncoupling. J Trauma. 2007;63(3):503–10. doi: 10.1097/TA.0b013e3181271c34. [DOI] [PubMed] [Google Scholar]
- 30.Schroeppel TJ, Fischer PE, Zarzaur BL, Magnotti LJ, Clement LP, Fabian TC, Croce MA. β-adrenergic blockade and traumatic brain injury: protective? J Trauma. 2010;69(4):776–82. doi: 10.1097/TA.0b013e3181e981b8. [DOI] [PubMed] [Google Scholar]
- 31.Naredi S, Eden E, Zall S, Stephensen H, Rydenhag B. A standardized neurosurgical neurointensive therapy directed toward vasogenic edema after severe traumatic brain injury: clinical results. Intensive Care Med. 1998;24:446–51. doi: 10.1007/s001340050594. [DOI] [PubMed] [Google Scholar]
- 32.Hou S, Robchevsky AG. Autonomic consequences of spinal cord injury. Compr Physiol. 2014;4(4):1419–53. doi: 10.1002/cphy.c130045. [DOI] [PubMed] [Google Scholar]
- 33.Bravo G, Hong E, Rojas G, Guízar-Sahagún G. Sympathetic blockade significantly improves cardiovascular alterations immediately after spinal cord injury in rats. Neurosci Lett. 2002;319(2):95–8. doi: 10.1016/s0304-3940(01)02557-5. [DOI] [PubMed] [Google Scholar]
- 34.Beril Gok H, Solaroglu I, Okutan O, Cimen B, Kaptanoglu E, Palaoglu S. Metoprolol treatment decreases tissue myeloperoxidase activity after spinal cord injury in rats. J Clin Neurosci. 2007;14(2):138–42. doi: 10.1016/j.jocn.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 35.Ilhan A, Yilmaz H, Armutcu F, Gurel A, Akyol O. The protective effect of nebivolol on ischemia/reperfusion injury in rabbit spinal cord. Prog Neuropsychopharmacol Biol Psychiatry. 2004 Nov;28(7):1153–60. doi: 10.1016/j.pnpbp.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 36.Taira Y, Marsala M. Effect of proximal arterial perfusion pressure on function, spinal cord flow, and histopathologic changes after increasing intervals of aortic occlusion in the rat. Stroke. 1996;27:1850–1858. doi: 10.1161/01.str.27.10.1850. [DOI] [PubMed] [Google Scholar]
- 37.James JH, Luchette FA, McCarter FD, Fischer JE. Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis. Lancet. 1999;354:505. doi: 10.1016/S0140-6736(98)91132-1. [DOI] [PubMed] [Google Scholar]
- 38.Luchette FA, Robinson BR, Friend LA, McCarter F, Frame SB, James JH. Adrenergic antagonists reduce lactic acidosis in response to hemorrhagic shock. J Trauma. 1999;46:873. doi: 10.1097/00005373-199905000-00017. [DOI] [PubMed] [Google Scholar]
- 39.McCarter FD, James JH, Luchette FA, Wang L, Friend LA, King JK, Evans JM, George MA, Fischer JE. Adrenergic blockade reduces skeletal muscle glycolysis and Na(+), K(+)-ATPase activity during hemorrhage. J Surg Res. 2001;99(2):235–44. doi: 10.1006/jsre.2001.6175. [DOI] [PubMed] [Google Scholar]
- 40.Levy B, Desebbe O, Montemont C, Gibot S. Increased aerobic glycolysis through β2 stimulation is a common mechanism involved in lactate formation during shock states. Shock. 2008;30(4):417–21. doi: 10.1097/SHK.0b013e318167378f. [DOI] [PubMed] [Google Scholar]
- 41.Brohi K, Cohen MJ. Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway. Ann Surg. 2007;245:812–18. doi: 10.1097/01.sla.0000256862.79374.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu L, Yu WK, Lin ZL, Tan SJ, Bai XW, Ding K, Li N. Impact of β-adrenoceptor blockade on systemic inflammation and coagulation disturbances in rats with acute traumatic coagulopathy. Med Sci Monit. 2015;21:468–76. doi: 10.12659/MSM.893544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johansson PI, Stensballe J, Rasmussen LS, Ostrowski SR. High circulating adrenaline levels at admission predict increased mortality after trauma. J Trauma Acute Care Surg. 2012;72:428–36. doi: 10.1097/ta.0b013e31821e0f93. [DOI] [PubMed] [Google Scholar]
- 44.van Zyl N, Reade MC, Fraser JF. Experimental Animal Models of Traumatic Coagulopathy: A Systematic Review. Shock. 2015;44(1):16–24. doi: 10.1097/SHK.0000000000000372. [DOI] [PubMed] [Google Scholar]
- 45.Xu L, Yu WK, Lin ZL, Tan SJ, Bai XW, Ding K, Li N. Chemical sympathectomy attenuates inflammation, glycocalyx shedding and coagulation disorders in rats with acute traumatic coagulopathy. Blood Coagul Fibrinolysis. 2015;26:152–160. doi: 10.1097/MBC.0000000000000211. [DOI] [PubMed] [Google Scholar]
- 46.Moore EE, Moore HB, Gonzalez E, Chapman MP, Hansen KC, Sauaia A, Silliman CC, Banerjee A. Postinjury fibrinolysis shutdown: Rationale for selective tranexamic acid. J Trauma Acute Care Surg. 2015;78(6 Suppl 1):S65–9. doi: 10.1097/TA.0000000000000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonten TN, Plaizier CE, Snoep JJ, Stijnen T, Dekkers OM, van der Bom JG. Effect of beta-blockers on platelet aggregation: a systematic review and meta-analysis. Br J Clin Pharmacol. 2014;78(5):940–9. doi: 10.1111/bcp.12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moore HB, Moore EE, Gonzalez E, Chapman MP, Chin TL, Silliman CC, Banerjee A, Sauaia A. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J Trauma Acute Care Surg. 2014;77(6):811–7. doi: 10.1097/TA.0000000000000341. discussion 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shander A, Gandhi NR, Goodnough LT. Anemia, erythropoietin, and the trauma patient. ITACCS. 2008;18:29–34. [Google Scholar]
- 50.Vincent JL, Baron JF, Reinhart K, Gattinoni L, Thijs L, Webb A, Meier-Hellmann A, Nollet G, Peres-Bota D. Anemia and blood transfusion in critically ill patients. JAMA. 2002;288:1499–1507. doi: 10.1001/jama.288.12.1499. [DOI] [PubMed] [Google Scholar]
- 51.Malone DL, Dunne J, Tracy JK, Putnam A, Scalea TM, Napolitano LM. Blood transfusion, independent of shock severity, is associated with worse outcome in trauma. J Trauma. 2003;54(5):898–905. doi: 10.1097/01.TA.0000060261.10597.5C. [DOI] [PubMed] [Google Scholar]
- 52.Charles A, Shaikh AA, Walters M, Huehl S, Pomerantz R. Blood transfusion is an independent predictor of mortality after blunt trauma. The American Surgeon. 2007;73(1):1–5. doi: 10.1177/000313480707300101. [DOI] [PubMed] [Google Scholar]
- 53.Pieracci FM, Stovall RT, Jaouen B, Rodil M, Cappa A, Burlew CC, Holena DN, Maier R, Berry S, Jurkovich J, Moore EE. A multicenter, randomized clinical trial of IV iron supplementation for anemia of traumatic critical illness. Crit Care Med. 2014;42(9):2048–57. doi: 10.1097/CCM.0000000000000408. [DOI] [PubMed] [Google Scholar]
- 54.Corwin HL, Gettinger A, Fabian TC, May A, Pearl RG, Heard S, An R, Bowers PJ, Burton P, Klausner MS. Efficacy and safety of epoetin alfa in critically ill patients. N Engl J Med. 2007;357(10):965–76. doi: 10.1056/NEJMoa071533. [DOI] [PubMed] [Google Scholar]
- 55.Baranski GM, Pasupuleti LV, Sifri ZC, Cook KM, Alzate WD, Rameshwar P, Livingston DH, Mohr AM. β Blockade Protection of Bone Marrow Following Injury: A Critical Link between Heart Rate and Immunomodulation. J Bone Marrow Res. 2013;1 doi: 10.4172/2329-8820.1000124. pii: 1000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bible LE, Pasupuleti LV, Alzate WD, Gore AV, Song KJ, Sifri ZC, Livingston DH, Mohr A. Early propranolol administration to severely injured patients can improve bone marrow dysfunction. J Trauma Acute Care Surg. 2014;77(1):54–60. doi: 10.1097/TA.0000000000000264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fonseca RB, Mohr AM, Wang L, Clinton E, Sifri ZC, Rameshwar P, Livingston DH. Adrenergic modulation of erythropoiesis following severe injury is mediated through bone marrow stroma. Surg Inf. 2004;5:385–393. doi: 10.1089/sur.2004.5.385. [DOI] [PubMed] [Google Scholar]
- 58.Livingston DH, Anjaria D, Wu J, Hauser CJ, Chang V, Deitch EA, Rameshwar P. Bone marrow failure following severe injury in humans. Ann Surg. 2003;238:748–753. doi: 10.1097/01.sla.0000094441.38807.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fonseca RB, Mohr AM, Wang L, Sifri ZC, Rameshwar P, Livingston DH. The impact of a hypercatecholamine state on erythropoiesis following severe injury and the role of IL-6. J Trauma. 2005;59:884–890. doi: 10.1097/01.ta.0000187653.64300.f5. [DOI] [PubMed] [Google Scholar]
- 60.Penn A, Mohr AM, Shah SG, Sifri ZC, Kaiser VL, Rameshwar P, Livingston DH. Dose-response relationship between Norepinephrine and Erythropoiesis: Evidence for a critical threshold. J Surg Res. 2010;163:85–90. doi: 10.1016/j.jss.2010.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, Frenette PS. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–21. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 62.Xiang M, Yuan Y, Fan L, Li Y, Li A, Yin L, Scott MJ, Xiao G, Billiar TR, Wilson MA, Fan J. Role of macrophages in mobilization of hematopoietic progenitor cells from bone marrow after hemorrhagic shock. Shock. 2012;37(5):518–23. doi: 10.1097/SHK.0b013e318249b81d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mendez-Ferrer S, Battista M, Frenette PS. Cooperation of beta(2)- and beta(3)-adrenergic receptors in hematopoietic progenitor cell mobilization. Ann N Y Acad Sci. 2010;1192:139–44. doi: 10.1111/j.1749-6632.2010.05390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gomes AC, Gomes MS. Hematopoietic niches, erythropoiesis and anemia of chronic infection. Exp Hematol. 2015 doi: 10.1016/j.exphem.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 65.Jenei-Lanzl Z, Grassel S, Pongratz G, Kees F, Miosge N, Angele P, Straub RH. Norepinephrine inhibition of mesenchymal stem cell and chondrogenic progenitor cell chondrogenesis and acceleration of chondrogenic hypertrophy. Arthritis Rheumatol. 2014;66(9):2472–81. doi: 10.1002/art.38695. [DOI] [PubMed] [Google Scholar]
- 66.Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood. 2003;102(3):783–8. doi: 10.1182/blood-2003-03-0672. [DOI] [PubMed] [Google Scholar]
- 67.Deitch EA, Sittig KM. A serial study of the erythropoietic response to thermal injury. Ann Surg. 1993;217:293–299. doi: 10.1097/00000658-199303000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bible LE, Pasupuleti LV, Gore AV, Sifri ZC, Kannan KB, Mohr AM. Chronic restraint stress after injury and shock is associated with persistent anemia despite prolonged elevation in erythropoietin levels. J Trauma Acute Care Surg. 2015;79(1):91–7. doi: 10.1097/TA.0000000000000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mohr AM, ElHassan IO, Hannoush EJ, Sifri ZC, Offin MD, Alzate WD, Rameshwar P, Livingston DH. Does β blockade postinjury prevent bone marrow suppression? J Trauma. 2011;70(5):1043–9. doi: 10.1097/TA.0b013e3182169326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.John GW, Doxey JC, Walter DS, Reid JL. The role of α and β adrenoreceptor subtypes in mediating the effects of catecholamines on fasting glucose and insulin concentrations in the rat. Br J Pharmacol. 1990;100:669–704. doi: 10.1111/j.1476-5381.1990.tb14078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McGuinness OP, Shau V, Benson EM, Lewis M, Snowden RT, Greene JE, Neal DW, Cherrington AD. Role of epinephrine and norepinephrine in the metabolic response to stress hormone infusion in the conscious dog. Am J Physiol. 1997;273:674–681. doi: 10.1152/ajpendo.1997.273.4.E674. [DOI] [PubMed] [Google Scholar]
- 72.Norbury WB, Jeschke MG, Herndon DN. Metabolism modulators in sepsis: propranolol. Crit Care Med. 2007;35:616–20. doi: 10.1097/01.CCM.0000278599.30298.80. [DOI] [PubMed] [Google Scholar]
- 73.Dickerson RN, Fried RC, Bailey PM, Stein TP, Mullen JL, Buzby GP. Effect of propranolol on nitrogen and energy metabolism in sepsis. J Surg Res. 1990;48:38–41. doi: 10.1016/0022-4804(90)90142-o. [DOI] [PubMed] [Google Scholar]
- 74.Herndon DN, Barrow RE, Rutan TC, Minifee P, Jahoor F, Wolfe RR. Effect of propranolol administration on hemodynamic and metabolic responses of burned pediatric patients. Ann Surg. 1988;17:484–492. doi: 10.1097/00000658-198810000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Belzberg H, Wo CC, Demetriades D, Shoemaker WC. Effects of age and obesity on hemodynamics, tissue oxygenation, and outcome after trauma. J Trauma. 2007;62(5):1192–200. doi: 10.1097/01.ta.0000219701.07295.b8. [DOI] [PubMed] [Google Scholar]
- 76.Brown CV, Neville AL, Rhee P, Salim A, Velmahos GC, Demetriades D. The impact of obesity on the outcomes of 1,153 critically injured blunt trauma patients. J Trauma. 2005;59(5):1048–51. doi: 10.1097/01.ta.0000189047.65630.c5. [DOI] [PubMed] [Google Scholar]
- 77.Rovlias A, Kotsou S. The influence of hyperglycemia on neurological outcome in patients with severe head injury. Neurosurgery. 2000;46(2):335–42. doi: 10.1097/00006123-200002000-00015. [DOI] [PubMed] [Google Scholar]
- 78.Sperry JL, Frankel HL, Vanek SL, Nathens AB, Moore EE, Maier RV, Minei JP. Early hyperglycemia predicts multiple organ failure and mortality but not infection. J Trauma. 2007;63(3):487–93. doi: 10.1097/TA.0b013e31812e51fc. [DOI] [PubMed] [Google Scholar]
- 79.Xiang L, Dearman J, Abram SR, Carter C, Hester RL. Insulin resistance and impaired functional vasodilation in obese Zucker rats. Am J Physiol Heart Circ Physiol. 2008;294(4):1658–66. doi: 10.1152/ajpheart.01206.2007. [DOI] [PubMed] [Google Scholar]
- 80.Xiang L, Lu S, Mittwede PN, Clemmer JS, Husband GW, Hester RL. β(2)-Adrenoreceptor blockade improves early posttrauma hyperglycemia and pulmonary injury in obese rats. Am J Physiol Heart Circ Physiol. 2014;307(4) doi: 10.1152/ajpheart.00208.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Magnotti LJ, Upperman JS, Xu DZ, Lu Q, Deitch EA. Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and promotes lung injury after hemorrhagic shock. Ann Surg. 1998;228:518–27. doi: 10.1097/00000658-199810000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Claridge JA, Enelow RI, Young JS. Hemorrhage and resuscitation induce delayed inflammation and pulmonary dysfunction in mice. J Surg Res. 2000;92:206–13. doi: 10.1006/jsre.2000.5899. [DOI] [PubMed] [Google Scholar]
- 83.Baranski GM, Sifri ZC, Cook KM, Alzate WD, Livingston DH, Mohr AM. Is the sympathetic system involved in shock-induced gut and lung injury? J Trauma Acute Care Surg. 2012;73:343–350. doi: 10.1097/TA.0b013e31825a785a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.ARDS Definition Task Force. Ranieri V, Rubenfeld G, Thompson B, Ferguson N, Caldwell E, Fan E, Camporota L, Slutsky A. Acute respiratory distress syndrome: the Berlin definition. JAMA. 2012;307:2526–33. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 85.Phua J, Badia J, Adhikari NK, Friedrich JO, Fowler RA, Singh JM, Scales DC, Stather DR, Li A, Jones A, Gattas DJ, Hallett D, Tomlinson G, Stewart TR, Ferguson NF. Has mortality from acute respiratory distress syndrome decreased over time? A systematic review. Am J Respir Crit Care Med. 2009;179:220–7. doi: 10.1164/rccm.200805-722OC. [DOI] [PubMed] [Google Scholar]
- 86.Le Tulzo Y, Shenkar R, Kaneko D, Moine P, Fantuzzi G, Dinarello CA, Abraham E. Hemorrhage increases cytokine expression in lung mononuclear cells in mice: involvement of catecholamines in nuclear factor-kappaB regulation and cytokine expression. J Clin Invest. 1997;99(7):1516–24. doi: 10.1172/JCI119314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suter PM, Suter S, Girardin E, Roux-Lombard P, Grau GE, Dayer JM. High bronchoalveolar levels of tumor necrosis factor and its inhibitors, interleukin-1, interferon, and elastase, in patients with adult respiratory distress syndrome after trauma, shock, or sepsis. Am Rev Respir Dis. 1992;145(5):1016–22. doi: 10.1164/ajrccm/145.5.1016. [DOI] [PubMed] [Google Scholar]
- 88.Moss G, Stein AA. The centrineurogenic etiology of the respiratory distress syndrome: induction by isolated cerebral hypoxemia and prevention by unilateral pulmonary denervation. Am J Surg. 1976;132(3):352–7. doi: 10.1016/0002-9610(76)90392-5. [DOI] [PubMed] [Google Scholar]
- 89.Hagiwara S, Iwasaka H, Maeda H, Noguchi T. Landiolol, an ultrashort-acting β1-adrenoceptor antagonist, has protective effects in an LPS-induced systemic inflammation model. Shock. 2009;31(5):515–20. doi: 10.1097/SHK.0b013e3181863689. [DOI] [PubMed] [Google Scholar]
- 90.Aboab J, Sebille V, Jourdain M, Mangalaboyi J, Gharbi M, Mansart A, Annane D. Effects of esmolol on systemic and pulmonary hemodynamics and on oxygenation in pigs with hypodynamic endotoxin shock. Intensive Care Med. 2011;37:1344–51. doi: 10.1007/s00134-011-2236-y. [DOI] [PubMed] [Google Scholar]
- 91.Novotny NM, Lahm T, Markel TA, Crisostomo PR, Wang M, Wang Y, Ray R, Tan J, Al-Azzawi D, Meldrum DR. β-blockers in sepsis: reexamining the evidence. Shock. 2009;31(2):113–9. doi: 10.1097/SHK.0b013e318180ffb6. [DOI] [PubMed] [Google Scholar]
- 92.Feinman R, Deitch EA, Aris V, Chu HB, Abungu B, Caputo FJ, Galante A, Xu D, Lu Q, Colorado I, Streck D, Dermody J, Soteropoulos P. Molecular signatures of trauma-hemorrhagic shock-induced lung injury: hemorrhage- and injury-associated genes. Shock. 2007;28(3):360–8. doi: 10.1097/shk.0b013e318048565b. [DOI] [PubMed] [Google Scholar]
- 93.Ballard-Croft C, Maass DL, Sikes P, White J, Horton J. Activation of stress-responsive pathways by the sympathetic nervous system in burn trauma. Shock. 2002;18(1):38–45. doi: 10.1097/00024382-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 94.Kulp GA, Herndon DN, Lee JO, Suman OE, Jeschke MG. Extent and magnitude of catecholamine surge in pediatric burned patients. Shock. 2010;33(4):369–74. doi: 10.1097/SHK.0b013e3181b92340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ali A, Herndon DN, Mamachen A, Hasan S, Andersen CR, Grogans RJ, Brewer JL, Lee JO, Heffernan J, Suman OE, Finnerty CC. Propranolol attenuates hemorrhage and accelerates wound healing in severely burned adults. Crit Care. 2015;19:217. doi: 10.1186/s13054-015-0913-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Herndon DN, Hart DW, Wolf SE, Chinkes DL, Wolfe RR. Reversal of catabolism by β-blockade after severe burns. N Engl J Med. 2001;345:1223–1229. doi: 10.1056/NEJMoa010342. [DOI] [PubMed] [Google Scholar]
- 97.Horton JW. A model of myocardial inflammation and dysfunction in burn complicated by sepsis. Shock. 2007;28(3):326–33. doi: 10.1097/01.shk.0000238064.54332.c8. [DOI] [PubMed] [Google Scholar]
- 98.Muthu K, Deng J, Romano F, He LK, Gamelli R, Shankar R, Jones SB. Thermal injury and sepsis modulates β-adrenergic receptors and cAMP responses in monocyte-committed bone marrow cells. J Neuroimmunol. 2005;165(1–2):129–38. doi: 10.1016/j.jneuroim.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 99.Brooks NC, Song J, Boehning D, Kraft R, Finnerty CC, Herndon DN, Jeschke MG. Propranolol improves impaired hepatic phosphatidylinositol 3-kinase/akt signaling after burn injury. Mol Med. 2012;18:707–11. doi: 10.2119/molmed.2011.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Romana-Souza B, Nascimento AP, Monte-Alto-Costa A. Low-dose propranolol improves cutaneous wound healing of burn-injured rats. Plast Reconstr Surg. 2008;122(6):1690–9. doi: 10.1097/PRS.0b013e31818cbf67. [DOI] [PubMed] [Google Scholar]
- 101.Zhang X, Meng C, Chinkes D, Finnerty C, Aarsland A, Jeschke M, Herndon D. Acute propranolol infusion stimulates protein synthesis in rabbit skin wound. Surgery. 2009;145(5):558–67. doi: 10.1016/j.surg.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 102.Pullar CE, Manabat-Hidalgo CG, Bolaji RS, Isseroff RR. beta-Adrenergic receptor modulation of wound repair. Pharmacol Res. 2008;58(2):158–64. doi: 10.1016/j.phrs.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 103.Gosain A, Jones SB, Shankar R, Gamelli RL, DiPietro LA. orepinephrine modulates the inflammatory and proliferative phases of wound healing. J Trauma. 2006;60(4):736–44. doi: 10.1097/01.ta.0000196802.91829.cc. [DOI] [PubMed] [Google Scholar]
- 104.Souza BR, Santos JS, Costa AM. Blockade of beta1- and beta2-adrenoceptors delays wound contraction and re-epithelialization in rats. Clin Exp Pharmacol Physiol. 2006;33(5–6):421–30. doi: 10.1111/j.1440-1681.2006.04383.x. [DOI] [PubMed] [Google Scholar]
- 105.Oberbeck R, van Griensven M, Nickel E, Tschernig T, Wittwer T, Pape H. Influence of β-adrenoceptor antagonists on hemorrhage-induced cellular immune suppression. Shock. 2002;18:331–335. doi: 10.1097/00024382-200210000-00007. [DOI] [PubMed] [Google Scholar]
- 106.Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve–an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52(4):595–638. [PubMed] [Google Scholar]
- 107.Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE. Differential expression of the β2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help. J Immunol. 1997;158(9):4200–10. [PubMed] [Google Scholar]
- 108.Tait SM, Wang P, Ba ZF, Chaudry IH. Downregulation of hepatic β-adrenergic receptors after trauma and hemorrhagic shock. Am J Physiol. 1995;268(5):749–53. doi: 10.1152/ajpgi.1995.268.5.G749. [DOI] [PubMed] [Google Scholar]
- 109.Benshop RJ, Nijkamp FP, Ballieuz RE, Heijen CJ. The effects of beta-adrenoceptor stimulation on adhesion of human natural killer cells to cultured endothelium. Br J Pharmacol. 1994;113(4):1311–6. doi: 10.1111/j.1476-5381.1994.tb17141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76(2):301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 111.Bukur M, Lustenberger T, Cotton B, Arbabi S, Talving P, Salim A, Ley EJ, Inaba K. β-blocker exposure in the absence of significant head injuries is associated with reduced mortality in critically ill patients. Am J Surg. 2012;204:697–703. doi: 10.1016/j.amjsurg.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 112.Huang L, Weil MH, Cammarata G, Sun S, Tang W. Nonselective β-blocking agent improves the outcome of cardiopulmonary resuscitation in a rat model. Crit Care Med. 2004;32(9):378–80. doi: 10.1097/01.ccm.0000134266.65164.7c. [DOI] [PubMed] [Google Scholar]
- 113.Friese RS, Barber R, McBride D, Bender J, Gentilello LM. Could β blockade improve outcome after injury by modulating inflammatory profiles? J Trauma. 2008;64:1061–8. doi: 10.1097/TA.0b013e3181684cf0. [DOI] [PubMed] [Google Scholar]
- 114.Deng J, Muthu K, Gamelli R, Shankar R, Jones SB. Adrenergic modulation of splenic macrophage cytokine release in polymicrobial sepsis. Am J Physiol Cell Physiol. 2004;287(3):730–6. doi: 10.1152/ajpcell.00562.2003. [DOI] [PubMed] [Google Scholar]
- 115.Heneka MT, Gavrilyuk V, Landreth GE, O’Banion MK, Weinberg G, Feinstein DL. Noradrenergic depletion increases inflammatory responses in brain: effects on IκB and HSP70 expression. J Neurochem. 2003;85:387–398. doi: 10.1046/j.1471-4159.2003.01694.x. [DOI] [PubMed] [Google Scholar]
- 116.Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW, 3rd, Bland KI, Chaudry IH. Cecal ligation and puncture. Shock. 2005;24(1):52–7. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 117.Sekut L, Champion BR, Page K, Menius JA, Jr, Connolly KM. Anti-inflammatory activity of salmeterol: down-regulation of cytokine production. Clin Exp Immunol. 1995;99(3):461–6. doi: 10.1111/j.1365-2249.1995.tb05573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.van der Poll T, Coyle SM, Barbosa K, Braxton CC, Lowry SF. Epinephrine inhibits tumor necrosis factor-[α] and potentiates interleukin 10 production during human endotoxemia. J Clin Invest. 1996;97:713–719. doi: 10.1172/JCI118469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ertel W, Kremer JP, Kenney J, Steckholzer U, Jarrar D, Trentz O, Schildberg FW. Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood. 1995;85(5):1341–7. [PubMed] [Google Scholar]
- 120.Bergmann M, Gornikiewicz A, Sautner T, Waldmann E, Weber T, Mittlböck M, Roth E, Függer R. Attenuation of catecholamine-induced immunosuppression in whole blood from patients with sepsis. Shock. 1999;12(6):421–7. doi: 10.1097/00024382-199912000-00002. [DOI] [PubMed] [Google Scholar]
- 121.Ayala A, Chaudry IH. Immune dysfunction in murine polymicrobial sepsis: mediators, macrophages, lymphocytes and apoptosis. Shock. 1996;6(1):27–38. [PubMed] [Google Scholar]
- 122.Le Tulzo Y, Pangault C, Gacouin A, Guilloux V, Tribut O, Amiot L, Tattevin P, Thomas R, Fauchet R, Drénou B. Early circulating lymphocyte apoptosis in human septic shock is associated with poor outcome. Shock. 2002;18(6):487–94. doi: 10.1097/00024382-200212000-00001. [DOI] [PubMed] [Google Scholar]
- 123.Chopra M, Das P, Golden H, Dostal DE, Watson LE, Sharma AC. Norepinephrine induces systolic failure and inhibits antiapoptotic genes in a polymicrobial septic rat model. Life Sci. 2010;87(23–26):672–8. doi: 10.1016/j.lfs.2010.09.029. [DOI] [PubMed] [Google Scholar]
- 124.Takahashi H, Kobayashi M, Tsuda Y, Hendon DN, Suzuki F. Contribution of the sympathetic nervous system on the burn-associated impairment of CCL3 production. Cytokine. 2005;29(5):208–14. doi: 10.1016/j.cyto.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 125.Rough J, Engdahl R, Opperman K, Yerrum S, Monroy MA, Daly JM. β2 Adrenoreceptor blockade attenuates the hyperinflammatory response induced by traumatic injury. Surgery. 2009;145(2):235–42. doi: 10.1016/j.surg.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 126.Wilson J, Higgins D, Hutting H, Serkova N, Baird C, Khailova L, Queensland K, Vu Tran Z, Weitzel L, Wischmeyer PE. Early propranolol treatment induces lung heme-oxygenase-1, attenuates metabolic dysfunction, and improves survival following experimental sepsis. Crit Care. 2013;17(5):195. doi: 10.1186/cc12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sanders VM, Munson AE. Role of α adrenoceptor activation in modulating the murine primary antibody response in vitro. J Pharmacol Exp Ther. 1985;232(2):395–400. [PubMed] [Google Scholar]
- 128.Spengler R, Allen R, Remick D, Strieter R, Kunkel S. Stimulation of α-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. J Immunol. 1990;145(5):1430–4. [PubMed] [Google Scholar]
- 129.Spengler RN, Chensue SW, Giacherio DA, Blenk N, Kunkel SL. Endogenous norepinephrine regulates tumor necrosis factor-α production from macrophages in vitro. J Immunol. 1994;152(6):3024–31. [PubMed] [Google Scholar]
- 130.Severn A, Rapson NT, Hunter CA, Liew FY. Regulation of tumor necrosis factor production by adrenaline and β-adrenergic agonists. J Immunol. 1992;148(11):3441–5. [PubMed] [Google Scholar]
- 131.Davies MG, Hagen PO. Systemic inflammatory response syndrome. Br J Surg. 1997;84(7):920–35. doi: 10.1002/bjs.1800840707. [DOI] [PubMed] [Google Scholar]
- 132.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24(7):1125–8. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 133.Macchia A, Romero M, Comignani PD, Mariani J, D’Ettorre A, Prini N, Santopinto M, Tognoni G. Previous prescription of β-blockers is associated with reduced mortality among patients hospitalized in intensive care units for sepsis. Crit Care Med. 2012;40(10):2768–72. doi: 10.1097/CCM.0b013e31825b9509. [DOI] [PubMed] [Google Scholar]
- 134.Morelli A, Ertmer C, Westphal M, Rehberg S, Kampmeier T, Ligges S, Orecchioni A, D’Egidio A, D’Ippoliti F, Raffone C, Venditti M, Guarracino F, Girardis M, Tritapepe L, Pietropaoli P, Mebazaa A, Singer M. Effect of heart rate control with esmolol on hemodynamic and clinical outcomes in patients with septic shock: a randomized clinical trial. JAMA. 2013;310:683–691. doi: 10.1001/jama.2013.278477. [DOI] [PubMed] [Google Scholar]