

Abstract
Human Cytomegalovirus (HCMV) infection is compromised in cells lacking p53, a transcription factor that mediates cellular stress responses. In this study we have investigated compromised functional virion production in cells with p53 knocked out (p53KOs). Infectious center assays found most p53KOs released functional virions. Analysis of electron micrographs revealed modestly decreased capsid production in infected p53KOs compared to wt. Substantially fewer p53KOs displayed HCMV-induced infoldings of the inner nuclear membrane (IINMs). In p53KOs, fewer capsids were found in IINMs and in the cytoplasm. The deficit in virus-induced membrane remodeling within the nucleus of p53KOs was mirrored in the cytoplasm, with a disproportionately smaller number of capsids re-enveloped. Reintroduction of p53 substantially recovered these deficits. Overall, the absence of p53 contributed to inhibition of the formation and function of IINMs and re-envelopment of the reduced number of capsids able to reach the cytoplasm.
Keywords: p53, Human Cytomegalovirus, capsids, nuclear egress, infoldings of the inner nuclear membrane (IINM), secondary envelopment, transmission electron microscopy (TEM)
INTRODUCTION
The HCMV lifecycle commences with the virion’s entrance into the cell via fusion of the viral envelope with the cellular membrane (Adler and Sinzger, 2013; Boyle and Compton, 1998; Compton, 2004; Compton et al., 1993; Feire and Compton, 2013; Isaacson and Compton, 2009; Kari and Gehrz, 1992, 1993; Ryckman et al., 2008a; Ryckman et al., 2006; Ryckman et al., 2008b; Vanarsdall et al., 2008; Wang and Shenk, 2005). Following entry, the capsid is transported along microtubules toward the nuclear pores, through which the 235 kB double-stranded DNA viral genome is deposited into the nucleus (Ogawa-Goto et al., 2003) and as reviewed in (Kalejta, 2008). The viral lifecycle is defined by several stages, which can be loosely defined by immediate early (IE), early (E) and late (L) protein expression. Various physiological and morphological effects have been noted concomitant with these stages, including p53 protein stabilization and strong association of this tumor suppressor protein with the viral replication centers (RCs) at E times pi (Fortunato and Spector, 1998; Muganda et al., 1998; Muganda et al., 1994).
The reproductive component of the virus lifecycle begins with replication and encapsidation of its genome in discrete viral RCs within the nucleus. This is followed by initial tegumentation of the capsid and transit of the encapsidated particles across the nuclear membrane (which includes primary envelopment and de-envelopment) (as reviewed in (Johnson and Baines, 2011; Mettenleiter et al., 2009)). Completion of tegumentation and re-envelopment of capsids occurs in the cytoplasm (Sanchez et al., 2000a; Sanchez et al., 2000b; Schauflinger et al., 2013). Viral reproduction concludes with these particles being shed in secretory vesicles via fusion with the plasma membrane (reviewed in reference Colberg-Poley and Williamson, 2013).
Ultra-structural analysis of sections of the nucleus of infected cells has identified three capsid types, A, B and C. These are characteristic of herpesvirus replication and represent stages in virion morphogenesis. The A capsid is empty, containing no scaffolding or viral genome. The B capsid has a translucent core composed of the scaffolding assembly protein (AP) not yet removed by proteolytic digestion and lacking a viral genome. C capsids have a dark electron dense core composed of the packed viral genome (Tandon et al., 2015) and as reviewed in (Mocarski, 2001).
All three types of capsids can be tegumented. The first layers of tegument protein are acquired inside the nucleus. pp150, the tegument layer closest to the capsid proper, is thought to provide stability to the capsid structure during packaging and passage through the nuclear envelope (AuCoin et al., 2006; Baxter and Gibson, 2001; Dai et al., 2013; Tandon and Mocarski, 2011). Tegumented capsids bud through the inner nuclear membrane into the perinuclear space, in the process acquiring a primary envelope. Infoldings of the inner nuclear membrane (IINMs), structures that have been observed by several groups to contain enveloped capsids (Buser et al., 2007; Dal Monte et al., 2002; Gilloteaux and Nassiri, 2000; Papadimitriou et al., 1984; Ruebner et al., 1964; Severi et al., 1988; Villinger et al., 2015), have been proposed as the principal site of transit through the nuclear membrane for the cytomegaloviruses (Buser et al., 2007). The primary enveloped capsids fuse with the outer nuclear membrane, in the process becoming de-enveloped, and are deposited into the cytoplasm (as reviewed in references Johnson and Baines, 2011; Mettenleiter et al., 2009).
The viral assembly complex (VAC) begins to appear within the cytoplasm of infected cells by approximately 48 h pi (Buchkovich et al., 2010; Buchkovich et al., 2009; Das and Pellett, 2011; Das et al., 2007; Indran et al., 2010). Within the VAC, capsids become fully tegumented and acquire their final envelope (re-envelopment) (Sanchez et al., 2000a; Sanchez et al., 2000b; Schauflinger et al., 2013). Another viral particle, the dense body (DB), is also enveloped in the VAC. DBs are enveloped “bags” of viral tegument proteins, largely composed of pp65 (Gibson, 1984; Sarov and Abady, 1975). In lab-adapted strains of HCMV, e.g. Towne and AD169, DBs constitute a large proportion of the viral particles within the cytoplasm. Golgi-derived vesicles containing viral particles are transported to the plasma membrane and their contents are released into the extra-cellular space via fusion with the cell membrane (reviewed in references in Johnson and Baines, 2011; Kalejta, 2008). The entire array of virus-derived particles including DBs, non-infectious enveloped particles (NIEPs), and functional/non-functional virions, are released from the infected cell and comprise the viral components of infected supernatant. Once peak virion release begins at approximately 72 h pi, infected cells continue to produce virus for several days until they are ultimately lysed.
Transcription of the tumor suppressor protein p53 is not increased in HCMV-infected fibroblasts, however by 4 h pi steady state levels of the protein begin to increase via stabilization of the protein (Fortunato and Spector, 1998; Muganda et al., 1998; Muganda et al., 1994). Despite the increased steady-state levels of p53 in an infected cell, its cellular targets, including p21, are not activated and the infected cell does not undergo apoptosis, nor does p53 trigger a successful DNA damage response (Bresnahan et al., 1996; Castillo et al., 2000; Chen et al., 2001; Fortunato and Spector, 1998; Hsu et al., 2004; Luo et al., 2007; Muganda et al., 1994; Murphy et al., 2000; Savaryn et al., 2013; Speir et al., 1994; Tsai et al., 1996; Zhu et al., 1995). p53 is tightly associated with the viral RCs as soon as they are established (Fortunato and Spector, 1998). p53’s normal function as a transcription factor suggests it may also be activating or inhibiting the expression of viral genes. In support of this supposition, earlier work in our laboratory found 21 p53-binding sites in the HCMV genome, several of which were directly bound (Rosenke et al., 2006), and that several viral proteins were down-regulated in the absence of p53 (Hannemann et al., 2009). Recent work has shown that p53 also plays a role in regulating expression of HSV-1 gene products (Maruzuru et al., 2013). These findings indicate that p53 plays an important role in the dynamics of both HCMV and HSV infection.
The availability of p53 knockout cells (p53KO), in which the protein is totally absent, has facilitated study of p53’s role during HCMV infection (Casavant et al., 2006; Hannemann et al., 2009; Rosenke et al., 2006). IE gene expression in these cells was near normal, while the accumulation of many E and L viral proteins was delayed by 24–48 h. Intriguingly, in the majority of p53KO cells the most abundant of the tegument proteins, pp65, did not display wt-like relocation from the nucleus to the cytoplasm, even at late times pi. While expression of the majority of viral proteins in the p53KO cells had reached near wild type (wt) levels by 120 h pi, functional virion production remained reduced by 25-fold until 144 h pi (Casavant et al., 2006). This indicated these cells were inherently inefficient viral hosts and that even additional infection duration could not overcome this deficit. The presence of even vanishingly low levels of wt p53 protein, as demonstrated by its exceedingly limited expression in a reintroduction cell line (see Materials and Methods and Supplemental Figure 1 of accompanying manuscript), reversed this trend (Casavant et al., 2006).
In this study we have identified structural shortcomings responsible for the compromised production of functional virus in cells lacking p53. The absence of p53 from the cellular environment correlated with a modest (~1.6-fold), but statistically significant, decrease in nuclear capsid production. Substantial reductions in the formation and size of virus-induced IINMs, correlated with a statistically significantly reduced rate of cytoplasmic trafficking to and secondary envelopment of capsids in the cytoplasm. These parameters combined to largely recapitulate the ~25-fold reduction in functional virion production. Re-introduction of very low levels of p53 largely rescued capsid nuclear egress and secondary envelopment, indicating a role, direct or indirect, for this regulatory protein in HCMV capsid maturation.
MATERIALS AND METHODS
Cells and virus growth
p53KO telomerase-immortalized human fibroblasts, their wt parental cell line, LOX (Bunz et al., 1998; Wei et al., 2001) (both kind gifts from Dr. John Sedivy, Brown University), and WTG (p53-reconstituted p53KO cells) were maintained in complete medium composed of Dulbecco’s Modified Eagle Media: Nutrient Mixture F-12 (Ham) 1:1 (DMEM/F-12) supplemented with 10% heat-inactivated fetal bovine serum (FBS), L-glutamine (2mM), penicillin (200 U/ml), and streptomycin (200 μg/ml). Cells were grown in incubators maintained at 37°C and 5% CO2. The Towne strain of HCMV was obtained from the ATCC (#VR 977), propagated under standard procedures (Tamashiro et al., 1982), and used at a multiplicity of infection (MOI) of 5 for all experiments. It should be noted that multiple labs have found HCMV virion release from fibroblast cultures is unaffected by telomerase immortalization (Bresnahan et al., 2000).
Background regarding the complete absence of p53 versus low-level expression
The p53KO cells (Bunz et al., 1998; Wei et al., 2001) used in this, and our previous studies (Casavant et al., 2006; Hannemann et al., 2009; Rosenke et al., 2006), were genetically ablated for p53 and expressed absolutely no protein. Earlier characterization of these cells showed that they were genetically stable and not prone to aneuploidy upon continuous passaging (Bunz et al., 2002). These cells were telomerase immortalized, however, in our laboratory they have not been passaged for long periods of time (rarely exceeding 30 and never more than 38 passages from original derivation). In order to ensure consistency, frozen earlier passage stocks were frequently returned to as experiments progressed. We have previously determined that cells from passage 8 or passage 38 display essentially identical phenotypes with respect to viral growth parameters (Casavant et al., 2006; Hannemann et al., 2009; Rosenke et al., 2006). The genetic ablation of p53 undoubtedly introduced additional changes into these cells’ genome; however, these cells were viable, susceptible/permissive to/for HCMV-infection and met many normal benchmarks at early times pi (Casavant et al., 2006). The major difference in viral life cycle parameters that we have noted was the shedding of infectious virus at substantially reduced rates. Given the relatively normal viral life cycle in the p53KO cells, these cells are an ideal candidate for studying p53’s role in HCMV-infection.
As also described in our previous work (Casavant et al., 2006), p53 was transfected into the p53KO cells to establish several independent p53-expressing stable clones. Four p53-expressing lines (designated WT D-G) were studied extensively. All produced similar, and quite low, levels of p53 (see Supplemental Figure 1 in the companion manuscript). Semi-quantitation of p53 level by WB estimated the infected p53KO cells produced approximately 5% of uninfected wt-levels of the protein. Whatever compensating mutations existed in the p53KO cells, which allowed them to survive in the absence of p53, appeared to render expression of p53 above very low levels lethal to these cells (Casavant et al., 2006). This was not surprising given that reintroduction of wt p53 into tumor cells has been documented to cause rapid arrest (as reviewed in (Levine et al., 1991)). All the stable wt p53-expressing lines behaved similarly with respect to all viral parameters tested and returned toward wt in each case. Most relevant, infectious titers from these cell lines at late times pi were only 4–5-fold lower than wt and were increased ~10-fold over their p53KO parental line (Casavant et al., 2006). In sharp contrast selected pools, expressing either of two p53 mutant proteins (the DNA binding domain mutants R273H and H179Q), behaved identically to the p53KO cells, and produced no increases in titers, indicating the presence of a wt protein was necessary to aid in viral growth parameters (Casavant et al., 2006). It should be noted that HSV-1 studies using a different p53KO system (HCT116 cells) also recovered slightly lower than wt titers (~4-fold), even when much higher levels of wt p53 could be reintroduced (Maruzuru et al., 2013).
Cell cycle synchronization and infection
G0 synchronization was carried out via serum starvation as previously described (Casavant et al., 2006). The cells were then trypsinized and reseeded onto plates or flasks in complete medium. After allowing 1–2 h for attachment, cells were infected. After 4 h, medium was replaced with fresh complete medium to remove the excess virus that had not entered the cells.
Assays for infectious virion shedding cells
The shedding of infectious virus was assessed using two protocols. First, we used conventional focus forming assays as previously described (Luo and Fortunato, 2007). Briefly, infected cell samples (3 × 105), were harvested at 120 h pi. Infected cells were washed with phosphate-buffered saline (PBS) three times before trypsinization and centrifugation to pellet. The cell pellet was washed and re-pelleted three additional times with complete medium to fully remove any viral particles potentially adhering to the outside of the infected cells. The medium from the final wash was reserved for later use as a negative control and to ensure removal of all extra-cellular virus. The cell pellet was then resuspended in 1 ml of complete culture medium and cells were counted and diluted as necessary. Immediately following dilution, 50 cells were inoculated (in triplicate) onto a monolayer of human foreskin fibroblasts (HFFs). Cells were overlaid with agar and resulting foci were counted after 7 d. The mean of the replicate samples were expressed as percent focus-forming cells (focus-forming units/50 cells × 100). The assays were performed five times and the mean plus one standard error is presented.
In a second assay infected cells were assessed for infectious centers via immunofluorescent staining for IE1 and UL44 foci. First, monolayers of HFFs were seeded and grown on coverslips in 12-well dishes. Approximately 40 infected cells were then seeded in each well. Three duplicate wells were seeded to provide technical replicates for each cell type and timepoint. The entire experiment was performed twice with similar results. Quantitation was performed on one experiment. Medium was replaced daily to as much as possible contain the spread of infection to only directly adjacent cells. Coverslips were harvested each day for three days. Immunofluorescence staining assessed HCMV shedding capability from each of the infected cells. Seeded cells were identified as the cells with large UL44 foci amidst surrounding infected (IE1+) cells.
Antibodies
Primary mouse monoclonal antibodies (mAb) used were anti-IE1 (IgG2A; a kind gift from Dr. Bill Britt, University of Alabama, Birmingham) and anti-UL44 (1202S [IgG1]; Goodwin Institute). Secondary antibodies used were goat anti-mouse IgG2A TRITC-coupled Ab (Southern Biotech) and goat anti-mouse IgG1 Alex Fluor 488-coupled Ab (Molecular Probes).
Immunofluorescence (IF) Assays
All coverslips were fixed in 3% formaldehyde followed by permeabilization in 1% TritonX-100 as previously described (Rosenke et al., 2006). Nuclei were counterstained with Hoechst dye. Epifluorescence analysis was performed on a Nikon Eclipse E800 flourescence microscope equipped with a Nikon DS-Ri1 high-resolution color camera and Nikon NIS Elements Basic Research (Br) imaging software.
Transmission electron microscopy (TEM)
Cells were serum starved as described above, then reseeded, infected and harvested at 120 h pi. Cells were then fixed in 2% paraformaldehyde/2% glutaraldehyde in 0.1 M PBS, rinsed extensively in 0.1M Cacodylate buffer with 3.5% sucrose, post fixed in 2% OsO4 in 0.1M Cacodylate buffer for 2 h at RT, and then rinsed extensively in de-ionized distilled (dd) H2O. Optimal membrane contrast was obtained by staining cells with 1% tannic acid for 1h at RT and then rinsing twice with dd H2O. Samples were dehydrated through an increasing ethanol series (30–100%), followed by two 100% acetone dehydrations. To prepare for embedding, cells were infiltrated in acetone and Spurr’s Resin (1:1 ratio) overnight. The resin was then replaced three times with fresh 100% Spurr’s Resin for 1 h. Samples were stored in Spurr’s Resin overnight at RT before they were embedded at 70°C for 48 h. After embedding, samples were sectioned at ~100 nm using a diamond knife. Sections were contrast enhanced by staining with 4% uranyl acetate for 10 min, followed by Reynold’s lead citrate staining for 5 min in a CO2 free environment. Samples were examined with an FEI Technai G2 20 Twin transmission electron microscope (FEI TEM) equipped with a 200 kV LaB6 electron source and a 4K Eagle camera. Images were captured using TEM Imaging Analysis (TIA) software. Images for analysis were captured at a magnification of 2500x and 5000x. As necessary higher magnification images were stitched together, using image-editing software (PhotoShop), to form mosaic images of the individual captures from an individual cell section. Capsids were enumerated using these mosaic images.
Statistical analysis
No correlations between the number of capsids in one cellular compartment, e.g. sections of nuclei, IINMs, or cytoplasm, and any other compartment of an individual cell section within any experimental population (LOX, p53KO or WTG) were observed. This indicated the number of capsids in one compartment, at least within the microtome sections that were analyzed, did not influence the contents of an adjacent compartment. Consequently relative comparisons of the movement from one compartment to another would not have been valid within individual cell sections, between sections of different cells, or cell populations. Therefore only capsid numbers within a given cellular compartment have been compared.
Analysis of variance for differences among infected and uninfected LOX and p53KO cells in Figure 6 were conducted using SAS (Cary, NC). Because these data were proportions, the values were first transformed using the arcsin of the square roots of each proportion (Ott and Longnecker, 2010). Since the overall test was found statistically significant (p=0.034), Tukey’s Studentized Range Test was used for pairwise comparisons of the transformed percentages (values are reported in the figure).
Statistical tests for differences among the EM sections of the LOX, p53KO and WTG cells (all graphs in Figures 2–5 and Supplemental Figures) were conducted using the Kruskal-Wallace test as implemented in SAS (Cary, NC). This test is designed for the analysis of data that does not assume a normal distribution of errors. If the overall tests were found statistically significant (p-values reported in the Figure Legends), then pairwise comparisons among the three cell lines were conducted using the same test. In Figure 3G, a χ2 test, implemented in SAS, was used to determine differences between LOX, p53KO and WTG in the proportion of cell sections with IINMs. In this analysis, an overall test was performed first, followed by pairwise analysis. For all analyses, a p-value of 0.05 or less was considered statistically different.
The large range of capsid numbers in the EM cell sections in all parameters examined, including substantially elevated outliers and null entries in all cell types, could markedly influence the calculation of mean values. Therefore, in all graphs in Figures 2–5 and Supplemental Figures, the median value for each experiment was selected as the most representative of a prototypical cell. The median values for each cell type from each experiment were compared to generate a fold-change for that experiment and, in order to facilitate the evaluation of the magnitude of changes, the mean (and range) of these fold-changes has been reported in the text. If the median for a parameter was zero (Figure 3H, I and J in p53KO cell sections only), the fold-change calculation excluded these values; therefore the reported fold-changes represent the minimum increases. These fold-changes are not directly comparable with the Kruskal-Wallace test p-values nor readily tested for significance independently, nevertheless, we believe the fold-changes reported provide a useful gauge to compare magnitude of changes between the experimental groups.
Quantitation of lipid droplet consumption
Mock- and virus-infected LOX and p53KO coverslips from four separate experiments were harvested at 120 hpi, fixed in 3% formaldehyde as described above and stored at 4°C in PBS. Cells were stained in 1% of the lipophilic dye Sudan IV (dissolved in 70% EtOH) for 5 minutes and then rinsed extensively in ddH2O. Quantitation of cells containing lipid droplets in the cytoplasm was accomplished by counterstaining with haematoxylin to more readily view the nucleus, followed by mounting the coverslips in 70% glycerol. At least 175 cells were scored per coverslip (except for one sample of infected LOX cells). Results are reported as means of four experiments. Error bars represent one standard error of the mean.
RESULTS
The majority of p53KO cells released functional virions
Titration of the virus produced by the p53KOs found an ~25-fold reduction at 120 h pi compared to LOX, their parental counterpart (Casavant et al., 2006). Conventional focus forming assays were performed to determine the percentage of infected p53KO cells harvested at 120 h pi that were capable of forming plaques. Figure 1A displays the percentage of seeded cells producing recognizable plaques at 7 days (d) post plating. Essentially 100% of seeded LOX cells, produced plaques, while only ~23% of the p53KO cells generated identifiable plaques, even at 9 d post seeding. These cells were likely the ~30% of cells found in our earlier studies to traffic pp65 into the cytoplasm and contain large VAC formations at late times pi (Casavant et al., 2006). Were the remaining ~70–75% of cells also shedding virus, but too few functional virions to generate identifiable plaques?
Figure 1.

Quantitation of virion shedding cells in wt LOX cells compared to p53KO cells. Infected LOX and p53KO cells were harvested for seeding onto monolayers at 120 h pi. (A) The mean percentage of cells generating visible plaques in conventional plaque forming assays. Error bars represent one standard error calculated from five experiments. (B) Infectious center assays to more sensitively detect virion shedding. Coverslips were stained for UL44 to detect seeded cells (arrows) and IE1 to detect newly infected neighboring cells (arrow heads). (C) Mean percentage of shedding positive cells detected by immunofluorescent staining of infectious center assays. Error bars represent one standard error calculated from three technical replicates. (D) Quantitation of newly infected neighboring cells at day 1 and 2 post seeding. Mean numbers of infected neighboring cells were binned at 0, 1–10, and more than 10. Error bars represent one standard error calculated from three technical replicates.
This question was addressed using a more sensitive infectious center assay. Infected cells were harvested at 120 h pi. Cells were counted and seeded in known quantities onto monolayers of HFFs grown on coverslips. To allow further processing of these cells no agarose overlays were used. Coverslips were harvested for three successive days. The coverslips were stained for UL44 and IE1 to examine the shedding capability of the seeded cells. The majority of LOX and p53KO cells harvested at 120 h pi contained advanced stage replication centers (RCs) ((Casavant et al., 2006) and Kuan and Fortunato, unpublished) and could therefore be identified by staining for the viral processivity factor UL44. Coverslips were co-stained with IE1 to identify any newly infected cells neighboring the seeded cells. Figure 1B shows examples of the UL44+ seeded cells (arrows), surrounded by newly infected cells stained for IE1 (arrow heads). Greater than 95% of the LOX cells shed functional virus to an adjacent cell on d1. Approximately 60% of the p53KO cells shed functional virus on d1. This percentage increased to ~80% by d3 (Figure 1C). The remaining p53KO cells displayed no adjacent IE1 staining (Figure 1B - no shedding panels). The three-day timecourse in the p53KO cells was possible due to the delayed development of large single focus RCs in the newly infected cells compared to LOX (Casavant et al., 2006). The LOX cells appeared to release more functional virions. Twice as many LOX cells, ~73%, infected greater than ten neighboring cells compared to ~36% of the p53KO cells (Figure 1D). Due to second round shedding from newly infected cells, these assays could only be carried out for two days following seeding. These results suggested that the ~35% of p53KOs with greater than ten IE+ adjacent cells 2 d pi, were likely the same sub-population which formed identifiable plaques in the initial assay (Figure 1A). These assays demonstrated that a large majority of the p53KO cells were shedding functional virus, although it was perhaps only a single functional virion to an adjacent cell.
A detailed analysis of the nucleus, including cells with reconstituted p53
We captured ultrastructural EM images of infected LOX and p53KO cell sections from three infections (experiments 1, 2 and 3). Two of these experiments (experiments 2 and 3) also included WTG cell sections. The p53KO cells have been reconstituted with a very low level of wt p53 (for additional information on these cells see the Materials and Methods and Supplemental Figure 1 of the accompanying manuscript). These images were analyzed for total capsid production, distribution of capsid types, and the presence of capsids within or adjacent to the perinuclear space. Representative images of the cross section of infected cells at magnification 1,700X are shown in Figure 2A, C and E. Images captured at higher magnification (5000x) were stitched together to form a mosaic image for each cell section. Nuclear capsids in these mosaic images were quantitated (see Materials and Methods for more detailed explanation). Figures 2B, D and F show examples of nuclear capsids present within the boxed areas in 2A, C and E, as visualized in the 5,000x images. The mean of the fold changes of the median values of nuclear capsids in LOX cell sections compared to p53KO cell sections was ~1.6-fold (0.5–2.1X) higher and compared to WTG cell sections was ~1.4-fold (0.9–1.9x) higher, with virtually no differences between p53KO and WTG cell sections (Figure 2G). Closer examination of the results revealed quite similar (and large) ranges of capsid production in all cell types. There was little evidence that the absence of p53 in the p53KO cells (or the presence of a small amount of p53 in the WTG cells) affected capsid production (e.g. in EXP 2 p53KO cell sections contained ~two-fold more capsids than LOX cell sections). While the decrease in capsid production in the p53KO cell sections compared to LOX was statistically significant, an ~1.6-fold (0.5–2.1 X) decrease in capsid production could not account for the ~25-fold decrease in functional virions shed by these cells. The capsids present within each nucleus section were also enumerated by type (A, B or C) and no statistically significant differences were observed between the p53KOs and LOX (or another wt fibroblast line)(Supplemental Figure 1). In addition, there were no statistically significant differences between cell type sections in the numbers of capsids found directly adjacent to the inner and outer nuclear membranes or immediately between them, i.e. trafficking through the peripheral perinuclear space (as defined in (Buser et al., 2007)) (Supplemental Figure 2).
Figure 2.

Quantitation of capsid production in the nucleus in wt cell sections compared to p53KO cell sections. TEM micrographs of infected cell sections showed RCs with numerous HCMV capsids at 120 h pi. Cross sections of entire cell sections were captured at a magnification of 1,700X (A, C and E). Examples of nuclear capsids present in the boxed areas in A, C and E as visualized in images captured at 5,000x are shown for LOX (panel B), p53KO (panel D) and WTG (panel F). As necessary, images captured at higher magnification in this and all subsequent figures were stitched together to form a mosaic image of the individual captures from an individual cell section. Capsids were enumerated using these mosaic images. Statistical tests for differences among the EM sections of the LOX, p53KO and WTG cells (all graphs in Figures 2–5 and Supplemental Figures) were conducted using the Kruskal-Wallace test as described in Materials and Methods. If the overall tests were found statistically significant (p-values reported in the figure legends), then pairwise comparisons among the three cell lines were conducted using the same test (and reported within the figures). For all analyses, a p-value of 0.05 or less was considered statistically different. (G) Quantitation of capsids in the nucleus in wt LOX cell sections compared to p53KO and WTG cell sections. Each symbol represents an individual cell section in an experiment in this, and all following figures. Experiment 1 (EXP 1) used LOX cell sections (n=20) compared to p53KO cell sections (n=10). Experiment 2 (EXP 2) used LOX cell sections (n=11) compared to p53KO cell sections (n=11) and WTG cell sections (n=19). Experiment 3 (EXP 3) used LOX cell sections (n=10) compared to p53KO cell sections (n=12) and WTG cell sections (n=11). Median capsid counts for each cell type in a given experiment are marked with a horizontal bar in this, and all following figures. Kruskal-Wallace overall test, p=0.0025.
Analysis of our data and of the existing literature brought to our attention an additional portion of the nuclear capsid population, which should be included in the actively trafficking category. This population included all capsids within infoldings of the inner nuclear membrane (IINMs), structures that have been observed by others to contain primary-enveloped capsids (Buser et al., 2007; Dal Monte et al., 2002; Gilloteaux and Nassiri, 2000; Papadimitriou et al., 1984; Ruebner et al., 1964; Severi et al., 1988; Villinger et al., 2015). Buser et al. specifically suggested that these tubular infolding structures were the sites where the majority of trafficking capsids in the cytomegaloviruses exited the nucleus (Buser et al., 2007). These structures (pictured in Figure 3A–F) were present in a greater proportion of the LOX cell sections (a mean of ~70%) than in the p53KO cell sections (~40%)(p= 0.044). Notably, ~60% of WTG cell sections contained IINMs (Figure 3G). As seen by others (Buser et al., 2007; Villinger et al., 2015) and confirmed by the secretion of NIEPs from herpesvirus infected cells (as reviewed in Mocarski, 2001; Tandon et al., 2015), all three types of capsids were seen within the IINMs (Figures 3B, D and F). Interestingly, in all cell types the IINMs were all almost entirely full of capsids (as pictured in Figure 3B, D and F and (Buser et al., 2007; Villinger et al., 2015)), with very little unoccupied space within them. This was true of the largest and smallest IINMs.
Figure 3.

Identification of capsids exiting the nucleus via infoldings of the inner nuclear membrane (IINMs). (A, C and E) Whole cell sections were captured at a magnification of 1,700X. (B, D and F) Examples of nuclear capsids present within the boxed areas in 2A, C and E, as visualized in images captured at 5,000x. In Figure 3G, a χ2 test, implemented in SAS, was used to determine differences between LOX, p53KO and WTG in the proportion of cell sections with IINMs. In this analysis, an overall test was performed first, followed by pairwise analysis. (G) Percentage of cell sections containing IINMs in LOX, p53KO and WTG. Error bars represent one standard error calculated from three (LOX and p53KO) or two (WTG) experiments. χ2 overall test, p=0.0509. As this overall test was just outside statistical significance, pairwise comparisons were still reported. (H) Number of IINMs per cell section. All cell sections in each experiment are represented whether they contain IINMs or not. Kruskal-Wallace overall test, p=0.13 (not significant). (I) Total number of capsids found within IINMs per cell section for LOX, p53KO and WTG. Kruskal-Wallace overall test, p=0.016. (J) The number of capsids in IINMs was divided by the number of IINMs in that cell section to determine the mean content/size of each cell section’s IINMs. Kruskal-Wallace overall test, p<0.0001. Nu = nucleus; Cyto = cytoplasm.
The number of IINMs per cell section was enumerated (Figure 3H). In some instances due to the relatively small numbers of IINMs, large distribution, and considerable number of cell sections in all cell types containing zero IINMs (largest in p53KOs), these comparisons were less than statistically significant. The median numbers of IINMs were higher in LOX and WTG than in p53KO cell sections, but did not reach statistical significance. The median number of total capsids found within IINMs per cell section in each cell type was also compared (Figure 3I). IINMs within the LOX cell sections contained statistically significantly more capsids (p=0.0155). LOX cell sections contained ~six-fold (4.5–7.4 X) more capsids within IINMs than p53KO cell sections. The quantity of capsids within IINMs in p53KO and WTG cell sections was not statistically significantly different. The number of capsids in IINMs was divided by the number of IINMs in that cell section to determine the mean content/size of each cell section’s IINMs (Figure 3J). By this metric the IINMs in LOX compared to p53KO cell sections were statistically significantly larger (p=0.0016). The median IINM in a LOX cell section was ~4.75-fold (4.0–5.5 X) larger than its p53KO counterpart. The IINMs were slightly larger in WTG compared to p53KO cell sections in one experiment and approximately the same in a second, a difference that was statistically significant. Although not all statistically significant, the reintroduction of p53 correlated with a relatively large increase in the proportion of cell sections containing IINMs (~50%, not statistically significant), and with smaller increases in IINMs per cell section (~30%, not statistically significant), the total number of capsids in IINMs per cell section (~15%, not statistically significant), and the size of IINMs per cell section (incalculable percent change, but statistically significant), which together demonstrated a modest recovery of IINM formation and, presumably, function.
Capsids were scarce in the cytoplasm of p53KO cells
Large numbers of capsids were found in the cytoplasm in the wt LOX cell sections (Figure 4A and B). Cytoplasmic capsids are indicated by the arrow heads in Figure 4B. There were many fewer capsids in the cytoplasm of p53KO cell sections (Figures 4C and D; notice the lack of arrow heads). At 120 h pi, noticeably more capsids were present in the cytoplasm of the WTG cell sections than in the p53KOs (Figure 4E and F; compare arrow heads in D and F). By 120 h pi, the median number of capsids found in the cytoplasm of infected LOX cell sections was ~65 (Figure 4G). In contrast, a median of approximately ~12 capsids were found in the cytoplasm of infected p53KO cell sections. This difference was highly statistically significant (p<0.0001). The ~5.2- fold (3.4–6.9 X) differential was very similar in scale to the differential of the median number of total capsids found within IINMs (~six-fold) between LOX and p53KO cell sections. The median number of capsids in the cytoplasm of the WTG cell sections was ~27, and was statistically significantly increased over that found in the KO cell sections (p=0.0004). There were ~2.1-fold (1.8–2.5 X) more capsids in the cytoplasm of WTG compared to p53KO cell sections, indicating that reintroduction of p53 to the cellular environment enhanced capsid egress from the nucleus.
Figure 4.

Quantitation of cytoplasmic capsids. Whole cell sections were captured at a magnification of 1,700X (A, C, and E). Images captured at higher magnification (2500x) were used to quantitate cytoplasmic capsids. Arrow heads in B, D and F show examples of cytoplasmic capsids present within the boxed areas in A, C and E, as visualized in 2,500x images. (G) Quantitation of capsids in the cytoplasm in LOX, p53KO and WTG cell sections. Kruskal-Wallace overall test, p<0.0001.
Fewer of the capsids present in the cytoplasm of p53KO cells were enveloped
Secondary envelopment of capsids was determined by the presence of a void ringing the tegumented particle (Figure 5 – B, D and F show examples of secondary enveloped capsids present within the boxed areas in A, C and E, as visualized in 2,500x images). Such voids, similar to those seen in (Granzow et al., 2001; Grefte et al., 1993; Tandon et al., 2015), were identifiable around capsids and dense bodies and particularly apparent when these particles were adjacent to non-enveloped viral inclusions. Higher magnification images (6500x in 5G and 5000x in 5H) were examined to verify the identification of the membranes (see arrows in Figures 5G and H). The proportion and quantity of secondary-enveloped capsids in the cytoplasm was higher in the LOX compared to p53KO cell sections (Figure 5I). The mean ratio of total cytoplasmic capsids to re-enveloped cytoplasmic capsids (Figure 4G medians compared to Figure 5I medians) in LOX and WTG cell sections was approximately two to one (2:1), whereas this ratio for p53KO cell sections was approximately seven to one (7:1). A mean of ~16-fold (7.7–24 X) more re-enveloped capsids were present in the cytoplasm of the LOX cell sections than the p53KO cell sections. We believe this ~16-fold differential was representative of the secretion of functional virus, since there were no observable differences in the distribution of capsid types in the nucleus. The ~16-fold differential largely recapitulated the ~25-fold LOX/p53KO titer differential. In WTG cell sections the number of secondary-enveloped capsids found in the cytoplasm was statistically significantly (p=0.0001) increased compared to p53KO cell sections. The ~seven-fold (2.1–12 X) increase in WTG over the p53KO cell sections largely replicated the increase in titers of WTG compared to the p53KO cell sections observed at late times pi (Casavant et al., 2006).
Figure 5.

Identification and quantitation of enveloped cytoplasmic capsids. (A–F) Whole cell sections were captured at a magnification of 1,700X (A, C, and E). Images captured at higher magnification (2500x) were used to quantitate enveloped and non-enveloped capsids. B, D and F show examples of enveloped (arrows) and non-enveloped (arrow heads) capsids present within the boxed areas in A, C and E, as visualized in 2,500x images. (G and H) LOX (G) and WTG (H) cell sections captured at higher magnification (6500x and 5000x, respectively), displayed secondary envelopes more distinctly (arrows). (I) Quantitation of enveloped capsids in the cytoplasm in LOX, p53KO, and WTG cell sections. Kruskal-Wallace overall test, p<0.0001.
Membrane synthesis was compromised in the absence of p53
The reduced secondary-envelopment in the p53KO cell sections indicated a potential reduction in either lipid or membrane synthesis or in utilization of lipid stores in the re-envelopment of capsids, which is necessary for final virion assembly. Recent literature has indicated that the formation of very long chain fatty acids (VLCAs) may be an important step in the secondary envelopment of virions and that the neutral lipids stored within existing lipid droplets provide the starting material for this synthesis and are thus depleted in cells post-infection (Koyuncu et al., 2013). Cells were stained with the histochemical stain Sudan IV, which directly stains neutral lipids, to determine whether the neutral lipids being made by the cells were consumed in the production of VLCAs as had been previously demonstrated (Koyuncu et al., 2013). Sudan IV staining revealed lipid droplets present in the cytoplasm of most uninfected LOX and p53KO cells (Figure 6A). These droplets were depleted in the large majority of infected LOX cells by 120 h pi (Figure 6B). In contrast, most infected p53KO cells remained full of lipid droplets. Quantitation of cells containing lipid droplets in mock- and virus-infected cells found a marked increase in consumption of neutral lipids in LOX compared to p53KO-infected cells, although this did not quite reach the level of statistical significance (p=0.062) (Figure 6C). This suggested that, while lipid synthesis functioned at near wt levels in the absence of p53, membrane synthesis, from their neutral lipid precursors, was under utilized in the p53KO cells.
Figure 6.

Consumption of lipid precursors was compromised in the absence of p53. Cells stained with Sudan IV. (A) Uninfected LOX and p53KO cells were largely full of lipid droplets. (B) Lipid droplets were depleted in infected LOX cells, but not in p53KO cells. (C) Quantitation of the percentage of cells in each cell type containing lipid droplets. Error bars represent one standard error calculated from four experiments. Analysis of variance for differences among infected and uninfected LOX and p53KO cells was found to be statistically significant (p=0.034), therefore Tukey’s Studentized Range Test was used for pairwise comparisons of the transformed percentages (values reported in (C)).
DISCUSSION
In our previous work, we observed that at 120 h pi virus titers of wt LOX cells were ~25-fold higher than p53KO cells and that virus entry and IE gene expression were essentially normal (Casavant et al., 2006). We have extended those studies to examine the cellular ultrastructure during the HCMV life cycle to determine if the absence of p53 affected specific parameters that could be associated with reduced functional virion production. The data presented here demonstrate that in the absence of p53 the failure of nucleocapsids to properly exit the nucleus was one of two substantial blockades to functional virus production. In particular, in the absence of p53, typical IINM formation was compromised. The paucity of these membrane-derived structures in the p53KO cell sections was paralleled by inefficient secondary envelopment of the capsids able to reach the cytoplasm. In total, LOX cell sections contained ~16-fold (7.7–24 X) more secondary enveloped capsids than p53KO cell sections. Re-introduction of p53 into the WTG cells recovered the number of re-enveloped capsids in cell sections by ~seven-fold (2.1–12 X) compared to p53KO cell sections, substantially recapitulating the recovery in titers found in our earlier studies (Casavant et al., 2006).
Our earlier titer data could not distinguish whether the entire population of p53KO cells produced low levels of virus or if a minority of the population produced wt-like titers and the majority little or no virus progeny. Standard plaque-forming assays found that ~30% of the p53KO cells expressed enough functional virions to form readily identifiable plaques (Figure 1A). If this one third of the population of cells produced wt-like levels of virus why were titers reduced by ~25-fold? More sensitive fluorescence-based focus forming assays allowed us to determine that the large majority of p53KO cells secreted virions (Figure 1B–D), however all p53KO cells appeared to produce less virus than their wt counterparts, with most producing extremely small amounts. This indicated that the absence of p53 led to inefficient secretion of functional virions. Where then were the blockades that could explain the decrease in functional virion release? Did the absence of p53 reduce capsid production or DNA packaging efficiency? Quantification of nuclear capsids in the p53KO cell sections found them decreased by a mean of only ~1.6-fold (0.5–2.1 X) compared to LOX sections (Figure 2G). Additionally, the ratio of conversion from B to C capsids at 120 h pi was very similar in LOX and p53KO populations (Supplemental Figure 1C), indicating equivalently efficient DNA packaging in the absence of p53 by this late time pi. The p53KO cells were not substantially compromised in their capacity to manufacture or package virus capsids.
When quantitative analysis of capsids in the cytoplasm was performed, ~5.2-fold (3.4–6.9 X) fewer of these particles in p53KO compared to LOX cell sections were found (Figure 4G). Each EM section served as a snapshot of activity at a given moment. If all systems were functioning properly, the presence of near wt-like numbers of capsids within the nucleus of a population of p53KO cells by 120 h pi would have been expected to elicit wt-like nuclear membrane transit, which has been suggested to be a rapid process (Buser et al., 2007). If nuclear egress were not inhibited we would have expected analysis of the EM snapshots to produce more similar medians of cytoplasmic capsid quantities in the two cell type populations by 120 h pi. We found that in the absence of p53, formation of infoldings of the INM was reduced. Our parallel protein localization study showed that the lamin structure in p53KO cells was less disrupted, likely due to failure of the nuclear egress complex components to assemble properly at the INM (Kuan et al., submitted).
Infoldings (or invaginations) of the INM have been reported in the cytomegaloviruses to be the site of the most prolific capsid nuclear exit (Buser et al., 2007; Dal Monte et al., 2002; Gilloteaux and Nassiri, 2000; Papadimitriou et al., 1984; Ruebner et al., 1964; Severi et al., 1988; Villinger et al., 2015). We observed that only ~40% of the p53KO cell sections contained IINMs (Figures 3G). It seems likely that this group was comprised of the similarly sized sub-populations noted previously, namely the ~1/3 of the p53KO population we had initially observed trafficking pp65 normally into the cytoplasm, the ~30% cells found expressing virus by conventional plaque assay (i.e.- the cells with the highest level of secretion), and the ~30% of cells found to infect modest numbers of neighboring cells in focus forming assays. The literature regarding the use of extended tubes or IINMs for trafficking of capsids is most abundant for the beta herpesviruses (Villinger et al., 2015) (Buser et al., 2007; Dal Monte et al., 2002; Gilloteaux and Nassiri, 2000; Papadimitriou et al., 1984; Ruebner et al., 1964; Severi et al., 1988), although there have also been indications of their presence in the alpha herpesvirus PrV (Hagen et al., 2015; Klupp et al., 2007). Much of the literature on HSV and PrV (both alpha herpesviruses) indicate that these viruses traffic directly through a remodeled INM without the formation of tubules (Fuchs et al., 2002; Klupp et al., 2000; Reynolds et al., 2001; Reynolds et al., 2002; Roller et al., 2000). In addition, it has been shown that the UL50/UL53 equivalents in these viruses are capable of distinct deformation of membranes, indicating that these components of the NEC can alone affect the primary envelopment process (Bigalke et al., 2014; Hagen et al., 2015; Klupp et al., 2007; Lorenz et al., 2015) and references within (Lye et al., 2015). Despite this possible difference, all the herpesviruses require remodeling of the lamina and utilize components of the NEC, and perhaps other cellular components, to accomplish this task (Camozzi et al., 2008; Dal Monte et al., 2002; Farina et al., 2005; Fuchs et al., 2002; Gonnella et al., 2005; Hamirally et al., 2009; Krosky et al., 2003; Marschall et al., 2005; Milbradt et al., 2007; Milbradt et al., 2009; Milbradt et al., 2014; Milbradt et al., 2010; Miller et al., 2010; Reim et al., 2013; Reynolds et al., 2001; Reynolds et al., 2002; Sam et al., 2009; Sharma et al., 2014). Lacking normal formation of the tubules, HCMV capsid trafficking through the INM became, at best, exceedingly inefficient and similar to the trafficking patterns reported for UL97, UL50 and UL53 knockout viruses (Azzeh et al., 2006; Krosky et al., 2003; Prichard et al., 2005; Prichard et al., 1999; Sharma et al., 2014; Wolf et al., 2001). It was noteworthy that the IINMs of all cell types were almost entirely full of capsids (as shown in Figure 3B, D and F). This suggested that these tubule-like structures formed and expanded in size as the result of the entrance of capsids into these extensions of the perinuclear space.
As mentioned above, approximately 5.2-fold (3.4–6.9 X) fewer capsids were identified in the cytoplasm of p53KO cell sections compared to LOX cell sections. Egress from the nucleus was substantially inhibited in the absence of cellular p53, but not to the degree of the ~25-fold reduced titers. The relative dearth of IINMs in the p53KO cell sections suggested that perhaps production of new membranes and/or utilization of available lipid components overall was less efficient in the p53KO cells. Re-enveloped cytoplasmic capsids were ~16-fold (7.7–24 X) more common in LOX cell sections compared to p53KO cell sections. It appeared that p53’s absence correlated not only with inhibited virus-mediated IINM formation within the nucleus and the enhanced capsid egress associated with these structures, but also compromised cytoplasmic capsids’ propensity for re-envelopment. The lack of utilization of the lipid found within cytoplasmic lipid droplets seen in the p53KO cells indicated that, although the p53KO cells were capable of synthesizing lipid precursors and storing them as neutral lipids in these droplets, in the absence of p53, these lipids were not incorporated into new membrane as efficiently. Koyuncu and colleagues (Koyuncu et al., 2013) indicate that these neutral lipids serve as the building blocks for the production of VLCAs within HCMV-infected cells and that the enzymes involved in converting these lipid droplet constituents into VLCAs are upregulated during infection. They also predicted a role for cytosolic carbonic anhydrase 7, an enzyme needed to make the substrates for fatty acid elongases. Intriguingly, both fatty acid elongase 1 and cytosolic carbonic anhydrase 7 have p53 binding sites within their promoters (Genecards information). Perhaps the absence of p53 within these cells prevents upregulation of transcription needed to produce the increased levels of these enzymes necessary for efficient conversion of neutral lipids to VLCAs. Future experiments will focus on determining the mechanism responsible for this low level of secondary envelopment and whether it is directly related to the absence of p53.
Reintroduction of p53 into the p53KO cellular environment recovered nearly all the stages of the viral life cycle examined, including the prevalence of IINMs in the WTG population. However, three exceptions were only minor increases to the prevalence of IINMs (~1.5X), the number of IINMs formed (~1.25X), and the total number of capsids in IINMs (~1.15X) (Figure 3H, I and J) in the WTG cell sections. The lack of statistical significance to the increases noted above in WTG compared to p53KO cell sections renders any combination of these parameters purely speculative. However, together, these small increases could account for the modest, but statistically significant, ~2.1-fold (3.4–6.9 X) increase in capsids trafficked into the cytoplasm of WTG cell sections.
Reintroducing a very small amount of p53 allowed increased trafficking of nucleocapsids into the cytoplasm, but was not sufficient to fully reconstitute the formation of IINMs to wt numbers, sizes or trafficking capabilities. This suggested that an increased supply of p53 was needed to accomplish IINM synthesis; a hypothesis examined in the companion paper demonstrating that cells’ levels of p53 expression was highly correlative with these parameters. The ~16-fold (7.7–24 X) reduction in re-enveloped cytoplasmic capsids in p53KO cell sections compared to LOX cell sections and the ~6-fold (2.1–12 X) recovery of WTG cell sections compared to their parental knockout line, largely reconstituted our initial virus titer data and we believe, combined with our other findings, suggests a critical role for p53 in the IINM-mediated egress of HCMV capsids from their nuclear factory and the subsequent re-envelopment of those particles able to transit to their cytoplasmic distribution hub.
Supplementary Material
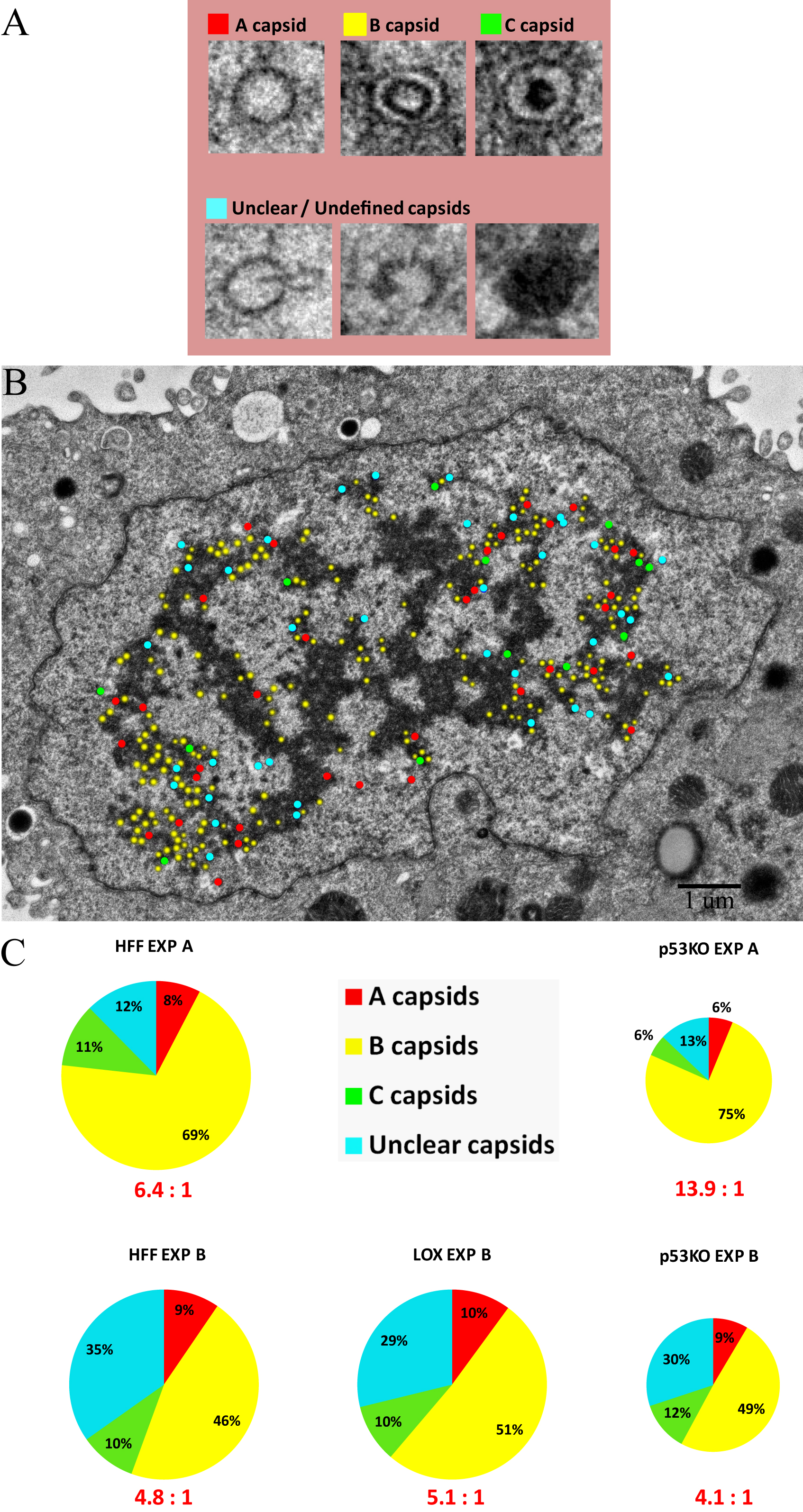
Supplemental Figure 1. Identification and quantitation of capsid types in wt fibroblast cell sections (HFF and LOX) compared to p53KO cell sections. (A) Examples of A, B, C and U (Unclear) capsid types as identified. Capsids were labeled red, yellow, green, and blue, respectively. (B) Example of capsid type analysis. 5,000X magnification was used. Note mosaic images were used to quantitate capsids as described in Figure 2. (C) Quantitation of A, B, C and U capsids in HFFs and LOX cell sections compared to p53KO cell sections. The size of each pie chart represents the medians of the total number of capsids found in the nucleus (see Figure 2G). The ratios below each graph represent the mean B:C ratio for a given cell type in an experiment, determined by dividing the number of B capsids by the number of C capsids for each cell section within that experiment. Kruskal-Wallace overall test, p=0.23.
{kind=link}
Supplemental Figure 2. Quantitation of capsids moving through the nuclear membrane. Whole cell sections were captured at a magnification of 1,700X (A and C). Images captured at higher magnification (5000x) were used to quantitate capsids trafficking into and out of the perinuclear space. B and D show examples of trafficking capsids present in the boxed areas in A and C in LOX cell sections at 120 h pi as visualized in the 5000x images. Perinuclear capsids budded through the inner nuclear membrane (panel B; arrow head). Perinuclear capsids were also located within the perinuclear space (panel B; arrow) or immediately adjacent to the outer nuclear membrane (panel D; arrow). Nu = nucleus; Cyto = cytoplasm in all images. (E) Quantitation of capsids in the perinuclear space in all cell types. Kruskal-Wallace overall test, p=0.197.
{kind=link}
Highlights.
The majority of p53KO cells release fewer functional virions than wt cells.
Nucleocapsids do not efficiently exit the nucleus in p53KO cells.
Infoldings of the inner nuclear membrane are not efficiently formed in p53KO cells.
Cytoplasmic capsids are not efficiently re-enveloped in p53KO cells.
Reintroduction of p53 largely ameliorates these phenotypes.
Acknowledgments
FUNDING INFORMATION
This work was supported by NIH grants RO1 AI051563, COBRE program P20 RR015587 and INBRE program P20 GM103408 to EAF. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank Dr. John Sedivy (Brown University, USA) for providing the p53KO fibroblasts and LOX cells, Dr. Chris Davitt and the WSU Franchesci Microscopy Center for TEM support and advice and Drs. Jennifer Watts, Anamaria Zavala and Amit Kulkarni for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler B, Sinzger C. Cytomegalovirus Interstrain Variance in Cell Type Tropism. In: Reddehase MJ, Lemmermann NAW, editors. Cytomegaloviruses: From Molecular Pathogenesis to Intervention. Caister Academic Press; Norfolk, UK: 2013. pp. 297–321. [Google Scholar]
- AuCoin DP, Smith GB, Meiering CD, Mocarski ES. Betaherpesvirus-conserved cytomegalovirus tegument protein ppUL32 (pp150) controls cytoplasmic events during virion maturation. J Virol. 2006;80:8199–8210. doi: 10.1128/JVI.00457-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzeh M, Honigman A, Taraboulos A, Rouvinski A, Wolf DG. Structural changes in human cytomegalovirus cytoplasmic assembly sites in the absence of UL97 kinase activity. Virology. 2006;354:69–79. doi: 10.1016/j.virol.2006.05.037. [DOI] [PubMed] [Google Scholar]
- Baxter MK, Gibson W. Cytomegalovirus basic phosphoprotein (pUL32) binds to capsids in vitro through its amino one-third. J Virol. 2001;75:6865–6873. doi: 10.1128/JVI.75.15.6865-6873.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigalke JM, Heuser T, Nicastro D, Heldwein EE. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun. 2014;5:4131. doi: 10.1038/ncomms5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle KA, Compton T. Receptor-binding properties of a soluble form of human cytomegalovirus glycoprotein B. J Virol. 1998;72:1826–1833. doi: 10.1128/jvi.72.3.1826-1833.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnahan WA, Boldogh I, Thompson EA, Albrecht T. Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology. 1996;224:150–160. doi: 10.1006/viro.1996.0516. [DOI] [PubMed] [Google Scholar]
- Bresnahan WA, Hultman GE, Shenk T. Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. Journal of virology. 2000;74:10816–10818. doi: 10.1128/jvi.74.22.10816-10818.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchkovich NJ, Maguire TG, Alwine JC. Role of the endoplasmic reticulum chaperone BiP, SUN domain proteins, and dynein in altering nuclear morphology during human cytomegalovirus infection. J Virol. 2010;84:7005–7017. doi: 10.1128/JVI.00719-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchkovich NJ, Maguire TG, Paton AW, Paton JC, Alwine JC. The endoplasmic reticulum chaperone BiP/GRP78 is important in the structure and function of the human cytomegalovirus assembly compartment. J Virol. 2009;83:11421–11428. doi: 10.1128/JVI.00762-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Bunz F, Fauth C, Speicher MR, Dutriaux A, Sedivy JM, Kinzler KW, Vogelstein B, Lengauer C. Targeted inactivation of p53 in human cells does not result in aneuploidy. Cancer research. 2002;62:1129–1133. [PubMed] [Google Scholar]
- Buser C, Walther P, Mertens T, Michel D. Cytomegalovirus primary envelopment occurs at large infoldings of the inner nuclear membrane. J Virol. 2007;81:3042–3048. doi: 10.1128/JVI.01564-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camozzi D, Pignatelli S, Valvo C, Lattanzi G, Capanni C, Dal Monte P, Landini MP. Remodelling of the nuclear lamina during human cytomegalovirus infection: role of the viral proteins pUL50 and pUL53. J Gen Virol. 2008;89:731–740. doi: 10.1099/vir.0.83377-0. [DOI] [PubMed] [Google Scholar]
- Casavant NC, Luo MH, Rosenke K, Winegardner T, Zurawska A, Fortunato EA. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J Virol. 2006;80:8390–8401. doi: 10.1128/JVI.00505-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo JP, Yurochko AD, Kowalik TF. Role of human cytomegalovirus immediate-early proteins in cell growth control. J Virol. 2000;74:8028–8037. doi: 10.1128/jvi.74.17.8028-8037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Knutson E, Kurosky A, Albrecht T. Degradation of p21cip1 in cells productively infected with human cytomegalovirus. J Virol. 2001;75:3613–3625. doi: 10.1128/JVI.75.8.3613-3625.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colberg-Poley AM, Williamson CD. Intracellular Sorting and Trafficking of Cytomegalovirus Proteins during Permissive Infection. In: Reddehase MJ, Lemmermann NAW, editors. Cytomegaloviruses: From Molecular Pathogenesis to Intervention. Caister Academic Press; Norfolk, UK: 2013. pp. 196–229. [Google Scholar]
- Compton T. Receptors and immune sensors: the complex entry path of human cytomegalovirus. Trends Cell Biol. 2004;14:5–8. doi: 10.1016/j.tcb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Compton T, Nowlin DM, Cooper NR. Initiation of human cytomegalovirus infection requires initial interaction with cell surface heparan sulfate. Virology. 1993;193:834–841. doi: 10.1006/viro.1993.1192. [DOI] [PubMed] [Google Scholar]
- Dai X, Yu X, Gong H, Jiang X, Abenes G, Liu H, Shivakoti S, Britt WJ, Zhu H, Liu F, Zhou ZH. The smallest capsid protein mediates binding of the essential tegument protein pp150 to stabilize DNA-containing capsids in human cytomegalovirus. PLoS Pathog. 2013;9:e1003525. doi: 10.1371/journal.ppat.1003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Monte P, Pignatelli S, Zini N, Maraldi NM, Perret E, Prevost MC, Landini MP. Analysis of intracellular and intraviral localization of the human cytomegalovirus UL53 protein. J Gen Virol. 2002;83:1005–1012. doi: 10.1099/0022-1317-83-5-1005. [DOI] [PubMed] [Google Scholar]
- Das S, Pellett PE. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J Virol. 2011;85:5864–5879. doi: 10.1128/JVI.00155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Vasanji A, Pellett PE. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol. 2007;81:11861–11869. doi: 10.1128/JVI.01077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina A, Feederle R, Raffa S, Gonnella R, Santarelli R, Frati L, Angeloni A, Torrisi MR, Faggioni A, Delecluse HJ. BFRF1 of Epstein-Barr virus is essential for efficient primary viral envelopment and egress. J Virol. 2005;79:3703–3712. doi: 10.1128/JVI.79.6.3703-3712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feire AL, Compton T. Virus Entry and Activation of Innate Defence. In: Reddehase MJ, Lemmermann NAW, editors. Cytomegaloviruses: From Molecular Pathogenesis to Intervention. Caister Academic Press; Norfolk, UK: 2013. pp. 125–140. [Google Scholar]
- Fortunato EA, Spector DH. p53 and RPA are sequestered in viral replication centers in the nuclei of cells infected with human cytomegalovirus. J Virol. 1998;72:2033–2039. doi: 10.1128/jvi.72.3.2033-2039.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J Virol. 2002;76:364–378. doi: 10.1128/JVI.76.1.364-378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson W. Synthesis, structure, and function of cytomegalovirus major nonvirion nuclear protein. UCLA Symp Mol Biol. 1984;21:423–440. [Google Scholar]
- Gilloteaux J, Nassiri MR. Human bone marrow fibroblasts infected by cytomegalovirus: ultrastructural observations. J Submicrosc Cytol Pathol. 2000;32:17–45. [PubMed] [Google Scholar]
- Gonnella R, Farina A, Santarelli R, Raffa S, Feederle R, Bei R, Granato M, Modesti A, Frati L, Delecluse HJ, Torrisi MR, Angeloni A, Faggioni A. Characterization and intracellular localization of the Epstein-Barr virus protein BFLF2: interactions with BFRF1 and with the nuclear lamina. J Virol. 2005;79:3713–3727. doi: 10.1128/JVI.79.6.3713-3727.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzow H, Klupp BG, Fuchs W, Veits J, Osterrieder N, Mettenleiter TC. Egress of alphaherpesviruses: comparative ultrastructural study. Journal of virology. 2001;75:3675–3684. doi: 10.1128/JVI.75.8.3675-3684.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grefte A, Blom N, van der Giessen M, van Son W, The TH. Ultrastructural analysis of circulating cytomegalic cells in patients with active cytomegalovirus infection: evidence for virus production and endothelial origin. J Infect Dis. 1993;168:1110–1118. doi: 10.1093/infdis/168.5.1110. [DOI] [PubMed] [Google Scholar]
- Hagen C, Dent KC, Zeev-Ben-Mordehai T, Grange M, Bosse JB, Whittle C, Klupp BG, Siebert CA, Vasishtan D, Bauerlein FJ, Cheleski J, Werner S, Guttmann P, Rehbein S, Henzler K, Demmerle J, Adler B, Koszinowski U, Schermelleh L, Schneider G, Enquist LW, Plitzko JM, Mettenleiter TC, Grunewald K. Structural Basis of Vesicle Formation at the Inner Nuclear Membrane. Cell. 2015;163:1692–1701. doi: 10.1016/j.cell.2015.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamirally S, Kamil JP, Ndassa-Colday YM, Lin AJ, Jahng WJ, Baek MC, Noton S, Silva LA, Simpson-Holley M, Knipe DM, Golan DE, Marto JA, Coen DM. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 2009;5:e1000275. doi: 10.1371/journal.ppat.1000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannemann H, Rosenke K, O’Dowd JM, Fortunato EA. The presence of p53 influences the expression of multiple human cytomegalovirus genes at early times postinfection. J Virol. 2009;83:4316–4325. doi: 10.1128/JVI.02075-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CH, Chang MD, Tai KY, Yang YT, Wang PS, Chen CJ, Wang YH, Lee SC, Wu CW, Juan LJ. HCMV IE2-mediated inhibition of HAT activity downregulates p53 function. EMBO J. 2004;23:2269–2280. doi: 10.1038/sj.emboj.7600239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indran SV, Ballestas ME, Britt WJ. Bicaudal D1-dependent trafficking of human cytomegalovirus tegument protein pp150 in virus-infected cells. J Virol. 2010;84:3162–3177. doi: 10.1128/JVI.01776-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson MK, Compton T. Human cytomegalovirus glycoprotein B is required for virus entry and cell-to-cell spread but not for virion attachment, assembly, or egress. J Virol. 2009;83:3891–3903. doi: 10.1128/JVI.01251-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Baines JD. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol. 2011;9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- Kalejta RF. Tegument proteins of human cytomegalovirus. Microbiol Mol Biol Rev. 2008;72:249–265. doi: 10.1128/MMBR.00040-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari B, Gehrz R. A human cytomegalovirus glycoprotein complex designated gC-II is a major heparin-binding component of the envelope. J Virol. 1992;66:1761–1764. doi: 10.1128/jvi.66.3.1761-1764.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari B, Gehrz R. Structure, composition and heparin binding properties of a human cytomegalovirus glycoprotein complex designated gC-II. J Gen Virol. 1993;74:255–264. doi: 10.1099/0022-1317-74-2-255. [DOI] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci USA. 2007;104:7241–7246. doi: 10.1073/pnas.0701757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Mettenleiter TC. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J Virol. 2000;74:10063–10073. doi: 10.1128/jvi.74.21.10063-10073.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyuncu E, Purdy JG, Rabinowitz JD, Shenk T. Saturated very long chain fatty acids are required for the production of infectious human cytomegalovirus progeny. PLoS pathogens. 2013;9:e1003333. doi: 10.1371/journal.ppat.1003333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krosky PM, Baek MC, Coen DM. The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J Virol. 2003;77:905–914. doi: 10.1128/JVI.77.2.905-914.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- Lorenz M, Vollmer B, Unsay JD, Klupp BG, Garcia-Saez AJ, Mettenleiter TC, Antonin W. A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. The Journal of biological chemistry. 2015;290:6962–6974. doi: 10.1074/jbc.M114.627521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo MH, Fortunato EA. Long-term infection and shedding of human cytomegalovirus in T98G glioblastoma cells. J Virol. 2007;81:10424–10436. doi: 10.1128/JVI.00866-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo MH, Rosenke K, Czornak K, Fortunato EA. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J Virol. 2007;81:1934–1950. doi: 10.1128/JVI.01670-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye MF, Sharma M, El Omari K, Filman DJ, Schuermann JP, Hogle JM, Coen DM. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. Embo J. 2015;34:2937–2952. doi: 10.15252/embj.201592651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschall M, Marzi A, aus dem Siepen P, Jochmann R, Kalmer M, Auerochs S, Lischka P, Leis M, Stamminger T. Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J Biol Chem. 2005;280:33357–33367. doi: 10.1074/jbc.M502672200. [DOI] [PubMed] [Google Scholar]
- Maruzuru Y, Fujii H, Oyama M, Kozuka-Hata H, Kato A, Kawaguchi Y. Roles of p53 in herpes simplex virus 1 replication. Journal of virology. 2013;87:9323–9332. doi: 10.1128/JVI.01581-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettenleiter TC, Klupp BG, Granzow H. Herpesvirus assembly: an update. Virus Res. 2009;143:222–234. doi: 10.1016/j.virusres.2009.03.018. [DOI] [PubMed] [Google Scholar]
- Milbradt J, Auerochs S, Marschall M. Cytomegaloviral proteins pUL50 and pUL53 are associated with the nuclear lamina and interact with cellular protein kinase C. J Gen Virol. 2007;88:2642–2650. doi: 10.1099/vir.0.82924-0. [DOI] [PubMed] [Google Scholar]
- Milbradt J, Auerochs S, Sticht H, Marschall M. Cytomegaloviral proteins that associate with the nuclear lamina: components of a postulated nuclear egress complex. J Gen Virol. 2009;90:579–590. doi: 10.1099/vir.0.005231-0. [DOI] [PubMed] [Google Scholar]
- Milbradt J, Kraut A, Hutterer C, Sonntag E, Schmeiser C, Ferro M, Wagner S, Lenac T, Claus C, Pinkert S, Hamilton ST, Rawlinson WD, Sticht H, Coute Y, Marschall M. Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol Cell Proteomics. 2014;13:2132–2146. doi: 10.1074/mcp.M113.035782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbradt J, Webel R, Auerochs S, Sticht H, Marschall M. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J Biol Chem. 2010;285:13979–13989. doi: 10.1074/jbc.M109.063628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MS, Furlong WE, Pennell L, Geadah M, Hertel L. RASCAL is a new human cytomegalovirus-encoded protein that localizes to the nuclear lamina and in cytoplasmic vesicles at late times postinfection. J Virol. 2010;84:6483–6496. doi: 10.1128/JVI.02462-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocarski ES, Courcelle CT. Cytomegaloviruses and Their Replication. In: Knippe DM, Howley PM, editors. Fields Virology. 4. Lippincott Williams & Wilkins; 2001. pp. 2629–2673. [Google Scholar]
- Muganda P, Carrasco R, Qian Q. The human cytomegalovirus IE2 86 kDa protein elevates p53 levels and transactivates the p53 promoter in human fibroblasts. Cell Mol Biol (Noisy-le-grand) 1998;44:321–331. [PubMed] [Google Scholar]
- Muganda P, Mendoza O, Hernandez J, Qian Q. Human cytomegalovirus elevates levels of the cellular protein p53 in infected fibroblasts. J Virol. 1994;68:8028–8034. doi: 10.1128/jvi.68.12.8028-8034.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy EA, Streblow DN, Nelson JA, Stinski MF. The human cytomegalovirus IE86 protein can block cell cycle progression after inducing transition into the S phase of permissive cells. J Virol. 2000;74:7108–7118. doi: 10.1128/jvi.74.15.7108-7118.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa-Goto K, Tanaka K, Gibson W, Moriishi E, Miura Y, Kurata T, Irie S, Sata T. Microtubule network facilitates nuclear targeting of human cytomegalovirus capsid. J Virol. 2003;77:8541–8547. doi: 10.1128/JVI.77.15.8541-8547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott RL, Longnecker M. An Introduction to Statistical Methods and Data Analysis. Brooks and Cole; Belmont, CA: 2010. [Google Scholar]
- Papadimitriou JM, Shellam GR, Robertson TA. An ultrastructural investigation of cytomegalovirus replication in murine hepatocytes. J Gen Virol. 1984;65(Pt 11):1979–1990. doi: 10.1099/0022-1317-65-11-1979. [DOI] [PubMed] [Google Scholar]
- Prichard MN, Britt WJ, Daily SL, Hartline CB, Kern ER. Human cytomegalovirus UL97 Kinase is required for the normal intranuclear distribution of pp65 and virion morphogenesis. J Virol. 2005;79:15494–15502. doi: 10.1128/JVI.79.24.15494-15502.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MN, Gao N, Jairath S, Mulamba G, Krosky P, Coen DM, Parker BO, Pari GS. A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J Virol. 1999;73:5663–5670. doi: 10.1128/jvi.73.7.5663-5670.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim NI, Kamil JP, Wang D, Lin A, Sharma M, Ericsson M, Pesola JM, Golan DE, Coen DM. Inactivation of retinoblastoma protein does not overcome the requirement for human cytomegalovirus UL97 in lamina disruption and nuclear egress. J Virol. 2013;87:5019–5027. doi: 10.1128/JVI.00007-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol. 2001;75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol. 2002;76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roller RJ, Zhou Y, Schnetzer R, Ferguson J, DeSalvo D. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J Virol. 2000;74:117–129. doi: 10.1128/jvi.74.1.117-129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenke K, Samuel MA, McDowell ET, Toerne MA, Fortunato EA. An intact sequence-specific DNA-binding domain is required for human cytomegalovirus-mediated sequestration of p53 and may promote in vivo binding to the viral genome during infection. Virology. 2006;348:19–34. doi: 10.1016/j.virol.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Ruebner BH, Miyai K, Slusser RJ, Wedemeyer P, Medearis DN., Jr Mouse Cytomegalovirus Infection. An Electron Microscopic Study of Hepatic Parenchymal Cells. Am J Pathol. 1964;44:799–821. [PMC free article] [PubMed] [Google Scholar]
- Ryckman BJ, Chase MC, Johnson DC. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc Natl Acad Sci USA. 2008a;105:14118–14123. doi: 10.1073/pnas.0804365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol. 2006;80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckman BJ, Rainish BL, Chase MC, Borton JA, Nelson JA, Jarvis MA, Johnson DC. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J Virol. 2008b;82:60–70. doi: 10.1128/JVI.01910-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam MD, Evans BT, Coen DM, Hogle JM. Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J Virol. 2009;83:2996–3006. doi: 10.1128/JVI.02441-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez V, Greis KD, Sztul E, Britt WJ. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J Virol. 2000a;74:975–986. doi: 10.1128/jvi.74.2.975-986.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez V, Sztul E, Britt WJ. Human cytomegalovirus pp28 (UL99) localizes to a cytoplasmic compartment which overlaps the endoplasmic reticulum-golgi-intermediate compartment. J Virol. 2000b;74:3842–3851. doi: 10.1128/jvi.74.8.3842-3851.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarov I, Abady I. The morphogenesis of human cytomegalovirus. Isolation and polypeptide characterization of cytomegalovirions and dense bodies. Virology. 1975;66:464–473. doi: 10.1016/0042-6822(75)90218-4. [DOI] [PubMed] [Google Scholar]
- Savaryn JP, Reitsma JM, Bigley TM, Halligan BD, Qian Z, Yu D, Terhune SS. Human cytomegalovirus pUL29/28 and pUL38 repression of p53-regulated p21CIP1 and caspase 1 promoters during infection. Journal of virology. 2013;87:2463–2474. doi: 10.1128/JVI.01926-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauflinger M, Villinger C, Mertens T, Walther P, von Einem J. Analysis of human cytomegalovirus secondary envelopment by advanced electron microscopy. Cell Microbiol. 2013;15:305–314. doi: 10.1111/cmi.12077. [DOI] [PubMed] [Google Scholar]
- Severi B, Landini MP, Govoni E. Human cytomegalovirus morphogenesis: an ultrastructural study of the late cytoplasmic phases. Arch Virol. 1988;98:51–64. doi: 10.1007/BF01321005. [DOI] [PubMed] [Google Scholar]
- Sharma M, Kamil JP, Coughlin M, Reim NI, Coen DM. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J Virol. 2014;88:249–262. doi: 10.1128/JVI.02358-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir E, Modali R, Huang ES, Leon MB, Shawl F, Finkel T, Epstein SE. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science. 1994;265:391–394. doi: 10.1126/science.8023160. [DOI] [PubMed] [Google Scholar]
- Tamashiro JC, Hock LJ, Spector DH. Construction of a cloned library of the EcoRI fragments from the human cytomegalovirus genome (strain AD169) J Virol. 1982;42:547–557. doi: 10.1128/jvi.42.2.547-557.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon R, Mocarski ES. Cytomegalovirus pUL96 is critical for the stability of pp150-associated nucleocapsids. J Virol. 2011;85:7129–7141. doi: 10.1128/JVI.02549-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon R, Mocarski ES, Conway JF. The A, B, Cs of herpesvirus capsids. Viruses. 2015;7:899–914. doi: 10.3390/v7030899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HL, Kou GH, Chen SC, Wu CW, Lin YS. Human cytomegalovirus immediate-early protein IE2 tethers a transcriptional repression domain to p53. J Biol Chem. 1996;271:3534–3540. [PubMed] [Google Scholar]
- Vanarsdall AL, Ryckman BJ, Chase MC, Johnson DC. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J Virol. 2008;82:11837–11850. doi: 10.1128/JVI.01623-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villinger C, Neusser G, Kranz C, Walther P, Mertens T. 3D Analysis of HCMV Induced-Nuclear Membrane Structures by FIB/SEM Tomography: Insight into an Unprecedented Membrane Morphology. Viruses. 2015;7:5686–5704. doi: 10.3390/v7112900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Shenk T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci USA. 2005;102:18153–18158. doi: 10.1073/pnas.0509201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Hemmer RM, Sedivy JM. Role of p14(ARF) in replicative and induced senescence of human fibroblasts. Mol Cell Biol. 2001;21:6748–6757. doi: 10.1128/MCB.21.20.6748-6757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DG, Courcelle CT, Prichard MN, Mocarski ES. Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation. Proc Natl Acad Sci USA. 2001;98:1895–1900. doi: 10.1073/pnas.98.4.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Shen Y, Shenk T. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol. 1995;69:7960–7970. doi: 10.1128/jvi.69.12.7960-7970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Identification and quantitation of capsid types in wt fibroblast cell sections (HFF and LOX) compared to p53KO cell sections. (A) Examples of A, B, C and U (Unclear) capsid types as identified. Capsids were labeled red, yellow, green, and blue, respectively. (B) Example of capsid type analysis. 5,000X magnification was used. Note mosaic images were used to quantitate capsids as described in Figure 2. (C) Quantitation of A, B, C and U capsids in HFFs and LOX cell sections compared to p53KO cell sections. The size of each pie chart represents the medians of the total number of capsids found in the nucleus (see Figure 2G). The ratios below each graph represent the mean B:C ratio for a given cell type in an experiment, determined by dividing the number of B capsids by the number of C capsids for each cell section within that experiment. Kruskal-Wallace overall test, p=0.23.
Supplemental Figure 2. Quantitation of capsids moving through the nuclear membrane. Whole cell sections were captured at a magnification of 1,700X (A and C). Images captured at higher magnification (5000x) were used to quantitate capsids trafficking into and out of the perinuclear space. B and D show examples of trafficking capsids present in the boxed areas in A and C in LOX cell sections at 120 h pi as visualized in the 5000x images. Perinuclear capsids budded through the inner nuclear membrane (panel B; arrow head). Perinuclear capsids were also located within the perinuclear space (panel B; arrow) or immediately adjacent to the outer nuclear membrane (panel D; arrow). Nu = nucleus; Cyto = cytoplasm in all images. (E) Quantitation of capsids in the perinuclear space in all cell types. Kruskal-Wallace overall test, p=0.197.