

Abstract
Our electron microscopy study found HCMV nuclear capsid egress was significantly reduced in p53 knockout cells (p53KOs), correlating with inhibited formation of infoldings of the inner nuclear membrane (IINMs). Molecular examination of these phenomena has found p53KOs expressed UL97 and phosphorylated lamins, however the lamina failed to remodel. The nuclear egress complex (NEC) protein UL50 was expressed in almost all cells. UL50 re-localized to the inner nuclear membrane (INM) in ~90% of wt cells, but only ~35% of p53KOs. UL53 expression was significantly reduced in p53KOs, and cells lacking UL50 nuclear staining, expressed no UL53. Re-introduction of p53 into p53KOs largely recovered UL53 positivity and UL50 nuclear re-localization. Nuclear rim located UL50/53 puncta, which co-localized with the major capsid protein, were largely absent in p53KOs. We believe these puncta were IINMs. In the absence of p53, UL53 expression was inhibited, disrupting formation of the NEC/IINMs, and reducing functional virion secretion.
Keywords: p53, Human Cytomegalovirus, nuclear egress complex, infoldings of inner nuclear membrane, confocal microscopy, UL50 and UL53
INTRODUCTION
Human Cytomegalovirus (HCMV) is a linear double stranded DNA virus ubiquitous in most human populations. HCMV is an opportunistic pathogen that causes medical problems in immunocompromised individuals such as AIDS patients and transplant recipients (Fornara et al., 2011; Shi et al., 2011; Zhang et al., 1995). Some studies also report that HCMV may contribute to certain types of cancers including malignant glioblastomas, prostate carcinomas, and colorectal cancers (Cobbs et al., 2002; Dziurzynski et al., 2012; Harkins et al., 2002; Samanta et al., 2003; Soderberg-Naucler, 2006). Perhaps most problematic is that HCMV is a major cause of congenital birth defects. Approximately 8,000 infants are negatively affected each year in the United States. Half of these cases have severe neurological defects at birth that include blindness, deafness, mental retardation, microencephaly, and cerebral calcification. The remaining cases manifest sequelae within 1–2 years after birth (Boppana and Britt, 1996; Boppana et al., 1999; Britt and Alford, 1996; Cinque, 1997).
HCMV infection can lead not only to significant negative consequences for its host, but also highly disrupts individual cellular functions including normal cell cycling. Virus-induced re-localization of certain cellular proteins into the viral replication centers (RCs) may, at least in part, be responsible for these disruptions. The RCs are non-membrane bound structures located within the nucleus of infected cells and are the site of viral DNA replication (de Bruyn Kops and Knipe, 1988; Liptak et al., 1996; Lukonis et al., 1997; Lukonis and Weller, 1996; Quinlan et al., 1984; Rixon et al., 1983; Uprichard and Knipe, 1997). One of the tightly RC-associated cellular proteins is the cell cycle regulatory protein p53 (Fortunato and Spector, 1998). Amongst a myriad of functions, p53 is a specific transcription factor that mediates the cellular damage response. Activation of p53 can lead to either p53-mediated cell cycle arrest (presumably for repair of damaged DNA) or apoptosis (as reviewed in Beckerman and Prives, 2010). These p53-mediated responses do not occur during HCMV infection.
We have previously shown that cellular p53 was required for several viral functions to occur at wild type (wt) levels of activity. Rosenke et al. reported 21 viral genes have canonical p53 binding sites (Rosenke et al., 2006). p53 was differentially bound at many of these sites during the first 48 h post infection (h pi). p53 was found to influence viral gene expression, through either direct or indirect interaction, at early times of infection (Hannemann et al., 2009). Most significantly, the presence of p53 in the infected cell was found to be necessary for wt levels of functional virion secretion (Casavant et al., 2006). Secretion of functional virions was reduced by approximately 25-fold at late times pi in knockout cells completely absent wt p53 (p53KOs).
During the normal course of infection, input pp65, the most abundant tegument protein, transits rapidly to the nucleus via its nuclear localization signal (NLS) (Schmolke et al., 1995). Prior to 48 h pi, this protein’s staining pattern is predominantly nuclear, however, after this timepoint, the vast majority of pp65 is found in the cytoplasm (Sanchez et al., 2000; Sanchez et al., 2007) and, therefore, has been used as a proxy for virion nuclear maturation (Britt and Vugler, 1987; Casavant et al., 2006; Sanchez et al., 2002). Undoubtedly at least some pp65 protein is still present in the nucleus at late times pi during infection of a wt cell, where it participates in capsid tegumentation. However, in the p53KOs pp65 remained largely confined to the nucleus throughout infection (Casavant et al., 2006). The absence of pp65 from the cytoplasm suggested that nuclear capsids might also be unable to exit the nucleus. Our parallel electron microscopy study found that the absence of p53 disrupted the efficient egress of HCMV capsids from the nucleus (Kuan et al, submitted).
Understanding of the precise molecular mechanism of HCMV virion nuclear egress is limited. The nuclear pore complex, capable of transporting molecules up to 40 nm (Pante and Kann, 2002), does not have the capacity to transport a ~130 nm diameter capsid. Recently it has been proposed that a Nuclear Egress Complex (NEC) facilitates HCMV virion egress from the nucleus by destabilization of the nuclear lamins (Bjerke and Roller, 2006; Camozzi et al., 2008; Cano-Monreal et al., 2009; Hamirally et al., 2009; Leach and Roller, 2010; Marschall et al., 2005; Milbradt et al., 2010; Muranyi et al., 2002; Park and Baines, 2006; Sharma et al., 2014) and as reviewed in (Johnson and Baines, 2011; Mettenleiter et al., 2009). The nuclear lamina is a dense meshwork that mechanically supports the shape of the nucleus. This meshwork is composed of lamins and their associated membrane proteins (reviewed in Dechat et al., 2008; Goldman et al., 2002). The lamina is dissociated during efficient egress of HCMV capsids from the nucleus.
Many studies have defined two HCMV proteins as key to the NEC, UL50 and UL53. UL50 re-locates from the cytoplasm into the nucleus, and to the nuclear rim in particular, beginning at 48 h pi. How UL50 and its homologues are transported to the nucleus is the subject of speculation (Reynolds et al., 2002; Schmeiser et al., 2013). UL50 lacks its own NLS and it has been suggested that translocation of this protein is accomplished via membrane bound diffusion along the pore membrane (Milbradt et al., 2012; Schmeiser et al., 2013). UL50 directly recruits UL53, and indirectly recruits other potential NEC components, to the nuclear membrane (Camozzi et al., 2008; Lye et al., 2015; Marschall et al., 2005; Milbradt et al., 2009; Milbradt et al., 2010; Miller et al., 2010; Sam et al., 2009; Sharma et al., 2015; Sharma et al., 2014; Sonntag et al., 2016). The C-terminus of UL50 is embedded in the inner nuclear membrane (INM) and secures the NEC to the INM (Camozzi et al., 2008; Milbradt et al., 2007; Milbradt et al., 2012; Milbradt et al., 2009; Milbradt et al., 2014; Miller et al., 2010; Muranyi et al., 2002; Sam et al., 2009; Schmeiser et al., 2013; Sharma et al., 2014; Sonntag et al., 2016). UL50’s principal binding partner in the NEC is UL53. UL53 localizes to the nucleus beginning at 48 h pi, presumably via its nuclear localization signal (NLS) (Milbradt et al., 2012; Schmeiser et al., 2013). From the nuclear rim, UL50 recruits UL53 to that location (Camozzi et al., 2008; Milbradt et al., 2012; Milbradt et al., 2014; Sam et al., 2009; Schmeiser et al., 2013; Sonntag et al., 2016). Several cellular proteins have been identified as potential components of the NEC, including the lamin B receptor (LBR), protein kinase C (PKC), p32, peptidyl-prolyl cis-trans isomerase (pin1), and emerin (Camozzi et al., 2008; Hamirally et al., 2009; Krosky et al., 2003; Marschall et al., 2005; Milbradt et al., 2009; Milbradt et al., 2014; Milbradt et al., 2010; Miller et al., 2010; Sam et al., 2009; Sharma et al., 2014). p32 is recruited to the lamina through interaction with the LBR (Milbradt et al., 2009) and in turn recruits UL97 (Marschall et al., 2005). Of some note, the UL97 gene was found to have a p53 binding site and be bound during the course of infection (Rosenke et al., 2006). UL97 has been found to contribute to phosphorylating lamin A/C (Hamirally et al., 2009; Milbradt et al., 2010) and has very recently been reported to phosphorylate the key NEC components, UL50 and UL53 (Sharma et al., 2015). Phosphorylation of the lamins generates a binding site for pin1, which in turn may promote conformational changes of the lamins, and lead to their localized depletion (Milbradt et al., 2010). Infoldings of the inner nuclear membrane (IINMs), structures that have been observed by several groups to contain enveloped capsids (Buser et al., 2007; Dal Monte et al., 2002; Gilloteaux and Nassiri, 2000; Papadimitriou et al., 1984; Ruebner et al., 1964; Severi et al., 1988; Villinger et al., 2015), have been proposed as the principal site of transit through the nuclear membrane for the CMV family of viruses (Buser et al., 2007; Villinger et al., 2015). These tubule-like structures are reported to facilitate capsid transport into the perinuclear space and subsequently through the outer nuclear membrane.
Our study has focused on isolating which viral and cellular mechanisms failed to allow normal nuclear egress of capsids in the absence of p53. The expression and function of critical viral proteins was examined using a variety of methods. We believe we have isolated a molecular pathway elucidating the role of the viral protein UL53 in promoting wt-like nuclear capsid egress and, ultimately, functional virion production. We believe the cell cycle protein, p53, either directly or indirectly, transactivated the expression of UL53, which was greatly curtailed in p53’s absence. The absence of p53 from the cellular environment precluded efficient formation of the NEC, and their associated IINMs, thereby inhibiting nuclear capsid egress.
MATERIALS AND METHODS
Cells and virus growth
p53KO telomerase-immortalized human fibroblasts, their parental cell line, LOX (Bunz et al., 1998; Wei et al., 2001) (both kind gifts from Dr. John Sedivy, Brown University), and the p53 re-introduced WT clones (Casavant et al., 2006) were maintained in complete medium composed of Dulbecco’s Modified Eagle Media: Nutrient Mixture F-12 (Ham) 1:1 (DMEM/F-12) supplemented with 10% heat-inactivated fetal bovine serum (FBS), L-glutamine (2mM), penicillin (200 U/ml), and streptomycin (200 µg/ml). Cells were grown in incubators maintained at 37°C and 5% CO2. The Towne strain of HCMV was obtained from the ATCC (#VR 977), propagated under standard procedures (Tamashiro et al., 1982), and used at a multiplicity of infection (MOI) of 5 for all experiments. To inhibit viral replication and test for UL97 functionality, cells were infected in the presence of 270 µM Ganciclovir and harvested at the indicated timepoints pi.
Cell cycle synchronization and infection
Confluent cells were synchronized by serum starvation and infected in G0 as previously described (Casavant et al., 2006) to allow for completely synchronous initiation of viral antigen expression. Infection at high MOI ensured that all cells were infected. As was observed previously (Casavant et al., 2006), >90% of all cells (LOX, p53KO and WT clones) were positive for IE1 expression by the 24 h pi timepoint. Cells were then trypsinized and reseeded 1) into plates containing coverslips for immunofluorescence (IF) analysis at a density of 5 × 105 cells per plate and 2) at 1 × 106 cells per plate for Western blot analysis, in complete medium. After 1–2 h to allow for attachment, cells were infected with HCMV Towne supernatant. Supernatant was removed after 4 h and cells were rinsed with PBS to remove any remaining unadsorbed virus, and finally re-fed with complete medium.
Antibodies (Abs)
Primary mouse monoclonal Abs used included anti-lamin A/C (636 [IgG2B]; Santa Cruz Biotechnology), anti-lamin A (133A2 [IgG3]; Abcam), anti-UL97 (IgG2A; a kind gift from Dr. Mark Prichard, University of Alabama School of Medicine, Birmingham)(Gill et al., 2012; Milbradt et al., 2014), anti-UL44 (1202S [IgG1]; Goodwin Institute), anti-UL53 (IgG1; a kind gift from Dr. Stipan Jonjic, University of Rijeka, Croatia)(Milbradt et al., 2014), anti-MCP (a kind gift from Dr. Bill Britt, University of Alabama, Birmingham) (Lai and Britt, 2003; Sanchez et al., 2000), anti-Pan Actin (Ab-5; NeoMarkers) and anti-Flag (9A3 [IgG1]; Cell Signaling). Rabbit Abs used included anti-UL50 (a kind gift from Dr. James Alwine, University of Pennsylvania, Philadelphia) (Buchkovich et al., 2010; Milbradt et al., 2014; Schmeiser et al., 2013), anti-Phospho-Rb (Ser780; 9307; Cell Signaling), anti-HA (C29F4; Cell Signaling), anti-phospho-lamin A/C (Ser22; Cell Signaling) and anti-lamin B1 (Proteintech). Secondary Abs used for IF were goat anti-mouse IgG1 Alexa Fluor 488-coupled Ab (Molecular Probes), goat anti-mouse IgG 2A TRITC-coupled Ab (Southern Biotech), goat anti-mouse IgG2B Alex Fluor 488-coupled Ab (Molecular Probes), goat anti-mouse IgG3 Alex Fluor 488-coupled Ab (Molecular Probes), and donkey anti-rabbit TRITC-coupled Ab (Jackson Immunoresearch). Western blot analysis used sheep anti-mouse secondary Ab (Jackson Immunoresearch) or donkey anti-rabbit secondary Ab (GE Healthcare), both conjugated with horseradish peroxidase.
Immunofluorescence (IF) staining assays
Coverslips were harvested and washed in PBS. In this study a 3% formaldehyde fixation (fix-first) method was used for investigating all proteins (as described in (Luo and Fortunato, 2007; Rosenke et al., 2006), except as noted below. When staining for total lamin A/C, a mix of the two monoclonal Abs listed above was used to obtain the strongest signal. Nuclei were counterstained with Hoechst dye. Note that if a rabbit primary Ab was used, 10% human IgG (20 mg/ml) was used for initial blocking.
Detection of UL50 (either alone or in combination with other proteins) in its anchored state at the nuclear rim was performed using an extraction first (extract-first) methodology as previously described (Cardoso et al., 1993; Lombard and Guarente, 2000; Luo et al., 2007; Marciniak et al., 1998), unless otherwise noted. This methodology was also used to detect lamin A/C and lamin B for confocal analysis. Briefly, coverslips were extracted in a CSK buffer solution {10 mM PIPES [piperazine-N-N-bis (2-ethanesulfonic acid)], 100mM NaCl, 300 mM sucrose, and 3 mM MgCl2} containing 0.5% Triton X-100 (Lombard and Guarente, 2000). Cells were then rinsed in CSK twice and fixed with 3% formaldehyde in PBS (with 0.5 mM MgCl2 and 0.5 mM CaCl2) for 10 min. This extract-first procedure removes proteins that are not attached to the chromatin or scaffolding substructure of the nucleus before fixation, providing an unobstructed view of protein localization within this compartment. This method also extracts proteins from the cytoplasm that are not localized within cytoplasmic organelles, or tightly associated with these membranes, providing exceptionally clear visualization of the nuclear rim. After extraction and fixation, cells were blocked and incubated with primary and secondary Abs as described in (Luo and Fortunato, 2007; Rosenke et al., 2006).
Epifluorescence analysis was performed on a Nikon Eclipse E800 fluorescence microscope equipped with a Nikon DS-Ri1 high-resolution color camera and Nikon NIS Elements Basic Research imaging software. Confocal analysis was performed using one of two microscopes: 1) an Olympus FluoView 1000 confocal microscope with FluoView ASW software or 2) a Nikon/Andor spinning disk confocal microscope with NIS Elements acquisition software. Samples were excited using lasers at 488 nm for Alexafluor 488, 559 nm for TRITC, and 405 nm for Hoechst. Confocal images were analyzed using Imaris software version 8.1.1.
Western blot analysis
Cells were seeded and infected as described above, and harvested over a timecourse. Cells were trypsinized, washed, counted, and prepared as described previously (Kulkarni and Fortunato, 2011; Luo et al., 2007). Equivalent amounts of cell lysates (105 cell equivalents unless otherwise noted) were run on SDS-polyacrylamide gels, and then transferred to a Protran membrane (GE Healthcare Life Sciences). Proteins were blocked in 5% milk in TBS with 0.1% Tween 20, and probed with primary Abs. Membranes were washed extensively and then probed with secondary Abs as listed above. Proteins were visualized with enhanced-chemiluminescence reagents (Pierce SuperSignal West Pico), with the exception of the UL50 and UL53 dilution series blots, which were detected using Advansta Westernbright Quantum. In the case of p53 detection in Supplemental Figure 1 (see below), maximum sensitivity reagent (Advansta Westernbright Quantum) and an extended exposure time were necessary to visualize the protein in the WT clone cells. Although not quantitative, NIH Image J software was used to roughly estimate the level of protein expressed in the virus-infected WT clones (normalizing to the levels of actin in each sample).
Nucleofection
LOX and p53KO nucleofections used 1×106 cells and 10 µg of UL50-HA and/or UL53-FLAG constructs (kind gifts from Dr. Laura Hertel, Children’s Hospital of Oakland Research Institute, CA USA). Nucleofections were carried out in an Amaxa Nucleofector II, using program A-033 for LOX and U-023 for p53KO cells. Basic fibroblast nucleofection solution was used per manufacturer’s instructions (Amaxa Biosystems). Nucleofected cells were incubated overnight and the medium was changed the following day. Coverslips were harvested at 24 and 96 h post nucleofection using fix-first methodology as described in (Luo and Fortunato, 2007; Rosenke et al., 2006).
Background regarding the complete absence of p53 versus low-level expression
The p53KO cells (Bunz et al., 1998; Wei et al., 2001) used in this, and our previous studies (Casavant et al., 2006; Hannemann et al., 2009; Rosenke et al., 2006), were genetically ablated for p53 and expressed absolutely no protein. Earlier characterization of these cells showed that they were genetically stable and not prone to aneuploidy upon continuous passaging (Bunz et al., 2002). These cells were telomerase immortalized, however, in our laboratory they have not been passaged for long periods of time (rarely exceeding 30 and never more than 38 passages from original derivation). In order to ensure consistency, frozen earlier passage stocks were frequently returned to as experiments progressed. We have previously determined that cells from passage 8 or passage 38 display essentially identical phenotypes with respect to viral growth parameters (Casavant et al., 2006; Hannemann et al., 2009; Rosenke et al., 2006). The genetic ablation of p53 undoubtedly introduced additional changes into these cells’ genome; however, these cells were viable, susceptible/permissive to/for HCMV-infection and met many normal benchmarks at early times pi (Casavant et al., 2006). The p53KO cells’ principal variation from a wt viral life cycle was the substantially reduced shedding of infectious virus. The relatively normal viral life cycle in the p53KO cells recommends these cells as an ideal candidate for studying p53’s role in HCMV-infection.
Also described in our previous work (Casavant et al., 2006), p53 was transfected into the p53KO cells to establish several independent p53-expressing stable clones. Four p53-expressing lines (designated WT D–G) were studied extensively. All produced similar, and quite low, levels of p53 (Supplemental Figure 1). Although not quantitative, Image J analysis of the Western blot estimated the level of p53 in the virus-infected WT clone cells at approximately 5% of mock-infected wt-levels of the protein. Whatever compensating mutations existed in the p53KO cells, allowing them to survive in the absence of p53, rendered expression of p53 above very low levels lethal to these cells (Casavant et al., 2006). This was unsurprising, as reintroduction of wt p53 into tumor cells has been documented to cause rapid arrest (as reviewed in (Levine et al., 1991)). All the stable wt p53-expressing lines behaved similarly with respect to all viral parameters tested and returned toward wt in each case. Most relevant, infectious titers from these cell lines at late times pi were only 4–5-fold lower than wt and were increased ~10-fold over their p53KO parental line (Casavant et al., 2006). In sharp contrast selected pools, expressing either of two p53 mutant proteins (the DNA binding domain mutants R273H and H179Q), behaved identically to the p53KO cells, and produced no increases in titers, indicating the presence of a wt protein was necessary to aid in viral growth parameters (Casavant et al., 2006). It should be noted that HSV-1 studies using a different p53KO system (HCT116 cells) also recovered slightly lower than wt titers (~4-fold), even when much higher levels of wt p53 could be reintroduced (Maruzuru et al., 2013).
Statistical Analysis
Analysis of variance for differences among the three different cell types LOX, p53KO and WTG were conducted using SAS (Cary, NC). Because these data are proportions, the values were first transformed using the arcsin of the square roots of each proportion (Ott and Longnecker, 2010). The overall tests were found statistically significant in all cases (p<0.0001). Subsequent pairwise comparisons of the transformed percentages were tested using Tukey’s Studentized Range Test. These are reported in the figures.
RESULTS
Infection-initiated remodeling of the nuclear lamina was much less substantial in p53KO cells
It has been shown that the nuclear lamina is substantially remodeled when HCMV capsid egress occurs (Camozzi et al., 2008; Hamirally et al., 2009; Leigh et al., 2015; Marschall et al., 2005; Milbradt et al., 2010; Sharma et al., 2014). LOX and p53KOs were infected at an MOI of 5 and harvested over a timecourse. Cells were fixed and stained for lamin A/C as described in the Materials and Methods. Epifluorescence analysis of nuclear lamina staining found large regions of gaps/remodeling in both cell types beginning at 24 h pi (Figure 1A; arrows – 120 h pi is shown). Leigh et al have described this as "ruffling and thinning" of the lamina (Leigh et al., 2015). These large gaps/regions of remodeling occurred much less frequently in the p53KO cells. Confocal imaging of lamin A/C-stained cells (using extract-first methodology as described in the Materials and Methods) distinguished much greater detail of the gaps/remodeling of the lamina. When viewed as a single “slice”, the lamin ring was not fully continuous in either mock or virus-infected cells of either cell type (see arrows in Figure 1B inset). The normal lamin staining could be more accurately described as a "string of beads" with recurrent small regions of at least reduced expression at the nuclear rim. We suspect these small gaps could be nuclear pores, which have been shown to lack lamination (reviewed in Goldman et al., 2005). Most notably, in the LOX cells, substantial gaps in the lamina ring (Figure 1B- arrow head), amounting to large fractions of the nuclear circumference, were easily identifiable. Staining of cells for Lamin B with extract-first procedures confirmed that the extract-first procedure did not alter the lamina itself, as no large gaps were observed for this protein (Figure 1B – right hand images).
Figure 1. Lamin rearrangement was induced by HCMV infection.

(A) Immunofluorescent staining displayed extensive remodeling of the lamin A/C ring in infected LOX and p53KO cells (arrows) using 3% formaldehyde fixation. This remodeling was present in only a minority of p53KO cells. A smooth, intact lamin ring was seen in mock- and the majority of infected p53KO cells. (B) Confocal imaging of lamin A/C staining using extract-first methodology. The inset (~13× magnification, mock shown) shows the “string of beads” appearance of lamin A/C (arrows) observed in both mock- and infected LOX and p53KO cells, not easily observable by epifluorescence. This was different than the large gaps in infected LOX cells (arrow head). Right panels show Lamin B1 staining of mock and infected LOX cells to confirm that extract-first methodology does not disrupt the cellular lamin ring. All size bars in IF images represent 5 µm. Nuclei in this and all other images are counterstained with Hoechst. (C) The mean percentage of cells displaying remodeling (ruffling/thinning) in LOX cells compared to p53KO cells over a time course of infection. Error bars represent one standard error calculated from at least two experiments. Significance calculations (for this and all other figures) represent p-values from pairwise comparisons of the transformed percentages using Tukey’s Studentized Range Test as described in Materials and Methods. A p value of less than 0.05 was considered statistically significant.
Cells were scored for the presence of remodeling of the Lamin A/C staining (Figure 1C) as visualized in Figure 1A. The percentage of mock-infected cells with Lamin A/C remodeling was always very low (<8%), and most probably represented cells traversing through mitosis, where the nuclear lamina must be disassembled. In LOX cells the percentage of remodeling steadily increased from a low of 17% at 24 h pi to a high of 71% at 144 h pi. The p53KO cells displayed a similar low of 14% at 24 h pi, but increased to only 33% by 96 h pi and not beyond 41% of the population by 144 h pi. The differential between LOX and p53KO cells at very late times pi was statistically significant.
UL97 was present and functional in the majority of p53KO cells
A functioning viral protein UL97 must be present for normal capsid egress from the nucleus to occur (Azzeh et al., 2006; Hamirally et al., 2009; Krosky et al., 2003; Milbradt et al., 2010; Prichard et al., 2005; Reim et al., 2013; Sharma et al., 2015; Sonntag et al., 2016). UL97 KO viruses fail to traffic capsids efficiently from the nucleus and produce dramatically lower titers (Azzeh et al., 2006; Prichard et al., 1999). Western blot analysis of the steady state level of UL97 found it present in both cell types starting at 24 h pi (Figure 2A). Epifluorescent imaging of UL97 found nuclear localization of the protein in both LOX and p53KO cells beginning at 24 h pi and extending throughout the timecourse (Figure 2B). Enumeration of cells with UL97+ nuclei found no sizeable difference in the number of positive cells at any timepoint (Figure 2C). Only cells with staining primarily in the nucleus are displayed in this graph. An additional 10–20% of each cell type contained equivalent nuclear and cytoplasmic staining. Essentially all cells were UL97 positive.
Figure 2. UL97 was present and functional in p53KO cells.

(A) UL97 timecourse of expression by Western blot analysis. An actin loading control is included in this and all other blots, unless otherwise noted. (B) IF staining at 24 and 120 h pi. (C) Quantitation of UL97 expression in LOX and p53KO cells by IF staining. Only cells with staining primarily in the nucleus are displayed. An additional 10–20% of each cell type showed equivalent nuclear and cytoplasmic staining. Error bars represent one standard error calculated from six experiments. (D) IF staining of p-lamin A/C at 120 h pi and (E) enumeration of percentage of 120 h pi mock- and infected LOX and p53KO cells displaying p-lamin A/C phosphorylation. Error bars represent one standard error calculated from two experiments. (F) Western blot analysis of the steady state level of p-lamin A/C in LOX and p53KO cells over a representative timecourse of infection.
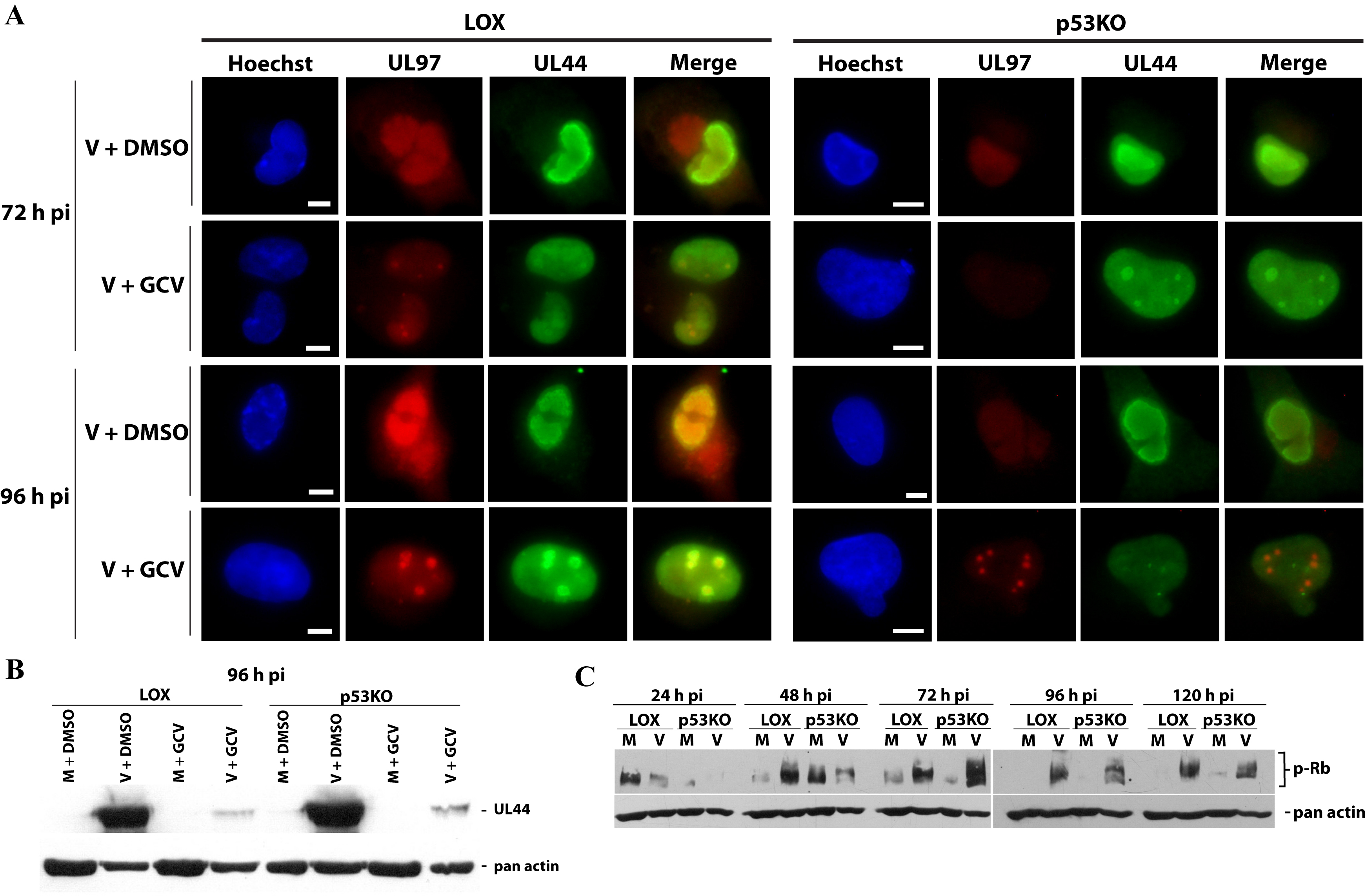
UL97 was present in p53KO cells, but was it functional? Ganciclovir (GCV) is used as an antiviral agent against HCMV infection (reviewed in Biron, 2006). In order for GCV to work effectively, it must be phosphorylated by UL97 (Littler et al., 1992; Sullivan et al., 1992). Phosphorylated GCV is added to the growing viral DNA chain, arresting its extension and terminating viral DNA replication (reviewed in Biron, 2006; McGavin and Goa, 2001). The termination of viral DNA replication can be visualized by examining the viral replication centers (RCs). UL44 is the viral processivity factor for HCMV’s DNA polymerase and therefore highly associated with the RCs. During HCMV infection RCs progress from multiple small foci at early times pi to a single large focus (frequently occupying the majority of the nucleus) at late times pi (Penfold and Mocarski, 1997). IF analysis of late times pi (72 and 96 h pi) found that GCV treatment inhibited formation of advanced stage foci in both cell types (Supplemental Figure 2A). He and colleagues found UL44 expression was suppressed after GCV treatment of wt-infected cells (He et al., 2013). Dr. Mark Prichard has confirmed this behavior (personal communication and data presented at IHW 2010, Salt Lake City). Western blot analysis of UL44 after GCV treatment showed clear inhibition of UL44 expression in both cell types at 96 h pi (Supplemental Figure 2B). UL97 also phosphorylated Rb, one of its nuclear cellular targets, in both cell types (Hume et al., 2008)(Supplemental Figure 2C). This indicated that UL97 was functional for exogenously introduced molecules and one of its normal cellular targets.
Previous studies have shown that HCMV-induced nuclear lamina remodeling only occurs following their phosphorylation, particularly phosphorylation of lamin A/C on serine residue 22 (Ser 22) (Hamirally et al., 2009; Milbradt et al., 2010). Further, reports suggest UL97 is necessary for this phosphorylation to occur (Hamirally et al., 2009; Milbradt et al., 2010; Reim et al., 2013). Phospho-lamin A/C (p-lamin A/C) Ab was used for IF analysis of infected cells. As expected, mock-infected cells displayed very little p-lamin staining (Figure 2D). Infected cells of both types were almost all (>92%) positive for p-lamin A/C staining at 120 h pi (Figure 2E). Western blot analysis of p-lamin A/C found these proteins modified in both cells types (Figure 2F). Total lamin A/C levels did not change over the course of infection in either LOX or p53KO cells (data not shown). UL97 was present in the large majority (~90%) of p53KO cells and functional for both exogenous and endogenous proteins. Further, UL97, perhaps in conjunction with other cellular kinases, was phosphorylating the lamina in the p53KO cells. However, in the absence of p53, despite being phosphorylated, the lamins were not substantially remodeled.
UL50, a required component of the NEC, was present in the p53KO cells, but failed to re-localize properly
Several studies report that UL50 is crucial for efficient capsid nuclear egress during HCMV infection (Camozzi et al., 2008; Dal Monte et al., 2002; Leigh et al., 2015; Milbradt et al., 2007; Muranyi et al., 2002; Sharma et al., 2014; Sonntag et al., 2016). UL50 is a key component of the NEC, which directly recruits UL53, and indirectly recruits other NEC components, to the nuclear membrane (Camozzi et al., 2008; Lye et al., 2015; Marschall et al., 2005; Milbradt et al., 2009; Milbradt et al., 2010; Miller et al., 2010; Sam et al., 2009; Sharma et al., 2015; Sharma et al., 2014; Sonntag et al., 2016). Although UL50 appeared slightly less robustly expressed in the p53KO cells at earlier timepoints by Western blot, levels appeared comparable to LOX cells by 96–120 h pi (Figure 3A). The comparable level was confirmed at 120 h pi using a dilution series of LOX and p53KO lysates (Figure 3B). Epifluorescent IF analysis using fix-first techniques found UL50 present in the large majority (>80%) of both infected LOX and p53KO cell populations at late times pi (Figures 3C and D). UL50 IF signal intensity in the p53KO cells was generally lower than in the LOX cells, therefore in Figures 3C (as well as, 3E and 3F) UL50 is additionally shown as "Adjusted Contrast", so that the similarities/differences in the staining patterns can be readily discerned. Staining patterns were variable in both LOX and p53KO cells (see Supplemental Figure 3 for more examples at 72 and 120 h pi). A large proportion of LOX cells displayed clear nuclear rim staining, accompanied by varying levels of cytoplasmic staining. These varying levels of cytoplasmic staining, in addition to staining at the nuclear rim, have been reported previously (Milbradt et al., 2014; Schmeiser et al., 2013; Sonntag et al., 2016). Surprisingly, many p53KO cells displayed no nuclear rim staining.
Figure 3. UL50 protein presence and localization in p53KO cells.

(A) Western blot analysis of UL50 in LOX and p53KO cells over a representative timecourse of infection. (B) Western blot showing a dilution series of LOX and p53KO cells at 120 h pi. Undiluted sample used lysate derived from an equivalent cell number as samples in (A). (C) Fix-first IF of total (nuclear and cytoplasmic) UL50 protein staining of both cell types at 120 h pi. Note the UL50 signal was less robust in the p53KO cells and therefore both cell types are shown with enhanced contrast (+50) for ease of visualization. (D) Quantitation of the percentage of cells displaying overall UL50 protein expression at late times pi. Error bars represent one standard error calculated from at least 2 experiments. (E) Confocal IF images of UL50 staining in LOX and p53KO cells at 72 and 120 h pi using a fix-first protocol. UL50 signal was concentrated at the nuclear rim in the majority of LOX cells and in the cytoplasm of the majority of p53KO cells. p53KO cells are again shown with enhanced contrast (+80). (F) UL50 localization was analyzed by IF using an extract-first protocol in all cell types at 120 h pi. The minority UL50-expressing p53KO cells required image contrast enhancement (+75) in order to clearly display its expression and localization. More WT-clone cells exhibited UL50 nuclear rim staining, but the signal was weaker and fewer puncta were present compared to LOX cells. Enhanced contrast (+75) is also shown for WT clone cells. (G) Quantitation of extract-first IF UL50 signal embedded into the INM for each cell type over a timecourse of infection. Error bars represent one standard error calculated from at least two experiments.
Functional capsid nuclear egress requires the components of the NEC to re-localize to the nuclear rim/inner nuclear membrane (Camozzi et al., 2008; Leigh et al., 2015; Milbradt et al., 2007; Milbradt et al., 2012; Milbradt et al., 2009; Milbradt et al., 2014; Miller et al., 2010; Muranyi et al., 2002; Sam et al., 2009; Schmeiser et al., 2013; Sharma et al., 2015; Sharma et al., 2014; Sonntag et al., 2016). Such rim staining was apparent in the LOX cells above the cytoplasmic background (see Figure 3C and Supplemental Figure 3); however, it was difficult to distinguish in most p53KO cells. Fix-first LOX cells visualized by confocal IF were seen to be distinctly UL50+ at the nuclear rim at both 72 and 120 h pi, with minimal staining in the cytoplasm, while in the large majority of p53KO cells UL50 was localized exclusively within the cytoplasm (Figure 3E). Nuclear-localized UL50 was accurately quantified by employing an extract-first methodology (as described in Materials and Methods). UL50 is anchored into the inner nuclear membrane in a functional NEC complex (Camozzi et al., 2008; Milbradt et al., 2007; Milbradt et al., 2012; Milbradt et al., 2009; Milbradt et al., 2014; Miller et al., 2010; Muranyi et al., 2002; Sam et al., 2009; Schmeiser et al., 2013; Sharma et al., 2014). Because UL50 re-localization was affected by the absence of p53, we included the wt p53 reintroduction clones (WT clone) from this point forward in our investigation.
In all three cell types, when UL50 localization was examined by epifluorescence under extract-first conditions, the remaining signal, when present, was concentrated at the nuclear rim (Figure 3F). Additionally, in the majority of LOX cells at very late times pi, numerous UL50 puncta were observed (see below for more data and discussion regarding these puncta). The p53KO and WT clone cells displayed very little UL50 nuclear rim staining prior to 96 h pi (Figure 3G). The nuclear UL50 signal, when present, was roughly equivalent in the p53KO and WT clone cells, but always weaker than LOX cells. Most prominently, UL50 was re-localized to the nucleus of ~90% of LOX cells, but only ~35% of the p53KO cells at 120 h pi, a statistically significant difference. The small amount of p53 expressed by the WT clone cells allowed recovery of UL50 nuclear re-localization to ~65% of the population, which was also statistically different (Figure 3G). What was preventing proper re-localization of UL50 in the absence of p53?
UL53, an essential component of the NEC, was largely absent from p53KO cells
UL50’s principal binding partner in the NEC is UL53. The presence of the UL50-UL53 complex is crucial for facilitating capsid nuclear egress during HCMV infection (Camozzi et al., 2008; Dal Monte et al., 2002; Milbradt et al., 2007; Muranyi et al., 2002; Sharma et al., 2014). UL53 expression commenced later pi and was expressed at considerably lower levels in p53KO cells than LOX (Figure 4A), even at 120 h pi. To more accurately estimate the extent of decreased expression, a dilution series was performed at 120 h pi with LOX and p53KO lysates (Figure 4B). It should be noted that the timing of the appearance of UL53 prior to 72 h pi was variable in the LOX cells. The immunoblot in Figure 4C showed that the WT clone cells expressed less UL53 than the LOX cells, but substantially more than the p53KO cells at late times pi. Fix-first IF analysis of LOX and WT clone cells found the majority were UL53+ (~91% and ~80%, respectively) at late times pi (Figure 4D and E). In most UL53+ nuclei the staining was principally seen at the nuclear rim, and included some punctate staining, by 120 h pi (see below). In contrast a minority (~47%) of infected p53KO cells were UL53+. These differences were statistically significant.
Figure 4. UL53 protein presence and localization in p53KO cells.

(A) Western blot analysis of UL53 in LOX and p53KO cells over a representative timecourse of infection. (B) Western blot showing a dilution series of LOX and p53KO cells at 120 h pi. Undiluted sample used lysate derived from an equivalent cell number as samples in (A). Lamin B1 is used as a loading control. (C) Western blot analysis of UL53 in LOX, p53KO and WT-clone cells at 120 h pi. Note that due to a lower number of cell equivalents (2.5×103) being used per lane in this blot, Westernbright quantum substrate was used for development. (D) Fix-first IF epifluorescent images of LOX, p53KO and WT-clone cells at 120 h pi. The majority of p53KO cells show no UL53 within the nucleus. (E) Quantitation of IF staining of UL53+ for each cell type over a timecourse of infection. Error bars represent one standard error calculated from at least two experiments. WT clone cells were only quantitated once for earlier timepoints (24–96 h pi). (F) UL50-HA and/or UL53-FLAG constructs were nucleofected into LOX and p53KO cells as described in Materials and Methods. Rabbit anti-HA antibody (mAb-HA) was used to detect UL50-HA and mouse anti-FLAG antibody (mAb-FLAG) was used to detect UL53-FLAG in nucleofected LOX and p53KO cells. Coverslips were harvested at 24 and 96 h post nucleofection using fix-first methodology. Inset shows magnified view (~150×) of boxed areas.
The literature indicated that UL50 was capable of nuclear trafficking in the absence of other viral proteins (Camozzi et al., 2008; Milbradt et al., 2007; Milbradt et al., 2012). UL53 encodes a nuclear localization signal (NLS). To ensure that the p53KO cells were capable of trafficking UL50 and UL53 properly, these proteins were transiently expressed and co-expressed in LOX and p53KO cells. In both cell types UL50 and UL53 localized to the nucleus (Figure 4F, top and second row), as previously observed. UL50 was principally localized to the nuclear rim when transfected alone. UL53 was found diffusely throughout the nucleoplasm when transfected independently. When the two proteins were co-expressed both proteins localized at the nuclear rim (Figure 4F, third row), as had also been previously observed (Camozzi et al., 2008; Milbradt et al., 2012; Sam et al., 2009; Schmeiser et al., 2013; Sonntag et al., 2016). Closer inspection revealed the two proteins were immediately adjacent to one another, and frequently co-localized, at the rim of transfected cells (Figure 4F, insets). The uninfected p53KO cells displayed this wt-phenotype and were capable of trafficking transiently expressed UL50 and UL53 to the nucleus alone and in combination.
UL50 nuclear re-localization required UL53 expression
The low level of UL53 expression in the p53KO cells and the similarity in percentages of UL50+ and UL53+ nuclei staining within the population prompted us to suspect a dependent relationship between these two proteins’ nuclear re-localization. Using fix-first confocal IF, to allow examination of the cytoplasmic component of UL50, cells were visualized at 120 h pi. Our suspicions were confirmed. In Figure 5A, three cells in a field exemplify our findings (p53KO cells shown). UL50 and UL53 were present in the cell at the top of the image. Both proteins were localized to the rim of the nucleus. In the cell at the bottom right of the image no UL53 was expressed and UL50 was found exclusively in the cytoplasm. The cell at the bottom left of the image expressed neither protein (seen in a small proportion of cells).
Figure 5. UL50 nuclear re-localization required UL53 expression.

(A) Confocal IF fix-first images of dual UL53 (green) and UL50 (red) staining from infected cells, p53KO cells shown. (B) Epifluorescent IF images of dual UL53 (green) and UL50 (red) staining from infected cells using extract-first protocol, LOX cell shown. (C) Quantitation of IF staining for each cell type of dual UL50+ and UL53+ intersecting the INM at 120 h pi. Error bars represent one standard error calculated from at least two experiments.
The above staining was repeated for epifluorescent IF analysis using extract-first conditions. UL50, UL53 and dual UL50/53 localization were quantified. An example of dual UL50/53 rim staining is found in Figure 5B at both 72 and 120 h pi (LOX cell shown). At 120 h pi, LOX cells were dual-positive in ~94% of the population, while p53KO cells were only dual-positive in 30% of cells. WT clone cells recovered dual-positivity to ~69% of the population (Figure 5C). The drop in dual-positive rim staining from LOX to p53KO cells and increase from p53KO to WT clone cells were both statistically significant. There were no UL53− cells with UL50+ nuclei. These results closely replicated our analysis of the proteins separately and confirmed that in the absence of UL53, UL50 failed to re-localize to the nucleus.
The NEC did not form in the large majority of p53KO cells
IF analysis using the extract-first protocol found that between 48 and 72 h pi UL50 staining formed a ring adjacent to the INM. The UL50 signal then became punctate in a steadily increasing proportion of the population (Figure 6A). By 120 h pi ~84% of LOX cells contained puncta, whereas only ~27% p53KO cells displayed this pattern. A statistically significantly increased proportion (~45%) of WT clone cells contained puncta compared to p53KO cells (Figure 6B – red bars). The p53KO and WT clone UL50+ sub-populations were delayed in conversion to punctate staining by ~24–48 h and contained fewer puncta (Figure 6A and B). The puncta of the NEC components, UL50 and UL53, were examined more closely by confocal IF using the extract-first staining protocol. Imaging of the puncta determined them to be elongated projections extending from the nuclear rim into the interior of the nucleus (see Figure 7 insets). Were these puncta of NEC components sites of nuclear capsid egress and the IINMs identified in our EM investigation?
Figure 6. UL50 staining progressed to a punctate pattern.

(A) Epifluorescent IF extract-first images of UL50 staining for all three cell types over a timecourse of infection. p53KO and WT-clone cells are additionally shown with enhanced contrast (+75) to aid in visualization. (B) Quantitation of IF staining for each cell type of UL50+ intersecting the INM (rim) and UL50-puncta+ (with NEC puncta) over a timecourse of infection. Blue portion of bars represents percentage of cells with UL50 intersecting the INM of the nucleus (at Rim). Red portion of the bars represents the percentage of cells with UL50 at the rim and with additional puncta (with NEC puncta). Error bars represent one standard error for the red portion of each bar only calculated from at least two experiments. Significance calculations represent p-values for the red portions of the bars only, which represent at least five experiments.
Figure 7. Co-localization of UL50 and UL53 puncta.

Confocal IF extract-first images of dual UL50 (red) and UL53 (green) staining for all three cell types at 120 h pi. Larger images in each row are compressed stacks of confocal slices. Cross-dashed lines delineate the plane of the individual slices shown to the right (vertical dashed line) and below (horizontal dashed line) each stack. Insets represent digital magnification (~14×) of the boxed areas immediately to their left and contained within the plane of that slice. Note that the large majority of KO cells did not contain puncta.
To determine if the UL50 puncta were IINMs, extract-first cells were dual-stained with Abs for UL50 and the HCMV major capsid protein, MCP. If the UL50/53 puncta were tubular IINMs, they would be expected to co-localize with fully mature capsids. Figure 8A displays UL50 puncta co-localized with MCP. There were many small MCP foci, as would be expected from individual capsids. There was also diffuse MCP staining in RCs (seen in the confocal image Figure 8B), which would be expected in these capsid-rich and packaging-intensive compartments (Lai and Britt, 2003; Sanchez et al., 2000). MCP is intimately associated with transiting capsids; therefore, if the UL50 puncta were nuclear egress pathways, there would be co-localization with MCP in these areas. As shown in Figure 8, all the UL50 puncta appeared co-localized to some extent with MCP. Single slices at the top, middle and bottom planes of the stack are shown to emphasize the extent of co-localization (Figure 8B). Neither the magnification nor the resolution of our confocal images were sufficient for precise measurement of these structures, but by approximating their widths using the scale bars they compared closely with the diameter of IINMs identified by EM, ranging from the smallest IINM (containing a single capsid, ~120nm in diameter) to the largest (containing 20–30 capsids, ~5 capsids in cross-section or 500nm to 1 micron in diameter). Using the confocal information we constructed a 3-D model (Figure 8C) of capsid location (the green MCP signal was thresholded and assigned the spherical volume of a capsid at each locus) in relation to the UL50 signal (shown in red as a reconstruction of its volume). The model indicated large numbers of free-floating capsids in the nucleus and cytoplasm. The model also indicated distinct associations between capsids and the red UL50 volumes, with capsids both interior to the red volumes and only partially within. We believed these formations were IINMs, which have been proposed as the principal sites of HCMV capsid nuclear egress (Buser et al., 2007; Villinger et al., 2015).
Figure 8. Co-localization of UL50-puncta and HCMV major capsid protein (MCP).

(A) Confocal IF extract-first image of dual UL50 (red) and MCP (green) staining of infected cell, LOX shown. Large image is a compressed stack of confocal slices. Cross-dashed lines delineate the plane of the individual slices shown to the right (vertical dashed line) and below (horizontal dashed line) each stack. (B) Individual confocal planes from the top, middle and bottom of the above cell. Insets are digital magnifications of the boxed region (~10×) showing colocalization of the two proteins within the foci. (C) 3-D reconstruction of the above cell. Nucleus is blue volume. Red volumes represent UL50 volumes. Green spheres (not to scale) represent MCP loci defined by thresholding to maximal values. Black plane provides reference for relative locations. Inset is zoomed view of boxed area (~50×).
DISCUSSION
p53, the “guardian-of-the-genome”, plays many roles within the cell. It is unsurprising that HCMV has evolved to both circumvent and exploit some of this ubiquitous cellular protein’s functions during its lifecycle within the cellular environment. Perhaps the most obvious sign that the virus alters normal p53 function is the near complete association of the protein with the viral RCs (Fortunato and Spector, 1998). In unstressed cells the protein is expressed at low levels and present in both the nucleus and cytoplasm. In a stressed cell, which virus infection could easily be considered, p53 is generally stabilized and principally accumulates in the nucleus, although in certain situations it can be found in the cytoplasm near the mitochondria (Moll et al., 2005). The virus-induced removal of p53 from these locations to the site of its own DNA replication suggests that, not only is p53 unavailable to perform its normal cellular functions, but also that the virus may be coopting the protein to its own ends. We have previously shown that p53 can bind specifically to viral DNA and its presence appears to be necessary to transactivate several viral gene products (Hannemann et al., 2009; Rosenke et al., 2006). Recent work has shown that p53 also plays a role in regulating expression of HSV-1 gene products (Maruzuru et al., 2013). p53’s cellular roles interacting with, and as a transactivator of, the DNA repair machinery suggest the virus may be utilizing this function in the replication and maintenance of fidelity of its own genome. Our earlier finding that the HCMV genome was selectively repaired in preference to the cellular genome following insult support this supposition (O’Dowd et al., 2012).
To ensure a relevant comparison to wt in these experiments we have utilized the p53KOs’ parental cells, LOX lung fibroblasts. We have also included partially reconstituted p53 lines generated from the p53KOs, WT clone cells. The p53KO cells were extremely sensitive to the presence of reintroduced p53. Only clones expressing extraordinarily small amounts of p53 (as shown in Supplemental Figure 1) were viable. Reintroduction of this small amount of p53 into WT clone cells recovered virus titers to within fourfold of LOX, while introduction of the same levels of non-functional DNA binding domain mutant p53 (R273H and H179Q) did not (Casavant et al., 2006). It should be noted that HSV-1 studies using a different p53KO system (HCT116 cells) also saw slightly less (~4-fold) than wt titers, even when a much higher level of reintroduced wt p53 could be achieved (Maruzuru et al., 2013). This current study, and its parallel EM analysis, were undertaken to further elucidate particular breakdown/s in the viral lifecycle in the absence of p53 to better understand the role of the protein in a functional wt infection.
Here, the molecular basis of the poor trafficking of capsids was investigated at a number of stages and for accepted molecular markers. The literature indicated that without remodeling of the nuclear lamina, capsids were not efficiently trafficked into the cytoplasm (Hamirally et al., 2009; Leigh et al., 2015; Milbradt et al., 2010; Reim et al., 2013; Sharma et al., 2014). The nuclear lamina is a dense meshwork that mechanically supports the nuclear architecture. Since HCMV infection primarily arrests the cell cycle at G1/S phase (Bresnahan et al., 1996; Dittmer and Mocarski, 1997; Lu and Shenk, 1996), HCMV cannot utilize the nuclear lamina breakdown that occurs at mitosis to traffic capsids out of the nucleus, nor can capsids transit through the nuclear pores due their size. It has been demonstrated that HCMV utilizes phosphorylation-dependent lamina remodeling to facilitate exiting the nucleus (Hamirally et al., 2009; Krosky et al., 2003; Milbradt et al., 2010; Reim et al., 2013). The viral protein UL97 plays a critical role in nuclear capsid egress, at least in part, via phosphorylation of lamin A/C and the ensuing lamina dissociation (Hamirally et al., 2009; Krosky et al., 2003; Milbradt et al., 2010; Reim et al., 2013; Sharma et al., 2015). This phosphorylation is thought to “unzip” the structure and make it more malleable, promoting dissociation. UL97 also contributes to various other functions during HCMV infection (Baek et al., 2004; Bigley et al., 2013; Kamil and Coen, 2007; Krosky et al., 2003; Marschall et al., 2003; Marschall et al., 2005; Sharma et al., 2015; Silva et al., 2011; Tran et al., 2008). Absence of UL97 produces a dramatic (up to 1,000-fold) growth defect and trafficking of nuclear capsids is greatly diminished (Prichard et al., 1999; Wolf et al., 2001). Experiments performed here found that in the absence of p53, UL97 was able to incorporate the chain-terminating nucleoside analog Ganciclovir into the growing viral DNA chain (Littler et al., 1992; Sullivan et al., 1992) and phosphorylate another cellular target, Rb (Hume et al., 2008). Most relevant, virtually all p53KO cells displayed phosphorylated lamin A/C, indicating this UL97 function (perhaps in concert with other cellular kinases) had been accomplished (Figure 2 and Supplemental Figure 2). However, while in LOX cells large sections of the lamin structure were frequently extensively remodeled much less frequently were such large segments of the nuclear rim equivalently modified in the p53KO cells (Figure 1).
What was preventing or not causing these substantial rearrangements of the lamins? In order for efficient nuclear capsid egress to occur the nuclear egress complex must function correctly. Two key NEC components are conserved across the herpesvirus family and are required for efficient primary envelopment and subsequent nuclear egress. In HCMV these two proteins are UL50 and UL53 (Camozzi et al., 2008; Dal Monte et al., 2002; Milbradt et al., 2007), in HSV-1 and psuedorabies virus (PrV) UL34 and UL31 (Fuchs et al., 2002; Klupp et al., 2007; Klupp et al., 2000; Reynolds et al., 2001; Reynolds et al., 2002), and BFLF1 and BFRF2 in Epstein-Barr virus (Farina et al., 2005; Gonnella et al., 2005; Lake and Hutt-Fletcher, 2004).
The presence and function of the NEC has been demonstrated as critical to productive CMV infections (Granato et al., 2008; Liang and Baines, 2005; Sharma et al., 2014). UL50 null mutations are lethal to replicative HCMV infection (Sharma et al., 2014). The viral constituents of the NEC begin to be expressed at early stages of infection. UL50 is a transmembrane protein and when properly localized its C-terminus is anchored into the INM (Camozzi et al., 2008; Lye et al., 2015; Milbradt et al., 2007; Milbradt et al., 2012; Milbradt et al., 2009; Milbradt et al., 2014; Miller et al., 2010; Muranyi et al., 2002; Sam et al., 2009; Schmeiser et al., 2013; Sharma et al., 2014). Extraction of un- and loosely bound UL50 from p53KO cells prior to fixation and staining revealed only 40% were positive for UL50 in the nucleus. The presence of p53 in WTG cells nearly doubled the proportion of the population containing properly re-localized UL50 compared to p53KOs.
In the absence of UL50, the NEC would not be formed and unable to facilitate capsid nuclear egress. Were other components of the NEC affected by the absence of p53? UL53 is UL50’s principal NEC binding partner. Transient expression assays have shown that UL50 is required for re-localization of UL53 to the nuclear rim (Camozzi et al., 2008; Milbradt et al., 2012; Sam et al., 2009; Schmeiser et al., 2013; Sonntag et al., 2016). Our observation confirmed this UL50/UL53 organizing strategy; in all cases, UL50+ nuclei displayed nuclear rim staining, where it was co-localized with UL53. We found that in the absence of p53, UL53 protein expression from the population was considerably reduced and, most significantly, that it was expressed in only ~47% of individual cells at late times pi (Figure 4E). We verified through transient expression assays that in p53KO cells UL50 re-localized to the nucleus outside the context of infection, thereby eliminating an inherent defect in these cells’ ability to traffic the protein, or any role for p53 in trafficking UL50.
We had seen UL50 present in only ~40% of the p53KO cells’ nuclei, and that UL53 was expressed in only ~47% of these cells. Were the cells containing UL50− nuclei the cells not expressing UL53? Dual staining for these proteins found that the absence of UL53, from any of the cell types used here, precluded the presence of UL50 in the nucleus. We have previously identified UL53 as a viral protein containing p53 binding sites nearby (located in the UL54 and UL55 loci)(Rosenke et al., 2006) and speculate that the virus utilizes p53 to transactivate UL53 expression. The dependence of UL50 nuclear trafficking on UL53 expression has been intimated in two earlier papers (Schmeiser et al., 2013; Sharma et al., 2014). Using alternately tagged viruses, Schmeiser et al. (Schmeiser et al., 2013) were able to show that UL50 nuclear re-localization was dependent upon UL53’s prior presence in that compartment, with UL50 staining very strongly in the cytoplasm prior to UL53’s entry/expression in the nucleus. Sharma et al. (Sharma et al., 2014) found that during a UL53KO BAC transfection, decidedly less UL50 accumulated at the nuclear rim. p53KO cells not expressing UL53 displayed a more dramatic phenotype, being completely absent UL50 nuclear rim staining. We are unaware of any prior reports implicating p53 in the expression of UL53, which is the primary finding of our work. This co-dependent nuclear-relocalization phenomenon has been described in the alpha herpesviruses, PRV and HSV-1. The UL50/53 homologues in these two viruses, UL34/31, have been shown via knockout viruses to display the same phenotype (Fuchs et al., 2002; Reynolds et al., 2001).
In the absence of p53, UL53 expression was diminished, leading to reduced NEC formation and, presumably, function. Was this disruption the same we had noted in our EM study, i.e. were the NEC and IINMs one in the same? Our EM study (Kuan et al., submitted) found a reduced number of IINMs in infected p53KO cells correlated closely with the reduced virus titers produced by these cells. Many ultrastructural studies of CMV-infected cells have found enlargements of the perinuclear space (variously termed intranuclear pseudoinclusions, invaginations or IINMs). These structures always contained primary-enveloped capsids (Buser et al., 2007; Dal Monte et al., 2002; Gilloteaux and Nassiri, 2000; Papadimitriou et al., 1984; Ruebner et al., 1964; Severi et al., 1988; Villinger et al., 2015). Buser et al reported that these infoldings of the INM accounted for approximately 5% of the total INM surface, but that the majority of nuclear trafficking capsids (86%) budded into these sites compared to those exiting at the peripheral INM (Buser et al., 2007). This behavior appears to be a beta herpesvirus phenomenon, although there is some indication of similar behavior in psuedorabies virus (Klupp et al., 2007). There are reports that transiently co-expressed UL50 and UL53 (and their homologues) are capable of deforming the INM (Gonnella et al., 2005; Hagen et al., 2015; Klupp et al., 2007; Lorenz et al., 2015; Milbradt et al., 2012; Passvogel et al., 2015; Reynolds et al., 2001; Sharma et al., 2014; Zeev-Ben-Mordehai et al., 2015), which has begun to be explained through structural analysis of these proteins (as reviewed in (Bigalke and Heldwein, 2015)). Our results (see Figure 4F) confirmed this behavior in the absence of p53 during transient expression.
At late times pi in this study a punctate pattern of UL50/UL53 was observed. Miller et al. indicated that these punctate patterns were IINMs (Miller et al., 2010). Confocal analysis determined that these puncta were elongated structures extending from the INM into the interior of the nucleus and were found in a range of sizes that correlated with the size of IINMs identified by EM. In support of our immunofluorescent localization, Dalmonte et al. used immuno-EM at late times pi to determine that UL53 was located within large invaginations (IINMs) within the nucleus (Dal Monte et al., 2002). In addition, transient expression of the equivalent PrV proteins (UL31/UL34) produces "speckles", which have been determined to be "vesicle formation in the perinuclear space" (Hagen et al., 2015; Passvogel et al., 2015; Zeev-Ben-Mordehai et al., 2015). We found that the majority of LOX cells formed abundant puncta at the late stages of infection. The ~40% of p53KO cells positive for UL50 in the nucleus also progressed to a punctate UL50/53 staining pattern. Re-introduction of p53 in the WT clone cells nearly doubled the percentage of the population progressing to the punctate pattern/IINMs compared to p53KO. These two populations were proportionally equivalent in progression to punctate staining, with ~70–75% of the UL50+ p53KO or WT clone populations displaying this pattern at late times (see Figure 6B). Qualitatively, the number and size of IINMs in the LOX cells appeared considerably greater compared to either p53KO or WT clone cells. The above measures all agreed with the assertion of Miller et al. that the NEC puncta were IINMs (Miller et al., 2010). Reinforcing this we found dual staining of UL50 puncta and MCP always produced some degree of co-localization (Figure 8A and B). We concluded that the puncta were capsid-containing tubular infoldings of the inner nuclear membrane.
Two recent papers have reported the presence of large UL50/53 protein agglomerations/foci at the rim of infected nuclei when a nonfunctional UL97 was present in the cells (Sharma et al., 2015; Sonntag et al., 2016). Nuclear egress and viral shedding were decreased in these infected cells when these foci were present. LOX cells produced more and increasingly distinct, UL50/53 puncta over the course of infection. However, the wt-LOX cells trafficked more capsids into the cytoplasm than the p53KO cells and shed substantially more virus. Therefore, if these puncta, which we believe to be IINMs, were the same as seen in the Sharma and Sonntag papers, they would be less prevalent/absent from the wt-LOX cells and more prevalent in the KO cells. This is precisely opposite to our observations. We accept that in the absence of UL97, UL50/53 protein agglomeration might well continue and IINMs might fail to form. Our observations of UL50/53 puncta co-localized with MCP in wt-level virus expressing cells supports our contention that these formations are IINMs.
The absence of p53 correlated closely with the total expression of UL53 protein. Cellular expression of UL53 correlated precisely with nuclear import of the critical NEC protein UL50. The manner of UL50’s normal import into the nucleus has not been conclusively defined, we suggest that, for lack of a better description, UL53 "jams its foot in the door" facilitating UL50 entry into the nucleus. The absence from the nucleus of the two principal NEC proteins precluded efficient capsid nuclear egress. The high agreement between measures of NEC puncta and IINMs, as visualized by EM, suggest these are a single phenomenon. We believe, and intend to pursue establishing, that HCMV utilizes cellular p53 to transactivate expression of UL53 to, at least in part, aid in the formation and function of the NEC/IINMs, thereby promoting efficient capsid nuclear egress.
Supplementary Material
Three different exposures of a Western blot showing the level of reintroduced p53 in WT clones compared to p53KO and wt LOX cells as controls. Note p53KO V sample is overloaded with eightfold more cells to confirm the absence of p53. Top and second panels show short and longer exposures, respectively, using mid-range chemiluminescence substrate (Pierce SuperSignal West Pico) for development. Third panel shows the same blot, with a long exposure after development with a maximum sensitivity chemiluminescence substrate (Advansta Westernbright Quantum). Bottom panel shows an actin loading control.
{kind=link}
(A) Investigation of the functionality of UL97 after GCV treatment. GCV-treated and DMSO-control treated cells were co-stained for UL97 and UL44. UL97 functionality was indirectly determined by lack of development of large viral RCs. (B) UL44 Western blots showed a dramatic decrease in the concentration of UL44 within the cell after GCV treatment in both cell types. (C) Phosphorylation of cellular Rb was observed in both cell types throughout the timecourse of infection.
{kind=link}
Fix-first IF of total (nuclear and cytoplasmic) UL50 protein staining of both cell types at 72 and 120 h pi. Note the UL50 signal was less robust in the p53KO cells and therefore cells are additionally shown with enhanced contrast (+50) for ease of visualization. Note also that the majority of p53KO cells show only cytoplasmic staining at both timepoints, but a minority of these cells does show UL50 nuclear rim staining.
{kind=link}
Highlights.
Phosphorylated nuclear lamins were inefficiently remodeled in p53KO cells.
p53KO cells expressed UL50, but it was not efficiently targeted to the nuclear rim.
UL53 was not expressed in the large majority of p53KO cells.
Cells failing to express UL53 did not localize UL50 to the nucleus.
NEC puncta/infoldings of the inner nuclear membrane were scarce in p53KO cells.
Acknowledgments
FUNDING INFORMATION
This work was supported by NIH grants RO1 AI051563, COBRE program P20 RR015587 and INBRE program P20 GM103408 to EAF and by a fellowship to MK provided by the University of Idaho College of Science. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank Dr. John Sedivy (Brown University, USA) for providing the p53KO fibroblasts, Dr. James Alwine (University of Pennsylvania, USA) for providing the UL50 antibody, Dr. Laura Hertel (Children’s Hospital of Oakland Research Institute, USA) for providing UL50-HA and UL53-FLAG constructs, Dr. Mark Prichard (University of Alabama at Birmingham, USA) for providing UL97 antibody, Dr. Stipan Jonjic (University of Rijeka, Croatia) for providing the UL53 antibody. We also thank Ann Norton and the Optical Imaging Core at UI for confocal imaging support. Lastly, we thank Drs. Amit Kulkarni and Anamaria Zavala for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Azzeh M, Honigman A, Taraboulos A, Rouvinski A, Wolf DG. Structural changes in human cytomegalovirus cytoplasmic assembly sites in the absence of UL97 kinase activity. Virology. 2006;354:69–79. doi: 10.1016/j.virol.2006.05.037. [DOI] [PubMed] [Google Scholar]
- Baek MC, Krosky PM, Pearson A, Coen DM. Phosphorylation of the RNA polymerase II carboxyl-terminal domain in human cytomegalovirus-infected cells and in vitro by the viral UL97 protein kinase. Virology. 2004;324:184–193. doi: 10.1016/j.virol.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010;2:a000935. doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigalke JM, Heldwein EE. The Great (Nuclear) Escape: New Insights into the Role of the Nuclear Egress Complex of Herpesviruses. Journal of Virology. 2015;89:9150–9153. doi: 10.1128/JVI.02530-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigley TM, Reitsma JM, Mirza SP, Terhune SS. Human cytomegalovirus pUL97 regulates the viral major immediate early promoter by phosphorylation-mediated disruption of histone deacetylase 1 binding. J. Virol. 2013;87:7393–7408. doi: 10.1128/JVI.02825-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron KK. Antiviral drugs for cytomegalovirus diseases. Antiviral Res. 2006;71:154–163. doi: 10.1016/j.antiviral.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Bjerke SL, Roller RJ. Roles for herpes simplex virus type 1 UL34 and US3 proteins in disrupting the nuclear lamina during herpes simplex virus type 1 egress. Virology. 2006;347:261–276. doi: 10.1016/j.virol.2005.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boppana SB, Britt WJ. Recognition of human cytomegalovirus gene products by HCMV-specific cytotoxic T cells. Virology. 1996;222:293–296. doi: 10.1006/viro.1996.0424. [DOI] [PubMed] [Google Scholar]
- Boppana SB, Fowler KB, Britt WJ, Stagno S, Pass RF. Symptomatic congenital cytomegalovirus infection in infants born to mothers with preexisting immunity to cytomegalovirus. Pediatrics. 1999;104:55–60. doi: 10.1542/peds.104.1.55. [DOI] [PubMed] [Google Scholar]
- Bresnahan WA, Boldogh I, Thompson EA, Albrecht T. Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology. 1996;224:150–160. doi: 10.1006/viro.1996.0516. [DOI] [PubMed] [Google Scholar]
- Britt W, Alford C. Cytomegalovirus, Fields Virology. Philadelphia: Lippincott-Raven Publishers; 1996. pp. 2493–2523. [Google Scholar]
- Britt WJ, Vugler L. Structural and immunological characterization of the intracellular forms of an abundant 68,000 Mr human cytomegalovirus protein. J. Gen. Virol. 1987;68(Pt 7):1897–1907. doi: 10.1099/0022-1317-68-7-1897. [DOI] [PubMed] [Google Scholar]
- Buchkovich NJ, Maguire TG, Alwine JC. Role of the endoplasmic reticulum chaperone BiP, SUN domain proteins, and dynein in altering nuclear morphology during human cytomegalovirus infection. J. Virol. 2010;84:7005–7017. doi: 10.1128/JVI.00719-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Bunz F, Fauth C, Speicher MR, Dutriaux A, Sedivy JM, Kinzler KW, Vogelstein B, Lengauer C. Targeted inactivation of p53 in human cells does not result in aneuploidy. Cancer research. 2002;62:1129–1133. [PubMed] [Google Scholar]
- Buser C, Walther P, Mertens T, Michel D. Cytomegalovirus primary envelopment occurs at large infoldings of the inner nuclear membrane. J. Virol. 2007;81:3042–3048. doi: 10.1128/JVI.01564-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camozzi D, Pignatelli S, Valvo C, Lattanzi G, Capanni C, Dal Monte P, Landini MP. Remodelling of the nuclear lamina during human cytomegalovirus infection: role of the viral proteins pUL50 and pUL53. J. Gen. Virol. 2008;89:731–740. doi: 10.1099/vir.0.83377-0. [DOI] [PubMed] [Google Scholar]
- Cano-Monreal GL, Wylie KM, Cao F, Tavis JE, Morrison LA. Herpes simplex virus 2 UL13 protein kinase disrupts nuclear lamins. Virology. 2009;392:137–147. doi: 10.1016/j.virol.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso MC, Leonhardt H, Nadal-Ginard B. Reversal of terminal differentiation and control of DNA replication: cyclin A and Cdk2 specifically localize at subnuclear sites of DNA replication. Cell. 1993;74:979–992. doi: 10.1016/0092-8674(93)90721-2. [DOI] [PubMed] [Google Scholar]
- Casavant NC, Luo MH, Rosenke K, Winegardner T, Zurawska A, Fortunato EA. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J. Virol. 2006;80:8390–8401. doi: 10.1128/JVI.00505-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinque P, Marenzi R, Ceresa D. Cytomegalovirus infections of the nervous system. InterVirology. 1997;40:85–97. doi: 10.1159/000150536. [DOI] [PubMed] [Google Scholar]
- Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, Nabors LB, Cobbs CG, Britt WJ. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002;62:3347–3350. [PubMed] [Google Scholar]
- Dal Monte P, Pignatelli S, Zini N, Maraldi NM, Perret E, Prevost MC, Landini MP. Analysis of intracellular and intraviral localization of the human cytomegalovirus UL53 protein. J. Gen. Virol. 2002;83:1005–1012. doi: 10.1099/0022-1317-83-5-1005. [DOI] [PubMed] [Google Scholar]
- de Bruyn Kops A, Knipe DM. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell. 1988;55:857–868. doi: 10.1016/0092-8674(88)90141-9. [DOI] [PubMed] [Google Scholar]
- Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22:832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer D, Mocarski ES. Human cytomegalovirus infection inhibits G1/S transition. J. Virol. 1997;71:1629–1634. doi: 10.1128/jvi.71.2.1629-1634.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziurzynski K, Chang SM, Heimberger AB, Kalejta RF, McGregor Dallas SR, Smit M, Soroceanu L, Cobbs CS, Hcmv, Gliomas S. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 2012;14:246–255. doi: 10.1093/neuonc/nor227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina A, Feederle R, Raffa S, Gonnella R, Santarelli R, Frati L, Angeloni A, Torrisi MR, Faggioni A, Delecluse HJ. BFRF1 of Epstein-Barr virus is essential for efficient primary viral envelopment and egress. J. Virol. 2005;79:3703–3712. doi: 10.1128/JVI.79.6.3703-3712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornara C, Lilleri D, Revello MG, Furione M, Zavattoni M, Lenta E, Gerna G. Kinetics of effector functions and phenotype of virus-specific and gammadelta T lymphocytes in primary human cytomegalovirus infection during pregnancy. J. Clin. Immunol. 2011;31:1054–1064. doi: 10.1007/s10875-011-9577-8. [DOI] [PubMed] [Google Scholar]
- Fortunato EA, Spector DH. p53 and RPA are sequestered in viral replication centers in the nuclei of cells infected with human cytomegalovirus. J. Virol. 1998;72:2033–2039. doi: 10.1128/jvi.72.3.2033-2039.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 2002;76:364–378. doi: 10.1128/JVI.76.1.364-378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill RB, James SH, Prichard MN. Human cytomegalovirus UL97 kinase alters the accumulation of CDK1. The Journal of general Virology. 2012;93:1743–1755. doi: 10.1099/vir.0.039214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilloteaux J, Nassiri MR. Human bone marrow fibroblasts infected by cytomegalovirus: ultrastructural observations. J. Submicrosc. Cytol. Pathol. 2000;32:17–45. [PubMed] [Google Scholar]
- Goldman RD, Goldman AE, Shumaker DK. Nuclear lamins: building blocks of nuclear structure and function. Novartis Found. Symp. 2005;264:3–16. discussion 16–21, 227–230. [PubMed] [Google Scholar]
- Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK, Spann TP. Nuclear lamins: building blocks of nuclear architecture. Genes Dev. 2002;16:533–547. doi: 10.1101/gad.960502. [DOI] [PubMed] [Google Scholar]
- Gonnella R, Farina A, Santarelli R, Raffa S, Feederle R, Bei R, Granato M, Modesti A, Frati L, Delecluse HJ, Torrisi MR, Angeloni A, Faggioni A. Characterization and intracellular localization of the Epstein-Barr virus protein BFLF2: interactions with BFRF1 and with the nuclear lamina. J. Virol. 2005;79:3713–3727. doi: 10.1128/JVI.79.6.3713-3727.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granato M, Feederle R, Farina A, Gonnella R, Santarelli R, Hub B, Faggioni A, Delecluse HJ. Deletion of Epstein-Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J. Virol. 2008;82:4042–4051. doi: 10.1128/JVI.02436-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen C, Dent KC, Zeev-Ben-Mordehai T, Grange M, Bosse JB, Whittle C, Klupp BG, Siebert CA, Vasishtan D, Bauerlein FJ, Cheleski J, Werner S, Guttmann P, Rehbein S, Henzler K, Demmerle J, Adler B, Koszinowski U, Schermelleh L, Schneider G, Enquist LW, Plitzko JM, Mettenleiter TC, Grunewald K. Structural Basis of Vesicle Formation at the Inner Nuclear Membrane. Cell. 2015;163:1692–1701. doi: 10.1016/j.cell.2015.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamirally S, Kamil JP, Ndassa-Colday YM, Lin AJ, Jahng WJ, Baek MC, Noton S, Silva LA, Simpson-Holley M, Knipe DM, Golan DE, Marto JA, Coen DM. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 2009;5:e1000275. doi: 10.1371/journal.ppat.1000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannemann H, Rosenke K, O’Dowd JM, Fortunato EA. The presence of p53 influences the expression of multiple human cytomegalovirus genes at early times postinfection. J. Virol. 2009;83:4316–4325. doi: 10.1128/JVI.02075-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkins L, Volk AL, Samanta M, Mikolaenko I, Britt WJ, Bland KI, Cobbs CS. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet. 2002;360:1557–1563. doi: 10.1016/S0140-6736(02)11524-8. [DOI] [PubMed] [Google Scholar]
- He R, Forman M, Mott BT, Venkatadri R, Posner GH, Arav-Boger R. Unique and highly selective anticytomegalovirus activities of artemisinin-derived dimer diphenyl phosphate stem from combination of dimer unit and a diphenyl phosphate moiety. Antimicrob Agents Chemother. 2013;57:4208–4214. doi: 10.1128/AAC.00893-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, Kalejta RF. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science. 2008;320:797–799. doi: 10.1126/science.1152095. [DOI] [PubMed] [Google Scholar]
- Johnson DC, Baines JD. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011;9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- Kamil JP, Coen DM. Human cytomegalovirus protein kinase UL97 forms a complex with the tegument phosphoprotein pp65. J. Virol. 2007;81:10659–10668. doi: 10.1128/JVI.00497-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc. Natl. Acad. Sci. USA. 2007;104:7241–7246. doi: 10.1073/pnas.0701757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Mettenleiter TC. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 2000;74:10063–10073. doi: 10.1128/jvi.74.21.10063-10073.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krosky PM, Baek MC, Coen DM. The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J. Virol. 2003;77:905–914. doi: 10.1128/JVI.77.2.905-914.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni AS, Fortunato EA. Stimulation of homology-directed repair at I-SceI-induced DNA breaks during the permissive life cycle of human cytomegalovirus. J. Virol. 2011;85:6049–6054. doi: 10.1128/JVI.02514-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L, Britt WJ. The interaction between the major capsid protein and the smallest capsid protein of human cytomegalovirus is dependent on two linear sequences in the smallest capsid protein. Journal of Virology. 2003;77:2730–2735. doi: 10.1128/JVI.77.4.2730-2735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake CM, Hutt-Fletcher LM. The Epstein-Barr virus BFRF1 and BFLF2 proteins interact and coexpression alters their cellular localization. Virology. 2004;320:99–106. doi: 10.1016/j.virol.2003.11.018. [DOI] [PubMed] [Google Scholar]
- Leach NR, Roller RJ. Significance of host cell kinases in herpes simplex virus type 1 egress and lamin-associated protein disassembly from the nuclear lamina. Virology. 2010;406:127–137. doi: 10.1016/j.virol.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh KE, Sharma M, Mansueto MS, Boeszoermenyi A, Filman DJ, Hogle JM, Wagner G, Coen DM, Arthanari H. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:9010–9015. doi: 10.1073/pnas.1511140112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- Liang L, Baines JD. Identification of an essential domain in the herpes simplex virus 1 UL34 protein that is necessary and sufficient to interact with UL31 protein. J. Virol. 2005;79:3797–3806. doi: 10.1128/JVI.79.6.3797-3806.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liptak LM, Uprichard SL, Knipe DM. Functional order of assembly of herpes simplex virus DNA replication proteins into prereplicative site structures. J. Virol. 1996;70:1759–1767. doi: 10.1128/jvi.70.3.1759-1767.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler E, Stuart AD, Chee MS. Human cytomegalovirus UL97 open reading frame encodes a protein that phosphorylates the antiviral nucleoside analogue ganciclovir. Nature. 1992;358:160–162. doi: 10.1038/358160a0. [DOI] [PubMed] [Google Scholar]
- Lombard DB, Guarente L. Nijmegen breakage syndrome disease protein and MRE11 at PML nuclear bodies and meiotic telomeres. Cancer Res. 2000;60:2331–2334. [PubMed] [Google Scholar]
- Lorenz M, Vollmer B, Unsay JD, Klupp BG, Garcia-Saez AJ, Mettenleiter TC, Antonin W. A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. The Journal of biological chemistry. 2015;290:6962–6974. doi: 10.1074/jbc.M114.627521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Shenk T. Human cytomegalovirus infection inhibits cell cycle progression at multiple points, including the transition from G1 to S. J. Virol. 1996;70:8850–8857. doi: 10.1128/jvi.70.12.8850-8857.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukonis CJ, Burkham J, Weller SK. Herpes simplex virus type 1 prereplicative sites are a heterogeneous population: only a subset are likely to be precursors to replication compartments. J. Virol. 1997;71:4771–4781. doi: 10.1128/jvi.71.6.4771-4781.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukonis CJ, Weller SK. Characterization of nuclear structures in cells infected with herpes simplex virus type 1 in the absence of viral DNA replication. J. Virol. 1996;70:1751–1758. doi: 10.1128/jvi.70.3.1751-1758.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo MH, Fortunato EA. Long-term infection and shedding of human cytomegalovirus in T98G glioblastoma cells. J. Virol. 2007;81:10424–10436. doi: 10.1128/JVI.00866-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo MH, Rosenke K, Czornak K, Fortunato EA. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J. Virol. 2007;81:1934–1950. doi: 10.1128/JVI.01670-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye MF, Sharma M, El Omari K, Filman DJ, Schuermann JP, Hogle JM, Coen DM. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. Embo J. 2015;34:2937–2952. doi: 10.15252/embj.201592651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak RA, Lombard DB, Johnson FB, Guarente L. Nucleolar localization of the Werner syndrome protein in human cells. Proc. Natl. Acad. Sci. USA. 1998;95:6887–6892. doi: 10.1073/pnas.95.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschall M, Freitag M, Suchy P, Romaker D, Kupfer R, Hanke M, Stamminger T. The protein kinase pUL97 of human cytomegalovirus interacts with and phosphorylates the DNA polymerase processivity factor pUL44. Virology. 2003;311:60–71. doi: 10.1016/s0042-6822(03)00147-8. [DOI] [PubMed] [Google Scholar]
- Marschall M, Marzi A, aus dem Siepen P, Jochmann R, Kalmer M, Auerochs S, Lischka P, Leis M, Stamminger T. Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J. Biol. Chem. 2005;280:33357–33367. doi: 10.1074/jbc.M502672200. [DOI] [PubMed] [Google Scholar]
- Maruzuru Y, Fujii H, Oyama M, Kozuka-Hata H, Kato A, Kawaguchi Y. Roles of p53 in herpes simplex virus 1 replication. Journal of Virology. 2013;87:9323–9332. doi: 10.1128/JVI.01581-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGavin JK, Goa KL. Ganciclovir: an update of its use in the prevention of cytomegalovirus infection and disease in transplant recipients. Drugs. 2001;61:1153–1183. doi: 10.2165/00003495-200161080-00016. [DOI] [PubMed] [Google Scholar]
- Mettenleiter TC, Klupp BG, Granzow H. Herpesvirus assembly: an update. Virus Res. 2009;143:222–234. doi: 10.1016/j.virusres.2009.03.018. [DOI] [PubMed] [Google Scholar]
- Milbradt J, Auerochs S, Marschall M. Cytomegaloviral proteins pUL50 and pUL53 are associated with the nuclear lamina and interact with cellular protein kinase C. J. Gen. Virol. 2007;88:2642–2650. doi: 10.1099/vir.0.82924-0. [DOI] [PubMed] [Google Scholar]
- Milbradt J, Auerochs S, Sevvana M, Muller YA, Sticht H, Marschall M. Specific residues of a conserved domain in the N terminus of the human cytomegalovirus pUL50 protein determine its intranuclear interaction with pUL53. J. Biol. Chem. 2012;287:24004–24016. doi: 10.1074/jbc.M111.331207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbradt J, Auerochs S, Sticht H, Marschall M. Cytomegaloviral proteins that associate with the nuclear lamina: components of a postulated nuclear egress complex. J. Gen. Virol. 2009;90:579–590. doi: 10.1099/vir.0.005231-0. [DOI] [PubMed] [Google Scholar]
- Milbradt J, Kraut A, Hutterer C, Sonntag E, Schmeiser C, Ferro M, Wagner S, Lenac T, Claus C, Pinkert S, Hamilton ST, Rawlinson WD, Sticht H, Coute Y, Marschall M. Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol. Cell. Proteomics. 2014;13:2132–2146. doi: 10.1074/mcp.M113.035782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbradt J, Webel R, Auerochs S, Sticht H, Marschall M. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 2010;285:13979–13989. doi: 10.1074/jbc.M109.063628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MS, Furlong WE, Pennell L, Geadah M, Hertel L. RASCAL is a new human cytomegalovirus-encoded protein that localizes to the nuclear lamina and in cytoplasmic vesicles at late times postinfection. J. Virol. 2010;84:6483–6496. doi: 10.1128/JVI.02462-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll UM, Wolff S, Speidel D, Deppert W. Transcription-independent pro-apoptotic functions of p53. Curr. Opin. Cell Biol. 2005;17:631–636. doi: 10.1016/j.ceb.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science. 2002;297:854–857. doi: 10.1126/science.1071506. [DOI] [PubMed] [Google Scholar]
- O’Dowd JM, Zavala AG, Brown CJ, Mori T, Fortunato EA. HCMV-infected cells maintain efficient nucleotide excision repair of the viral genome while abrogating repair of the host genome. PLoS Pathog. 2012;8:e1003038. doi: 10.1371/journal.ppat.1003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott RL, Longnecker M. An Introduction to Statistical Methods and Data Analysis. Belmont, CA: Brooks and Cole; 2010. [Google Scholar]
- Pante N, Kann M. Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol. Biol. Cell. 2002;13:425–434. doi: 10.1091/mbc.01-06-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadimitriou JM, Shellam GR, Robertson TA. An ultrastructural investigation of cytomegalovirus replication in murine hepatocytes. J. Gen. Virol. 1984;65(Pt 11):1979–1990. doi: 10.1099/0022-1317-65-11-1979. [DOI] [PubMed] [Google Scholar]
- Park R, Baines JD. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J. Virol. 2006;80:494–504. doi: 10.1128/JVI.80.1.494-504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passvogel L, Klupp BG, Granzow H, Fuchs W, Mettenleiter TC. Functional characterization of nuclear trafficking signals in pseudorabies virus pUL31. Journal of Virology. 2015;89:2002–2012. doi: 10.1128/JVI.03143-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penfold ME, Mocarski ES. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology. 1997;239:46–61. doi: 10.1006/viro.1997.8848. [DOI] [PubMed] [Google Scholar]
- Prichard MN, Britt WJ, Daily SL, Hartline CB, Kern ER. Human cytomegalovirus UL97 Kinase is required for the normal intranuclear distribution of pp65 and virion morphogenesis. J. Virol. 2005;79:15494–15502. doi: 10.1128/JVI.79.24.15494-15502.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MN, Gao N, Jairath S, Mulamba G, Krosky P, Coen DM, Parker BO, Pari GS. A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J. Virol. 1999;73:5663–5670. doi: 10.1128/jvi.73.7.5663-5670.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan MP, Chen LB, Knipe DM. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell. 1984;36:857–868. doi: 10.1016/0092-8674(84)90035-7. [DOI] [PubMed] [Google Scholar]
- Reim NI, Kamil JP, Wang D, Lin A, Sharma M, Ericsson M, Pesola JM, Golan DE, Coen DM. Inactivation of retinoblastoma protein does not overcome the requirement for human cytomegalovirus UL97 in lamina disruption and nuclear egress. J. Virol. 2013;87:5019–5027. doi: 10.1128/JVI.00007-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 2001;75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 2002;76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rixon FJ, Atkinson MA, Hay J. Intranuclear distribution of herpes simplex virus type 2 DNA synthesis: examination by light and electron microscopy. J. Gen. Virol. 1983;64(Pt 9):2087–2092. doi: 10.1099/0022-1317-64-9-2087. [DOI] [PubMed] [Google Scholar]
- Rosenke K, Samuel MA, McDowell ET, Toerne MA, Fortunato EA. An intact sequence-specific DNA-binding domain is required for human cytomegalovirus-mediated sequestration of p53 and may promote in vivo binding to the viral genome during infection. Virology. 2006;348:19–34. doi: 10.1016/j.virol.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Ruebner BH, Miyai K, Slusser RJ, Wedemeyer P, Medearis DN., Jr Mouse Cytomegalovirus Infection. An Electron Microscopic Study of Hepatic Parenchymal Cells. Am. J. Pathol. 1964;44:799–821. [PMC free article] [PubMed] [Google Scholar]
- Sam MD, Evans BT, Coen DM, Hogle JM. Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J. Virol. 2009;83:2996–3006. doi: 10.1128/JVI.02441-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta M, Harkins L, Klemm K, Britt WJ, Cobbs CS. High prevalence of human cytomegalovirus in prostatic intraepithelial neoplasia and prostatic carcinoma. J. Urol. 2003;170:998–1002. doi: 10.1097/01.ju.0000080263.46164.97. [DOI] [PubMed] [Google Scholar]
- Sanchez V, Clark CL, Yen JY, Dwarakanath R, Spector DH. Viable human cytomegalovirus recombinant virus with an internal deletion of the IE2 86 gene affects late stages of viral replication. J. Virol. 2002;76:2973–2989. doi: 10.1128/JVI.76.6.2973-2989.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez V, Greis KD, Sztul E, Britt WJ. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J. Virol. 2000;74:975–986. doi: 10.1128/jvi.74.2.975-986.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez V, Mahr JA, Orazio NI, Spector DH. Nuclear export of the human cytomegalovirus tegument protein pp65 requires cyclin-dependent kinase activity and the Crm1 exporter. J. Virol. 2007;81:11730–11736. doi: 10.1128/JVI.02760-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeiser C, Borst E, Sticht H, Marschall M, Milbradt J. The cytomegalovirus egress proteins pUL50 and pUL53 are translocated to the nuclear envelope through two distinct modes of nuclear import. J. Gen. Virol. 2013;94:2056–2069. doi: 10.1099/vir.0.052571-0. [DOI] [PubMed] [Google Scholar]
- Schmolke S, Drescher P, Jahn G, Plachter B. Nuclear targeting of the tegument protein pp65 (UL83) of human cytomegalovirus: an unusual bipartite nuclear localization signal functions with other portions of the protein to mediate its efficient nuclear transport. J. Virol. 1995;69:1071–1078. doi: 10.1128/jvi.69.2.1071-1078.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severi B, Landini MP, Govoni E. Human cytomegalovirus morphogenesis: an ultrastructural study of the late cytoplasmic phases. Arch. Virol. 1988;98:51–64. doi: 10.1007/BF01321005. [DOI] [PubMed] [Google Scholar]
- Sharma M, Bender BJ, Kamil JP, Lye MF, Pesola JM, Reim NI, Hogle JM, Coen DM. Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. Journal of Virology. 2015;89:523–534. doi: 10.1128/JVI.02426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Kamil JP, Coughlin M, Reim NI, Coen DM. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J. Virol. 2014;88:249–262. doi: 10.1128/JVI.02358-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lu H, He T, Yang Y, Liu L, Zhang R, Zheng Y, Shen Y, Zhang Y, Zhang Z. Prevalence and clinical management of cytomegalovirus retinitis in AIDS patients in Shanghai, China. BMC Infect. Dis. 2011;11:326. doi: 10.1186/1471-2334-11-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LA, Strang BL, Lin EW, Kamil JP, Coen DM. Sites and roles of phosphorylation of the human cytomegalovirus DNA polymerase subunit UL44. Virology. 2011;417:268–280. doi: 10.1016/j.virol.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg-Naucler C. Does cytomegalovirus play a causative role in the development of various inflammatory diseases and cancer? J. Intern. Med. 2006;259:219–246. doi: 10.1111/j.1365-2796.2006.01618.x. [DOI] [PubMed] [Google Scholar]
- Sonntag E, Hamilton ST, Bahsi H, Wagner S, Jonjic S, Rawlinson WD, Marschall M, Milbradt J. Cytomegalovirus pUL50 is the multi-interacting determinant of the core nuclear egress complex (NEC) that recruits cellular accessory NEC components. The Journal of general Virology. 2016 doi: 10.1099/jgv.0.000495. [DOI] [PubMed] [Google Scholar]
- Sullivan V, Talarico CL, Stanat SC, Davis M, Coen DM, Biron KK. A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature. 1992;358:162–164. doi: 10.1038/358162a0. [DOI] [PubMed] [Google Scholar]
- Tamashiro JC, Hock LJ, Spector DH. Construction of a cloned library of the EcoRI fragments from the human cytomegalovirus genome (strain AD169) J. Virol. 1982;42:547–557. doi: 10.1128/jvi.42.2.547-557.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran K, Mahr JA, Choi J, Teodoro JG, Green MR, Spector DH. Accumulation of substrates of the anaphase-promoting complex (APC) during human cytomegalovirus infection is associated with the phosphorylation of Cdh1 and the dissociation and relocalization of APC subunits. J. Virol. 2008;82:529–537. doi: 10.1128/JVI.02010-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uprichard SL, Knipe DM. Assembly of herpes simplex virus replication proteins at two distinct intranuclear sites. Virology. 1997;229:113–125. doi: 10.1006/viro.1996.8430. [DOI] [PubMed] [Google Scholar]
- Villinger C, Neusser G, Kranz C, Walther P, Mertens T. 3D Analysis of HCMV Induced-Nuclear Membrane Structures by FIB/SEM Tomography: Insight into an Unprecedented Membrane Morphology. Viruses. 2015;7:5686–5704. doi: 10.3390/v7112900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Hemmer RM, Sedivy JM. Role of p14(ARF) in replicative and induced senescence of human fibroblasts. Mol. Cell. Biol. 2001;21:6748–6757. doi: 10.1128/MCB.21.20.6748-6757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DG, Courcelle CT, Prichard MN, Mocarski ES. Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation. Proc. Natl. Acad. Sci. USA. 2001;98:1895–1900. doi: 10.1073/pnas.98.4.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeev-Ben-Mordehai T, Weberruss M, Lorenz M, Cheleski J, Hellberg T, Whittle C, El Omari K, Vasishtan D, Dent KC, Harlos K, Franzke K, Hagen C, Klupp BG, Antonin W, Mettenleiter TC, Grunewald K. Crystal Structure of the Herpesvirus Nuclear Egress Complex Provides Insights into Inner Nuclear Membrane Remodeling. Cell reports. 2015;13:2645–2652. doi: 10.1016/j.celrep.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LJ, Hanff P, Rutherford C, Churchill WH, Crumpacker CS. Detection of human cytomegalovirus DNA, RNA, and antibody in normal donor blood. J. Infect. Dis. 1995;171:1002–1006. doi: 10.1093/infdis/171.4.1002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Three different exposures of a Western blot showing the level of reintroduced p53 in WT clones compared to p53KO and wt LOX cells as controls. Note p53KO V sample is overloaded with eightfold more cells to confirm the absence of p53. Top and second panels show short and longer exposures, respectively, using mid-range chemiluminescence substrate (Pierce SuperSignal West Pico) for development. Third panel shows the same blot, with a long exposure after development with a maximum sensitivity chemiluminescence substrate (Advansta Westernbright Quantum). Bottom panel shows an actin loading control.
(A) Investigation of the functionality of UL97 after GCV treatment. GCV-treated and DMSO-control treated cells were co-stained for UL97 and UL44. UL97 functionality was indirectly determined by lack of development of large viral RCs. (B) UL44 Western blots showed a dramatic decrease in the concentration of UL44 within the cell after GCV treatment in both cell types. (C) Phosphorylation of cellular Rb was observed in both cell types throughout the timecourse of infection.
Fix-first IF of total (nuclear and cytoplasmic) UL50 protein staining of both cell types at 72 and 120 h pi. Note the UL50 signal was less robust in the p53KO cells and therefore cells are additionally shown with enhanced contrast (+50) for ease of visualization. Note also that the majority of p53KO cells show only cytoplasmic staining at both timepoints, but a minority of these cells does show UL50 nuclear rim staining.