

Abstract
Rationale
The molecular mechanism by which ABCA1 mediates cellular binding of apolipoprotein A-I (apoA1) and nascent HDL assembly is not well understood.
Objective
To determine the cell surface lipid that mediates apoA1 binding to ABCA1 expressing cells and the role it plays in nascent HDL assembly.
Methods and Results
Using multiple biochemical and biophysical methods, we found that apoA1 binds specifically to phosphatidylinositol (4,5) bis-phosphate (PIP2). Flow cytometry and PIP2 reporter binding assays demonstrated that ABCA1 led to PIP2 redistribution from the inner to the outer leaflet of the plasma membrane. Enzymatic cleavage of cell surface PIP2 or decreased cellular PIP2 by knockdown of phosphatidylinositol-5-phosphate 4-kinase impaired apoA1 binding and cholesterol efflux to apoA1. PIP2 also increased the spontaneous solubilization of phospholipid liposomes by apoA1. Using site directed mutagenesis; we found that ABCA1's PIP2 and phosphatidylserine translocase activities are independent from each other. Furthermore, we discovered that PIP2 is effluxed from cells to apoA1, where it is associated with HDL in plasma, and that PIP2 on HDL is taken up by target cells in a scavenger receptor-BI (SR-BI) dependent manner. Mouse plasma PIP2 levels are apoA1 gene dosage dependent and are > 1 μM in apoA1 transgenic mice.
Conclusions
ABCA1 has a PIP2 floppase activity, which increases cell surface PIP2 levels that mediate apoA1 binding and lipid efflux during nascent HDL assembly. We found that PIP2 itself is effluxed to apoA1 and it circulates on plasma HDL, where it can be taken up via the HDL receptor SR-BI.
Keywords: ABC transporter, apolipoprotein, high-density lipoprotein, cholesterol
HDL plays a role in many cellular pathways via diverse mechanisms, including anti-thrombotic, vasoprotective, anti-inflammatory, and cholesterol efflux activities.1, 2 HDL assembly involves the cellular lipidation of extracellular apolipoprotein A-I (apoA1) by the membrane protein ABCA1.3 The importance of the ABCA1 pathway in generating nascent HDL (nHDL) is demonstrated in human patients carrying mutations in ABCA1 (Tangier disease) who have extremely low levels of plasma HDL.4 These patients have increased accumulation of cholesterol in peripheral tissues, resulting in premature atherosclerotic vascular disease. 5, 6 Although recent trials of HDL-cholesterol (HDL-C) raising drugs have not appeared to prevent cardiovascular events, a consensus is building that it is HDL function in reverse cholesterol transport (RCT), rather than the levels of HDL-C, that is protective against cardiovascular disease. 7, 8 For example, cholesterol efflux capacity of apoB-depleted serum is inversely associated with both prevalent and incident cardiovascular disease, independent of HDL-C levels.7, 8
The mechanism of cellular lipidation of apoA1 by ABCA1 is not understood at the molecular level with various models discussed in recent reviews.9, 10 ABCA1 has two well-established intermediate activities leading to apoA1 lipidation: 1) the outward translocation or “flopping” of phosphatidylserine (PS) to cell surface, and 2) apoA1 binding to the cell surface. We recently characterized a third activity, the unfolding of N-terminal hairpin of apoA1 on the cell surface.11 Interestingly, apoA1 binding to the cell surface is independent of the PS floppase activity of ABCA1, as the W590S-ABCA1 Tangier disease mutation is defective in PS floppase but not in apoA1 binding, while the C1477R-ABCA1 Tangier disease mutant is defective in apoA1 binding but not in PS floppase activity.11-14 It is important to note that both W590S and C1477R have impaired apoA1 lipidation, indicating that PS floppase and apoA1 cell surface binding are both required for efficient transfer of cellular lipids to apoA1 during nHDL biogenesis.13
Several models have been proposed to explain the mechanism responsible for the specific binding of apoA1 to ABCA1-expressing cells: a) apoA1 binding to cell surface phosphatidylserine (PS) due to ABCA1 PS floppase activity;15, 16 b) direct interaction between apoA1 and ABCA1 as demonstrated by protein cross-linking;3, 17 c) low-capacity binding of apoA1 to ABCA1 and high-capacity binding of apoA1 to membrane lipids;18, 19 d) apoA1 interaction with membrane protrusions due to ABCA1 bulk phospholipid outward translocase (floppase) activity.20 Recent solid-phase binding studies from the Molday lab showed no direct binding between apoA1 and purified ABCA1 in the presence or absence of several classes of phospholipids including PS.21 Since these experiments were carried using immobilized ABCA1, the possibility of apoA1 and ABCA1 direct interaction on cell surface cannot be ruled out.
The major phospholipid constituents of HDL are phosphatidylcholine (PC), PS, phosphatidylethanolamine (PE), and phosphatidylinositol (PI).22 Unlike other structural phospholipids, phosphatidylinositol phosphates (PIPs) are minor components of cellular membranes, but they serve as critical integral signaling molecules for multiple pathways. PI(4,5)bis-phosphate (PIP2) is the major cellular PIP species and it is predominantly found on the inner leaflet of the plasma membrane where it play roles in many cellular processes such as membrane ruffling, endocytosis, exocytosis, protein trafficking and receptor mediated signaling.23-26 PIP2 binds to various effector proteins through interacting with pleckstrin homology (PH) domains thereby regulating the cellular localization and activity of effector proteins.27-29 PIP2 synthesis is tightly regulated by Pl-kinases, such as PIP-5-phosphate 4 kinase (PIP5K), and PIP phosphatases, such as PTEN.26, 30
Here, we show that apoA1 binds to various PIPs including PIP2, and that ABCA1 has PIP2 floppase activity independent of its PS floppase activity. ABCA1 increases cell surface PIP2 facilitating apoA1 binding and lipidation. Furthermore, PIP2 itself is effluxed to apoA1 during nHDL formation and it is found on human and mouse HDL, where it can be delivered to target cells in an scavenger receptor-BI (SR-BI) dependent manner. We propose a new model for apoA1-ABCA1 mediated HDL biogenesis highlighting the essential role of PIP2 in this process.
Methods
A brief description of the methods is provided below. Detailed description of the materials and methods are available at the Online Data Supplement.
Protein-lipid overlay assays
The PIP strip and sphingo strip membrane assays (Echelon Biosciences) were performed according to the manufacturer's protocol.
Surface Plasmon resonance
Binding kinetics of PIP2 with different apolipoproteins were analyzed using a Biacore3000 instrument. Either biotinylated apoA1 or biotinylated PIP2 was immobilized on streptavidin sensor chips. For comparing binding kinetics of PIP2 with apoA1, apoA2 and apoE, these proteins were immobilized by covalent coupling on a CM5 sensor chip.
Fluorescence anisotropy
Increasing concentrations of apoA1 were incubated with fatty acid-labeled bodipy PIP2 in a quartz cuvette at 25°C. Relative anisotropy was determined using polarized filters with excitation at 503 nm and emission at 513 nm in a Perkin Elmer spectrofluorimeter.
Liposome clearance assay
Multilamellar vesicles (MLV) were prepared from 1,2-dimyristoyl-sn-glycero-3-phos-phocholine (DMPC) or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) with or without 5 mole % PIP2. These MLVs were incubated with human apoA1 at 25 °C. MLV solubilization by human apoA1 was monitored by measuring sample turbidity (absorbance) at 325 nm. Liposome floatation assay was performed as described. 31
ApoA1 oligomerization
ApoA1was incubated in the presence or absence of PIP2 or POPC at 1:4 mole ratio and then cross linked by the addition of bis(sulfosuccinimidyl) suberate (BS3, Pierce) for 30 minutes at room temperature. Oligomerization was visualized by SDS-PAGE and apoA1 western blot.
ABCA1 induction and cholesterol efflux
ABCA1 induction in RAW264.7 cells by 0.3 mM 8Br-cAMP and methods for cholesterol efflux were previously described.32 ABCA1 was induced in stably transfected BHK cells by 16-24 hr incubation with 10 nM mifepristone.
Inositol containing lipid efflux
Cells were labeled with 40 μCi/mL of [3H]myo-inositol for 24 hr followed by ABCA1 induction in serum-free medium as indicated. The cells were washed and chased for 4-6 hr in serum-free medium in the presence or absence of 5 μg/ml apoA1. The chase media and cellular lipids were subjected to acidic solvent extraction yielding the fraction containing PI and PIPs. Radioactivity was determined by scintillation counting for efflux calculations.
Reverse PIP2 transport in vivo
Bone-marrow derived macrophages from C57BL/6 mice were labeled with 40 μci/ml of [3H]myo-inositol for 24h. ~1.8 × 106 dpm of labeled macrophages were injected s.c. into the back of each mouse. 3 days later, plasma was collected, followed by acidic extraction of lipids. The lipid fraction was incubated with PH-PLC δ-GST tagged protein. The PIP2 bound to GST tagged protein was separated using glutathione-beads. The % PIP2 efflux to plasma was determined by calculating 100 × PIP2 dpm in total plasma divided by the injected inositol lipid dpm.
PIP2 intracellular localization
RAW264.7 macrophages and ABCA1-inducible BHK cells were stably transfected with the 2XPH-PLCδ-GFP PIP2 reporter plasmid. PIP2 localization in cells with or without ABCA1 induction was visualized by epifluorescent microscopy.
Cell surface PS, PIP2, and apoA1 binding
Cell surface PS levels and apoA1 binding were determined by flow cytometry. PIP2 antibody was either labeled with FITC or Alexa-647 for determination of cell surface PIP2 levels by flow cytometry.
PIP2 ELISA
Cells, conditioned media, and plasma samples were solvent extracted, dried, and resuspended in aqueous buffer. PIP2 was quantified by using the PI(4,5)P2 Mass ELISA kit from Echelon Biosciences, following the protocol provided.
Plasma analyses
0.5 ml of fresh human plasma (obtained under informed consent in an IRB approved protocol) was separated by fast protein liquid chromatography. Total cholesterol and PIP2 levels were measured in mouse plasma or human FPLC fractions. LC-MS/MS was used for PIP2 profiling in human HDL as previously described.33
SR-BI mediated PIP2 uptake
Mifepristone SR-BI-inducible BHK cells were treated with or without 10 nM mifepristone for 14 hr.34 Human HDL was labeled with [3H]PIP2 and incubated at 100 μg/ml with cells in serum free media for 4 hr at 37°C. Cellular lipids were extracted and 3H was determined by scintillation counting, and normalized to cellular protein.
Statistical analyses
Data are shown as mean ± SD. Comparisons of 2 groups were performed by a 2-tailed t test, and comparisons of 3 or more groups were performed by one-way ANOVA with Bonferroni posttest. For these multiple comparison tests, all columns were compared against each other and the letter above a bar show significant difference vs. all other columns marked with different letters. All statistics were performed using Prism software (GraphPad).
Results
Lipid-free apoA1 binds to PIPs but not to other major phospholipids
ABCA1 is required for HDL biogenesis; it remodels the plasma membrane, translocating PS to the cell surface and promoting apoA1 binding. To determine the lipid-binding profile of lipid-free apoA1, lipid-protein overlay assays were performed using phospholipid/PIP and sphingolipid membrane strips. As PC is the major cellular and HDL phospholipid and previous studies implicated PS in apoA1 binding to the cell surface, we expected apoA1 might bind to these lipid species. However, lipid-free apoA1 was directly bound only to PIPs containing 2 or 3 head-group phosphates and not to other lipids including PC, PS, or PI (Figure 1A). ApoA1 did not bind to PI3P, PI4P, PI5P or sphingosine 1-phosphate, indicating that mere presence of phosphate in the head-group is not sufficient for apoA1 binding. Furthermore, lipid-free apoA1 did not bind to any lipids on the sphingolipid strip, including sphingomyelin, ceramide, and cholesterol (Online Figure I). Thus, apoA1 binds specifically to PIP2 isoforms and PIP3. Since PI(4,5)P2 is a major cellular PIP2 species that is particularly enriched at the cell surface, we performed further experiments using this PIP2 species. Binding of apoA1 to immobilized PIP2 was demonstrated by surface plasmon resonance (SPR) (Figure 1B). In addition, PIP2, but not PC, showed direct binding to immobilized apoA1 by SPR in dose-dependent manner (Figure 1C). To ensure that apoA1 binding to PIP2 was not due to some unintended artefactual due to immobilization on membrane, we performed an apoA1-PIP2 solution binding assays using fluorescence anisotropy, which demonstrated high affinity binding of apoA1 to Bodipy-fatty acid labeled PIP2 (kd= 93 nM, Figure 1D). To determine if apoA1 can bind to PIP2 in a membrane environment, we performed a liposome floatation assay. We confirmed apoA1 binding to PIP2 (5 mole %) in POPC MLVs. We observed increased co-migration of apoA1 with the PIP2 MLVs vs. control MLVs in the top 0% sucrose gradient fraction (Figure 1E).
Figure 1. ApoA1 binds PIP2.

A. Lipid-protein overlay assay using a PIP strip for detection of apoA1 binding to cellular lipids. B. SPR assay showing direct binding of apoA1 (550 nM) (blue line) to biotinylated PIP2 immobilized on an SPR sensor chip (green and red lines are buffer controls). C. SPR assay showing PIP2 binding (red and dark blue lines) to immobilized biotinylated apoA1, while a higher dose of PC showed no binding (purple line). D. Fluorescent anisotropy of bodipy-lableled PIP2 binding to varying concentrations of apoA1. E. Liposome floatation assay showing increased apoA1 floatation with POPC MLVs (0% sucrose fraction; top layer of gradient) containing 5 mol% PIP2 vs. those without PIP2.
Other amphipathic apolipoproteins bind PIP2
Many proteins bind PIP2 through their conserved PH domains35; however, recent studies have shown that proteins lacking a PH domain can bind to PIP2 by adapting a cationic grip dimeric configuration that specifically accommodate the head group of PIP2.36, 37 ApoA1 does not contain a PH domain, but its class A amphipathic helical structure contains a surface lined with positively charged lysine and arginine residues, which we postulate to be responsible for its PIP2 binding activity. In support of this hypothesis, we tested PIP2 binding to apoA2 and apoE, other ABCA1 acceptors with similar class A amphipathic helical structures. We immobilized apoA1, apoA2, and apoE to SPR membranes and found that PIP2 reversibly bound to each of these proteins with Kd values ranging from ~60 nM to 1.3 μM (Online Figure II A-C). The Kd for apoA1 binding to PIP2 was140 nM, with Kon=2.23×104 M−1s−1 and Koff=1.93×10−3 M−1s−1 (Online Figure IIA), similar to the Kd obtained by fluorescence anisotropy (Figure 1D).
PIP2 promotes apoA1 monomer formation and lipid solubilization
Lipid-free apoA1 exists in equilibrium between its monomeric and oligomeric forms, and the lipid-free monomer is postulated to mediate the initial interaction with the cell membrane and act as the primary ABCA1 acceptor.38, 39 We hypothesized that PIP2 binding to apoA1 may promote apoA1 monomerization, leading to increased membrane insertion. We found that pre-incubating PIP2, but not PS, with lipid-free apoA1 shifted the equilibrium towards the monomeric form, as assessed by SDS-PAGE after addition of the chemical crosslinker BS3 (Figure 2A). Thus, PIP2 both recruits apoA1 to the lipid surface and promotes its monomeric structure favored for lipid solubilization. To test the hypothesis that PIP2 can indeed promote apoA1 mediated lipid solubilization, we performed liposome clearance assays. Lipid-free apoA1 was added to DMPC MLVs with or without 5 mole % PIP2. The PIP2 containing MLVs were solubilized faster and to a greater extent than the DMPC-only MLVs (Figure 2B). POPC MLVs are resistant to apoA1 solubilization, but POPC:PS:cholesterol MLVs were previously found to be slowly solubilized by apoA1 at acidic pH.40 Here, we found that the addition of 5% PIP2 to POPC: PS: cholesterol: (70:20:10) MLVs increased the rate of their solubilization by lipid-free apoA1 at pH = 5.0 (Figure 2C). Thus, in cell-free systems the presence of PIP2 led to increased apoA1 monomerization, and liposome solubilization, which implies increased apoA1 insertion into the liposome membrane.
Figure 2. PIP2 promotes apoA1 mediated lipid solubilization.
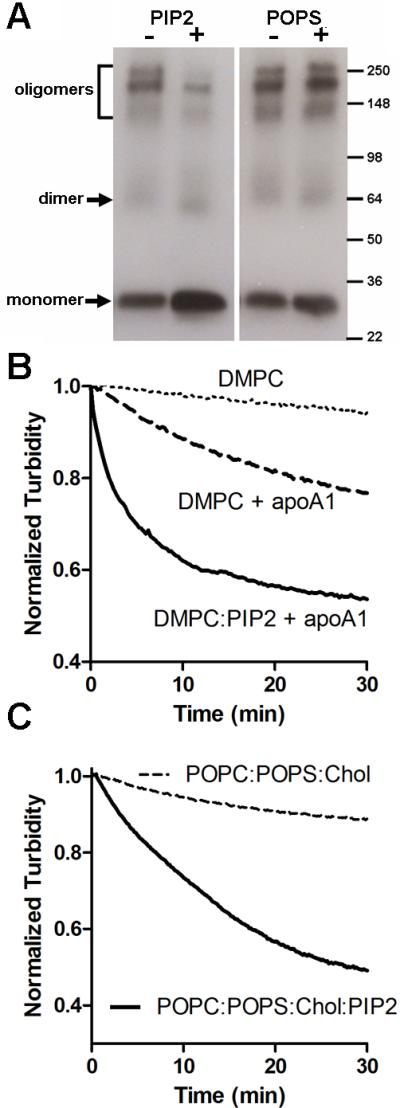
A. PIP2 incubation promoted a shift to apoA1 monomer conformation. Lipid free apoA1 was incubated with or without PIP2 or POPS and subjected to BS3 mediated cross linking followed by SDS-PAGE and apoA1 western blot. B. PIP2 (5 mole %) promoted DMPC MLV solubilization when incubated with apoA1 (100:1 lipid:apoA1 mole ratio) at 25°C with lipid solubilization determined by measuring turbidity at 325 nm. C. PIP2 (5 mole %) promoted solubilization of MLVs composed of POPC:POPS:cholesterol (70:20:10 mole ratio) incubated with apoA1 (100:1 lipid:apoA1 mole ratio) at 25°C, pH 5.0.
ABCA1 expression exposes PIP2 at the cell surface
PIP2 is thought to be localized at the inner leaflet of the plasma membrane where it plays important roles in targeting proteins to the membrane, membrane trafficking, and signal transduction.24, 26 PIP2 exposure on the cell surface would be required to interact with lipid-free apoA1, but cell surface PIP2 has not been previously described. As ABCA1 has well defined PS floppase activity,16 we considered the possibility that ABCA1 might also act as a PIP2 floppase. Using a PI(4,5)P2 specific monoclonal antibody, we detected increased levels of cell surface PIP2 after induction of ABCA1 in RAW264.7 cells by 8Br-cAMP (Figure 3A). The enhanced level of cell surface PIP2 was decreased to the baseline level by treatment with exogenous phosphatidylinositol specific phospholipase C (PI-PLC), without altering ABCA1 expression (Figure 3A, B). To determine if increased cell surface PIP2 was a cAMP effect independent of ABCA1, we knocked down ABCA1 expression using siRNA and observed that this treatment markedly decreased cell surface PIP2 levels and cholesterol efflux to apoA1 in 8Br-cAMP treated RAW264.7 cells (Online Figure III A-C). To confirm the role of ABCA1 in translocating PIP2 to the cell surface, we stably transfected RAW264.7 cells with an intracellular PIP2-binding reporter protein (2X-PH-PLCδ-eGFP) that does not bind to other PIP species.29 This reporter was localized mainly to the plasma membrane in RAW264.7 without ABCA1 induction, consistent with PIP2 localization in the inner leaflet of the membrane; however, upon ABCA1induction, the PIP2 reporter redistributed with increased cytosolic and less prominent plasma membrane localization (Figure 3C), which we attribute to PIP2 translocation to the outer leaflet of plasma membrane. We quantified the redistribution of this PIP2 reporter and found its inner leaflet membrane association was decreased from 42 to 27% upon ABCA1 induction (Figure 3D, p<0.0001). All of these findings were independently reproduced in stably transfected BHK cells in which ABCA1 is inducible by Mifepristone (Online Figure IV A-D). Thus, in addition to ABCA1's well-known PS floppase activity, we demonstrated its PIP2 floppase activity, which both promote cell surface remodeling.
Figure 3. ABCA1 flops PIP2.

A. Cell surface PIP2 assessed by flow cytometry in RAW264.7 cells ± ABCA1 induction that were pretreated ± PI-PLC (RFI, relative fluorescence intensity; mean ± SD; different letters show p<0.001 by ANOVA Bonferroni posttest, n=3). B. Western blot of RAW264.7 cell extracts showing expression of ABCA1was not altered by PI-PLC treatment. C. ABCA1 expression redistributed the PIP2 reporter, 2XPH-PLCδ-eGFP, away from the plasma membrane in live stably transfected RAW264.7 cells. D. Percentage of PIP2 reporter association with plasma membrane ± ABCA1 induction (n=10 and 20 for control and ABCA1 expressing cells, respectively).
PIP2 and PS floppase activities of ABCA1 are independent of each other
The PS floppase and apoA1 cellular binding activities of ABCA1 can be distinguished from each other using naturally occurring Tangier disease-associated mutations in the first and second large extracellular domains of ABCA1.5, 13 Cells expressing the W590S ABCA1 isoform are deficient in PS floppase activity but display normal apoA1 binding activity, while cells expressing the C1477R ABCA1 isoform have normal PS floppase activity but are deficient in apoA1 binding.11-13 To evaluate if the PS and PIP2 floppase activities of ABCA1 are independent of each other, stably transfected HEK293 cells with equal expression of WT-ABCA1-GFP, W590SABCA1-GFP, or C1477R-ABCA1-GFP were analyzed for cholesterol efflux, cell surface exposure of PIP2 and PS, as well as apoA1 binding (Figure 4A-D). Cells expressing WT ABCA1 had all of these activities induced vs. control HEK cells. Cells expressing W590SABCA1 had defective cholesterol efflux and PS exposure, but had normal PIP2 exposure and apoA1 binding activity, while cells expressing C1477R-ABCA1 had defective cholesterol efflux, apoA1 binding, and PIP2 exposure, but had normal PS exposure. We were able to rescue the decreased cholesterol efflux activity of the C1477R-ABCA1 expressing HEK293 cells by enriching these cells with PIP2 via pre-incubation with MLVs containing POPC:PIP2 (90:10 mole ratio, Figure 4E). Combining these results with earlier studies, we conclude that a specific mutation in first large extracellular domain of ABCA1 abrogates PS floppase activity, an activity that remodels the plasma membrane and increases cholesterol extractability,14 while a specific mutation in the second large extracellular domain of ABCA1 abrogates PIP2 floppase activity, an activity that we show below is required for apoA1 binding. Thus, these two phospholipid floppase activities of ABCA1 are independent of each other and mediated by distinct domains.
Figure 4. ABCA1 flops PIP2 and PS independently.

Cholesterol efflux to apoA1 (A), cell surface PIP2 (B), cell surface PS (C), and apoA1 cellular binding (D) were measured in control HEK cells and those stably transfected with either ABCA1, W590S-ABCA1, or C1477R-ABCA1 isoforms. Decreased cell surface PIP2 and apoA1 binding were only observed for the C1447R isoform, while decreased cell surface PS was only observed for the W590S isoform. For panels A-D, mean ± SD; different letters show p<0.001 by ANOVA Bonferroni posttest, n=3. E. Cholesterol efflux to apoA1 from HEK cells (black bars) and stably transfected cells with either ABCA1 (red bars) or C1477R-ABCA1 (green bars), each untreated (control, open bars) or pretreated with a final concentration of 20 μg/ml POPC MLVs (diagonal striped bars) or POPCPIP2 (90:10 mole ratio) MLVs (horizontal striped bars). Mean ± SD; different letters show p<0.05 by ANOVA Bonferroni posttest, n=3.
Cell-surface PIP2 promotes apoA1 binding and cholesterol acceptor activity
To probe the role of the ABCA1-mediated increase in cell surface PIP2, we determined the effect of a PI-PLC dose response treatment on apoA1 binding and cholesterol efflux. PI-PLC treatment of RAW264.7 and HEK293 cells greatly diminished ABCA1-dependent apoA1 binding and cholesterol efflux in a dose dependent manner (Figure 5A, B, Online Figure V A, B). The limited cholesterol efflux activity of the PIP2 floppase competent W590S ABCA1 isoform was completely eliminated upon treatment with PI-PLC in stably transfected HEK293 cells (Online Figure V C). We blocked PIP2 exposure with an anti-PIP2 monoclonal antibody on ABCA1-induced RAW264.7 cells and this also resulted in decreased apoA1 binding to the cell surface (Figure 5C), confirming that PIP2 plays a role in apoA1 binding. Pre-incubation of apoA1 with PIP2 decreased apoA1 binding and cholesterol efflux in ABCA1-induced RAW264.7 cells (Figure 5C, D, with additional controls in Online Figure VI). Thus, cell surface PIP2 is required for ABCA1-mediated apoA1 binding and cholesterol efflux. Furthermore, exogenous PIP2 added to lipid-free apoA1 appeared to compete against cell surface PIP2, diminishing apoA1 binding and cholesterol efflux. Instead of adding PIP2 to apoA1, when PIP2 was enriched on the cell membrane by treating RAW264.7 cells with POPC MLVs containing 10% PIP2, we observed increased apoA1 cellular binding in the absence of ABCA1 expression, but not to the extent seen in ABCA1 expressing cells (Figure 5E). POPC only MLVs failed to induce apoA1 binding (Figure 5E). However, this liposome-mediated cell enrichment with PIP2 failed to support cholesterol efflux in the absence of ABCA1 expression (Figure 5F), indicating the requirement of ABCA1 for cellular lipid efflux activity, even in conditions where apoA1 binding is induced.
Figure 5. PIP2 required for ABCA1-mediated apoA1 binding and cholesterol efflux.

ApoA1 binding to (A) and cholesterol efflux from (B) RAW264.7 cells ± ABCA1 expression and treated with a PI-PLC dose response (0.4, 0.8, 1.2, and 2 U/ml, with 2 U/ml also used for cells without ABCA1). C. ApoA1 binding to ABCA1 expressing RAW264.7 cells ± exogenous PIP2 preincubated with apoA1 prior to adding to cells (PIP2:apoA1, 5:1 mole ratio), or ± PIP2 specific monoclonal antibody (2 μg/ml, PIP2 antibody was added to cells along with apoA1). D. Cholesterol efflux from ABCA1-expressing RAW264.7 cells to apoA1 (5 μg/ml) that was pre-incubated ± exogenous PIP2 (PIP2:apoA1, 5:1mole ratio). ApoA1 binding (E) and cholesterol efflux (F) from RAW264.7 cells ± ABCA1 expression, untreated (control, open bars for panel E only) or pretreated with a final concentration of 20 μg/ml POPC MLVs (diagonal striped bars) or POPC-PIP2 (90:10 mole ratio) MLVs (horizontal striped bars). PIP5Kα siRNA mediated knockdown (KD) decreased apoA1 binding (G), cholesterol efflux to apoA1 (H), and cellular PIP2 levels (I). For all panels, mean ± SD; n=3, for multiple comparisons different letters show p<0.05, by ANOVA Bonferroni posttest.
Alteration of cellular PIP2 metabolism affects cellular apoA1 binding and cholesterol efflux.
To further demonstrate the central role of PIP2 in ABCA1-mediated apoA1 binding and cholesterol efflux, we performed siRNA knockdown of PIP5K, the enzyme that converts PI5P to PIP2. We found that the PIP5Kα isoform was highly expressed in both RAW264.7 and HEK293 cells and susceptible to protein knockdown by siRNA (Online Figure VII A, B), while the PIP5Kβ protein was not detected by Western blot. PIP5Kα knockdown significantly decreased ABCA1 mediated apoA1 binding and cholesterol efflux in both RAW264.7 and HEK293 cells (Figure 5 G, H, Online Figure VII C, D). We confirmed that PIP5Kα knockdown significantly decreased whole cell PIP2 levels by 30% in RAW264.7 cells (Figure 5 I). Conversely, we knocked down Tmem55b, a specific PIP2 phosphatase that converts PIP2 to PI5P and has a role in cellular cholesterol metabolism 41, whose expression we detected in RAW264.7 cells (Online Figure VIII A). This knockdown modestly, but significantly, increased cellular apoA1 binding, cholesterol efflux to apoA1, and whole cell PIP2 levels (Online Figure VIII B-D). These gene knockdown studies support the central role of cellular PIP2 in ABCA1-mediated apoA1 binding and cholesterol efflux.
PIP2 efflux to apoA1
To determine if PIP2 could be effluxed from cells along with other phospholipids and cholesterol during HDL biogenesis we labeled cells with [3H]myo-inositol, and after chasing with apoA1, the conditioned media radioactivity in extracted lipids was measured. Efflux of inositol labeled lipids was increased upon ABCA1 induction in RAW264.7 cells (Figure 6A). However, the inositol lipid fraction can contain PI and other PIP species. Thus, we performed a protein-lipid overlay assay of lipids extracted from apoA1-containing conditioned media derived from cells with or without ABCA1 expression; and, the presence of PIP2 or PI4P was detected using tagged PIP2 or PI4P binding proteins, respectively. The conditioned media obtained from RAW264.7 cells contained elevated PIP2 only in the ABCA1-induced cells (Figures 6B). In contrast, PI4P in the conditioned media was not increased by ABCA1 induction in RAW264.7 cells (Figure 6B). An ELISA assay was used to quantify the amount of PIP2 in the conditioned media. RAW264.7 cells expressing ABCA1 effluxed ~20-fold more PIP2 to apoA1 vs. control cells (Figure 6C). Similar results were observed for ABCA1-mediated inositol labeled lipid efflux and PIP2 levels in the conditioned media in ABCA1-inducible BHK cells (Online Figure IX A, B).
Figure 6. PIP2 effluxed to apoA1.

A. ABCA1 mediates efflux of [3H]inositol labeled lipids to apoA1 from RAW264.7 cells (mean ± SD; ***, p<0.001, by two-tailed t-test, n=3). B. PIP2 and PI4P in lipids from RAW264.7 cells and in apoA1-containing conditioned media visualized by lipid-protein overlay assays using tagged PIP2 or PI4P binding proteins. C. ABCA1 dependent efflux of PIP2 to apoA1 in conditioned media assessed by ELISA, normalized to cell protein (mean ± SD; ***, p<0.001, by two-tailed t-test, n=3).
PIP2 is effluxed to apoA1 in vivo and carried on HDL
To confirm the role of apoA1 as a PIP2 acceptor, we determined levels of PIP2 in fresh mouse plasma. Plasma from apoA1 knockout (A1 KO), wild type (WT), and human apoA1 transgenic (A1-Tg) mice contained apoA1-gene dosage dependent levels of both cholesterol and PIP2, with 64-fold higher PIP2 levels in the A1-Tg vs. A1 KO mice (Figure 7A). WT mice had plasma levels of ~0.4 μM PIP2. The low level of plasma PIP2 in A1 KO plasma (~0.03 μM) implies that most PIP2 is carried on HDL and not bound with albumin or found free in plasma. To determine if PIP2 can be reverse transported from macrophages to the plasma, we performed a modified reverse cholesterol transport study, where macrophages were labeled in culture with [3H]myoinositol and implanted s.c. into A1 KO and WT mice. Plasma was collected 3 days post implantation, and radioactivity in PIP2 was determined after pull-down with a tagged PIP2 binding protein. ~3-fold more of the injected radioactivity in PIP2 was recovered in the plasma of the WT hosts vs. the A1 KO hosts (Figure 7B, p<0.01). Thus, similar to cell-based assays, PIP2 can be effluxed to apoA1 in vivo.
Figure 7. PIP2 circulates on plasma HDL.

A. PIP2 (ELISA assay, blue bars) and cholesterol (open bars) levels in freshly prepared plasma derived from apoA1 KO, WT, and apoA1 transgenic mice (mean ± SD). B. Plasma PIP2 radioactivity in apoA1 KO and WT recipients 3 d after s.c. implantation of bone marrow macrophages labeled with [3H]myo-inositol (mean ± SD; **, p<0.01, by two-tailed t-test, n=3). C. PIP2 (ELISA assay, blue circles) and cholesterol (open circles) levels in fresh human plasma separated by FPLC. D. Human HDL analyzed by liquid chromatography mass spectrometry to identify endogenous PIP2 fatty acid species. E. Uptake of [3H]PIP2 labeled HDL by BHK cells ± SR-BI induction (mean ± SD; **, p<0.01, by two-tailed t-test, n=3).
FPLC separation of fresh human plasma determined that almost all of the PIP2 was found in the HDL fractions (Figure 7C). In human HDL, two PIP2 species were detected by liquid chromatography tandem mass spectrometry consistent with either 18:0, 20:4 fatty acids or 16:0, 20:4 fatty acids (Figure 7D). Therefore, PIP2 is effluxed from cells and is carried on HDL, implying that HDL may serve as a vehicle to deliver PIP2 to target tissues. We labeled HDL with [3H]PIP2 and incubated it with SR-BI-inducible BHK cells. We found ~2-fold higher cellular uptake of [3H]PIP2 after SR-BI induction (Figure 7E), indicating that HDL can deliver PIP2 to target cells via SR-BI.
Discussion
Several models have been proposed for the mechanism by which apoA1 binds to ABCA1 expressing cells to initiate nHDL assembly.2, 9, 10 Here we demonstrated that ABCA1 is a PIP2 floppase leading to increased cell surface PIP2 and redistributing a cellular PIP2 reporter away from the inner leaflet of the plasma membrane. We then demonstrated using several independent lines of evidence that apoA1 binding to ABCA1 expressing cells and efflux to apoA1 is mediated by PIP2: 1) apoA1 pre-incubation with PIP2 diminished cellular apoA1 binding and cholesterol efflux presumably due to competition with cell surface PIP2; 2) direct addition of PIP2-containing liposomes could rescue the cholesterol efflux activity of HEK293 cells expressing the C1477R ABCA1 isoform that is deficient in PIP2 floppase activity; 3) direct addition of PIP2-containing liposomes to RAW264.7 cells could promote apoA1 cellular binding, but not cholesterol efflux, in the absence of ABCA1 expression; 4) Treatment of RAW264.7 or HEK cells with PI-PLC decreased cell surface PIP2, apoA1 binding, and cholesterol efflux; 5) knockdown of PIP5Kα, a PIP2 synthesizing enzyme, led to reduced cellular PIP2 levels, apoA1 binding, and cholesterol efflux; and, 6) knockdown of Tmem55b, a PIP2 degrading enzyme, led to increased cellular PIP2 levels, apoA1 binding, and cholesterol efflux. Since ABCA1 flops PIP2, the PIP2 levels will be highest in proximity to ABCA1, and PIP2 may serve as a bridge to account for the previously observed specificity of apoA1crosslinked to ABCA1.3, 12, 19 Reboul et al. purified ABCA1 and did not observe apoA1 binding in the absence or presence of PC and PE.21 However, since PIP2 was not used in their studies, they could not assess if PIP2 could serve as bridge between ABCA1 and apoA1. Quazi et al. assessed the floppase activity of liposome reconstituted ABCA1 using several different phospholipid substrates, and they observed ABCA1 floppase of PC, PS, and SM, but not PE or PG.42 PIP2 floppase activity was not assessed in their study.
We now propose a revised model for the ABCA1-mediated biogenesis of nHDL that incorporates the role of PIP2 in this process (Figure 8). The PS floppase activity, disrupted by mutations in the first large extracellular domain of ABCA1, promotes membrane remodeling that makes the membrane more susceptible to detergents such as sodium taurocholate or amphipathic proteins such as apoA1 12, 13, 15, 16. The PIP2 floppase activity, disrupted by mutations in the second large extracellular domain of ABCA1, promotes apoA1 binding to the cell surface. Once bound to the cell, the PIP2-apoA1 interaction may favor apoA1 monomerization, which may promote our previously observed partial unfolding of the apoA1 N-terminal helical hairpin on the cell surface.11. Gursky et al. recently proposed that N-terminal unfolded apoA1 can re-dimerize through domain swapping around repeat 5 assuming the conformation that can insert into the cell membrane, microsolubilize cellular lipids, and assemble nHDL particles that are released from the cell.39 We suspect that the unfolded apoA1 on the cell surface may be able to form dimers, trimers, or tetramers in the conformation that allows membrane insertion and microsolubilization resulting in the release of several distinct size classes of nHDL, as previously observed by Mulya et al.43 Nagata et al. showed that ABCA1 forms immobile dimers on the plasma membrane which are dissociated into monomers after apoA1 addition.44 Our model is compatible with ABCA1 dimers serving as the active PIP2 floppase and the suggestion by Nagata et al. that apoA1 dimers (we suggest these are in the unfolded N-terminal confirmation) interact with ABCA1 dimers.44 Thus, membrane lipid remodeling by ABCA1 flops both PS and PIP2 to the cell surface, and both of these activities are required for maximal cholesterol efflux.
Figure 8. Model for ABCA1 mediated HDL biogenesis.

Model demonstrates that PS and PIP2 floppase activities, mediated by distinct extracellular loops of ABCA1, remodel the plasma membrane with PIP2 floppase mediating apoA1 binding. After binding to cell surface PIP2, apoA1 monomerizes, unfolds at the N-terminal hairpin, forms dimers or oligomers in a conformation that can insert into the membrane to assemble nHDL, which is released from the cell surface.
PIP2, found prevalently on the inner leaflet of the plasma membrane, plays a role in protein targeting and many aspects of cell biology. Several actin-binding proteins bind to PIP2, which promotes actin polymerization and actin-membrane linkages.23 In addition, increasing cellular PIP2 leads to increased recruitment of the endocytic adaptor protein AP-2 to the plasma membrane and increased rates of receptor-mediated endocytosis.27 In fact, the observation that ABCA1-defective Tangier disease fibroblasts have increased rates of receptor-mediated endocytosis can now be interpreted in light of ABCA1's PIP2 floppase activity, such that defective ABCA1 may increase PIP2 in inner leaflet resulting in increased endocytic vesicle formation.45
ABCA1 deficiency in macrophages is associated with free cholesterol accumulation and increased expression of inflammatory genes in response to toll like receptor (TLR) ligands.46 This appears to be mediated by an ABCA1-dependent depletion of lipid-raft cholesterol that inhibits the trafficking of Myd88 associated TLRs to the plasma membrane.47 The anti-inflammatory effects of LXR agonists in macrophages are mediated by the ABCA1 action on Myd88, TRAF6, and TLR recruitment.48 In addition to ABCA1's cholesterol-mediated lipid raft remodeling, the PIP2 floppase activity of ABCA1 may also play a role in TLR trafficking. Kagan and Medzhitov demonstrated that the PIP2-binding adaptor TIRAP recruits MyD88 and TLR to the plasma membrane to initiate TLR signaling.49 Thus, the ABCA1-mediated flop of PIP2 out of the inner leaflet would be expected to decrease TLR trafficking to the plasma membrane, consistent with the observed effects of ABCA1 expression. In addition, ABCA1 may play an important role in AKT signaling, as Landry et al. demonstrated that ABCA1 expression decreased lipid-raft cholesterol and inhibited EGF-mediated AKT phopshorylation.50 AKT phosphorylation is dependent upon PI(3,4,5)tris-phosphate (PIP3), which is generated via phosphorylation of PIP2. We can now reinterpret the ABCA1 inhibition of AKT phosphorylation in light of the ABCA1 PIP2 floppase activity, which would decrease the substrate for PIP3 generation.
We demonstrated here that apoA1, apoA2, and apoE can all bind to PIP2 (Online Figure II), with apoA1 binding with a Kd ~100 nM (Figure 1E). None of these proteins have a consensus PH domain, associated with PIP binding; however, other domains including the PX domain of a PI3-kinase, the ENTH domain of Epsin, the ANTH domain of AP180, the TRAF domain of TRAF6, and the dimeric cationic grip domain of tomato defensin TPP3 also bind to PIP2.36, 51, 52 The common feature of PIP2 binding proteins is the coordination of PIP headgroup phosphates by positively charged amino acid residues. The class A amphipathic helical secondary structure of apoA1, apoA2, and apoE all contain a positively charged surface adjacent to the hydrophobic surface,53 and we postulate that the tertiary structure of these apolipoproteins, along with short synthetic class A amphipathic helical peptides, can effectively coordinate PIPs through their positively charged residues. The PIP binding property of class A amphipathic helical peptide would not demonstrate stereo-specificity, and this could explain why these peptides made entirely of D-amino acids function efficiently as ABCA1-depedent cholesterol acceptors.54, 55 As apoA1 is the most abundant apolipoprotein in plasma with normal levels of 1 – 2 mg/ml, any weak detergent activity of apoA1 could be detrimental to the host. We speculate that the ABCA1 PIP2 floppase activity may have co-evolved with the PIP2 binding activity of apoA1 and other amphipathic apolipoproteins as a mechanism to prevent the promiscuous detergent activity of these proteins, allowing apoA1 to solubilize lipids from cells under tight control by ABCA1 expression.
Our finding that PIP2 is effluxed by ABCA1 to nHDL and then circulates in the plasma on HDL has implications for the pleiotropic activity of HDL. HDL carries a wide array of signaling molecules including sphingosine 1-phosphate, which is carried on ApoM containing HDL and plays many biological roles.56 PIP2 carried on circulating HDL might also serve as a signaling molecule by the delivery of PIP2 to target cells via SR-BI. In wildtype mice we observed plasma PIP2 levels at ~0.4 μM; however, with plasma apoA1 of ~ 32 uM 57, and assuming on average 2.5 apoA1 molecules per HDL, the final HDL concentration is ~13 μM. Thus, it appears that there is less than one PIP2 molecule per HDL particle in circulation. This may be due to PIP2 uptake by cells via SR-BI (as shown) or other pathways, PIP2 degradation in the circulation, or a PIP2 under estimate due to loss during lipid extraction.
Supplementary Material
Novelty and Significance.
What is known
Nascent HDL is assembled by lipidation of exogenous apolipoprotein A-I (apoA1) by the cell membrane protein ABCA1, in the process of cholesterol and phospholipid efflux from cells.
Two intermediate activities of ABCA1 are its translocation of phosphatidylserine (PS) from the inner to the outer leaflet of the plasma membrane and the increased cellular binding of apoA1.
What new information does this article contribute
ApoA1 binds tightly and specifically to phosphatidylinositol (4.5) bis phosphate (PIP2). • ABCA1 transfers PIP2 from the inner to the outer leaflet of the plasma membrane, and this is required for the ABCA1-mediated induction of cellular binding of apoA1.
PIP2 is effluxed along with cholesterol from ABCA1 expressing cells to nascent HDL, and PIP2 is found on circulating HDL.
The molecular mechanism by which ABCA1 assembles exogenous apoA1 into nascent HDL is not known. This article demonstrates for the first time that apoA1 binds specifically to the phospholipid PIP2. This was followed by the discovery that ABCA1 has PIP2 floppase (outward translocase) activity independent of its well-defined PS floppase activity, leading to PIP2 exposure on cell surface. PIP2 has not previously been characterized on the cell surface. Cell surface PIP2 serves as ligand for apoA1 to mediate apoA1 binding to ABCA1-expressing cells, and this cell surface PIP2 promotes cholesterol efflux to apoA1. PIP2 itself is effluxed to apoA1 from ABCA1-expressing cells and this is the first demonstration that PIP2 is found on plasma HDL in mice and humans. This HDL-associated PIP2 can be taken up by target cells expressing the HDL receptor SR-BI. We suggest a new model for the mechanism of ABCA1 mediated lipidation of apoA1 in the assembly of nascent HDL, which incorporates the PIP2 floppase activity of ABCA1 as well as apoA1 conformational changes. It was not clear if lipid-poor apoA1 binds directly to ABCA1 or to membrane lipids. Our data suggests that apoA1 binds directly to PIP2 in the membrane lipids.
Acknowledgments
Funding: This work was supported by NIH grants R01 HL130085 and HL128268 to J.D.S., HL PO1 HL0958055 to J.D.S and S.L.H., and American Heart Association National Scientist Development Grant 15SDG25710128 to K.G.
Non-standard abbreviations and acronyms
- apoA1
apolipoprotein A-I
- apo2
apolipoprotein A-II
- apoE
apolipoprotein E
- ABCA1
ATP-binding cassette transporter A1
- DMPC
1,2-dimyristoyl-sn-glycero-3-phos-phocholine
- GST
glutathione-S-transferase
- HDL-C
high-density lipoprotein-cholesterol
- MLVs
multilamellar vesicles
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PG
phosphatidylglycerol
- PI
phosphatidylinositol
- PIP5K
phosphatidylinositiol-5-phosphate 4-kinase
- PIP
phosphatidylinositol phosphate
- PIP2
phosphatidylinositol (4,5) bis-phosphate
- PIP3
phosphatidylinositol (3,4,5) tris-phosphate
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPS
1-palmitoyl-2-oleoyl-sn-glycero-3-serine
- PS
phosphatidylserine
- SPR
surface plasmon resonance
- SR-BI
scavenger receptor-BI
- TLR
toll like receptor
Footnotes
Disclosures: None.
References
- 1.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–55. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–75. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Wang N, Silver DL, Thiele C, Tall AR. ATP-binding cassette transporter A1 (ABCA1) functions as a cholesterol efflux regulatory protein. J Biol Chem. 2001;276:23742–7. doi: 10.1074/jbc.M102348200. [DOI] [PubMed] [Google Scholar]
- 4.Bodzioch M, Orso E, Klucken J, Langmann T, Bottcher A, Diederich W, Drobnik W, Barlage S, Buchler C, Porsch-Ozcurumez M, Kaminski WE, Hahmann HW, Oette K, Rothe G, Aslanidis C, Lackner KJ, Schmitz G. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999;22:347–51. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 5.Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denefle P, Assmann G. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet. 1999;22:352–5. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 6.Oram JF. Molecular basis of cholesterol homeostasis: lessons from Tangier disease and ABCA1. Trends Mol Med. 2002;8:168–73. doi: 10.1016/s1471-4914(02)02289-x. [DOI] [PubMed] [Google Scholar]
- 7.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–35. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med. 2012;18:1344–6. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- 9.Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. 2014;289:24020–9. doi: 10.1074/jbc.R114.583658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang S, Smith JD. ABCA1 and nascent HDL biogenesis. Biofactors. 2014;40:547–54. doi: 10.1002/biof.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang S, Gulshan K, Brubaker G, Hazen SL, Smith JD. ABCA1 mediates unfolding of apolipoprotein AI N terminus on the cell surface before lipidation and release of nascent high-density lipoprotein. Arterioscler Thromb Vasc Biol. 2013;33:1197–205. doi: 10.1161/ATVBAHA.112.301195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzgerald ML, Morris AL, Rhee JS, Andersson LP, Mendez AJ, Freeman MW. Naturally occurring mutations in the largest extracellular loops of ABCA1 can disrupt its direct interaction with apolipoprotein A-I. J Biol Chem. 2002;277:33178–87. doi: 10.1074/jbc.M204996200. [DOI] [PubMed] [Google Scholar]
- 13.Nagao K, Zhao Y, Takahashi K, Kimura Y, Ueda K. Sodium taurocholate-dependent lipid efflux by ABCA1: effects of W590S mutation on lipid translocation and apolipoprotein A-I dissociation. J Lipid Res. 2009;50:1165–72. doi: 10.1194/jlr.M800597-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gulshan K, Brubaker G, Wang S, Hazen SL, Smith JD. Sphingomyelin Depletion Impairs Anionic Phospholipid Inward Translocation and Induces Cholesterol Efflux. J Biol Chem. 2013 doi: 10.1074/jbc.M113.512244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chambenoit O, Hamon Y, Marguet D, Rigneault H, Rosseneu M, Chimini G. Specific docking of apolipoprotein A-I at the cell surface requires a functional ABCA1 transporter. J Biol Chem. 2001;276:9955–60. doi: 10.1074/jbc.M010265200. [DOI] [PubMed] [Google Scholar]
- 16.Alder-Baerens N, Muller P, Pohl A, Korte T, Hamon Y, Chimini G, Pomorski T, Herrmann A. Headgroup-specific exposure of phospholipids in ABCA1-expressing cells. J Biol Chem. 2005;280:26321–9. doi: 10.1074/jbc.M413993200. [DOI] [PubMed] [Google Scholar]
- 17.Fitzgerald ML, Morris AL, Chroni A, Mendez AJ, Zannis VI, Freeman MW. ABCA1 and amphipathic apolipoproteins form high-affinity molecular complexes required for cholesterol efflux. J Lipid Res. 2004;45:287–94. doi: 10.1194/jlr.M300355-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Vedhachalam C, Ghering AB, Davidson WS, Lund-Katz S, Rothblat GH, Phillips MC. ABCA1-induced cell surface binding sites for ApoA-I. Arterioscler Thromb Vasc Biol. 2007;27:1603–9. doi: 10.1161/ATVBAHA.107.145789. [DOI] [PubMed] [Google Scholar]
- 19.Hassan HH, Denis M, Lee DY, Iatan I, Nyholt D, Ruel I, Krimbou L, Genest J. Identification of an ABCA1-dependent phospholipid-rich plasma membrane apolipoprotein A-I binding site for nascent HDL formation: implications for current models of HDL biogenesis. J Lipid Res. 2007;48:2428–42. doi: 10.1194/jlr.M700206-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat GH, Lund-Katz S, Phillips MC. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J Biol Chem. 2007;282:25123–30. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- 21.Reboul E, Dyka FM, Quazi F, Molday RS. Cholesterol transport via ABCA1: new insights from solid-phase binding assay. Biochimie. 2013;95:957–61. doi: 10.1016/j.biochi.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Sorci-Thomas MG, Owen JS, Fulp B, Bhat S, Zhu X, Parks JS, Shah D, Jerome WG, Gerelus M, Zabalawi M, Thomas MJ. Nascent high density lipoproteins formed by ABCA1 resemble lipid rafts and are structurally organized by three apoA-I monomers. J Lipid Res. 2012;53:1890–909. doi: 10.1194/jlr.M026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doughman RL, Firestone AJ, Wojtasiak ML, Bunce MW, Anderson RA. Membrane ruffling requires coordination between type Ialpha phosphatidylinositol phosphate kinase and Rac signaling. J Biol Chem. 2003;278:23036–45. doi: 10.1074/jbc.M211397200. [DOI] [PubMed] [Google Scholar]
- 24.Doughman RL, Firestone AJ, Anderson RA. Phosphatidylinositol phosphate kinases put PI4,5P(2) in its place. J Membr Biol. 2003;194:77–89. doi: 10.1007/s00232-003-2027-7. [DOI] [PubMed] [Google Scholar]
- 25.Yin HL, Janmey PA. Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol. 2003;65:761–89. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 26.Sun Y, Thapa N, Hedman AC, Anderson RA. Phosphatidylinositol 4,5-bisphosphate: targeted production and signaling. Bioessays. 2013;35:513–22. doi: 10.1002/bies.201200171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Padron D, Wang YJ, Yamamoto M, Yin H, Roth MG. Phosphatidylinositol phosphate 5-kinase Ibeta recruits AP-2 to the plasma membrane and regulates rates of constitutive endocytosis. J Cell Biol. 2003;162:693–701. doi: 10.1083/jcb.200302051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abel K, Anderson RA, Shears SB. Phosphatidylinositol and inositol phosphate metabolism. J Cell Sci. 2001;114:2207–8. doi: 10.1242/jcs.114.12.2207. [DOI] [PubMed] [Google Scholar]
- 29.Varnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143:501–10. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takenawa T, Itoh T. Phosphoinositides, key molecules for regulation of actin cytoskeletal organization and membrane traffic from the plasma membrane. Biochim Biophys Acta. 2001;1533:190–206. doi: 10.1016/s1388-1981(01)00165-2. [DOI] [PubMed] [Google Scholar]
- 31.Manneville JB, Leduc C, Sorre B, Drin G. Studying in vitro membrane curvature recognition by proteins and its role in vesicular trafficking. Methods Cell Biol. 2012;108:47–71. doi: 10.1016/B978-0-12-386487-1.00003-1. [DOI] [PubMed] [Google Scholar]
- 32.Le Goff W, Zheng P, Brubaker G, Smith JD. Identification of the cAMP-responsive enhancer of the murine ABCA1 gene: requirement for CREB1 and STAT3/4 elements. Arterioscler Thromb Vasc Biol. 2006;26:527–33. doi: 10.1161/01.ATV.0000201042.00725.84. [DOI] [PubMed] [Google Scholar]
- 33.Wenk MR, Lucast L, Di Paolo G, Romanelli AJ, Suchy SF, Nussbaum RL, Cline GW, Shulman GI, McMurray W, De Camilli P. Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat Biotechnol. 2003;21:813–7. doi: 10.1038/nbt837. [DOI] [PubMed] [Google Scholar]
- 34.Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13:423–33. doi: 10.1038/ncb2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harlan JE, Hajduk PJ, Yoon HS, Fesik SW. Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature. 1994;371:168–70. doi: 10.1038/371168a0. [DOI] [PubMed] [Google Scholar]
- 36.Baxter AA, Richter V, Lay FT, Poon IK, Adda CG, Veneer PK, Phan TK, Bleackley MR, Anderson MA, Kvansakul M, Hulett MD. The tomato defensin TPP3 binds phosphatidylinositol(4,5)-bisphosphate via a conserved dimeric cationic grip conformation to mediate cell lysis. Mol Cell Biol. 2015 doi: 10.1128/MCB.00282-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poon I, Baxter AA, Lay FT, Mills GD, Adda CG, Payne JA, Phan TK, Ryan GF, White JA, Veneer PK, van der Weerden NL, Anderson MA, Kvansakul M, Hulett MD. Phosphoinositide-mediated oligomerization of a defensin induces cell lysis. Elife. 2014;3:e01808. doi: 10.7554/eLife.01808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jayaraman S, Abe-Dohmae S, Yokoyama S, Cavigiolio G. Impact of self-association on function of apolipoprotein A-I. J Biol Chem. 2011;286:35610–23. doi: 10.1074/jbc.M111.262485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gursky O, Jones MK, Mei X, Segrest JP, Atkinson D. Structural basis for distinct functions of the naturally occurring Cys mutants of human apolipoprotein A-I. J Lipid Res. 2013;54:3244–57. doi: 10.1194/jlr.R037911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukuda M, Nakano M, Miyazaki M, Tanaka M, Saito H, Kobayashi S, Ueno M, Handa T. Conformational change of apolipoprotein A-I and HDL formation from model membranes under intracellular acidic conditions. J Lipid Res. 2008;49:2419–26. doi: 10.1194/jlr.M800287-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Medina MW, Bauzon F, Naidoo D, Theusch E, Stevens K, Schilde J, Schubert C, Mangravite LM, Rudel LL, Temel RE, Runz H, Krauss RM. Transmembrane protein 55B is a novel regulator of cellular cholesterol metabolism. Arterioscler Thromb Vasc Biol. 2014;34:1917–23. doi: 10.1161/ATVBAHA.113.302806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quazi F, Molday RS. Differential Phospholipid Substrates and Directional Transport by ATP-binding Cassette Proteins ABCA1, ABCA7, and ABCA4 and Disease-causing Mutants. J Biol Chem. 2013;288:34414–26. doi: 10.1074/jbc.M113.508812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mulya A, Lee JY, Gebre AK, Thomas MJ, Colvin PL, Parks JS. Minimal lipidation of pre-beta HDL by ABCA1 results in reduced ability to interact with ABCA1. Arterioscler Thromb Vasc Biol. 2007;27:1828–36. doi: 10.1161/ATVBAHA.107.142455. [DOI] [PubMed] [Google Scholar]
- 44.Nagata KO, Nakada C, Kasai RS, Kusumi A, Ueda K. ABCA1 dimer-monomer interconversion during HDL generation revealed by single-molecule imaging. Proc Natl Acad Sci U S A. 2013;110:5034–9. doi: 10.1073/pnas.1220703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zha X, Genest J, Jr., McPherson R. Endocytosis is enhanced in Tangier fibroblasts: possible role of ATP-binding cassette protein A1 in endosomal vesicular transport. J Biol Chem. 2001;276:39476–83. doi: 10.1074/jbc.M105067200. [DOI] [PubMed] [Google Scholar]
- 46.Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–47. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ, Hiltbold EM, Fessler MB, Parks JS. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res. 2010;51:3196–206. doi: 10.1194/jlr.M006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, Gratton E, Parks J, Tontonoz P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife. 2015;4:e08009. doi: 10.7554/eLife.08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125:943–55. doi: 10.1016/j.cell.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 50.Landry YD, Denis M, Nandi S, Bell S, Vaughan AM, Zha X. ATP-binding cassette transporter A1 expression disrupts raft membrane microdomains through its ATPase-related functions. J Biol Chem. 2006;281:36091–101. doi: 10.1074/jbc.M602247200. [DOI] [PubMed] [Google Scholar]
- 51.Ford MG, Mills IG, Peter BJ, Vallis Y, Praefcke GJ, Evans PR, McMahon HT. Curvature of clathrin-coated pits driven by epsin. Nature. 2002;419:361–6. doi: 10.1038/nature01020. [DOI] [PubMed] [Google Scholar]
- 52.Rousseau A, McEwen AG, Poussin-Courmontagne P, Rognan D, Nomine Y, Rio MC, Tomasetto C, Alpy F. TRAF4 is a novel phosphoinositide-binding protein modulating tight junctions and favoring cell migration. PLoS Biol. 2013;11:e1001726. doi: 10.1371/journal.pbio.1001726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–66. [PubMed] [Google Scholar]
- 54.Mendez AJ, Anantharamaiah GM, Segrest JP, Oram JF. Synthetic amphipathic helical peptides that mimic apolipoprotein A-I in clearing cellular cholesterol. J Clin Invest. 1994;94:1698–705. doi: 10.1172/JCI117515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Remaley AT, Thomas F, Stonik JA, Demosky SJ, Bark SE, Neufeld EB, Bocharov AV, Vishnyakova TG, Patterson AP, Eggerman TL, Santamarina-Fojo S, Brewer HB. Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. J Lipid Res. 2003;44:828–36. doi: 10.1194/jlr.M200475-JLR200. [DOI] [PubMed] [Google Scholar]
- 56.Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M, Egerer-Sieber C, Muller YA, Hla T, Nielsen LB, Dahlback B. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A. 2011;108:9613–8. doi: 10.1073/pnas.1103187108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berisha SZ, Brubaker G, Kasumov T, Hung KT, DiBello PM, Huang Y, Li L, Willard B, Pollard KA, Nagy LE, Hazen SL, Smith JD. HDL from ApoA1 Transgenic Mice Expressing the 4WF Isoform Is Resistant to Oxidative Loss of Function. J Lipid Res. 2015;56:653–64. doi: 10.1194/jlr.M056754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.