

Abstract
Nuclear replication of DNA viruses activates DNA damage repair (DDR) pathways, which are thought to detect and inhibit viral replication. However, many DNA viruses also depend on these pathways in order to optimally replicate their genomes. We investigated the relationship between murine polyomavirus (MuPyV) and components of DDR signaling pathways including CHK1, CHK2, H2AX, ATR, and DNAPK. We found that recruitment and retention of DDR proteins at viral replication centers was independent of H2AX, as well as the viral small and middle T-antigens. Additionally, infectious virus production required ATR kinase activity, but was independent of CHK1, CHK2, or DNAPK signaling. ATR inhibition did not reduce the total amount of viral DNA accumulated, but affected the amount of virus produced, indicating a defect in virus assembly. These results suggest that MuPyV may utilize a subset of DDR proteins or non-canonical DDR signaling pathways in order to efficiently replicate and assemble.
Keywords: Polyomavirus, DNA damage repair, viral replication, ATR, checkpoint signaling
Introduction
Robust replication and repair of DNA is required to maintain the integrity of the cellular genome. DNA damage repair (DDR) pathways protect the genome from mutation by repairing lesions from endogenous and exogenous DNA damage. In mammalian cells, ataxia-telangiectasia mutated (ATM) kinase, ataxia-telangiectasia and Rad3 related (ATR) kinase and DNA dependent protein kinase (DNA-PK) are the phosphatidylinositol 3-kinase-related kinases (PIKKs) that activate DDR proteins and induce checkpoint signaling when cellular DNA damage occurs (Ciccia and Elledge 2010). The ATM and DNA-PK pathways are activated primarily in response to double strand breaks, which are detected and bound by the Mre11-Rad50-Nbs1 (MRN) or Ku70/80 complex (Lee and Paull 2004, Mahaney, et al. 2009, Stracker and Petrini 2011). ATR kinase is activated in response to single stranded DNA lesions bound by the replication protein A (RPA) complex (Cimprich and Cortez 2008, Zou and Elledge 2003). ATM, ATR and DNA-PK also phosphorylate H2AX (γH2AX), to maintain repair enzymes around break sites, and checkpoint kinases, CHK1 and CHK2, to prevent progression of the cell cycle while repair occurs (Bassing, et al. 2002, Celeste, et al. 2003, Cimprich and Cortez 2008, Matsuoka, et al. 2007).
In addition to their role in cellular DNA damage, these DDR pathways are often activated by viruses that replicate and assemble in the nucleus, resulting in a complex interplay between DDR pathways and viral replication. These cellular DDR pathways can both promote and inhibit viral replication and assembly. Herpes Simplex Virus 1 (HSV-1) utilizes a subset of DDR proteins, including ATM kinase and Mre11, to replicate its viral genome, while degrading others such as RNF8 and RNF168 (Lilley, et al. 2005, Lilley, et al. 2011). DDR pathways have also been implicated as a host cell defense that distinguishes between viral genomes and cellular DNA. For example, during adenovirus (Ad5) infection, the cellular MRN complex acts as a potent inhibitor of viral DNA replication, but specific viral gene products, E4ORF3 and E1B55k, counteract this inhibition by degrading the MRN complex (Carson, et al. 2003, Evans and Hearing 2005, Stracker, et al. 2002). The different interactions between DNA viruses and DDR proteins are related both to the structure of the viral genome (Circular versus linear) and the phase of the cell cycle during which these viruses replicate (Chaurushiya and Weitzman 2009, Lilley, et al. 2007).
Polyomaviruses (PyVs) are small dsDNA viruses with circular genomes that replicate in the nucleus. The murine polyomavirus (MuPyV) genome has six gene products, three early T-antigen proteins (small, middle, and large) and three late proteins (VP1, VP2, and VP3). The T-antigen (Tag) proteins prime the cell for viral DNA replication through interactions with host proteins. Small T-antigen (ST) binds to PP2A and modulates the phosphorylation of cellular proteins, including mitogen activated kinase (MAPK) and AKT (Andrabi, et al. 2007, Arroyo and Hahn 2005). Middle T-antigen (MT) is a membrane bound protein that activates signaling pathways through interactions with phosphatidylinositide 3-kinase (PI3K) and src family kinases (Courtneidge and Smith 1983, Fluck and Schaffhausen 2009). Large T-antigen (LT) drives cell cycle progression into S-phase through interactions with host proteins, such as phosphorylated retinoblastoma protein (Rb) (Sheng, et al. 1997, Zalvide, et al. 1998). Additionally, it is thought that LT binds to DDR proteins, including a component of the MRN complex, Nbs1, and the RPA complex to manipulate the host cell DDR to enhance viral DNA replication (Banerjee, et al. 2013, Wu, et al. 2004).
DDR pathways are activated when PyVs replicate in the nucleus. For example, SV40, BKPyV, and MuPyV activate and utilize ATM kinase signaling for efficient replication (Dahl, et al. 2005, Jiang, et al. 2012, Sowd, et al. 2013). In cells infected by PyVs, DDR proteins co-localize at nuclear sites of viral DNA replication (Erickson, et al. 2012, Jiang, et al. 2012, Orba, et al. 2010, Tsang, et al. 2014). MuPyV utilizes ATM signaling to regulate the cell cycle and maintain a prolonged S-phase (Dahl, et al. 2005). SV40 requires both ATM and ATR kinase activity to replicate circular, monomeric viral genomes. Following inhibition of ATM or ATR, SV40 infected cells accumulate concatemerized and damaged viral genomes that cannot be packaged into virions (Sowd, et al. 2013, Sowd, et al. 2014). SV40 LT induces the proteasome-dependent degradation of the MRN complex, an activator of ATM kinase (Zhao, et al. 2008). These findings suggest that some proteins in the ATM pathway are required, while others may inhibit PyV replication.
The roles of DDR pathways and proteins during viral replication are complex because each virus may have particular requirements for replication and assembly. We investigated specific components of the ATM and ATR pathways in order to further elucidate the role of DDR proteins during MuPyV infection. We analyzed the recruitment of DDR proteins, such as Mre11and RPA32, to nuclear viral replication centers, as well as the functional requirement for ATR and checkpoint signaling in MuPyV virus production. Finally, we evaluated DDR signaling and viral replication center formation in cells infected with mutant viruses that do not express ST or MT.
Methods
Cell Lines and Infections
C57BL/6 mouse embryonic fibroblasts (MEF) were obtained from ATCC (SCRC-1008; Manassas, VA) and served as a wild-type (WT) MEF. MEFs were grown in DMEM (D6429, Sigma) supplemented with 10% fetal bovine calf serum (FBS; F0926, Sigma), and 55 μM β-mercaptoethanol (βME) at 37°C with 5% CO2. Virus strain NG59RA (Feunteun, et al. 1976) was used for all WT virus infections. Cells were plated for 24 hrs, then cultured in DMEM supplemented with 0.5% serum for 24 hrs, before infections were carried out in adsorption buffer (Hanks Balanced Salt Solution (HBSS) with 10 mM HEPES, pH 5.6 and 0.5% BCS) as previously described (Cripe, et al. 1995). Cells were infected for 2 hrs at a multiplicity of infection of approximately 10-15 pfu/cell, and then cultured in DMEM supplemented with serum for the remainder of the experiment. CHK2-null and syngeneic control cells were a gift from Tak Mak. H2AX-null and syngeneic WT controls were a gift from Andre Nussenzweig. CHK2 and H2AX-null cells and their syngeneic WT cells were grown in DMEM supplemented with 10% bovine calf serum (BCS), 55 μM βME and penicillin/streptomycin at 37°C with 5% CO2.
Immunofluorescence
Cells were plated on acid-etched coverslips and infected as described above. Coverslips were washed twice with 4 °C phosphate buffered saline (PBS), pre-extracted with 0.5% Triton X-100 in CSK Buffer (10 mM piperazine-1,4-bis(2-ethanesulfonic acid) (PIPES), pH 6.8, 100 mM NaCl, 300 mM sucrose, 1 mM MgCl2, 1 mM EGTA) on ice for 5 mins, washed twice with 4 °C PBS, and fixed with 4% paraformaldehyde (PFA) for 10 mins at room temperature (RT). Following an overnight block in 10% BCS, coverslips were incubated with primary antibody at 37°C for 1 hr, washed three times with 10% BCS in PBS, then incubated for 1 hr at RT with secondary antibodies. Slides were washed three times in PBS and then mounted in ProLong antifade reagent containing DAPI (Invitrogen) and allowed to cure at 22 °C for 24-48 hrs.
Immunofluorescence antibodies
Primary antibodies used for immunostaining were rat anti-Tag (E1, a gift from Tom Benjamin) (Goldman and Benjamin 1975) 1:5000; rabbit anti-VP1(Montross, et al. 1991) 1:5000, rabbit anti-pCHK1 (s345, Cell Signaling) 1:1000; mouse anti-pATM (ser1981, Abcam) 1:1000; rabbit anti-Mre11a (B1447; LSBio) 1:1000; rat anti-RPA32 (4E4, a gift from Heinz Nasheuer) 1:5; and rat anti-pSer23 RPA32 (a gift from Heinz Nasheuer) 1:20. For RPA32 co-staining with rat anti-Tag antibody, the E1 Tag antibody was conjugated with AlexaFluor 647 (Invitrogen) and was used at a 1:1000 dilution. All other primary antibodies were detected using secondary antibodies, conjugated with AlexaFluor 488, AlexaFluor 546, or AlexaFluor 647 (Invitrogen), diluted 1:2000. All primary and secondary antibodies were diluted in 10% BCS in PBS.
Fluorescent in Situ Hybridization (FISH) of MuPyV DNA
The entire MuPyV genome (NG59RA) was cloned into pUC18 and a FISH probe was generated by nick translation of pUC18-MuPyV plasmid using Promofluor-550 NT Labeling Kit (PK-PF550-NTLK) according to manufacturer's instructions. FISH analysis was performed as described previously (Jul-Larsen, et al. 2004), with modifications. Briefly, cells grown on coverslips were infected, fixed and immunostained for viral or host proteins, as described above. Immunostained cells were fixed a second time with 4% PFA in PBS to crosslink bound antibodies, followed by treatment with 0.2 mg/ml RNase Type III (Sigma) in 2X SSC at 37°C for 15 min and washed three times in 2× SSC. The FISH probe was diluted in cDenHyb (InSitus). The probe was hybridized with samples for 3 min at 90°C, for 2 min each at 80°C, 70°C, 60°C, 50°C, 45°C, and finally overnight at 37°C. Coverslips were washed at 45°C with 1.5× SSC, 50% formamide/1.5× SSC for 5 mins and 1.5× SSC for 5 mins. Coverslips were washed twice for 5 min in PBS at RT. Stained cells were mounted onto glass slides with ProLong antifade reagent containing Dapi (Invitrogen) and allowed to cure at 22 °C for 24-48 hrs.
EdU labeling
EdU was added to media at final concentration of 10 μM at 28 HPI and allowed to incubate for 15 min at 37°C. Coverslips were washed once in PBS and immediately fixed and stained, as described above. The fluorescent dye conjugation reaction was performed according to manufacturer's protocol (Invitrogen, C10340).
Microscopy
Confocal images were acquired on the Nikon A1R laser scanning confocal microscope, using a 1.45NA 100× objective and 405, 488, 561 and 638 laser lines. All images are a single z-plane through the center of the nucleus. Image processing and analysis were completed using ImageJ analysis software (NIH).
Western Blot Analysis
Cells were lysed with 2X SDS lysis buffer with phosphatase inhibitors (25 mM Tris, pH 6.8 / 1% SDS / 6.25 mM EDTA with 1 mM Na3VO4 and 25 mM NaF) on ice for 15 mins followed by a brief sonication to shear the DNA. The total protein concentration of each lysate was determined by BioRad DC Protein Assay. Samples were prepared by dilution in 4× SDS sample buffer (1 M Tris, pH 6.8 / 2% SDS / 12.5 mM EDTA / 40% glycerol / 600 mM βME / 0.02% Bromophenol Blue) and heated at 95 °C for 5 mins. 25 μg total protein was resolved on 8% SDS-polyacrylamide gels and proteins were electrophoretically transferred to PVDF membranes. Membranes were blocked in 5% powered milk in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.1% Tween-20) with 1 mM Na3VO4 and 25 mM NaF. All antibodies except for pCHK1 were incubated at 37°C with rocking for 1 hr followed by three 20 min washes in TBST with rocking. The pCHK1 antibody was incubated at 4°C with rocking overnight, and then followed the same wash and secondary antibody protocol. Membranes were incubated with HRP-conjugated secondary antibodies (Promega) diluted 1:20,000 in TBST and washed as described for the primary antibodies. Proteins were detected using enhanced chemiluminescence (ECL, Pierce) detection and imaged on the LAS4000 Imager. Image processing and integrated density analysis were completed using ImageJ analysis software (NIH).
Western Blot Antibodies
Primary antibodies used for western blot were: rabbit anti-Rad50 (GeneTex, GTX119731) 1:1000; rabbit anti- pCHK1 S317 (Cell Signaling, 12302P) 1:200; rabbit anti-pCHK1 S296 (Cell Signaling, 2349) 1:1000, rabbit anti-GAPDH (Abcam, 37168) 1:1000; mouse anti-Tag, PN116 (a gift from Brian Schaffhausen), 1:250; rabbit anti-VP1 (I58), 1:5000 (Montross, et al. 1991).
Small molecule inhibitors
All small molecule inhibitors were dissolved in DMSO and diluted in DMEM with 0.5% serum to working concentrations and added to cells at indicated time points: NU7441 (Tocris), 2 μM; PF477736 (Tocris), 1μM; and ATR inhibitor IV (VE-821, EMD Millipore), 5 μM.
Virus Supernatant Harvest
Cells (4×104 per well) were seeded on a six well dish (9.5cm2 well) for 24 hrs, then starved in 0.5%FBS or BCS for 24 hrs before infection. At this time, the cells were 60-70% confluent. Virus preparation and infection was carried out as described above. After a 2 hr incubation with virus, 0.5% serum in DMEM was added for the remainder of the experiment. At times indicated, supernatants were transferred to 15 ml conical tubes and saved. Remaining cells on the plate were treated with 2 units of neuraminidase (NA) Type V (Sigma) that was diluted in NA buffer (10 mM Hepes, pH 5.6/1 mM CaCl2/1 mM MgCl2/5 mM KCl) for 30 min at 37°C. The NA supernatant was collected and combined with the original supernatant. The plates were washed three times with PBS and each wash was collected and combined with supernatants. The combined supernatants and washes were stored at −20°C.
Immunoplaque Assay
WT C57 cells were plated on a 96-well optical imaging dish (Costar, 9603) for 24 hrs, then cultured in 0.5% FBS for an additional 24 hrs. Cells were infected with viral supernatants diluted in adsorption buffer (as described above) at 1:100-1:1000 and infected in triplicate. Virus was removed after 2 hrs and DMEM with 0.5% FBS was added to cells. At 28-30 HPI, cells were fixed with 4% PFA/PBS at RT for 10 min, permeabilized with 0.5% Triton X-100/ PBS. Samples were then incubated with anti-Tag and anti-VP1 antibodies at dilutions described for immunofluorescence, followed by AlexaFluor 488 anti-rabbit, and AlexaFluor 546 anti-rat secondary antibodies. Nuclei were stained with Hoescht dye diluted in PBS. Plates were imaged on a Molecular Devices ImageXpress Micro XL High-Content Screener with a 10× objective. Tag and VP1 positive nuclei were determined based on the average pixel intensity within the nuclei, as defined by Hoescht dye staining. Image analysis was completed using ImageJ analysis software (NIH). An example of the analysis outlined in Figure S1.
Cell Line Normalization
For CHK2 control and knockout, and H2AX WT and knockout comparisons, viral titers and viral DNA accumulation were normalized for the infection level of each individual cell line. Cell line normalization is described in Figure S2.
DNA Isolation
Viral DNA was isolated by Hirt extraction (Hirt 1967), with modifications. Briefly, cellular DNA was precipitated in high salt and remaining supernatants were treated with 10 μg RNase, followed by 25 μg of proteinase k. Viral DNA was purified by phenol-chloroform extraction and sodium acetate/ethanol precipitation. The DNA pellet was resuspended in 50 μL Tris-EDTA, diluted 1:200, and analyzed by qPCR.
Quantitative PCR (qPCR)
Primer Express 3.0 software (Applied Biosystems, Warrington, UK) was used to design primers that amplified of a 67-bp region of the VP1 gene of the MuPyV genome (NCBI accession # NC_001515). Primers used for qPCR were: MuPyV VP1 forward primer, 5′TGGGAGGCAGTCTCAGTGAAA3′; MuPyV VP1 reverse primer, 5′TGAACCCATGCACATCTAACAGT3′. qPCR reactions were prepared in 96-well optical plates (Applied Biosystems) in a volume of 25 μl. Each reaction contained 450 nM of each forward and reverse primer, 12.5 μl FastSYBR Master Mix (Applied Biosystems) and 5 μl purified viral DNA or DNA standards. DNA amplification was carried out using a Biorad CFX96 thermocycler using cycling conditions of 50°C for 2 min, 95°C for 10 min followed by 40 cycles of 95°C for 15 sec, 60°C for 1 min. For each run, triplicates of five dilutions of the viral standard DNA (from 0.5 ng to 8×10-4 ng; pGEX-VP1 plasmid DNA), viral DNA samples and no template controls were simultaneously analyzed.
Statistical analysis
All error bars are standard error of the mean of three biological replicates. Statistical significance was calculated using a two-tailed student's t-test assuming unequal variance. p values of <0.05 indicated with *. Non-significant differences marked “n.s.”
Results
MuPyV recruits DDR proteins to viral DNA replication centers in the nucleus of infected cells
JCPyV, BKPyV, and SV40 activate and recruit host DDR proteins to sites of viral replication in the nucleus (Jiang, et al. 2012, Orba, et al. 2010, Zhao, et al. 2008). Therefore, we characterized nuclear MuPyV viral replication centers in infected mouse embryonic fibroblasts (MEFs). The MuPyV LT formed “tracks” throughout the nucleus that co-localized with replicating viral DNA (Fig. 1A). We analyzed the localization of DNA repair proteins during infection and found many that were recruited to these viral replication centers, including RPA32, and Mre11 (Fig. 1B, E). Additionally, DDR signaling was active at replication centers, as evidenced by phosphorylated RPA32 (pSer23 RPA32), CHK1 (pCHK1), ATM (pATM) and H2AX (γH2AX) (Fig. 1C, D, F, G). DDR proteins do not form similar replication centers in the nucleus of uninfected MEFs (Fig S3). These data suggest that MuPyV activates and recruits DDR proteins similar to other PyVs.
Figure 1. MuPyV activates and reorganizes host DDR proteins in the nucleus.
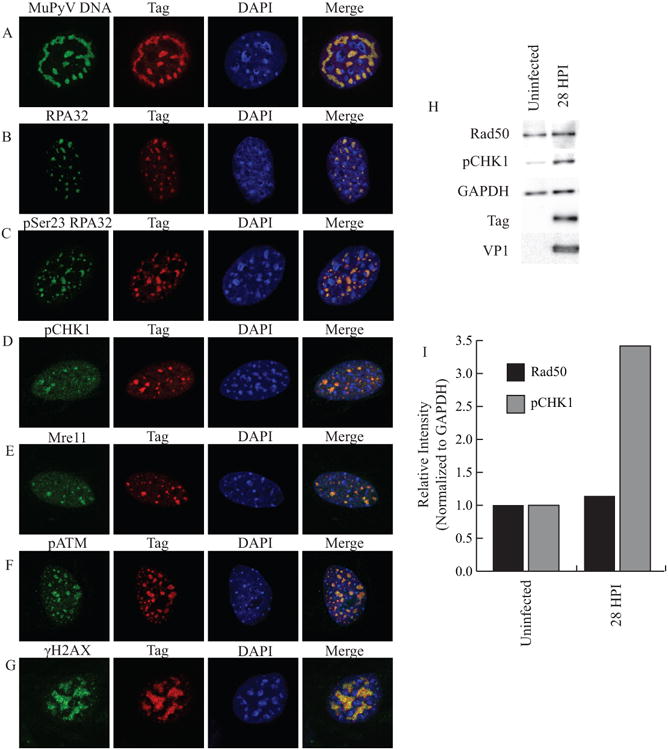
MEFs were infected with WT MuPyV (NG59RA), and fixed, permeabilized, and immunostained at 28 hrs post infection (HPI). Single z-plane confocal immunofluorescence images show cells stained for Tag and A) MuPyV DNA (FISH) B) RPA32 C) pSer23 RPA32 D) pSer345 CHK1 E) Mre11 F) pATM or G) γH2AX. H) Total proteins were isolated at 28 HPI and analyzed by immunoblot for pSer317 CHK1 and Rad50 protein levels. I) Quantification of integrated density of pSer317 CHK1 and Rad50 levels in control and infected MEFs normalized to GAPDH loading control. All samples were normalized to the value of the uninfected control cells.
It has been previously reported that SV40 infection activates DDR signaling and induces the degradation of the MRN complex (Zhao, et al. 2008). We therefore analyzed immuno-blots of infected cell lysates to determine if levels of MRN proteins were affected during MuPyV infection. In contrast to SV40, we found that expression levels of Rad50 and Mre11, members of the MRN complex, were comparable in mock and infected samples (Fig. 1H, Fig. S4). However, similar to SV40 there was a striking increase in the amount of pCHK1 in MuPyV infected cells (Fig. 1H). Quantification of the immuno-blots revealed an approximately three-fold increase in pCHK1, but no change in Rad50 levels (Fig. 1I). These data suggest that MuPyV activates and recruits DDR proteins, but does not affect their overall expression or stability, in contrast to observations during SV40 infection.
Recruitment of DDR proteins is not dependent on ST or MT
Recruitment of DDR proteins to sites of SV40 viral DNA replication was previously found to depend on the viral LT (Zhao, et al. 2008). Thus, we determined whether MuPyV LT was sufficient for DDR protein recruitment to replication centers, by analyzing the formation of viral replication centers in cells infected with MuPyV mutant viruses lacking ST and MT. NG18 is a mutant virus with a deletion that abrogates the expression of both ST and MT, and 808A is a mutant virus with a mutation in the MT splice acceptor, preventing expression of only MT, while allowing ST expression (Benjamin 1982, Garcea and Benjamin 1983, Garcea, et al. 1989). Interestingly, NG18 replication centers (Fig. 2A) were smaller than either 808A (Fig. 2A) or WT virus (Fig. 1A). These data suggest that ST has a role in the expansion of these centers, possibly through activation of other signaling pathways that aid DNA replication. However, recruitment of phosphorylated RPA32 (Fig. 2B) and Mre11 (Fig. 2C) were maintained during infection by 808A and NG18 mutant viruses, suggesting that LT is likely the viral protein responsible for recruitment of DDR proteins to viral replication centers.
Figure 2. ST and MT are not required for localization of DDR proteins to viral replication centers.
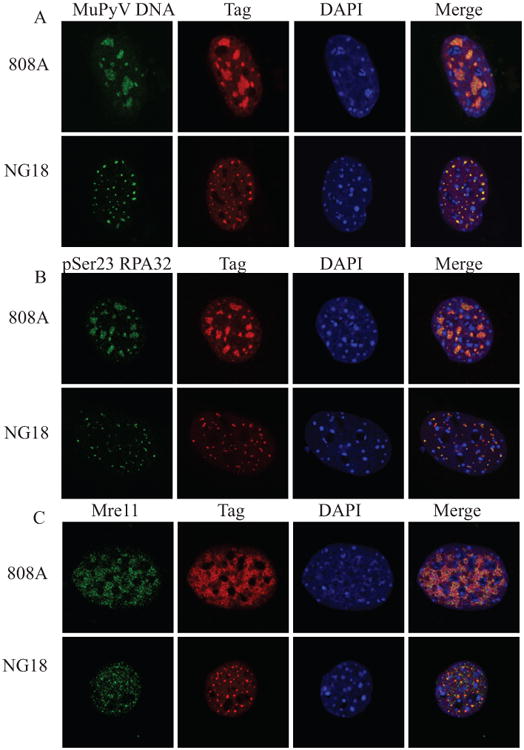
MEFs were infected with the MT mutant virus (808A) or the ST/MT mutant virus (NG18). At 28 HPI cells were fixed, permeabilized, and immunostained. Single z-plane confocal immunofluorescence images showing cells stained for A) Tag and MuPyV DNA (FISH), B) Tag and pSer23 RPA32, and C) Tag and Mre11.
Mre11 is preferentially recruited to replicating viral DNA
Many DDR proteins, such as the MRN complex, have roles in both the DNA damage response and cellular DNA replication (Stracker and Petrini 2011). The MRN complex normally stabilizes replication forks at sites of cellular DNA replication (Maser, et al. 2001, Tittel-Elmer, et al. 2009). In order to distinguish replicating viral DNA from replicating cellular DNA we labeled replicating DNA in infected cells with EdU, and a fluorescent in-situ hybridization (FISH) probe specific to the MuPyV genome (Fig. 3A). To determine whether Mre11 was recruited to all actively replicating DNA, we also immunostained for Mre11 and analyzed its recruitment to replicating host or viral DNA (Fig. 3B). Host cell DNA replicated during early times after infection and could be visualized along with replicating viral DNA. During infection, Mre11 specifically localized to replicating viral DNA and not replicating cellular DNA (Fig 3C, D, Fig. S5). Thus, Mre11 is preferentially recruited to viral replication centers.
Figure 3. Mre11 is preferentially recruited to replicating MuPyV DNA during infection.
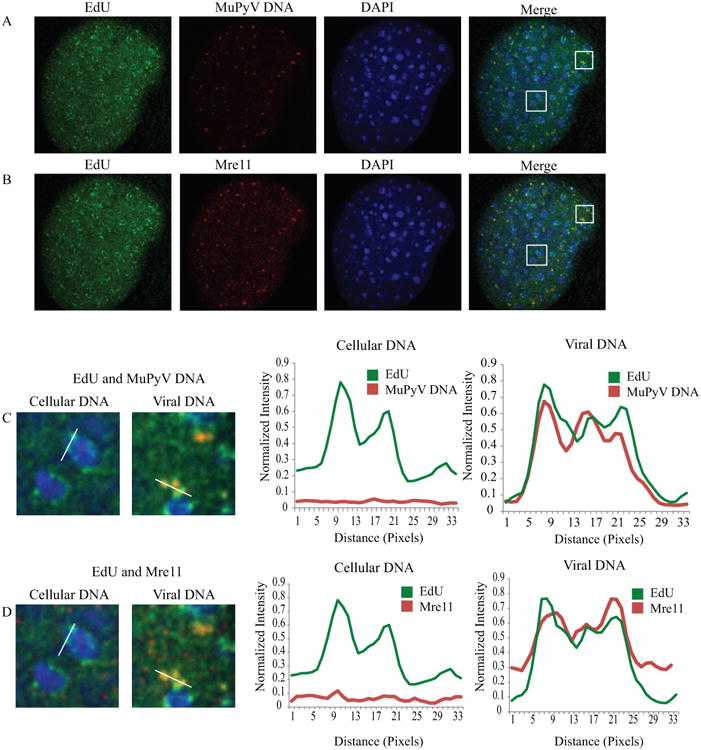
MEFs were infected with NG59RA, at 28 HPI. 10 μM EdU was added to the media. Samples were then fixed, permeabilized, and stained for EdU, MuPyV DNA, and Mre11. Single z-plane confocal image shows A) EdU and MuPyV DNA or B) EdU and Mre11 (same cell, pseudo-colored for clarity). C) Insets show EdU-labeled cellular DNA (left) or EdU-labeled viral DNA (right) with corresponding line scan intensity plots, indicated in each panel. D) Insets show Mre11 localization with cellular DNA (left) and viral DNA (right), with corresponding line scan intensity plots, indicated in each panel. Intensity plots were normalized to image maximum for each label.
Checkpoint Kinases, CHK1 and CHK2, are not required for MuPyV replication
Activation of CHK1 and CHK2 has been observed during JCPyV and SV40 infection (Boichuk, et al. 2010, Orba, et al. 2010). It has been suggested that intra S-phase signaling stalls MuPyV-infected cells in S or G2 phase, allowing viral replication to proceed for an extended time (Dahl, et al. 2007, Dahl, et al. 2005). To assess the potential requirement for CHK1 and CHK2 during MuPyV infection, we utilized a CHK1 inhibitor (PF477736) or CHK2-null fibroblasts (Fig. S6). As expected, PF477736 treatment significantly reduced CHK1 autophosphorylation at Ser296 during infection (Fig. S7). However, inhibition of CHK1 using PF477736 did not affect viral titers (Fig. 4A) or viral DNA replication (Fig. 4B). Additionally, CHK2-null fibroblasts produced infectious viral progeny (Fig. 4A) and replicated viral DNA at similar levels to syngeneic control (CHK2 +/−) cells (Fig. 4B). These results suggest that, despite robust CHK1 activation during infection, neither CHK1 nor CHK2 kinases are required for efficient MuPyV replication and assembly.
Figure 4. CHK2 and CHK1 are not required for MuPyV virus production or viral DNA accumulation.

Cells were infected with NG59RA. CHK1 inhibitor or DMSO control was added to media for indicated samples at 2 HPI. A) Viral titers at 48 HPI quantified by immunoplaque assay, normalized to the value of the control. B) Viral DNA accumulation at 32 HPI quantified by qPCR, normalized to the value of the control.
H2AX is not required for MuPyV Replication
The histone marker γH2AX is required for maintenance of DDR proteins at sites of cellular DNA damage and for full activation of downstream signaling (Bassing, et al. 2002, Celeste, et al. 2002, Rogakou, et al. 1999). Since γH2AX is present at viral replication centers (Fig. 1G), we used H2AX-null fibroblasts to determine whether H2AX is required for viral DNA replication, virus assembly, or DDR signaling during MuPyV infection (Fig. S6) (Celeste, et al. 2002). We found that viral replication centers that formed in H2AX-null fibroblasts were similar to those observed in WT cells (Fig. 5A). Additionally, H2AX was dispensable for recruitment of Mre11 and pCHK1 to viral replication centers (Fig. 5B, C). Interestingly, H2AX-null fibroblasts produced similar outputs of virus to syngeneic WT cells (Fig. 5D), but replicated approximately two-fold more viral DNA than WT (Fig. 5E). These data suggest that although H2AX may affect accumulation of viral DNA, overall it is not required for MuPyV genome replication and virus assembly.
Figure 5. H2AX is not required for recruitment of DDR proteins, virus production, or viral DNA accumulation during MuPyV infection.
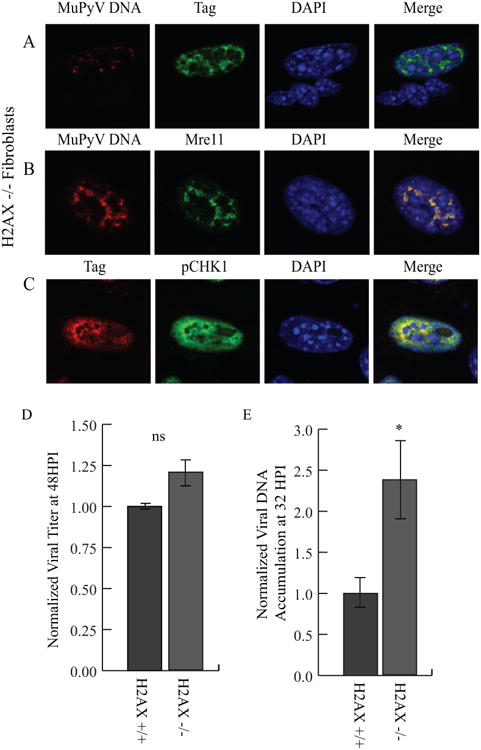
H2AX-null (H2AX-/-) fibroblasts were infected with NG59RA, and at 28 HPI cells were fixed and immunostained. Single z-plane confocal images of cells stained for A) Viral DNA and Tag, B) Viral DNA and Mre11, or C) Tag and pCHK1. D) H2AX WT (H2AX +/+) and null (H2AX-/-) cells were infected with NG59RA. Virus supernatants were harvested at 48 HPI and viral titers were quantified by immunoplaque assay, normalized to the value of the control or E) viral DNA was harvested at 32 HPI and viral DNA accumulation was quantified by qPCR, normalized to the value of the control.
ATR signaling is required for virus production, but not viral DNA replication
During SV40 infection, ATR kinase activity stabilizes and resolves viral DNA replication intermediates (Sowd, et al. 2013). Following DNA damage, CHK1 is directly phosphorylated by ATR and can activate checkpoint signaling (Zhao and Piwnica-Worms 2001). In contrast, DNA-PK, the DDR kinase associated with non-homologous end-joining (NHEJ), is not required during SV40 DNA replication (Sowd, et al. 2013). To determine whether ATR activity or DNA-PK activity are required for MuPyV replication, we utilized small molecule inhibitors of ATR (VE-821) and DNA-PKcs (NU7441). As expected, inhibition of ATR resulted in an approximate 50-70% decrease in CHK1 phosphorylation in both control and infected MEFs (Fig 6. A, B).
Figure 6. ATR signaling is required for virus production, but not viral DNA replication.
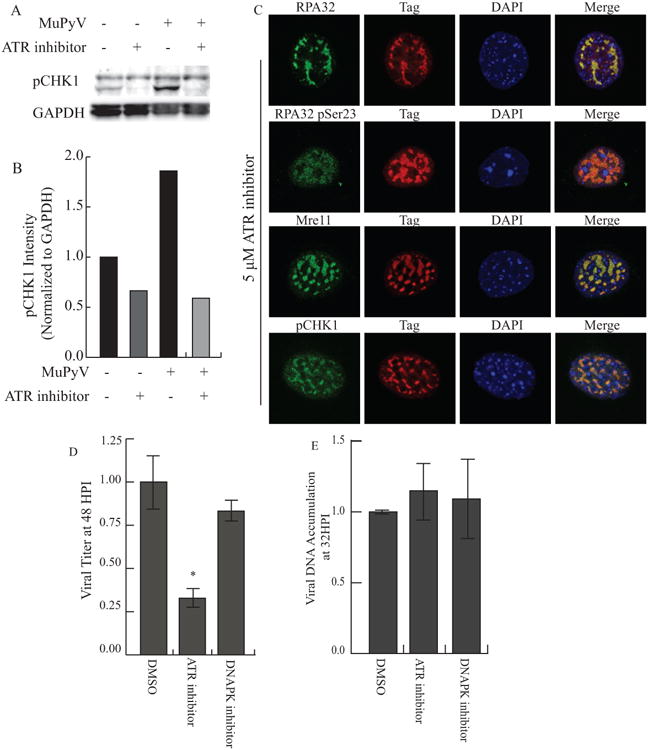
MEFs were infected with NG59RA. At 16 HPI DMSO control or inhibitors were added and the infection proceeded until analysis. A) At 28 HPI total protein from DMSO or ATR inhibitor-treated cells was isolated and analyzed. Immunoblot of pCHK1 (S317) and GAPDH in infected and control MEFs. B) Quantification of integrated density of pCHK1 immunoblot, normalized to GAPDH loading control. All samples were normalized to the value of the untreated control cells. C) Single z-plane confocal immunofluorescence images showing cells stained for Tag and RPA32, pSer23 RPA32, Mre11 or pCHK1. D) Viral titers from MEFs treated with DMSO, 2 μM DNA-PK inhibitor (NU7441), or 5 μM ATR inhibitor, quantified by immunoplaque assay. E) Viral DNA accumulation from MEFs treated with DMSO, 2 μM DNA-PK inhibitor (NU7441), or 5 μM ATR inhibitor, quantified by qPCR. All samples were normalized to the value of the control.
However, ATR inhibitor treatment did not affect the recruitment of DDR proteins such as RPA32, Mre11, and pCHK1, to viral replication centers (Fig. 6C). Interestingly, ATR inhibition reduced virus production (Fig. 6D), but did not affect replication of viral DNA (Fig. 6E). In contrast, inhibition of DNA-PK during MuPyV infection did not affect either viral titer (Fig. 6D) or viral DNA accumulation (Fig. 6E), similar to observations for SV40. The reduction in viral titer in ATR inhibitor treated samples was not due to decreased cell viability or a reduction in the number of cells expressing Tag (Fig. S8). These results suggest that ATR signaling, but not DNA-PK activity, may aid MuPyV in resolving viral DNA replication intermediates, similar to its role in SV40 DNA replication.
Discussion
Viruses and host DDR signaling have a complex relationship that varies significantly between different viruses. PyVs have been shown to inhibit or degrade members of DDR pathways, such as components of the MRN complex (Wu, et al. 2004, Zhao, et al. 2008). Conversely, DDR signaling through the ATM pathway seems to be ubiquitously required for PyV replication and assembly (Dahl, et al. 2005, Jiang, et al. 2012, Shi, et al. 2005, Sowd, et al. 2013, Tsang, et al. 2014, Zhao, et al. 2008). In order to interrogate the relationship between MuPyV and DDR pathways we utilized knockout cell lines, small molecule inhibitors, and confocal microscopy to visualize viral replication centers. We found that MuPyV MT and ST are dispensable for DDR protein recruitment to replication centers, and that efficient virus production requires ATR signaling. Several roles for DDR pathways during PyV infection have been suggested, including maintenance of the S-phase of the cell cycle, repair of damaged viral DNA, resolution of replication intermediates, or a host defense (Dahl, et al. 2005, Jiang, et al. 2012, Orba, et al. 2010, Sowd, et al. 2013, Zhao, et al. 2008). Our results suggest that checkpoint kinase signaling and H2AX phosphorylation may be dispensable for MuPyV replication, but ATR activity is required.
MuPyV ST and MT interact with PP2A and other host proteins to activate cellular signaling pathways. Mutant viruses lacking ST and MT have significant defects in viral DNA replication and associated reductions in virus outputs. However, viruses with mutations in MT alone have close to WT levels of DNA accumulation, but substantially reduced viral titers (Chen, et al. 2006, Garcea and Benjamin 1983, Garcea, et al. 1989). Cells infected by the ST and MT mutant virus, NG18, had smaller viral replication centers than cells infected by either WT virus or the MT mutant virus, 808A. Despite the smaller size, DDR proteins, such as Mre11 and RPA32, were recruited to WT and mutant virus replication centers. This observation suggests that recruitment of DDR proteins to replication centers is mediated through LT. The MuPyV LT may facilitate recruitment and activation of DDR protein through an interaction with the RPA complex or NBS1 (Banerjee, et al. 2013, Wu, et al. 2004). However, the increased viral DNA accumulation and the expansion of viral replication centers observed in WT and 808A infected cells is likely dependent on signaling pathways regulated by ST, including AKT or MAPK.
Biochemical analysis of SV40 infected cells revealed that inhibition of either ATM or ATR increased the accumulation of incorrectly resolved viral genomes (Sowd, et al. 2013). Specifically, inhibition of ATM was observed to increase unidirectional fork and rolling circle replication, while inhibition of ATR showed a slight increase in these aberrant replication products along with an increase in broken and unrepaired viral genomes (Sowd, et al. 2013, Sowd, et al. 2014). Previous reports have shown a requirement for ATM kinase in MuPyV replication (Dahl, et al. 2005). We found an additional requirement for ATR signaling during MuPyV infection. ATR inhibition significantly reduced viral titers, but not total viral DNA accumulation, suggesting a defect in assembly or egress. It is likely that ATR inhibitor treatment leads to the formation of viral DNA concatemers or other viral DNA intermediates that cannot be packaged into virions, similar to observations for SV40. However, inhibition of CHK1, a downstream target of ATR, did not affect viral titers or DNA accumulation indicating that checkpoint signaling from the ATR pathway is not required for MuPyV replication and assembly. The defects in viral titers we observed are therefore due to another function of ATR, such as recruitment or activation of repair proteins. SV40 LT induces the degradation of the MRN complex during infection; however, levels of the MRN complex remained constant during MuPyV infection. This finding suggests that although SV40 and MuPyV require ATM and ATR signaling, there are differences in their interaction with DDR proteins.
MuPyV may utilize signaling through ATM to induce a prolonged S-phase in order to efficiently replicate (Dahl, et al. 2005). CHK2 is a direct target of ATM kinase and can activate checkpoint signaling following DNA damage (Ahn, et al. 2004). To determine if activation of checkpoint signaling by CHK2 was required for MuPyV infection, we analyzed CHK2-null MEFs (Hirao, et al. 2000) and found that MuPyV did not require CHK2 in order to efficiently replicate. Additionally, we found that CHK1, a downstream target of ATR kinase, was not necessary for efficient MuPyV virus production. Although we did not observe a requirement for either CHK1 or CHK2 signaling during infection, it is possible that either kinase is sufficient to compensate for the other and a combinatorial inhibition or knockdown would be necessary to observe a phenotype.
H2AX is a histone variant present in a subset of nucleosomes and is thought to act as a signal of DNA damage and chromatin abnormalities. Phosphorylation of H2AX at serine 139 generates γH2AX, which activates and recruits downstream DDR proteins. The loss of H2AX during cellular DNA damage results in reduced DDR mediated checkpoint signaling and reduced retention of DDR proteins at DNA break sites (Celeste, et al. 2003, Fernandez-Capetillo, et al. 2002). Interestingly, during MuPyV infection, H2AX is not required to activate checkpoint kinase CHK1 or to retain DDR proteins such as Mre11 at viral DNA replication centers. Additionally, we found that while H2AX null fibroblasts accumulated higher levels of viral DNA, there was not a significant increase in viral titer produced by these cells. It has been reported that when extreme levels of cellular DNA damage occur, H2AX is not required to maintain downstream signaling (Fernandez-Capetillo, et al. 2002). Therefore, it is possible viral replication induces an “extreme” level of DNA damage, since MuPyV replication activates CHK1 in the absence of H2AX. However, a more intriguing explanation is that a different mechanism of activation or recruitment of DDR proteins occurs during viral infection than during cellular DNA damage.
During adenovirus infection, non-canonical DDR pathways are activated in response to viral DNA replication. A “localized” activation of DDR signaling occurs at adenovirus (Ad5) replication centers, which does not induce H2AX phosphorylation (Shah and O'Shea 2015). We observed a similar activation of DDR signaling that was independent of H2AX. This finding is consistent with models proposing “global” signaling following cellular DNA damage, and “local” DDR signaling at viral replication centers. These varying types of DDR signaling may aid the cell in distinguishing viral DNA from host DNA damage. Our data support the hypothesis that these local signaling events may not require the same signaling molecules as global cellular DDR. Although MuPyV exploits some DDR proteins in order to replicate, it does not require many components of the ATM and ATR pathways, including checkpoint signaling, a hallmark of canonical cellular DDR signaling. A more thorough understanding of the mechanism of activation and recruitment of DDR proteins by viral DNA replication is required to better understand these non-canonical signaling pathways.
Supplementary Material
Figure S1: Quantification of viral titers using an immunoplaque assay. A) DAPI (blue) and Tag (green) staining of cells used in virus quantification protocol. B) DAPI image with threshold applied to define outline of nucleus and Tag image with Regions of Interest (ROIs) outlined in white. C) Standard curve showing a linear correlation between virus added and percent of cells infected.
Figure S2: Strategy for assessing MuPyV replication across cell types. To address the requirement for CHK2 and H2AX during MuPyV replication and assembly, we utilized knockout and syngeneic control fibroblasts. We found differences in the number of permissive cells (i.e. infection efficiency) between cell lines. We normalized for these differences in infection efficiency across cell lines using an imaging based assay to quantify the number of cells infected during any given experiment To assess the role of DDR proteins in MuPyV replication and assembly, cell lines were normalized for infection efficiency. A) Timeline of viral titer and viral DNA accumulation assays. B) Equation used to calculate the normalized viral titer and DNA accumulation between cell lines of variable infection levels. C) Infection levels for cell lines quantified by high throughput imaging of Tag staining. D) Raw values for viral titer, quantified by immunoplaque assay and viral DNA accumulation, quantified by qPCR. E) Calculated reported values for viral titer and viral DNA accumulation for each cell line, normalized for infection efficiency.
Figure S3: Localization of DDR proteins in uninfected MEFs. MEFs were fixed, permeabilized and immunostained for A) RPA32 B) pSer23 RPA32 C) pCHK1 D) Mre11 E) pATM or F) γH2AX.
Figure S4: Mre11 is not degraded during MuPyV infection. A) MEFs were mock infected or infected with NG59RA. Total proteins were isolated at 24 and 32 HPI and analyzed by immunoblot for Mre11 levels. B) Quantification of integrated density of Mre11 immunoblot, normalized to actin loading control. All samples were normalized to the value of the mock treated cells.
Figure S5: Quantification of Mre11 at MuPyV replication centers and replicating cellular DNA. MEFs were mock infected or infected for 28 hrs, 10 μM EdU was then added to the media. Samples were then fixed, permeabilized, and stained for EdU, Tag, and Mre11. 2.5 μ m z-stacks were acquired on a spinning disc confocal using a 100X objective, NA 1.45. A) Single z-plane confocal images show DAPI, Tag, EdU, and MRE11 in uninfected and infected cells. B) The region of interest (ROI) for “total DNA” was defined by DAPI staining. The ROI that marked the viral replication centers, or “viral DNA”, was defined by Tag staining. The ROI that marked cellular DNA in infected cells or “cellular DNA (infected)” was defined by subtracting the Tag ROI from the DAPI ROI. Uninfected cells were analyzed using the “total DNA” ROI, only. All ROIs used for quantification are shown in white. C) Merged image of the EdU (green) and Mre11 (red) showing localization in ROIs of an infected nucleus and an uninfected nucleus. D) Co-localization analysis showing the percent of EdU co-localized with Mre11 in infected cells in the “total DNA” region, “viral DNA” region and “cellular DNA” region and uninfected cells.
Figure S6: Genotype validation for control and null cell lines. A) CHK2 control and null cell lines were analyzed using PCR primers specific for WT and neomycin alleles. WT PCR primers amplified a 440 bp product for the WT allele and no product for the null allele. Neo primers amplified a 900 bp product for the null allele and no product for the WT allele. B) H2AX control and null cell lines were analyzed using PCR primers specific for WT and null alleles. WT PCR primers amplified a 547 bp product for the WT allele and no product for the null allele. Null primers amplified a 424 bp product for the null allele and no product for the WT allele.
Figure S7: CHK1 autophosphorylation is inhibited by PF477736 treatment during infection. A) MEFs were mock infected or infected with NG59RA and at 2 HPI DMSO or 1 μM PF477736 were added to the media. Total proteins were isolated at 28 HPI and analyzed by immunoblot for pSer296 CHK1 levels. B) Quantification of integrated density of pSer296 CHK1 levels in control and infected MEFs normalized to GAPDH loading control. All samples were normalized to the value of the uninfected control cells.
Figure S8: Cell viability and infection levels following treatment with ATR inhibitor (VE-821) A) MEFs were treated with DMSO or ATR inhibitor for 32 hrs, and cell viability was measured by propidium iodide uptake using high throughput microscopy. The data were normalized to DMSO control, with live cell counts shown in black and dead cell counts shown in gray. B) MEFs were infected with NG59RA. At 16 HPI DMSO or the ATR inhibitor was added and the infection proceeded until 32 HPI. The number of infected cells in DMSO and ATR inhibitor treated samples at 32 HPI was measured by Tag positive immunofluorescence. All samples were normalized to the value of the DMSO control.
Highlight.
- Murine polyomavirus activates and recruits DNA damage repair (DDR) proteins to replication centers
- Large T-antigen mediates recruitment of DDR proteins to viral replication centers
- Inhibition or knockout of CHK1, CHK2, DNA-PK or H2AX do not affect viral titers
- Inhibition of ATR activity reduces viral titers, but not viral DNA accumulation
Acknowledgments
The authors would like to thank Andre Nussenzweig and Tak Mak for kindly sharing cell lines, Heinz Nasheuer for RPA32 antibodies, and Tom Benjamin and Brian Schaffhausen for the Tag antibodies. We would like to thank the CU Boulder Biochemistry Shared Instrument Core Facility for instrument use; Joe Dragavon and BioFrontiers Advanced Light Microscopy Core Facility for instrument use and microscopy support; Tom Cech, Roy Parker, and the Howard Hughes Medical Institute for use of the spinning disc confocal microscope; The BioFrontiers Institute and NIST-CU Cooperative Agreement award number 70NANB15H226 for use of the Nikon A1R microscope and NIH grant 1S10RR026680- 01A1 for the ImageXpress microscope and Imaris software.
This work was funding by NIH/NCI grant R01 CA37667. The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Conceived and designed the experiments: KH, CN and RLG. Performed the experiments: KH and CN. Analyzed the data: KH, CN RLG. Wrote the paper: KH and RLG.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ahn J, Urist M, Prives C. The Chk2 protein kinase. DNA Repair (Amst) 2004;3:1039–1047. doi: 10.1016/j.dnarep.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 2.Andrabi S, Gjoerup OV, Kean JA, Roberts TM, Schaffhausen B. Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc Natl Acad Sci U S A. 2007;104:19011–19016. doi: 10.1073/pnas.0706696104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arroyo JD, Hahn WC. Involvement of PP2A in viral and cellular transformation. Oncogene. 2005;24:7746–7755. doi: 10.1038/sj.onc.1209038. [DOI] [PubMed] [Google Scholar]
- 4.Banerjee P, DeJesus R, Gjoerup O, Schaffhausen BS. Viral interference with DNA repair by targeting of the single-stranded DNA binding protein RPA. PLoS Pathog. 2013;9:e1003725. doi: 10.1371/journal.ppat.1003725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, Livingston DM, Ferguson DO, Scully R, Alt FW. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benjamin TL. The hr-t gene of polyoma virus. Biochim Biophys Acta. 1982;695:69–95. doi: 10.1016/0304-419x(82)90018-x. [DOI] [PubMed] [Google Scholar]
- 7.Boichuk S, Hu L, Hein J, Gjoerup OV. Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T antigen. J Virol. 2010;84:8007–8020. doi: 10.1128/JVI.00334-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003;22:6610–6620. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nature Cell Biology. 2003;5:675–U651. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 10.Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaurushiya MS, Weitzman MD. Viral manipulation of DNA repair and cell cycle checkpoints. DNA Repair (Amst) 2009;8:1166–1176. doi: 10.1016/j.dnarep.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Wang X, Fluck MM. Independent contributions of polyomavirus middle T and small T to the regulation of early and late gene expression and DNA replication. J Virol. 2006;80:7295–7307. doi: 10.1128/JVI.00679-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Courtneidge SA, Smith AE. Polyoma virus transforming protein associates with the product of the c-src cellular gene. Nature. 1983;303:435–439. doi: 10.1038/303435a0. [DOI] [PubMed] [Google Scholar]
- 16.Cripe TP, Delos SE, Estes PA, Garcea RL. In vivo and in vitro association of hsc70 with polyomavirus capsid proteins. J Virol. 1995;69:7807–7813. doi: 10.1128/jvi.69.12.7807-7813.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dahl J, Chen HI, George M, Benjamin TL. Polyomavirus small T antigen controls viral chromatin modifications through effects on kinetics of virus growth and cell cycle progression. J Virol. 2007;81:10064–10071. doi: 10.1128/JVI.00821-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahl J, You J, Benjamin TL. Induction and utilization of an ATM signaling pathway by polyomavirus. J Virol. 2005;79:13007–13017. doi: 10.1128/JVI.79.20.13007-13017.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erickson KD, Bouchet-Marquis C, Heiser K, Szomolanyi-Tsuda E, Mishra R, Lamothe B, Hoenger A, Garcea RL. Virion assembly factories in the nucleus of polyomavirus-infected cells. PLoS Pathog. 2012;8:e1002630. doi: 10.1371/journal.ppat.1002630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans JD, Hearing P. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J Virol. 2005;79:6207–6215. doi: 10.1128/JVI.79.10.6207-6215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, Carpenter PB, Bonner WM, Chen J, Nussenzweig A. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol. 2002;4:993–997. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- 22.Feunteun J, Sompayrac L, Fluck M, Benjamin T. Localization of gene functions in polyoma virus DNA. Proc Natl Acad Sci U S A. 1976;73:4169–4173. doi: 10.1073/pnas.73.11.4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fluck MM, Schaffhausen BS. Lessons in signaling and tumorigenesis from polyomavirus middle T antigen. Microbiol Mol Biol Rev. 2009;73:542–563. doi: 10.1128/MMBR.00009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcea RL, Benjamin TL. Host range transforming gene of polyoma virus plays a role in virus assembly. Proc Natl Acad Sci U S A. 1983;80:3613–3617. doi: 10.1073/pnas.80.12.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcea RL, Talmage DA, Harmatz A, Freund R, Benjamin TL. Separation of host range from transformation functions of the hr-t gene of polyomavirus. Virology. 1989;168:312–319. doi: 10.1016/0042-6822(89)90271-7. [DOI] [PubMed] [Google Scholar]
- 26.Goldman E, Benjamin TL. Analysis of host range of nontransforming polyoma virus mutants. Virology. 1975;66:372–384. doi: 10.1016/0042-6822(75)90210-x. [DOI] [PubMed] [Google Scholar]
- 27.Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 28.Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 29.Jiang M, Zhao L, Gamez M, Imperiale MJ. Roles of ATM and ATR-mediated DNA damage responses during lytic BK polyomavirus infection. PLoS Pathog. 2012;8:e1002898. doi: 10.1371/journal.ppat.1002898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jul-Larsen A, Visted T, Karlsen BO, Rinaldo CH, Bjerkvig R, Lonning PE, Boe SO. PML-nuclear bodies accumulate DNA in response to polyomavirus BK and simian virus 40 replication. Exp Cell Res. 2004;298:58–73. doi: 10.1016/j.yexcr.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 31.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 32.Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci U S A. 2005;102:5844–5849. doi: 10.1073/pnas.0501916102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lilley CE, Chaurushiya MS, Boutell C, Everett RD, Weitzman MD. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 2011;7:e1002084. doi: 10.1371/journal.ppat.1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lilley CE, Schwartz RA, Weitzman MD. Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol. 2007;15:119–126. doi: 10.1016/j.tim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maser RS, Mirzoeva OK, Wells J, Olivares H, Williams BR, Zinkel RA, Farnham PJ, Petrini JHJ. Mre11 Complex and DNA Replication: Linkage to E2F and Sites of DNA Synthesis. Molecular and Cellular Biology. 2001;21:6006–6016. doi: 10.1128/MCB.21.17.6006-6016.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 38.Montross L, Watkins S, Moreland RB, Mamon H, Caspar DL, Garcea RL. Nuclear assembly of polyomavirus capsids in insect cells expressing the major capsid protein VP1. J Virol. 1991;65:4991–4998. doi: 10.1128/jvi.65.9.4991-4998.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orba Y, Suzuki T, Makino Y, Kubota K, Tanaka S, Kimura T, Sawa H. Large T antigen promotes JC virus replication in G2-arrested cells by inducing ATM- and ATR-mediated G2 checkpoint signaling. J Biol Chem. 2010;285:1544–1554. doi: 10.1074/jbc.M109.064311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shah GA, O'Shea CC. Viral and Cellular Genomes Activate Distinct DNA Damage Responses. Cell. 2015;162:987–1002. doi: 10.1016/j.cell.2015.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sheng Q, Denis D, Ratnofsky M, Roberts TM, DeCaprio JA, Schaffhausen B. The DnaJ domain of polyomavirus large T antigen is required to regulate Rb family tumor suppressor function. J Virol. 1997;71:9410–9416. doi: 10.1128/jvi.71.12.9410-9416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Y, Dodson GE, Shaikh S, Rundell K, Tibbetts RS. Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication in vivo. J Biol Chem. 2005;280:40195–40200. doi: 10.1074/jbc.C500400200. [DOI] [PubMed] [Google Scholar]
- 44.Sowd GA, Li NY, Fanning E. ATM and ATR activities maintain replication fork integrity during SV40 chromatin replication. PLoS Pathog. 2013;9:e1003283. doi: 10.1371/journal.ppat.1003283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sowd GA, Mody D, Eggold J, Cortez D, Friedman KL, Fanning E. SV40 utilizes ATM kinase activity to prevent non-homologous end joining of broken viral DNA replication products. PLoS Pathog. 2014;10:e1004536. doi: 10.1371/journal.ppat.1004536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 47.Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12:90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tittel-Elmer M, Alabert C, Pasero P, Cobb JA. The MRX complex stabilizes the replisome independently of the S phase checkpoint during replication stress. EMBO J. 2009;28:1142–1156. doi: 10.1038/emboj.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsang SH, Wang X, Li J, Buck CB, You J. Host DNA damage response factors localize to merkel cell polyomavirus DNA replication sites to support efficient viral DNA replication. J Virol. 2014;88:3285–3297. doi: 10.1128/JVI.03656-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu X, Avni D, Chiba T, Yan F, Zhao Q, Lin Y, Heng H, Livingston D. SV40 T antigen interacts with Nbs1 to disrupt DNA replication control. Genes Dev. 2004;18:1305–1316. doi: 10.1101/gad.1182804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zalvide J, Stubdal H, DeCaprio JA. The J domain of simian virus 40 large T antigen is required to functionally inactivate RB family proteins. Mol Cell Biol. 1998;18:1408–1415. doi: 10.1128/mcb.18.3.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao X, Madden-Fuentes RJ, Lou BX, Pipas JM, Gerhardt J, Rigell CJ, Fanning E. Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J Virol. 2008;82:5316–5328. doi: 10.1128/JVI.02677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Quantification of viral titers using an immunoplaque assay. A) DAPI (blue) and Tag (green) staining of cells used in virus quantification protocol. B) DAPI image with threshold applied to define outline of nucleus and Tag image with Regions of Interest (ROIs) outlined in white. C) Standard curve showing a linear correlation between virus added and percent of cells infected.
Figure S2: Strategy for assessing MuPyV replication across cell types. To address the requirement for CHK2 and H2AX during MuPyV replication and assembly, we utilized knockout and syngeneic control fibroblasts. We found differences in the number of permissive cells (i.e. infection efficiency) between cell lines. We normalized for these differences in infection efficiency across cell lines using an imaging based assay to quantify the number of cells infected during any given experiment To assess the role of DDR proteins in MuPyV replication and assembly, cell lines were normalized for infection efficiency. A) Timeline of viral titer and viral DNA accumulation assays. B) Equation used to calculate the normalized viral titer and DNA accumulation between cell lines of variable infection levels. C) Infection levels for cell lines quantified by high throughput imaging of Tag staining. D) Raw values for viral titer, quantified by immunoplaque assay and viral DNA accumulation, quantified by qPCR. E) Calculated reported values for viral titer and viral DNA accumulation for each cell line, normalized for infection efficiency.
Figure S3: Localization of DDR proteins in uninfected MEFs. MEFs were fixed, permeabilized and immunostained for A) RPA32 B) pSer23 RPA32 C) pCHK1 D) Mre11 E) pATM or F) γH2AX.
Figure S4: Mre11 is not degraded during MuPyV infection. A) MEFs were mock infected or infected with NG59RA. Total proteins were isolated at 24 and 32 HPI and analyzed by immunoblot for Mre11 levels. B) Quantification of integrated density of Mre11 immunoblot, normalized to actin loading control. All samples were normalized to the value of the mock treated cells.
Figure S5: Quantification of Mre11 at MuPyV replication centers and replicating cellular DNA. MEFs were mock infected or infected for 28 hrs, 10 μM EdU was then added to the media. Samples were then fixed, permeabilized, and stained for EdU, Tag, and Mre11. 2.5 μ m z-stacks were acquired on a spinning disc confocal using a 100X objective, NA 1.45. A) Single z-plane confocal images show DAPI, Tag, EdU, and MRE11 in uninfected and infected cells. B) The region of interest (ROI) for “total DNA” was defined by DAPI staining. The ROI that marked the viral replication centers, or “viral DNA”, was defined by Tag staining. The ROI that marked cellular DNA in infected cells or “cellular DNA (infected)” was defined by subtracting the Tag ROI from the DAPI ROI. Uninfected cells were analyzed using the “total DNA” ROI, only. All ROIs used for quantification are shown in white. C) Merged image of the EdU (green) and Mre11 (red) showing localization in ROIs of an infected nucleus and an uninfected nucleus. D) Co-localization analysis showing the percent of EdU co-localized with Mre11 in infected cells in the “total DNA” region, “viral DNA” region and “cellular DNA” region and uninfected cells.
Figure S6: Genotype validation for control and null cell lines. A) CHK2 control and null cell lines were analyzed using PCR primers specific for WT and neomycin alleles. WT PCR primers amplified a 440 bp product for the WT allele and no product for the null allele. Neo primers amplified a 900 bp product for the null allele and no product for the WT allele. B) H2AX control and null cell lines were analyzed using PCR primers specific for WT and null alleles. WT PCR primers amplified a 547 bp product for the WT allele and no product for the null allele. Null primers amplified a 424 bp product for the null allele and no product for the WT allele.
Figure S7: CHK1 autophosphorylation is inhibited by PF477736 treatment during infection. A) MEFs were mock infected or infected with NG59RA and at 2 HPI DMSO or 1 μM PF477736 were added to the media. Total proteins were isolated at 28 HPI and analyzed by immunoblot for pSer296 CHK1 levels. B) Quantification of integrated density of pSer296 CHK1 levels in control and infected MEFs normalized to GAPDH loading control. All samples were normalized to the value of the uninfected control cells.
Figure S8: Cell viability and infection levels following treatment with ATR inhibitor (VE-821) A) MEFs were treated with DMSO or ATR inhibitor for 32 hrs, and cell viability was measured by propidium iodide uptake using high throughput microscopy. The data were normalized to DMSO control, with live cell counts shown in black and dead cell counts shown in gray. B) MEFs were infected with NG59RA. At 16 HPI DMSO or the ATR inhibitor was added and the infection proceeded until 32 HPI. The number of infected cells in DMSO and ATR inhibitor treated samples at 32 HPI was measured by Tag positive immunofluorescence. All samples were normalized to the value of the DMSO control.