

Summary
Interferons (IFNs) exert their anti-viral effects by inducing the expression of hundreds of IFN-stimulated genes (ISGs). The activity of known ISGs is insufficient to account for the antiretroviral effects of IFN, suggesting that ISGs with antiretroviral activity are yet to be described. We constructed an arrayed library of ISGs from rhesus macaques and tested the ability of hundreds of individual macaque and human ISGs to inhibit early and late replication steps for 11 members of the retroviridae from various host species. These screens uncovered numerous ISGs with antiretroviral activity at both the early and late stages of virus replication. Detailed analyses of two antiretroviral ISGs indicate that indoleamine 2,3-dioxygenase 1 (IDO1) can inhibit retroviral replication by metabolite depletion while tripartite motif-56 (TRIM56) accentuates ISG induction by IFNα and inhibits the expression of late HIV-1 genes. Overall, these studies reveal numerous host proteins that mediate the antiretroviral activity of IFNs.
Graphical Abstract
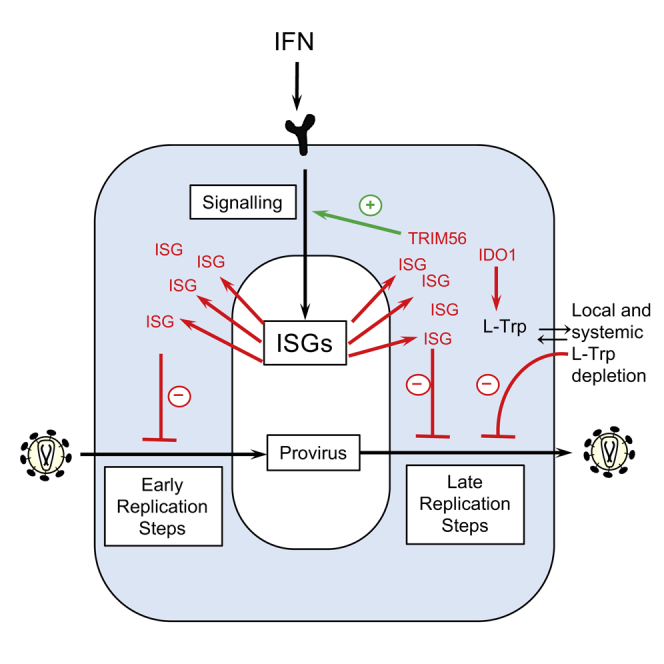
Highlights
-
•
ISG screening identifies direct and indirect antiretroviral proteins
-
•
Interferon-γ inhibits HIV-1 through IDO1-mediated tryptophan depletion
-
•
TRIM56 enhances the antiretroviral potential of interferon-α
Screening of interferon-stimulated genes for antiretroviral activity reveals numerous genes that directly or indirectly inhibit retroviral replication. Detailed analyses of two antiretroviral effectors indicate that IDO1 inhibits retroviral replication via nutrient depletion while TRIM56 increases the antiretroviral potential of IFNα.
Introduction
Interferons (IFNs) are a component of an early response to invading pathogens and induce the expression of hundreds of IFN-stimulated genes (ISGs) (Schoggins et al., 2011, Stetson and Medzhitov, 2006). The IFN response can ameliorate viral disease by facilitating clearance of acute infections, or by reducing the volume of chronic virus replication. Additionally, a genetic barrier imposed by species-dependent variation in antiviral ISGs can prevent interspecies transmission of viruses.
Retroviruses are a diverse family that includes human and simian immunodeficiency viruses (HIV and SIV). Multiple observations suggest that IFNs influence retroviral replication, transmission, and pathogenesis. For example, an IFNα antagonist can increase acute SIV replication and accelerate disease progression, while IFNα treatment can prevent SIV infection of macaques (Sandler et al., 2014) and reduce HIV-1 viremia in humans (Asmuth et al., 2010). Moreover, IFN-resistance may be selected during HIV-1 transmission (Fenton-May et al., 2013).
Some proteins with direct antiretroviral activity, such as APOBEC3 proteins, TRIM5α, tetherin, SAMHD1, ZAP, CNP, Mov10, and Mx2 (Doyle et al., 2015, Hatziioannou and Bieniasz, 2011), are encoded by ISGs. Additionally, some ISGs, namely TRIM5 and tetherin, can also exert antiviral effects by signaling to induce other antiviral genes (Galão et al., 2012; Pertel et al., 2011). Thus, response to IFN includes directly antiviral proteins and signal amplifiers that are induced by IFNs (Schoggins et al., 2014, Stetson and Medzhitov, 2006). However, the activity of known ISGs is insufficient to account for the antiretroviral effect of type I IFN, suggesting that some ISGs with antiretroviral activity are yet to be described.
Rapid evolution at host-pathogen interfaces means that ISG evasion or antagonism strategies employed by viruses are often only effective in the native host (reviewed in Doyle et al., 2015). Therefore, antiviral protein activity is sometimes revealed by using viral mutants or non-native viral hosts. HIV-1, for example, evades or antagonizes human APOBEC3, TRIM5α, and tetherin but is fully sensitive to these proteins in non-hominid species (Doyle et al., 2015). Species-dependent variation in antiretroviral proteins can constitute a profound genetic barrier to interspecies retroviral transmission. Accordingly, type I IFN is a significantly more potent inhibitor of HIV-1 and SIV infection in cells of non-native primates (Bitzegeio et al., 2013). Nevertheless, some ISGs exert antiviral effects without evidence for evasion or antagonism by the retroviral target (Doyle et al., 2015).
These concepts shaped our approach to reveal ISGs that inhibit retroviridae. We describe an arrayed library of ISGs from a non-human species (M. mulatta) and a series of screens in which we tested the ability of hundreds of individual ISGs from humans and macaques to inhibit the replication of eleven different retroviruses. This approach revealed that IFN’s antiretroviral activity is mediated by numerous candidate antiviral factors. We also describe detailed studies of two ISGs (IDO1 and TRIM56) that were revealed by our screens to exhibit antiretroviral activity.
Results
ISG Expression Screening to Identify Genes That Inhibit Retrovirus Replication
We first generated arrayed libraries of hundreds of ISGs in mammalian expression and lentiviral vectors and measured the impact of each ISG on retroviral replication. To expand the utility of a human ISG library (Schoggins et al., 2011) that we previously used to identify pathogen sensors and antiviral effectors (Dittmann et al., 2015, Schoggins et al., 2014, Wilson et al., 2012) and capture the ability of nonhuman ISGs to inhibit retroviruses, we constructed a library of ISGs from rhesus macaques (M. mulatta). This arrayed library contained 344 cDNAs from unique macaque ISGs as well as >90 additional ORFs that captured some of the allelic and splicing-generated diversity of macaque ISGs. Together, the two libraries include 488 different ISGs with 252 genes represented by both human and macaque variants (Figure 1A).
Figure 1.

ISG Screening Strategies and Effects of ISGs on HIV-1 and HIV-2
(A) Diagrammatic representations of genes present in the ISG libraries.
(B) Schematic representation of the pSCRPSY lentiviral vector. D, splice donor; A, splice acceptor. See Supplemental Experimental Procedures for details.
(C and D) Screening strategy to identify candidate genes that protect cells from incoming retroviral infection (B) or reduce infectious virion yield from infected cells (C).
(E and F) Effect of ISGs in incoming screens with HIV-1 (D) and HIV-2 (E) in MT4 and THP-1 cells and in production screens with HIV-1 (D) and HIV-2 (E) in 293T cells. Black data points, human ISGs; blue data points, macaque ISGs. Hits that appear twice represent variant transcripts. All values were normalized to the screen average (100 a.u.).
To identify human and macaque ISGs with antiretroviral activity, we carried out 25 screens of 11 different retroviruses (Table S1). We conducted these screens in two different ways. First, we used an HIV-1-based lentiviral vector (SCRPSY, Figure 1B) to express each ISG along with TagRFP in target cells that were then challenged with GFP-encoding retroviruses or retroviral vectors. Infection was then measured using two-color flow cytometry. This “incoming screening” strategy (Figure 1C) identifies ISGs that inhibit steps in the retroviral life cycle prior to GFP expression. Second, we co-transfected an ISG-expression plasmid (pDEST40) with a plasmid(s) that generates retroviral particles, then measured the yield of infectious virions by transduction of a GFP reporter gene, or using a reporter cell line. This “production screening” strategy (Figure 1D) identifies genes that inhibit the latter stages of the retroviral life cycle. For each ISG, the fraction of infected, ISG/TagRFP-expressing cells (incoming screens) or the yield of infectious virions (production screens) was expressed as a percentage of the mean value across all wells in a given screen (Figures 1E, 1F, 2A–2G, S1B–S1E, S2, S3, and S4).
Figure 2.

Effects of Human and Macaque ISGs on Diverse Retroviruses
(A–F) The effect of ISGs on lentiviruses, FIV (A) and EIAV (B); betaretroviruses, MPMV (C) and HERV-K (D); gammaretroviruses, MLV (E) and CERV2/MLV (F); and a spumaretrovirus, PFV (G). Screen details are as in Figures 1E and 1F except that the indicated cell lines were used.
Many ISGs Are Capable of Inhibiting Retroviral Replication
The screens identified numerous ISGs that were apparently capable of inhibiting the retroviral life cycle. A number of ISGs that were known to have antiretroviral activity, including several APOBEC3 proteins, TRIM5, tetherin, MOV10, SAMHD1, CNP, and Mx2, were identified in the appropriate incoming or production screens (Figures 1, 2, S1, S2, and S3; Table S5), suggesting that this approach is a powerful way to identify antiviral genes. Many additional ISGs whose expression conferred either resistance to incoming retroviral infection or reduced the production of infectious retroviral particles were identified. Some ISGs appeared to be specific inhibitors of one or a few retroviruses, while others were broadly inhibitory. As examples, OASL inhibited the production of infectious prototypic foamy virus (PFV) virions, and ULK4 inhibited the production of feline immunodeficiency virus (FIV), but OASL and ULK4 had no activity against other retroviruses. Conversely, macaque SLFN12 inhibited the production of all retroviruses (Figures 1, 2, S1, S2, and S3; Table S5). Overall, ∼60% of ISGs were hits against a single retrovirus, and the remainder targeted multiple retroviruses. In general, a greater number of ISGs were more potently inhibitory in production screens than in the incoming screens. This finding may result from potentially higher ISG protein levels associated with transient transfection, or may be due to the greater number and complexity of events associated with the late, as opposed to the early, steps of retroviral replication. A number of ISGs appeared to exert antiretroviral activity in a species-dependent manner. For example, human, but not macaque, ANGTPL1 inhibited SIVmac production, while macaque, but not human, IRF2 inhibited HIV-1 infection in human MT4 cells (Figures S1E–S1F and S4; Table S5). More than one-third of the “hits” were identified exclusively using our macaque ISG library (Table S5), but further work is required to determine which of these candidates have genuine species-dependent antiretroviral activity.
The use of lentiviral vectors in incoming screens enabled the effect of ISGs to be determined in different cellular backgrounds. We conducted incoming screens using human CD4+ T cell (MT4) and human CD4+ monocyte (THP-1) cell lines as targets for HIV-1, HIV-2, and SIVmac infection. Interestingly, more ISGs were protective in THP-1 cells than in MT4 cells, even though MT4 cells were more efficiently transduced with the ISG-encoding vectors (Figures 1 and S1; Table S2). Ten ISGs conferred >10-fold protection against infection by at least one primate lentivirus in THP-1 cells but conferred minimal or no protection in MT4 cells (Figures 1, S1, S2, and S3). These genes (cGAS, RIPK2, TLR7, IRF7, TRIM5, TRIM38, MYD88, IL1R, LGALS9, and IFI16) included ISGs known to mediate pattern recognition and inflammatory signaling. Thus, the differential protection conferred by certain ISGs likely reflects the differential ability of THP-1 and MT4 cells to transduce signals.
While ISG screening experiments are a powerful way to identify candidate antiretroviral ISGs, they do not distinguish between genes that act directly on viruses from those that serve a regulatory function. Overexpression screening can also lead to inhibition resulting from unnatural perturbation of cell physiology. ISGs that inhibit one or a few retroviruses are less likely to exert their effects through regulatory or nonspecific mechanisms. However, ISGs with broad activity (e.g., tetherin) can also have important effects. Validation and characterization of all the candidate antiretroviral ISGs identified herein would take many years. We therefore adopted a targeted approach to identify ISGs that (1) act directly to inhibit viral replication or (2) act as regulators of the antiviral state. Importantly, we sought to identify ISGs that affected retrovirus replication at native expression levels.
Some ISGs Inhibit Retroviral Replication by Activating Type I IFN or ISG Expression
To help identify ISGs that inhibited retroviral infection by facilitating the induction of an antiviral state, we determined which ISGs could stimulate expression of type I IFN or other ISGs. We generated 293T and THP-1 cell lines that expressed reporter genes driven by the human IFNβ promoter or an IFN-stimulated response element (ISRE). These cells were transduced with the ISG libraries and reporter gene expression recorded (Figures 3A and 3B; Tables S3 and S4). The ISRE reporter screens were analyzed soon after transduction to minimize potential indirect effects of IFN induction.
Figure 3.

Putatively Indirect and Direct Effects of ISGs on Incoming HIV-1 and HIV-2 Infection
(A) Activation of integrated IFNβ promoter driven luciferase reporter genes in HEK293T and THP-1 cells following transduction with lentiviral vectors encoding macaque and human ISGs.
(B) Activation of integrated ISRE-driven GFP reporter gene in HEK293T cells following transduction with lentiviral vectors encoding macaque and human ISGs.
(C and D) Infectious titers of HIV-1-GFP and HIV-2-GFP in MT4 cells (C) and THP-1 cells (D) transduced with SCRPSY vectors expressing selected ISGs. Titers are mean + SD.
Multiple ISGs that stimulated ISRE or IFNβ promoter activity were identified using this approach. An example was TRIM38, whose expression activated the IFNβ promoter (Figure 3A; Tables S3 and S4). Both human and macaque variants of TRIM38 conferred substantial protection against infection with HIV-1, HIV-2, and SIVmac when expressed in THP-1 cells (5- to 10-fold), but not in MT4 cells (Figures 1, S1, S2, and S3). These findings suggest that TRIM38 may participate in pattern recognition or signaling pathways that lead to type I IFN expression and consequently to inhibition of retroviral infection. As such, this and other proteins identified in this screen are worthy of further investigation as proteins that may regulate IFN expression.
Candidate Directly Acting Inhibitors of Incoming Retroviral Infection
We next elected to pursue ISGs that were likely to directly inhibit incoming HIV-1 or HIV-2 infection. From a list of ISGs that conferred >2-fold protection in any incoming HIV-1 or HIV-2 screen, we selected genes that did not stimulate IFNβ-promoter or ISRE driven reporter expression (Figures 3A and 3B; Tables S3 and S4). Thereafter, we determined infectious titers of HIV-1 and HIV-2 on MT4 and THP-1 cells expressing these ISGs (Figures 3C and 3D). This analysis confirmed the activity of ISGs known to inhibit incoming HIV-1 and HIV-2 infection (such as TRIM5 and Mx2) as well as a number of ISGs for which anti HIV-1 or HIV-2 activity was not previously described (e.g., IDO1, BCL3, LGALS9, and C5orf39) (Figures 3C and 3D). Despite our attempts to focus this sub-screen on directly acting ISGs, several genes that only conferred protection in THP-1 cells, such as cGAS, human TRIM5, and TLRs 3 and 7, likely act by inducing an antiviral state, even though they did not activate IFNβ or ISRE reporters (Figures 3A and 3B). Nevertheless, one ISG that had anti-HIV-1 activity only in THP-1 cells, CDKN1A, may exert its effect by decreasing SAMHD1 phosphorylation (Pauls et al., 2014). Thus, certain ISGs may exert antiretroviral activity, contingent on cofactors that are expressed in a cell-type-dependent manner.
IDO1 Inhibits Retroviral Replication
Of the candidate directly acting, not previously reported, inhibitors of incoming retrovirus infection (Figures 3C and 3D; Table S5), we selected IDO1 for further investigation. IDO1 encodes indoleamine 2,3-dioxygenase 1, which catalyzes the initial rate-limiting step in the conversion of L-tryptophan (L-Trp) to kynurenine (Hayaishi, 1976). Although IDO1 expression is upregulated by type I IFNs, the magnitude of its induction by IFNγ is clearly greater (Hassanain et al., 1993). Notably, IDO1 expression is profoundly upregulated during HIV-1 infection (Favre et al., 2010). IDO1 was active in incoming screens against a number of retroviruses but appeared most potent against HIV-2 (Figures 1F, 2, S1, S2, and S3; Table S5). Interestingly, it reduced both the intensity and the number of EGFP-positive cells (Figures 4A and 4B) and was effective in MT4 cells, but not THP-1 cells (that were less efficiently transduced) (Figure 1).
Figure 4.

IDO1 Inhibits HIV-1 and HIV-2 Replication
(A and B) FACS plots (A) and quantitation of mean fluorescence intensity (MFI, B) depicting GFP expression following standard or low-dose HIV-2-GFP infection of MT4 cells transduced with vectors expressing no ISG or IDO1.
(C) Western blot (WB) analysis of IDO1 expression with or without induction by IFNγ (A549 cells) or doxycycline (Dox) (MT4-IDO1 cells).
(D) Infectious titer of HIV-1-GFP (NHG) or HIV-2-GFP in target MT4 cells containing Dox-inducible IDO1, rhT5α, or TagRFP.
(E) HIV-1-GFP (NHG) replication in MT4 cells containing Dox inducible IDO1, rhesus TRIM5α (rhT5α), or TagRFP.
(F) Production of infectious HIV-1-GFP (NHG) or HIV-2 (VSV-G pseudotyped ROD10) during a single cycle of replication in MT4 cells containing Dox inducible IDO1, rhT5α, or TagRFP. Where indicated (5×), 5-fold higher input was used to negate the “incoming” effect.
(G) WB analysis of Gag expression in the same HIV-1 (NHG) infected cells as in (F).
(H) Infectious titer of HIV-1 produced and WB analysis of Gag, Env, and Nef expression in MT4 cells containing Dox-inducible IDO1 during a single cycle of HIV-1 (NL4.3) replication.
(I) Yield of infectious virions and cell lysate and particulate capsid during a single replication cycle of mixed (in the indicated ratios) unmodified MT4 cells and MT4 cells containing Dox-inducible IDO1.
(J and K) Effects of 1-MT (J) or L-Trp (K) on the yield of infectious HIV-1 particles and WB analysis of expression and release of viral Gag proteins during a single cycle of HIV-1 replication in MT4 cells containing Dox-inducible IDO1. Titers are mean + SD.
We generated MT4 cells that inducibly expressed IDO1 (Busnadiego et al., 2014) at levels similar to those in IFNγ-treated A549 cells (in which IFNγ is known to stimulate IDO1 expression) (Figure 4C). In single-cycle infection assays, IDO1 expression in target cells inhibited incoming HIV-2 infection by ∼5-fold but did not inhibit HIV-1 infection (Figure 4D). However, in spreading replication assays, IDO1 reduced the number of HIV-1-infected cells by >10-fold for several days (Figure 4E). Correspondingly, the yield of infectious HIV-1 or HIV-2 progeny virions, harvested 44 hr after infection, was reduced by 50- to 100-fold, by IDO1, similar to the reduction conferred by rhTRIM5α over a single replication cycle (Figure 4F). Western blot analyses revealed that IDO1 caused a substantial reduction in HIV-1 Gag, Env, and Nef protein levels, and a corresponding reduction in extracellular particulate CA (Figures 4G and 4H). Thus, IDO1 appears to act by inhibiting viral protein production, and its identification in an incoming screen was the result of reduced reporter gene expression.
IDO1-driven generation of kynurenine can have immune regulatory effects (Opitz et al., 2011) and deplete L-Trp (Pfefferkorn, 1984, Schmidt and Schultze, 2014). Single-cycle HIV-1 replication was inhibited when cells MT4 expressing inducible IDO1 were mixed with unmodified cells, even when <50% cells expressed IDO1 (Figure 4I). This finding suggested that IDO1 inhibits HIV-1 through L-Trp catabolites or by L-Trp depletion, rather than the direct action of the IDO1 protein. Moreover, 1-methyl-L-tryptophan (1-MT), a competitive inhibitor of IDO1 enzymatic activity, substantially reversed the anti-HIV-1 effect of IDO1 (Figure 4J). Crucially, L-Trp supplementation fully restored IDO1-inhibited HIV-1 Gag expression and infectious particle generation (Figure 4K). In the absence of IDO1, treatment with 1-MT or additional L-Trp had little influence on infectious progeny virion yield (Figure S5A and S5B). Because L-Trp supplementation should not prevent the formation of IDO1-specific catabolites, IDO1 appears to inhibit HIV-1 through L-Trp depletion.
IDO1 Can Constitute a Major Component of IFNγ-Mediated Inhibition of HIV-1
We next considered whether endogenous IDO1 contributes to the anti-HIV-1 activity of IFNγ (Hammer et al., 1986). IDO1 is induced in antigen-presenting cells (APCs) following HIV-1 infection in vivo or IFNγ stimulation in vitro (Favre et al., 2010), but most immortalized cell lines do not respond to IFNγ in this way. Therefore, we used A549 cells, which, unusually, exhibit robust IDO1 induction following IFNγ treatment (Figure 5A). IFNγ treatment of A549 cells conferred only 2- to 3-fold protection from incoming pseudotyped HIV-1 infection (Figure 5B). In contrast, IFNγ caused up to 100-fold reduction in HIV-1 infectious virion yield from infected A549 cells in a single cycle of replication (Figure 5C). Addition of L-Trp or 1-MT increased the yield of infectious HIV-1 from IFNγ-treated A549 cells by ∼10-fold (Figure 5D) but did not have this effect in the absence of IFNγ (Figure S4C). A similar L-Trp induced rescue of infectious virion yield occurred with IFNγ–treated TZM-bl cells, a commonly used HIV-1 indicator cell line (Figures 5D–5F and S5D). Thus, IDO1-induced nutrient (L-Trp) depletion is responsible for a substantial fraction of the anti-HIV-1 activity of IFNγ in A549 and TZM-bl cells.
Figure 5.

Endogenous IDO1 Inhibits HIV-1
(A) WB analysis of IDO1 expression in IFNγ-treated A549 cells.
(B) Titer of VSV-G pseudotyped replication competent HIV-1-GFP (NHG) on A549 cells following treatment with increasing doses of IFNγ.
(C) The yield of infectious progeny virions from a single cycle of HIV-1 replication in A549 cells treated with increasing doses of IFNγ.
(D) The yield of infectious progeny virions from a single cycle of HIV-1 replication in A549 cells treated with IFNγ in the presence of 50 μg/ml L-Trp (D and E) or 100 μg/ml 1-MT (D and F).
(E) WB analysis of IDO1 expression in IFNγ-treated TZM-bl cells.
(F) As in D using TZM-bl cells. Titers are mean + SD.
TRIM56 Expression Inhibits HIV-1 Replication
As an alternative approach to identify ISGs that could mediate the antiretroviral activity of IFNα, several genes that were hits in outgoing HIV-1 screens (>3-fold inhibition) were tested for their ability to inhibit spreading HIV-1 replication when stably expressed in GHOSTX4 indicator cells. HIV-1 replication in GHOSTX4 cells is resistant to inhibition by IFNα (our unpublished data), so this strategy could, in principle, capture ISGs that regulate the antiviral state, or directly inhibit late HIV-1 replication steps. Most of the selected ISGs modestly inhibited HIV-1 replication GHOSTX4 cells (Figures 6A and 6B), but one ISG, TRIM56, substantially delayed HIV-1 spread (Figure 6A). TRIM56 has previously been reported to inhibit the replication of some, but not all, RNA viruses through mechanisms that are unclear (Liu et al., 2014, Wang et al., 2011). In our screens, TRIM56 greatly inhibited the production of infectious HIV-1 and HERV-K virions and exhibited weak activity against HIV-2 in incoming screens (Figures 1E, 1F, and 2D).
Figure 6.

TRIM56 Overexpression Can Inhibit HIV-1 Replication
(A and B) HIV-1 replication in populations of GHOST cells transduced with lentiviral vectors (CCIB) expressing the indicated human (A) or macaque (B) ISGs.
(C) WB analysis of GHOST cell clones overexpressing TRIM56.
(D) Single round HIV-1 infection of GHOST cells transduced with empty vector (CCIB, filled symbol) or clones overexpressing TRIM56.
(E) Spreading replication of HIV-1 in GHOST cells transduced with empty vector (CCIB, red symbol) or the clones overexpressing TRIM56 described in (C).
(F) WB analysis of Gag, Nef, Env, and TRIM56 expression and particle release in GHOST vector and GHOST-TRIM56#2 cells during a single cycle of HIV-1 infection.
(G) The yield of infectious progeny virions from a single cycle of HIV-1 replication in GHOST-vector and GHOST-TRIM56#2 cells.
(H) Adaptation of HIV-1 to GHOST-TRIM56#2 cells via spreading replication. At each discontinuity, virions harvested from the GHOST-TRIM56#2 cells were passaged onto GHOST-vector and GHOST-TRIM56#2 cells.
(I) Schematic representation and sequence analysis of an HIV-1 clone (TRIM56R) adapted to replicate in GHOST cells expressing TRIM56.
(J) WB analysis of Gag expression and particle release in GHOST vector and GHOST-TRIM56 cells, during a single cycle of HIV-1 and HIV-1/TRIM56R replication.
We confirmed the apparent specificity of the effect of TRIM56 by cotransfecting varying amounts of TRIM56 expression plasmid with plasmids generating infectious HIV-1 or MLV particles. TRIM56 reduced the yield of infectious HIV-1 particles by up to 50-fold (Figures S6A and S6B) but had little effect on MLV particle yield (Figures S6C and S6D). Western blot analysis revealed that TRIM56 reduced the levels of cell- and virion-associated HIV-1 Gag but did not affect MLV Gag expression. Thus, exogenous TRIM56 appeared to specifically inhibit HIV-1 gene expression.
We next generated nine single-cell clones of GHOSTX4 cells expressing TRIM56 at levels that were only modestly higher than unmodified GHOSTX4 cells (Figure 6C) and significantly lower than the transiently transfected 293T cells used in the cotransfection and screening experiments (Figures S6A and S6C). When the GHOSTX4-TRIM56 clones were challenged with HIV-1, viral spread was significantly delayed (four clones) or apparently abolished (five clones; Figures 6D and 6E). Despite this dramatic inhibition of viral spread, approximately equivalent numbers of control and TRIM56-expressing cells were infected in the initial infection cycle (Figure 6D). This finding is consistent with the notion that TRIM56 inhibits late rather than early steps of HIV-1 replication (Figure 1E).
Western blot analysis of a single cycle of HIV-1 replication in one GHOSTX4-TRIM56 cell clone (#2) revealed that the product of an early HIV-1 gene (Nef) was not affected by TRIM56 (Figure 6F). Conversely, Gag and Env expression and the yield of viral particles were significantly reduced in GHOSTX4-TRIM56#2 cells (Figure 6F). The reduction in yield of HIV-1 particles as measured by quantitative western blotting or using infectivity assays was similar (Figures 6G and S6E), indicating that TRIM56 reduced the number but not the infectiousness of virions generated by infected cells.
TRIM56-Induced Inhibition of HIV-1 Involves Multiple Viral and Host Genes
To identify viral determinants of TRIM56 sensitivity, we passaged HIV-1 in GHOSTX4-TRIM56 cells. Replication was eventually detected in some GHOST-TRIM56 clones (Figure 6E), and virions harvested from these cells were used to initiate an iterative passage series in GHOSTX4-TRIM56#2 cells (Figure 6H). After eight passages, replication was only modestly delayed in GHOSTX4-TRIM56#2 cells. Sequence analysis of a full-length molecular clone of the adapted virus (termed HIV-1/TRIM56R) revealed numerous mutations including a frameshift in Vif and missense mutations in Gag, Pol, Vpr, Tat, Rev, and Env (Figure 6I). In single-cycle replication experiments, the yield of WT HIV-1 virions was diminished in the TRIM56-expressing clones, while HIV-1/TRIM56R yielded similar levels of virions from control and TRIM56-expressing cells (Figure 6J).
Unlike WT HIV-1, the cloned HIV-1/TRIM56R virus replicated robustly in GHOSTX4-TRIM56#2 cells (Figure 7A) and with slightly faster kinetics than WT HIV-1 in unmodified GHOSTX4 cells. Analysis of chimeric proviruses containing either the 5′ or 3′ half of the HIV-1/TRIM56R genome (5′R and 3′R; Figure 6I) suggested that TRIM56 resistance mapped to the 3′R region (Figure 7B). Further analysis of chimeric viruses containing elements from 3′R indicated that the TRIM56 resistant phenotype could not be mapped to a single determinant (our unpublished data). Nevertheless, a single substitution in the HIV-1 Env gene (T6421A, Env N67K) present in 3′R enabled some level of replication in GHOSTX4-TRIM56 cells (Figure 7B). When the T6421A mutation was combined with mutations in 5′R (generating HIV-1 5′R-6421), the HIV-1/TRIM56R virus phenotype was reproduced. Attempts to map determinants within 5′R conferring TRIM56 resistance upon HIV-1 5′R-6421 again indicated that multiple determinants in 5′R contributed to the HIV-1/TRIM56R phenotype (our unpublished data). Thus, the ability of the HIV-1/TRIM56R virus to replicate in GHOSTX4-TRIM56#2 cells was conferred by the cumulative effect of multiple mutations and not governed by a single viral gene.
Figure 7.

Multiple Viral Determinants and ISGs Underlie Inhibition of HIV-1 Replication by Endogenous TRIM56
(A and B) Spreading replication of HIV-1 and HIV-1/TRIM56R in GHOSTX4-vector and GHOSTX4-TRIM56#2 cells (A), or chimeric viruses constructed using HIV-1 and HIV-1/TRIM56R (B).
(C) WB analysis of TRIM56 expression in clones of MT4-LTR-GFP cells transduced with lentiviral vectors expressing Cas9 only or Cas9 plus one of two TRIM56-targeted guide RNAs (CR1 or CR3).
(D and E) HIV-1 replication in the absence (D) or presence (E) of 25U/ml IFNα in clones of control (n = 4) or TRIM56 knockout (n = 7) MT4-LTR-GFP cells described in (C).
(F and G) Microarray analysis of the expression levels (in a.u.) for the ∼120 genes most highly induced by 25U/ml IFNα in clones of control (n = 4) or TRIM56 knockout (n = 7) MT4-LTR-GFP cells described in (C) in the presence (G) or absence (F) of IFN.
To determine whether endogenous TRIM56 could inhibit HIV-1 replication, we used CRISPR editing to disrupt TRIM56 in an MT4 cell clone that contains an HIV-1 LTR-driven GFP gene (MT4/LTR-GFP). We generated four Cas9-expressing MT4/LTR-GFP subclones in which TRIM56 was not perturbed, and seven expressing Cas9 and TRIM56-targeted guide RNAs. All control MT4/LTR-GFP clones expressed equivalent levels of TRIM56, while the TRIM56 targeted clones expressed greatly reduced or undetectable levels of TRIM56 (Figure 7C). HIV-1 replicated with nearly identical kinetics in each of the cell clones (Figure 7D), indicating that they were equally permissive and that endogenous TRIM56 does not inhibit HIV-1 replication (Figure 7D). Strikingly, however, in the presence of IFNα, which elevated the level of TRIM56 expression ∼2-fold (Figures S7A and S7B), replication was inhibited to a far greater extent in unedited clones compared to clones in which TRIM56 was disrupted.
Because resistance to TRIM56 involved multiple viral genes, and the antiviral effect of endogenous TRIM56 was evident only upon IFNα treatment, it was possible that TRIM56 acted by increasing the levels or effects of other antiviral proteins. A microarray analysis of the four control and sevevn TRIM56 knockout MT4 clones, focusing on the 120 most highly IFNα-induced ISGs, revealed that, despite variation among individual cell clones, basal ISG expression (i.e., in the absence of IFN) was generally unaffected by TRIM56 (Figure 7F). In contrast, following IFNα treatment, many ISGs were expressed at higher levels in control than in TRIM56 knockout clones (Figure 7G). These genes included several that were hits in our screens (Figure 1C), as well as known antiretroviral genes, including APOBEC3G, Mx2, MOV10, and IFITM3. Analysis of a larger set of 500 ISGs (Figures S7C and S7D) confirmed that TRIM56 knockout reduced ISG expression following IFNα treatment. Thus, TRIM56 likely enhances the antiretroviral activity of IFNα, at least in part, by enhancing cellular responsiveness to this cytokine.
Discussion
Here we report the most comprehensive screen of ISGs yet undertaken for antiviral activity against a given virus family. Previously, we conducted a narrowly focused incoming screen that tested the ability of human ISGs to inhibit HIV-1 (Schoggins et al., 2011). Notably, those results are generally consistent with this study, with five of seven ISGs re-identified in this study. However, in this study we identified many additional candidate anti-retroviral genes, a large number of which have not previously been reported to have antiretroviral activity. In general, our findings underscore the complexity of the interferon response with respect to inhibition of retroviral replication and suggest that the antiretroviral activity of type I IFN is mediated and regulated through the action of many ISGs (Table S5). Some ISGs appeared to act in a retrovirus-specific manner, while others had broad activity (Table S5). We also found some discrepancies in the antiretroviral activity between human and macaque variants of ISGs, which may contribute to the species-dependent barriers to retroviral replication that can be erected by type I IFN treatment (Bitzegeio et al., 2013). However, a caveat is that the screens were conducted in human cells, and it is possible that some ISGs may only function properly in cells of species from which they originate. Clearly, some of the genes identified herein act to enhance or modify signaling pathway rather than as directly antiviral proteins, while others appear to have direct antiviral activity. To characterize all of the candidate antiretroviral genes identified herein would take many years. Thus, we have focused on a small number of ISGs, as proof-of-principle of the usefulness of this strategy. Expression screens such as those reported herein are a powerful tool, but have weaknesses, including the potential for overexpression artifacts. However, we emphasize the fact that endogenous levels of two host factors we identify characterized herein can cause inhibition of retroviral replication. These two ISGs contribute to the anti-HIV-1 activity of IFNs via completely different mechanisms.
One antiretroviral ISG identified in our screens was IDO1. The observation that exogenous L-Trp can relieve inhibition by IDO1 and IFNγ strongly suggests that IDO1 inhibits HIV-1 through L-Trp depletion rather than through the generation of catabolites such as kynurenine. Nutrient-depletion is a recognized anti-pathogen strategy and similar effects of IDO1 have been noted for other pathogens including measles virus and hepatitis B virus (Mao et al., 2011, Obojes et al., 2005). Indeed, SAMHD1, an IFN-inducible deoxynucleoside triphosphate (dNTP) triphosphohydrolase that is also active against HIV-1, acts by depleting essential substrates required for retrovirus replication (Goldstone et al., 2011).
A simple explanation for the effects of IDO1 is that L-Trp depletion could inhibit HIV-1 by limiting the availability of L-Trp for nascent viral protein synthesis. However, it is also true that T cells can actively respond to L-Trp depletion by activating GCN2 and mTOR signaling pathways (Cobbold et al., 2009, Munn et al., 2005), which could potentially impact viral gene expression. Importantly, it is plausible that L-Trp could be sufficiently depleted in T cells in vivo to exert an antiretroviral effect. IDO1 is expressed at very high levels in APCs in HIV-1-infected patients (Favre et al., 2010), and abundant IDO1 is observed in the lymph nodes of SIV-infected macaques (Estes et al., 2006). Efficient local depletion of L-Trp by APCs could therefore inhibit HIV-1 replication in neighboring CD4+ T cells. Notably, IDO1 is sufficiently active during chronic HIV-1 infection that serum L-Trp can be 50% lower than in healthy controls (reviewed in Murray, 2003). To impact serum L-Trp levels, extreme local depletion of L-Trp likely occurs in the microenvironments (i.e., lymphoid tissues) that support viral replication. Crucially, such local depletion of L-Trp is observed in vivo. GCN2 is activated by L-Trp depletion to levels around 20-fold lower than in normal serum and this IDO-dependent GCN2 activation is readily observed in vivo (Munn et al., 2005). Thus, tissue microenvironments likely experience L-Trp depletion to an extent that could suppress HIV-1 replication.
Although L-Trp depletion has antiviral effects, elevated IDO1 activity has additional consequences in chronic HIV-1 infection (Murray, 2003, Schmidt and Schultze, 2014), as it suppresses T cell responses and promotes tolerance (Friberg et al., 2002, Munn et al., 1998, Sakurai et al., 2002). IDO1 activity has been reported to skew CD4+ T cell differentiation to regulatory T cells at the expense of Th17 cells, thereby exacerbating CD4+ Th17 depletion, perhaps leading to increased microbial translocation and systemic immune activation (Brenchley et al., 2006, Favre et al., 2010). Thus, IDO1 activity could potentially mediate inhibition of HIV-1 replication while simultaneously promoting immunosuppression.
Our screens uncovered two antiretroviral TRIM proteins, TRIM38 and TRIM56, that appear to act by regulating the establishment of an antiviral state. Other TRIM proteins can stimulate cell responses to pathogens, including TRIM25, which induces ubiquitination of RIG-I (Gack et al., 2007), and TRIM5, which has been implicated in promoting inflammatory transcription (Pertel et al., 2011). Overexpression screens have suggested that other TRIM proteins have antiretroviral activity, through unknown mechanisms (Uchil et al., 2008). While overexpressed TRIM56 did not appear to function through IFNβ or ISRE promoter elements, endogenous TRIM56 both increased the expression of many ISGs in response to IFNα and enhanced the establishment of an anti-HIV-1 state. Moreover, modest TRIM56 overexpression caused dramatic inhibition of HIV-1 replication. It is likely that these findings are causally related, although we cannot exclude the possibility that these reflect distinct activities of TRIM56, as the signaling and directly antiviral activities of tetherin and TRIM5 are clearly separable functions (Galão et al., 2012; Pertel et al., 2011). Previously, TRIM56 has been reported to inhibit the replication of several RNA viruses, while other RNA viruses are unaffected (Liu et al., 2014, Wang et al., 2011); it has also been reported to enhance cellular responsiveness to TLR3 signaling by binding to TRIF (Shen et al., 2012) and to enhance IFNβ production in response to dsDNA by binding to and ubiquitinating STING (Tsuchida et al., 2010). Our data are not consistent with an important role for STING-induced IFN expression as a key mediator of TRIM56 antiretroviral activity, because anti-HIV-1 activity of overexpressed TRIM56 was observed in cells that do not express STING and cGAS, and endogenous TRIM56 only exerted anti-HIV-1 activity and enhanced ISG expression following IFNα treatment. STING-independent TRIM56 antiviral activity has also been reported for influenza viruses (Liu et al., 2016). Further work will be required to determine precisely how TRIM56 functions to inhibit HIV-1 replication and whether any previously described properties are relevant to its antiretroviral activity.
The consequences of ISG expression (such as nutrient depletion) are expected to have fitness costs for the host. However, if those costs are temporary, and smaller than those imposed by viral infection, then ISG expression is ultimately beneficial to the host. Thus, these data highlight the need for caution when contemplating therapeutic interventions designed to modulate the consequences of HIV-1 infection. Multiple ISGs that are associated with immune activation, illness, and disease progression during chronic infection likely also mediate suppression of HIV-1 replication. While the antiretroviral activities of ISGs are unable to tip the balance in favor of the host during natural HIV-1 infection, it is clear the IFNs can slow or ameliorate disease in HIV-1 and other retroviral infections. The findings reported herein should inform future efforts to understand the molecular basis by which IFNs shape susceptibility to retroviral infection, disease progression, cross-species transmission, and retroviral emergence.
Experimental Procedures
Retroviruses and Cell Lines
All the common cell lines were maintained under standard culture conditions as detailed in the Supplemental Experimental Procedures. MT4-LTR-GFP indicator cells and IFNβ1/ISRE reporter cell lines represent cell clones modified using MLV-derived retroviral vectors. ISG-expressing MT4 and GHOSTX4 cell lines were modified using lentiviral vectors. Limiting dilution was also used to generate a panel of GHOSTX4 cells, modified to express TRIM56. Replication competent proviral clones encoding GFP (PFV) or pseudotyped envelope minus derivatives of HIV-1,-1 (NHG Δenv), HIV-2 (HIV-2 Δenv EGFP), SIVmac, (SIVmac Δnef Δenv EGFP), and SIVagmTAN (Δnef Δenv EGFP) or multi-plasmid vector systems for FIV, EIAV, HERV-K, MLV, and CERV-2 encoding GFP were used as described previously (Busnadiego et al., 2014, Kane et al., 2013). Intact proviral clones of HIV-1 (NL4-3) (M19921) and HIV-2 (ROD10), as well as GFP encoding (in place of nef) NHG, were used. Infectivity and replication assays were performed as described previously (Busnadiego et al., 2014, Kane et al., 2013) and as detailed in the Supplemental Experimental Procedures and Table S1.
ISG Screens
Using the same criteria used to select human ISGs (Dittmann et al., 2015, Schoggins et al., 2011), we attempted to clone ∼600 macaque ISGs using RT-PCR of IFN-treated Macaca mulatta cell lines. The resulting macaque and extended human ISG libraries were subcloned into the pSCRPSY vector (Accession KT368137) and pcDNA-DEST40 using Gateway (Invitrogen) technology. ISG screens were conducted in a 96-well plate format using SCRPSY lentiviral transduced target cells (incoming screens, ISRE and IFNβ promoter screens) or through cotransfection of 293T cells with ISGs encoded by pcDNA-DEST40 (outgoing screens) detailed in the Supplemental Experimental Procedures and Table S1. For each ISG, the fraction of infected, ISG/TagRFP-expressing cells (incoming screens) or the yield of infectious virions (production screens) was expressed as a percentage of the mean value across all wells for the respective library in a given screen, except the MPMV incoming screen, in which values are expressed as the mean value across each plate. ISGs that affected SCRPSY lentiviral titers are shown in Figure S1A and Table S2.
CRISPR-Mediated TRIM56 Knockout
A derivative of the HIV-based retroviral vector lentiCRISPR Version 2 (Addgene, Plasmid #52961) was constructed. MT4-LTR-GFP knockout cells were derived by transduction with lentiCRISPRV2-based viruses and single-cell clones transduced with TRIM56-targeting CR1 and CR3 vectors were derived by limiting dilution.
Quantitative Western Blotting
Western blotting was performed as described previously (Busnadiego et al., 2014, Kane et al., 2013) and detailed in the Supplemental Experimental Procedures using cell lysates and material pelleted through 20% sucrose. Antigens were visualized using secondary antibodies labeled with IRDye 800CW or IRDye 680RD (LI-COR Biosciences or Thermo Scientific) and the relevant primary antibody and scanned using a Li-Cor Odyssey Scanner.
Microarray Analysis
Total RNA was extracted, using the RNeasy Plus Mini Kit (QIAGEN), from MT4-LTR-GFP cells, control subclones, and CRISPR knockout subclones that were untreated or treated with 25 U/ml IFNα (Sigma) for 24 hr before harvest. cRNA was prepared and probed using Human HT12 Expression Beadchip (Illumina), containing ∼48,000 transcript probes, according to the manufacturer’s instructions.
Author Contributions
Conceptualization, P.D.B.; Library Construction, S.J.W., M.A., and J.S.; Screening, S.J.W., M.K., F.Z., M.A., and T.K.; IDO1, S.J.R.; TRIM56, T.M.Z.; Resources, J.S. and C.M.R.; Writing – Original Draft, S.J.W. and P.D.B.; Writing – Review & Editing, M.K., T.M.Z., S.J.R., F.Z., T.K., J.S., C.M.R., S.J.W., and P.D.B.; Funding Acquisition, P.D.B., S.J.W., M.K., J.S., and C.M.R.; Supervision, P.D.B. and S.J.W.
Acknowledgments
We thank Steven Soll for CERV2/MLV chimeric Gag-Pol, Nick J. Loman and Shalini Yadav for assistance, and the Developmental Studies Hybridoma Bank, University of Iowa, and NIH AIDS reagents program for antibodies. This work was supported by grants from the NIH R3764003 (to P.D.B.), 1F32AI116263-01 (to M.K.), AI091707 (to J.S. and C.M.R.), and UK MRC MR/K024752/1 (to S.J.W.). The content of this piece is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Published: September 14, 2016
Footnotes
Supplemental Information includes seven figures, five tables, and Supplemental Experimental Procedures and can be found with this article at http://dx.doi.org/10.1016/j.chom.2016.08.005.
Contributor Information
Sam J. Wilson, Email: sam.wilson@glasgow.ac.uk.
Paul D. Bieniasz, Email: pbienias@adarc.org.
Supplemental Information
References
- Asmuth D.M., Murphy R.L., Rosenkranz S.L., Lertora J.J., Kottilil S., Cramer Y., Chan E.S., Schooley R.T., Rinaldo C.R., Thielman N., AIDS Clinical Trials Group A5192 Team Safety, tolerability, and mechanisms of antiretroviral activity of pegylated interferon Alfa-2a in HIV-1-monoinfected participants: a phase II clinical trial. J. Infect. Dis. 2010;201:1686–1696. doi: 10.1086/652420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitzegeio J., Sampias M., Bieniasz P.D., Hatziioannou T. Adaptation to the interferon-induced antiviral state by human and simian immunodeficiency viruses. J. Virol. 2013;87:3549–3560. doi: 10.1128/JVI.03219-12. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Brenchley J.M., Price D.A., Schacker T.W., Asher T.E., Silvestri G., Rao S., Kazzaz Z., Bornstein E., Lambotte O., Altmann D. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- Busnadiego I., Kane M., Rihn S.J., Preugschas H.F., Hughes J., Blanco-Melo D., Strouvelle V.P., Zang T.M., Willett B.J., Boutell C. Host and viral determinants of Mx2 antiretroviral activity. J. Virol. 2014;88:7738–7752. doi: 10.1128/JVI.00214-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbold S.P., Adams E., Farquhar C.A., Nolan K.F., Howie D., Lui K.O., Fairchild P.J., Mellor A.L., Ron D., Waldmann H. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc. Natl. Acad. Sci. USA. 2009;106:12055–12060. doi: 10.1073/pnas.0903919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmann M., Hoffmann H.H., Scull M.A., Gilmore R.H., Bell K.L., Ciancanelli M., Wilson S.J., Crotta S., Yu Y., Flatley B. A serpin shapes the extracellular environment to prevent influenza A virus maturation. Cell. 2015;160:631–643. doi: 10.1016/j.cell.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle T., Goujon C., Malim M.H. HIV-1 and interferons: who’s interfering with whom? Nat. Rev. Microbiol. 2015;13:403–413. doi: 10.1038/nrmicro3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes J.D., Li Q., Reynolds M.R., Wietgrefe S., Duan L., Schacker T., Picker L.J., Watkins D.I., Lifson J.D., Reilly C. Premature induction of an immunosuppressive regulatory T cell response during acute simian immunodeficiency virus infection. J. Infect. Dis. 2006;193:703–712. doi: 10.1086/500368. [DOI] [PubMed] [Google Scholar]
- Favre D., Mold J., Hunt P.W., Kanwar B., Loke P., Seu L., Barbour J.D., Lowe M.M., Jayawardene A., Aweeka F. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci. Transl. Med. 2010;2:32ra36. doi: 10.1126/scitranslmed.3000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton-May A.E., Dibben O., Emmerich T., Ding H., Pfafferott K., Aasa-Chapman M.M., Pellegrino P., Williams I., Cohen M.S., Gao F. Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology. 2013;10:146. doi: 10.1186/1742-4690-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg M., Jennings R., Alsarraj M., Dessureault S., Cantor A., Extermann M., Mellor A.L., Munn D.H., Antonia S.J. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. International journal of cancer Journal international du cancer. 2002;101:151–155. doi: 10.1002/ijc.10645. [DOI] [PubMed] [Google Scholar]
- Gack M.U., Shin Y.C., Joo C.H., Urano T., Liang C., Sun L., Takeuchi O., Akira S., Chen Z., Inoue S., Jung J.U. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- Galão R.P., Le Tortorec A., Pickering S., Kueck T., Neil S.J. Innate sensing of HIV-1 assembly by Tetherin induces NFκB-dependent proinflammatory responses. Cell Host Microbe. 2012;12:633–644. doi: 10.1016/j.chom.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstone D.C., Ennis-Adeniran V., Hedden J.J., Groom H.C., Rice G.I., Christodoulou E., Walker P.A., Kelly G., Haire L.F., Yap M.W. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–382. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- Hammer S.M., Gillis J.M., Groopman J.E., Rose R.M. In vitro modification of human immunodeficiency virus infection by granulocyte-macrophage colony-stimulating factor and gamma interferon. Proc. Natl. Acad. Sci. USA. 1986;83:8734–8738. doi: 10.1073/pnas.83.22.8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassanain H.H., Chon S.Y., Gupta S.L. Differential regulation of human indoleamine 2,3-dioxygenase gene expression by interferons-gamma and -alpha. Analysis of the regulatory region of the gene and identification of an interferon-gamma-inducible DNA-binding factor. J. Biol. Chem. 1993;268:5077–5084. [PubMed] [Google Scholar]
- Hatziioannou T., Bieniasz P.D. Antiretroviral restriction factors. Curr. Opin. Virol. 2011;1:526–532. doi: 10.1016/j.coviro.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayaishi O. Properties and function of indoleamine 2,3-dioxygenase. J. Biochem. 1976;79:13P–21P. doi: 10.1093/oxfordjournals.jbchem.a131115. [DOI] [PubMed] [Google Scholar]
- Kane M., Yadav S.S., Bitzegeio J., Kutluay S.B., Zang T., Wilson S.J., Schoggins J.W., Rice C.M., Yamashita M., Hatziioannou T., Bieniasz P.D. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature. 2013;502:563–566. doi: 10.1038/nature12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Li N.L., Wang J., Shi P.Y., Wang T., Miller M.A., Li K. Overlapping and distinct molecular determinants dictating the antiviral activities of TRIM56 against flaviviruses and coronavirus. J. Virol. 2014;88:13821–13835. doi: 10.1128/JVI.02505-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Li N.L., Shen Y., Bao X., Fabrizio T., Elbahesh H., Webby R.J., Li K. The C-Terminal Tail of TRIM56 Dictates Antiviral Restriction of Influenza A and B Viruses by Impeding Viral RNA Synthesis. J. Virol. 2016;90:4369–4382. doi: 10.1128/JVI.03172-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao R., Zhang J., Jiang D., Cai D., Levy J.M., Cuconati A., Block T.M., Guo J.T., Guo H. Indoleamine 2,3-dioxygenase mediates the antiviral effect of gamma interferon against hepatitis B virus in human hepatocyte-derived cells. J. Virol. 2011;85:1048–1057. doi: 10.1128/JVI.01998-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn D.H., Zhou M., Attwood J.T., Bondarev I., Conway S.J., Marshall B., Brown C., Mellor A.L. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- Munn D.H., Sharma M.D., Baban B., Harding H.P., Zhang Y., Ron D., Mellor A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22:633–642. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Murray M.F. Tryptophan depletion and HIV infection: a metabolic link to pathogenesis. Lancet Infect. Dis. 2003;3:644–652. doi: 10.1016/s1473-3099(03)00773-4. [DOI] [PubMed] [Google Scholar]
- Obojes K., Andres O., Kim K.S., Däubener W., Schneider-Schaulies J. Indoleamine 2,3-dioxygenase mediates cell type-specific anti-measles virus activity of gamma interferon. J. Virol. 2005;79:7768–7776. doi: 10.1128/JVI.79.12.7768-7776.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz C.A., Litzenburger U.M., Sahm F., Ott M., Tritschler I., Trump S., Schumacher T., Jestaedt L., Schrenk D., Weller M. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- Pauls E., Ruiz A., Riveira-Muñoz E., Permanyer M., Badia R., Clotet B., Keppler O.T., Ballana E., Este J.A. p21 regulates the HIV-1 restriction factor SAMHD1. Proc. Natl. Acad. Sci. USA. 2014;111:E1322–E1324. doi: 10.1073/pnas.1322059111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertel T., Hausmann S., Morger D., Züger S., Guerra J., Lascano J., Reinhard C., Santoni F.A., Uchil P.D., Chatel L. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature. 2011;472:361–365. doi: 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferkorn E.R. Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc. Natl. Acad. Sci. USA. 1984;81:908–912. doi: 10.1073/pnas.81.3.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai K., Zou J.P., Tschetter J.R., Ward J.M., Shearer G.M. Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2002;129:186–196. doi: 10.1016/s0165-5728(02)00176-5. [DOI] [PubMed] [Google Scholar]
- Sandler N.G., Bosinger S.E., Estes J.D., Zhu R.T., Tharp G.K., Boritz E., Levin D., Wijeyesinghe S., Makamdop K.N., del Prete G.Q. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. 2014;511:601–605. doi: 10.1038/nature13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S.V., Schultze J.L. New Insights into IDO Biology in Bacterial and Viral Infections. Front. Immunol. 2014;5:384. doi: 10.3389/fimmu.2014.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W., Wilson S.J., Panis M., Murphy M.Y., Jones C.T., Bieniasz P., Rice C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W., MacDuff D.A., Imanaka N., Gainey M.D., Shrestha B., Eitson J.L., Mar K.B., Richardson R.B., Ratushny A.V., Litvak V. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Li N.L., Wang J., Liu B., Lester S., Li K. TRIM56 is an essential component of the TLR3 antiviral signaling pathway. J. Biol. Chem. 2012;287:36404–36413. doi: 10.1074/jbc.M112.397075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson D.B., Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Tsuchida T., Zou J., Saitoh T., Kumar H., Abe T., Matsuura Y., Kawai T., Akira S. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity. 2010;33:765–776. doi: 10.1016/j.immuni.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Uchil P.D., Quinlan B.D., Chan W.T., Luna J.M., Mothes W. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 2008;4:e16. doi: 10.1371/journal.ppat.0040016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Liu B., Wang N., Lee Y.M., Liu C., Li K. TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J. Virol. 2011;85:3733–3745. doi: 10.1128/JVI.02546-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S.J., Schoggins J.W., Zang T., Kutluay S.B., Jouvenet N., Alim M.A., Bitzegeio J., Rice C.M., Bieniasz P.D. Inhibition of HIV-1 particle assembly by 2′,3′-cyclic-nucleotide 3′-phosphodiesterase. Cell Host Microbe. 2012;12:585–597. doi: 10.1016/j.chom.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.