

Abstract
As the number of drug-resistant influenza viruses continues to increase, antivirals with novel mechanisms of action are urgently needed. Among the two classes of FDA-approved antiviral drugs, neuraminidase (NA) inhibitors, oseltamivir, zanamivir, and peramivir, are currently the only choice for the prevention and treatment of influenza virus infection. Due to the antigenic drift and antigenic shift, it will only be a matter of time before influenza viruses become completely resistant to these NA inhibitors. In pursuing the next generation of antiviral drugs with complementary mechanisms of action to those of the NA inhibitors, we have identified a natural product, cyclosporine A (CsA) (1), as a desired drug candidate. In this study, we discovered that CsA (1) and its analogs have broad-spectrum antiviral activity against multiple influenza A and B strains, including strains that are resistant to either NA or M2 inhibitors or both. Moreover, CsA (1) displays a high in vitro genetic barrier of drug resistance than oseltamivir carboxylate Mechanistic studies revealed that CsA (1) acts at the intermediate step of viral replication post viral fusion. Its antiviral mechanism is independent of inhibiting the isomerase activity of cyclophilin A (CypA), and CsA (1) has no effect on the viral polymerase activity The potent antiviral efficacy of CsA (1), coupled with the high in vitro genetic barrier of drug resistance and novel mechanism of action, renders CsA (1) a promising anti-influenza drug candidate for further development.
Keywords: Influenza virus, antiviral, CsA, broad spectrum, resistance, serial passage
Graphical Abstract
1. Introduction
Influenza viruses pose a persistent threat to global public health. Despite the existence of influenza vaccines and antiviral drugs, each seasonal influenza epidemic claims an estimated 250,000–500,000 lives worldwide. In addition, influenza pandemics caused by emerging or re-emerging influenza strains have more catastrophic impact, as witnessed by the 1918 Spanish flu (H1N1), the 1957 Asian flu (H2N2), the 1968 Hong Kong flu (H3N2), and the 2009 swine flu (H1N1) (Monto and Webster, 2013). The cornerstone in preventing influenza virus infection is vaccination (Krammer and Palese, 2015). However, influenza vaccines have to be reformulated every year in order to match with the antigens in the circulating viruses in that particular year (Lambert and Fauci, 2010; Wong and Webby, 2013). Influenza vaccines also have limited efficacy in immunocompromised patients and seniors over the age of 65 (Osterholm et al., 2012). Aside from flu vaccines, direct-acting antivirals are available for both prophylaxis and treatment of influenza infection. There are currently two classes of FDA-approved anti-influenza drugs: the M2 channel blockers amantadine and rimantadine and the neuraminidase (NA) inhibitors oseltamivir, zanamivir, and peramivir (Loregian et al., 2014). Since M2 channel blockers are no longer recommended for use in the United States as a result of widespread drug resistance (Fiore et al., 2008), NA inhibitors are currently the only line of defense against influenza virus infection. Although the majority of currently circulating influenza viruses remain sensitive to oseltamivir, it will be only a matter of time before NA-resistant viruses become predominant. In fact, NA-resistant influenza strains have been continuously isolated from human patients and animals (Hurt et al., 2009). The 2008–2009 seasonal H1N1 influenza strain in North America was completely resistant to oseltamivir (Hurt, 2014; Renaud et al., 2011), suggesting NA resistance does not compromise the fitness and transmissibility of the virus (Govorkova, 2013). Therefore, the next generation of antivirals is apparently needed (Lee and Yen, 2012; Li et al., 2015; Loregian et al., 2014; Wang, 2015; Wang et al., 2015).
Influenza viruses are negative-stranded, segmented RNA viruses that belong to the Orthomyxoviridae family. Influenza viruses are a quasispecies as a result of antigenic drift and antigenic shift (Nelson and Holmes, 2007). The heterogeneous genetic background of influenza viruses poses a great challenge in devising antiviral drugs. To complement the currently available NA inhibitors, the next generation of antiviral drugs should ideally meet the following criteria: (1) no cross resistance with NA inhibitors, (2) broad-spectrum antiviral activity, and (3) a high genetic barrier of drug resistance. It is essential for the next generation of antivirals to have different mechanisms of action than those of the NA inhibitors, such that they can be used either alone or in combination with NA inhibitors to achieve synergistic effects (Hayden, 2009). Broad-spectrum antiviral activity is also desired because current circulating influenza viruses among humans consist of at least two influenza A strains, A/California/7/2009 (H1N1) and A/Switzerland/9715293/2013 (H3N2), and two influenza B strains, B/Phuket/3073/2013 (Yamagata lineage) and B/Brisbane/60/2008 (Victoria lineage). Thus it would be ideal if a single antiviral agent could inhibit all four of these strains. In addition, the next generation of antiviral drugs should also have a high genetic barrier of drug resistance; otherwise, years of efforts in producing the antiviral drug will become futile once resistance emerges.
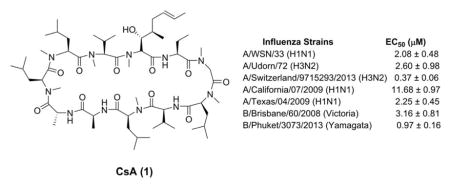
In pursuing such antiviral drugs, we have identified a lead compound, cyclosporine A (CsA) (1) (Fig. 1), which fulfills all three criteria. CsA (1) is a natural product produced by the fungus Tolypocladium inflatum. It is a known medicine used as an immunosuppressant in organ transplantation. As a cyclic peptide of 11 amino acids, CsA (1) contains three non-proteinogenic amino acids, a Bmt ((4R)-4-[(E)-2-butenyl]-4-methyl-L-threonine), an Abu (L-2-aminobutyric acid), and a D-alanine at positions 1, 2, and 8, respectively (Fig. 1). Seven out of the eleven amide bonds were methylated (residues 1, 3, 4, 6, 9, 10, and 11), which confers the oral bioavailability of CsA (1) (Chatterjee et al., 2013; Giordanetto and Kihlberg, 2014). The immunosuppressant activity of CsA is mediated through its intracellular receptors, cyclophilin A (CypA) and calcineurin. CsA binds to CypA and forms the binary complex CsA–CypA, which in turn binds to calcineurin and inhibits its phosphatase activity. As a result, T-cell activation is suppressed (Liu et al., 1991). As a protein chaperone with peptidyl-prolyl cis-trans isomerase activity, CypA has been involved in the replication of multiple viruses and represents a host factor for therapeutic intervention (Lin and Gallay, 2013; Peel and Scribner, 2013b; Sweeney et al., 2014). Re-design of CsA (1) has produced a number of non-immunosuppressive antiviral candidates in clinical trials, such as Alisporivir (Debio-025) and SCY-635 for HCV infection and NIM818 for HIV infection (Peel and Scribner, 2013b). Due to its promising antiviral efficacy, CsA (1) was also investigated in inhibiting influenza A virus replication and was found to inhibit two influenza A strains, A/WSN/33 (H1N1) and A/Puerto Rico/8/34 (H1N1), with low micromolar EC50 values (Hamamoto et al., 2013a; Liu et al., 2012). However, it is unknown whether CsA (1) has broad-spectrum antiviral activity against other influenza A or influenza B viruses, particularly the ones that are currently in circulation. Moreover, the genetic barrier of drug resistance of CsA (1) has not been addressed, and it is unknown whether viruses will rapidly evolve to become resistant to it. In this study we investigated the potential of CsA (1) and its analogs as the next generation of antiviral drugs by profiling their therapeutic scope, in vitro genetic barrier of resistance, and mechanism of action. CsA (1) was found to have broad-spectrum antiviral activity with a high in vitro genetic barrier of drug resistance. Mechanistic studies indicate CsA (1) inhibits influenza virus replication at the post fusion stage and its antiviral activity is independent of the inhibition of CypA’s isomerase activity. Collectively, these results suggest it is promising to further develop CsA (1) and its analogs as non-immunosuppressant anti-influenza drugs.
Figure 1.

Structures of CsA (1) and TMN355 (2).
2. Materials and methods
2.1. Chemical synthesis
All chemicals were purchased from commercial sources and used without further purification. CsA (1) was purchased from Biotang Inc. (cat # BC020). TMN355 (2) (cat # sc-361384), Cyclosporine C (CsC) (3) (cat # sc-203012), Cyclosporine D (CsD) (4) (cat # sc-204702), and Cyclosporine H (CsH) (5) (cat # sc-203013) were purchased from Santa Cruz Biotechnology. Details regarding the syntheses and characterizations of CsA analogs can be found in the Supplementary Data.
2.2. Biological experiments
2.2.1. Cell lines, viruses, and viral infection
Madin-Darby Canine Kidney (MDCK) cells were grown at 37°C in 5% CO2 atmosphere in DMEM media (high glucose, with L-glutamine) supplemented with 10% fetal bovine serum (FBS), 100 IU/ml penicillin and 100 μg/ml streptomycin. MDCK cells overexpressing ST6Gal I (Hatakeyama et al., 2005) were obtained from Dr. Yoshihiro Kawaoka at the University of Wisconsin at Madison through a material transfer agreement and were maintained in the presence of 7.5 μg/ml of puromycin, except when they were used for viral infection.
Influenza A virus strains, A/California/07/2009 (H1N1) and A/Texas/04/2009 (H1N1), and influenza B virus strains, B/Brisbane/60/2008 (Victoria lineage) and B/Wisconsin/1/2010 (Yamagata lineage), were obtained from Dr. James Noah at the Southern Research Institute. Influenza A virus strains A/Denmark/524/2009 (H1N1) and A/Denmark/528/2009 (H1N1) were obtained from Dr. Elena Govorkova at St. Jude Children’s Research Hospital. Influenza A virus, A/Switzerland/9715293/2013 (H3N2), FR-1368, and influenza B virus, B/Phuket/3073/2013 (Yamagata Lineage), FR-1364, were obtained through the Influenza Reagent Resource, Influenza Division, WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza, Centers for Disease Control and Prevention, Atlanta, GA, USA. Virus stocks were amplified in MDCK cells in the presence of 2 μg/ml N-acetyl trypsin. Two days post infection, the culture media were harvested and cell debris was removed by centrifugation at 3000 rpm for 30 minutes. Virus titers were determined by plaque reduction assay using MDCK cells expressing ST6Gal I.
2.2.2. Plaque reduction assay
Plaque reduction assay was carried out as previously described (Li et al., 2016; Wang et al., 2013) except MDCK cells expressing ST6Gal I were used instead of regular MDCK cells.
2.2.3. Cytotoxicity assay
Evaluation of the cytotoxicity of compounds was carried out using the neutral red uptake assay (Repetto et al., 2008). Briefly, 80,000 cells/mL MDCK cells in DMEM medium which was supplemented with 10% FBS and 100 U/mL of Penicillin-Streptomycin P/S were dispensed into clear 96-well cell culture plates (Cat #: CLS3362) at 100 μL/well. Twenty-four hours later, the growth medium was removed and washed with 100 μL PBS buffer; then 200 μL fresh DMEM (No FBS) medium contains serial diluted compounds was added to each well. After incubating for 48 hours at 37 °C with 5% CO2 in a CO2 incubator, the medium was removed and replaced with 100 μL DMEM medium contains 40 μg/mL neutral red for 4 hours at 37 °C. The amount of retained neutral red was determined by absorbance at 540 nm using a Multiskan FC Microplate Photometer (Fisher Scientific). The CC50 values were calculated from best-fit dose response curves with variable slope in Prism 5.
2.2.4. Serial passage experiment
Two sets of serial passage experiments were performed. The first set was done with A/WSN/33 (H1N1) virus. MDCK cells were infected with A/WSN/33 (H1N1) virus at MOI 0.001 for 1 h. Then the inoculum was removed and MDCK cells were incubated with 3 μM CsA (1). In each passage, the viruses were harvested when a significant cytopathic effect was observed, which usually takes 2–3 days after virus infection. The titers of harvested viruses were determined by plaque reduction assay. The CsA (1) sensitivity after passages 3, 6, and 10 were determined via plaque reduction assay as described previously (Li et al., 2016; Wu et al., 2014). The second set of serial passage experiment was performed with A/Switzerland/9715293/2013 (H3N2). Experiment conditions were the same as the first set except the drug concentration of CsA (1) was gradually increased from 0.5 μM to 4 μM from passages 1 to 4 and was kept constant at 4 μM at passages 5 and 6. Oseltamivir carboxylate was included as a control. The drug sensitivity of CsA (1) and oseltamivir at passages 3 and 6 were determined via plaque reduction assay.
2.2.5. Time-of-addition experiment
MDCK cells were seeded at 2X 105 cells/6 cm dish. 1 μM oseltamivir carboxylate or 6 μM CsA (1) was added at different time points as illustrated in Fig. 8. MDCK cells were infected 48 hours after seeding with A/WSN/33 (H1N1) virus at MOI 0.01. Viruses were harvested at 12 hours after infection. The virus titer was determined with plaque assay. Oseltamivir carboxylate was included as a control.
Figure 8.
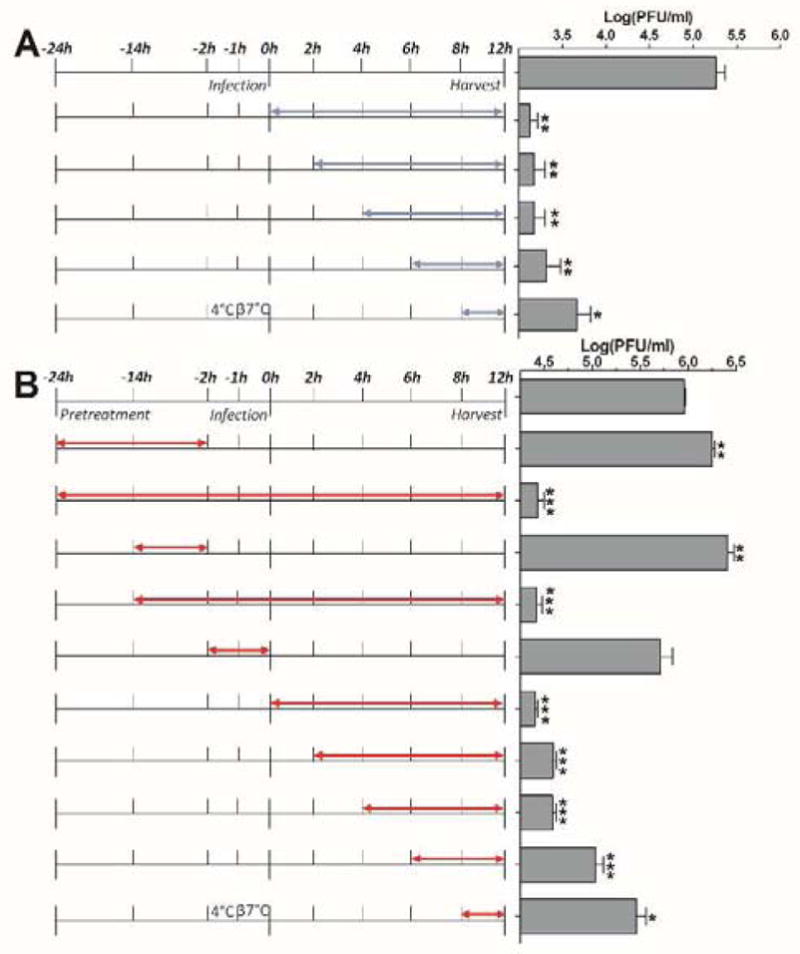
Time-of-addition experiments. Cells were infected with A/WSN/33 (H1N1) virus at −2 hour time point; viruses were first incubate at 4°C for 1 hour for attachment, then at 37°C for 1 hour for virus entry. At time point 0 hour, cells were washed with PBS buffer and progeny viruses were harvested at 12 hours post infection. The titer of harvested virus was determined with plaque assay. (A) Oseltamivir carboxylate time-of addition experiment. Blue arrows show the period in which 1 μM oseltamivir carboxylate is present. (B) CsA (1) time-of-addition experiment. Red arrows show the period in which 6 μM CsA (1) is present.
2.2.6. Influenza virus minigenome assay
HEK293 cells were seeded at 3 × 105 cells per well in 12-well plates and incubated overnight at 37 °C, with 5% CO2 in a CO2 cell culture incubator. The cells were transfected with pCDNA constructs for influenza A/WSN/33 virus PB1, PB2, and PA (100 ng each) and NP (200 ng), the RNA polymerase II driven Renilla luciferase reporter pRL-SV40 (Promega) (250 ng), and the influenza virus-specific RNA polymerase I driven firefly luciferase reporter (vRNA Luc) (250 ng). The transfection was performed with TransIT-293 (Mirus) in OptiMEM (Invitrogen). Two hours after transfection the media was supplemented with 1 μl compounds to their final concentrations. Twenty-four hours after incubation, cells were harvested, and firefly luciferase and Renilla luciferase expression was determined using the Dual Luciferase Assay Kit (Promega).
3. Results
3.1 CsA has broad-spectrum antiviral activity against clinical isolates of influenza A and B viruses
Previous studies have shown that CsA (1) inhibits the influenza A/Puerto Rico/8/34 (H1N1) and the A/WSN/33 (H1N1) with low micromolar EC50 values (Hamamoto et al., 2013a; Liu et al., 2012). To confirm the antiviral activity of CsA (1), the MDCK cells were infected with A/WSN/33 (H1N1) at about 100 pfu in the presence of increasing concentrations of CsA (1). Plaque numbers were counted after two days of incubation. CsA (1) inhibited A/WSN/33 (H1N1) replication in a dose-dependent manner with an EC50 of 2.08 μM (Table 1), which is consistent with previous results. Next, to investigate whether CsA (1) has broad-spectrum antiviral activity, we chose six influenza A strains and three influenza B strains. Table 1 summarizes the drug sensitivity of these influenza strains toward the approved antiviral drugs amantadine and oseltamivir, as well as the testing drug CsA (1). Representative plaque assay results were shown in Fig. 2. The viruses chosen cover both types of influenza viruses (A and B) as well as their subtypes including influenza A viruses from group 1 and group 2 hemagglutinin groups, and influenza B viruses from Yamagata and Victoria lineages. Viruses that are resistant to one or both classes of FDA-approved antiviral drugs, amantadine and oseltamivir, were also included. For example, A/Texas/04/2009 (H1N1) and A/Denmark/528/2009 (H1N1) are resistant to both amantadine and oseltamivir, which is conferred by the S31N and H275Y mutations, respectively. The appearance of such strains from clinics is of particular concern because they pose a great threat to public health (Hurt, 2014). Despite the variable drug sensitivity of these six influenza A strains, CsA (1) was found to inhibit the replication of all six strains at 10 μM in plaque reduction assays (Table 1 and Fig. 2). Of note, CsA (1) significantly decreased the plaque size but to a lesser extent the plaque number of A/California/07/2009 (H1N1) and A/Denmark/524/2009 (H1N1) when tested at 10 μM (Fig. 2A). This might due to intrinsic reduced drug sensitivity of these two viruses. This is possible as not all the viral genes are highly conserved across different influenza strains, thus it is expected that the interactions between various viral proteins and host factors might vary among different strains. As a result, they may have different drug sensitivity towards host-targeting antivirals. For the two influenza B strains, CsA (1) similarly had potent inhibition at 10 μM (Table 1 and Fig. 2). The efficacies of CsA (1) against A/Udorn/72 (H3N2), A/Switzerland/9715293/2013 (H3N2), A/California/07/2009 (H1N1), A/Texas/04/2009 (H1N1), B/Brisbane/60/2008 (Victoria lineage), and B/Phuket/3073/2013 (Yamagata lineage) were further evaluated by serial drug titrations. The EC50 values range from 0.37 to 11.68 μM (Table 1). Collectively, these results clearly demonstrated that CsA (1) has broad-spectrum antiviral activity against both influenza A and B viruses and has no cross resistance with the currently approved anti-influenza drugs, amantadine and oseltamivir.
Table 1.
Drug sensitivity of influenza strains toward CsA (1), amantadine, and oseltamivir carboxylate.
| Influenza Strains | Sensitivity to Amantadine | Sensitivity to Oseltamivir carboxylate | Sensitivity to CsA (1) | EC50 (μM) (mean ± S.E.) |
|---|---|---|---|---|
| A/WSN/33 (H1N1) | R (S31N) | S | S | 2.08 ± 0.48 |
| A/Udorn/72 (H3N2) | S | S | S | 2.60 ± 0.98 |
| A/Switzerland/9715293/2013 (H3N2) | R (S31N) | S | S | 0.37 ± 0.06 |
| A/California/07/2009 (H1N1) | R (S31N) | S | S | 11.68 ± 0.97 |
| A/Texas/04/2009 (H1N1) | R (S31N) | R (H275Y) | S | 2.25 ± 0.45 |
| A/Denmark/524/2009 (H1N1) | R(S31N) | S | S | N.D. |
| A/Denmark/528/2009 (H1N1) | R (S31N) | R (H275Y) | S | N.D. |
| B/Wisconsin/1/2010 (Yamagata) | R | S | S | N.D. |
| B/Brisbane/60/2008 (Victoria) | R | S | S | 3.16 ± 0.81 |
| B/Phuket/3073/2013 (Yamagata) | R | S | S | 0.97 ± 0.16 |
S: sensitive; R: resistant; N.D.: not determined.
Figure 2.

Plaque reduction assays of CsA (1) in inhibiting A/WSN/33 (H1N1) and clinically isolated influenza A and B viruses. The assay was carried out with MDCK cells expressing ST6Gal I gene. Plaque numbers were counted at each concentration, and the data was fit into dose-response curve with Prism 5, and the best fit EC50 values were shown in Table 1. (A) Plaque assays of CsA (1) against six influenza A and two influenza B strains at 10 μM. Quantification of the plaque size by ImageJ revealed that the plaque sizes of all eight viruses were significantly reduced in the presence of 10 μM CsA (1). The plaque area ratios with and without CsA (1) are 29.5 % for A/California/07/2009 (H1N1), 15.6 % for A/Texas/04/2009 (H1N1), 24.0 % for A/Denmark/524/2009 (H1N1), 9.4 % for A/Denmark/528/2009 (H1N1), 4.1 % for A/Udorn/72 (H3N2), 0.6 % for A/Switzerland/9715293/2013 (H3N2), 7.2 % for B/Wisconsin/1/2010, and 15.3 % for B/Brisbane/60/2008. (B) CsA (1) in inhibiting A/WSN/33 (H1N1) at different concentrations; (C) CsA (1) in inhibiting A/Switzerland/9715293/2013 (H3N2) at different concentrations; (D) CsA (1) in inhibiting A/Texas/04/2009 (H1N1) at different concentrations; (E) CsA (1) in inhibiting B/Brisbane/60/2008 (Victoria) at different concentrations; (F) CsA (1) in inhibiting B/Phuket/3073/2013 (Yamagata) at different concentrations.
3.2 CsA (1) has a high in vitro genetic barrier of drug resistance
To access the in vitro genetic barrier of drug resistance of CsA (1), CsA (1) was subjected to the serial viral passage experiments. Two sets of serial passage experiments were performed, one was with A/WSN/33 (H1N1), another was with A/Switzerland/9715293/2013 (H3N2). For the first set, the A/WSN/33 (H1N1) virus was replicated in the presence of 3 μM of CsA (1) (1.5 times the EC50 value). The amplified virus was then titrated and applied to the next cycle of viral replication with 3 μM of CsA (1) (Table 2). The drug sensitivities of resulting viruses at passages 3, 6, and 10 were tested. The EC50 values of CsA (1) against viruses selected at passages 3, 6, and 10 were 1.24, 2.08, and 0.84 μM, respectively, which is similar to the efficacy of CsA (1) in inhibiting the parent strain (EC50 = 2.08 μM). These results suggested that CsA (1) has a high in vitro genetic barrier of drug resistance and resistance to CsA (1) is difficult to evolve. To test whether the high in vitro genetic barrier of drug resistance of CsA (1) can be extended to other strains, we repeated the serial viral passage experiments with A/Switzerland/9715293/2013 (H3N2). A/Switzerland/9715293/2013 (H3N2) was one of the predominant circulating influenza A strains during the 2014–2015 flu season in North America. In this set of experiment, the drug concentration of CsA (1) was gradually increased from 0.5 μM to 4 μM from passages 1 to 4 and was kept constant from passages 5 and 6. To ensure the serial passage experiments were conducted properly, oseltamivir carboxylate was included as a control and its concentration was increased proportionally as that of CsA (1). As shown in Table 2, no resistance was observed for CsA (1) up to passage 6, while more than ten-fold of EC50 increase was observed for oseltamivir carboxylate. The serial viral passage results for oseltamivir carboxylate were consistent with literature reports (Ehrhardt et al., 2013; Shih et al., 2010). Thus, it can be concluded that CsA (1) has a higher in vitro genetic barrier of drug resistance than oseltamivir carboxylate.
Table 2.
Serial viral passages experiments with CsA (1) drug selection pressure.
| Set 1: A/WSN/33 (H1N1) | Set 2: A/Switzerland/9715293/2013 (H3N2) | |||||
|---|---|---|---|---|---|---|
| Passages | CsA drug concentration applied (μM) | (μM) EC50 | CsA drug concentration applied (μM) | EC50 (μM) (mean ± S.E.) | Oseltamivir drug concentration applied (nM) | EC50 (nM) (mean ± S.E.) |
| 0 | 0 | 2.08 ± 0.46 | 0 | 0.37 ± 0.06 | 0 | 12 ± 5 |
| 1 | 3 | N.D. | 0.5 | N.D. | 15 | N.D. |
| 2 | 3 | N.D. | 1.0 | N.D. | 30 | N.D. |
| 3 | 3 | 1.24 ± 0.45 | 2.0 | 0.36 ± 0.03 | 60 | 175 ± 53 |
| 4 | 3 | N.D. | 4.0 | N.D. | 120 | N.D. |
| 5 | 3 | N.D. | 4.0 | N.D. | 120 | N.D. |
| 6 | 3 | 2.08 ± 1.08 | 4.0 | 0.49 ± 0.09 | 120 | 260 ± 77 |
| 7 | 3 | N.D. | ||||
| 8 | 3 | N.D. | ||||
| 9 | 3 | N.D. | ||||
| 10 | 3 | 0.84 ± 0.81 | ||||
N.D.: not determined
3.3 Structure–activity relationships of CsA (1)
To study the structure-activity relationships of CsA (1), several natural CsA analogs, CsC (3), CsD (4), and CsH (5) (Fig. 3A), were tested for their antiviral efficacy against A/WSN/33 (H1N1) in plaque reduction assays. It was found that CsD (4) and CsH (5) had similar activity to that of CsA (1) while CsC (3) was less active (Fig. 3B and 3C). These results suggest hydrophobic residue is preferred at residue 2 (CsA (1), CsD (4) versus CsC (3)), while the stereochemistry at residue 11 is not critical for the antiviral effect (CsA (1) versus CsH (5)).
Figure 3.

Natural CsA analogs and their antiviral efficacy against A/WSN/33 (H1N1). (A) Molecular structures of CsA (1), CsC (3), CsD (4), and CsH (5). (B) Anti-viral efficacy of CsA analogs against A/WSN/33 (H1N1). WJ332 is a known M2-S31N channel inhibitor and was used as a positive control (A/WSN/33 contains M2-S31N mutant and it is sensitive to WJ332). CsD (4) and CsH (5) displayed similar efficacy against A/WSN/33 (H1N1) as CsA (1); CsC (3) is slightly less effective than CsA (1). *, p<0.05; **, p<0.01; ***, p<0.001 in two-tail t-test when compared with No Drug condition.
Aside from the natural CsA analogs (3–5) mentioned above, several CsA derivatives (6–20) were synthesized by semi-synthesis following established synthesis procedures (Fig. 4) (Peel and Scribner, 2015; Sweeney et al., 2014). The two functional groups, a hydroxyl and an alkene, in the side chains of Bmt at residue 1 allow easy access to CsA analogs by direct derivatization. For example, epoxidation of CsA (1) with meta-Chloroperoxybenzoic acid (m-CPBA) gave compound 6, which underwent ring opening with 1,3-diaminopropane to give compound 7 (Fig. 4A). Using Grubbs’ 2nd generation catalyst, CsA (1) metathesized with N-(Hydroxysuccinimidyl)-4-pentenoate to give compound 8 (Fig. 4B). The succinimidyl ester in 8, when reacted with 1, 3-diaminopropane, gave the amide 9. The alkene in CsA (1) was reduced to alkane 10 using palladium on carbon. The beta-hydroxyl group in CsA (1) was esterified using acetic anhydride, dimethylpyridine, and pyridine to give compound 11. The alkene in ester 11 was converted to epoxide 12 using m-CPBA. Residue 3 at CsA (1) could be directly alkylated using strong bases such as lithium bis(trimethylsilyl)amide (LiHMDS) or lithium diisopropylamide (LDA). Treatment of CsA (1) with LiHMDS and methyl iodide gave compound 13. Similarly, deprotonation of CsA (1) with LDA, followed by quenching with carbon dioxide gave compound 14. The carboxylic acid in 14 was esterified using methyl iodide and potassium carbonate to give the ester 15. Other than epoxidation and metathesis, the alkene in ester 11 was also transformed to aldehyde 18 and acid 16 under different oxidation conditions (Fig. 4C). The acid in compound 16 was esterified using methyl iodide and potassium carbonate to give compound 17. The aldehyde in 18 was condensed with 3,4-diaminotoluene to give compound 19 (Malesevic et al., 2013). Reduction of 18 using sodium borohydride gave the alcohol 20.
Figure 4.

Synthesis scheme of CsA analogs. Detailed synthesis procedures and compound characterization can be found in the Supporting Information.
When assayed at 10 μM drug concentration, compounds 10, 11, 12, 13, 15, and 18 had similar antiviral activity as that of CsA (1) (Fig. 5A), compounds 17 and 20 had reduced antiviral activity compared to CsA (1), compounds 7, 8, 9, and 16 were not active, and compounds 14 and 19 were cytotoxic. The cytotoxicity of potent compounds was profiled using the neutral red assay (Repetto et al., 2008). CsC (3) and CsD (4) were not cytotoxic up to 50 μM, while the CC50 values for other active compounds range from 6.3 μM to 38.6 μM (Fig. 5B). Combined with the SAR results from natural CsA analogs, the following conclusions can be drawn: hydrophobic substitutions are preferred at residues 1, 2, and 3, while polar and charged residues are not tolerated these residues; the stereochemistry at residue 11 is not critical for the anti-influenza activity (Fig. 6).
Figure 5.

Anti-viral efficacy of semi-synthesized CsA analogs against A/WSN/33 (H1N1) virus, and their cytotoxicity. (A) Anti-viral efficacy was evaluated with plaque reduction assay. All compounds were tested at 10 μM, and compounds (14) and (19) displayed severe cytopathic effect after 48 hours at 10 μM and no cells remained on the dishes. *, p<0.05; **, p<0.01; ***, p<0.001 in two-tail t-test when compared with No Drug condition. (B) Cytotoxicity of CsA (1) and its analogs. The cytotoxicity of compounds was evaluated using the neutral red uptake assay.
Figure 6.

Summary of structure–activity relationships of CsA.
3.4 The antiviral activity of CsA (1) is predominantly independent of CypA inhibition
To investigate whether the antiviral activity of CsA (1) is correlated with its inhibition of CypA isomerase activity, TMN355 (2) was chosen as a control compound. TMN355 (2) is a rationally designed CypA inhibitor that is 27 times more potent than CsA (1) in inhibiting the isomerase activity of CypA (Ni et al., 2009). If the enzymatic activity of CypA is essential for influenza viral replication, then TMN355 (2) is predicted to have potent anti-influenza activity. However, treatment with 10 μM TMN355 (2) had no effect on the replication of several influenza A and B strains (Fig. 7). Thus the antiviral activity of CsA (1) appears to be independent of CypA’s isomerase activity. This conclusion was further strengthened by the antiviral activity of a non-CypA binding CsA analog, CsH (5) (Jeffery, 1991; Peel and Scribner, 2013a), as shown in Fig. 3. CsH (5) is a natural analog of CsA (1) and differs from CsA (1) in residue 11. The residues at residue 11 are L-valine and D-valine for CsA (1) and CsH (5), respectively. CsH (5) does not bind to CypA and has no immunosuppressant activity as it does not inhibit the phosphatase activity of calcineurin (Jeffery, 1991). As shown in Fig. 3B, CsH (5) had similar antiviral activity to that of CsA (1) when tested at 10 μM, suggesting CypA is not essential for CsA’s anti-influenza activity. The antiviral results of TMN355 (2) and CsH (5) suggest that CsA (1) exerts its anti-influenza activity through a novel mechanism. This novel mechanism is distinct from that of HIV and HCV inhibition by CsA (1). In both cases of HIV and HCV inhibition by CsA (1), the affinity of CsA (1) binding to CypA is positively correlated with its antiviral potency (Liu et al., 2012).
Figure 7.

Anti-viral efficacy of CypA inhibitor TMN355 (2) against clinically isolated influenza A and B viruses in plaque assays. 10 μM TMN355 (2) were applied to overlay media after infection with virus. TMN355 (2) is a known CypA inhibitor.
3.5 CsA inhibits influenza virus replication at the intermediate stage of viral replication after viral fusion
To identify at which stage of the viral replication cycle CsA (1) exerts its inhibitory effect, a time-of-addition experiment was performed. In this experiment, CsA (1) was added at different time points throughout the viral replication cycle. If CsA (1) acts at the early stage of viral replication, adding CsA (1) at the late stage of viral replication will have less inhibitory effect. Likewise, if CsA (1) acts at the late stage of viral replication, adding CsA (1) at both early and late stages will result in a potent inhibitory effect. Oseltamivir carboxylate was included as a positive control. Oseltamivir carboxylate inhibits the last step of viral replication by preventing the release of progeny virion; thus it is expected to have potent antiviral activity even when added in the late stage of viral replication. Consistent with its mechanism of inhibition, oseltamivir carboxylate had a potent antiviral effect even when added 8 hours post initial viral infection in a single-cycle viral replication experiment (virus was harvested at 12 hours post infection) (Fig. 8A). In contrast, the antiviral efficacy of CsA (1) gradually decreased when added at the later stage of viral replication (Fig. 8B). When MDCK cells were pretreated with 6 μM CsA (1) for 12 h or 22 h, virus production was slightly increased (Fig. 8B). This result is consistent with the previous report (Hamamoto et al., 2013b), suggesting CypA might play an antiviral role during viral replication. Thus efforts on exploring CsA analogs as anti-influenza drugs should focus on those non-cyclophilin-binding analogs. Collectively, the time-of-addition experiments suggest CsA (1) inhibits influenza virus replication at the intermediate stage of viral replication post viral fusion.
3.6 CsA has no effect on the viral polymerase activity
To test whether CsA (1) inhibits the influenza viral polymerase activity, minigenome assay was performed (Hoffmann et al., 2011). In the minigenome assay, 293 T cells were transfected with six plasmids, four of which encode the viral polymerase complex NP, PA, PB1, and PB2, one plasmid encodes the influenza virus-specific RNA polymerase I driven firefly luciferase reporter (vRNA Luc), and one plasmid encodes the RNA polymerase II driven Renilla luciferase reporter pRL-SV40, which was used to normalize the transfection efficacy. Nucleozin (21) was included as a positive control. Nucleozin (21) is a known NP inhibitor and has been shown to inhibit the viral polymerase activity in the minigenome assay (Kao et al., 2010). As shown in Fig. 9, nucleozin (21) completely inhibited the viral polymerase activity, while both CsA (1) and CsH (5) had no effect on the normalized luciferase ratio, suggesting both compounds do not inhibit viral polymerase activity (Hamamoto et al., 2013b; Liu et al., 2012). This result, together with the time-of-addition experiments, suggests that CsA (1) inhibits the intermediate step at the viral replication cycle, probably through inhibiting viral protein trafficking, post-translational modifications, or budding. Further studies are ongoing to delineate its exact antiviral mechanism.
Figure 9.
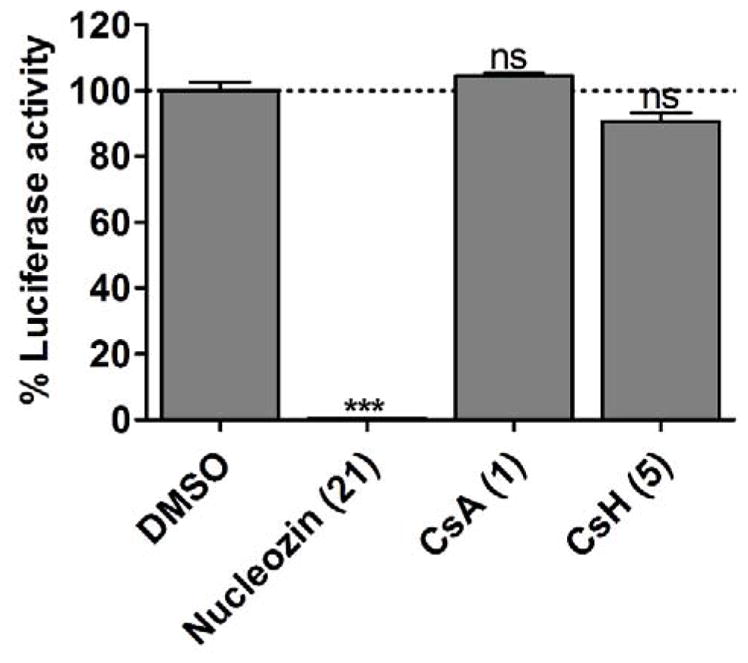
Influenza minigenome assay. CsA (1) and CsH (5) did not inhibit A/WSN/33 (H1N1) viral polymerase activity. HEK293 cells were transfected with protein expression plasmids for influenza A/WSN/33 virus polymerase subunits PB1, PB2, PA, and nucleoprotein NP. An influenza virus-specific firefly luciferase reporter and a Renilla luciferase expression plasmid were co-transfected. Two hours after transfection, 10 μM nucleozin (21), 6 μM CsA (1), or CsH (5) were added to media. Nucleozin (21) is a known NP inhibitor and was used as a positive control. The firefly luciferase activity was normalized against Renilla luciferase activity. ns, NOT significant; *, p<0.05; **, p<0.01; ***, p<0.001 in two-tail t-test when compared with DMSO condition.
4. Conclusion
In this study we have investigated the potential of CsA (1) and its analogs (3–20) as the next generation of anti-influenza drugs. We have found that CsA (1) has broad-spectrum antiviral activity against both influenza A and B viruses. More importantly, CsA (1) displays a higher in vitro genetic barrier of drug resistance than oseltamivir carboxylate and no resistance was selected for two influenza A strains, A/WSN/33 (H1N1) and A/Switzerland/9715293/2013 (H3N2). Furthermore, we provide convincing evidence that CsA (1) inhibits influenza replication via a different mechanism to that of HIV and HCV viruses.
The antiviral mechanisms of CsA (1) and its analogs in inhibiting HIV and HCV have been well studied. CypA is involved in multiple steps of the HIV replication cycle. The interaction between CypA and capsid protein in infected cells promotes viral infectivity (Braaten et al., 1996; Towers et al., 2003). CypA also interacts with other HIV-1 proteins, such as Vpr and p6, to regulate HIV infection (Solbak et al., 2012; Zander et al., 2003). In the replication of HCV, CypA also plays a positive role. The PPIase activity of CypA assists the replication of HCV (Fischer et al., 2010), and CypA binding to NS5B increases the affinity of the polymerase to viral RNA, therefore enhancing HCV replication (Liu et al., 2009). Moreover, the interaction between CypA and NS5A protein also aids HCV viral replication (Tellinghuisen et al., 2008). In summary, CsA inhibits HIV and HCV replication via binding to CypA. However, our results showed that TMN355 (2), a high affinity inhibitor of CypA, has no effect on influenza virus replication; CsH (5), a CsA (1) analog, which does not bind to CypA, displayed similar efficacy as CsA (1) in inhibiting influenza virus replication. Taken together, our study showed that CsA (1) inhibits influenza virus replication via a CypA-independent mechanism.
Previous studies regarding the mechanism of action of CsA (1) yielded controversial results (Hamamoto et al., 2013a; Liu et al., 2012). Liu et al. showed that CsA (1) inhibits influenza virus replication via CypA-dependent and -independent pathways (Liu et al., 2012). In the CypA-dependent pathway, CsA (1) enhanced the CypA-mediated influenza M1 protein degradation. However, Hamamoto et al. showed that CsA (1) treatment did not alter the M1 protein level (Hamamoto et al., 2013a). Nevertheless, both studies showed that in CypA knockout or knockdown conditions, CsA (1) treatment greatly reduced influenza replication, which is consistent with our observation that CsA (1) inhibition of influenza replication does not require its binding to CypA. Our time-of-addition experiment and minigenome assay indicate that CsA (1) acts at intermediate step(s) during influenza replication, probably through a novel mechanism that warrants further investigation. Overall, the broad-spectrum antiviral activity, coupled with the high in vitro genetic barrier of drug resistance and novel mechanism of action, highlights the great potential of natural products in drug discovery. As suggested by current results, the next step SAR studies should focus on designing non-cyclophilin-binding and non-calcinurin-inhibiting CsA analogs as anti-influenza drugs. These studies are ongoing and will be reported when they are available.
Supplementary Material
Highlights.
Cyclosporine A (CsA) (1) has broad-spectrum antiviral activity against current circulating influenza A and B viruses
CsA (1) displays a high in vitro genetic barrier of drug resistance than oseltamivir carboxylate
CsA (1) inhibits an intermediate stage of viral replication post viral fusion
CsH (5), an non-immunosuppressant analog of CsA (1), also has potent antiviral activity
Acknowledgments
This work was supported by the startup funds from the University of Arizona and the NIH grant AI119187 to J.W. R.M. was supported by the NIH training grant T32 GM008804. We thank David Bishop for proofreading and editing the manuscript. We thank Jiantao Zhang for help with editing the figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- http://www.who.int/influenza/vaccines/virus/recommendations/2015_16_north/en/
- Palese P, Shaw ML. Orthomyxoviridae: The Viruses and Their Replication. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 1647–1690. [Google Scholar]
- WHO. [Accessed October 7th 2015];Influenza fact sheet. updated March, 2014. http://www.who.int/mediacentre/factsheets/fs211/en/
- Braaten D, Aberham C, Franke EK, Yin L, Phares W, Luban J. Cyclosporine A-resistant human immunodeficiency virus type 1 mutants demonstrate that Gag encodes the functional target of cyclophilin A. J Virol. 1996;70:5170–5176. doi: 10.1128/jvi.70.8.5170-5176.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee J, Rechenmacher F, Kessler H. N-Methylation of Peptides and Proteins: An Important Element for Modulating Biological Functions. Angew Chem Int Ed Engl. 2013;52:254–269. doi: 10.1002/anie.201205674. [DOI] [PubMed] [Google Scholar]
- Ehrhardt C, Ruckle A, Hrincius ER, Haasbach E, Anhlan D, Ahmann K, Banning C, Reiling SJ, Kuhn J, Strobl S, Vitt D, Leban J, Planz O, Ludwig S. The NF-kappaB inhibitor SC75741 efficiently blocks influenza virus propagation and confers a high barrier for development of viral resistance. Cell Microbiol. 2013;15:1198–1211. doi: 10.1111/cmi.12108. [DOI] [PubMed] [Google Scholar]
- Fiore AE, Shay DK, Broder K, Iskander JK, Uyeki TM, Mootrey G, Bresee JS, Cox NS Centers for Disease Control, Prevention, Advisory Committee on Immunization Practices. Prevention and control of influenza: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2008. MMWR Recomm Rep. 2008;57:1–60. [PubMed] [Google Scholar]
- Fischer G, Gallay P, Hopkins S. Cyclophilin inhibitors for the treatment of HCV infection. Curr Opin Investig Drugs. 2010;11:911–918. [PubMed] [Google Scholar]
- Giordanetto F, Kihlberg J. Macrocyclic Drugs and Clinical Candidates: What Can Medicinal Chemists Learn from Their Properties? J Med Chem. 2014;57:278–295. doi: 10.1021/jm400887j. [DOI] [PubMed] [Google Scholar]
- Govorkova E. Consequences of resistance: in vitro fitness, in vivo infectivity, and transmissibility of oseltamivir-resistant influenza A viruses. Influenza Other Respir Viruses. 2013;7:50–57. doi: 10.1111/irv.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto I, Harazaki K, Inase N, Takaku H, Tashiro M, Yamamoto N. Cyclosporin A inhibits the propagation of influenza virus by interfering with a late event in the virus life cycle. Jpn J Infect Dis. 2013a;66:276–283. doi: 10.7883/yoken.66.276. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S, Sakai-Tagawa Y, Kiso M, Goto H, Kawakami C, Mitamura K, Sugaya N, Suzuki Y, Kawaoka Y. Enhanced Expression of an α 2,6-Linked Sialic Acid on MDCK Cells Improves Isolation of Human Influenza Viruses and Evaluation of Their Sensitivity to a Neuraminidase Inhibitor. J Clin Microbiol. 2005;43:4139–4146. doi: 10.1128/JCM.43.8.4139-4146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden F. Developing New Antiviral Agents for Influenza Treatment: What Does the Future Hold? Clin Infect Dis. 2009;48:S3–S13. doi: 10.1086/591851. [DOI] [PubMed] [Google Scholar]
- Hoffmann HH, Kunz A, Simon VA, Palese P, Shaw ML. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc Natl Acad Sci. 2011;108:5777–5782. doi: 10.1073/pnas.1101143108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt A, Holien J, Parker M, Barr I. Oseltamivir Resistance and the H274Y Neuraminidase Mutation in Seasonal, Pandemic and Highly Pathogenic Influenza Viruses. Drugs. 2009;69:2523–2531. doi: 10.2165/11531450-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Hurt AC. The epidemiology and spread of drug resistant human influenza viruses. Curr Opin Virol. 2014;8:22–29. doi: 10.1016/j.coviro.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Jeffery JR. Cyclosporine analogues. Clin Biochem. 1991;24:15–21. doi: 10.1016/0009-9120(91)90105-n. [DOI] [PubMed] [Google Scholar]
- Kao RY, Yang D, Lau LS, Tsui WHW, Hu L, Dai J, Chan MP, Chan CM, Wang P, Zheng BJ, Sun J, Huang JD, Madar J, Chen G, Chen H, Guan Y, Yuen KY. Identification of influenza A nucleoprotein as an antiviral target. Nat Biotech. 2010;28:600–605. doi: 10.1038/nbt.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer F, Palese P. Advances in the development of influenza virus vaccines. Nat Rev Drug Discov. 2015;14:167–182. doi: 10.1038/nrd4529. [DOI] [PubMed] [Google Scholar]
- Lambert LC, Fauci AS. Influenza Vaccines for the Future. N Engl J Med. 2010;363:2036–2044. doi: 10.1056/NEJMra1002842. [DOI] [PubMed] [Google Scholar]
- Lee SMY, Yen HL. Targeting the host or the virus: Current and novel concepts for antiviral approaches against influenza virus infection. Antiviral Res. 2012;96:391–404. doi: 10.1016/j.antiviral.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Ma C, DeGrado WF, Wang J. Discovery of highly potent inhibitors targeting the predominant drug-resistant S31N mutant of the influenza A virus M2 proton channel. J Med Chem. 2016;59:1207–1216. doi: 10.1021/acs.jmedchem.5b01910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Ma C, Wang J. Inhibitors Targeting the Influenza Virus Hemagglutinin. Curr Med Chem. 2015;22:1361–1382. doi: 10.2174/0929867322666150227153919. [DOI] [PubMed] [Google Scholar]
- Lin K, Gallay P. Curing a viral infection by targeting the host: The example of cyclophilin inhibitors. Antiviral Res. 2013;99:68–77. doi: 10.1016/j.antiviral.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhao Z, Li Z, Xu C, Sun L, Chen J, Liu W. Cyclosporin A inhibits the influenza virus replication through cyclophilin A-dependent and -independent pathways. PLoS One. 2012;7:e37277. doi: 10.1371/journal.pone.0037277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Yang F, Robotham JM, Tang H. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J Virol. 2009;83:6554–6565. doi: 10.1128/JVI.02550-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loregian A, Mercorelli B, Nannetti G, Compagnin C, Palù G. Antiviral strategies against influenza virus: towards new therapeutic approaches. Cell Mol Life Sci. 2014:1–25. doi: 10.1007/s00018-014-1615-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malesevic M, Gutknecht D, Prell E, Klein C, Schumann M, Nowak RA, Simon JC, Schiene-Fischer C, Saalbach A. Anti-inflammatory Effects of Extracellular Cyclosporins Are Exclusively Mediated by CD147. J Med Chem. 2013;56:7302–7311. doi: 10.1021/jm4007577. [DOI] [PubMed] [Google Scholar]
- Monto AS, Webster RG. Influenza pandemics: History and lessons learned, Textbook of Influenza. John Wiley & Sons, Ltd; 2013. pp. 20–34. [Google Scholar]
- Nelson MI, Holmes EC. The evolution of epidemic influenza. Nat Rev Genet. 2007;8:196–205. doi: 10.1038/nrg2053. [DOI] [PubMed] [Google Scholar]
- Ni S, Yuan Y, Huang J, Mao X, Lv M, Zhu J, Shen X, Pei J, Lai L, Jiang H, Li J. Discovering Potent Small Molecule Inhibitors of Cyclophilin A Using de Novo Drug Design Approach. J Med Chem. 2009;52:5295–5298. doi: 10.1021/jm9008295. [DOI] [PubMed] [Google Scholar]
- Osterholm M, Kelley N, Sommer A, Belongia E. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect Dis. 2012;12:36–44. doi: 10.1016/S1473-3099(11)70295-X. [DOI] [PubMed] [Google Scholar]
- Peel M, Scribner A. CHAPTER 11 Optimization of Cyclophilin Inhibitors for Use in Antiviral Therapy, Successful Strategies for the Discovery of Antiviral Drugs. The Royal Society of Chemistry; 2013a. pp. 384–418. [Google Scholar]
- Peel M, Scribner A. Cyclophilin inhibitors as antiviral agents. Bioorg Med Chem Lett. 2013b;23:4485–4492. doi: 10.1016/j.bmcl.2013.05.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel M, Scribner A. Semi-synthesis of cyclosporins. Biochimica et Biophysica Acta (BBA) - General Subjects. 2015;1850:2121–2144. doi: 10.1016/j.bbagen.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Renaud C, Kuypers J, Englund JA. Emerging oseltamivir resistance in seasonal and pandemic influenza A/H1N1. J Clin Virol. 2011;52:70–78. doi: 10.1016/j.jcv.2011.05.019. [DOI] [PubMed] [Google Scholar]
- Repetto G, del Peso A, Zurita JL. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc. 2008;3:1125–1131. doi: 10.1038/nprot.2008.75. [DOI] [PubMed] [Google Scholar]
- Shih SR, Horng JT, Poon LL, Chen TC, Yeh JY, Hsieh HP, Tseng SN, Chiang C, Li WL, Chao YS, Hsu JT. BPR2-D2 targeting viral ribonucleoprotein complex-associated function inhibits oseltamivir-resistant influenza viruses. J Antimicrob Chemother. 2010;65:63–71. doi: 10.1093/jac/dkp393. [DOI] [PubMed] [Google Scholar]
- Solbak SM, Reksten TR, Roder R, Wray V, Horvli O, Raae AJ, Henklein P, Henklein P, Fossen T. HIV-1 p6-Another viral interaction partner to the host cellular protein cyclophilin A. Biochim Biophys Acta. 2012;1824:667–678. doi: 10.1016/j.bbapap.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Sweeney ZK, Fu J, Wiedmann B. From Chemical Tools to Clinical Medicines: Nonimmunosuppressive Cyclophilin Inhibitors Derived from the Cyclosporin and Sanglifehrin Scaffolds. J Med Chem. 2014;57:7145–7159. doi: 10.1021/jm500223x. [DOI] [PubMed] [Google Scholar]
- Tellinghuisen TL, Foss KL, Treadaway JC, Rice CM. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J Virol. 2008;82:1073–1083. doi: 10.1128/JVI.00328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, Bieniasz PD. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat Med. 2003;9:1138–1143. doi: 10.1038/nm910. [DOI] [PubMed] [Google Scholar]
- Wang J. M2 as a target to combat influenza drug resistance: what does the evidence say? Future Virol. 2015;11:1–4. [Google Scholar]
- Wang J, Li F, Ma C. Recent progress in designing inhibitors that target the drug-resistant M2 proton channels from the influenza A viruses. Peptide Science. 2015;104:291–309. doi: 10.1002/bip.22623. [DOI] [PubMed] [Google Scholar]
- Wang J, Wu Y, Ma C, Fiorin G, Wang J, Pinto LH, Lamb RA, Klein ML, DeGrado WF. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proc Natl Acad Sci U S A. 2013;110:1315–1320. doi: 10.1073/pnas.1216526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S, Webby R. Traditional and New Influenza Vaccines. Clin Microbiol Rev. 2013;26:476–492. doi: 10.1128/CMR.00097-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Canturk B, Jo H, Ma C, Gianti E, Klein ML, Pinto LH, Lamb RA, Fiorin G, Wang J, DeGrado WF. Flipping in the Pore: Discovery of Dual Inhibitors That Bind in Different Orientations to the Wild-Type versus the Amantadine-Resistant S31N Mutant of the Influenza A Virus M2 Proton Channel. J Am Chem Soc. 2014;136:17987–17995. doi: 10.1021/ja508461m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander K, Sherman MP, Tessmer U, Bruns K, Wray V, Prechtel AT, Schubert E, Henklein P, Luban J, Neidleman J, Greene WC, Schubert U. Cyclophilin A interacts with HIV-1 Vpr and is required for its functional expression. J Biol Chem. 2003;278:43202–43213. doi: 10.1074/jbc.M305414200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.