

Abstract
Helicobacter pylori (H. pylori) infection was thought to be the main cause of gastric cancer, and its eradication showed improvement in gastric inflammation and decreased the risk of gastric cancer. Recently, a number of studies reported the occurrence of gastric cancer after successful eradication. Patients infected with H. pylori, even after eradication, have a higher risk for the occurrence of gastric cancer when compared with uninfected patients. Metachronous gastric cancer occurs frequently following the endoscopic removal of early gastric cancer. These data indicate that metachronous cancer leads to the occurrence of gastric cancer even after successful eradication of H. pylori. The pathogenesis of this metachronous cancer remains unclear. Further research is needed to identify biomarkers to predict the development of metachronous gastric cancer and methods for gastric cancer screening. In this article, we review the role of the H. pylori in carcinogenesis and the histological and endoscopic characteristics and risk factors for metachronous gastric cancer after eradication. Additionally, we discuss recent risk predictions and possible approaches for reducing the risk of metachronous gastric cancer after eradication.
Keywords: Helicobacter pylori, Eradication, Atrophic gastritis, Intestinal metaplasia, Metachronous gastric cancer
Core tip: Helicobacter pylori (H. pylori) eradication and endoscopic resection appeared to reduce the risk of gastric cancer. However, recent studies show that the risk of metachronous gastric cancer increases in the background of gastric mucosal atrophy even after successful eradication. Thus, curing H. pylori infections may not prevent metachronous gastric cancer in background mucosa with intestinal metaplasia. We review the risk factors and possible approaches for reducing the risk of metachronous gastric cancer.
INTRODUCTION
Gastric cancer is the fourth most common cancer in the world and the second leading cause of cancer deaths worldwide with more than 700000 deaths annually[1]. After the discovery of Helicobacter pylori (H. pylori) in 1983, the casual relationship between this bacterium and gastritis or gastric cancer has been steadily elucidated. In 1994, H. pylori was classified as a carcinogen by the International Agency for Research on Cancer of the World Health Organization (WHO). A 2009 meta-analysis showed that H. pylori eradication appeared to reduce the risk of gastric cancer[2]. In Japan, approximately 99% of gastric cancers are caused by H. pylori; thus, H. pylori-negative gastric cancer constitutes less than 1% of all cases[3].
H. pylori eradication has been one of the major therapeutic strategies to reduce gastric cancer incidence in healthy individuals and gastric cancer patients who have undergone endoscopic mucosal resection. The number of gastric cancers diagnosed and treated at an early stage increased after the development of endoscopic treatments. Ten years ago, more than 50% of early-stage gastric cancers were endoscopically treated, and the 5-year survival rate of early-stage gastric cancer patients after endoscopic treatment was 90%[4]. Large lesions can be resected en block, including both the mucosa and submucosa, by endoscopic submucosal resection (ESD), which has improved histopathological diagnoses and decreased tumor recurrence. The endoscopic removal of early-stage gastric tumors does not affect the overall cancer risk. Research on gastric cancers after H. pylori eradication has been conducted for more than a decade. In 2008, an open-label, randomized controlled trial indicated that the occurrence of metachronous gastric cancer is reduced by approximately 1/3 after eradication[5]. This study led to the recommendation of H. pylori eradication in patients with endoscopically treated gastric cancer. In Japan in 2013, health insurance covered H. pylori eradication as a treatment for gastritis, and this treatment was expected to reduce the incidence of gastric cancer[6]. However, a subsequent Japanese study indicated that even after H. pylori infection is cured and gastric inflammation is eliminated, the risk for developing gastric cancer remains; furthermore, this risk was dependent on the level of gastric mucosal atrophy present before eradication therapy[7]. Thus, the gastric mucosa endures continuous H. pylori-induced inflammation that increases the risk of metachronous gastric cancer even after treatment. Previous studies revealed that H. pylori eradication does not reduce the incidence of metachronous gastric cancer in patients who underwent endoscopic resection and recommended that eradication should be performed before the progression of gastric mucosal atrophy. Extensive atrophy in the stomach and intestinal metaplasia of multiple areas causes gastric cancer and may increase the risk for metachronous gastric cancer when compared with cases of chronic gastritis mucosa[8-11].
Currently, the success of a gastric cancer prevention strategy depends on timing because the treatment must be introduced before the progression of gastric carcinogenesis. However, recent studies on gastric cancer suppression suggested that critical features of gastric carcinogenesis can be reversed via molecular mechanisms.
Thus, monitoring patients for signs of gastric cancer after eradication is important. Here, we review the observed macroscopic and histological gastric mucosal changes, risks for metachronous gastric cancer, and possible approaches for reducing gastric cancer. We also discuss some of the potential molecular mechanisms for gastric cancer development after eradication.
H. PYLORI-INDUCED GASTRIC CANCER
A mechanism for carcinogenesis from H. pylori-triggered inflammation was first proposed by Correa[12,13]. Correa proposed that chronic inflammation causes superficial gastritis that progresses to multifocal atrophic gastritis, followed by intestinal metaplasia, wherein gastric epithelium undergoes an “epithelial-mesenchymal transition” and begins to exhibit an intestinal phenotype. The subsequent stage consists of dysplasia culminating in invasive carcinoma, thus completing the “pre-cancerous cascade”. This mechanism can be influenced by interactions between host and pathogen genotypes and environmental factors, such as socioeconomic indicators, a high-salt diet, low fruit/vegetable intake and smoking[14]. In particular, H. pylori is the sole bacterium to be classified by the WHO as a class I carcinogen[15].
Gastric cancer is an inflammation-associated cancer that occurs as a result of the infection which causes chronic, life-long, gastric mucosal inflammation. The pathogenesis of gastric cancer depends on the presence of genetic instability, that is, a consequence of H. pylori-induced acute and chronic inflammation, direct bacterial host interactions, and interactions with exogenous factors to produce carcinogens locally in the stomach[16]. However it should be added that H. pylori is recognized as the primary cause of gastric cancer, it is a necessary but insufficient cause of gastric cancer which is typical of infectious cause of cancer such as relation between hepatitis C virus and liver cancer or human papilloma virus and cervical cancer.
Recent studies have shown that H. pylori infection causes gastric cancer by inducing gene mutations, aberrant DNA methylation, and disturbance of intracellular signaling pathways. Point mutations and aberrant DNA methylation accumulated even in normal mucosa, leading to field cancerization[17,18]. We describe below a molecular mechanism of the gastric cancer.
Field cancerization
Field cancerization was first proposed by Slaughter et al[19] based on a study of oral stratified squamous epithelium in 1953. This phenomenon, also known as widespread carcinogenesis, can be caused by long-term exposure to common cancer inducers in several body areas. During field cancerization, abnormalities caused by exposure to cancer inducers can accumulate while the organism continues to appear normal. H. pylori infections contribute to carcinogenesis and field cancerization by causing point mutations and DNA methylation abnormalities in the gastric mucosa.
Gene mutations caused by activation-induced deaminase
Activation-induced deaminase (AID) is a member of the cytidine deaminase family, which includes DNA- and RNA-editing enzymes. AID expression is highly regulated, restricted to germinal center B cells and essential for somatic hypermutation and class-switch recombination in B cells[20]. Infection with cag pathogenicity island (cag PAI)-positive H. pylori ectopically induced the high expression of AID in human gastric epithelial cells, leading to multiple mutations in the TP53 tumor suppressor gene.
AID expression is induced by the activation of NF-κB caused by the H. pylori-induced inflammation in gastric epithelium cells. Additionally, TP53 mutations are induced by AID-producing gastric epithelium cells, which play an important role in stomach carcinogenesis.
AID upregulation in H. pylori-infected stomachs occurs via the introduction of bacterial macromolecules through the type IV secretion system encoded by cag PAI and inflammatory cytokines, such as tumor necrosis factor (TNF), that are produced by H. pylori-related gastric inflammation. Infection with cag PAI-positive H. pylori resulted in aberrant AID expression in gastric epithelial cells, leading to the generation of somatic mutations in the host genome, such as the TP53 gene. Cag PAI-positive H. pylori is more commonly associated with AID upregulation than cag PAI-negative H. pylori, and NF-κB activation provides evidence linking the pathogenic strain of H. pylori to the accumulation of nucleotide alterations and the subsequent development of gastric cancer[21].
DNA methylation abnormalities caused by chronic inflammation
H. pylori infection potently and temporarily induces methylation of multiple CpG islands (CGIs) in specific promoter regions, including tumor suppressor genes. Aberrant DNA methylation in gastric mucosa is associated with an increased risk of gastric cancer. Methylation levels in H. pylori-positive individuals were higher when compared with cases of H. pylori-negative gastric cancer[22]. The persistence of DNA methylation in the gastric mucosa decreases after H. pylori eradication[17]. Chronic inflammation causes H. pylori-induced DNA methylation and thus may be more important than the infection itself. The expression of certain inflammatory genes, such as TNF, IL-Iβ, and Nos2, increases DNA methylation[23]. These data suggest a relationship between inflammatory signals and DNA methylation.
Disruption of intracellular signaling by the direct action of the virulence factor CagA
CagA is a protein produced by H. pylori that is directly injected into gastric epithelium cells through a type IV secretion system. Next, CagA activates Ras-ErK signaling by binding to Src homology 2-containing protein tyrosine phosphatase[24,25]. Cell death in the gastric mucosa is inhibited by activating extracellular signal-regulated kinase and promoting myeloid cell leukemia-1 protein expression, which subsequently inhibits apoptotic cell death and delays the turnover of epithelial cells[26].
Influence of H. pylori on carcinogenesis
Accumulation of point mutations: Frequent TP53 mutations were discovered in the H. pylori-infected gastric mucosa of non-cancer patients using new sequencing technologies. Increased cytidine deaminase activity in these tissues appeared to increase these mutations and thus may promote gastric carcinogenesis in patients with H. pylori infection because most of the mutations were C: G to T: A[27].
Accumulation of DNA methylation: The DNA methylation induced by helicobacter infection remains at the stem cell level in non-infected mucosa after eradication, and the residual methylation level correlates with carcinogenic risk[17].
PREVENTION OF GASTRIC CANCER BY H. PYLORI ERADICATION
For one approach to inhibit gastric cancer, early prevention is important. H. pylori eradication stops the progression cancer risk and reverse some of mucosal damage. Multicenter clinical study results that the incidence of new gastric cancers who have a history of such disease and are thus at high risk for developing further gastric cancers was reduced by one-third among those with H. pylori eradication compared to no eradication therapy[5]. This study also added that eradication did not prevent development on gastric cancer completely, the risk for gastric cancer is directly related to the degree of atrophy. A large-scale cohort study from Taiwan followed 80000 patients with peptic ulcer for 10 years after H. pylori eradication therapy[28]. They reached the conclusions that the earlier eradication obviously reduce the incidence of gastric cancer. A meta-analysis of randomized controlled trials in 2014 showed that H. pylori eradication decreased the risk for gastric cancer in healthy asymptomatic infected individuals by 34%[29]. However it remains unclear whether H. pylori eradication among those at lower risk. On the other hand, when there is not H. pylori infection, and there is not inflammation of the stomach, it may be said that you do not need to worry about gastric cancer immediately.
GASTRIC CANCER DEVELOPMENT AFTER ERADICATION
Endoscopic resection is widely applied as a curative treatment for gastric cancer. However, metachronous gastric cancer following endoscopic resection is becoming a major problem due to gastric mucosa atrophy and chronic inflammation of the intestinal epithelium caused by H. pylori.
Metachronous gastric cancer means here that a new cancer is found separately from an initial cancer with or without H. pylori. The metachronous gastric cancer contains gastric cancer after H. pylori eradication.
Uemura et al[30] were the first to report that H. pylori eradication after endoscopic resection decreased the occurrence of metachronous cancer. A prospective randomized trial in Japan showed that H. pylori eradication reduced the risk of metachronous gastric cancer during a 3-year follow-up period. In contrast, a prospective randomized trial in South Korea showed that eradication after endoscopic resection did not reduce the incidence of metachronous gastric cancer[31]. The percentage of metachronous gastric cancers after endoscopic treatment was 8.5% during a follow-up period of up to 11.1 years, which did not significantly differ from the 14.3% cancer rate in the eradicated group[8]. And other studies conducted in Japan did not support eradication after endoscopic resection[6,9].
The efficacy of H. pylori eradication at preventing metachronous gastric cancer after endoscopic resection remains controversial. One of the causes which has no significant difference between eradicated group and not eradicated group, there exists nearly 30% overlooked cancers and those are often found by whole follow-up for 1 or 2 years. There is the report that cumulative incidence rate of metachronous gastric cancer was lower in the eradication group and the high risk of metachronous gastric cancer probably does not continue after 10 years[32]. Yoon et al[33] reviewed 13 studies and 6237 patients in a meta-analysis of the beneficial effects of H. pylori eradication and recommended that patients who received endoscopic treatment should also receive eradication therapy. Similarly, Yuhara et al[34] assessed 2 randomized control trials and 5 retrospective cohorts. There was variability in the observation period, atrophic degree of the background gastric mucosa, sanitization period and definition of the eradication group; however, the risk of metachronous gastric cancer in the non-eradication group was higher when compared with eradication group. Thus, eradication after endoscopic treatment may reduce the risk of metachronous gastric cancer.
There is one more thing to be added. That is, recurrent infection after successful H. pylori eradication. The causes to become positive again after H. pylori eradication was classified in “relapse” and “re-infection” by various kinds of judging methods. “Relapse” is the phenomenon that increases again after quantity of bacteria decreased in sensitivity or less of the testing concerned temporarily, and becomes positive by reexamination. “Re-infection” means H. pylori completely disappeared after eradication, they were infected with different helicobacter newly. The reinfection rate of H. pylori is reported 0.22%-11.5% and the variations date may reflect differences in the prevalence of H. pylori, hygienic environment, and false-negative eradication judgment[35,36]. Generally, it is not necessary to mind gastric cancer risk of re-infection because there are few re-infection.
Gastric mucosal changes after successful H. pylori eradication
Atrophic mucosa with intestinal metaplasia in differentiated gastric cancer and undifferentiated cancer of the gastric fundic gland mucosa are well-known examples of the relationship between gastric cancer and the background mucosa[37].
The continued study of H. pylori and gastric cancer has revealed a link between nodular gastritis in young women and undifferentiated gastric cancer[38]. Yagi et al[39] reported the regular arrangement of collecting venules (RAC) using a magnifying endoscope, and Nakajima[40,41] reported the RAC based on radiography. Both studies noted the importance of assessing background gastric mucosa with diagnostic imaging. H. pylori infected gastric mucosa presents with strong active gastritis that is immediately improved after eradication.
Kato et al[42] reported that spotty redness is significantly improved and related to eradication success in a multicenter study comparing unsuccessful and successful groups.
Using NBI magnifying observation, Okubo et al[43] showed that enlarged or elongated pits improved to small oval or pinhole-like round pits and the density of fine irregular vessels decreased after successful eradication without severe gastric atrophy or intestinal metaplasia. Kong et al[44] reported histological changes after successful eradication using a meta-analysis. This study illustrates a very strong correlation between the eradication of H. pylori infection and improvement in intestinal metaplasia in the gastric antrum but not in the corpus and between gastric atrophy in both the antrum and the corpus. After eradication, the neutrophilic infiltration of the lamina propria of the gastric mucosa and the lymphocytic infiltration of plasma cells were immediately improved[45,46].
Endoscopic detection of gastric cancer after successful eradication
Gastric cancer after successful H. pylori eradication is often difficult to diagnose by endoscope because of the indistinct borderline or disappearance of the characteristic surface structures of tumors.
In 2005, Ito et al[47] discovered that metachronous gastric cancers were difficult to identify endoscopically due to flattened and obscured tumor cells with an outer layer that lacked atypical columnar epithelium. Saka et al[48] described many characteristic changes in gastric cancer detection after successful H. pylori eradication: (1) a gastritis-like mucosal pattern that is often ill-delineated; (2) a portion of the gastritis-like gastric mucosa that contains a pattern distinct from the background mucosa; and (3) a mucosal pattern of a white zone that exhibits “morphological heterogeneity” and “direction diversity” using NBI magnifying endoscopy. These changes can impair the detection of gastric cancer after eradication; however, an area exhibiting “morphological heterogeneity” and “direction diversity” when compared with the background mucosa is relatively easy to diagnose as cancer[49].
With regard to the gastritis-like gastric mucosa, Kitamura et al[50] reported that “epithelium with low-grade atypia (ELA)” is frequently observed on the surface of gastric tumors after successful eradication therapy. Caudal-related homeobox 2 (CDX2) was not expressed, and neither p53- nor Ki67-positive cells were found in ELA, regardless of their expression in tumors. The presence of ELA positively correlated with the clinical interval between eradication and gastric cancer detection. Moreover, Yamamoto et al[51] reported that the average diameter of gastric tumors was smaller and the Ki-67 index was lower in patients who underwent H. pylori eradication. An analysis of macroscopic morphology revealed mainly depressed-type tumors and a high ratio of gastric differentiated-predominant mixed type lesions after a long eradication interval. H. pylori eradication may suppress growth, intestinalization, and acid hyposecretion during the development of gastric cancer.
These studies show a long-term risk for gastric cancer after successful eradication. Thus, endoscopic follow-ups must consider the distinct characteristics of gastric cancer after eradication.
Development and risk of metachronous gastric cancer
Various risks have been associated with the development of atrophic mucosa. Patients with precancerous changes in the gastric mucosa show an increased risk of gastric cancer. H. pylori eradication does not prevent the development of gastric cancer in all patients; furthermore the risk of cancer is higher in patients with precancerous changes prior to eradication[52-55].
There is continued speculation on gastric cancer diagnoses after eradication. Some researchers question the discovery of gastric cancer after eradication. Asaka et al[56] suggested a cancer diagnosis could be due to a preexisting tumor that was not detected endoscopically prior to eradication and the potential inhibitory effect of H. pylori eradication on tumor growth. That is, there are two kinds of the gastric cancer that discovery after eradication even if it occurs before eradication and the new cancer occurred after eradication truly. Take et al[55] found that the incidence of developing gastric cancer after amelioration of an H. pylori infection was 0.30% per year; furthermore, the cancer could develop as long as 10 years after H. pylori eradiation even without gastric inflammation. It was also reported that doubling time of the intramucosal carcinoma is approximately 16.6 mo[57] and it take more than 10 years from occurrence of gastric cancer cell until we can recognize visually endoscopically[48]. Thus, gastric cancer cannot be completely prevented by eradication of H. pylori.
Well-known risk factors of gastric cancer after successful eradication include another active H. pylori infection, increased age, and atrophic gastritis severity at the time of eradication therapy[11,58,59]. The independent risk factors of metachronous gastric cancer are male sex, severe gastric mucosal atrophy, and multiple gastric cancers prior to a successful H. pylori eradication[8,58,60,61].
Shiotani et al[62] showed that atrophy in biopsy specimens from the lesser curvature of the corpus was strongly associated with gastric cancer risk. A serum pepsinogen I level less than 25 ng/mL prior to eradication was significantly associated with subsequent tumor development.
Other reported risk factors include aberrant DNA methylation, microsatellite instability, aberrant expression of miRNAs, CD44v9 expression by tumor cells, and microscopic foci of intramucosal neoplasia elsewhere in the stomach.
The next chapter describes the prevention of metachronous gastric cancer through the use of predictive markers.
POSSIBLE APPROACHES FOR REDUCING CANCER RISK
Arginine adenosine-5’-diphosphate ribosylation inhibitors and prostaglandin E2
Patients presenting with atrophic gastritis, metaplasia, or dysplasia are routinely subjected to eradication therapy targeting the underlying infection; however, eradication is only partly effective at reversing atrophy and often fails to treat metaplasia and dysplasia[63].
Patients with any of these conditions have at least a 10-fold increased risk of developing gastric cancer; thus, a “watch-and-wait” strategy is not appropriate. For high-risk patients, improved treatments for metaplasia have been reported.
Recently, patients and animals taking tamoxifen have been shown to have regression in intestinal metaplasia[64]. Olaparib (Lynparza™), an Arginine adenosine-5’-diphosphate ribosylation (ADP-ribosylation) inhibitor used for ovarian cancer, has been shown to reverse intestinal metaplasia. ADP-ribosylation inhibitors may successfully prevent and cure helicobacter-induced gastric preneoplasia[65]. Prostaglandin E2 showed efficacy as a treatment during the early stages of Helicobacter-induced gastric carcinogenesis[66].
Interactions between H. pylori and CD44v9-positive cancer stem cells
Researchers have sought to understand why all H. pylori infected people do not develop gastric cancer.
CD44v9, one of the main surface marker proteins of cancer stem cells, prevents the accumulation of reactive oxygen species and contributes to the increased resistance of tumors to anticancer drugs[67]. CD44 overexpression, especially variant 9 (CD44v9), has been implicated in the local inflammatory response and metaplasia-carcinoma sequence in the human stomach[68].
As a long-term survival strategy for the stomach, the CagA released by H. pylori is degraded via VacA-mediated autophagy[69]. In contrast, CD44v9-expressing cancer stem cells accumulate intracellular CagA by suppressing autophagy. Therefore, the presence of CD44v9-expressing cancer stem cells is strongly associated with an increased risk of gastric carcinogenesis in the presence of H. pylori. CD44v9-positive cancer stem cells can appear after long-term inflammation. Chemopreventive treatments targeting this cancer protein may restore autophagy[69,70]. It would be important index to examine CD44v9-expressing when we evaluate a recurrence risk of the gastric cancer that occur after eradication of H. pylori.
Treating DNA methylation abnormalities with demethylating agents
Reversal of DNA methylation abnormalities may be effective for gastric cancer prevention. H. pylori infection induces DNA methylation abnormalities, which create the groundwork for cancerogenesis in the gastric mucosa. The carcinogenic risk can be assessed by measuring the extent of DNA methylation abnormalities that result in a “point of no return”. Nanjo et al[71] identified seven specific CGIs that show increased methylation levels after H. pylori infection. EMX1, NKX6-1, and NEFM were particularly influential, and the carcinogenic gastric cancer risk was 23.8 times higher in cases of increased EMX1 methylation. These cancer risks also apply to individuals with past infections.
A multicenter prospective cohort study by Asada et al[72] showed that the methylation level in the non-cancerous gastric mucosa of patients with gastric cancer was significantly (P = 0.042) associated with an increased risk of developing metachronous gastric cancers. Specifically, the methylation level of miR-124a-3 results in an elevated risk of metachronous gastric cancer; furthermore, similar trends were observed for EMX1 and NKX6-1. In conclusion, miR-124a-3 is an informative biomarker for predicting the risk of metachronous gastric cancer[73].
CDX2 is a transcriptional control factor that is indispensable for intestinal epithelium differentiation, and it functions as a tumor suppressing factor for tumors derived from intestinal epithelium. DNA methylation is frequently found in the CDX2 gene promoter region due to the development of intestinal gastric tumors that inactivate the gene. In addition, CDX2 is not expressed in stomachs uninfected by H. pylori. Early eradication prevents gastric cancer by inhibiting aberrant CDX2 expression. H. pylori eradication can reverse the gastric phenotype and diminish aberrant CDX2 expression in the early stages of intestinalization[11,74].
The technique used to determine the presence or absence of gastric cancer consists of harvesting DNA with an endoscope. The genetic DNA methylation rate of SRY-box containing gene 17 (SOX17) was assessed before and after endoscopic submucosal dissection. The pathological examination revealed that the DNA methylation rate of SOX17 was significantly decreased in all of the patients after endoscopic submucosal dissection. A decreased DNA methylation rate of SOX17 can be used to infer the extent of the resection. A decreased DNA methylation ratio after ESD should be observed during routine follow-ups. In contrast, an increased ratio indicates an incomplete resection and may suggest the need for additional surgery[75].
DNA demethylating agents are used for myelodysplastic syndrome patients. Demethylation of the gastric mucosa has been considered as a potential treatment. Additional research on treatment adaptation and side effects are needed to determine the applicability of preventing gastric cancer by inhibiting aberrant DNA methylation.
Chromosomal aberration of carcinoma tissue is not found in precancerous lesions; however, DNA methylation abnormalities can be used to identify both carcinomas and precancerous lesions. Examination of aberrant DNA methylation after eradication can be used to differentiate a high risk group from the sample population (Figure 1). Additional studies are needed to determine the relationships between aberrant DNA methylation and the invasion degree and cancer prognosis.
Figure 1.
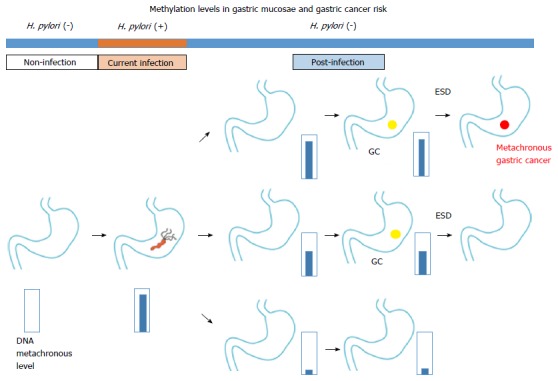
Relationship between gastric mucosae methylation levels and Helicobacter pylori infection/gastric cancer (modified from Maekita et al[22]). Residual aberrant methylation even after eradication is thought to reflect methylation in gastric gland stem cells. From endoscopically biopsied tissue, predicting GC risk based on the accumulation of aberrant DNA methylation in the gastric mucosae. ESD: Endoscopic submucosal resection; GC: Gastric cancer; H. pylori: Helicobacter pylori.
Utilization of pepsinogen serum levels and an H. pylori antibody titer
A novel and rapid diagnostic method has been introduced in Japan. This simple method, which consists of a pepsinogen serum level assay and helicobacter antibody titer, can be easily applied to large populations.
Yoshida et al[76] reported that altered DNA methylation levels in the stomach mucosa closely correlated with H. pylori-associated gastritis as assessed by serum pepsinogen II levels and a helicobacter antibody titer. Moreover, 4655 healthy asymptomatic subjects with no eradication treatment and who were followed-up for 16 years were divided into four groups based on the pepsinogen and antibody levels. This study showed a graded and significant rise in the hazard ratio for gastric cancer as chronic gastritis worsened. The mild atrophic gastritis group showed high gastric mucosa inflammation, which is a risk factor for diffuse-type cancer[77]. These results indicate that gastric cancer mainly develops from the gastritis-atrophy-metaplasia-cancer sequence and partly from active inflammation-based carcinogenesis. Notably, at-risk individuals require follow-up because this serum method should be used as a risk examination and not as a cancer screening. We previously reported that Congo-red chromoendoscopy methods can be used to identify high-risk areas after eradication. Biopsies of high-risk areas (non-acid-secreting area) revealed sustained hyperproliferation, accumulation of p53 protein and immunoreactivity for Ki-67[78]. Moreover, we demonstrated that a slow-releasing L-cysteine capsule effectively eliminated acetaldehyde from the gastric juice of PPI-treated aldehyde dehydrogenase 2 (ALDH2)-active and ALDH2-deficient subjects. These novel methods may aid in the prevention of gastric cancer, especially in established high-risk groups[79].
Eventually, we have got one method to make H. pylori may become extinct before the future children infect. However, we should take measures to cope with the gastric cancer which may occur after H. pylori eradication. Surveillance and follow-up based on the feature or the gastric cancer including remnant stomach after H. pylori eradication is important, and it can be said there is little invasive method more than a stomach is removed. Additional studies are needed to clarify these surveillance methods under multiple conditions and determine their reliability as biomarkers for metachronous gastric cancers.
Footnotes
Conflict-of-interest statement: No conflict of interests.
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Japan
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): D
Grade E (Poor): 0
Peer-review started: March 24, 2016
First decision: May 23, 2016
Article in press: July 18, 2016
P- Reviewer: Dar NA, Imaeda H, Liu YP S- Editor: Qi Y L- Editor: A E- Editor: Zhang FF
References
- 1.Melton SD, Genta RM, Souza RF. Biomarkers and molecular diagnosis of gastrointestinal and pancreatic neoplasms. Nat Rev Gastroenterol Hepatol. 2010;7:620–628. doi: 10.1038/nrgastro.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuccio L, Eusebi LH, Zagari RM, Bazzoli F. Helicobacter pylori eradication treatment reduces but does not abolish the risk of gastric cancer. Am J Gastroenterol. 2009;104:3100; author reply 3101–3102. doi: 10.1038/ajg.2009.516. [DOI] [PubMed] [Google Scholar]
- 3.Matsuo T, Ito M, Takata S, Tanaka S, Yoshihara M, Chayama K. Low prevalence of Helicobacter pylori-negative gastric cancer among Japanese. Helicobacter. 2011;16:415–419. doi: 10.1111/j.1523-5378.2011.00889.x. [DOI] [PubMed] [Google Scholar]
- 4.Kitano S, Shiraishi N. Current status of laparoscopic gastrectomy for cancer in Japan. Surg Endosc. 2004;18:182–185. doi: 10.1007/s00464-003-8820-7. [DOI] [PubMed] [Google Scholar]
- 5.Fukase K, Kato M, Kikuchi S, Inoue K, Uemura N, Okamoto S, Terao S, Amagai K, Hayashi S, Asaka M. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: an open-label, randomised controlled trial. Lancet. 2008;372:392–397. doi: 10.1016/S0140-6736(08)61159-9. [DOI] [PubMed] [Google Scholar]
- 6.Asaka M, Mabe K, Matsushima R, Tsuda M. Helicobacter pylori Eradication to Eliminate Gastric Cancer: The Japanese Strategy. Gastroenterol Clin North Am. 2015;44:639–648. doi: 10.1016/j.gtc.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 7.Take S, Ishiki K, Mizuno M. [Helicobacter pylori eradication therapy does not prevent gastric cancer development in all patients] Gan To Kagaku Ryoho. 2011;38:353–357. [PubMed] [Google Scholar]
- 8.Maehata Y, Nakamura S, Fujisawa K, Esaki M, Moriyama T, Asano K, Fuyuno Y, Yamaguchi K, Egashira I, Kim H, et al. Long-term effect of Helicobacter pylori eradication on the development of metachronous gastric cancer after endoscopic resection of early gastric cancer. Gastrointest Endosc. 2012;75:39–46. doi: 10.1016/j.gie.2011.08.030. [DOI] [PubMed] [Google Scholar]
- 9.Kato M, Nishida T, Yamamoto K, Hayashi S, Kitamura S, Yabuta T, Yoshio T, Nakamura T, Komori M, Kawai N, et al. Scheduled endoscopic surveillance controls secondary cancer after curative endoscopic resection for early gastric cancer: a multicentre retrospective cohort study by Osaka University ESD study group. Gut. 2013;62:1425–1432. doi: 10.1136/gutjnl-2011-301647. [DOI] [PubMed] [Google Scholar]
- 10.Kato M, Ono S, Mabe K, Sakamoto N, Asaka M. [After endoscopic treatment of early stage gastric cancer] Nihon Rinsho. 2013;71:1429–1435. [PubMed] [Google Scholar]
- 11.Jung DH, Kim JH, Lee YC, Lee SK, Shin SK, Park JC, Chung HS, Kim H, Kim H, Kim YH, et al. Helicobacter pylori Eradication Reduces the Metachronous Recurrence of Gastric Neoplasms by Attenuating the Precancerous Process. J Gastric Cancer. 2015;15:246–255. doi: 10.5230/jgc.2015.15.4.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Correa P. Human gastric carcinogenesis: a multistep and multifactorial process--First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992;52:6735–6740. [PubMed] [Google Scholar]
- 13.Correa P, Piazuelo MB. The gastric precancerous cascade. J Dig Dis. 2012;13:2–9. doi: 10.1111/j.1751-2980.2011.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khalifa MM, Sharaf RR, Aziz RK. Helicobacter pylori: a poor man’s gut pathogen? Gut Pathog. 2010;2:2. doi: 10.1186/1757-4749-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.IARC working group on the evaluation of carcinogenic risks to humans: some industrial chemicals. Lyon, 15-22 February 1994. IARC Monogr Eval Carcinog Risks Hum. 1994;60:1–560. [Google Scholar]
- 16.Shiotani A, Cen P, Graham DY. Eradication of gastric cancer is now both possible and practical. Semin Cancer Biol. 2013;23:492–501. doi: 10.1016/j.semcancer.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Nakajima T, Enomoto S, Yamashita S, Ando T, Nakanishi Y, Nakazawa K, Oda I, Gotoda T, Ushijima T. Persistence of a component of DNA methylation in gastric mucosae after Helicobacter pylori eradication. J Gastroenterol. 2010;45:37–44. doi: 10.1007/s00535-009-0142-7. [DOI] [PubMed] [Google Scholar]
- 18.Nakajima T, Maekita T, Oda I, Gotoda T, Yamamoto S, Umemura S, Ichinose M, Sugimura T, Ushijima T, Saito D. Higher methylation levels in gastric mucosae significantly correlate with higher risk of gastric cancers. Cancer Epidemiol Biomarkers Prev. 2006;15:2317–2321. doi: 10.1158/1055-9965.EPI-06-0436. [DOI] [PubMed] [Google Scholar]
- 19.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–968. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 20.Honjo T, Kinoshita K, Muramatsu M. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu Rev Immunol. 2002;20:165–196. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, Azuma T, Okazaki IM, Honjo T, Chiba T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470–476. doi: 10.1038/nm1566. [DOI] [PubMed] [Google Scholar]
- 22.Maekita T, Nakazawa K, Mihara M, Nakajima T, Yanaoka K, Iguchi M, Arii K, Kaneda A, Tsukamoto T, Tatematsu M, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 2006;12:989–995. doi: 10.1158/1078-0432.CCR-05-2096. [DOI] [PubMed] [Google Scholar]
- 23.Niwa T, Toyoda T, Tsukamoto T, Mori A, Tatematsu M, Ushijima T. Prevention of Helicobacter pylori-induced gastric cancers in gerbils by a DNA demethylating agent. Cancer Prev Res (Phila) 2013;6:263–270. doi: 10.1158/1940-6207.CAPR-12-0369. [DOI] [PubMed] [Google Scholar]
- 24.Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, Hatakeyama M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295:683–686. doi: 10.1126/science.1067147. [DOI] [PubMed] [Google Scholar]
- 25.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4:688–694. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 26.Mimuro H. [Strategy of Helicobacter pylori to enhance colonization of the stomach] Nihon Saikingaku Zasshi. 2009;64:311–317. doi: 10.3412/jsb.64.311. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu T, Marusawa H, Matsumoto Y, Inuzuka T, Ikeda A, Fujii Y, Minamiguchi S, Miyamoto S, Kou T, Sakai Y, et al. Accumulation of somatic mutations in TP53 in gastric epithelium with Helicobacter pylori infection. Gastroenterology. 2014;147:407–417.e3. doi: 10.1053/j.gastro.2014.04.036. [DOI] [PubMed] [Google Scholar]
- 28.Wu CY, Kuo KN, Wu MS, Chen YJ, Wang CB, Lin JT. Early Helicobacter pylori eradication decreases risk of gastric cancer in patients with peptic ulcer disease. Gastroenterology. 2009;137:1641–8.e1-2. doi: 10.1053/j.gastro.2009.07.060. [DOI] [PubMed] [Google Scholar]
- 29.Ford AC, Forman D, Hunt RH, Yuan Y, Moayyedi P. Helicobacter pylori eradication therapy to prevent gastric cancer in healthy asymptomatic infected individuals: systematic review and meta-analysis of randomised controlled trials. BMJ. 2014;348:g3174. doi: 10.1136/bmj.g3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uemura N, Okamoto S. Effect of Helicobacter pylori eradication on subsequent development of cancer after endoscopic resection of early gastric cancer in Japan. Gastroenterol Clin North Am. 2000;29:819–827. doi: 10.1016/s0889-8553(05)70149-7. [DOI] [PubMed] [Google Scholar]
- 31.Choi J, Kim SG, Yoon H, Im JP, Kim JS, Kim WH, Jung HC. Eradication of Helicobacter pylori after endoscopic resection of gastric tumors does not reduce incidence of metachronous gastric carcinoma. Clin Gastroenterol Hepatol. 2014;12:793–800.e1. doi: 10.1016/j.cgh.2013.09.057. [DOI] [PubMed] [Google Scholar]
- 32.Kobayashi M, Narisawa R, Sato Y, Takeuchi M, Aoyagi Y. Self-limiting risk of metachronous gastric cancers after endoscopic resection. Dig Endosc. 2010;22:169–173. doi: 10.1111/j.1443-1661.2010.00987.x. [DOI] [PubMed] [Google Scholar]
- 33.Yoon SB, Park JM, Lim CH, Cho YK, Choi MG. Effect of Helicobacter pylori eradication on metachronous gastric cancer after endoscopic resection of gastric tumors: a meta-analysis. Helicobacter. 2014;19:243–248. doi: 10.1111/hel.12146. [DOI] [PubMed] [Google Scholar]
- 34.Yuhara H, Nakae H, Nakamura J, Tsukune Y, Utida T, Koike J, Igarashi M, Suzuki T, Mine T. Examination by the protective efficacy meta-analysis of the H.pylori eradication for the metachronous gastric cancer after endoscopic treatment (Japanese) Jpn J Helicobacter Res. 2015;16:71–73. [Google Scholar]
- 35.Take S, Mizuno M, Ishiki K, Imada T, Okuno T, Yoshida T, Yokota K, Oguma K, Kita M, Okada H, et al. Reinfection rate of Helicobacter pylori after eradication treatment: a long-term prospective study in Japan. J Gastroenterol. 2012;47:641–646. doi: 10.1007/s00535-012-0536-9. [DOI] [PubMed] [Google Scholar]
- 36.Morgan DR, Torres J, Sexton R, Herrero R, Salazar-Martínez E, Greenberg ER, Bravo LE, Dominguez RL, Ferreccio C, Lazcano-Ponce EC, et al. Risk of recurrent Helicobacter pylori infection 1 year after initial eradication therapy in 7 Latin American communities. JAMA. 2013;309:578–586. doi: 10.1001/jama.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imai T, Kubo T, Watanabe H. Chronic gastritis in Japanese with reference to high incidence of gastric carcinoma. J Natl Cancer Inst. 1971;47:179–195. [PubMed] [Google Scholar]
- 38.Kamada T, Haruma K, Sugiu K, NagashimaY, Qian DM, Koga H, Takeda M, Kusunoki H, Honda K, Fujimura Y, et al. Case of early gastric cancer with nodular gastritis. Dig Endosc. 2004;16:39–43. [Google Scholar]
- 39.Yagi K, Nakamura A, Sekine A. Characteristic endoscopic and magnified endoscopic findings in the normal stomach without Helicobacter pylori infection. J Gastroenterol Hepatol. 2002;17:39–45. doi: 10.1046/j.1440-1746.2002.02665.x. [DOI] [PubMed] [Google Scholar]
- 40.Nakajima S. Reassessment of the serum pepsinogen method by the stomach X-ray examination [Japanese] Jpn J Helicobacter Res. 2013;14:19–22. [Google Scholar]
- 41.Nakajima S. [Roles of pepsinogen test, ABC method and barium X-ray examination on gastric cancer screening] Nihon Shokakibyo Gakkai Zasshi. 2013;110:225–233. [PubMed] [Google Scholar]
- 42.Kato M, Terao S, Adachi K, Nakajima S, Ando T, Yoshida N, Uedo N, Murakami K, Ohara S, Ito M, et al. Changes in endoscopic findings of gastritis after cure of H. pylori infection: multicenter prospective trial. Dig Endosc. 2013;25:264–273. doi: 10.1111/j.1443-1661.2012.01385.x. [DOI] [PubMed] [Google Scholar]
- 43.Okubo M, Tahara T, Shibata T, Nakamura M, Yoshioka D, Maeda Y, Yonemura J, Ishizuka T, Arisawa T, Hirata I. Changes in gastric mucosal patterns seen by magnifying NBI during H. pylori eradication. J Gastroenterol. 2011;46:175–182. doi: 10.1007/s00535-010-0335-0. [DOI] [PubMed] [Google Scholar]
- 44.Kong YJ, Yi HG, Dai JC, Wei MX. Histological changes of gastric mucosa after Helicobacter pylori eradication: a systematic review and meta-analysis. World J Gastroenterol. 2014;20:5903–5911. doi: 10.3748/wjg.v20.i19.5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tucci A, Biasco G, Paparo GF. Effect of eradication of Helicobacter pylori in patients with fundic atrophic gastritis. N Engl J Med. 1997;336:957–958. doi: 10.1056/NEJM199703273361313. [DOI] [PubMed] [Google Scholar]
- 46.Ohkusa T, Fujiki K, Takashimizu I, Kumagai J, Tanizawa T, Eishi Y, Yokoyama T, Watanabe M. Improvement in atrophic gastritis and intestinal metaplasia in patients in whom Helicobacter pylori was eradicated. Ann Intern Med. 2001;134:380–386. doi: 10.7326/0003-4819-134-5-200103060-00010. [DOI] [PubMed] [Google Scholar]
- 47.Ito M, Tanaka S, Takata S, Oka S, Imagawa S, Ueda H, Egi Y, Kitadai Y, Yasui W, Yoshihara M, et al. Morphological changes in human gastric tumours after eradication therapy of Helicobacter pylori in a short-term follow-up. Aliment Pharmacol Ther. 2005;21:559–566. doi: 10.1111/j.1365-2036.2005.02360.x. [DOI] [PubMed] [Google Scholar]
- 48.Saka A, Yagi K, Nimura S. Endoscopic and histological features of gastric cancers after successful Helicobacter pylori eradication therapy. Gastric Cancer. 2016;19:524–530. doi: 10.1007/s10120-015-0479-y. [DOI] [PubMed] [Google Scholar]
- 49.Yagi K, Saka A, Nozawa Y, Nakamura A, Nimura S. Endoscopic technique for qualitative diagnosis of gastric cancer and diagnosis of its expansion after successful H.pylori eradication therapy: focused on diagnosis by NBI magnifying endoscopy. JGES. 2015;57:1210–1218. [Google Scholar]
- 50.Kitamura Y, Ito M, Matsuo T, Boda T, Oka S, Yoshihara M, Tanaka S, Chayama K. Characteristic epithelium with low-grade atypia appears on the surface of gastric cancer after successful Helicobacter pylori eradication therapy. Helicobacter. 2014;19:289–295. doi: 10.1111/hel.12132. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto K, Kato M, Takahashi M, Haneda M, Shinada K, Nishida U, Yoshida T, Sonoda N, Ono S, Nakagawa M, et al. Clinicopathological analysis of early-stage gastric cancers detected after successful eradication of Helicobacter pylori. Helicobacter. 2011;16:210–216. doi: 10.1111/j.1523-5378.2011.00833.x. [DOI] [PubMed] [Google Scholar]
- 52.de Vries AC, Kuipers EJ, Rauws EA. Helicobacter pylori eradication and gastric cancer: when is the horse out of the barn? Am J Gastroenterol. 2009;104:1342–1345. doi: 10.1038/ajg.2008.15. [DOI] [PubMed] [Google Scholar]
- 53.Wong BC, Lam SK, Wong WM, Chen JS, Zheng TT, Feng RE, Lai KC, Hu WH, Yuen ST, Leung SY, et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA. 2004;291:187–194. doi: 10.1001/jama.291.2.187. [DOI] [PubMed] [Google Scholar]
- 54.Fock KM, Talley N, Moayyedi P, Hunt R, Azuma T, Sugano K, Xiao SD, Lam SK, Goh KL, Chiba T, et al. Asia-Pacific consensus guidelines on gastric cancer prevention. J Gastroenterol Hepatol. 2008;23:351–365. doi: 10.1111/j.1440-1746.2008.05314.x. [DOI] [PubMed] [Google Scholar]
- 55.Take S, Mizuno M, Ishiki K, Yoshida T, Ohara N, Yokota K, Oguma K, Okada H, Yamamoto K. The long-term risk of gastric cancer after the successful eradication of Helicobacter pylori. J Gastroenterol. 2011;46:318–324. doi: 10.1007/s00535-010-0347-9. [DOI] [PubMed] [Google Scholar]
- 56.Asaka M, Kato M, Graham DY. Prevention of gastric cancer by Helicobacter pylori eradication. Intern Med. 2010;49:633–636. doi: 10.2169/internalmedicine.49.3470. [DOI] [PubMed] [Google Scholar]
- 57.Haruma K, Suzuki T, Tsuda T, Yoshihara M, Sumii K, Kajiyama G. Evaluation of tumor growth rate in patients with early gastric carcinoma of the elevated type. Gastrointest Radiol. 1991;16:289–292. doi: 10.1007/BF01887370. [DOI] [PubMed] [Google Scholar]
- 58.Take S, Mizuno M, Ishiki K, Nagahara Y, Yoshida T, Yokota K, Oguma K. Baseline gastric mucosal atrophy is a risk factor associated with the development of gastric cancer after Helicobacter pylori eradication therapy in patients with peptic ulcer diseases. J Gastroenterol. 2007;42 Suppl 17:21–27. doi: 10.1007/s00535-006-1924-9. [DOI] [PubMed] [Google Scholar]
- 59.Kwon YH, Heo J, Lee HS, Cho CM, Jeon SW. Failure of Helicobacter pylori eradication and age are independent risk factors for recurrent neoplasia after endoscopic resection of early gastric cancer in 283 patients. Aliment Pharmacol Ther. 2014;39:609–618. doi: 10.1111/apt.12633. [DOI] [PubMed] [Google Scholar]
- 60.Mori G, Nakajima T, Asada K, Shimazu T, Yamamichi N, Maekita T, Yokoi C, Fujishiro M, Gotoda T, Ichinose M, et al. Incidence of and risk factors for metachronous gastric cancer after endoscopic resection and successful Helicobacter pylori eradication: results of a large-scale, multicenter cohort study in Japan. Gastric Cancer. 2016;19:911–918. doi: 10.1007/s10120-015-0544-6. [DOI] [PubMed] [Google Scholar]
- 61.Fock KM. Review article: the epidemiology and prevention of gastric cancer. Aliment Pharmacol Ther. 2014;40:250–260. doi: 10.1111/apt.12814. [DOI] [PubMed] [Google Scholar]
- 62.Shiotani A, Uedo N, Iishi H, Yoshiyuki Y, Ishii M, Manabe N, Kamada T, Kusunoki H, Hata J, Haruma K. Predictive factors for metachronous gastric cancer in high-risk patients after successful Helicobacter pylori eradication. Digestion. 2008;78:113–119. doi: 10.1159/000173719. [DOI] [PubMed] [Google Scholar]
- 63.Rokkas T, Pistiolas D, Sechopoulos P, Robotis I, Margantinis G. The long-term impact of Helicobacter pylori eradication on gastric histology: a systematic review and meta-analysis. Helicobacter. 2007;12 Suppl 2:32–38. doi: 10.1111/j.1523-5378.2007.00563.x. [DOI] [PubMed] [Google Scholar]
- 64.Goldenring JR. Gastric intestinal metaplasia and tamoxifen: can we reverse the inevitable? Dig Dis Sci. 2014;59:1078–1079. doi: 10.1007/s10620-014-3088-4. [DOI] [PubMed] [Google Scholar]
- 65.Toller IM, Altmeyer M, Kohler E, Hottiger MO, Müller A. Inhibition of ADP ribosylation prevents and cures helicobacter-induced gastric preneoplasia. Cancer Res. 2010;70:5912–5922. doi: 10.1158/0008-5472.CAN-10-0528. [DOI] [PubMed] [Google Scholar]
- 66.Toller IM, Hitzler I, Sayi A, Mueller A. Prostaglandin E2 prevents Helicobacter-induced gastric preneoplasia and facilitates persistent infection in a mouse model. Gastroenterology. 2010;138:1455–1467, 1467.e1-4. doi: 10.1053/j.gastro.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 67.Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19:387–400. doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 68.Ishimoto T, Izumi D, Watanabe M, Yoshida N, Hidaka K, Miyake K, Sugihara H, Sawayama H, Imamura Y, Iwatsuki M, et al. Chronic inflammation with Helicobacter pylori infection is implicated in CD44 overexpression through miR-328 suppression in the gastric mucosa. J Gastroenterol. 2015;50:751–757. doi: 10.1007/s00535-014-1019-y. [DOI] [PubMed] [Google Scholar]
- 69.Tsugawa H, Suzuki H, Saya H, Hatakeyama M, Hirayama T, Hirata K, Nagano O, Matsuzaki J, Hibi T. Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells. Cell Host Microbe. 2012;12:764–777. doi: 10.1016/j.chom.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 70.Hirata K, Suzuki H, Imaeda H, Matsuzaki J, Tsugawa H, Nagano O, Asakura K, Saya H, Hibi T. CD44 variant 9 expression in primary early gastric cancer as a predictive marker for recurrence. Br J Cancer. 2013;109:379–386. doi: 10.1038/bjc.2013.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nanjo S, Asada K, Yamashita S, Nakajima T, Nakazawa K, Maekita T, Ichinose M, Sugiyama T, Ushijima T. Identification of gastric cancer risk markers that are informative in individuals with past H. pylori infection. Gastric Cancer. 2012;15:382–388. doi: 10.1007/s10120-011-0126-1. [DOI] [PubMed] [Google Scholar]
- 72.Asada K, Nakajima T, Shimazu T, Yamamichi N, Maekita T, Yokoi C, Oda I, Ando T, Yoshida T, Nanjo S, et al. Demonstration of the usefulness of epigenetic cancer risk prediction by a multicentre prospective cohort study. Gut. 2015;64:388–396. doi: 10.1136/gutjnl-2014-307094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shimazu T, Asada K, Charvat H, Kusano C, Otake Y, Kakugawa Y, Watanabe H, Gotoda T, Ushijima T, Tsugane S. Association of gastric cancer risk factors with DNA methylation levels in gastric mucosa of healthy Japanese: a cross-sectional study. Carcinogenesis. 2015;36:1291–1298. doi: 10.1093/carcin/bgv125. [DOI] [PubMed] [Google Scholar]
- 74.Shiotani A, Nishi R, Uedo N, Iishi H, Tsutsui H, Ishii M, Imamura H, Kamada T, Hata J, Haruma K. Helicobacter pylori eradication prevents extension of intestinalization even in the high-risk group for gastric cancer. Digestion. 2010;81:223–230. doi: 10.1159/000264651. [DOI] [PubMed] [Google Scholar]
- 75.Oishi Y, Watanabe Y, Yoshida Y, Sato Y, Hiraishi T, Oikawa R, Maehata T, Suzuki H, Toyota M, Niwa H, et al. Hypermethylation of Sox17 gene is useful as a molecular diagnostic application in early gastric cancer. Tumour Biol. 2012;33:383–393. doi: 10.1007/s13277-011-0278-y. [DOI] [PubMed] [Google Scholar]
- 76.Yoshida T, Kato J, Maekita T, Yamashita S, Enomoto S, Ando T, Niwa T, Deguchi H, Ueda K, Inoue I, et al. Altered mucosal DNA methylation in parallel with highly active Helicobacter pylori-related gastritis. Gastric Cancer. 2013;16:488–497. doi: 10.1007/s10120-012-0230-x. [DOI] [PubMed] [Google Scholar]
- 77.Yoshida T, Kato J, Inoue I, Yoshimura N, Deguchi H, Mukoubayashi C, Oka M, Watanabe M, Enomoto S, Niwa T, et al. Cancer development based on chronic active gastritis and resulting gastric atrophy as assessed by serum levels of pepsinogen and Helicobacter pylori antibody titer. Int J Cancer. 2014;134:1445–1457. doi: 10.1002/ijc.28470. [DOI] [PubMed] [Google Scholar]
- 78.Iijima K, Abe Y, Koike T, Uno K, Endo H, Hatta W, Asano N, Asanuma K, Imatani A, Shimosegawa T. Gastric cancers emerging after H. pylori eradication arise exclusively from non-acid-secreting areas. Tohoku J Exp Med. 2012;226:45–53. doi: 10.1620/tjem.226.45. [DOI] [PubMed] [Google Scholar]
- 79.Maejima R, Iijima K, Kaihovaara P, Hatta W, Koike T, Imatani A, Shimosegawa T, Salaspuro M. Effects of ALDH2 genotype, PPI treatment and L-cysteine on carcinogenic acetaldehyde in gastric juice and saliva after intragastric alcohol administration. PLoS One. 2015;10:e0120397. doi: 10.1371/journal.pone.0120397. [DOI] [PMC free article] [PubMed] [Google Scholar]