

Abstract
Inflammation after stroke consists of activation of microglia/astrocytes in situ and infiltration of blood-borne leukocytes, resulting in brain damage and neurological deficits. Mounting data demonstrated that most natural components from medicinal plants had anti-inflammatory effects after ischemic stroke through inhibiting activation of resident microglia/astrocytes within ischemic area. However, it is speculated that this classical activity cannot account for the anti-inflammatory function of these natural components in the cerebral parenchyma, where they are detected at very low concentrations due to their poor membrane permeability and slight leakage of BBB. Could these drugs exert anti-inflammatory effects peripherally without being delivered across the BBB? Factually, ameliorating blood-borne neutrophil recruitment in peripheral circulatory system has been proved to reduce ischemic damage and improve outcomes. Thus, it is concluded that if drugs could achieve effective concentrations in the cerebral parenchyma, they can function via crippling resident microglia/astrocytes activation and inhibiting neutrophil infiltration, whereas the latter will be dominating when these drugs localize in the brain at a low concentration. In this review, the availability of some natural components crossing the BBB in stroke will be discussed, and how these drugs lead to improvements in stroke through inhibition of neutrophil rolling, adhesion, and transmigration will be illustrated.
1. Introduction
Stroke is the fourth leading cause of death [1]. Of all strokes, subarachnoid hemorrhage stroke and intracerebral hemorrhagic stroke account for 3% and 10%, respectively, while 87% are ischemic stroke [2]. Ischemic stroke starts with rapid reduction of blood flow and oxygen and disorder of glucose and energy metabolism within lesion area, followed by a series of pathophysiology events which are called “ischemic cascades,” including glutamate-induced excitotoxicity, calcium overload, inflammatory response, and cell death [3]. Due to accelerating neurovascular damage and promoting neurorestorative events, inflammatory response plays an extremely important role in ischemic stroke [4]. After cerebral ischemia, inflammation consists of two processes, described as activation of resident microglia/astrocytes and infiltration of peripheral leukocytes [5], resulting in microvessel obstruction, edema formation, cell death, and tissue infarction [6]. For several decades, clinical therapies and basic experiments were focused on neuroprotection, such as anti-inflammation, antioxidative injury, and antiapoptosis [7, 8]. However, the only effective pharmacological intervention to treat stroke is intravascular thrombolysis by administrating recombinant tissue plasminogen activator (r-tPA), which shows therapeutic effects when given within 4.5 hours after onset of symptoms in strictly selected patients. Nevertheless, accompanied with high incidence of intracerebral hemorrhage and narrow therapeutic window, application of r-tPA to clinical patients is limited [9]. Recent studies have revealed that alleviating inflammatory response in ischemic stroke could widen the therapeutic window [10]. Therefore, targeting inflammation could be a crucial and promising therapeutic strategy for preventing brain damage and improving outcomes.
Spencer mentioned that some polyphenols were absorbed to only a limited degree and detected at very low concentrations within the injured brain, so antioxidant activity of these agents failed to account for their neurological improvement ability [11, 12]. But it has become evident that they could protect vulnerable and existing neurons and stimulate neuronal regeneration and neurogenesis to mediate the ability [12]. Similarly, a large number of studies have displayed that anti-inflammatory effects of most natural components on cerebral ischemia have been attributed to their inhibiting resident microglia/astrocytes activation [13–16]. Nevertheless, some drug concentrations in most animal tissues are lower than the concurrent plasma level and the brain level is usually very low or even not detected, because of their poor membrane permeability determined by physicochemical properties [17] and slight leakage of blood-brain barrier (BBB). Therefore, how do they exert anti-inflammatory effects on the brain when they localize in the brain at low or no concentrations? It has been postulated that their anti-inflammatory activities on the brain were mainly mediated by potential to downregulate expression of inflammatory mediators and ameliorate leukocyte infiltration. As a matter of fact, emerging evidence has revealed that they were able to peripherally inhibit leukocyte infiltration at the level of inflammatory mediators. Some natural compounds applied commonly, like sodium β-aescin, alleviate neutrophils adhesion to endothelium by downregulating the expression of cellular adhesion molecules (CAMs) [18–20]. This present review will attempt to clarify the probable mechanisms in which certain natural compounds exert such effects on the brain.
Data have revealed that an increased number of leukocytes were detected in the microvessels and parenchyma in lesion area [21], mainly in the border zones of infarcts, ischemic penumbra, but not in the normal brain [22]. In the local ischemic brain, leukocyte (especially neutrophil) recruitment cascade is generally subjected to rolling, adhesion, and transmigration [23] (Figure 1). This cascade starts with release of specific inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), inducing the expression of CAMs on endothelium surface [24]. Hence, targeting neutrophil activation and recruitment has been evaluated as stroke remedies [25].
Figure 1.

The process of neutrophil infiltration into the cerebral parenchyma. Injured brain cells release DAMPs and cytokines within lesion area and into the vessel lumen. Subsequently, CAMs on the surface of endothelium and neutrophils are induced, participating in the series steps of neutrophil rolling, adhesion, and transmigration. ICAM-1, intercellular cell adhesion molecule-1; PSGL-1, P-selectin glycoprotein ligand 1; MAC-1, macrophage adhesion molecule 1; CCL, monocyte chemoattractant protein; CXCL, chemokine (C-X-C motif) ligand; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor-α; ILR, interleukin receptor; TNFR, tumor necrosis factor receptor; CCLR, monocyte chemoattractant protein receptor; CXCR, chemokine (C-X-C motif) receptor; DAMPs, damage-associated molecular patterns.
2. Impact of Experimental Stroke Models and the BBB Openings on Penetrability of Drugs
Forms of human stroke are various and complicated, and no single animal model can encompass all of the variables on behalf of the events in human ischemic stroke [26]. Reliable animal stroke models provide powerful evidence of efficacy and safety for these drugs to be applied to clinical patients [27]. In order to analyze mechanisms in ischemic stroke, researchers have established less than 10 rodent models of focal cerebral ischemia used in experiment, such as permanent or transient middle cerebral artery (p/t-MCAO) or distal middle cerebral artery occlusion (p/t-dMCAO) by ligating, clipping, photothrombosis, electrocoagulation, or thread [28]. The coagulation model, characterized as distal, permanent MCAO by transcranial electrocoagulation, fulfills criteria of clinical relevance, low mortality, and high reproducibility. Also, the resulting infarct of the model is located mainly in the cortex and the infarct volume corresponds to that of majority of clinical patients [29]. Published study of our laboratory revealed that BBB leakage in t-MCAO mice, given Evans blue (EB) via tail vein injection at 2 h before execution, was measured at 24 h after surgery and the extensive blue area indicating BBB disruption was corresponding to visualized infarct size [30]. Of interest is another data set unpublished in our laboratory, showing that the blue area in p-dMCAO mice, of which EB administration method was the same to that of t-MCAO mice, was not detected obviously owing to the permanent occlusion of the distal MCA and blocking partly EB delivery in the vessel. Therefore, many medicines that cannot pass through the BBB under physiology conditions, even if the brain undergoes ischemic injury, are most likely not to penetrate the BBB efficiently to function at early phase, which at least exists in the p-dMCAO models.
The BBB is formed by microvessel endothelial cells in the central nervous system (CNS) and acts as an interface keeping brain's internal milieu from the rest of the body structurally and functionally, regulating the migration of molecules, ions, and cells strictly [31–33]. It is possible for small molecules with molecular weight < 400 Da and forms < 8 hydrogen bonds to cross the BBB via free diffusion mediated by lipid in principle, while majority of small molecule drugs factually fail to pass, as well as all large-molecule drugs, especially products of biotechnology [34]. However, inflammatory response cripples BBB integrity, described as upregulation of number of endothelial caveolae and rate of transcytosis after t-MCAO, as well as disruption of tight junction (TJ) morphology and enhanced paracellular BBB permeability [35]. Ultimately, some drugs prevented physiologically are allowed to cross the BBB to function under pathological conditions. And there are two openings of BBB after MCAO [36]. The EB extravasation reached two peaks at 6 h and 48 h after cerebral ischemia/reperfusion and decreased at 24 h [37]. Another data set showed that there was a 200-fold increase in barrier permeability to FITC-albumin at 6 h and a further period of disruption around 24 h [38]. Many researches revealed that two openings of BBB permeability appeared at 3–6 h and 48 h or 72 h after ischemia/reperfusion, respectively [39–41]. In fact, changes in TJ permeability following ischemia may not be measured immediately but after continuous hours of reduction in cerebral blood flow, inducing an observable increase in paracellular permeability [42]. Knowland considered that crippling barrier function occurred as early as 6 hours after stroke, and TJs related to paracellular diffusion displayed significant structural defects only after 48 hours [35]. Many drugs with short half-life, administrated only once before the first opening or between the two openings of BBB for anti-inflammation, are absorbed rapidly and metabolized extensively before passing completely through BBB. Thus, single-dose administration of these drugs may not reach the effective concentration to exert anti-inflammation on the ischemic area except given mega doses and repetitive doses.
Equally importantly, the ability of drugs to localize in the brain at an efficient concentration also depends partly on their physicochemical properties, including molecular weight, hydrogen-bonding capacity, and number of rotation bonds (NROTB), of which the favorable values are <500 Da, <12, and <10, respectively. Drugs with poor membrane permeability (more than the favorable value) have poor intestinal absorption, leading to low concentration in most tissues of animals, especially in the brain [17].
3. Neutrophils and Inflammatory Mediators in Ischemic Stroke
Inflammation plays an important role in ischemic stroke pathophysiology, particularly in the cerebral ischemia/reperfusion [3]. Inflammatory cells in situ including microglia and astrocytes, as well as infiltrated macrophages, and neutrophils from peripheral blood are involved in poststroke inflammatory response [43]. Many studies showed that leukocytes increased in the microvessels and parenchyma of lesion area as early as 30 min after artery occlusion onset [44, 45]. In particular, neutrophils are among the primary cells in the blood responding to ischemia and associated with increased infarct volumes in ischemic stroke [46]. After stroke, necrotic lesion produces abundant damage-associated molecular patterns (DAMPs) [47], such as mitochondrial DNA [48], ATP [49], and carboxyalkylpyrroles [50], to which injured brain cells exposed secret inflammation mediators, such as platelet-activating factor (PAF), TNF-α and IL-1β, and chemokines [51, 52]. Subsequently, the expression of CAMs on the surface of endothelium is induced [53]. Neutrophil rolling depends mostly on selectins, while adhesion and transmigration are integrin-dependent [23]. Once present on the surface of endothelial cells, these CAMs interact with complementary receptors on neutrophils surface; for example, P-selectin binds to its ligands-P-selectin glycoprotein ligand 1 (PSGL1) [54, 55], resulting in neutrophil rolling on the surface of endothelium along the direction of blood flow [23], and the interactions between neutrophil macrophage adhesion molecule-1 (MAC-1) and endothelial intercellular adhesion molecule-1 (ICAM-1) contribute to neutrophil adhesion to activated endothelium [56]. Consequently, neutrophils transmigrate into the brain parenchyma via orderly adhering to the endothelium and crossing the microvessel wall. Also, macrophages and monocytes enter the ischemic region and promote the inflammatory response predominantly 5 to 7 days after ischemia [53]. Infiltrated neutrophils have a remarkably destructive potential in several ways. Activated neutrophils infiltrating into the parenchyma produce many inflammatory factors including proteases (matrix metalloproteinases and elastase), reactive oxygen species (ROS) [23], and inducible nitric oxide synthase (iNOS) [53], facilitating inflammatory response, BBB breakdown, and cell death, as well as affecting brain repair. Additionally, neutrophils release tissue factor and neutrophil extracellular traps (NETs) acting on coagulation and interact with platelets [57, 58], leading to enhanced platelet aggregation and thrombus formation [59]. Also, influx of neutrophils contributes to neurotoxicity from the release of decondensed DNA [60]. Recently, researchers have focused on targeting neutrophils specifically as cerebroprotective strategies through neutrophil depletion, inhibition of inflammatory mediators, or neutrophil function to reduce infarct size and improve outcomes [21].
Inflammatory mediators, including cytokines, chemokines, and CAMs, play pivotal roles in the process of neutrophil activation, rolling on and adherence to the activated endothelium of blood vessel walls, and transmigration into the cerebral parenchyma. Cytokines, such as TNF-α and IL-1β, contribute to neutrophils and endothelium activation via upregulating the expression of CAMs [61]. TNF-α mRNA elevated in low levels from 30 minutes to 6 hours after occlusion and a rapid increase of IL-1β were observed as soon as 1 to 2 hours after occlusion, and they both persisted for approximately 5 days [62]. Modulating the function of TNF-α and IL-1β has remarkable effects on evolution of the infarct in both experimental and human stroke at the early phase of stroke, corresponding to the therapeutic window (<4.5 h) [4, 62, 63]. Emerging evidence displayed that inflammatory cytokines increased both centrally and systemically after ischemic stroke and correlated with infarct volume and stroke severity, and the systemic cytokine response paralleled the events in the CNS, suggesting that serum TNF-α and IL-1β had a predictive value for stroke outcome [64, 65]. Intraventricular injection of recombinant IL-1β immediately after reperfusion remarkably aggravated brain edema in a dose-dependent manner, increased brain infarction volume, and facilitated neutrophils adherent to the endothelium and infiltrating into ischemic areas, which were reversed by injection of anti-IL-1β [66]. Consistently, IL-1β deficient mice presented smaller infarct volume [67]. Interestingly, peripheral administration of recombinant human interleukin-1 receptor antagonist significantly inhibited infarct size and cerebral edema formation by 46% and 49%, respectively, after MCAO [68]. In experimental stroke models, administration of neutralizing antibodies to TNF or TNF-binding protein had protective effects [62]. CAMs, mainly including selectins, integrins, and immunoglobulin, expressed on leukocytes and endothelium, contribute to the process of inflammatory cells rolling, adhesion, and transmigration [24, 69]. CAMs (Table 1), especially ICAM-l, P-selectin, and E-selectin, are induced by cytokines after stroke and are responsible for the interaction between leukocytes (particularly neutrophils) and vascular endothelium [61]. In p-MCAO rats, levels of ICAM-1 mRNA in the ischemic cortex increased significantly at 3 h (2.6-fold) and peaked at 6 to 12 h (6.0-fold), while those in t-MCAO rats were 1 h (6.3-fold) and 12 h (12-fold), respectively. And they both remained for 5 days [70]. In experimental stroke models, blocking ICAM-1 reduced infarct volume, decreased mortality, and improved outcomes via pharmaceutical intervention and genetic deletion, as well as immunodepletion of neutrophils volume [71, 72]. P-Selectin and E-selectin on the endothelial cell surface are upregulated in response to ischemia, which promotes neutrophil adhesion by binding PSGL-1 [52, 55]. Expression of E-selectin was upregulated in the ischemic cerebral vasculature within 4 hours of reperfusion and lasted for 24 hours [73]. In rodent ischemic stroke, blocking E-selectin or P-selectin with antibody or by knockout decreased infarct volume and improved functional outcomes [73–75]. Knockout of MAC-1 reduced neutrophil infiltration and infarct size after cerebral ischemia [76]. In rodent and rabbit stroke, treatment with a humanized MAC-1 monoclonal antibody (Hu23F2G) crippled neutrophil migration and reduced ischemic injury [25, 77]. In addition, free radicals generation leads to increased expression of selectins on leukocyte surface and hypoxia can directly stimulate the upregulation of ICAM-1 and neutrophil adherence to endothelium through interaction of MAC-1 with ICAM-1 [4]. Chemokines, lining the luminal part of endothelium, are able to induce changes of integrins on neutrophil surface [23] and guide the migration of blood-derived inflammatory cells towards the source of the chemokines, which are indispensable in inflammatory cells recruitment [61]. The level of chemokines, such as monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-1a (MIP-1a), has been found to increase in experimental stroke and low expression of chemokines is associated with reduced injury by inhibition or deficiency [78, 79], attributed to impairing leukocyte infiltration [80]. Data showed that MCP-1 mRNA is increased at 6 hours after MCAO [81] and remained for 5 days [82]. Injection of a chemokine-receptor antagonist into cerebral ventricular displayed a reduction of infarct volume [21]. As mentioned above, crippling neutrophil infiltration peripherally to reduce cerebral ischemic damage and improve neurological deficits can be carried out through several mechanisms including reducing neutrophil activation and recruitment, blocking neutrophil adhesion to endothelium, and transmigration at the level of cytokines, chemokines, and CAMs (receptors and CAMs on neutrophils are illustrated in Figure 4).
Table 1.
Major cell adhesion molecules (CAMs) involved in neutrophils infiltration.
| Categories | Cell adhesion molecules | Expressed on |
|---|---|---|
| Selectins | P-Selectin | Endothelium, platelet |
| E-Selectin | Endothelium, leukocyte | |
| L-Selectin | Endothelium, leukocyte | |
|
| ||
| Integrins | CD11b/CD18 (MAC-1) | Leukocyte |
| CD41 | Platelet | |
|
| ||
| Immunoglobulin superfamily | ICAM-1 | Endothelium, leukocyte |
| ICAM-2 | Endothelium, platelet | |
| VCAM-1 | Endothelium | |
|
| ||
| Mucin-like family | PSGL-1 | Leukocyte |
Figure 4.
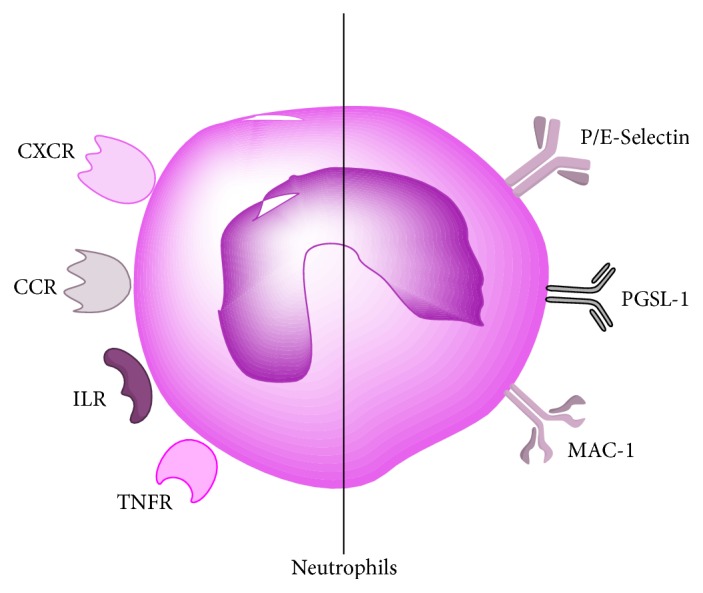
The receptors (the left) related to chemokines (CCR, CXCR) and cytokines (TNFR, ILR), and CAMs (the right) on neutrophils.
4. Natural Components from Medicinal Plants Inhibiting Neutrophil Infiltration
Astragaloside IV (AsIV) (Figure 2(a)), the major active ingredient extracted from the Chinese herb Astragalus membranaceus, has been widely applied to treat cardiovascular disease [83]. Luo et al. have demonstrated that postischemic treatment of AsIV significantly reduced infarct volume in t-MCAO mice, resulting partly from its antioxidant properties [84]. Recent study showed that the protective effects on focal cerebral ischemia of AsIV might be linked with the antioxidation, downregulating the expressions of iNOS and upregulating nerve growth factor (NGF) in t-MCAO rats [85]. Moreover, AsIV has potential to inhibit cytokine-stimulated expression of E-selectin and VCAM-1 on the surface of HUVECs and leukocytes adhesion to endothelial cells in a dose- and time-dependent manner [18, 86]. The molecular weight (785 Da), hydrogen-bonding capacity (23), and NROTB (16) of AsIV all exceed the favorable value <500 Da, <12, and <10, respectively, which result in the extremely low level of AsIV in the brain compared with that of other tissues [17]. In the rats given AsIV at 3.0 mg/kg intravenously, half-life (t 1/2) of the drug is 0.97 h, while first prominent enhancement of barrier permeability occurs at 3–6 hours after stroke. As a result, AsIV may not pass through BBB sufficiently to be delivered into the CNS and exert anti-inflammation on injured brain. Novel data revealed that AsIV was able to exert a neuroprotective effect and improve the outcome in short term, as a consequence of its inhibiting neutrophils adhesion and infiltration through attenuating upregulation of MAC-1 and ICAM-1, as well as suppressing proinflammatory factors (TNF-α and IL-1β) [86]. AsIV was also confirmed to attenuate TNF-α-induced upregulation of CAMs mRNA [87] and strongly inhibit neutrophil infiltration and activation [88]. In addition, AsIV could downregulate the level of ICAM-1 mRNA [89] and decrease the serum levels of TNF-α, MCP-1, and ICAM-1 [90].
Figure 2.

Structure of natural components: (a) Astragaloside IV, (b) Glycyrrhizin, (c) Beta-aescin, and (d) Tetrandrine.
Glycyrrhizin (GL) (Figure 2(b)), a triterpene present in the roots and rhizomes of Licorice, has been shown to have anti-inflammatory and antioxidative effects [91]. Due to large molecular weight (823 Da), high hydrogen-bonding capacity (24), and high molecular flexibility (NROTB: 12), pure GL administrated intravenously at 100 mg/kg to rats was not detected in the brain [17]. Another study also mentioned that GL demonstrated a negligible distribution in the brain [92]. Meanwhile, t 1/2 of GL was determined 1.8 h at the dose of intravenously 200 mg/kg [93] and GL bound highly (97–99%) to rat or human serum albumin [94, 95], probably contributing to difficulty to cross the BBB [17]. Treatment with GL significantly reduced the levels of ICAM-1 in TNF-α-stimulated HaCaT cells and inhibited subsequent monocytes adhesion to keratinocytes [96]. Research showed that administration of GL significantly improved neurological outcomes via decreasing inflammatory cytokines expression (TNF-α and IL-1β) induced by NF-κB and alleviating neutrophil infiltration in ischemic spinal cord [19]. In Japan, Stronger Neominophagen C (SNMC), of which GL is the major active ingredient, has been used to treat chronic hepatitis for more than 30 years. A recent study displayed that SNMC exerted neuroprotective effects through suppressing microglia activation and neutrophil infiltration in the ischemic brain in MCAO rats [91]. GL was potentially useful for inflammatory response by suppressing the adherence of neutrophils to the vascular endothelium [97–99], with evidence of reduced activity of myeloperoxidase (MPO) [100, 101].
Sodium β-aescin (Figure 2(c)), extracted from the seeds of Horse chestnut and divided into four types, has been widely used clinically as antiedema and antiexudation drug and all types are active ingredients. Sodium β-aescin significantly reduced cerebral infarct volume and water content and improved the neurological deficits through its antioxidant activity [102] with fewer side effects and complications [103]. Sodium β-aescin with large molecule weight and high hydrogen-bond capacity which are far more than the favorable values like AsIV is thought perhaps not to penetrate the BBB efficiently in principle, while there is no direct evidence to describe its tissue distribution, especially in the brain. Data showed that pretreatment with sodium β-aescin for 7 d could remarkably alleviate cerebral ischemia/reperfusion injury induced by MCAO, resulting from significantly reducing the cerebral infarct volume and ameliorating the neurological deficits through downregulating the protein expressions of ICAM-1 and E-selectin and inhibiting the migration of neutrophils after cerebral ischemia/reperfusion [20]. Several experiments also suggested that aescin treatment decreased neutrophil recruitment, adherence, and activation [104, 105], which could explain in part the potential benefit of the drug in the prevention of inflammatory response. Another data demonstrated that aescin influenced the later phases of inflammation (leukocyte migration) [106]. Likewise, although there is lack of evidence in brain distribution of Tetrandrine (TTD) (Figure 2(d)), a large-molecule natural chemical product purified from Fourstamen stefania root, it is certified to inhibit neutrophil recruitment by downregulating the expression of ICAM-1 and improve stroke outcomes [107]. Many data sets suggested that the protective effect of TTD against inflammation could be explained by significantly inhibiting neutrophil priming and activation, thereby abolishing subsequent infiltration [108, 109] via suppressing upregulation of MAC-1 [110–112].
Hydroxysafflor yellow A (HSYA) (Figure 3(a)), an active ingredient of Carthamus tinctorius L. flowers, has been reported to treat cerebrovascular and cardiovascular diseases because of its multiple biological activities. Zhang et al. demonstrated that HSYA significantly reduced the expression of the proinflammatory mediators such as IL-1β, IL-4, TNF-α, and iNOS, as well as inhibiting Ab1-42-induced neuroinflammation in vitro [113]. After injected intravenously in mice, HSYA was found existing in plasma, kidney, liver, lung, heart, and spleen but not in the brain [114], resulting partly from its large molecule weight and high hydrogen-bonding capacity. It has also been observed that HSYA had potential to inhibit the elevation of TNF-α, IL-6, ICAM-1, and VCAM-1 induced by lipopolysaccharide (LPS) to protect endothelial cells against inflammatory injury [115]. HSYA was effective to protect LPS-induced high expression of endothelium adhesive molecule, such as ICAM-1 and E-selectin [116–118]. HSYA significantly reduced the cell surface level of TNFR1 and inhibited TNF-α-induced adhesion of macrophage to endothelium [119]. However, there is little evidence that HSYA exerts anti-inflammation on brain injury through directly inhibiting neutrophil infiltrating; thus, additional researches are required to confirm the postulation. Naringin, a major antioxidant constituent in Citrus fruits and herbs, is similar to HSYA regarding the absent evidence of directly inhibiting neutrophil infiltrating after cerebral ischemia (Figure 3(b)). Naringin has been used as an effective anti-inflammatory medicine. One study reported that the free forms of Naringin and Naringenin were not detected in all the tissues assayed, and Naringenin glucuronides were present in liver and kidney but not in spleen, brain, and heart [120]. Naringin was tested to inhibit upregulation levels of proinflammatory mediators (ICAM-1, MIP-2) and downregulation of anti-inflammatory mediators [121]. Treatment of Naringin (50 and 100 mg/kg) for seven days significantly improved locomotor activity and reduced resistance to lateral push and transfer latency in cerebral ischemia/reperfusion [122]. Naringin prevented cigarette smoke-induced infiltration of neutrophils and activation of MPO and matrix metalloproteinase-9 [123]. Naringin dose-dependently suppressed TNF-α-induced expressions of CAMs and chemokines at both the mRNA and protein levels, as well as the adhesion of monocytes to the TNF-α-stimulated HUVECs [124]. Also, Naringin reduced the stimuli-induced ICAM-1 expression [125, 126], contributing to the inhibition of monocytes adhesion to endothelial cells [126, 127].
Figure 3.

Structure of natural components: (a) Hydroxysafflor yellow A, (b) Naringin, and (c) Morroniside.
Furthermore, Morroniside (Figure 3(c)) is an important constituent of the traditional Chinese medicine Fructus Corni with several bioactivities. Some findings demonstrated that Morroniside exerted neuroprotective effects on the brain damage induced by focal cerebral ischemia, related to its antioxidant and antiapoptotic properties in the brain [128]. Pharmacokinetic study revealed that Morroniside administrated intravenously and orally was absorbed and eliminated rapidly in rats and absolute bioavailability of Morroniside was lower, and no Morroniside was found in brain [129]. Morroniside promoted angiogenesis, in part through accelerating endothelial progenitor cells (EPCs) proliferation in ischemic stroke models [130]. EPCs derived from bone marrow mobilize into the blood and home to the site of ischemic region through adherence, differentiation, proliferation, and migration to participate in tissue vascularization [131]. Processes of EPCs mobilizing from bone marrow to ischemic area partly occur in the peripheral blood. In addition, insulin-like growth factor-1 (IGF-1) and Erythropoietin (EPO) are generated by peripheral organs like liver and kidney, respectively, and delivered into the brain through cerebrovasculature to participate in angiogenesis, which can be modulated peripherally by medicine that cannot penetrate the BBB [132, 133].
5. Conclusion
Although majority of agents showed anti-inflammation activity in cerebral parenchyma after stroke, we cannot ignore the problem that they were at very low concentrations or not detected in the brain tissue. Therefore, in the coagulation models, they may play an anti-inflammatory role due to their inhibiting neutrophil infiltration in peripheral circulation system. And such pathway was attributed to poor membrane permeability decided by physicochemical properties of agents and slight BBB leakage depending on type of stroke models. Additionally, administration time, frequency, and method of drugs influence the concentration in the brain. In summary, if drugs could achieve effective concentration in the cerebral parenchyma, they can function centrally and peripherally; otherwise, the latter may be dominating. Also, Kim et al. have reported that neutrophil counts were associated positively with the severity of stroke, while lower lymphocyte counts were linked with poor functional outcome [134]. When inhibiting neutrophils at the level of inflammatory mediators, will these drugs cripple the number and function of lymphocytes? There is no clear answer. Thus, much more work are required to do to explore inflammation process, the anti-inflammatory mechanisms about medicine localized in the brain at a very low concentration, and pharmacokinetics of drugs in various stroke models, as well as difference of BBB disruption in all types of models at a molecule level. Also, can different therapies be given as the infarct types of patients and time course of stroke progression?
Competing Interests
The authors declare that they have no competing interests.
References
- 1.Go A. S., Mozaffarian D., Roger V. L., et al. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation. 2014;129(3):e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Go A. S., Mozaffarian D., Roger V. L., et al. Heart disease and stroke statistics-2013 update: a report from the American Heart Association. Circulation. 2013;127(1):e6–e245. doi: 10.1161/cir.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xing C., Arai K., Lo E. H., Hommel M. Pathophysiologic cascades in ischemic stroke. International Journal of Stroke. 2012;7(5):378–385. doi: 10.1111/j.1747-4949.2012.00839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gu Y., Chen J., Shen J. Herbal medicines for ischemic stroke: combating inflammation as therapeutic targets. Journal of Neuroimmune Pharmacology. 2014;9(3):313–339. doi: 10.1007/s11481-014-9525-5. [DOI] [PubMed] [Google Scholar]
- 5.Campanella M., Sciorati C., Tarozzo G., Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;33(2):586–592. doi: 10.1161/hs0202.103399. [DOI] [PubMed] [Google Scholar]
- 6.Clark R. K., Lee E. V., Fish C. J., et al. Development of tissue damage, inflammation and resolution following stroke: an immunohistochemical and quantitative planimetric study. Brain Research Bulletin. 1993;31(5):565–572. doi: 10.1016/0361-9230(93)90124-t. [DOI] [PubMed] [Google Scholar]
- 7.Beck H., Plate K. H. Angiogenesis after cerebral ischemia. Acta Neuropathologica. 2009;117(5):481–496. doi: 10.1007/s00401-009-0483-6. [DOI] [PubMed] [Google Scholar]
- 8.Fan Y., Yang G.-Y. Therapeutic angiogenesis for brain ischemia: a brief review. Journal of Neuroimmune Pharmacology. 2007;2(3):284–289. doi: 10.1007/s11481-007-9073-3. [DOI] [PubMed] [Google Scholar]
- 9.Weintraub M. I. Thrombolysis (tissue plasminogen activator) in stroke: a medicolegal quagmire. Stroke. 2006;37(7):1917–1922. doi: 10.1161/01.str.0000226651.04862.da. [DOI] [PubMed] [Google Scholar]
- 10.Grønberg N. V., Johansen F. F., Kristiansen U., Hasseldam H. Leukocyte infiltration in experimental stroke. Journal of Neuroinflammation. 2013;10, article 115 doi: 10.1186/1742-2094-10-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tzounis X., Vulevic J., Kuhnle G. G. C., et al. Flavanol monomer-induced changes to the human faecal microflora. British Journal of Nutrition. 2008;99(4):782–792. doi: 10.1017/S0007114507853384. [DOI] [PubMed] [Google Scholar]
- 12.Spencer J. P. E. The impact of flavonoids on memory: physiological and molecular considerations. Chemical Society Reviews. 2009;38(4):1152–1161. doi: 10.1039/b800422f. [DOI] [PubMed] [Google Scholar]
- 13.Zhang G., Xia F., Zhang Y., et al. Ginsenoside Rd is efficacious against acute ischemic stroke by suppressing microglial proteasome-mediated inflammation. Molecular Neurobiology. 2016;53(4):2529–2540. doi: 10.1007/s12035-015-9261-8. [DOI] [PubMed] [Google Scholar]
- 14.Ruan Z., Wang H. M., Huang X. T., et al. A novel caffeoyl triterpene attenuates cerebral ischemic injury with potent anti-inflammatory and hypothermic effects. Journal of Neurochemistry. 2015;133(1):93–103. doi: 10.1111/jnc.13046. [DOI] [PubMed] [Google Scholar]
- 15.Zhu J., Jiang Y., Wu L., Lu T., Xu G., Liu X. Suppression of local inflammation contributes to the neuroprotective effect of ginsenoside Rb1 in rats with cerebral ischemia. Neuroscience. 2012;202:342–351. doi: 10.1016/j.neuroscience.2011.11.070. [DOI] [PubMed] [Google Scholar]
- 16.Girbovan C., Plamondon H. Resveratrol downregulates type-1 glutamate transporter expression and microglia activation in the hippocampus following cerebral ischemia reperfusion in rats. Brain Research. 2015;1608:203–214. doi: 10.1016/j.brainres.2015.02.038. [DOI] [PubMed] [Google Scholar]
- 17.Yu K., Chen F., Li C. Absorption, disposition, and pharmacokinetics of saponins from Chinese medicinal herbs: what do we know and what do we need to know more? Current Drug Metabolism. 2012;13(5):577–598. doi: 10.2174/1389200211209050577. [DOI] [PubMed] [Google Scholar]
- 18.Zhang W.-J., Hufnagl P., Binder B. R., Wojta J. Antiinflammatory activity of astragaloside IV is mediated by inhibition of NF-κB activation and adhesion molecule expression. Thrombosis and Haemostasis. 2003;90(5):904–914. doi: 10.1160/TH03-03-0136. [DOI] [PubMed] [Google Scholar]
- 19.Ni B., Cao Z., Liu Y. Glycyrrhizin protects spinal cord and reduces inflammation in spinal cord ischemia-reperfusion injury. International Journal of Neuroscience. 2013;123(11):745–751. doi: 10.3109/00207454.2013.796551. [DOI] [PubMed] [Google Scholar]
- 20.Hu X.-M., Zhang Y., Zeng F.-D. Effects of sodium β-aescin on expression of adhesion molecules and migration of neutrophils after middle cerebral artery occlusion in rats. Acta Pharmacologica Sinica. 2004;25(7):869–875. [PubMed] [Google Scholar]
- 21.Huang J., Upadhyay U. M., Tamargo R. J. Inflammation in stroke and focal cerebral ischemia. Surgical Neurology. 2006;66(3):232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 22.Krupinski J., Kaluza J., Kumar P., Kumar S., Wang J. M. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke. 1994;25(9):1794–1798. doi: 10.1161/01.STR.25.9.1794. [DOI] [PubMed] [Google Scholar]
- 23.Kolaczkowska E., Kubes P. Neutrophil recruitment and function in health and inflammation. Nature Reviews Immunology. 2013;13(3):159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 24.Mo X., Li T., Ji G., Lu W., Hu Z. Peripheral polymorphonuclear leukocyte activation as a systemic inflammatory response in ischemic stroke. Neurological Sciences. 2013;34(9):1509–1516. doi: 10.1007/s10072-013-1447-0. [DOI] [PubMed] [Google Scholar]
- 25.Jickling G. C., Liu D., Ander B. P., Stamova B., Zhan X., Sharp F. R. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. Journal of Cerebral Blood Flow and Metabolism. 2015;35(6):888–901. doi: 10.1038/jcbfm.2015.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Howells D. W., Porritt M. J., Rewell S. S. J., et al. Different strokes for different folks: the rich diversity of animal models of focal cerebral ischemia. Journal of Cerebral Blood Flow & Metabolism. 2010;30(8):1412–1431. doi: 10.1038/jcbfm.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morancho A., García-Bonilla L., Barceló V., et al. A new method for focal transient cerebral ischaemia by distal compression of the middle cerebral artery. Neuropathology and Applied Neurobiology. 2012;38(6):617–627. doi: 10.1111/j.1365-2990.2012.01252.x. [DOI] [PubMed] [Google Scholar]
- 28.Carmichael S. T. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx. 2005;2(3):396–409. doi: 10.1602/neurorx.2.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Llovera G., Roth S., Plesnila N., Veltkamp R., Liesz A. Modeling stroke in mice: permanent coagulation of the distal middle cerebral artery. Journal of Visualized Experiments. 2014;4(89) doi: 10.3791/51729.e51729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang N., Zhang X., Liu X., et al. Chrysophanol inhibits nalp3 inflammasome activation and ameliorates cerebral ischemia/reperfusion in mice. Mediators of Inflammation. 2014;2014:12. doi: 10.1155/2014/370530.370530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandoval K. E., Witt K. A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiology of Disease. 2008;32(2):200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Gloor S. M., Wachtel M., Bolliger M. F., Ishihara H., Landmann R., Frei K. Molecular and cellular permeability control at the blood-brain barrier. Brain Research Reviews. 2001;36(2-3):258–264. doi: 10.1016/S0165-0173(01)00102-3. [DOI] [PubMed] [Google Scholar]
- 33.Daneman R. The blood-brain barrier in health and disease. Annals of Neurology. 2012;72(5):648–672. doi: 10.1002/ana.23648. [DOI] [PubMed] [Google Scholar]
- 34.Pardridge W. M. Drug transport across the blood-brain barrier. Journal of Cerebral Blood Flow and Metabolism. 2012;32(11):1959–1972. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knowland D., Arac A., Sekiguchi K. J., et al. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron. 2014;82(3):603–617. doi: 10.1016/j.neuron.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuroiwa T., Ting P., Martinez H., Klatzo I. The biphasic opening of the blood-brain barrier to proteins following temporary middle cerebral artery occlusion. Acta Neuropathologica. 1985;68(2):122–129. doi: 10.1007/BF00688633. [DOI] [PubMed] [Google Scholar]
- 37.Wu X.-D., Du L.-N., Wu G.-C., Cao X.-D. Effects of electroacupuncture on blood-brain barrier after cerebral ischemia-reperfusion in rat. Acupuncture and Electro-Therapeutics Research. 2001;26(1-2):1–9. doi: 10.3727/036012901816356063. [DOI] [PubMed] [Google Scholar]
- 38.Dimitrijevic O. B., Stamatovic S. M., Keep R. F., Andjelkovic A. V. Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. Journal of Cerebral Blood Flow and Metabolism. 2006;26(6):797–810. doi: 10.1038/sj.jcbfm.9600229. [DOI] [PubMed] [Google Scholar]
- 39.Jiao H., Wang Z., Liu Y., Wang P., Xue Y. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. Journal of Molecular Neuroscience. 2011;44(2):130–139. doi: 10.1007/s12031-011-9496-4. [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg G. A., Estrada E. Y., Dencoff J. E. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29(10):2189–2195. doi: 10.1161/01.STR.29.10.2189. [DOI] [PubMed] [Google Scholar]
- 41.Başkaya M. K., Rao A. M., Doğan A., Donaldson D., Dempsey R. J. The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neuroscience Letters. 1997;226(1):33–36. doi: 10.1016/S0304-3940(97)00239-5. [DOI] [PubMed] [Google Scholar]
- 42.Betz A. L. Alterations in cerebral endothelial cell function in ischemia. Advances in Neurology. 1996;71:311–313. [PubMed] [Google Scholar]
- 43.Brambilla R., Couch Y., Lambertsen K. L. The effect of stroke on immune function. Molecular and Cellular Neuroscience. 2013;53:26–33. doi: 10.1016/j.mcn.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 44.del Zoppo G. J., Schmid-Schönbein G. W., Mori E., Copeland B. R., Chang C.-M. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22(10):1276–1283. doi: 10.1161/01.STR.22.10.1276. [DOI] [PubMed] [Google Scholar]
- 45.Del Z. G. Microvascular changes during cerebral ischemia and reperfusion. Cerebrovascular and Brain Metabolism Reviews Journal. 1994;6(1):47–96. [PubMed] [Google Scholar]
- 46.Segel G. B., Halterman M. W., Lichtman M. A. The paradox of the neutrophil's role in tissue injury. Journal of Leukocyte Biology. 2011;89(3):359–372. doi: 10.1189/jlb.0910538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iadecola C., Anrather J. The immunology of stroke: from mechanisms to translation. Nature Medicine. 2011;17(7):796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maeda A., Fadeel B. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death and Disease. 2014;5(7, article e1312) doi: 10.1038/cddis.2014.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang F., Wang Z., Wei X., et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. Journal of Cerebral Blood Flow & Metabolism. 2014;34(4):660–667. doi: 10.1038/jcbfm.2013.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.West X. Z., Malinin N. L., Merkulova A. A., et al. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467(7318):972–976. doi: 10.1038/nature09421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDonald B., Pittman K., Menezes G. B., et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330(6002):362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 52.Dimasi D., Sun W. Y., Bonder C. S. Neutrophil interactions with the vascular endothelium. International Immunopharmacology. 2013;17(4):1167–1175. doi: 10.1016/j.intimp.2013.05.034. [DOI] [PubMed] [Google Scholar]
- 53.Dirnagl U., Iadecola C., Moskowitz M. A. Pathobiology of ischaemic stroke: an integrated view. Trends in Neurosciences. 1999;22(9):391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 54.Ley K., Laudanna C., Cybulsky M. I., Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nature Reviews Immunology. 2007;7(9):678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 55.Zarbock A., Ley K., McEver R. P., Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. 2011;118(26):6743–6751. doi: 10.1182/blood-2011-07-343566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morrison H., McKee D., Ritter L. Systemic neutrophil activation in a mouse model of ischemic stroke and reperfusion. Biological Research for Nursing. 2011;13(2):154–163. doi: 10.1177/1099800410384500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Darbousset R., Thomas G. M., Mezouar S., et al. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood. 2012;120(10):2133–2143. doi: 10.1182/blood-2012-06-437772. [DOI] [PubMed] [Google Scholar]
- 58.von Brühl M.-L., Stark K., Steinhart A., et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. Journal of Experimental Medicine. 2012;209(4):819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jin R., Yu S., Song Z., et al. Soluble CD40 ligand stimulates CD40-dependent activation of the β2 integrin Mac-1 and protein kinase C Zeda (PKCζ) in neutrophils: implications for neutrophil-platelet interactions and neutrophil oxidative burst. PLoS ONE. 2013;8(6, article e64631) doi: 10.1371/journal.pone.0064631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allen C., Thornton P., Denes A., et al. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. The Journal of Immunology. 2012;189(1):381–392. doi: 10.4049/jimmunol.1200409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lakhan S. E., Kirchgessner A., Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. Journal of Translational Medicine. 2009;7, article 97 doi: 10.1186/1479-5876-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lambertsen K. L., Biber K., Finsen B. Inflammatory cytokines in experimental and human stroke. Journal of Cerebral Blood Flow and Metabolism. 2012;32(9):1677–1698. doi: 10.1038/jcbfm.2012.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lambertsen K. L., Gregersen R., Finsen B. Microglial-macrophage synthesis of tumor necrosis factor after focal cerebral ischemia in mice is strain dependent. Journal of Cerebral Blood Flow and Metabolism. 2002;22(7):785–797. doi: 10.1097/00004647-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 64.Zaremba J., Losy J. Cytokines in clinical and experimental ischemic stroke. Neurologia I Neurochirurgia Polska. 2004;38(1, supplement 1):S57–S62. [PubMed] [Google Scholar]
- 65.Chang L., Chen Y., Li J., et al. Cocaine-and amphetamine-regulated transcript modulates peripheral immunity and protects against brain injury in experimental stroke. Brain, Behavior, and Immunity. 2011;25(2):260–269. doi: 10.1016/j.bbi.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 66.Yamasaki Y., Matsuura N., Shozuhara H., et al. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke. 1995;26(4):676–681. doi: 10.1161/01.STR.26.4.676. [DOI] [PubMed] [Google Scholar]
- 67.Boutin H., LeFeuvre R. A., Horai R., Asano M., Iwakura Y., Rothwell N. J. Role of IL-1alpha and IL-1beta in ischemic brain damage. The Journal of Neuroscience. 2001;21(15):5528–5534. doi: 10.1523/JNEUROSCI.21-15-05528.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barone F. C., Feuerstein G. Z. Inflammatory mediators and stroke: new opportunities for novel therapeutics. Journal of Cerebral Blood Flow and Metabolism. 1999;19(8):819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 69.Kozuka K., Kohriyama T., Nomura E., Ikeda J., Kajikawa H., Nakamura S. Endothelial markers and adhesion molecules in acute ischemic stroke—sequential change and differences in stroke subtype. Atherosclerosis. 2002;161(1):161–168. doi: 10.1016/s0021-9150(01)00635-9. [DOI] [PubMed] [Google Scholar]
- 70.Wang X., Siren A.-L., Liu Y., Yue T.-L., Barone F. C., Feuerstein G. Z. Upregulation of intercellular adhesion molecule 1 (ICAM-1) on brain microvascular endothelial cells in rat ischemic cortex. Molecular Brain Research. 1994;26(1-2):61–68. doi: 10.1016/0169-328X(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 71.Zhang R. L., Chopp M., Jiang N., et al. Anti-intercellular adhesion molecule-1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the Wistar rat. Stroke. 1995;26(8):1438–1443. doi: 10.1161/01.str.26.8.1438. [DOI] [PubMed] [Google Scholar]
- 72.Kitagawa K., Matsumoto M., Mabuchi T., et al. Deficiency of intercellular adhesion molecule 1 attenuates microcirculatory disturbance and infarction size in focal cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism. 1998;18(12):1336–1345. doi: 10.1097/00004647-199812000-00008. [DOI] [PubMed] [Google Scholar]
- 73.Huang J., Choudhri T. F., Winfree C. J., et al. Postischemic cerebrovascular E-selectin expression mediates tissue injury in murine stroke. Stroke. 2000;31(12):3047–3053. doi: 10.1161/01.str.31.12.3047. [DOI] [PubMed] [Google Scholar]
- 74.Mocco J., Choudhri T., Huang J., et al. HuEP5C7 as a humanized monoclonal anti-E/P-selectin neurovascular protective strategy in a blinded placebo-controlled trial of nonhuman primate stroke. Circulation Research. 2002;91(10):907–914. doi: 10.1161/01.RES.0000042063.15901.20. [DOI] [PubMed] [Google Scholar]
- 75.Jin A. Y., Tuor U. I., Rushforth D., et al. Reduced blood brain barrier breakdown in P-selectin deficient mice following transient ischemic stroke: a future therapeutic target for treatment of stroke. BMC Neuroscience. 2010;11, article 12 doi: 10.1186/1471-2202-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Soriano S. G., Coxon A., Wang Y. F., et al. Mice deficient in Mac-1 (CD11b/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke. 1999;30(1):134–139. doi: 10.1161/01.STR.30.1.134. [DOI] [PubMed] [Google Scholar]
- 77.Yenari M. A., Kunis D., Sun G. H., et al. Hu23F2G, an antibody recognizing the leukocyte CD11/CD18 integrin, reduces injury in a rabbit model of transient focal cerebral ischemia. Experimental Neurology. 1998;153(2):223–233. doi: 10.1006/exnr.1998.6876. [DOI] [PubMed] [Google Scholar]
- 78.Hughes P. M., Allegrini P. R., Rudin M., Perry V. H., Mir A. K., Wiessner C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. Journal of Cerebral Blood Flow and Metabolism. 2002;22(3):308–317. doi: 10.1097/00004647-200203000-00008. [DOI] [PubMed] [Google Scholar]
- 79.Kumai Y., Ooboshi H., Takada J., et al. Anti-monocyte chemoattractant protein-1 gene therapy protects against focal brain ischemia in hypertensive rats. Journal of Cerebral Blood Flow and Metabolism. 2004;24(12):1359–1368. doi: 10.1097/00004647-200412000-00005. [DOI] [PubMed] [Google Scholar]
- 80.Kim J. S., Gautam S. C., Chopp M., et al. Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. Journal of Neuroimmunology. 1995;56(2):127–134. doi: 10.1016/0165-5728(94)00138-E. [DOI] [PubMed] [Google Scholar]
- 81.Wang X., Yue T.-L., Barone F. C., Feuerstein G. Z. Monocyte chemoattractant protein-1 messenger RNA expression in rat ischemic cortex. Stroke. 1995;26(4):661–666. doi: 10.1161/01.str.26.4.661. [DOI] [PubMed] [Google Scholar]
- 82.Gourmala N. G., Buttini M., Limonta S., Sauter A., Boddeke H. W. G. M. Differential and time-dependent expression of monocyte chemoattractant protein-1 mRNA by astrocytes and macrophages in rat brain: effects of ischemia and peripheral lipopolysaccharide administration. Journal of Neuroimmunology. 1997;74(1-2):35–44. doi: 10.1016/s0165-5728(96)00203-2. [DOI] [PubMed] [Google Scholar]
- 83.Qin H., Liu P., Lin S. Effects of astragaloside IV on the SDF-1/CXCR4 expression in atherosclerosis of apoE−/− mice induced by hyperlipaemia. Evidence-Based Complementary and Alternative Medicine. 2015;2015:8. doi: 10.1155/2015/385154.385154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Luo Y., Qin Z., Hong Z., et al. Astragaloside IV protects against ischemic brain injury in a murine model of transient focal ischemia. Neuroscience Letters. 2004;363(3):218–223. doi: 10.1016/j.neulet.2004.03.036. [DOI] [PubMed] [Google Scholar]
- 85.Yin Y.-Y., Li W.-P., Gong H.-L., Zhu F.-F., Li W.-Z., Wu G.-C. Protective effect of astragaloside on focal cerebral ischemia/reperfusion injury in rats. American Journal of Chinese Medicine. 2010;38(3):517–527. doi: 10.1142/S0192415X10008020. [DOI] [PubMed] [Google Scholar]
- 86.Li M., Qu Y. Z., Zhao Z. W., et al. Astragaloside IV protects against focal cerebral ischemia/reperfusion injury correlating to suppression of neutrophils adhesion-related molecules. Neurochemistry International. 2012;60(5):458–465. doi: 10.1016/j.neuint.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 87.Liu Q.-S., Wang H.-F., Sun A.-K., et al. A comparative study on inhibition of total astragalus saponins and astragaloside IV on TNFR1-mediated signaling pathways in arterial endothelial cells. PLoS ONE. 2014;9(7) doi: 10.1371/journal.pone.0101504.e101504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang W.-J., Frei B. Astragaloside IV inhibits NF-κB activation and inflammatory gene expression in LPS-treated mice. Mediators of Inflammation. 2015;2015:11. doi: 10.1155/2015/274314.274314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huang X.-P., Ding H., Lu J.-D., Tang Y.-H., Deng B.-X., Deng C.-Q. Effects of the combination of the main active components of Astragalus and Panax notoginseng on inflammation and apoptosis of nerve cell after cerebral ischemia-reperfusion. American Journal of Chinese Medicine. 2015;43(7):1419–1438. doi: 10.1142/s0192415x15500809. [DOI] [PubMed] [Google Scholar]
- 90.Gui D., Huang J., Guo Y., et al. Astragaloside IV ameliorates renal injury in streptozotocin-induced diabetic rats through inhibiting NF-κB-mediated inflammatory genes expression. Cytokine. 2013;61(3):970–977. doi: 10.1016/j.cyto.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 91.Kim S., Lim C., Lee H., Lee J. The use of Stronger Neo-Minophagen C, a glycyrrhizin-containing preparation, in robust neuroprotection in the postischemic brain. Anatomy & Cell Biology. 2011;44(4):304–313. doi: 10.5115/acb.2011.44.4.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ishida S., Sakiya Y., Ichikawa T., Taira Z., Awazu S. Prediction of glycyrrhizin disposition in rat and man with liver failure by a physiologically based pharmacokinetic model. Journal of Pharmacobio-Dynamics. 1990;13(2):142–157. doi: 10.1248/bpb1978.13.142. [DOI] [PubMed] [Google Scholar]
- 93.Tsai T.-H., Liao J.-F., Shum A. Y.-C., Chen C.-F. Pharmacokinetics of glycyrrhizin after intravenous administration to rats. Journal of Pharmaceutical Sciences. 1992;81(9):961–963. doi: 10.1002/jps.2600810925. [DOI] [PubMed] [Google Scholar]
- 94.Ishida S., Ichikawa T., Sakiya Y. Binding of glycyrrhetinic acid to rat plasma, rat serum albumin, human serum, and human serum albumin. Chemical and Pharmaceutical Bulletin. 1988;36(1):440–443. doi: 10.1248/cpb.36.440. [DOI] [PubMed] [Google Scholar]
- 95.Ishida S., Sakiya Y., Ichikawa T., Kinoshita M., Awazu S. Binding of glycyrrhizin to human serum and human serum albumin. Chemical and Pharmaceutical Bulletin. 1989;37(1):226–228. doi: 10.1248/cpb.37.226. [DOI] [PubMed] [Google Scholar]
- 96.Xiong H., Xu Y., Tan G., et al. Glycyrrhizin ameliorates imiquimod-induced psoriasis-like skin lesions in BALB/c Mice and inhibits TNF-α-induced ICAM-1 expression via NF-κB/MAPK in HaCaT Cells. Cellular Physiology and Biochemistry. 2015;35(4):1335–1346. doi: 10.1159/000373955. [DOI] [PubMed] [Google Scholar]
- 97.Huang W., Chen X., Li Q., et al. Inhibition of intercellular adhesion in herpex simplex virus infection by glycyrrhizin. Cell Biochemistry and Biophysics. 2012;62(1):137–140. doi: 10.1007/s12013-011-9271-8. [DOI] [PubMed] [Google Scholar]
- 98.Nakata N., Kira Y., Yabunaka Y., Takaoka K. Prevention of venous thrombosis by preoperative glycyrrhizin infusion in a rat model. Journal of Orthopaedic Science. 2008;13(5):456–462. doi: 10.1007/s00776-008-1259-x. [DOI] [PubMed] [Google Scholar]
- 99.Mabuchi A., Wake K., Marlini M., Watanabe H., Wheatley A. M. Protection by glycyrrhizin against warm ischemia-reperfusioninduced cellular injury and derangement of the microcirculatory blood flow in the rat liver. Microcirculation. 2009;16(4):364–376. doi: 10.1080/10739680902796917. [DOI] [PubMed] [Google Scholar]
- 100.Ni Y.-F., Kuai J.-K., Lu Z.-F., et al. Glycyrrhizin treatment is associated with attenuation of lipopolysaccharide-induced acute lung injury by inhibiting cyclooxygenase-2 and inducible nitric oxide synthase expression. Journal of Surgical Research. 2011;165(1):e29–e35. doi: 10.1016/j.jss.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 101.Nakata N., Takaoka K. Use of glycyrrhizin in prevention of tissue damage caused by ischemia-reperfusion in rabbit hind limbs. Journal of Orthopaedic Science. 2006;11(4):375–379. doi: 10.1007/s00776-006-1027-8. [DOI] [PubMed] [Google Scholar]
- 102.Hu X.-M., Zeng F.-D. Protective effects of sodium beta-aescin on ischemia-reperfusion injury in rat brain. Yao Xue Xue Bao. 2004;39(6):419–423. [PubMed] [Google Scholar]
- 103.Liu J., Li Y., Yuan X., Yang Z., Lin Z. Sodium β-aescin may be an effective therapeutic agent for Bell's palsy. Medical Hypotheses. 2008;71(5):762–764. doi: 10.1016/j.mehy.2008.07.037. [DOI] [PubMed] [Google Scholar]
- 104.Bougelet C., Roland I. H., Ninane N., Arnould T., Remacle J., Michiels C. Effect of aescine on hypoxia-induced neutrophil adherence to umbilical vein endothelium. European Journal of Pharmacology. 1998;345(1):89–95. doi: 10.1016/S0014-2999(97)01616-6. [DOI] [PubMed] [Google Scholar]
- 105.Zhang X., Zheng T., Sang L., et al. Otitis media induced by peptidoglycan-polysaccharide (PGPS) in TLR2-deficient (Tlr2−/−) mice for developing drug therapy. Infection, Genetics and Evolution. 2015;35:194–203. doi: 10.1016/j.meegid.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rothkopf-Ischebeck M. Phenyl isothiocyanate pleurisy in rats-a model for the evaluation of anti-exudative substances. Agents and Actions. 1978;8(6):610–617. doi: 10.1007/BF01998890. [DOI] [PubMed] [Google Scholar]
- 107.Liu S.-J., Zhou S.-W., Xue C.-S. Effect of tetrandrine on neutrophilic recruitment response to brain ischemia/reperfusion. Acta Pharmacologica Sinica. 2001;22(11):971–975. [PubMed] [Google Scholar]
- 108.Shen Y.-C., Chen C.-F., Sung Y.-J. Tetrandrine ameliorates ischaemia-reperfusion injury of rat myocardium through inhibition of neutrophil priming and activation. British Journal of Pharmacology. 1999;128(7):1593–1601. doi: 10.1038/sj.bjp.0702958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Seow W. K., Si-Ying L., Thong Y. H. Inhibitory effects of tetrandrine on human neutrophil and monocyte adherence. Immunology Letters. 1986;13(1-2):83–88. doi: 10.1016/0165-2478(86)90131-8. [DOI] [PubMed] [Google Scholar]
- 110.Shen Y.-C., Chen C.-F., Wang S.-Y., Sung Y.-J. Impediment to calcium influx and reactive oxygen production accounts for the inhibition of neutrophil Mac-1 up-regulation and adhesion by tetrandrine. Molecular Pharmacology. 1999;55(1):186–193. doi: 10.1124/mol.55.1.186. [DOI] [PubMed] [Google Scholar]
- 111.Chen Y., Tsai Y.-H., Tseng S.-H. The potential of tetrandrine as a protective agent for ischemic stroke. Molecules. 2011;16(9):8020–8032. doi: 10.3390/molecules16098020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shen Y.-C., Chou C.-J., Chiou W.-F., Chen C.-F. Anti-inflammatory effects of the partially purified extract of radix Stephaniae tetrandrae: comparative studies of its active principles tetrandrine and fangchinoline on human polymorphonuclear leukocyte functions. Molecular Pharmacology. 2001;60(5):1083–1090. [PubMed] [Google Scholar]
- 113.Zhang Z., Wu Z., Zhu X., Hui X., Pan J., Xu Y. Hydroxy-safflor yellow A inhibits neuroinflammation mediated by Aβ 1–42 in BV-2 cells. Neuroscience Letters. 2014;562:39–44. doi: 10.1016/j.neulet.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 114.Liu Y.-Q., Zhou H.-T., Bi K.-S. Study on distribution of safflor yellow A in tissues of mice. Yao Xue Xue Bao. 2004;39(3):217–219. [PubMed] [Google Scholar]
- 115.Wang X., Zang B., Wu W., Tong J., Jin M. Protective effect of hydroxysafflor yellow A on endothelial cell injury induced by lipopolysaccharide. Zhongguo Zhong Yao Za Zhi. 2011;36(12):1650–1653. doi: 10.4268/cjcmm20111224. [DOI] [PubMed] [Google Scholar]
- 116.Jin M., Sun C.-Y., Zang B.-X. Hydroxysafflor yellow A attenuate lipopolysaccharide-induced endothelium inflammatory injury. Chinese Journal of Integrative Medicine. 2016;22(1):36–41. doi: 10.1007/s11655-015-1976-x. [DOI] [PubMed] [Google Scholar]
- 117.Song L., Zhu Y., Jin M., Zang B. Hydroxysafflor yellow a inhibits lipopolysaccharide-induced inflammatory signal transduction in human alveolar epithelial A549 cells. Fitoterapia. 2013;84(1):107–114. doi: 10.1016/j.fitote.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 118.Wang X.-F., Jin M., Tong J., Wu W., Li J.-R., Zang B.-X. Protective effect of hydroxysafflor yellow A against acute lung injury induced by oleic acid and lipopolysaccharide in rats. Yao Xue Xue Bao. 2010;45(7):940–944. [PubMed] [Google Scholar]
- 119.Wang H., Liu J., Yang Y., et al. Hydroxy-safflower yellow a inhibits the TNFR1-mediated classical pathway by inducing shedding of TNFR1. Phytotherapy Research. 2016;30(5):790–796. doi: 10.1002/ptr.5579. [DOI] [PubMed] [Google Scholar]
- 120.Lin S.-P., Hou Y.-C., Tsai S.-Y., Wang M.-J., Lee Chao P.-D. Tissue distribution of naringenin conjugated metabolites following repeated dosing of Naringin to rats. BioMedicine. 2014;4, article 16 doi: 10.7603/s40681-014-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ahmad S. F., Attia S. M., Bakheet S. A., et al. Naringin attenuates the development of carrageenan-induced acute lung inflammation through inhibition of NF-κb, STAT3 and pro-inflammatory mediators and enhancement of IκBα and anti-inflammatory cytokines. Inflammation. 2015;38(2):846–857. doi: 10.1007/s10753-014-9994-y. [DOI] [PubMed] [Google Scholar]
- 122.Gaur V., Aggarwal A., Kumar A. Protective effect of naringin against ischemic reperfusion cerebral injury: possible neurobehavioral, biochemical and cellular alterations in rat brain. European Journal of Pharmacology. 2009;616(1–3):147–154. doi: 10.1016/j.ejphar.2009.06.056. [DOI] [PubMed] [Google Scholar]
- 123.Nie Y.-C., Wu H., Li P.-B., et al. Anti-inflammatory effects of naringin in chronic pulmonary neutrophilic inflammation in cigarette smoke-exposed rats. Journal of Medicinal Food. 2012;15(10):894–900. doi: 10.1089/jmf.2012.2251. [DOI] [PubMed] [Google Scholar]
- 124.Hsueh T., Sheen J., Pang J., et al. The anti-atherosclerotic effect of naringin is associated with reduced expressions of cell adhesion molecules and chemokines through NF-κB pathway. Molecules. 2016;21(2, article 195) doi: 10.3390/molecules21020195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Choe S.-C., Kim H.-S., Jeong T.-S., Bok S.-H., Park Y.-B. Naringin has an antiatherogenic effect with the inhibition of intercellular adhesion molecule-1 in hypercholesterolemic rabbits. Journal of Cardiovascular Pharmacology. 2001;38(6):947–955. doi: 10.1097/00005344-200112000-00017. [DOI] [PubMed] [Google Scholar]
- 126.Kim S.-W., Kim C. E., Kim M. H. Flavonoids inhibit high glucose-induced up-regulation of ICAM-1 via the p38 MAPK pathway in human vein endothelial cells. Biochemical and Biophysical Research Communications. 2011;415(4):602–607. doi: 10.1016/j.bbrc.2011.10.115. [DOI] [PubMed] [Google Scholar]
- 127.Chanet A., Milenkovic D., Deval C., et al. Naringin, the major grapefruit flavonoid, specifically affects atherosclerosis development in diet-induced hypercholesterolemia in mice. Journal of Nutritional Biochemistry. 2012;23(5):469–477. doi: 10.1016/j.jnutbio.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 128.Wang W., Xu J., Li L., et al. Neuroprotective effect of morroniside on focal cerebral ischemia in rats. Brain Research Bulletin. 2010;83(5):196–201. doi: 10.1016/j.brainresbull.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 129.Li X., Wang Q., Zhang X., et al. HPLC study of pharmacokinetics and tissue distribution of morroniside in rats. Journal of Pharmaceutical and Biomedical Analysis. 2007;45(2):349–355. doi: 10.1016/j.jpba.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 130.Sun F.-L., Wang W., Cheng H., et al. Morroniside improves microvascular functional integrity of the neurovascular unit after cerebral ischemia. PLoS ONE. 2014;9(6) doi: 10.1371/journal.pone.0101194.e101194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lapergue B., Mohammad A., Shuaib A. Endothelial progenitor cells and cerebrovascular diseases. Progress in Neurobiology. 2007;83(6):349–362. doi: 10.1016/j.pneurobio.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 132.Digicaylioglu M. Erythropoietin in stroke: quo vadis. Expert Opinion on Biological Therapy. 2010;10(6):937–949. doi: 10.1517/14712598.2010.481435. [DOI] [PubMed] [Google Scholar]
- 133.Torres-Aleman I. Toward a comprehensive neurobiology of IGF-I. Developmental Neurobiology. 2010;70(5):384–396. doi: 10.1002/dneu.20778. [DOI] [PubMed] [Google Scholar]
- 134.Kim J., Song T.-J., Park J. H., et al. Different prognostic value of white blood cell subtypes in patients with acute cerebral infarction. Atherosclerosis. 2012;222(2):464–467. doi: 10.1016/j.atherosclerosis.2012.02.042. [DOI] [PubMed] [Google Scholar]