


Abstract
Dimerization is a unique and vital characteristic of retroviral genomes. It is commonly accepted that genomic RNA (gRNA) must be dimeric at the plasma membrane of the infected cells to be packaged during virus assembly. However, where, when and how HIV-1 gRNA find each other and dimerize in the cell are long-standing questions that cannot be answered using conventional approaches. Here, we combine two state-of-the-art, multicolor RNA labeling strategies with two single-molecule microscopy technologies to address these questions. We used 3D-super-resolution structured illumination microscopy to analyze and quantify the spatial gRNA association throughout the cell and monitored the dynamics of RNA-RNA complexes in living-cells by cross-correlation fluctuation analysis. These sensitive and complementary approaches, combined with trans-complementation experiments, reveal for the first time the presence of interacting gRNA in the cytosol, a challenging observation due to the low frequency of these events and their dilution among the bulk of other RNAs, and allow the determination of the subcellular orchestration of the HIV-1 dimerization process.
INTRODUCTION
HIV-1, like most retroviruses, packages two identical copies of its genomic RNA (gRNA) in the form of a non-covalently linked gRNA dimer (1). The presence of two copies of gRNA in a single virion has two evolutionary advantages. Firstly, it provides a backup template, in case one strand is damaged. Secondly, elements of both copies are retro-transcribed into proviral DNA, via RNA template switching, by the viral reverse transcriptase promoting frequent recombination and, thus, genetic diversity, a major evolutionary driving force for HIV-1 (2). The proviral DNA is integrated into the host chromosomes, from where it is transcribed into a single full-length RNA precursor. Some of the RNA precursor molecules are spliced to generate mRNAs encoding the envelope glycoproteins as well as the accessory and regulatory proteins. Others are exported from the nucleus as full-length RNA that serve as mRNA for the synthesis of the Gag and GagPol polyproteins, or as gRNA that are packaged into viruses as dimers.
Packaging of the gRNA dimer requires its specific binding to the nucleocapsid (NC) domain of the Gag structural protein (3). The initial recognition of the HIV-1 gRNA by Gag has recently been reported to occur in the cytosol, although the low oligomerization of Gag on gRNA has challenged its detection (4,5). The Gag-RNA complexes then traffic to the plasma membrane (PM), where thousands of Gag molecules are efficiently recruited for virion assembly (6). Selection of full-length gRNA from the bulk of cellular and spliced viral RNAs is mediated by direct interaction between NC and highly structured sequences located in the 5′ untranslated region (5′ UTR) of the gRNA (7,8). In particular, the stem-loop 1 (SL1) has received major attention, since it constitutes the main packaging determinant of gRNA through specific binding of Gag (9).
The molecular mechanisms underlying dimerization have been well characterized in vitro. Dimerization initiates via kissing-loop interactions between the highly conserved palindromic sequences of the SL1 apical loop, named the dimerization initiation site (DIS) (10–15). Extended basepairing then stabilizes the dimer structure in a process chaperoned by NC (16,17). The role of the DIS in selecting the gRNA partner for co-packaging has been studied in vivo through recombination analyses (18) and by two-color gRNA labeling within virions (19). However, other determinants besides the DIS also contribute to gRNA dimerization and packaging. These include the trans-activation response element (TAR), the primer binding site (PBS) and the unique sequence in 5′ site (U5) in the 5′ UTR or sequences within the ORF of Gag. Interestingly, these determinants of dimerization and packaging map to the same region (20), suggesting that RNA dimerization and packaging are two linked processes. Surprisingly, in contrast to this wealth of information in vitro, our understanding of gRNA dimerization within the cell has remained a ‘black box’. The major hurdles in these studies were the presence of low and transient amounts of dimers and their low stability that makes their investigation difficult by classical techniques. With the emergence of high-performance RNA imaging approaches, the cell biological aspects of HIV-1 genome dimerization can now be addressed. Indeed, fluorescent RNA labeling techniques have been used to study HIV-1 RNA trafficking in the living cell (21–25) and its interaction with Gag at the PM (26,27). Here, we used two-color RNA labeling approaches combined with advanced fluorescence microscopic techniques to uncover gRNA dimerization in the three-dimensionality of the cell. We show, for the first time, that dimerization first occurs in the cytosol of HeLa cells and demonstrate that Gag and SL1/DIS sequences are key players in this process.
MATERIALS AND METHODS
Plasmids and constructs used in this study are described in supplementary information
Cell culture, transfections and viral particle preparation
HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 1% PenStrep (Sigma), at 5% CO2, 37°C. For virus particle preparations, 2 × 106 HeLa cells were seeded on 10 cm dishes and transfected the following day with 2 μg of HIV-1 or non-viral (NV) tagged constructs at equimolar ratios using jetPEI® transfection reagent (Polyplus). Cells were then incubated for 24 h at 37°C/5% CO2 and the culture medium collected for virus concentration. Briefly, culture medium was centrifuged at 1500 × g for 5 min at 4°C and clarified through a 0.45 μm pore-size filter to eliminate cell debris before ultracentrifugation on a 20% sucrose/posphate-buffered saline (PBS) cushion at 100 000 × g for 1.5 h. Virus-containing pellets were then resuspended in medium for fluorescence in situ hybridization (FISH) using microscopy and biochemical analyses.
Western-blotting
Analysis of protein expression of the viral constructs used in the study was performed by western-blotting. Cells were collected by scraping and lysed in the presence of protease inhibitor cocktail with the Complete lysis-M kit (Sigma) according to the manufacturer's instructions. Total protein concentration was determined by Bradford assay using a BSA standard set (Fermentas) and virions were purified from culture medium as described above. Proteins (100 μg of cell lysate or 12.5% of virus lysate) were loaded on 12% SDS-PAGE and were electro-transferred onto nitrocellulose membrane. Gag was detected with a mouse anti-capsid (CA) antibody (1/150, NIH AIDS Reagent Program, hybridoma H183, from B. Chesebro) and Vpu with a rabbit anti-Vpu polyclonal antiserum (1/10 000, NIH AIDS Reagent Program, from K. Strebel). Actin was detected with an anti-actin (1/500, Sigma). After incubation with a peroxidase-conjugated (HRP) secondary antibody, ECL fluorescence was recorded by a CCD chemiluminescence camera system (Gnome, Syngene).
RNA extraction and analysis by reverse transcription polymerase chain reaction (RT-PCR)
Some aliquots of cells and viral particle preparations (half of total) were subjected to RNA analysis by RT-PCR. RNA was extracted from cells with TriReagent (MRC) according to the manufacturer's instructions. Viral RNA was extracted in the presence of 20 μg of tRNA carrier. All RNA samples were treated with RQ1 DNase (Promega) in the presence of RNasin (Promega) for 25 min at 37°C. RNA was extracted with phenol–chloroform then chloroform and finally precipitated with 100% ethanol. RNA pellets were washed with 70% ethanol and dissolved in water. RNAs were quantified by measuring optical absorption at 260 nm. For RT-PCR, RNAs were reverse transcribed with Expand reverse transcriptase (Roche) using oligo (dT) primer for NV RNAs or specific primers targeting viral sequences for the viral RNAs. PCR was performed with 1/10th or 1/500th of the RT reaction of viral and NV RNAs, respectively. Primers were designed to specifically amplify viral MS2-RNA, viral lacZ-RNA and NV-MS2 RNA. All primer sequences and detailed PCR conditions will be provided on request.
Single molecule FISH
FISH was performed on cells and virions to detect MS2- and lacZ-tagged RNA (preparation of the coverslips for FISH is described in Supplementary Information). Fixed cells were washed in PBS and permeabilized with 0.5% Triton-X-100/PBS for 1 min at room temperature. After two washes in PBS, they were incubated in 1× SSC/20% formamide (Sigma) for 15 min at room temperature and then hybridized overnight at 37°C with the hybridization mix containing fluorescent probes (1× SSC, 20% formamide, 10% dextran sulphate, 0.02% RNA grade BSA, 2 mM Vanadyl-R-C, 40 μg tRNA, MS2-Alexa488 probe and a mix of 12 LacZ-Cy3 probes). The oligonucleotide probes were briefly denatured in formamide for 1 min at 80°C and then added to the hybridization mix. After overnight incubation, cells were washed twice for 30 min at 37°C in 1× SSC/20% formamide and then in PBS. Oligonucleotide sequences were described previously (MS2 probes (22) and lacZ probes (28)). Labeling of aminoallyl-T modified oligonucleotides was performed with Alexa Fluor® 488 Reactive Dye Decapack (Thermo Fisher Scientific) or Cy3 mono-Reactive Dye Pack (GE Healthcare) following the manufacturers’ instructions.
For cell preparations, coverslips were further counterstained with DAPI (Thermo Fisher Scientific, diluted 1/2000) and a fluorescently labeled wheat germ agglutinin (WGA-AF647) (Thermo Fisher Scientific, diluted 1/200) for 20 min at room temperature to visualize the nucleus and cell membranes, respectively. In some cases (indicated in the main text), immunofluorescence was performed after FISH to detect Gag by incubating with a rabbit anti-p17 polyclonal antiserum (NIH AIDS Reagent Program, diluted 1/500) followed by incubation with a donkey anti-Rabbit-AF647 (Molecular Probes, diluted 1/2000). Finally, coverslips were mounted in VECTASHIELD® Mounting Media (Vectorlabs).
Epifluorescence microscopy analysis of RNA in virions
FISH-stained viral particles were imaged using a wide-field Leica DM6000 upright microscope equipped with a 100x PL APO 1.4–0.7 oil-objective (Leica) and a Coolsnap HQ charge-coupled device camera (Roper Scientific) at Montpellier Rio Imaging (MRI) facilities. Dual-color images were acquired with the excitation and emission filter sets of green fluorescent protein (GFP) and Cy3. Around 10 fields of view were captured for each preparation. The center position of fluorescent spots was identified by their local maxima intensity using a manually defined threshold in ImageJ software. Red-green spots were considered colocalized if the distance between their centers was below three pixels (∼200 nm). To calculate the percentage of WT-lacZ gRNA (red spots) that colocalized with an MS2-tagged RNA (green spots), the number of red spots that colocalized with a green spot was divided by the total number of WT-lacZ gRNA, assuming that each virion contains two copies of viral RNA (19).
Total internal reflection fluorescence microscopy (TIRFM) of cells
TIRFM of FISH-stained cells was achieved using a TIRF microscope (based on an inverted TE Eclipse microscope (Nikon)) equipped with a Plan Fluor 100×/1.49 NA TIR objective (Nikon) and an EMCCD camera (ImagEM) and controlled using MetaMorph software (MRI facilities). Dual-color imaging of MS2-AF488 and lacZ-Cy3 labeled RNA was achieved by exciting with 491 nm and 561 nm lasers, respectively, and appropriate filters were selected to separate green and red emissions (500–520 nm and 600–660 nm, respectively). The green and red channels were aligned prior to imaging using two-color fluorescent beads of 200 nm diameter (Thermo Fisher Scientific). Imaging was performed on cells that exhibited similar fluorescence intensities in the red and green channels upon episcopic illumination, to ensure similar expression of both plasmids. Illumination was then switched to TIRF mode to excite selectively molecules within ∼100 nm of the cover glass. Image acquisition was performed by illuminating with each wavelength in the TIRF mode for 6 s using a 300 ms integration time. Each set of images was then stacked using the average intensity and corrected for background using the rolling ball background subtraction algorithm (29) in ImageJ, with a radius of five pixels according to the size of spots.
For colocalization analysis, the center positions of fluorescent spots were identified by their local maxima using a manually defined threshold in ImageJ. The colocalization of red-green spots was determined by calculating the distances between spots of one color to their mutual closest neighboring spot of the other color using R software (http://www.r-project.org), with distances below 360 nm considered as colocalized. The percentage of colocalized spots was calculated by dividing the number of colocalized red spots by the total number of red spots ×100. To discriminate between specific and random colocalization for each cell, the center positions of the green spots were shifted successively in the x-axis from 0 to ±12 pixels relative to the other color and the number of colocalized spots between the shifted and the original image was calculated at each shift position as before. The values where the decay of colocalization reached a plateau (shifts of 8–12 pixels) were averaged to estimate randomness. This way, a specific degree of colocalization was calculated by subtracting the random value from the original colocalization value.
3D-structured illumination microscopy (3D-SIM)
3D-SIM of FISH-labeled cells was performed using an OMX-V3 microscope (General Electrics) equipped with a 100× oil-objective, 405, 488 and 561 nm lasers and the corresponding dichroic and filter sets and four EMCCD cameras (MRI facilities). The far red channel used to detect WGA-AF647 was excited using the 561 nm laser line, which excites the absorption tail of the dye. This allowed us to profit from the 3D structured illumination pattern provided by our 561 nm laser line which is currently not available on our 642 nm laser line and, therefore, obtain 3D-SIM images in the far red channel. Emission was optimally detected using a 684/40 nm bandpass filter. The multi-camera design of our OMX allows this unusual experimental setup. Due to the intense signal obtained with WGA staining, image acquisition in these conditions provided enough fluorescence signals for our purpose, which was used to delimitate the plasma membrane in images. Image stacks of the whole cell depth (10 μm) were acquired with a Z-step size of 125 nm. Reconstruction and alignment of the 3D-SIM images was carried out with softWoRx v 5.0 (General Electrics). 100 nm green fluorescent beads (Life Technologies) were used to measure the optical transfer function (otf) used for the 405 and 488 channels, and 100 nm red fluorescent beads (Life Technologies) were used to measure the otf used for the 561 channel. 170 nm TetraSpeck beads (Life Technologies) were employed to measure the offsets in the axial direction and the supplied target slide was used to calibrate X–Y image registration. All SIM data were checked to ensure they were of sufficient quality for analysis using the SIMCheck plugin (30).
3D-SIM image analysis
Segmentation of FISH spots and 3D colocalization analysis
3D-SIM images were scaled to 16-bit prior to segmentation. Segmentation of FISH spots in the green and red channels was achieved using the 3D Spot Segmentation plugin in ImageJ (31). The center positions of segmented spots in each channel were determined with subpixel precision using the 3D Objects Counter plugin and were then used to calculate the Euclidian 3D distances between spots of one color and their mutual nearest neighboring spot of the other color using R software (http://www.r-project.org). The spots of one color within a distance ≤160 nm to their mutual nearest neighboring spot of the other color were considered as colocalized, based on the distribution of nearest neighboring 3D distances (NNdist) obtained for an RNA that contained both tags on the same molecule (NV-lacZ-MS2 in Supplementary Figure S2) and in accordance with the resolution limits of the microscope (32). More details on spot segmentation, colocalization analysis and computer simulations are provided in Supplementary Information.
Attribution of spots to different regions of interest (ROIs)
The total number of spots of each color, as well as the number of colocalized spots, were enumerated within different regions of interest (plasma membrane region (PMr), cytosol and nucleus) using three sequential 2D masks through substacks of 15 slices (1.8 μm depth) beginning 1.8 μm away from the coverslip. By limiting the measurements to this cell depth and adapting the ROIs at each set of five slices, we ensured confinement of cell compartments within the defined regions of interest. To create a 2D mask containing the PMr, individual cells were manually delineated and their contour identified with the help of WGA staining. First, a mask of the cell contour was generated by applying an automated threshold to the maximum intensity projection of the WGA image stack, followed by repeated closing of the binary image. The outline of the cell contour was then enlarged five pixels outward and shrunk 45 pixels inward from the cell contour to create a band of ∼2 μm width containing the PM. The cell nucleus was delimited with the help of DAPI staining and the cytosol region was obtained by subtracting the PM and nucleus masks to the cell mask image.
Cross-number and brightness (N&B) analysis
HeLa cells, cultured in DMEM/F12 medium without phenol red 15 mM Hepes (non-fluorescent medium) and supplemented with 10% FBS and 1% PenStrep (Sigma), were transfected using jetPEI® reagent (Polyplus) and seeded in a six-well plate containing 25 mm diameter #1.5 glass coverslips pre-treated with collagen-I. 4 × 105 HeLa cells were co-transfected with 400 ng of MS2-/BoxB-tagged HIV-1 and/or NV plasmids and 20 ng of the plasmids encoding the corresponding fluorescent coat proteins (MS2-cherry and λN-GFP) with an RNA:fluorescent protein ratio of 1:10. Cells were imaged 24 h post-transfection (pt) in a cell chamber with 1 mL of non-fluorescent medium supplemented with 1% FBS.
We used two-photon excitation to simultaneously excite the two fluorophores using a single femtosecond pulsed infrared laser tuned to 980 nm (MAI-TAI, Spectra-Physics, Santa Clara, CA, USA). Laser scanning and fluorescence detection were performed using an ALBA 5 system (ISS, Champain, IL, USA), a Zeiss Axiovert 200 microscope stage, and a 60× Plan-Apochromat 1.4NA oil immersion objective. The emitted fluorescence was detected on two separate avalanche photodiode detectors with an appropriate dichroic 580LP (chroma, AHF analysentechnik, Tuebingen; DE)) and emission filters to separate the green (eGFP) (530/43, Chroma) and red (mCherry) (653/95, Chroma) emissions. The excitation volume was calibrated as diffraction-limited two-photon PSF using 60 nM Fluorescein in Tris pH9 with a w0 value of 340 nm and a z0 value of 1.4 μm. For Cross N&B data acquisition, we collected 50 frames of 20×20 μm size (256 Pxl X 256 Pxl). The laser power was 16 mW and the laser dwell time 32 μs. During the 50 images stack acquisition, photobleaching was always below 10%. The data were analyzed using in house software written in IgorPro.
Scanning cross number and brightness extends fluorescence cross-correlation spectroscopy (FCCS) to two-dimensional cellular images by a covariance analysis of the raster-scanned fluorescence fluctuations in two channels (33,34) (extended information is provided in Supplementary Information). In two-color mode (here using GFP and mCherry fluorescent protein fusions for each of two interacting partners), calculation of the covariance between the intensity fluctuations in the green and red detection channels (ch1 and ch2, respectively) allows evaluation of the cross-brightness (Bcc) or covariance of the intensities (34).
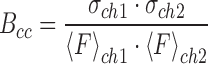 |
where σ and 〈F〉 are the variance and the average of the fluorescence intensity signal in each channel, respectively. Non-zero Bcc is only observed in the case of co-diffusion of the fluorescent molecules present in the sample at the observed pixels, and hence is an absolute quantitative measure of protein hetero-complex formation. Because shot noise between detectors does not co-vary, the Bcc is a particularly sensitive and reliable indicator of heterologous protein interactions.
Statistics
The statistical significance of data was evaluated using the Student's-t-test in GraphPad Prism. P values are represented as follows: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
RESULTS
Two-color RNA labeling system to study HIV-1 gRNA dimerization
First, we used fluorescence in situ hybridization (FISH) to visualize gRNA heterodimers in cells and virions. To detect single HIV-1 gRNA molecules in two colors, the 5′-end of the lacZ gene (Figure 1A, WT-lacZ) or 24 repeats of the bacteriophage MS2 stem-loop (Figure 1A, WT-MS2), were introduced within the pol gene of HIV-1 clone pNL4-3, replacing the reverse transcriptase and the beginning (∼40%) of integrase. Thereby, the introduced tags are present in unspliced RNAs only. For safety consideration, pNL4-3 also harbors an inactivating deletion in the envelope gene (env) (Supplementary Materials and Methods). These constructs express Gag and all the accessory and regulatory proteins including Vpu, essential for efficient virus release from cells (35). However, despite the fact that the protease sequence of pol was intact, Western blot analysis revealed intracellular accumulation of Gag intermediates (p41MACAp2 and p25CAp2) and no evidence for virion-associated p24CA (Figure 1B). Thus, Gag protein processing was significantly impaired by the introduced sequences (Figure 1B). The production of tagged gRNAs was first monitored in cells and virions by RT-PCR at 24 h pt. Co-expression of both constructs in HeLa cells produced similar amounts of WT-lacZ and WT-MS2 gRNA and both gRNAs were packaged into viruses with similar efficiencies (Figure 1C). We also generated a control plasmid encoding a non-viral RNA tagged with 24xMS2 (NV-MS2, Figure 1A) that does not code for viral proteins (Figure 1B, lane 1) and is not encapsidated into viruses (Figure 1C, right).
Figure 1.

Dual-RNA labeling system and FISH analysis of heterodimeric RNA in virions. (A) Schematic representation of the constructions used in the study. HIV-1 tagged genomes were constructed by replacing a portion of the pol gene in NL4-3Δenv plasmid with 5′-lacZ (WT-lacZ) or 24xMS2 (WT-MS2) sequences to allow specific detection of unspliced RNA (positions and numbers indicated in gray refer to HIV-1 DNA nucleotides). NV-MS2 encodes a non-viral RNA containing 24xMS2 repeats from a pcDNA3 backbone. (B) Western-blotting of protein extracts from HeLa cells and purified virions from culture supernatants at 24 h pt with WT-lacZ, WT-MS2 or NV-MS2 plasmids. The expression of Gag (55kDa), Vpu (16 kDa) and Actin (42 kDa) is shown. The quantification of three independent blots revealed that cells transfected with the LacZ construct produced 84 ± 30% of Gag produced with the MS2 construct (P = 0.4). (C) RT-PCR analysis of tagged RNAs in the cells and viruses used for FISH analyses. For dual-WT RNA detection, the RT was initiated with the same primers followed by a PCR step with two sense primers (one specific for lacZ and one for MS2) together with a common antisense primer in the same PCR reaction, generating two amplicons with specific sizes. Lanes 1 and 2 in the agarose gel correspond to the amplification of WT-lacZ and WT-MS2 plasmids, respectively (n ≥ 3). NV-MS2 is detected by another RT-PCR system using specific primers (n = 2). (D) Representative images of lacZ- (red) and MS2-tagged (green) RNAs visualized by single-molecule FISH of virus preparations. Images were obtained with a 100×-oil objective in a wide-field fluorescence microscope. Scale bar is 10 μm. The lower images show the center positions of identified spots. (E) Ratio between the number of green and red fluorescent spots. (F) Percentage of WT-lacZ RNA colocalized with WT-MS2 or NV-MS2 RNA. The fraction of bars filled in red represent the remaining proportion of WT-lacZ gRNA engaged in homodimers. Data are the mean ± SEM of three independent transfections (counting ≥1000 red spots per sample) and the significance of differences with control (NV-WT) was assessed using an unpaired Student's t-test (****P ≤ 0.0001).
Detection of MS2- and lacZ-tagged RNAs by single molecule FISH was achieved using fluorescent DNA probes that specifically hybridize to each tag (MS2-AF488 and lacZ-Cy3, respectively; see Materials and Methods). A good signal-to-noise ratio with bright detection of fluorescence spots representing labeled RNAs and no cross-reactivity and/or spectral cross-talk was observed (Supplementary Figure S1).
A previous study showed that most HIV-1 virus particles contain two copies of gRNA (19). To determine the frequency of dual-RNA co-packaging, aliquots of the viruses produced by cells expressing similar amounts of WT-lacZ and WT-MS2 gRNA (as used in Figure 1C) were examined by FISH (Figure 1D). We quantified the colocalization between red and green spots, indicative of MS2/lacZ–RNA heterodimers. Consistent with RT-PCR (Figure 1C), similar amounts of each tagged gRNA were detected in virus preparations, whereas NV-MS2 (green) was mostly absent (Figure 1E). We found 47 ± 0.9% of WT-lacZ gRNA colocalized with WT-MS2 gRNA (Figure 1F). As expected, colocalization of both gRNAs occurred with a frequency similar to that predicted by the Hardy–Weinberg equilibrium (experimental/predicted = 0.9 ± 0.1), supporting random assortment of the two tagged HIV-1 genomes into viral particles (19).
Colocalization of gRNAs at the plasma membrane visualized by TIRFM
Convincing evidence suggests that gRNAs are selected for packaging as pre-formed dimers, indicating that dimers should already be present at virus assembly sites (5,26,36). Thus, we first studied the colocalization of MS2-/lacZ-tagged gRNAs at the plasma membrane (PM), the main assembly site of HIV-1. To this end, HeLa cells co-transfected with WT-lacZ and WT-MS2 (or NV-MS2) plasmids were labeled by FISH and specific fluorescent signals near the cell surface were examined by TIRFM (Figure 2A). Similar numbers of red and green spots were found at the PM of cells co-expressing HIV-1 plasmids (Figure 2B), whereas the non-viral RNA localized there less efficiently. To measure specific colocalization of red and green spots, we developed a quantitative analysis based on van Steensel's methodology (37) to remove random colocalization due to spot density. To this end, successive shifts of the green channel, relative to the other color, were applied for each cell and the values where the decay of colocalization reached a plateau (shifts of 8–12 pixels) were averaged to estimate a threshold for random colocalization (Figure 2C). Of note, ∼14% of the WT-lacZ gRNA specifically colocalized with WT-MS2 gRNA at the PM, a ∼9-fold increase compared to that obtained with a NV RNA (Figure 2D). In view of the higher colocalization rates found in virions (47%, Figure 1F), these results support the notion that both monomeric and dimeric gRNAs localized at the PM and the selective packaging of dimeric gRNAs leads to an enrichment of the heterodimer frequency in virions.
Figure 2.

Analysis of gRNA colocalization at the PM by two-color TIRFM. (A) Representative TIRFM images of HeLa cells co-transfected with WT-lacZ (red) and WT-MS2 or NV-MS2 (green) plasmids for 24 h, obtained by TIRFM. The lower images show the centers of red spots (red) and those that colocalized with a green spot in yellow. Scale bars are 10 μm. (B) Ratio of green to red spots (**P ≤ 0.01). (C) Plot showing the decay of colocalized WT-lacZ RNA (red) with increasing shifts of the green image in both directions of the x-axis. The asymptotic part of the curve is used to define unspecific colocalization for each cell. (D) Percentage of specific colocalization (with respect to WT-lacZ RNA) calculated by subtracting the unspecific colocalization rate (averaged from x-shifts of ±8–12 pixels) to the original one for each image. All data correspond to the mean ± SEM of at least 30 cells from two independent transfections. The significance of differences with control (NV-WT) was assessed using an unpaired Student's t-test (****P ≤ 0.0001).
3D-super-resolution microscopy reveals colocalized gRNAs in the cytosol
We next examined whether dimerization occurs before the gRNA reaches the PM by performing a compartmentalized 3D colocalization analysis of the cell using super-resolution microscopy. 3D Structured Illumination Microscopy (3D-SIM) was performed on an OMX-V3 microscope (see Materials and Methods). The resolution of this system is twice that of a state-of-the-art wide-field microscope in each dimension, resulting in an 8-fold effective reduction of the observation volume (32). It is therefore well suited for the study of such small structures in the 3D space.
Cells were labeled by FISH, as above, and the nucleus and membranes were counterstained with DAPI and WGA-AF647, respectively. Super-resolution images (Figure 3A) were analyzed over a cell depth of 2 μm, where different regions of interest (ROI) were defined: the PM region (PMr), the cytosol and the nucleus (see Materials and Methods). By adapting the different ROIs in the 3D-space, we ensured a specific compartmentalized analysis. The colocalization of red-green spots was estimated by calculating the mutual nearest neighboring 3D distances (NNdist) between the centers of spots of different colors for the entire cell and two spots were considered colocalized if their NNdist was ≤160 nm, corresponding to the resolution limits of the microscope (32). Moreover, as described for TIRF images, the specific colocalization was calculated by removing the random colocalization relative to signal density for each cell (see Materials and Methods for details). The robustness of the analysis was demonstrated by comparing a NV RNA harboring the two tags on the same molecule (NV-lacZ-MS2, positive control) with two singly tagged NV RNAs (NV-lacZ+NV-MS2, negative control), which reports the biological probability of two RNAs to encounter each other in the different compartments or to be imaged within the same voxel due to resolution limits (Supplementary Figure S2A–C). Of note, the value of random colocalization estimated by this method was similar to that predicted by the theoretical probability formula and by computer simulation of random spots in two-color stacks, given the number of spots and volume of the cell (Supplementary Figure S2D).
Figure 3.

3D-SIM study of HIV-1 gRNA colocalization in cells. (A–D) Representative 3D-SIM images of HeLa cells co-transfected with WT-lacZ+WT-MS2 (A, C) or WT-lacZ+NV-MS2 (B, D) for 24 h (A, B) or 16 h (C, D) and labeled by FISH with MS2 (green) and lacZ probes (red). The nucleus and membranes are stained with DAPI (blue) and WGA (magenta), respectively. Image stacks of the whole cell depth were acquired with a Z-step size of 125 nm, reconstructed and shown as maximal intensity projections of 3–5 Z-slices. The orthogonal views shown for the first image (A, upper left) illustrate the cell depth considered for quantitative analyses (dotted white lines). Blow ups correspond to zoom in areas of the image (white squares). For each cell, the center positions of red spots and those that colocalized with a green spot are shown in red and yellow, respectively. The PMr, cytosol (C) and nucleus (N) were defined (white lines) for analysis. Scale bars are 10 μm. (E) Specific colocalization values normalized to the negative control WT-lacZ+NV-MS2 RNA are presented as the mean ± SEM of 30 cells from at least two independent transfections. The significance of differences with controls (NV-WT or NV-NV) was assessed using an unpaired Student's t-test (****P ≤ 0.0001).
At 24 h pt, HIV-1 gRNA mainly localized at the PMr and cytosol of the cell and similar amounts of red and green fluorescent spots could be detected (Figure 3A and Supplementary Figure S3A). Colocalization analysis of 3D-SIM images revealed that WT-lacZ gRNA specifically colocalized ∼14-times more with the WT-MS2 than with the NV-MS2 RNA at the PMr (Figure 3E). Interestingly, a lower but specific and significant level of gRNA colocalization was detected in the cytosol, with a ∼6-fold increase in WT-WT gRNA colocalization compared to NV-WT RNA. The higher frequencies observed at the PMr versus the cytosol could be explained by selective targeting of dimeric gRNA to the PM by Gag leading to dimer accumulation and/or stabilization at the PM, or by additional dimer formation at this site. Furthermore, we cannot exclude the possibility that some dimers formed at the PM are subsequently released into the cytosol.
There is some indirect evidence suggesting that the gammaretrovirus, murine leukemia virus (MLV), could form gRNA dimers in the nucleus (38,39). Therefore, we further performed 3D-SIM analyses at a shorter time (16 h pt), when most RNA is still localized in the nucleus before its Rev-mediated nuclear export (Figure 3C and D and Supplementary Figure S3B). No significant differences were observed between WT-WT, WT-NV or NV-NV specific colocalization rates in the nucleus (Figure 3E), suggesting that dimerization is not initiated in this compartment. Thus, our results provide direct evidence of spatial association of HIV-1 gRNA in the cytosol, suggesting that dimerization already takes place in this compartment.
Cross-number and brightness imaging of gRNAs in the cytosol of live cells
To further elucidate whether gRNA interactions could be detected in living cells, we used fluorescence cross-correlation spectroscopy, a two-color imaging-based technique that reports on molecular interactions of diffusing molecules (33,34). The number and brightness (N&B) modality uses the variance of the pixel-by-pixel fluorescence signal in a stack of raster-scanned images to deconvolve this signal into the number N of fluorescent particles (i.e. their concentration) and their individual molecular brightness ϵ (that reports on their stoichiometry) (see Materials and Methods). When extended to the two-color mode, the covariance of fluorescence intensities in two channels allows the calculation of the cross-brightness (Bcc), a direct measure of interaction between two fluorescently labeled fusion proteins of different colors. As compared to classical (point-by-point) Fluorescence Correlation Spectroscopy (FCS), scanning N&B allows for a strong reduction of the photobleaching, while providing spatially-resolved information of the molecular concentrations and interactions, but does not report on the diffusion coefficient of the fluorescent species.
To enable real-time two-color visualization of gRNA in living cells, we used the MS2 (40) and λN-BoxB (41) systems, which take advantage of the specific recognition of target sequences in the RNA (MS2 or BoxB) by their respective fluorescently fused RNA-binding protein (MCP-mCherry or λN-GFP, respectively). Both systems have been used successfully together without exhibiting cross-reactivity (42,43). Both fusion proteins contain a nuclear localization signal that confines the unbound chimera to the nucleus, facilitating visualization of RNA–protein complexes in the cytosol. Concomitant with WT-MS2 (described above), we created the WT-BoxB construct by inserting 16 repeats of the BoxB sequence into the NL4-3 HIV-1 clone and we also generated NV plasmids to serve as controls: NV-BoxB and NV-MS2 as negative controls and NV-BoxB-MS2 to be used as a positive control for cross-correlation (co-diffusion) (Figure 4A). Thus, HeLa cells were co-transfected with WT-MS2 and WT-BoxB, together with MCP-mCherry and λN-GFP, and the virus production by these constructs in the presence or the absence of coat proteins was verified by western-blotting (Supplementary Figure S4). Live cells were imaged by two-photon fluorescence microscopy at 24 h pt. A stack of 50 images with a pixel dwell time of 32 μs was taken (Figure 4B) and a pixel-based analysis was performed in the cytosol of 30 cells, over 5 days of independent experiments. We chose to represent the distribution of individual brightness (ϵGFP and ϵmCherry) obtained for all the pixels (Figure 4C and D) to account for the inherent molecular stoichiometry heterogeneity of the sample due to: unbound coat proteins, saturation of RNA binding sites (i.e. up to 48 MCP-mCherry proteins can bind to the 24xMS2 repeats) or oligomerization of RNA-protein complexes. Interestingly, cross-brightness analysis, which only reports on fluorescent proteins that are simultaneously detected in both channels, allowed us to detect a large amount of diffusing complexes of high brightness (Figure 4D). This contribution to the cross-brightness signal was probably due to the presence in the cytosol of WT-MS2+WT-BoxB gRNA complexes. To verify this hypothesis, the same analysis was applied to NV-MS2+NV-BoxB transfected cells, and to NV-BoxB-MS2 transfected cells (Figure 4C and D). For the negative control, the Bcc value was significantly reduced compared to WT-WT (Figure 4D), especially for its highest values reporting mainly the fluorescent proteins bound to RNA, while the individual ϵGFP and ϵmCherry values did not change significantly (Figure 4C). In striking contrast, the Bcc histogram obtained for the positive control was similar to that measured for WT-WT (as well as the ϵGFP and ϵmCherry histograms, as expected). Although the fraction of interacting species could not be quantified due to the broad distribution of brightness observed, our results indicate that, in the case of HIV-1 constructs, a significant number of gRNA dimers or higher order oligomers are detected in the cytosol of living cells, significantly more than when a non-viral construct is used. Thus, by using a fluorescence cross-correlation approach, we could confirm the presence of direct interactions between HIV-1 gRNAs in the cytosol of living cells.
Figure 4.

Cross-N&B analysis of HIV-1 gRNA dimerization in the cytosol of living cells. (A) Schematic representation of the constructions used in this study. (B) Representative image of a HeLa cell transfected with WT-MS2, WT-Box B, MCP-mCherry and λN-GFP, obtained by 2-photon laser scanning microscopy, and used for N&B analysis (top : GFP channel, bottom : mCherry channel). Scale bar is 2 μm. Apparent brightness (ϵGFP and ϵmCherry) (C) and Cross Brightness (Bcc) (D) pixel frequency histograms for WT-MS2+WT-BoxB (red disks), NV-MS2+NV-BoxB (negative control, black squares) and NV-BoxB-MS2 (positive control, blue triangles). The high frequency of pixels with a high Bcc for the WT-MS2+WT-Box B construct reflects the interactions between these two HIV-1 gRNAs in the cytosol. Experiments were performed over five independent days and on average measurements on 30 different cells.
Intact SL1 is required for gRNA colocalization in virions and cells
The SL1 structure of the gRNA plays a central role in the dimerization-dependent packaging and its apical loop (DIS) initiates gRNA dimerization (18,44–46). Therefore, we generated a series of HIV-MS2 mutants with deleted SL1 (delSL1-MS2), deleted DIS (delDIS-MS2) or abrogated DIS palindrome (mutDIS-MS2) (Figure 5A) that retain the same coding ability as WT-MS2 (Supplementary Figure S5A). We then challenged the capacity of WT gRNA to associate with a gRNA lacking SL1 or the DIS palindrome. To this end, WT-lacZ was co-transfected with delSL1-MS2, delDIS-MS2 or mutDIS-MS2 plasmids for 24 h and FISH performed as above. As expected, FISH analysis of virions revealed a severe defect in the encapsidation of delSL1-MS2 gRNA and to a lesser extent, of DIS mutants (Figure 5B), whereas both gRNAs were similarly expressed in cells (Supplementary Figure S5B). Importantly, the colocalization of WT-lacZ with delSL1-MS2 gRNA was strongly impaired, leading to preferential encapsidation of WT-lacZ homodimers (Figure 5C). Alterations in the DIS palindrome also decreased, but to a lesser extent, the percentage of colocalized gRNA in virions (Figure 5C). Altogether, these results support that dimers with intact SL1 are preferentially packaged.
Figure 5.

Role of SL1-DIS in gRNA colocalization in virions and cells. (A) Schematic representation of the mutants used in this study. In WT-lacZ, SL1 is shown with its palindromic DIS loop (GCGCGC). The complete SL1 was deleted in delSL1-MS2 while only the DIS was deleted in delDIS-MS2. The DIS was changed to CCCCCC sequence in mutDIS-MS2 mutant. (B, C) Quantitative FISH analyses of purified virions produced by HeLa cells co-transfected with WT-lacZ and delSL1-MS2, delDIS-MS2 or mutDIS-MS2 plasmids for 24 h. The ratio of green to red spots (B) and the percentage of colocalized WT-lacZ RNA (C) are averaged from three independent transfections with ≥1000 red spots/sample. (D) Representative 3D-SIM images of cells. The lower images show the centers of red spots (red) and those that colocalized with a green spot in yellow. Scale bars are 10μm. (E) The graph shows the specific colocalization as the percentage of WT-WT at the PMr and cytosol. Bars are the mean ± s.e.m. of 30 cells. In all graphs, the significance of differences with control (NV-WT) was assessed using an unpaired Student's t-test (*P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001). At the PMr, a significant difference of delSL1 with the other mutants was found and is therefore indicated.
To further determine whether the mutations impaired gRNA heterodimerization in the cell, we investigated the colocalization of WT and SL1/DIS mutants in cells by 3D-SIM (Figure 5D). Similar numbers of red/green spots were found in the cytoplasm (Supplementary Figure S6). Colocalization analyses of delSL1-MS2-, delDIS-MS2- or mutDIS-MS2- with WT-lacZ gRNA revealed strong impairments at the PMr with colocalization scores at 18%, 30% and 31% of WT-MS2+WT-lacZ, respectively (Figure 5E). Interestingly, the mutations also decreased colocalization levels in the cytosol with levels for delSL1-MS2, delDIS-MS2 or mutDIS-MS2 with WT-LacZ gRNA at 40%, 58% and 51% of WT-WT, respectively (Figure 5E). These results strongly support that specifically colocalized gRNAs resulted from the dimerization process, and reveal the importance of SL1/DIS in gRNA pairing in the cytosol. Moreover, the fact that mutations were more deleterious on gRNA colocalization at the PMr than in the cytosol suggests that homodimers with an intact SL1 (WT-lacZ) may be preferentially targeted to the PM diluting the MS2-RNA mutants there. In addition, in line with previous observations (26), this stronger effect at the PM could result from a cumulative effect on the de novo dimer formation at this site. However, additional sequences other than DIS in SL1 could also contribute to targeting dimers to the PM.
Gag promotes gRNA colocalization in the cytosol and at the PM
Gag promotes gRNA packaging during virus assembly (27). While its role in gRNA dimerization was well-accepted (47,48), however, whether Gag picks already formed dimers for packaging remains unclear. To decipher the role of Gag in dimerization, we performed gRNA colocalization analyses in the absence of Gag. The expression of Gag in tagged gRNA was inactivated by inserting the tag at the 5′-end of the gag gene, resulting in GagKO-MS2 and GagKO-lacZ mutants (Figure 6A and Supplementary Figure S7A, lane 1). HeLa cells were co-transfected as before with GagKO-lacZ and GagKO-MS2 (or NV-MS2) plasmids and the subcellular RNA colocalization was assessed by 3D-SIM (Figure 6B). As with WT gRNA, GagKO gRNA mainly localized in the cytosol at 24 h pt (Supplementary Figure S8). We found that GagKO-lacZ colocalized 3-fold more efficiently with GagKO-MS2 than with NV-MS2 RNA at the PMr and 4-fold more efficiently in the cytosol (Figure 6B). Even if some gRNA dimers could be observed in the absence of Gag, the frequency of two GagKO gRNAs colocalized at the PMr was strongly impaired compared to that observed with two WT gRNAs encoding Gag (∼5-fold decrease, Figure 3E), though this was less pronounced in the cytosol (∼1.5-fold decrease, Figure 3E). To further understand the role of Gag, we performed trans-complementation experiments by providing Gag protein in trans (GagWT) with a 1:1.5 molar ratio between GagKO and GagWT plasmids. The expression of plasmids and the production of virus-like particles (VLPs) were controlled by western blotting (Supplementary Figure S7A, lanes 2–3) and the relative levels of the two tagged GagKO gRNAs in total cells were checked by RT-PCR assays (Supplementary Figure S7B). FISH analysis of the VLPs formed by Gag in trans showed that both tagged GagKO gRNAs colocalized with a similar frequency to that found for WT-WT (Supplementary Figure S7C), indicating that the tag in gag did not disturb pairing abilities of the GagKO gRNAs.
Figure 6.

3D-SIM analysis of the role of Gag in HIV-1 gRNA colocalization. (A) Schematic representation of the constructs used in this study. To generate GagKO mutants, lacZ or MS2 tags were inserted within the capsid domain of the gag gene (position indicated by a dashed gray line). The insertion prevents the synthesis of Gag and Gag-Pol polyproteins but maintains the expression of all the regulatory and accessory proteins. Representative 3D-SIM images of cells co-transfected with GagKO-lacZ (red) and GagKO-MS2 (green) in the absence (B) or the presence (C) of Gag (GagWT or GagdelNC). The lower images show the centers of red spots (red) and those that colocalized with a green spot in yellow. Scale bars are 10μm. The graphs show the specific colocalization normalized to the control (GagKO-lacZ+NV-MS2 in B) or to GagKO-MS2+GagKO-lacZ (C). Data are the mean ± SEM of 30 cells shown for each condition. The significance of differences with control (NV-GagKO in B) or with GagKO-GagKO (C) was assessed using an unpaired Student's t-test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). No significant difference was found between the trans-complementation with GagWT or GagdelNC in the cytosol.
Importantly, within cells, the expression of Gag in trans increased the colocalization rates of GagKO gRNAs by 3.2-fold at the PMr and 2.5-fold in the cytosol (Figure 6C). Because Gag binds the gRNA via its NC domain (9,49–53), we next examined the trans-complementing ability of a Gag lacking its NC domain. To this end, trans-complementation experiments were performed with a Gag deleted of the entire NC domain (GagdelNC), which was unable to release VLPs (Supplementary Figure S7A, lanes 4 and 5). Immunofluorescence (IF) analysis of Gag in cells showed that GagdelNC, unlike GagWT, appeared diffuse throughout the cytoplasm with sparse oligomerization signals at the PM, as previously reported (23,54) (Supplementary Figure S7D). Unexpectedly, 3D-SIM analyses of the cytosol revealed that GagdelNC could partially restore the colocalization rates of GagKO gRNAs by 1.8-fold (Figure 6C), indicating that NC is not the sole determinant for Gag function in gRNA dimerization. In contrast, deletion of NC was detrimental on the capability of Gag to rescue GagKO gRNA colocalization at the PM (Figure 6C), which is consistent with the deficiency of GagdelNC signals observed at the PM by IF. Taken together, these results demonstrate a major role of Gag in gRNA dimerization in the cytosol and in the recruitment of gRNA dimers, via the NC domain, and their targeting to the PM.
DISCUSSION
Where, when, and how the viral RNA find each other and dimerize remains unknown. Many assumptions about these long-standing questions remain untested. Up to now, the prevailing answers have been mainly based on biochemical data showing the importance of Gag and the DIS RNA signal in dimerization mechanism. Convincing evidence indicates that HIV-1 genomes are selected for packaging as pre-formed dimers in cells (5,36,55–59), resulting in the release of virions that contain mainly dimeric gRNA (19).
Here, we have combined advanced imaging technologies to obtain a three-dimensional vision of the subcellular orchestration of HIV-1 gRNA dimerization. We used a single molecule dual-color RNA labeling approach to allow the study of dimerization by means of RNA colocalization. As demonstrated in virions, tagged gRNAs were co-packaged in random proportions, as determined previously for HIV-1 (19), confirming their dimerization and packaging abilities. Then, we performed multicolor 3D-SIM imaging of FISH-labeled cells to analyze at super-resolution the spatial relationship between these tagged RNAs. Since fortuitous RNA interactions are dependent on the density of molecules, we followed a rigorous methodology to discriminate specific from random colocalization and the biological relevance of specifically colocalized gRNAs was further reinforced using non-viral RNA controls. Although at this scale one cannot determine whether RNA colocalization resulted from direct interactions, the fact that gRNA colocalization frequencies depended on the integrity of the principal RNA elements (SL1/DIS) involved in in vitro and in vivo dimerization strongly suggests that colocalizing signals correspond to dimeric gRNA. In addition, our cross-N&B data showed the correlated diffusion of gRNA molecules in a sub-femtoliter excitation volume in living cells, demonstrating direct physical interaction(s) between gRNAs.
Our 3D study of gRNA colocalization throughout its traffic in the cell provides novel insights to the long-standing question of where HIV-1 gRNA dimerization takes place. We found no significant colocalization of gRNAs in the nucleus, suggesting that HIV-1 RNA partner selection may occur outside of the nucleus, which is consistent with a previous study based on recombination analyses between heterozygous gRNAs in fused cells (56). It is of note that a different mechanism has been purposed for other retroviruses, such as MLV, in which dimerization appears to occur in the nucleus in a co-transcriptional manner (39).
In striking contrast, a significant percentage of colocalized gRNAs appeared in the cytosol where dimerizable gRNA molecules are likely in minority among full-length RNA molecules, since the RNA is exported in the form of monomers to the cytosol. Close examination by 3D-SIM and N&B microscopies exclude the presence of dimeric gRNAs in large cytosolic foci. The lack of a specific subcytosolic confinement of gRNA would be plausible with the low frequencies of dimers observed, since interactions between genomes could then depend on random encounters of RNAs diffusing in the cytosol. Indeed, most viral gRNA diffuses in a random manner in the cytosol (23). Importantly, direct interactions between dynamic genomes in the cytosol were confirmed by cross-N&B imaging of living cells. Taken together, these results strongly suggest that the cytosol is the primary site for HIV-1 RNA dimerization and we hypothesize that dimerization in the cytosol occurs at low frequencies provoked by random encounters between diffusing genomes.
Moreover, we found that the frequency of dimers was enriched near the PM. This is consistent with a recent study reporting that ∼13–34% of the HIV-1 RNAs that reach the PM are recruited into assembling particles (60).This enrichment likely results from the accumulation of gRNA dimers at the PM during virus assembly, which lasts ∼30 min (36,61,62). Thus, dimerization could initiate in the cytosol and gRNA dimers be then specifically targeted to the PM. It is likely that additional dimer formation takes place partially at the PM, as reported by a very recent study, where the authors imaged the interaction of gRNAs at the PM supporting that some monomeric gRNAs were targeted to the PM and dimerized there (26). However, our results correlated with imaging studies performed by Jouvenet et al. on the dynamics of MS2-tagged gRNAs by TIRFM, revealing that most gRNAs arrived at the PM in a single peak of fluorescence (27).
Despite the large number of studies, our current understanding of the role of Gag in intracellular dimerization and how it selects HIV-1 gRNA dimers for packaging remains limited. Previous studies have shown that NC stabilizes the dimer through extended base pairing of the kissing-loop sequence (17) and that this process probably occurs during viral particle maturation (46). Transformation of immature dimers into mature dimers requires processing of Pr55gag/Pr160gag-pol into NCp9 or NCp7 protein that contributes via the proximal zinc finger and linker sequences of NC (48,63). In the present context of impaired Gag processing, the presence of mature dimers is unlikely, since there may be too few of these proteins in the cytosol. It is difficult to compare the nature of the fluorescent dimers with those previously observed by more standard techniques into newly released virions (46,48), since fragile dimers may be detectable by fluorescence assays but not after RNA has been extracted from virions and electrophoresed on agarose gels.
Within cells, Gag-gRNA interactions have been demonstrated in the cytosol (4,5) and at perinuclear sites (64) but without discriminating whether Gag interacts with monomeric or dimeric RNA. Our imaging studies reveal for the first time that Gag plays a role in facilitating RNA dimerization in the cytosol, since its expression in trans significantly enhanced the colocalization frequencies of gRNAs. Dimer accumulation at the PM would then be favoured by coating with Gag proteins that likely stabilize the RNA–RNA interaction during virus assembly. Although a possible role for the NC in facilitating the conformational RNA switch that leads to dimerization and encapsidation has been proposed (48,65), our findings suggest that the contribution of the NC domain (if any) in initiating gRNA dimerization is limited. Conspicuously, an intact NC domain is strongly required for the localization of gRNA dimers at the PM, suggesting a role for the NC domain in the specific targeting of dimers at the PM. This is consistent with the role of GagNC in the recruitment of the gRNA to the PM, enabling the oligomerization and assembly of Gag proteins (23,51). Indeed, a recent study by Kutluay et al. using crosslinking-immunoprecipitation (CLIP) sequencing has revealed a dramatic change in Gag-RNA binding specificity, coincident with high-order multimerization at the plasma membrane, that contributes to selective packaging of gRNA (66). Interestingly, a new role in targeting Gag to the PM has been ascribed to the GagMA. By its transient binding of tRNA, which blocks Gag recruitment to intracellular membranes, the MA domain facilitates the recruitment of Gag to the PM (66). Gag must select the viral gRNA among the bulk of other RNAs in cells. An original in vitro study with the entire Gag protein indicated that Gag specifically recognizes the internal loop and the lower part of SL1 motif in the gRNA, which acts as the primary binding site (9). Indeed, deleting the entire SL1 produced larger effects on gRNA packaging than deletion or substitutions of its apical loop (DIS) (this study and (8,44,57)). The highly structured leader gRNA is sensitive to structural changes. Thus, it is probable that complete deletion of SL1 hairpin has a more global impact on the structure of the gRNA than smaller deletion or substitutions of the external loop. However, we have found that an intact DIS palindrome is required for optimal pairing abilities of gRNAs in the cytosol. This correlates with previous results indicating that DIS plays similar roles in formation of immature dimers in protease-inactive and WT-HIV-1, as if these immature dimers were characterized by identical intermolecular interactions (48).
These results suggest that DIS contributes to gRNA dimerization while other sequences in SL1 preferentially promote trafficking of gRNA to the PM. Consistent with this notion, the GagNC domain, that is known to bind SL1, appeared more critical for gRNA colocalization at the PM than in the cytosol, supporting a role for GagNC in recruiting and targeting gRNA dimers to the PM. Finally, it is noteworthy to mention that neither the absence of Gag nor the deletion of the DIS/SL1 signals were sufficient to completely abrogate dimerization in cells, suggesting that redundant sequences or currently unidentified mechanisms may act with less efficiency, as has been previously suggested by others (67–70). In the literature, we found that it is difficult to block HIV-1 RNA dimerization. Mainly because our understanding of dimerization is still clouded by a lack of consensus about precise requirements of the cis- and trans-acting elements. Indeed, in several hands, inactivating SL1 left fully dimeric the gRNA isolated from virions (55,59,67,71). While the role of SL1 is well-accepted, it is admitted that other dimerization sites exist on the gRNA. Similarly, Gag is not the unique RNA partner. In vitro studies showed that like NC, Vif chaperones the gRNA dimerization (72). Furthermore, since the production of gRNA dimers is cell-dependent (67,73), cellular factors are likely implicated. For instance, Staufen, which is incorporated into virions, binds gRNA and facilitates its packaging and thus has been proposed to be implicated in gRNA dimerization (74,75).
In conclusion, our report of cytosolic HIV-1 gRNA dimers does not support the classical notion of gRNA dimerization initiating in sites of high concentration of RNA molecules, for example at transcription sites as proposed for MLV (39), at perinuclear/centrosomal sites (64) or at the PM where all the viral components converge for virus assembly (26). Our data are consistent with an HIV-1 gRNA dimerization process initiating in the cytosol, preferentially via DIS sequences and orchestrated by Gag. This is supported by the existence of RNA-interacting Gag oligomers in the cytosol (4,5). Then, dimeric genomes would be selected by GagNC via SL1 and targeted to the PM to be packaged into virions. Nevertheless, our study also suggests that further, unidentified determinants, in addition to NC and SL1, are implicated in the dimerization process. The roles of these putative elements need future investigation.
Supplementary Material
Acknowledgments
We are grateful to the Montpellier RIO Imaging staff and in particular to J. Matteos-Langerak for technical assistance. Many thanks to Dr I. Robbins for critical reading of the manuscript. We acknowledge France-BioImaging infrastructure supported by the French National Research Agency (ANR-10-INSB-04-01, « Investments for the future ») and the GIS « IBiSA : Infrastructures en Biologie Sante et Agronomie ».
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Grant of French National Agency for Research on AIDS and Viral Hepatitis (ANRS) to M.M and ANRS fellowhip to M.F.
Conflict of interest statement. None declared.
REFERENCES
- 1.Höglund S., Ohagen A., Goncalves J., Panganiban A.T., Gabuzda D. Ultrastructure of HIV-1 genomic RNA. Virology. 1997;233:271–279. doi: 10.1006/viro.1997.8585. [DOI] [PubMed] [Google Scholar]
- 2.Temin H.M. Sex and recombination in retroviruses. Trends Genet. 1991;7:71–74. doi: 10.1016/0168-9525(91)90272-R. [DOI] [PubMed] [Google Scholar]
- 3.Kafaie J., Song R., Abrahamyan L., Mouland A.J., Laughrea M. Mapping of nucleocapsid residues important for HIV-1 genomic RNA dimerization and packaging. Virology. 2008;375:592–610. doi: 10.1016/j.virol.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Hendrix J., Baumgartel V., Schrimpf W., Ivanchenko S., Digman M.A., Gratton E., Krausslich H.-G., Muller B., Lamb D.C. Live-cell observation of cytosolic HIV-1 assembly onset reveals RNA-interacting Gag oligomers. J. Cell Biol. 2015;210:629–646. doi: 10.1083/jcb.201504006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kutluay S.B., Bieniasz P.D. Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathog. 2010;6:e1001200. doi: 10.1371/journal.ppat.1001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jouvenet N., Neil S.J.D., Bess C., Johnson M.C., Virgen C.A., Simon S.M., Bieniasz P.D. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006;4:e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Didierlaurent L., Racine P.J., Houzet L., Chamontin C., Berkhout B., Mougel M. Role of HIV-1 RNA and protein determinants for the selective packaging of spliced and unspliced viral RNA and host U6 and 7SL RNA in virus particles. Nucleic Acids Res. 2011;39:8915–8927. doi: 10.1093/nar/gkr577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houzet L., Paillart J.C., Smagulova F., Maurel S., Morichaud Z., Marquet R., Mougel M. HIV controls the selective packaging of genomic, spliced viral and cellular RNAs into virions through different mechanisms. Nucleic Acids Res. 2007;35:2695–2704. doi: 10.1093/nar/gkm153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abd El-Wahab E.W., Smyth R.P., Mailler E., Maurel S., Morichaud Z., Marquet R., Bernacchi S., Vivet-Boudou V., Hijnen M., Jossinet F., Mak J., Paillart J.C., Marquet R. Specific recognition of the HIV-1 genomic RNA by the Gag precursor. Nat Commun. 2014;5:4304. doi: 10.1038/ncomms5304. [DOI] [PubMed] [Google Scholar]
- 10.Laughrea M., Jette L. HIV-1 genome dimerization: Kissing-loop hairpin dictates whether nucleotides downstream of the 5′ splice junction contribute to loose and tight dimerization of human immunodeficiency virus RNA. Biochemistry. 1997;36:9501–9508. doi: 10.1021/bi970862l. [DOI] [PubMed] [Google Scholar]
- 11.Paillart J.C., Marquet R., Skripkin E., Ehresmann B., Ehresmann C. Mutational analysis of the bipartite dimer linkage structure of human immunodeficiency virus type 1 genomic RNA. J. Biol. Chem. 1994;269:27486–27493. [PubMed] [Google Scholar]
- 12.Skripkin E., Paillart J.C., Marquet R., Ehresmann B. Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc. Natl. Acad. Sci. U.S.A. 1994;91:4945–4949. doi: 10.1073/pnas.91.11.4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jossinet F., Paillart J.C., Westhof E., Hermann T., Skripkin E., Lodmell J.S., Ehresmann C., Ehresmann B., Marquet R. Dimerization of HIV-1 genomic RNA of subtypes A and B: RNA loop structure and magnesium binding. RNA. 1999;5:1222–1234. doi: 10.1017/s1355838299990982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paillart J.C., Westhof E., Ehresmann C., Ehresmann B., Marquet R. Non-canonical interactions in a kissing loop complex: the dimerization initiation site of HIV-1 genomic RNA. J. Mol. Biol. 1997;270:36–49. doi: 10.1006/jmbi.1997.1096. [DOI] [PubMed] [Google Scholar]
- 15.Weixlbaumer A., Werner A., Flamm C., Westhof E., Schroeder R. Determination of thermodynamic parameters for HIV DIS type loop-loop kissing complexes. Nucleic Acids Res. 2004;32:5126–5133. doi: 10.1093/nar/gkh841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ennifar E., Paillart J.-C., Bernacchi S., Walter P., Pale P., Decout J.-L., Marquet R., Dumas P. A structure-based approach for targeting the HIV-1 genomic RNA dimerization initiation site. Biochimie. 2007;89:1195–1203. doi: 10.1016/j.biochi.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Muriaux D., Fosse P., Paoletti J. A kissing complex together with a stable dimer is involved in the HIV-1Lai RNA dimerization process in vitro. Biochemistry. 1996;35:5075–5082. doi: 10.1021/bi952822s. [DOI] [PubMed] [Google Scholar]
- 18.Moore M.D., Fu W., Nikolaitchik O., Chen J., Ptak R.G., Hu W.S. Dimer initiation signal of human immunodeficiency virus type 1: its role in partner selection during RNA copackaging and its effects on recombination. J. Virol. 2007;81:4002–4011. doi: 10.1128/JVI.02589-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J., Nikolaitchik O., Singh J., Wright A., Bencsics C.E., Coffin J.M., Ni N., Lockett S., Pathak V.K., Hu W.-S. High efficiency of HIV-1 genomic RNA packaging and heterozygote formation revealed by single virion analysis. Proc. Natl. Acad. Sci. U.S.A. 2009;106:13535–13540. doi: 10.1073/pnas.0906822106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paillart J.C., Shehu-Xhilaga M., Marquet R., Mak J. Dimerization of retroviral RNA genomes: an inseparable pair. Nat. Rev. Microbiol. 2004;2:461–472. doi: 10.1038/nrmicro903. [DOI] [PubMed] [Google Scholar]
- 21.Bann D.V., Parent L.J. Application of live-cell RNA imaging techniques to the study of retroviral RNA trafficking. Viruses. 2012;4:963–979. doi: 10.3390/v4060963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boireau S., Maiuri P., Basyuk E., de la Mata M., Knezevich A., Pradet-Balade B., Bäcker V., Kornblihtt A., Marcello A., Bertrand E. The transcriptional cycle of HIV-1 in real-time and live cells. J. Cell Biol. 2007;179:291–304. doi: 10.1083/jcb.200706018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J., Grunwald D., Sardo L., Galli A., Plisov S., Nikolaitchik O.a., Chen D., Lockett S., Larson D.R., Pathak V.K., et al. Cytoplasmic HIV-1 RNA is mainly transported by diffusion in the presence or absence of Gag protein. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E5205–E5213. doi: 10.1073/pnas.1413169111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kemler I., Meehan A., Poeschla E.M. Live-cell coimaging of the genomic RNAs and Gag proteins of two lentiviruses. J. Virol. 2010;84:6352–6366. doi: 10.1128/JVI.00363-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levesque K., Halvorsen M., Abrahamyan L., Chatel-Chaix L., Poupon V., Gordon H., DesGroseillers L., Gatignol A., Mouland A.J. Trafficking of HIV-1 RNA is Mediated by Heterogeneous Nuclear Ribonucleoprotein A2 Expression and Impacts on Viral Assembly. Traffic. 2006;7:1177–1193. doi: 10.1111/j.1600-0854.2006.00461.x. [DOI] [PubMed] [Google Scholar]
- 26.Chen J., Rahman S.A., Nikolaitchik O.A., Grunwald D., Sardo L., Burdick R.C., Plisov S., Liang E., Tai S., Pathak V.K., et al. HIV-1 RNA genome dimerizes on the plasma membrane in the presence of Gag protein. Proc. Natl. Acad. Sci. U.S.A. 2015;113:E201–E208. doi: 10.1073/pnas.1518572113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jouvenet N., Simon S.M., Bieniasz P.D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. U.S.A. 2009;106:19114–19119. doi: 10.1073/pnas.0907364106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt U., Basyuk E., Robert M.C., Yoshida M., Villemin J.P., Auboeuf D., Aitken S., Bertrand E. Real-time imaging of cotranscriptional splicing reveals a kinetic model that reduces noise: implications for alternative splicing regulation. J. Cell Biol. 2011;193:819–829. doi: 10.1083/jcb.201009012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sternberg S.R. Biomedical image-processing. Computer. 1983;16:22–34. [Google Scholar]
- 30.Ball G., Demmerle J., Kaufmann R., Davis I., Dobbie I.M., Schermelleh L. SIMcheck: a Toolbox for Successful Super-resolution Structured Illumination Microscopy. Sci. Rep. 2015;5:15915. doi: 10.1038/srep15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ollion J., Cochennec J., Loll F., Escude C., Boudier T. TANGO: a generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics. 2013;29:1840–1841. doi: 10.1093/bioinformatics/btt276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schermelleh L., Heintzmann R., Leonhardt H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010;190:165–175. doi: 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Digman M.A., Dalal R., Horwitz A.F., Gratton E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 2008;94:2320–2332. doi: 10.1529/biophysj.107.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Digman M.A., Stakic M., Gratton E. Raster image correlation spectroscopy and number and brightness analysis. Methods Enzymol. 2013;518:121–144. doi: 10.1016/B978-0-12-388422-0.00006-6. [DOI] [PubMed] [Google Scholar]
- 35.Neil S.J., Eastman S.W., Jouvenet N., Bieniasz P.D. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006;2:e39. doi: 10.1371/journal.ppat.0020039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jouvenet N., Bieniasz P.D., Simon S.M. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature. 2008;454:236–240. doi: 10.1038/nature06998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Steensel B., van Binnendijk E.P., Hornsby C.D., van der Voort H.T., Krozowski Z.S., de Kloet E.R., van Driel R. Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J. Cell Sci. 1996;109:787–792. doi: 10.1242/jcs.109.4.787. [DOI] [PubMed] [Google Scholar]
- 38.Kharytonchyk S.a., Kireyeva A.I., Osipovich A.B., Fomin I.K. Evidence for preferential copackaging of Moloney murine leukemia virus genomic RNAs transcribed in the same chromosomal site. Retrovirology. 2005;2:3. doi: 10.1186/1742-4690-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maurel S., Mougel M. Murine leukemia virus RNA dimerization is coupled to transcription and splicing processes. Retrovirology. 2010;7:64. doi: 10.1186/1742-4690-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bertrand E., Chartrand P., Schaefer M., Shenoy S.M., Singer R.H., Long R.M. Localization of ASH1 mRNA particles in living yeast. Mol. Cell. 1998;2:437–445. doi: 10.1016/s1097-2765(00)80143-4. [DOI] [PubMed] [Google Scholar]
- 41.Daigle N., Ellenberg J. LambdaN-GFP: an RNA reporter system for live-cell imaging. Nat. Methods. 2007;4:633–636. doi: 10.1038/nmeth1065. [DOI] [PubMed] [Google Scholar]
- 42.Lange S., Katayama Y., Schmid M., Burkacky O., Brauchle C., Lamb D.C., Jansen R.P. Simultaneous transport of different localized mRNA species revealed by live-cell imaging. Traffic. 2008;9:1256–1267. doi: 10.1111/j.1600-0854.2008.00763.x. [DOI] [PubMed] [Google Scholar]
- 43.Martin R.M., Rino J., Carvalho C., Kirchhausen T., Carmo-Fonseca M. Live-cell visualization of pre-mRNA splicing with single-molecule sensitivity. Cell Rep. 2013;4:1144–1155. doi: 10.1016/j.celrep.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clever J.L., Wong M.L., Parslow T.G. Requirements for kissing-loop-mediated dimerization of human immunodeficiency virus RNA. J. Virol. 1996;70:5902–5908. doi: 10.1128/jvi.70.9.5902-5908.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Russell R.S., Roldan A., Detorio M., Hu J., Wainberg M.A., Liang C. Effects of a single amino acid substitution within the p2 region of human immunodeficiency virus type 1 on packaging of spliced viral RNA. J. Virol. 2003;77:12986–12995. doi: 10.1128/JVI.77.24.12986-12995.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song R., Kafaie J., Yang L., Laughrea M. HIV-1 viral RNA is selected in the form of monomers that dimerize in a three-step protease-dependent process; the DIS of stem-loop 1 initiates viral RNA dimerization. J. Mol. Biol. 2007;371:1084–1098. doi: 10.1016/j.jmb.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 47.Feng Y.X., Campbell S., Harvin D., Ehresmann B., Ehresmann C., Rein A. The human immunodeficiency virus type 1 Gag polyprotein has nucleic acid chaperone activity: possible role in dimerization of genomic RNA and placement of tRNA on the primer binding site. J. Virol. 1999;73:4251–4256. doi: 10.1128/jvi.73.5.4251-4256.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jalalirad M., Laughrea M. Formation of immature and mature genomic RNA dimers in wild-type and protease-inactive HIV-1: differential roles of the Gag polyprotein, nucleocapsid proteins NCp15, NCp9, NCp7, and the dimerization initiation site. Virology. 2010;407:225–236. doi: 10.1016/j.virol.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 49.D'Souza V., Summers M.F. How retroviruses select their genomes . Nat Rev Microbiol. 2005;3:643–655. doi: 10.1038/nrmicro1210. [DOI] [PubMed] [Google Scholar]
- 50.Darlix J.L., Godet J., Ivanyi-Nagy R., Fosse P., Mauffret O., Mely Y. Flexible nature and specific functions of the HIV-1 nucleocapsid protein. J. Mol. Biol. 2011;410:565–581. doi: 10.1016/j.jmb.2011.03.037. [DOI] [PubMed] [Google Scholar]
- 51.de Rocquigny H., El Meshri S.E., Richert L., Didier P., Darlix J.-L., Mély Y. Role of the nucleocapsid region in HIV-1 Gag assembly as investigated by quantitative fluorescence-based microscopy. Virus Res. 2014;193:78–88. doi: 10.1016/j.virusres.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 52.Heng X., Kharytonchyk S., Garcia E.L., Lu K., Divakaruni S.S., LaCotti C., Edme K., Telesnitsky A., Summers M.F. Identification of a minimal region of the HIV-1 5′-leader required for RNA dimerization, NC binding, and packaging. J. Mol. Biol. 2012;417:224–239. doi: 10.1016/j.jmb.2012.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y., Barklis E. Nucleocapsid protein effects on the specificity of retrovirus RNA encapsidation. J. Virol. 1995;69:5716–5722. doi: 10.1128/jvi.69.9.5716-5722.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grigorov B., Decimo D., Smagulova F., Pechoux C., Mougel M., Muriaux D., Darlix J.L. Intracellular HIV-1 Gag localization is impaired by mutations in the nucleocapsid zinc fingers. Retrovirology. 2007;4:54. doi: 10.1186/1742-4690-4-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berkhout B., van Wamel J.L. Role of the DIS hairpin in replication of human immunodeficiency virus type 1. J. Virol. 1996;70:6723–6732. doi: 10.1128/jvi.70.10.6723-6732.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moore M.D., Nikolaitchik O.A., Chen J., Hammarskjold M.L., Rekosh D., Hu W.S. Probing the HIV-1 genomic RNA trafficking pathway and dimerization by genetic recombination and single virion analyses. PLoS Pathog. 2009;5:e1000627. doi: 10.1371/journal.ppat.1000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paillart J.C., Berthoux L., Ottmann M., Darlix J.L., Marquet R., Ehresmann B., Ehresmann C. A dual role of the putative RNA dimerization initiation site of human immunodeficiency virus type 1 in genomic RNA packaging and proviral DNA synthesis. J. Virol. 1996;70:8348–8354. doi: 10.1128/jvi.70.12.8348-8354.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Russell R.S., Liang C., Wainberg M.A. Is HIV-1 RNA dimerization a prerequisite for packaging? Yes, no, probably? Retrovirology. 2004;1:23. doi: 10.1186/1742-4690-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sakuragi J., Ueda S., Iwamoto A., Shioda T. Possible role of dimerization in human immunodeficiency virus type 1 genome RNA packaging. J. Virol. 2003;77:4060–4069. doi: 10.1128/JVI.77.7.4060-4069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sardo L., Hatch S.C., Chen J., Nikolaitchik O., Burdick R.C., Chen D., Westlake C.J., Lockett S., Pathak V.K., Hu W.S. Dynamics of HIV-1 RNA near the plasma membrane during virus assembly. J Virol. 2015;89:10832–10840. doi: 10.1128/JVI.01146-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ivanchenko S., Godinez W.J., Lampe M., Kräusslich H.-G., Eils R., Rohr K., Bräuchle C., Müller B., Lamb D.C. Dynamics of HIV-1 assembly and release. PLoS Pathog. 2009;5:e1000652. doi: 10.1371/journal.ppat.1000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ku P.I., Miller A.K., Ballew J., Sandrin V., Adler F.R., Saffarian S. Identification of pauses during formation of HIV-1 virus like particles. Biophys J. 2013;105:2262–2272. doi: 10.1016/j.bpj.2013.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ohishi M., Nakano T., Sakuragi S., Shioda T., Sano K., Sakuragi J. The relationship between HIV-1 genome RNA dimerization, virion maturation and infectivity. Nucleic Acids Res. 2011;39:3404–3417. doi: 10.1093/nar/gkq1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poole E., Strappe P., Mok H.P., Hicks R., Lever A.M. HIV-1 Gag-RNA interaction occurs at a perinuclear/centrosomal site; analysis by confocal microscopy and FRET. Traffic. 2005;6:741–755. doi: 10.1111/j.1600-0854.2005.00312.x. [DOI] [PubMed] [Google Scholar]
- 65.Huthoff H., Berkhout B. Two alternating structures of the HIV-1 leader RNA. RNA. 2001;7:143–157. doi: 10.1017/s1355838201001881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kutluay S.B., Zang T., Blanco-Melo D., Powell C., Jannain D., Errando M., Bieniasz P.D. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell. 2014;159:1096–1109. doi: 10.1016/j.cell.2014.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hill M.K., Shehu-Xhilaga M., Campbell S.M., Poumbourios P., Crowe S.M., Mak J. The dimer initiation sequence stem-loop of human immunodeficiency virus type 1 is dispensable for viral replication in peripheral blood mononuclear cells. J. Virol. 2003;77:8329–8335. doi: 10.1128/JVI.77.15.8329-8335.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chamanian M., Purzycka K.J., Wille P.T., Ha J.S., McDonald D., Gao Y., Le Grice S.F.J., Arts E.J. A cis-acting element in retroviral genomic RNA links Gag-Pol ribosomal frameshifting to selective viral RNA encapsidation. Cell Host Microbe. 2013;13:181–192. doi: 10.1016/j.chom.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Das A.T., Vrolijk M.M., Harwig A., Berkhout B. Opening of the TAR hairpin in the HIV-1 genome causes aberrant RNA dimerization and packaging. Retrovirology. 2012;9:59. doi: 10.1186/1742-4690-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nikolaitchik O.A., Dilley K.A., Fu W., Gorelick R.J., Tai S.H., Soheilian F., Ptak R.G., Nagashima K., Pathak V.K., Hu W.S. Dimeric RNA recognition regulates HIV-1 genome packaging. PLoS Pathog. 2013;9:e1003249. doi: 10.1371/journal.ppat.1003249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sakuragi J.I., Panganiban A.T. Human immunodeficiency virus type 1 RNA outside the primary encapsidation and dimer linkage region affects RNA dimer stability in vivo. J. Virol. 1997;71:3250–3254. doi: 10.1128/jvi.71.4.3250-3254.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Henriet S., Sinck L., Bec G., Gorelick R.J., Marquet R., Paillart J.C. Vif is a RNA chaperone that could temporally regulate RNA dimerization and the early steps of HIV-1 reverse transcription. Nucleic Acids Res. 2007;35:5141–5153. doi: 10.1093/nar/gkm542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Laughrea M., Shen N., Jette L., Darlix J.L., Kleiman L., Wainberg M.A. Role of distal zinc finger of nucleocapsid protein in genomic RNA dimerization of human immunodeficiency virus type 1; no role for the palindrome crowning the R-U5 hairpin. Virology. 2001;281:109–116. doi: 10.1006/viro.2000.0778. [DOI] [PubMed] [Google Scholar]
- 74.Mouland A.J., Mercier J., Luo M., Bernier L., DesGroseillers L., Cohen E.A. The double-stranded RNA-binding protein Staufen is incorporated in human immunodeficiency virus type 1: evidence for a role in genomic RNA encapsidation. J. Virol. 2000;74:5441–5451. doi: 10.1128/jvi.74.12.5441-5451.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chatel-Chaix L., Clement J.F., Martel C., Beriault V., Gatignol A., DesGroseillers L., Mouland A.J. Identification of Staufen in the human immunodeficiency virus type 1 Gag ribonucleoprotein complex and a role in generating infectious viral particles. Mol. Cell. Biol. 2004;24:2637–2648. doi: 10.1128/MCB.24.7.2637-2648.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.