


Abstract
Allele-specific copy number analysis (ASCN) from next generation sequencing (NGS) data can greatly extend the utility of NGS beyond the identification of mutations to precisely annotate the genome for the detection of homozygous/heterozygous deletions, copy-neutral loss-of-heterozygosity (LOH), allele-specific gains/amplifications. In addition, as targeted gene panels are increasingly used in clinical sequencing studies for the detection of ‘actionable’ mutations and copy number alterations to guide treatment decisions, accurate, tumor purity-, ploidy- and clonal heterogeneity-adjusted integer copy number calls are greatly needed to more reliably interpret NGS-based cancer gene copy number data in the context of clinical sequencing. We developed FACETS, an ASCN tool and open-source software with a broad application to whole genome, whole-exome, as well as targeted panel sequencing platforms. It is a fully integrated stand-alone pipeline that includes sequencing BAM file post-processing, joint segmentation of total- and allele-specific read counts, and integer copy number calls corrected for tumor purity, ploidy and clonal heterogeneity, with comprehensive output and integrated visualization. We demonstrate the application of FACETS using The Cancer Genome Atlas (TCGA) whole-exome sequencing of lung adenocarcinoma samples. We also demonstrate its application to a clinical sequencing platform based on a targeted gene panel.
INTRODUCTION
Large-scale sequencing studies including The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) projects have generated tens of thousands whole-genomes (WGS) and whole-exomes (WES) of tumor-normal sample pairs. Allele-specific copy number analysis can greatly extend the utility of sequencing data beyond the identification of mutations. We present FACETS (Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing), an allele-specific copy number analysis (ASCN) pipeline and open-source software for next generation sequencing (NGS) data.
ASCN analysis has several major advantages over conventional total copy number analysis. First, it provides a much more comprehensive identification of copy number aberrations including copy-neutral loss-of-heterozygosity (LOH) events not detectable by analyzing total copy number alone. Thus genome-wide LOH pattern can be systematically evaluated. In addition, while conventional analysis typically converts total copy number ratio into qualitative copy number states (high versus low level gains, shallow versus deep losses, normal), ASCN analysis can be used to precisely annotate the genome for the detection of homozygous deletions, heterozygous deletions, copy-neutral LOH, allele-specific gains and amplifications with corresponding integer copy number. Furthermore, ASCN analysis provides more accurate estimates of tumor purity and ploidy. The output can be used for enhanced clonal heterogeneity analyses of somatic point mutations.
Early ASCN methods were primarily developed for copy number array platforms (1–4). More recently, a number of ASCN methods have been developed for next generation sequencing data, building on different analytical strategies. Patchwork (5) segments the genome based on total read count and then estimates the allele-specific copy number within each segment. The limitation lies in that segmenting total read count alone does not provide the complete picture and will inevitably miss certain events such as copy neutral LOH (Figure 1). Falcon (6) provides a joint segmentation procedure using a Binomial process for the allelic read count from heterozygous SNP loci. Several other methods including TITEN (7) further considered tumor purity and clonal heterogeneity to enhance the accuracy of copy number analysis by using various probabilistic modeling approaches including Bayesian mixture model (8), Hidden Markov Model (7) or other maximum likelihood methods (9,10).
Figure 1.

Joint segmentation identifies copy number neutral loss-of-heterozygosity (LOH) event. Top panel shows copy number log-ratio of total sequence read count in the tumor to that in the normal along genomic positions on chromosome 6 from a whole-exome sequencing of a lung cancer patient sample. Second panel shows the allelic log-odds-ratio of the variant allele read counts in the tumor/normal pair revealing a copy-neutral LOH event on 6p.
FACETS provides several unique contributions over existing methods. For one, we employ a non-parametric joint segmentation approach based on a Hotelling T2 statistic by directly combining the total and allele-specific read counts which does not depend on any model assumption and provides a fast implementation to search for change points in the genome.
ASCN analysis typically uses a SNP-based approach as allelic imbalance can only be measured at heterozygous sites. Nearly all ASCN methods for sequencing data uses read count information from heterozygous sites only. However, heterozygous sites are subject specific and sparse which leads to information loss on total copy number. Thus, a systematic enumeration of allele specific read counts from all SNPs, be it heterozygous or homozygous, provides full information on both total and allele specific copy numbers. Furthermore we also use read counts from a set of pseudo-SNPs (non polymorphic loci) along the target intervals so that regions with large gaps between consecutive SNPs are represented in total copy number analysis. In total copy number analysis, a moving window approach in which the read depths are averaged over all the loci within the window is used commonly. However, since the independent units of measurement are DNA fragments this leads to serial correlation as the same fragment contributes to read depth at several loci. Our approach of using read counts at SNPs that are sufficiently spaced from one another provide a way of obtaining information that have negligible serial correlation since each fragment is usually mapped to only one SNP locus. To address the imbalance in the number of loci used for total and allele specific copy numbers we introduce a weighting scheme that is inversely proportional to the overall heterozygous rate in the patient's genome which further enhances the detection of allele-specific alterations.
In addition, the current sequencing analysis methods for allelic imbalances based on B-allele frequency (BAF) has some inherent biases due to differential mapping affinity between the reference and the variant allele. To address this issue, we show that the allelic log-odds-ratio (logOR) metric provides an unbiased estimate of the allelic ratio by leveraging the paired tumor-normal sequencing design that cancels out the mapping bias. To obtain allele-specific copy number calls, we devised a Gaussian-non-central χ2 mixture model. Tumor purity, ploidy and clonal heterogeneity are factored in the model to obtain accurate ASCN output and facilitates the identification of subclonal events.
FACETS provides a complete analysis pipeline that include BAM file post-processing steps including library size and GC-normalization, joint segmentation of total and allele-specific signals, and integer copy number calls taking into account of tumor purity, ploidy and clonal heterogeneity, all seamlessly integrated in a single workflow with comprehensive output, integrated visualization, with fast computation to facilitate large-scale application. Figure 2 shows FACETS analysis of a TCGA chromophobe renal cell carcinoma (chRCC) sample (TCGA-KL-8331), revealing multiple chromosomal losses including chromosomes 1, 2, 6, 10, 13, 17, 21 which are signatures of chRCC genome alteration as characterized in the TCGA chRCC study (11). In addition, two major subclonal clusters of losses unique in this tumor sample were further identified that included chr 11, 18 and 22 representing events occurring later in time.
Figure 2.

Integrated visualization of FACETS analysis of whole-exome sequencing data from a TCGA chromophobe renal cell carcinoma sample (TCGA-KL-8331). The top panel displays total copy number log-ratio (logR), and the second panel displays allele-specific log-odds-ratio data (logOR) with chromosomes alternating in blue and gray. The third panel plots the corresponding integer (total, minor) copy number calls. The overall tumor ploidy is estimated to be 1.6, revealing a hypodiploid tumor genome due to the whole-chromosomal losses of multiple chromosomes. The tumor sample purity is estimated to be 0.89. The estimated cellular fraction (cf) profile is plotted at the bottom, revealing both clonal and subclonal copy number events.
Most existing methods are designed for WGS or WES. As targeted panel sequencing is increasingly used in clinical settings to detect ‘actionable’ mutations and copy number alterations toward precision medicine, robust copy number and clonal heterogeneity analysis tools such as FACETS for targeted panel sequencing are needed to further increase the clinical utility of NGS. The software is available at https://sites.google.com/site/mskfacets/.
In this paper, we benchmark our tumor purity and ploidy estimates using the TCGA whole-exome sequencing data in 286 lung adenocarcinoma samples and compared with the estimates from the ABSOLUTE algorithm (12). We show that FACETS can enhance the sensitivity of identifying aneuploid tumors by joint modeling of total and allele-specific pattern. In addition, as shown in Figure 2, FACETS facilitates systematic identification of clonal and subclonal copy number events through a cellular fraction feature in the model. Moreover, accurate, purity-, ploidy- and clonal heterogeneity-adjusted, integer copy number calls will be essential to reliably interpret NGS-based gene copy number calls in clinical sequencing panels. We will demonstrate that using a clinical sequencing sample profiled by the MSK-IMPACT platform (13).
MATERIALS AND METHODS
In the next sections, we discuss our approach for sequencing bias corrections, joint segmentation of total and allelic copy ratio, and methods for integer copy number calls correcting for tumor purity, ploidy and intratumor heterogeneity.
Total copy number log-ratio (logR)
Sequence read count information are first parsed form paired tumor-normal BAM files (Figure 3A). A normalizing constant is calculated for each tumor/normal pair to correct for total library size. Subsampling within 150–250 bp intervals is applied to reduce hypersegmentation in SNP-dense regions of the genome (Figure 3B). logR is then computed from the total read count in the tumor versus normal for all SNPs that have a minimum depth of coverage in the normal. logR provides information on total copy number ratio. Specifically, the expected value of logR can be expressed as
 |
where m* = mΦ + (1 − Φ) and p* = pΦ + (1 − Φ) are parental copy number in the tumor sample rising from a mixed normal (1,1) and aberrant (m,p) copy number genotype with mixing proportion Φ. We term Φ as the cellular fraction associated with the aberrant genotype, which is a function of tumor purity and clonal frequency (for subclonal alterations). The term  (·) denotes systematic bias. Here, we explicitly consider GC-content and use loess regression of logR over GC in 1 kb windows along the genome to estimate the GC-effect on read counts and subtract it from logR. In addition, we note that logR quantifies relative copy number, hence we introduce a constant λ for absolute copy number conversion which will be described in detail later.
(·) denotes systematic bias. Here, we explicitly consider GC-content and use loess regression of logR over GC in 1 kb windows along the genome to estimate the GC-effect on read counts and subtract it from logR. In addition, we note that logR quantifies relative copy number, hence we introduce a constant λ for absolute copy number conversion which will be described in detail later.
Figure 3.

Pre-processing and joint segmentation. (A) Parsing reference and variant allele count for SNP sites from tumor-nomal sequencing BAM files. All SNP sites contribute to total copy log-ratio (logR), and heterozygous sites contribute to allelic logOR. (B) Interval-sampling to reduce local serial dependencies in SNP-dense regions. (C) Joint segmentation logR and logOR and the detection of copy number aberrant regions of the genome. (D) Segment clustering to form groups with the same latent copy number states.
Allelic copy number logOR
Allelic imbalance analysis has been typically based on B-allele (or variant allele) frequency (BAF) in the tumor which informs m*/(m* + p*). In sequencing data, it has been observed that there is a significant bias toward higher mapping rates for the reference allele compared to those for the variant allele at heterozygous loci (14). Such bias can significantly impact allele-specific copy number inference if not corrected. To illustrate, let r denote the relative mapping affinity of the variant allele to the reference allele, and typically r < 1 (mapping biased in favor of the reference allele). As a result, the normal genotype becomes (1,r) instead of (1,1) and the aberrant genotype becomes (m*, rp*) or (p*, rm*) (Table 1). Therefore, it is easy to see that in sequencing data, BAF in fact informs m*/(m* + rp*), which is a biased estimate of B-allele frequency when r ≠ 1. To address this issue, we propose to use the logOR of the variant-allele count in tumor versus normal, which is an unbiased estimate of allelic copy ratio. In particular,
 |
depending on which parental copy the variant allele resides on. Since we do not have phased data, squared logOR is used to infer log2(m*/p*).
Table 1. Illustration of how differential mapping bias affects copy number inference.
| Reference allele on maternal copy | Reference allele on paternal copy | |||
|---|---|---|---|---|
| Reference | Variant | Reference | Variant | |
| Normal | 1 | r | 1 | r |
| Tumor | m* | rp* | p* | rm* |
Joint segmentation
Segmentation analysis identifies regions of the genome that have constant copy number using change point detection methods. Conventional methods (e.g. BIC-seq (15), ExomeCNV (16)) typically perform one-dimensional segmentation using logR alone, or separate application of one-dimensional segmentation to logR and BAF. Yet a truly joint segmentation can significantly improve the precision for change point detection and downstream analysis for estimating tumor purity, ploidy and allele-specific calls.
To address this challenge, we extended the circular binary segmentation (CBS) algorithm (17,18) to a joint segmentation of logR and logOR based on a bivariate Hotelling T2 statistic:
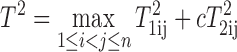 |
where T1ij is the Mann–Whitney statistic comparing the set of observed logR denoted as {X1k: i < k ≤ j} and its complement {X1k: 1 < k ≤ iorj < k ≤ n}. and similarly T2ij is the Mann–Whitney statistic comparing the set of observed logOR denoted as {X2k: i < k ≤ j} and its complement {X2k: 1 < k ≤ iorj < k ≤ n}. In the above, c is a scaling factor that is inversely proportional to the heterozygous rate which will be discussed shortly.
Here, if the maximal statistic is greater than a pre-determined critical value, we declare that a change exists and estimate the change-points as i, j that maximize the statistic. The algorithm iteratively searches for change points between any possible pair of breakpoints and its complement to identify regions of the genome that have constant allele-specific copy number. For each segment, the logR data are summarized using the median of the logR values  and the logOR data are summarized by
and the logOR data are summarized by  which takes the form
which takes the form  where s2 is the estimated variance of logOR.
where s2 is the estimated variance of logOR.
We point out that while logR is defined for all SNPs (both homozygous and heterozygous loci), logOR is only defined for heterozygous loci (het-loci or het-SNPs). This creates a large imbalance between the two in the combined statistic. To address this issue, we introduce a weight that is inversely proportional to the heterozygous rate to increase the het-SNP contributions in subsequent segmentation analysis. Specifically, a scaling factor c is introduced in the T2 statistic. This is empirically set at  where γ is the proportion of het-SNPs in the patient sample. Up-weighing the contribution of logOR for het-SNPs increases the power of detecting allelic imbalances for regions with low frequency of het-SNPs. We denote this a ‘full model’ approach.
where γ is the proportion of het-SNPs in the patient sample. Up-weighing the contribution of logOR for het-SNPs increases the power of detecting allelic imbalances for regions with low frequency of het-SNPs. We denote this a ‘full model’ approach.
Our ‘full model’ approach is distinct from the conventional method in which both logR and BAF are computed for het-loci only. For the whole-exome data we have analyzed, the genome-wide heterozygous rate typically ranges from 10–15%. As such, the het-SNP approach can lead to substantial information loss and reduced power for detecting alterations across the genome. To illustrate, we conducted a down-sampling experiment using two whole-exome samples with high and low tumor purity to assess the sensitivity of detecting genome alterations between the full model and the typical het-SNP approach (Supplementary Figure S1). For low purity tumor samples, the het-SNP approach shows reduced sensitivity at the outset. As genome coverage decreases by down-sampling, the sensitivity of het-SNP only approach for detecting all the altered copy number segments quickly deteriorates while the full model holds up substantially better.
After segmentation, we cluster the segments into groups of the same underlying genotype. Figure 3D shows an example in which a total of 27 segments resulting from the joint segmentation were clustered into four distinct genotype groups. Such clustering reduces the number of latent copy number and cellular fraction states needed in subsequent modeling.
Determine the 2-copy state
As mentioned earlier, logR estimates are proportional to the absolute total copy number up to a location constant λ. For diploid genome, logR = 0 (library size normalized logR) is the location for the 2-copy state. However, aneuploidy can lead to a location shift in the tumor. Therefore, we need to first determine the 2-copy state in a tumor genome and quantify the location shift λ. Without adjusting for the location shift, absolute copy number calls are not possible.
Let us denote the copy number states using total and minor integer copy number (e.g. 1-0 denotes monosomy with total copy number 1 and minor copy number 0). The estimate of λ should correspond to the logR level at which the segments are in 2-1 (normal diploid) or 2-0 (copy-neutral LOH) state. In order to estimate λ, we first note that normal diploid segments should be allelically balanced. Thus, candidate value for λ (referred to as λc) will be obtained from  for segment clusters that have
for segment clusters that have  values close to zero.
values close to zero.
However, note that homozygous deletions (0-0) and balanced gains (4-2, 6-3 etc.) are also allelically balanced and hence will have small  . Since large scale homozygous deletions of multiple genes will not be conducive to cell survival we can eliminate non-focal segments with small
. Since large scale homozygous deletions of multiple genes will not be conducive to cell survival we can eliminate non-focal segments with small  as being homozygous deletions. In addition, for the sake of simplicity we do not consider higher order balanced gains states (6-3, 8-4 etc.) spanning a large part of the genome. Finally, samples in which segments with allelic balance are a small fraction of targeted regions will be flagged and will require a manual review for their λ estimates.
as being homozygous deletions. In addition, for the sake of simplicity we do not consider higher order balanced gains states (6-3, 8-4 etc.) spanning a large part of the genome. Finally, samples in which segments with allelic balance are a small fraction of targeted regions will be flagged and will require a manual review for their λ estimates.
In samples that have large allelically balanced segments, there can be several  values from which λc can be chosen. The samples in Figures 2 and 3C have several balanced segments with
values from which λc can be chosen. The samples in Figures 2 and 3C have several balanced segments with  values with small variation around a single level. The samples in Figures 4A and B have segments with allelic balance at two distinct
values with small variation around a single level. The samples in Figures 4A and B have segments with allelic balance at two distinct  levels (chr11q and chr18 in Figure 4A and parts of chr1 and chr8 in Figure 4B). We group the balanced segments into either one or two distinct levels. For the single level scenario the choice of λc is obvious whereas in the two distinct levels scenario the higher level cannot be normal diploid (since it would imply the lower level is large scale homozygous deletion) and thus λc should the lower one.
levels (chr11q and chr18 in Figure 4A and parts of chr1 and chr8 in Figure 4B). We group the balanced segments into either one or two distinct levels. For the single level scenario the choice of λc is obvious whereas in the two distinct levels scenario the higher level cannot be normal diploid (since it would imply the lower level is large scale homozygous deletion) and thus λc should the lower one.
Figure 4.

Joint analysis of total and allelic copy number pattern to more accurately estimate tumor purity, ploidy and the precise genotypes of the copy number alterations. Two examples (A and B) are presented here to illustrate the use of allelically balanced segments (logR close to zero) to determine the 2-copy state (purple line) and location shift λ in total copy number log-ratio (logR) due to aneuploidy of the tumor. (C) The expected value of logR and logOR as a function of total and minor copy number and cellular fraction Φ are plotted to show the degree of separability among different copy number genotype and cellular fraction. Each line traces the cellular fraction from low (0.1) at the original point close to (0.0) to high (0.9) on the other end of the line. Triangles mark the cellular fraction of 0.5 on each line. The colors represent the minor copy number: 0 is black, 1 is red, 2 is green and 3 is blue. Line types change by total copy number.
We proceed by evaluating whether λc represents normal diploid state or balanced 4 copy state using all segments that are relative losses, i.e. segments with  smaller than the candidate level. If it represents the 2-1 state then the losses are at 1-0 state whereas if it represents the 4-2 state then the losses can be any of 3-0, 3-1, 2-0 or 1-0. We find the best m, p, Φ that fits
smaller than the candidate level. If it represents the 2-1 state then the losses are at 1-0 state whereas if it represents the 4-2 state then the losses can be any of 3-0, 3-1, 2-0 or 1-0. We find the best m, p, Φ that fits  and
and  Segments at 3-1 and 2-0 states in relation to 4-2 level with a clonal Φ is indistinguishabe from segments at 1-0 in relation to 2-1 level with different ϕ. On the other hand single copy loss from 2-1 cannot mimic the relationship between 4-2 and 3-0 or 1-0 states. Thus, λc will be considered to represent 4-2 state if best fit copy numbers for some segments are at 3-0 or 1-0. If all the segments are assigned 3-1 or 2-0 then it will be considered to represent 4-2 if a clonal fit with single ϕ fits as well as subclonal 1 copy loss from diploid with a single subclone fraction. If λc represents 2-1 state then we set λ = λc and if it represents 4-2 state then λ is estimated as the
Segments at 3-1 and 2-0 states in relation to 4-2 level with a clonal Φ is indistinguishabe from segments at 1-0 in relation to 2-1 level with different ϕ. On the other hand single copy loss from 2-1 cannot mimic the relationship between 4-2 and 3-0 or 1-0 states. Thus, λc will be considered to represent 4-2 state if best fit copy numbers for some segments are at 3-0 or 1-0. If all the segments are assigned 3-1 or 2-0 then it will be considered to represent 4-2 if a clonal fit with single ϕ fits as well as subclonal 1 copy loss from diploid with a single subclone fraction. If λc represents 2-1 state then we set λ = λc and if it represents 4-2 state then λ is estimated as the  value corresponding to the 2-0 state.
value corresponding to the 2-0 state.
In Figure 4A, the balanced segments at chr11q and chr18 represent the 2-1 and 4-2 states and although there are several losses and gains the average copy number of the sample is 2 and thus λ is estimated close to zero. In Figure 4B, however, 2-1 state in parts of chromosome 1 is a small fraction of the genome and chr8 at 4-2 is the dominant location of allelic balance. Even ignoring the 2-1 segments in chr1 the procedure can estimate λ at the 2-0 state represented by chr10q since chr9 is at 3-0 state compare to 4-2 level in chr8. The 2-copy state for this sample is significantly shifted below zero due high average copy number of the tumor.
Integer copy number call
In the next step, we obtain integer copy number (major and minor) and the associated cellular fraction estimates for each segment cluster by modeling the expected values of logR and logOR given total (t), and each parental (m,p) copy as a function of a cf parameter Φ, using a combination of parametric and non-parametric methods. This allows us to model both clonal and subclonal events. Figure 4C demonstrates the expected value of logR and logOR as a function of (m,p) and Φ. Note that the curves for most combinations of m and p are distinct and well separated indicating that they can be estimated well provided the cellular fraction is high.
The procedure starts by first obtaining a moment estimate of  , the total copy number for segment cluster i, by
, the total copy number for segment cluster i, by  , where
, where  denote the median logR for segment cluster i corrected for sequence bias and tumor ploidy (λ-normalized). Once the total number is obtained we calculate the allele specific copy numbers m and p and the cellular fraction Φ using the fact that the logOR summary measure
denote the median logR for segment cluster i corrected for sequence bias and tumor ploidy (λ-normalized). Once the total number is obtained we calculate the allele specific copy numbers m and p and the cellular fraction Φ using the fact that the logOR summary measure  is a moment estimate of μ2 which equals log2({mΦ + (1 − Φ)}/{pΦ + (1 − Φ)}).
is a moment estimate of μ2 which equals log2({mΦ + (1 − Φ)}/{pΦ + (1 − Φ)}).
To further refine the initial estimates, we employed a Gaussian-non-central χ2 model with error terms to account for the noise with a clonal structure imposed on the cellular fraction Φ. Specifically, let X1ij denote the logR for SNP loci j in segment cluster i (corrected for sequence bias and location shift) and follow a normal distribution:
 |
where νig is the expected value of logR given the underlying copy number state g taking the form
 |
where tg = mg + pg denotes the total copy number (sum of the two parental copy number) given the underlying copy number state g, Φk denotes the cellular fraction for clonal cluster k, and  is an independent variance parameter. In practice, it is quite reasonable to assume homoscedasticity and set
is an independent variance parameter. In practice, it is quite reasonable to assume homoscedasticity and set  .
.
Furthermore, let X2ij denote the logOR for SNP loci j in segment cluster i and (X2ij/σij)2 follow a non-central chi-squared distribution:
 |
where  is the variance parameter for logOR and
is the variance parameter for logOR and  is the non-centrality parameter in which
is the non-centrality parameter in which
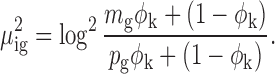 |
Assuming X1ij and X2ij are independent random variables given the underlying copy number state g, the joint data likelihood can then be written as
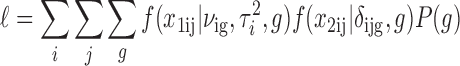 |
where P(g) is the prior probability of the latent copy number state g.
We apply an expectation-maximization (EM) algorithm to maximize the joint data likelihood. It can be viewed as an estimation problem with the latent copy number states as ‘missing’ data. In the E-step of the EM procedure, Bayes theorem is used to compute the posterior probability of segment cluster i being assigned copy number state g given the parameter estimates at the tth iteration:
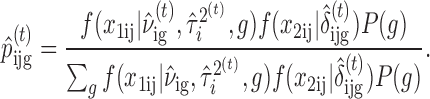 |
In the M-step, we first update the normal and non-central Chi-square distribution parameters
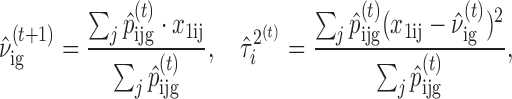 |
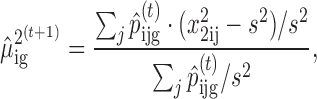 |
where s2 is the sample variance estimate of logOR. After obtaining the estimates of ν and then update the cellular fraction parameter  given
given
 |
where g* is the most likely genotype (with highest posterior probability) given the data and current parameter estimates in the tth iteration. The E-step and M-step are iterated until convergence.
A clonal structure is imposed on the cellular fraction Φk. This is done in a sequential approach where the algorithm starts with a single clonal cluster (k = 1) with cellular fraction parameter Φ1. We then identify segment clusters for which segment cluster-specific estimates is non-trivially lower (at least by 0.05) from the clonally constrained estimates that result in a suboptimal fit under k = 1. These segment clusters with discordant cellular fraction estimates then form a candidate subclonal cluster of events at a lower cellular fraction Φ2, and a model is fitted with the joint likelihood optimized under k = 2. This procedure is iterated until no additional discordance in cellular fraction estimates are found, or a specified maximum k is reached. In the default parameter setting, a maximum k = 5 is allowed although user can change it to a higher number if greater intratumor heterogeneity is expected. In the output,  is the cellular fraction estimate for the clonal events and also the tumor purity by definition, and
is the cellular fraction estimate for the clonal events and also the tumor purity by definition, and  for any subclonal clusters identified in the tumor sample.
for any subclonal clusters identified in the tumor sample.
Figure 5 plots the kernel density of the FACETS estimates of cellular fraction for the copy number alterations detected in the chRCC sample TCGA-KL-8831, revealing three major subclonal clusters. In this tumor sample,  capturing the clonal alterations (losses of chromosomes 1, 2, 6, 10, 13, 17 and 21). A subclonal cluster captured the subsequent loss of chromosomes 11 at
capturing the clonal alterations (losses of chromosomes 1, 2, 6, 10, 13, 17 and 21). A subclonal cluster captured the subsequent loss of chromosomes 11 at  , followed by additional losses of 18 and 22 at
, followed by additional losses of 18 and 22 at  .
.
Figure 5.

Kernel density plot of estimated cellular fraction reveals clonal and subclonal events.
RESULTS
Sequencing data source
We applied FACETS to 268 TCGA lung adenocarcinoma whole-exomes. The sequencing bam files were downloaded from the Cancer Genomics Hub (https://cghub.ucsc.edu/). Each bam file is about 15 GB in size. A pre-processing module that generates sequence count matrix from the sequencing bam file uses samtools/perl/c++ scripts to ensure scaleable and parallelizable implementation. Model fitting, analysis and visualization is done in R statistical programming language which provides a unified front end for analysis and visualization. The ABSOLUTE calls from SNP6.0 array profiling data for the same set of tumor samples published in Zack et al. (2013) (19) were obtained from Synapse (https://www.synapse.org/#!Synapse:syn1703335). The MSK-IMPACT targeted panel sequencing data are obtained from Paik et al. (2015) (20).
Data pre-processing
The input data for FACETS analysis pipeline are aligned sequence bam file with standard base and mapping quality filter. Reference and variant allele read counts were extracted from the bam file for germline polymorphic sites catalogued in the dbSNP and 1000 genome database (∼1.9 million polymorphic positions). For whole-exome seq, we include SNPs in target intervals expanded 50-bases on each side (target overhang). Positions with total read count below a lower depth threshold (e.g. <25 in 50x coverage experiment) or exceed an upper threshold (>1000) (excessive coverage) in the matched normal were removed.
Analysis of the data from HapMap project has revealed that SNPs are not distributed at random across the human genome, but are clustered. Regions with increased local variability and SNP clustering has been associated with recombination hotspots. In high-throughput genotyping arrays, such variation has been correlated with elevated rates of genotype failure and allele dropout (21). In high-throughput sequencing, we show that SNP-dense regions in the genome can cause strong local dependencies in read counts and lead to hyper-segmentation of the genome (Figure 1). To address this issue, we scan all positions by 150–250 bp interval to space out SNP-dense regions and effectively avoid local patterns of strong dependencies. This serial correlation in read counts can cause hyper-segmentation in the downstream steps if not removed.
Read depth ratio between tumor and normal gives information on total copy number. The variant (non-reference) allele frequency at heterozygous loci (germline variant allele frequency >0.25 or <0.75) contain information on allelic imbalance. This pre-processing procedure on average yields ∼350 000 SNP loci that pass these quality filters, and ∼10−15% of them are heterozygous. Homozygous positions will be kept in our analysis to inform total copy number which increases the precision for genotype calls. The MSK-IMPACT platform target all exons and selected introns of 410 cancer genes (<1 million bases) with high uniformity of coverage across targets. The pre-processing procedure yields on average ∼15 000 SNP loci with a similar ∼10–15% heterozygous rate.
Application to TCGA whole-exome sequencing data
Previous TCGA projects have utilized the ABSOLUTE algorithm (12) to determine tumor ploidy and purity. This paradigm works by combining segmented copy number output, together with pre-computed models of recurrent cancer karyotypes, and allelic fraction values for somatic point mutations. We compared FACETS output with the ABSOLUTE output reported in the original TCGA studies (19).
We first looked at the concordance of the segmentation analysis. Here, platform and method differences need to be taken into consideration. First, SNP6.0 array has more even coverage across the genome while whole-exome sequencing may be more sensitive for detecting intragenic changes. The coverage differences have the most effect on the detection of focal changes. Therefore in this analysis we excluded segments less than 1 MB. Secondly, CBS segmentation which segments total copy number was applied in the Zack et al. study (19) for ABSOLUTE input, whereas FACETS implements a joint segmentation of total and allele-specific copy ratios. Bivariate segmentation is more comprehensive and can detect events such as partial chromosomal cn-neutral LOH events that may be missed by a total copy number segmentation approach.
Figure 6A shows the number of segments per tumor sample is relatively comparable between the two methods. Figure 6B further shows the segments are over 90% concordant for segments over 10 MB in length and less so for smaller segments due to platform and method differences as discussed earlier. In this analysis, we define a segment is concordantly detected by both methods if there is more than 70% overlap between the stat and end positions of two segments.
Figure 6.

FACETS analysis of whole-exome sequencing of 286 TCGA lung adenocarcinoma samples. (A) total number of segments per sample from standard CBS segmentation of total copy number versus FACETS joint segmentation of total and allele-specific copy ratios. (B) Proportion of concordantly detected segments between two methods. (C) Comparing FACETS and ABSOLUTE tumor purity estimates. (D) Comapring FACETS and ABSOLUTE ploidy estimates. (E) Bubble plot of FACETS and ABSOLUTE integer copy number calls. The number of concordant (diagonal) and discordant (off diagonal) alterations called are indicated inside each bubble.
Figure 6C and D show that purity and ploidy estimates are highly concordant between the two methods. FACETS identified additional cases of aneuploidy in about 6% of the tumors (green) by incorporating LOH pattern in determining ploidy. Figure 4B is one of such cases where the total and allelic copy ratio together provide evidence for an aneuploidy tumor that was not identified in the original study based on total copy ratio alone. For a small fraction of tumors that FACETS called lower ploidy than that called by ABSOLUTE (orange), they tend to be lower purity samples.
To compare the integer copy number calls, we focused on samples with concordant ploidy calls (difference in ploidy estimates less than 0.5), tumor purity greater than 30%, and segments length greater than 10 MB. Figure 6E shows a high concordance of the integer copy number calls.
Application to targeted cancer gene panel sequencing
Figure 7 shows a FACETS application to the MSK-IMPACT clinical sequencing platform, a hybridization capture-based next-generation sequencing assay for targeted deep sequencing of all exons and selected introns of 410 key cancer genes in FFPE tumor samples (13). This is a stage IV lung squamous cell carcinoma (LUSC) patient sample. This patient genome is highly altered. Some key events include homozygous deletion of CDKN2A, copy-neutral LOH of chromosomes 9, 11 and 17p. Notably from the FACETS output, high level amplification of known oncogenes including CCND1 and PPM1D, both are druggable targets, are annotated with estimated integer copy number. This tumor also showed aneuploidy with an average ploidy estimated at 3.0.
Figure 7.

FACETS analysis of a lung squamous cell carcinoma from MSKCC profiled by MSK-IMPACT targeted cancer gene panel sequencing revealed several putative oncogenic drivers and druggable targets. Tumor purity-, ploidy-corrected FACETS analysis provides more accurate integer copy number calls for the driver genes. Integer copy number above 10 are plotted in log10 scale.
The FACETS estimate of integer copy number (purity-, ploidy-corrected) for PPM1D is 10. By contrast, a conventional PPM1D copy number call based on logR ratio (in this case logR = 1.3) without adjusting for purity and ploidy would be around 5. This difference is potentially clinically significant as to unambiguously identify amplified cancer genes to guide treatment decisions.
DISCUSSION
Comprehensive identification of allele-specific copy number alterations will be invaluable in the search for genomic correlates of clinical outcome and therapeutic targets. In this study, we present FACETS, a unified analysis pipeline and software for joint segmentation and allele-specific copy number analysis with broad applications to NGS platforms. Our method has a number of unique features. We point out that the conventional B-allele-frequency based on sequencing read counts has inherent bias due to mapping affinity toward reference allele. We propose the logOR metric which overcomes such reference bias to provide unbiased estimates of the allelic ratio. The joint segmentation of logR and logOR we developed allows more accurate identification of change points in the genome by directly combing the total and allele-specific read counts. Existing methods use read counts information from heterozygous SNP sites only. We included all SNPs sites. with a weighting scheme that is inversely proportional to the overall heterozygous rate in the patient genome. The combined approach increases the sensitivity and precision for detecting copy number aberrations in the genome especially in low purity samples. Clonal heterogeneity is explicitly considered in our method by introducing a cellular fraction feature associated with segment clusters to allow more accurate inference of ASCNs and facilitate the identification of subclonal events. A normal-non-central χ2 mixture model is used to jointly model the total and allelic copy ratio that iterates between imputing the underlying copy number genotype for each segment clusters and updating the model parameters.
FACETS provides a complete ASCN analysis pipeline. This is distinct from most existing methods which often require separate software packages for GC-normalization, sequencing bias adjustment and/or segmentation analysis. An integrated analysis pipeline from start to finish will provide more consistent results.
Supplementary Table S1 provides a feature-by-feature comparison between FACETS and other ASCN methods for sequencing data including TITAN and FALCON. Here we highlight several important differences. First, TITAN and FALCON are both based on heterozygous SNP loci which can lead to more rapid loss of sensitivity for detecting copy number alterations when applied to low resolution data (e.g. targeted panel sequencing) and/or low purity tumor samples as we demonstrated in Supplementary Figure S2 using down-sampling approach of whole-exome samples. The output of TITAN and FALCON are presented in Supplementary Figure S1, along with the FACETS output for the chromophobe sample (TCGA-KL-8331) whole-exome.
Average FACETS running time for a whole-exome sample takes ∼20 min for parsing read counts from each pair of tumor-normal BAM files, and 1–3 min for subsequent steps including GC-normalzation, joint segmentation and ASCN analysis on a single Intel Xeon E5-2640 core processor. The fast computation facilitates large-scale application. Finally, an application to targeted panel sequencing of clinical samples is also demonstrated. Accurate, purity- and ploidy-corrected, integer copy number calls provided by FACETS will be essential to more reliably interpret NGS-based cancer gene copy number data in the context of clinical sequencing. This may pave the way for the incorporation of NGS-based copy number calls into future updates of these clinical guidelines.
Supplementary Material
Acknowledgments
The authors thank Dr. Nicholas D. Socci, Dr. Barry S. Taylor, and Dr. Charlotte K.Y. Ng for their valuable input. This work is supported in part by funds from the P30-CA008748 Cancer Center Support Grant from the National Cancer Institute to Memorial Sloan Kettering Cancer Center.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Supported in part by National Cancer Institute grants CA124514, CA163251, CA195365, P01-CA129243, Susan G. Komen for the Cure foundation grant IIR12221291, and NCI [(Cancer Center Support) P30-CA008748 to Memorial Sloan Kettering Cancer Center, in part]. Funding for open access charge: National Cancer Institute [(Cancer Center Support) P30-CA008748].
Conflict of interest statement. None declared.
REFERENCES
- 1.Sun W., Wright F.A., Tang Z., Nordgard S.H., Van Loo P., Yu T., Kristensen V.N., Perou C.M. Integrated study of copy number states and genotype calls using high-density SNP arrays. Nucleic Acids Res. 2009;37:5365–5377. doi: 10.1093/nar/gkp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Loo P., Nordgard S.H., Lingjærde O.C., Russnes H.G., Rye I.H., Sun W., Weigman V.J., Marynen P., Zetterberg A., Naume B., et al. Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. U.S.A. 2010;107:16910–16915. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yau C., Mouradov D., Jorissen R.N., Colella S., Mirza G., Steers G., Harris A., Ragoussis J., Sieber O., Holmes C.C., et al. A statistical approach for detecting genomic aberrations in heterogeneous tumor samples from single nucleotide polymorphism genotyping data. Genome Biol. 2010;11:R92. doi: 10.1186/gb-2010-11-9-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rasmussen M., Sundstrom M., Goransson Kultima H., Botling J., Micke P., Birgisson H., Glimelius B., Isaksson A. Allele-specific copy number analysis of tumor samples with aneuploidy and tumor heterogeneity. Genome Biol. 2011;12:R108. doi: 10.1186/gb-2011-12-10-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mayrhofer M., DiLorenzo S., Isaksson A. Patchwork: allele-specific copy number analysis of whole-genome sequenced tumor tissue. Genome Biol. 2013;14:R24. doi: 10.1186/gb-2013-14-3-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen H., Bell J.M., Zavala N.A., Ji H.P., Zhang N.R. Allele-specific copy number profiling by next-generation DNA sequencing. Nucleic Acids Res. 2014;42:e23. doi: 10.1093/nar/gku1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ha G., Roth A., Khattra J., Ho J., Yap D., Prentice L.M., Melnyk N., McPherson A., Bashashati A., Laks E., et al. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 2014;24:1881–1893. doi: 10.1101/gr.180281.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen M., Gunel M., Zhao H. SomatiCA: identifying, characterizing and quantifying somatic copy number aberrations from cancer genome sequencing data. PloS One. 2013;8:e78143. doi: 10.1371/journal.pone.0078143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oesper L., Mahmoody A., Raphael B.J. THetA: inferring intra-tumor heterogeneity from high-throughput DNA sequencing data. Genome Biol. 2013;14:R80. doi: 10.1186/gb-2013-14-7-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y., Xie X. Deconvolving tumor purity and ploidy by integrating copy number alterations and loss of heterozygosity. Bioinformatics. 2014;30:2121–2129. doi: 10.1093/bioinformatics/btu174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis C.F., Ricketts C.J., Wang M., Yang L., Cherniack A.D., Shen H., Buhay C., Kang H., Kim S.C., Fahey C.C., et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–330. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carter S.L., Cibulskis K., Helman E., McKenna A., Shen H., Zack T., Laird P.W., Onofrio R.C., Winckler W., Weir B.A., et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng D.T., Mitchell T.N., Zehir A., Shah R.H., Benayed R., Syed A., Chandramohan R., Liu Z.Y., Won H.H., Scott S.N., et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Degner J.F., Marioni J.C., Pai A.A., Pickrell J.K., Nkadori E., Gilad Y., Pritchard J.K. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics. 2009;25:3207–3212. doi: 10.1093/bioinformatics/btp579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xi R., Luquette J., Hadjipanayis A., Kim T.-M., Park P.J. BIC-seq: a fast algorithm for detection of copy number alterations based on high-throughput sequencing data. Genome Biol. 2010;11(Suppl 1):O10. [Google Scholar]
- 16.Sathirapongsasuti J.F., Lee H., Horst B.A., Brunner G., Cochran A.J., Binder S., Quackenbush J., Nelson S.F. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011;27:2648–2654. doi: 10.1093/bioinformatics/btr462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olshen A., Venkatraman E., Lucito R., Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:657–672. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 18.Venkatraman E., Olshen A. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- 19.Zack T.I., Schumacher S.E., Carter S.L., Cherniack A.D., Saksena G., Tabak B., Lawrence M.S., Zhang C.-Z., Wala J., Mermel C.H., et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paik P.K., Shen R., Won H., Rekhtman N., Wang L., Sima C.S., Arora A., Venkatraman S., Ladanyi M., Berger M.F., et al. Next generation sequencing of stage IV squamous cell lung cancers reveals an association of PI3K aberrations and evidence of clonal evolution in patients with brain metastases. Cancer Discov. 2015;5:610–621. doi: 10.1158/2159-8290.CD-14-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koboldt D.C., Miller R.D., Kwok P.-Y. Distribution of human SNPs and its effect on high-throughput genotyping. Hum. Mutat. 2006;27:249–254. doi: 10.1002/humu.20286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.