

Abstract
Objective:
To gain further insight into cortical superficial siderosis (cSS), a new hemorrhagic neuroimaging marker of cerebral amyloid angiopathy (CAA), and to investigate the clinical, neuroimaging, genetic, and CSF biomarker profile of cSS in a large, consecutive memory clinic series.
Methods:
We included 1,504 memory clinic patients undergoing dementia investigation including a brain MRI in our center. Routine CSF biomarker analysis was performed in 1,039 patients and APOE genotyping in 520 patients. MRIs were systematically evaluated for presumed marker of small vessel disease: cSS, cerebral microbleeds, enlarged perivascular spaces, white matter hyperintensities, and lacunes.
Results:
cSS was detected in 40 patients (2.7%; 95% confidence interval [CI] 1.9–3.6); cSS was focal in 33 cases (2.2%; 95% CI 1.5–3.1) and disseminated in 7 (0.5%; 95% CI 0.2–1). Vascular dementia had the highest cSS prevalence (13%; 95% CI 5.4–24.9), followed by Alzheimer disease (5%; 95% CI 3.1–7.5). The most commonly affected area was the occipital lobe (70%; 95% CI 53.5–83.4). cSS was associated with lobar cerebral microbleeds (odds ratio [OR] 7.9; 95% CI 3.4–18.1; p < 0.001), high-degree centrum semiovale perivascular spaces (OR 1.7; 95% CI 1.2–2.6; p = 0.008), and white matter hyperintensities (OR 1.5; 95% CI 1.0–2.2; p = 0.062). APOE ε4/4 genotype was more common in cSS cases compared to those without. CSF β-amyloid 42 was lower in patients with cSS (coefficient −0.09; 95% CI −0.15 to −0.03; p = 0.004).
Conclusions:
Our large series of memory clinic patients provides evidence that cSS is related to cerebrovascular disease and may be a manifestation of severe CAA, even in patients without intracerebral hemorrhage.
Sporadic cerebral amyloid angiopathy (CAA) is a common cerebral small vessel disease and an increasingly recognized contributor to cognitive dysfunction in the elderly. Recently, neurodegenerative and microvascular processes have been considered to cross-talk in producing cognitive impairment. Hence, neuroimaging markers of small vessel disease such as cerebral microbleeds (CMBs), white matter hyperintensities (WMHs), and lacunes are suggested to be of clinical importance in cognitive impairment.1–3
Cortical superficial siderosis (cSS), representing subpial deposits of hemosiderin in the brain favoring cerebral convexities, has recently emerged as another key hemorrhagic MRI manifestation of CAA,4–9 along with multiple lobar CMBs and lobar intracerebral hemorrhage. cSS may cause transient focal neurologic episodes and be a warning sign for future intracranial hemorrhage.10–12 cSS can potentially identify specific subgroups of patients in a memory clinic setting, presumably with more severe underlying CAA. Despite recent interest in the field,5,13,14 large-scale studies in this setting remain, to our knowledge, limited, and the pathophysiologic mechanisms of cSS are still elusive.15
The aims of this study were therefore to systematically analyze the clinical, neuroimaging, genetic, and CSF biomarker profiles associated with cSS in a large series of consecutive memory clinic patients, across the spectrum of different diagnoses. We hypothesized that cSS would be a marker of severe CAA in this memory clinic setting and thus show associations with other characteristic markers of small vessel disease and CAA-related brain injury, including lobar CMBs, perivascular spaces, and WMHs, as well as the APOE ε4 allele and low CSF amyloid levels.
METHODS
Standard protocol approvals, registrations, and patient consents.
Informed consent was obtained from each patient according to the Declaration of Helsinki, and ethics approval was obtained from the regional ethics board, Stockholm, Sweden.
Patients.
This study is part of the Karolinska Imaging Dementia Study, a memory clinic–based cross-sectional study on small vessel disease and cognitive impairment.16,17 We included all consecutive patients (n = 1,509) undergoing dementia investigation and an MRI brain scan with hemosiderin-sensitive sequences at the Memory Clinic and Radiology Department of the Karolinska University Hospital from January 1, 2006, to January 1, 2012, for cross-sectional analysis. Exclusion criteria for all patients were insufficient MRI scan quality (n = 3), contusions in typical traumatic brain localizations, and history of hospitalization following head trauma (n = 2), as well as cSS caused by potential neoplasm (n = 0) or previously treated ruptured aneurysms and arteriovenous malformations (n = 0). After exclusions, the final eligible series consisted of 1,504 patients. Diagnosis was defined on the basis of the ICD-10 in multidisciplinary conferences as described previously.16 Clinicians responsible for patient care, and consequently final diagnosis, were blinded to the study hypothesis and imaging ratings.
MRI protocol.
Three MRI scanners (Siemens Medical Systems, Erlangen, Germany) at the Radiology Department, Karolinska University Hospital, were used. Details on sequences and distribution on scanners have been given elsewhere.16 For each patient, axial susceptibility-weighted imaging (SWI) or T2*–gradient recalled echo (GRE) sequences, as well as conventional structural MRI sequences such as T1, T2, and fluid-attenuated inversion recovery, were obtained as previously described in detail.
Image analysis.
A senior consultant neuroradiologist (J.M.) and a trained physician (S.S.) analyzed all images jointly (i.e., by consensus) according to the recently proposed Standards for Reporting Vascular Changes on Neuroimaging (version 1)18 and cSS according to recent proposed criteria.15 Both raters were blinded to all patient data. cSS was defined as linear gyriform hypointensities on axial T2*-GRE/SWI sequences.19 The severity of cSS was classified as focal (restricted to ≤3 sulci) or disseminated (≥4 sulci). The distribution of cSS per lobe was noted. CMBs were rated on axial T2* or SWI as previously described in detail.16,17 Care was taken to avoid CMB mimics as described elsewhere.16 For instance, T2 images were analyzed to differentiate between cavernomas and CMBs. Furthermore, any CMB-like structure in the vicinity of a deep venous anomaly was excluded since these may represent cavernomas. No cSS was noted in patients with evident cavernomas. In addition, a modification of the Boston criteria with CMBs instead of macroscopic hemorrhage was used. Patients with CMBs in strictly lobar or lobar with cerebellar locations were classified as probable CAA, whereas patients with CMBs in deep brain regions and the brainstem were considered not to have CAA.20,21 In patients with cSS, we noted if at least 1 CMB was present in the vicinity of cSS foci, defined as ≤1 cm, as previously suggested in similar studies.14 WMHs and lacunes were rated as previously described.17 Enlarged perivascular spaces were rated on axial T2 sequences and in the centrum semiovale and basal ganglia as follows: 0 = none, 1 = 1 to 10, 2 = 11 to 20, 3 = 21 to 40, 4 = >40, according to the validated enlarged perivascular space rating scale.22–24 All neuroradiologic analyses were made on a radiologic PACS workstation.
APOE genotyping and CSF analysis.
APOE genotyping (n = 520) was performed on coded genomic DNA samples, and CSF samples were available for 1,039 patients at the time of the MRI scan and analyzed for β-amyloid 42 (Aβ42), total tau, and tau phosphorylated at threonine 181 (ng/L), as described previously.17 All analyses were performed at the Department of Clinical Chemistry, Karolinska University Hospital. The team involved in the analysis was unaware of the dementia diagnosis and MRI findings.
Statistical analysis.
Categorical variables were analyzed with the Pearson χ2 or Fisher exact test, and continuous variables were analyzed by the 2-sample t test (for normal distributions) and Wilcoxon rank-sum test (for nonnormal distributions) as appropriate. We compared demographic, clinical, and neuroimaging characteristics, as well as APOE and CSF data, of patients with vs without cSS. Logistic regression analysis with cSS as a dependent variable was used to assess the relationship between cSS presence or burden and all other imaging markers of small vessel disease, as well as clinical variables. When looking at associations with the Mini-Mental State Examination (MMSE) in the whole series, we controlled for age, sex, and diagnostic group. We further constructed a multivariate logistic regression model, additionally adjusting for other potential confounders such as age, sex, and hypertension (see Results). In sensitivity analyses, the logistic regression models were repeated with diagnostic groups and MRI parameters (including field strength, i.e., 1.5 vs 3T, and SWI vs T2*-GRE) as additional covariates.
The association between CSF biomarkers and cSS presence was investigated in linear regression analyses. CSF biomarkers values were log-transformed for normality and put as the dependent variables in the models. The analyses were repeated for the whole series and Alzheimer disease (AD) and mild cognitive impairment (MCI) groups combined (because of the larger sample size). Significance level was set at 0.05. Values below the 0.10 level were considered indicative of a tendency. Stata software (version 13.0, StataCorp, College Station, TX) was used.
RESULTS
Our memory clinic series consisted of 1,509 patients, 1,504 total after exclusions. The distribution of patients over the different diagnostic categories and basic baseline characteristics are summarized in table 1.
Table 1.
Baseline characteristic and cortical superficial siderosis (cSS) prevalence and burden per diagnostic group in our memory clinic population
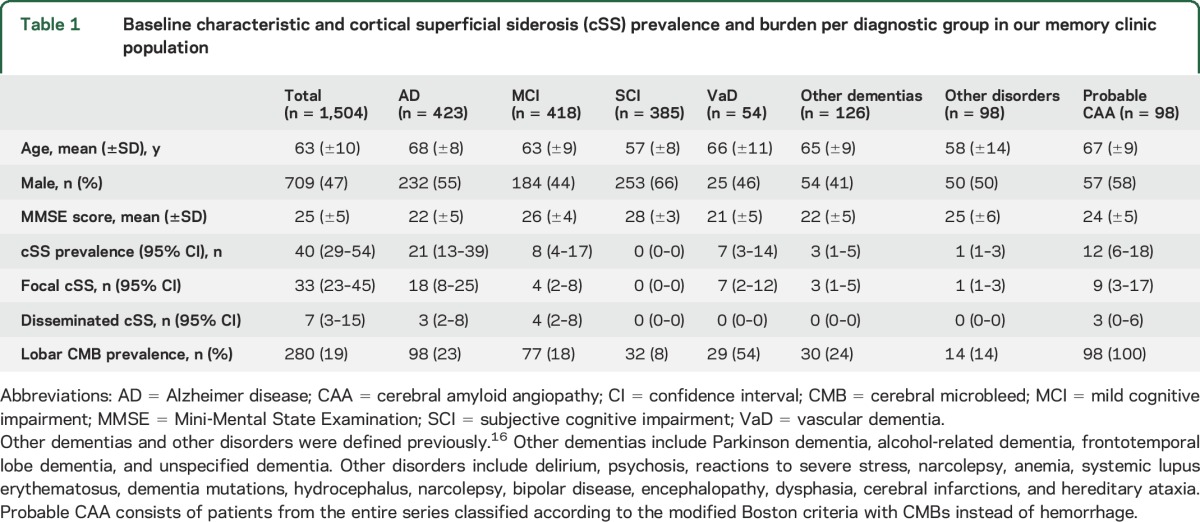
Prevalence, burden, and topography of cSS.
cSS was detected in 40 of 1,504 patients (2.7%; 95% confidence interval [CI] 1.9–3.6); in 33 (2.2%; 95% CI 1.5–3.1) cases, it was focal and in 7 (0.5%; 95% CI 0.2–1) cases disseminated. The prevalence was higher in patients with probable CAA (12%; 95% CI 5.6–18.4), classified according to the modified Boston criteria, than in patients without probable CAA (2%; p < 0.001). Patients with and without cSS, as well as diagnostic groups, were evenly distributed across MRI scanners at 1.5 vs 3T and SWI vs T2*-GRE (table 2).
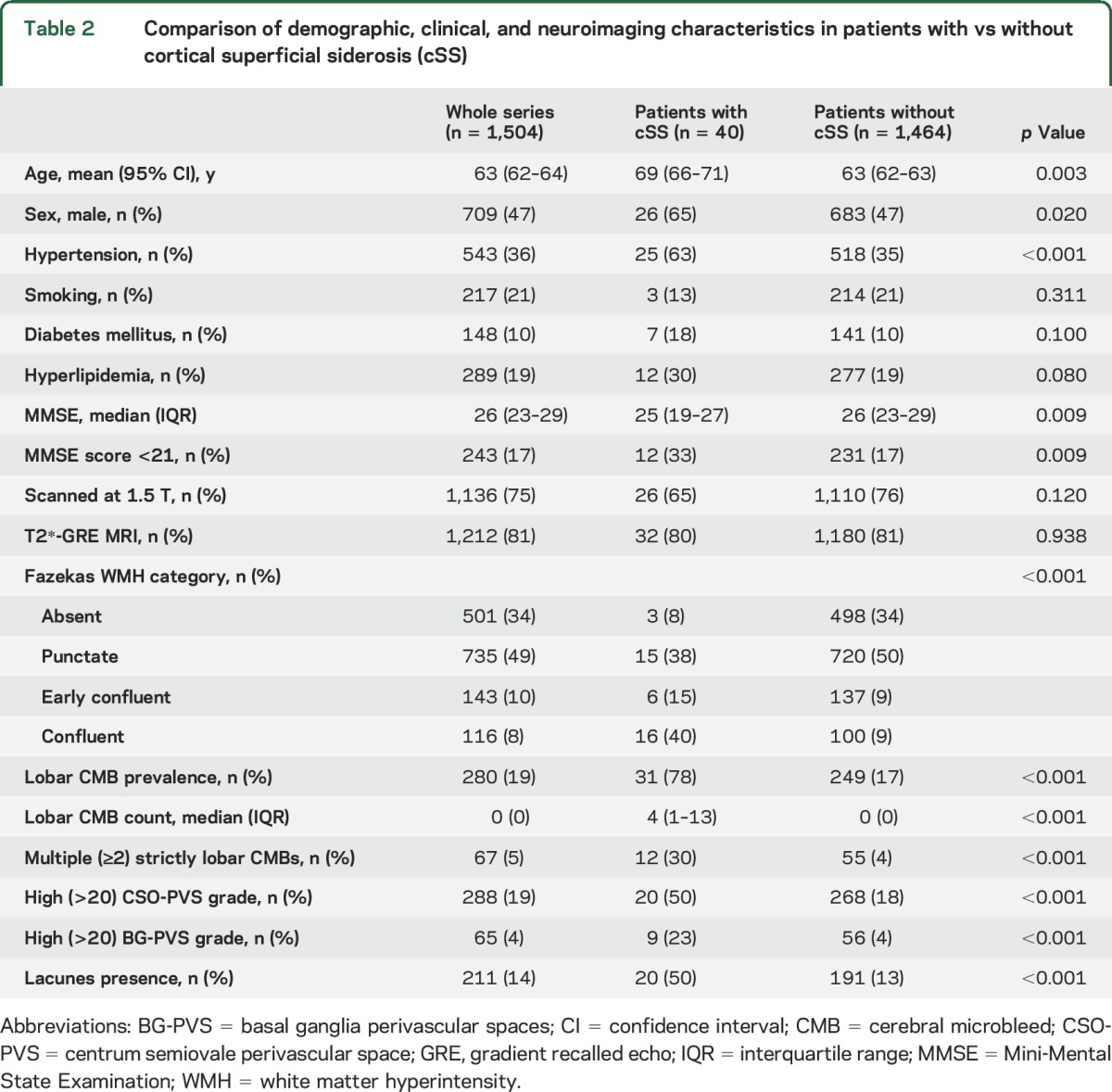
Table 2.
Comparison of demographic, clinical, and neuroimaging characteristics in patients with vs without cortical superficial siderosis (cSS)
The most commonly affected areas with cSS were the occipital lobes (70%; 95% CI 54–83), followed by the parietal lobes (38%; 95% CI 23–54) and frontal and temporal lobes (25%; 95% CI 13–41, and 23%; 95% CI 11–39, respectively). The topographic predilection of cSS was similar between patients with and without probable CAA. The prevalence of cSS differed across subtype of cognitive impairment (Fisher exact test p < 0.001; table 1). Vascular dementia had the highest prevalence of cSS (13%; 95% CI 5–25), followed by AD (5%; 95% CI 3–8) and MCI (2%; 95% CI 1–4). There were no cSS cases in the group with subjective cognitive impairment. Among other dementias, 3 patients had cSS: 1 patient in alcohol-related dementia, 1 patient in frontotemporal dementia, and 1 patient in unspecified dementia. In other disorders, 1 patient had cSS; the patient was undergoing genetic counseling and had hereditary frontotemporal lobe dementia (C9orf72 mutation), scoring 28 of 30 on the MMSE.
Demographic, clinical, and neuroimaging findings, and predictors of cSS.
Comparisons of demographic, clinical, and imaging characteristics between patients with and those without cSS are shown in table 2. Patients with cSS were older, were more often male and hypertensive, and had lower MMSE scores compared with patients without cSS. After adjustment for age, sex, and diagnostic category, there was only a tendency of an association between the presence of cSS and lower MMSE score (coefficient −1.5; 95% CI −3.1 to 0.1; p = 0.069 in linear regression). There was no difference in MMSE score in simple univariable analysis between patients with and without cSS after exclusion of patients with subjective cognitive impairment. In univariable analysis, the presence of cSS was associated with most of the other markers of small vessel disease, including WMH severity, lobar CMB presence, number of CMBs, high degree (>20) of MRI-visible perivascular spaces in the centrum semiovale and basal ganglia, and presence of lacunes (table 2).
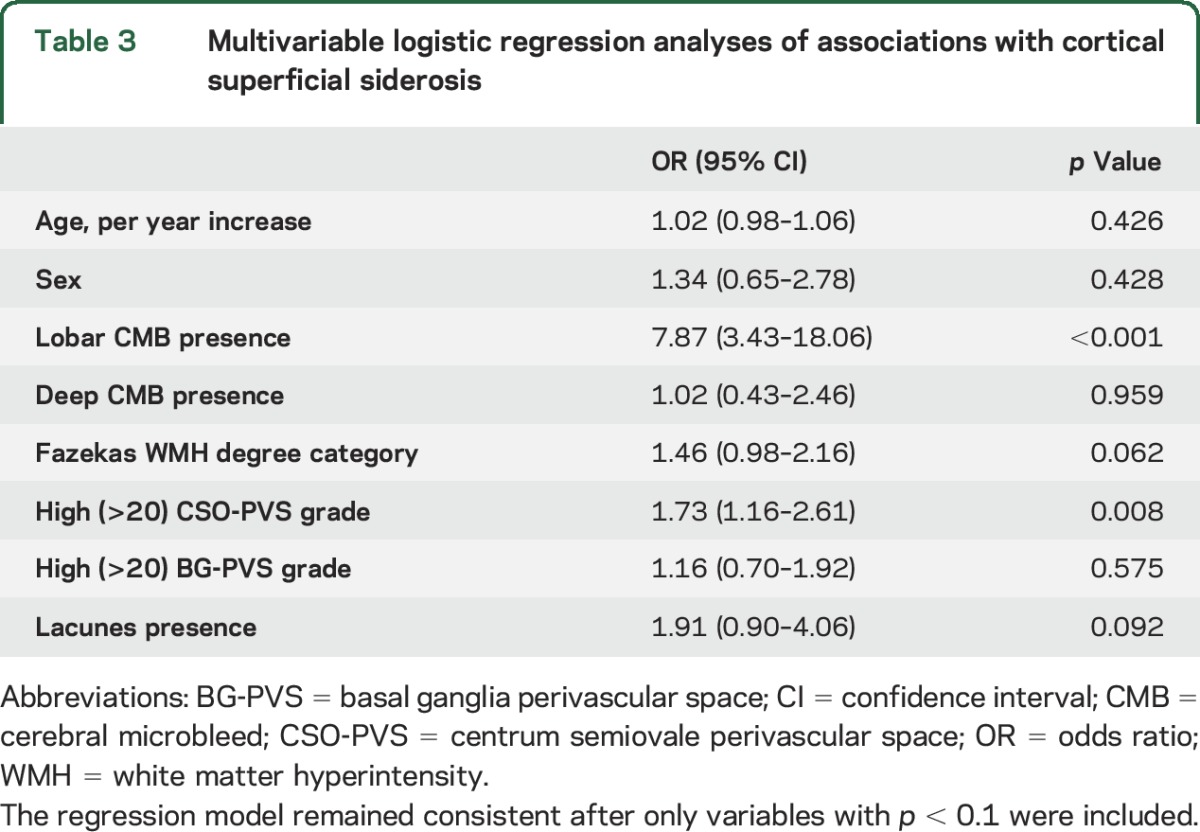
In multivariable logistic regression analysis, after adjustment for age and sex, the presence of cSS was associated with lobar (but not deep) CMB presence, with high degree (>20) of MRI-visible perivascular spaces in the centrum semiovale (but not in the basal ganglia), and marginally with WMHs (for every step progression in the Fazekas score; table 3). The results were consistent and of similar effect size in separate sensitivity analyses further including hypertension and MRI parameters (including magnetic field strength, SWI vs T2*-GRE) in the models. These associations were not influenced by the clinical diagnosis of the cognitive syndrome except for the attenuation of the association with WMH. We repeated these analyses in the AD and MCI subgroups as a further exploratory analysis. All associations remained consistent except for the association between cSS and WMH, which was no longer significant.
Table 3.
Multivariable logistic regression analyses of associations with cortical superficial siderosis
Lobar microbleeds in the vicinity of cSS.
Thirty-one of the 40 patients with cSS had lobar CMBs, 19 (61%) of whom had at least one CMB in the vicinity (i.e., within 1 cm) of cSS foci. Patients with occipital cSS similarly had CMBs in the vicinity in 57% of cases.
APOE genotype and CSF biomarkers.
Among patients with APOE data available, the only association with cSS was between APOE ε4 homozygosity: APOE ε4/4, 30.8% vs 12.5% in those with vs without cSS (Fisher exact test p = 0.053). No patient with cSS had the APOE ε2/2 genotype. Differences in APOE genotype associations between patients with and without cSS are given in table e-1 at Neurology.org.
CSF samples were available for analysis in 1,039 patients. The presence of cSS was associated with lower Aβ42 levels and higher total and phosphorylated tau levels, not with CSF/serum albumin ratios (figure). After adjustment for age, sex, and diagnostic subgroup in linear regression analysis (CSF biomarkers as a dependent variable), cSS was associated only with lower CSF Aβ42 levels (table 4). These results were consistent in the AD and MCI groups combined (n = 589), even after adjustment for age and sex (table 4). There were no differences in CSF markers between patients with focal and those with disseminated cSS.
Figure. CSF biomarkers and cortical superficial siderosis (cSS); cSS on MRI.

(A) Boxplots show different CSF marker levels in patients with vs without cSS in the entire memory clinic series. Colors represent a way of referencing different panels. (B) Characteristic examples of cSS (arrows) seen as linear gyriform hypointensities on blood-sensitive MRI sequences (right, susceptibility-weighted images; middle and left, T2*–gradient recalled echo). Focal cSS is seen on the example in the left. The middle and right images are from 2 patients with Alzheimer disease and disseminated cSS, affecting multiple sulci. Aβ42 = β-amyloid 42; p-tau = phosphorylated tau; t-tau = total tau.
Table 4.
Univariable and multivariable linear regression analyses with the CSF biomarkers as a dependent variable and the presence of cortical superficial siderosis as the main independent variable

DISCUSSION
Our findings in a large consecutive series provide further support for the notion that cSS is associated with cerebrovascular disease and may be a manifestation of advanced CAA in memory clinic patients, without major symptomatic lobar intracerebral hemorrhage. We show that cSS is associated with low CSF Aβ42 levels independently of cognitive impairment diagnostic subtype and that CSF total tau and phosphorylated tau are high with cSS in the whole series. APOE ε4 homozygosity is associated with patients with cSS. Putative markers of small vessel disease and CAA, and specifically lobar CMBs and perivascular spaces in the centrum semiovale, show strong independent associations with cSS, providing insights into potential underlying pathophysiologic mechanisms.
Previous data on cSS in large consecutive memory clinic series are relatively limited. Prevalence of cSS in memory clinic setting ranges from 2.1% to 6.1%,5,13,14,25 in line with our prevalence of 3.0%, with a 0.7% prevalence in healthy populations.26 cSS occurred in varying diagnoses throughout our series, with the highest prevalence observed in vascular dementia, followed by AD. The 5% prevalence of cSS in AD found in our study equals the prior reported prevalence.13,14 At the same time, it is important to note that imaging reports were available to the clinicians, which may have affected the final diagnosis.
cSS was predominantly occipital, which accords with pathologic data showing that CAA often favors the occipital lobe and, when present in that region, often has a more severe presentation.27,28 Our finding is also in line with our previous report of CMBs being most frequent in the occipital lobe,16 although another study reported the frontal lobe as the most frequent location of cSS.14
The associations with other markers of small vessel disease, as shown in our work and 2 prior studies,5,14 suggest that cSS may reflect cerebrovascular disease and CAA. In our study, cSS was independently associated with WMH severity, a high degree of MRI-visible perivascular spaces in the centrum semiovale, and lobar CMBs, a putative marker of CAA presence and severity. The relation with perivascular spaces in the centrum semiovale is novel and of particular interest as it may support the hypothesis that enlarged perivascular spaces in the centrum semiovale, but not in the basal ganglia, are related to CAA. Since both cSS and enlarged perivascular spaces in the centrum semiovale are proposed to represent CAA, this would be in line with the more lobar predilection of CAA, as well as previous findings in CAA-related intracerebral hemorrhage cohorts.29 Furthermore, cSS was related to APOE ε4 homozygosity,14 supporting the potential involvement and interrelationship of CAA and markers of CAA in dementia.
The routine CSF biomarker profile in memory clinic patients with cSS has not been analyzed before. The association of cSS with lower Aβ42 levels further supports the fact that cSS may be a marker of cerebrovascular amyloid accumulation. It may also support the fact that patients with cSS have brain amyloid deposition to a higher degree. Amyloid subtyping and using the Aβ 40 peptide would add important data for further understanding and differentiation of associations with brain and vascular amyloid. The lack of an association after adjustments is in favor of cSS being purely a marker of an amyloid-related process rather than neurodegeneration. cSS and relations with Pittsburgh compound B amyloid PET scans have shown higher global amyloid burden in patients with cSS compared to patients without, which is in line with the current CSF findings.13
The tendency for lower MMSE in patients with cSS might highlight the potential of this marker in identifying a more aggressive CAA phenotype interacting with AD and neurodegenerative pathology to cause more severe cognitive decline or lowering the threshold for cognitive deficits. Similarly, studies have indicated that the synergism between vascular pathology and AD is associated with more severe cognitive decline compared with pure AD, without significant vascular involvement.2,30 It is also possible that cSS may directly cause brain tissue injury, especially in the superficial cortical areas. Superficial astrocytes, microglia, and cerebellar Bergmann cells are thought to pick up blood and to break down heme to free iron.31–33 Free iron may cause injury through the production of reactive oxygen species, in turn leading to neurodegeneration.31–33 These hypotheses require further investigation in clinical and basic science studies.
Strengths of our study include the comprehensive assessment of the whole range of imaging markers of small vessel disease on structural MRI (including imaging signatures of CAA) according to current consensus guidelines, as well as the availability of APOE and CSF biomarker analysis. Furthermore, the large sample size (the largest to date focusing on cSS in a memory clinic setting) resulted in substantial statistical power to build robust multivariable models and to adjust for potential confounders and increases the external validity of our study. Important limitations include the use of different scanners, field strengths, and blood-sensitive sequences for both the T2*-GRE and SWI, although this was adjusted for in all our analyses. A limitation is the lack of autopsies and brain biopsies confirming CAA. Hippocampal atrophy ratings and detailed neuropsychological testing would have provided interesting aspects to our study. The fact that imaging (but not study hypothesis and research ratings) was available to clinicians setting the final diagnosis raises the possibility for circular diagnostic reasoning, making it hard to derive definitive conclusions on the basis of cSS prevalence and diagnosis.
Our study validates previous findings and adds new important data to the increasing evidence that cSS may be a marker of advanced CAA in a memory clinic population. Future prospective studies should explore cSS and the direct effects on cognition, brain parenchymal injury, and disease progression.
Supplementary Material
GLOSSARY
- Aβ42
β-amyloid 42
- AD
Alzheimer disease
- CAA
cerebral amyloid angiopathy
- CMB
cerebral microbleed
- cSS
cortical superficial siderosis
- GRE
gradient recalled echo
- ICD-10
International Classification of Diseases, 10th Revision
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- SWI
susceptibility-weighted imaging
- WMH
white matter hyperintensities
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
S.S. provided the study concept and design, image analysis, data acquisition and analysis, drafting of the manuscript, and critical revision. J.M. provided the study concept and design, image analysis, and critical revision of the manuscript. A.C. contributed to the study concept and design, data analysis, drafting of the manuscript, and critical revision. L.C. performed image analysis. T.G. contributed to the study concept, data acquisition and analysis, and critical revision of the manuscript. M.S. provided data acquisition and critical revision of the manuscript. Y.F. performed critical revision of the manuscript. P.A. provided the study concept and design, critical revision of manuscript, and study supervision. M.K.-W. contributed to the study concept and design, data acquisition, and critical revision of manuscript, study supervision. L.-O.W. contributed to study concept and design, data acquisition, critical revision of manuscript, and study supervision.
STUDY FUNDING
Stockholm County Council, Karolinska Institutet, the Swedish Dementia Association, and the Swedish Stroke Association.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer's disease: innocent observation or key player? Brain 2011;134:335–344. [DOI] [PubMed] [Google Scholar]
- 2.Cavalieri M, Enzinger C, Petrovic K, et al. . Vascular dementia and Alzheimer's disease: are we in a dead-end road? Neurodegener Dis 2010;7:122–126. [DOI] [PubMed] [Google Scholar]
- 3.Gorelick PB, Scuteri A, Black SE, et al. . Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011;42:2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charidimou A, Jäger RH, Fox Z, et al. . Prevalence and mechanisms of cortical superficial siderosis in cerebral amyloid angiopathy. Neurology 2013;81:626–632. [DOI] [PubMed] [Google Scholar]
- 5.Wollenweber FA, Buerger K, Mueller C, et al. . Prevalence of cortical superficial siderosis in patients with cognitive impairment. J Neurol 2014;261:277–282. [DOI] [PubMed] [Google Scholar]
- 6.Raposo N, Viguier A, Cuvinciuc V, et al. . Cortical subarachnoid haemorrhage in the elderly: a recurrent event probably related to cerebral amyloid angiopathy. Eur J Neurol 2011;18:597–603. [DOI] [PubMed] [Google Scholar]
- 7.Beitzke M, Enzinger C, Wünsch G, Asslaber M, Gattringer T, Fazekas F. Contribution of convexal subarachnoid hemorrhage to disease progression in cerebral amyloid angiopathy. Stroke 2015;46:1533–1540. [DOI] [PubMed] [Google Scholar]
- 8.Kumar S, Goddeau RP, Selim MH, et al. . Atraumatic convexal subarachnoid hemorrhage: clinical presentation, imaging patterns, and etiologies. Neurology 2010;74:893–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linn J, Halpin A, Demaerel P, et al. . Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010;74:1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linn J, Wollenweber FA, Lummel N, et al. . Superficial siderosis is a warning sign for future intracranial hemorrhage. J Neurol 2013;260:176–181. [DOI] [PubMed] [Google Scholar]
- 11.Charidimou A, Peeters A, Fox Z, et al. . Spectrum of transient focal neurological episodes in cerebral amyloid angiopathy: multicentre magnetic resonance imaging cohort study and meta-analysis. Stroke 2012;43:2324–2330. [DOI] [PubMed] [Google Scholar]
- 12.Greenberg SM, Vonsattel JP, Stakes JW, Gruber M, Finklestein SP. The clinical spectrum of cerebral amyloid angiopathy: presentations without lobar hemorrhage. Neurology 1993;43:2073–2079. [DOI] [PubMed] [Google Scholar]
- 13.Na HK, Park JH, Kim JH, et al. . Cortical superficial siderosis: a marker of vascular amyloid in patients with cognitive impairment. Neurology 2015;84:849–855. [DOI] [PubMed] [Google Scholar]
- 14.Zonneveld HI, Goos JDC, Wattjes MP, et al. . Prevalence of cortical superficial siderosis in a memory clinic population. Neurology 2014;82:698–704. [DOI] [PubMed] [Google Scholar]
- 15.Charidimou A, Linn J, Vernooij MW, et al. . Cortical superficial siderosis: detection and clinical significance in cerebral amyloid angiopathy and related conditions. Brain 2015;138:2126–2139. [DOI] [PubMed] [Google Scholar]
- 16.Shams S, Martola J, Granberg T, et al. . Cerebral microbleeds: different prevalence, topography, and risk factors depending on dementia diagnosis: the Karolinska Imaging Dementia Study. AJNR Am J Neuroradiol 2015;36:661–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shams S, Granberg T, Martola J, et al. . Cerebrospinal fluid profiles with increasing number of cerebral microbleeds in a continuum of cognitive impairment. J Cereb Blood Flow Metab 2016;36:621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wardlaw JM, Smith EE, Biessels GJ, et al. . Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013;12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feldman HH, Maia LF, Mackenzie IRA, Forster BB, Martzke J, Woolfenden A. Superficial siderosis: a potential diagnostic marker of cerebral amyloid angiopathy in Alzheimer disease. Stroke 2008;39:2894–2897. [DOI] [PubMed] [Google Scholar]
- 20.Akoudad S, Portegies MLP, Koudstaal PJ, et al. . Cerebral microbleeds are associated with an increased risk of stroke: the Rotterdam study. Circulation 2015;132:509–516. [DOI] [PubMed] [Google Scholar]
- 21.Martinez-Ramirez S, Romero JR, Shoamanesh A, et al. . Diagnostic value of lobar microbleeds in individuals without intracerebral hemorrhage. Alzheimers Dement 2015;11:1480–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Potter GM, Chappell FM, Morris Z, Wardlaw JM. Cerebral perivascular spaces visible on magnetic resonance imaging: development of a qualitative rating scale and its observer reliability. Cerebrovasc Dis 2015;39:224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maclullich AMJ, Wardlaw JM, Ferguson KJ, Starr JM, Seckl JR, Deary IJ. Enlarged perivascular spaces are associated with cognitive function in healthy elderly men. J Neurol Neurosurg Psychiatry 2004;75:1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doubal FN, MacLullich AMJ, Ferguson KJ, Dennis MS, Wardlaw JM. Enlarged perivascular spaces on MRI are a feature of cerebral small vessel disease. Stroke 2010;41:450–454. [DOI] [PubMed] [Google Scholar]
- 25.Inoue Y, Nakajima M, Uetani H, et al. . Diagnostic significance of cortical superficial siderosis for Alzheimer disease in patients with cognitive impairment. AJNR Am J Neuroradiol 2016;37:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vernooij MW, Ikram MA, Hofman A, Krestin GP, Breteler MMB, van der Lugt A. Superficial siderosis in the general population. Neurology 2009;73:202–205. [DOI] [PubMed] [Google Scholar]
- 27.Rosand J, Muzikansky A, Kumar A, et al. . Spatial clustering of hemorrhages in probable cerebral amyloid angiopathy. Ann Neurol 2005;58:459–462. [DOI] [PubMed] [Google Scholar]
- 28.Dierksen GA, Skehan ME, Khan MA, et al. . Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Ann Neurol 2010;68:545–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charidimou A, Jäger RH, Peeters A, et al. . White matter perivascular spaces are related to cortical superficial siderosis in cerebral amyloid angiopathy. Stroke 2014;45:2930–2935. [DOI] [PubMed] [Google Scholar]
- 30.Esiri MM, Nagy Z, Smith MZ, Barnetson L, Smith AD. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer's disease. Lancet 1999;354:919–920. [DOI] [PubMed] [Google Scholar]
- 31.Nanda S, Sharma SG, Longo S. Superficial siderosis—mechanism of disease: an alternative hypothesis. Ann Clin Biochem 2010;47:275–278. [DOI] [PubMed] [Google Scholar]
- 32.Kumar N. Neuroimaging in superficial siderosis: an in-depth look. AJNR Am J Neuroradiol 2010;31:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koeppen AH, Dickson AC, Chu RC, Thach RE. The pathogenesis of superficial siderosis of the central nervous system. Ann Neurol 1993;34:646–653. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.