

Abstract
Cystic Fibrosis (CF) is an autosomal recessive disease caused by mutations in CF transmembrane conductance regulator (CFTR), resulting in defective anion transport. Regardless of the disease-causing mutation, gene therapy is a strategy to restore anion transport to airway epithelia. Indeed, viral vector–delivered CFTR can complement the anion channel defect. In this proof-of-principle study, functional in vivo CFTR channel activity was restored in the airways of CF pigs using a feline immunodeficiency virus–based (FIV-based) lentiviral vector pseudotyped with the GP64 envelope. Three newborn CF pigs received aerosolized FIV-CFTR to the nose and lung. Two weeks after viral vector delivery, epithelial tissues were analyzed for functional correction. In freshly excised tracheal and bronchus tissues and cultured ethmoid sinus cells, we observed a significant increase in transepithelial cAMP-stimulated current, evidence of functional CFTR. In addition, we observed increases in tracheal airway surface liquid pH and bacterial killing in CFTR vector–treated animals. Together, these data provide the first evidence to our knowledge that lentiviral delivery of CFTR can partially correct the anion channel defect in a large-animal CF model and validate a translational strategy to treat or prevent CF lung disease.
An integrating lentivirus vector is used to deliver a CFTR expression cassette to the airway epithelium, correcting anion transport and host defense defects in vivo.
Introduction
Cystic fibrosis (CF) is a common autosomal recessive disorder caused by mutations in the CF transmembrane conductance regulator (CFTR) gene (1, 2). The most common CFTR mutation is a phenylalanine deletion at amino acid position 508 (ΔF508), however there are over 2,000 different disease-associated CFTR mutations. CF affects multiple organ systems, yet progressive lung disease, characterized by recurrent bacterial infections and inflammation, is the leading cause of CF morbidity and mortality (3). CFTR mutations result in impaired anion transport. This contributes to lung disease, in part, through reduced airway surface liquid pH (ASL pH), defective bacterial killing, and reduced mucociliary transport (MCT) (4–6). While new CFTR potentiator (7, 8) and corrector (9) therapies are now in the clinic, there remains a great need to develop treatments for all people with CF.
Gene therapy is a mutation agnostic approach to restore CFTR activity. Lentiviral vectors transduce both dividing and nondividing cells, integrate into the host genome, and provide long-term transgene expression (10, 11). A feline immunodeficiency virus–based (FIV-based) viral vector pseudotyped with the baculovirus envelope protein GP64 transduces epithelial cells at the apical surface and persistently expresses a transgene of interest in mouse airways (12, 13). We previously demonstrated that FIV pseudotyped with the GP64 envelope from baculovirus efficiently transduces both human and pig airway epithelia (12, 14). Here, we use a lentiviral vector to deliver a CFTR expression cassette to pig airways in vivo.
At birth, CF pigs manifest physiologic defects associated with loss of CFTR function, including a reduced ASL pH, impaired bacterial killing, and reduced MCT (15–18). In this single time point pilot study, we hypothesized that delivery of GP64-FIV-CFTR to the sinuses and lower respiratory tract of newborn CF pigs would correct the anion channel defect. We show that delivery of GP64 pseudotyped FIV-CFTR to newborn CF pigs (19) can achieve partial physiological correction of the defective anion transport and its host defense consequences in vivo.
Results
FIV-CFTR corrects the anion channel defect in vitro.
Multiple studies have established that CFTR complementation restores the anion channel defect in vitro (20–23). Before we delivered vector to CF pigs, we first confirmed that lentiviral-mediated porcine CFTR delivery corrects the anion defect in primary cultures of CF airway epithelia. Well-differentiated airway epithelial cultures derived from CF pig ethmoid sinuses were transduced apically with 4 × 107 transducing units (TU) of GP64-pseudotyped FIV-CFTR (MOI = 5). Four days after transduction, the bioelectric properties of epithelia were analyzed in Ussing chambers. FIV-CFTR–treated cells demonstrated a significant increase in transepithelial Cl– current in response to forskolin and 3-isobutyl-1-methylxanthine (IBMX, F&I). (Figure 1, A and B). The current was inhibited by the CFTR channel blocker, GlyH-101 (Figure 1, A and C). CFTR subcellular localization was examined using IHC. CFTR protein localized to the apical membrane in both ciliated and nonciliated epithelial cells (Figure 1D) and was absent in the untreated CF epithelia (Figure 1E). These data suggest that apical delivery of FIV-CFTR to primary airway epithelia can correct the anion channel defect in vitro.
Figure 1. Correction of chloride (Cl–) transport in cystic fibrosis (CF) pig primary epithelial with a lentiviral vector.

Feline immunodeficiency virus–based viral vector expressing cystic fibrosis transmembrane conductance regulator (FIV-CFTR; MOI = 5) was delivered to the apical surface of well-differentiated primary cultures of airway epithelial cells from CF pigs. Transepithelial Cl– currents were measured in Ussing chambers. (A) An example of CFTR-dependent Cl– current is shown. The average change in Cl– current upon addition of forskolin/3-isobutyl-1-methylxanthine (F&I) (B) and GlyH-101 (C) in treated and naive cultures are shown. n = 8 epithelial sheets/treatment (collected from 3 donor pigs). *P < 0.01, Mann-Whitney nonparametric t test. (D) IHC using a CFTR antibody reveals apical localization of CFTR protein in ciliated cells (arrows). c, ciliated cells; nc, nonciliated cells. (E) IHC using a CFTR antibody on untreated CF primary airway epithelia. Scale bar: 250 μM (D and E).
FIV-CFTR rescues the anion channel defect in CF pig trachea and bronchus.
To test the efficacy of FIV-CFTR in vivo, we aerosolized 6 × 109 TU of concentrated FIV-CFTR formulated with methylcellulose (24) into the ethmoid sinus and trachea of 3 newborn gut-corrected CF pigs. Gut-corrected CF pigs express CFTR in intestinal tissues but lack CFTR expression in the airways (19). Two weeks after vector delivery, tissues were collected and analyzed for CFTR correction. Tissues from untreated CF pigs showed little response to either F&I or GlyH-101 (Figure 2, A–D). In freshly excised tracheal tissues from the CF pigs treated with FIV-CFTR, a significant increase in cAMP-activated Cl– current was observed to near WT levels (Figure 2A). This current decreased in response to GlyH-101 (Figure 2B). A similar result was obtained for freshly excised bronchus tissue (Figure 2, C and D). We also quantified CFTR mRNA abundance in treated and untreated animals. In FIV-CFTR–treated trachea and bronchus tissues, we observed CFTR mRNA levels that were increased significantly over the average background levels detected in untreated CF pig tissue (Figure 2E). Together, these findings confirm that delivery of a CFTR expression cassette by a lentiviral vector can partially correct the anion defect in the trachea and bronchus of CF pigs in vivo.
Figure 2. Anion channel correction in tissue explants.

Feline immunodeficiency virus–based viral vector expressing cystic fibrosis transmembrane conductance regulator (FIV-CFTR) was delivered to the lung of newborn cystic fibrosis (CF) pigs. Two weeks after infection, freshly excised tracheal tissues were mounted into Ussing chambers to measure a change in transepithelial current in response to (A) forskolin/3-isobutyl-1-methylxanthine (F&I) or (B) GlyH-101, and freshly excised bronchus tissues were also tested for their response to (C) F&I or (D) GlyH-101. Diamonds indicate tracheal tissue, and circles indicate bronchus tissue from individual animals. n = 3 pigs; each data point represents 3 replicates/pig. *P < 0.05, one-way ANOVA comparison test. (E) RNA was harvested from trachea and bronchus to measure levels of CFTR by real-time PCR. n = 10 or 8 lung tissue samples from 3 treated pigs. The dotted line indicates the background CFTR mRNA level in CF tissue. *P < 0.05, Mann-Whitney nonparametric t test.
We collected genomic DNA from tracheal and bronchial tissues from FIV-CFTR–treated and untreated animals. The tissues included multiple cell types, including surface epithelia, basal cells, submucosal glands, basal lamina, and smooth muscle. Thus, not all cells sampled were surface epithelial cells. Using quantitative PCR (qPCR), we attempted to quantify the levels of DNA integration; however, the levels were below the limit of detection, i.e., less than 1 copy per 10 cells (data not shown). Because the tissues represent a mixed cell population, including many that received no vector, detecting CFTR transgene copy number in airway epithelial cells is challenging. However, these results suggest that, in the conducting airways, partial CFTR anion channel correction can be achieved by integrating a transgene in a small percentage of cells. We postulate that overexpressing CFTR with a heterologous promoter in a small subset of airway cells may help compensate for low overall gene transfer efficiency.
Partial rescue of ASL pH and bacterial killing in CF pigs.
The loss of bicarbonate transport through CFTR results in acidification of the ASL and subsequent inhibition of antimicrobial factors, thereby impairing bacterial killing (16). Therefore, we next determined if CFTR complementation by FIV-CFTR increased the ASL pH and improves ASL antimicrobial activity. Two weeks after vector delivery, we measured tracheal pH (16, 25). Compared with untreated animals, average tracheal ASL pH increased from 7.0 to 7.2 (Figure 3A); however, this trend did not reach statistical significance. The average ASL pH of the FIV-CFTR–treated group was nearly identical to the non-CF pig group.
Figure 3. Tracheal pH and bacterial killing ability in cystic fibrosis (CF) pigs.
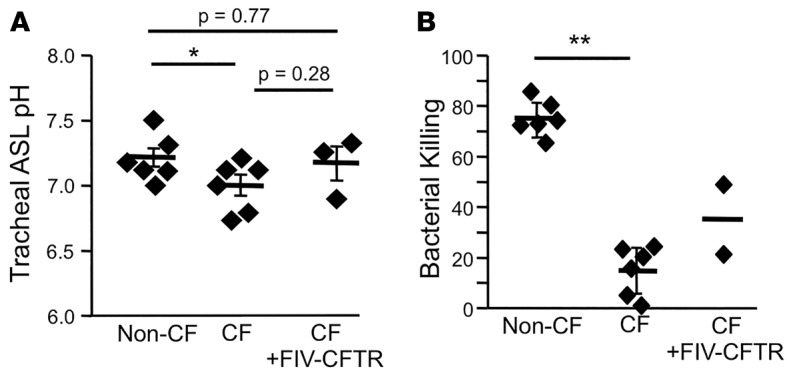
Feline immunodeficiency virus–based viral vector expressing cystic fibrosis transmembrane conductance regulator (FIV-CFTR) was delivered to the lung of newborn CF pigs. (A) Tracheal pH measurements were taken prior to performing the bacterial killing assay. The tracheal window was removed, and tracheal pH was measured in situ using the noninvasive dual lifetime referencing planar optode. (B) Bacterial-coated grids were placed on the airway surface of the trachea and incubated for 1 minute. Immediately following, grids were subjected to a live/dead stain, imaged by confocal microscopy, and quantified using Image J. Diamonds represent data from individual pigs. *P < 0.05, **P < 0.0001, one-way ANOVA comparison test. n = 2–6 pigs. The CF and non-CF control pigs (A and B) were shared with a companion manuscript by Steines, et al. (32).
To assess ASL antimicrobial activity, we interrogated individual bacteria attached to gold grids, as previously described (16). As shown in Figure 3B, the ASL from CF pigs that received FIV-CFTR had an increased bacterial killing ability as compared with untreated CF pigs. Bacterial killing activity measurements in a third CF pig that received FIV-CFTR failed due to technical complications; thus, statistical comparisons were not performed. We concluded that complementation of the anion channel defect in vivo by lentiviral vector transduction has the potential to increase ASL pH and bacterial killing.
Cultured ethmoid sinus epithelia from FIV-CFTR–treated CF pigs show rescue of anion current and ASL pH.
The CF pigs in this study received vector to both the nasal and pulmonary airways. Two weeks after delivery, the ethmoid sinuses were harvested and epithelia were enzymatically dispersed, cultured on collagen-coated filters, and grown at an air-liquid interface. After differentiation in culture, ethmoid sinus epithelia were mounted in Ussing chambers to measure CFTR anion channel activity. Culturing ethmoid epithelial cells was necessary because ethmoid tissue cannot be directly mounted in the Ussing chambers. As shown in Figure 4, A and B, in cells from FIV-CFTR–treated animals, we observed an increase in transepithelial CFTR-dependent Cl– conductance. This current was blocked by GlyH-101 (Figure 4, A and C). No change in current was observed in cells from untreated animals. In addition to functional Cl– channel correction, we observed a significant increase in ASL pH in epithelia cultured from the pigs that received FIV-CFTR (Figure 4D). Together, these results provide evidence of functional correction of CF anion transport and ASL pH defects by a lentiviral vector in the ethmoid sinus in vivo.
Figure 4. FIV-CFTR corrects cystic fibrosis (CF) pig ethmoid sinuses.

(A) Well-differentiated ethmoid cultures from CF pigs treated with feline immunodeficiency virus–based viral vector expressing cystic fibrosis transmembrane conductance regulator (FIV-CFTR) were mounted into Ussing chambers, and bioelectric properties were measured. The change in transepithelial current was measured in response to low chloride (Cl–), (B) forskolin/3-isobutyl-1-methylxanthine (F&I), or (C) GlyH-101. Black diamonds indicate individual animals. n = cultured epithelial cells from 3 pigs; each data point represents 3 replicates/pig. *P < 0.05, Mann-Whitney nonparametric t test. (D) Cultured ethmoid sinuses from CF, non-CF, and CF pigs that received FIV-CFTR were treated with seminaphtharhodafluor (SNARF) + dextran and imaged by confocal microscopy to measure airway surface liquid (ASL) pH. **P < 0.0001, n = 9, one-way ANOVA comparison.
Discussion
Here, we show functional CFTR complementation in a large animal model. Two weeks after aerosolized FIV-CFTR delivery to CF pig lungs, freshly excised tracheal and bronchial tissues exhibited partial restoration of cAMP-stimulated anion conductance. The response was blocked by the CFTR inhibitor, GlyH-101. We also observed an increase in CFTR mRNA levels in treated tissues. Loss of CFTR function leads to the development of chronic bacterial lung infections (26, 27). In CF pigs, loss of CFTR-mediated bicarbonate transport leads to acidic airways and impaired bacterial killing (16). The CF pigs that received FIV-CFTR in our experiments showed a trend toward an increase in both tracheal ASL pH and bacterial killing. Cultured nasal epithelial cells from the pigs receiving FIV-CFTR also demonstrated partial restoration of anion channel activity and a significant increase in ASL pH comparable with WT levels, even after 2 weeks in culture. Together, these data provide the first evidence of CFTR correction in a large-animal model by a lentiviral vector.
Three well-studied candidate viral vector platforms for CF gene therapy include adenovirus, adeno-associated virus, and lentivirus (28). In addition, nonviral approaches are under study and in clinical trials (29, 30). Each viral vector has its own unique attributes and limitations. In the presence of reagents that disrupt tight junctions, adenovirus robustly transduces airway epithelia. However, adenoviral-mediated transgene expression wanes quickly due to the robust immune-mediated response (31). Adeno-associated virus persists long-term but has a relatively small carrying capacity for a transgene as large as CFTR. In a companion manuscript by Steines and colleagues (32), this limitation is circumvented by using a previously described partial R-domain deleted CFTR (CFTRΔR) (33).
Lentiviral vectors are promising candidates for CF gene therapy because they can persistently express a transgene of interest in the lung and potentially be readministered without immune suppression (34, 35). Lentiviral vectors evaluated for CF gene therapy include human immunodeficiency virus (HIV), simian immunodeficiency virus (SIV), and FIV, as well as equine infectious anemia virus (EIAV) (12, 34, 36–39). Typically, retroviral and lentiviral vectors are pseudotyped with vesicular stomatitis virus (VSV-G), but its preference for basolateral entry requires disruption of the tight junctions for efficient transduction (40–42). Alternate pseudotyping envelope proteins that confer tropism to the airway include baculovirus GP64 (24), influenza hemagglutinin (HA) (39), and the Sendai F and HN proteins (38). Lentiviruses pseudotyped with these envelope glycoproteins efficiently transduce airway epithelia in vivo and persist through the lifetime of a mouse. Integrating vectors that transduce airway epithelial cells and express long-term are favorable candidates to achieve efficient and persistent phenotypic correction after a single administration.
Progress for CF gene therapy has been hindered in part due to the lack of animal models that share similar lung disease phenotypes as humans. Though preclinical CF gene therapy studies show correction of CFTR anion channel defects both ex vivo and in vivo in mice (43, 44), phenotypic correction of animal models that recapitulate human CF lung disease is desirable to evaluate the potential of viral vectors as therapeutics. The development of the CF pig and ferret has opened the door to study CF disease pathogenesis, perform translational studies, and develop a standard of metrics to quantify endpoints for CF gene therapy.
The sinuses are nearly universally involved in CF (45, 46). Sinus disease pathogenesis has been characterized in the CF pig model (22, 47). In well-differentiated primary airway epithelial cultures from CF pig sinuses, delivery of CFTR by an adenoviral vector corrects the defective anion transport phenotype, suggesting that the CF pig is a good preclinical model for sinus gene therapy studies (22). In the present studies, we show that lentiviral-mediated delivery of CFTR in vivo complements the anion channel defect in sinus epithelia and increases the ASL pH of epithelial cells cultured from a FIV-CFTR–treated pig.
We acknowledge that this study has limitations. We emphasize that this is a short-term proof-of-principle study that does not assess the ability to treat or prevent disease, and we have not yet tested the ability to readminister the vector in pigs (34, 48). However, evidence of partial in vivo CFTR correction in this pilot study is encouraging. These findings set the stage for future studies of lentiviral vector gene therapy efficacy, persistence, and safety. Goals for future studies will include efforts to increase the transduction efficiency (e.g., modify envelope pseudotype and vector production), engineer the transgene cassette, and investigate the feasibility of repeated vector administration. Studies of longer duration are needed to assess the persistence of gene expression and the ability to target cells with progenitor capacity. This study achieved important milestones regarding the feasibility of lentiviral-mediated gene therapy for CF.
The CF pig spontaneously develops lung disease similarly to humans with CF, experiencing the development of acidic ASL pH, bacterial infections, inflammation, impaired MCT, and airway remodeling (15). This allows us to measure CFTR correction in a large-animal model that recapitulates key features of CF in humans. Sinus and airway epithelia are easily accessible for vector delivery, and these tissues are anatomically available for in vivo and ex vivo analysis. Importantly, the endpoints that we developed for this study allowed us to identify and quantify CFTR-dependent gene transfer events. Historically, CFTR function has been assessed by measuring anion transport. The assays of ASL pH and bacterial killing provide additional end points for phenotypic correction. Future directions will also focus on safety. Study goals include the measurement of innate and adaptive immune responses to vector application and transgene expression, the development of neutralizing antibodies, mapping of integration sites, and dose-escalation studies. This study and a companion study by Steines and colleagues using AAV to deliver CFTR to CF pigs are the first gene therapy studies to our knowledge to quantify anion transport, ASL pH, and bacterial killing in a large-animal CF model.
Methods
Pigs.
CF pigs were generated by homologous recombination in fibroblasts as previously described (49). Gut-corrected CF pigs were generated by somatic cell nuclear transfer cloning (19). These CF pigs were housed at the University of Iowa throughout the study. For viral vector delivery, newborn pigs were anesthetized using 2% isofluorane while oxygen levels, pulse, and respiratory rate were monitored. For the bacterial killing assay, pigs were i.m. anesthetized with ketamine (20 mg/kg) and xylazine (both Akorn Animal Health; 2 mg/kg), and anesthesia was maintained with propofol (Fresenius Kabi USA;1 mg/kg) i.v. Animals were euthanized via i.v. Euthasol (Virbac AH Inc.; 90 mg/kg) after pH and bacterial killing were measured.
Vector production.
The FIV vector used in this study was produced by the Indiana University Vector Production Facility in collaboration with the NHLBI Gene Therapy Resource Program. FIV expressed a codon-optimized porcine CFTR cDNA (accession no. KT184306) under control of the respiratory syncytial virus (RSV) promoter, and viral vector particles were pseudotyped with the baculovirus Autographa californica multicapsid nucleopolyhedrovirus GP64 envelope by transient transfection as described previously (50). Virus was concentrated 250-fold for in vitro studies and 1,000-fold for in vivo studies by overnight centrifugation at 9,000 g and resuspended in α-lactose buffer. Virus was titered by real-time PCR by the University of Iowa Viral Vector Core (51) (www.medicine.uiowa.edu/vectorcore).
Primary cultures of airway epithelia.
Airway epithelial cells from CF pigs were isolated by enzymatic digestion, seeded onto semipermeable filters, and grown at the air-liquid interface as previously described (52). Cultures were maintained in media supplemented with Ultro-ser G (USG) and the following antibiotics: penicillin (50 units/ml), streptomycin (50 μg/μl), gentamicin (50 μg/ml), fluconazole (2 μg/ml), and amphotericin B (1.25 μg/ml). To transduce cells in vitro, FIV-CFTR concentrated lentiviral vector supernatants were applied to the apical surface of cultured primary airway epithelia overnight.
In vivo viral vector administration.
FIV-CFTR was concentrated 1,000-fold, and 1.5 ml of vector was mixed at a 1:1 ratio with 2% methylcellulose (Methocel A4C; Dow Chemical Company) (24). Vector (1 ml) was delivered to nasal passageways by a bolus dose using a 24G Jelco catheter (Smiths Medical). For intratracheal delivery, a MADgic atomizer (LMA) was passed through the vocal cords under direct observation, and 2 ml of vector formulated with 1% methylcellulose was instilled.
Electrophysiology of CFTR anion channel.
CFTR correction was measured in Ussing chambers. Well-differentiated primary cultures or tissue explants were mounted into Ussing chambers (53). The apical and basolateral chambers were bathed in symmetrical Ringers solution (135 mM NaCl, 5 mM HEPES, 0.6 mM KH2PO4, 2.4 mM K2HPO4, 1.2 mM MgCl2, 1.2 mM CaCl2, 5 mM Dextrose). CFTR Cl– current was measured using a previously described protocol (53). After baseline transepithelial currents were measured, Amiloride (Sigma Aldrich) (100 μM) was used to inhibit Na+ channels, followed by 4,4’-Dilsothiocyano-2,2’-stilbenedifulonic acid (DIDS) (Sigma Aldrich) (100 μM) to inhibit calcium-activated Cl– channels. Once current stabilized, we replaced the apical solution with a low Cl– solution as previously described (54). We next applied the cAMP agonists Forskolin (Cayman Chemical) (10 μM) and 3-isobutyl-1-methylxanthine (IBMX) (Sigma Aldrich) (100 μM). After the current stabilized, GlyH-101 was added to block CFTR-mediated Cl– current. Transepithelial voltage (Vt) was maintained at 0 to measure transepithelial current (I) and maintained under the voltage clamp.
Localization of CFTR.
Primary airway epithelia were fixed in 10% Neutral Buffered Formalin (Leica Biosystems), paraffin embedded, and sectioned vertically. CFTR IHC was performed using mouse anti-CFTR monoclonal antibody (769, CFFT) as previously described (55).
qPCR.
CFTR mRNA abundance was quantified by qPCR using SYBR green (Thermo Fisher Scientific). Total RNA was collected from freshly excised tissue using the Direct-zol RNA MiniPrep protocol (Zymo Research) and reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Primer sequences: codon-optimized pig CFTR: Forward: 5′-ACAGGTTCAGCAAAGACATCG-3′, Reverse: 5′- CAGTGGCGAGGAAGATGTAAG-3′. RPL4 was used as a housekeeping gene: Forward: 5′- AGCGCTGGTCATGTCTAAAG-3′, Reverse: 5′- TTCCAGGCCTTAAGCTTCTTC-3′. Cycle conditions: 48°C for 30 min; 95°C for 10 min; 40 × 95°C for 15 sec, and 60°C for 1 min.
pH and bacterial killing assays.
Tracheal ASL pH was measured by placing a noninvasive dual lifetime referencing planar optode, a pH-sensitive foil, directly onto the tracheal surface as previously described (16, 25). Bacterial killing assays were performed as previously described (16). As reported in these studies, S. aureus isolate SA43 was cultured to log-phase growth and conjugated to gold electron microscopy grids, which were coated with streptavidin and biotin. Bacterial-coated grids were then placed on the airway surface for 1 minute, rinsed with PBS, and immersed in SYTO9 and propidium iodide (Invitrogen) to determine bacterial viability (Live/Dead Bacterial Viability Assay, Invitrogen). Grids were analyzed via confocal microscopy and quantified by Image J.
Statistics.
Statistically significant differences were calculated as indicated using one-way ANOVA comparison or Mann-Whitney nonparametric t test (Graphpad Prism). All data are presented as mean and ±SEM. P < 0.05 was considered statistically significant.
Study approval.
All animal procedures were reviewed and approved by the University of Iowa IACUC, Iowa City, Iowa and in accordance with the United States Department of Agriculture and National Institutes of Health guidelines.
Author contributions
ALC designed research studies, conducted experiments, acquired data, analyzed data, and wrote the manuscript. MHAA conducted experiments, acquired data, and analyzed data. VSS conducted experiments, acquired data, and analyzed data. DCB, MRS, and LSP acquired data and monitored animal management. NDG acquired data, analyzed data, and monitored animal management. DKM acquired data, analyzed data, and provided reagents. DAS conducted the study design and manuscript editing. MJW conducted the study design, data interpretation, and manuscript editing. PLS designed research studies, analyzed data, and wrote the manuscript. PBMJ designed research studies, analyzed data, and wrote the manuscript.
Acknowledgments
We acknowledge the work of Randall Prather of The University of Missouri (Columbia, Missouri, USA) and his help in generating the CF pig. We thank our lab members who volunteered their time to feed and care for the animals during the duration of the experiments. We thank the University of Iowa Office of Animal Resources and the animal caretakers. We thank Chris Wohlford Lenane and Peter Taft for their assistance through these studies. We thank Sarah Ernst for her expertise in the Ussing chamber studies. We thank Joseph Zabner for his collaboration and insight during these studies. We also thank Phil Karp and the In Vitro Models and Cell Culture Core for providing the primary cell cultures and the Viral Vector Core for titering the virus. This work was supported by the NIH (P01 HL-51670, P01 HL-091842, and R01 HL-105821) and the Center for Gene Therapy of Cystic Fibrosis (P30 DK-54759). We thank Ken Cornetta and Daniela Bischof of the NIH National Heart, Lung, and Blood Institute Gene Therapy Resource Program for their support and consultation.
Footnotes
Conflict of interest: MJW is a cofounder of Exemplar Genetics. DAS, MJW, and the University of Iowa Research Foundation have applied for a patent related to genetically modified pigs. MJW and PBM are founders of and hold equity in Talee Bio.
Reference information:JCI Insight. 2016;1(14):e88730. doi:10.1172/jci.insight.88730.
References
- 1.Riordan JR, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352(19):1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 3.Farrell PM, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr. 2008;153(2):S4–S14. doi: 10.1016/j.jpeds.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015;372(16):1574–1575. doi: 10.1056/NEJMc1502191. [DOI] [PubMed] [Google Scholar]
- 5.Abou Alaiwa MH, et al. Neonates with cystic fibrosis have a reduced nasal liquid pH; a small pilot study. J Cyst Fibros. 2014;13(4):373–377. doi: 10.1016/j.jcf.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoegger MJ, et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science. 2014;345(6198):818–822. doi: 10.1126/science.1255825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Accurso FJ, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363(21):1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogan MP, Stoltz DA, Hornick DB. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest. 2011;139(6):1480–1490. doi: 10.1378/chest.10-2077. [DOI] [PubMed] [Google Scholar]
- 9.Wainwright CE, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naldini L, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272(5259):263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 11.Delenda C. Lentiviral vectors: optimization of packaging, transduction and gene expression. J Gene Med. 2004;6 Suppl 1:S125–S138. doi: 10.1002/jgm.501. [DOI] [PubMed] [Google Scholar]
- 12.Sinn PL, et al. Lentiviral vector gene transfer to porcine airways. Mol Ther Nucleic Acids. 2012;1:e88730. doi: 10.1038/mtna.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oakland M, Maury W, McCray PB, Sinn PL. Intrapulmonary Versus Nasal Transduction of Murine Airways With GP64-pseudotyped Viral Vectors. Mol Ther Nucleic Acids. 2013;2:e88730. doi: 10.1038/mtna.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinn PL, Burnight ER, Hickey MA, Blissard GW, McCray PB. Persistent gene expression in mouse nasal epithelia following feline immunodeficiency virus-based vector gene transfer. J Virol. 2005;79(20):12818–12827. doi: 10.1128/JVI.79.20.12818-12827.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoltz DA, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2(29):e88730. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pezzulo AA, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487(7405):109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welsh MJ, Rogers CS, Stoltz DA, Meyerholz DK, Prather RS. Development of a porcine model of cystic fibrosis. Trans Am Clin Climatol Assoc. 2009;120:149–162. [PMC free article] [PubMed] [Google Scholar]
- 18.Hoegger MJ, et al. Assessing mucociliary transport of single particles in vivo shows variable speed and preference for the ventral trachea in newborn pigs. Proc Natl Acad Sci U S A. 2014;111(6):2355–2360. doi: 10.1073/pnas.1323633111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoltz DA, et al. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clin Invest. 2013;123(6):2685–2693. doi: 10.1172/JCI68867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drumm ML, et al. Correction of the cystic fibrosis defect in vitro by retrovirus-mediated gene transfer. Cell. 1990;62(6):1227–1233. doi: 10.1016/0092-8674(90)90398-X. [DOI] [PubMed] [Google Scholar]
- 21.Olsen JC, et al. Correction of the apical membrane chloride permeability defect in polarized cystic fibrosis airway epithelia following retroviral-mediated gene transfer. Hum Gene Ther. 1992;3(3):253–266. doi: 10.1089/hum.1992.3.3-253. [DOI] [PubMed] [Google Scholar]
- 22.Potash AE, et al. Adenoviral gene transfer corrects the ion transport defect in the sinus epithelia of a porcine CF model. Mol Ther. 2013;21(5):947–953. doi: 10.1038/mt.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang G, et al. Feline immunodeficiency virus vectors persistently transduce nondividing airway epithelia and correct the cystic fibrosis defect. J Clin Invest. 1999;104(11):R55–R62. doi: 10.1172/JCI8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinn PL, Shah AJ, Donovan MD, McCray PB. Viscoelastic gel formulations enhance airway epithelial gene transfer with viral vectors. Am J Respir Cell Mol Biol. 2005;32(5):404–410. doi: 10.1165/rcmb.2004-0410OC. [DOI] [PubMed] [Google Scholar]
- 25.Blossfeld S, Gansert D. A novel non-invasive optical method for quantitative visualization of pH dynamics in the rhizosphere of plants. Plant Cell Environ. 2007;30(2):176–186. doi: 10.1111/j.1365-3040.2006.01616.x. [DOI] [PubMed] [Google Scholar]
- 26.Paganin P, et al. Changes in cystic fibrosis airway microbial community associated with a severe decline in lung function. PLoS One. 2015;10(4):e88730. doi: 10.1371/journal.pone.0124348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cullen L, et al. Phenotypic characterization of an international Pseudomonas aeruginosa reference panel: strains of cystic fibrosis (CF) origin show less in vivo virulence than non-CF strains. Microbiology (Reading, Engl) 2015;161(10):1961–1977. doi: 10.1099/mic.0.000155. [DOI] [PubMed] [Google Scholar]
- 28.Sinn PL, Anthony RM, McCray PB. Genetic therapies for cystic fibrosis lung disease. Hum Mol Genet. 2011;20(R1):R79–R86. doi: 10.1093/hmg/ddr104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alton EW, et al. A Phase I/IIa Safety and Efficacy Study of Nebulized Liposome-mediated Gene Therapy for Cystic Fibrosis Supports a Multidose Trial. Am J Respir Crit Care Med. 2015;192(11):1389–1392. doi: 10.1164/rccm.201506-1193LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alton EW, et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med. 2015;3(9):684–691. doi: 10.1016/S2213-2600(15)00245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y, Li Q, Ertl HC, Wilson JM. Cellular and humoral immune responses to viral antigens create barriers to lung-directed gene therapy with recombinant adenoviruses. J Virol. 1995;69(4):2004–2015. doi: 10.1128/jvi.69.4.2004-2015.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steines B, et al. CFTR gene-transfer with AAV improves early cystic fibrosis pig phenotypes. JCI Insight. 2016;1(14):e88730. doi: 10.1172/jci.insight.88728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ostedgaard LS, et al. CFTR with a partially deleted R domain corrects the cystic fibrosis chloride transport defect in human airway epithelia in vitro and in mouse nasal mucosa in vivo. Proc Natl Acad Sci U S A. 2002;99(5):3093–3098. doi: 10.1073/pnas.261714599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinn PL, Arias AC, Brogden KA, McCray PB. Lentivirus vector can be readministered to nasal epithelia without blocking immune responses. J Virol. 2008;82(21):10684–10692. doi: 10.1128/JVI.00227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griesenbach U, et al. Assessment of F/HN-pseudotyped lentivirus as a clinically relevant vector for lung gene therapy. Am J Respir Crit Care Med. 2012;186(9):846–856. doi: 10.1164/rccm.201206-1056OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Limberis M, Anson DS, Fuller M, Parsons DW. Recovery of airway cystic fibrosis transmembrane conductance regulator function in mice with cystic fibrosis after single-dose lentivirus-mediated gene transfer. Hum Gene Ther. 2002;13(16):1961–1970. doi: 10.1089/10430340260355365. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi M, Iida A, Ueda Y, Hasegawa M. Pseudotyped lentivirus vectors derived from simian immunodeficiency virus SIVagm with envelope glycoproteins from paramyxovirus. J Virol. 2003;77(4):2607–2614. doi: 10.1128/JVI.77.4.2607-2614.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitomo K, et al. Toward gene therapy for cystic fibrosis using a lentivirus pseudotyped with Sendai virus envelopes. Mol Ther. 2010;18(6):1173–1182. doi: 10.1038/mt.2010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel M, Giddings AM, Sechelski J, Olsen JC. High efficiency gene transfer to airways of mice using influenza hemagglutinin pseudotyped lentiviral vectors. J Gene Med. 2013;15(1):51–62. doi: 10.1002/jgm.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sinn PL, et al. Lentivirus vectors pseudotyped with filoviral envelope glycoproteins transduce airway epithelia from the apical surface independently of folate receptor alpha. J Virol. 2003;77(10):5902–5910. doi: 10.1128/JVI.77.10.5902-5910.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cmielewski P, Anson DS, Parsons DW. Lysophosphatidylcholine as an adjuvant for lentiviral vector mediated gene transfer to airway epithelium: effect of acyl chain length. Respir Res. 2010;11:e88730. doi: 10.1186/1465-9921-11-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang G, et al. Increasing epithelial junction permeability enhances gene transfer to airway epithelia In vivo. Am J Respir Cell Mol Biol. 2000;22(2):129–138. doi: 10.1165/ajrcmb.22.2.3938. [DOI] [PubMed] [Google Scholar]
- 43.Griesenbach U, Alton EW, UK Cystic Fibrosis Gene Therapy Consortium Gene transfer to the lung: lessons learned from more than 2 decades of CF gene therapy. Adv Drug Deliv Rev. 2009;61(2):128–139. doi: 10.1016/j.addr.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 44.Alton EW, et al. Non-invasive liposome-mediated gene delivery can correct the ion transport defect in cystic fibrosis mutant mice. Nat Genet. 1993;5(2):135–142. doi: 10.1038/ng1093-135. [DOI] [PubMed] [Google Scholar]
- 45.Robertson JM, Friedman EM, Rubin BK. Nasal and sinus disease in cystic fibrosis. Paediatr Respir Rev. 2008;9(3):213–219. doi: 10.1016/j.prrv.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 46.Chang EH, et al. Sinus hypoplasia precedes sinus infection in a porcine model of cystic fibrosis. Laryngoscope. 2012;122(9):1898–1905. doi: 10.1002/lary.23392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang EH. New insights into the pathogenesis of cystic fibrosis sinusitis. Int Forum Allergy Rhinol. 2014;4(2):132–137. doi: 10.1002/alr.21252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alton EW, et al. The safety profile of a cationic lipid-mediated cystic fibrosis gene transfer agent following repeated monthly aerosol administration to sheep. Biomaterials. 2013;34(38):10267–10277. doi: 10.1016/j.biomaterials.2013.09.023. [DOI] [PubMed] [Google Scholar]
- 49.Rogers CS, et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321(5897):1837–1841. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnston JC, et al. Minimum requirements for efficient transduction of dividing and nondividing cells by feline immunodeficiency virus vectors. J Virol. 1999;73(6):4991–5000. doi: 10.1128/jvi.73.6.4991-5000.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sinn PL, et al. Enhanced gene expression conferred by stepwise modification of a nonprimate lentiviral vector. Hum Gene Ther. 2007;18(12):1244–1252. doi: 10.1089/hum.2006.127. [DOI] [PubMed] [Google Scholar]
- 52.Karp PH, et al. An in vitro model of differentiated human airway epithelia. Methods for establishing primary cultures. Methods Mol Biol. 2002;188:115–137. doi: 10.1385/1-59259-185-X:115. [DOI] [PubMed] [Google Scholar]
- 53.Chen JH, et al. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143(6):911–923. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cooney AL, Singh BK, Sinn PL. Hybrid nonviral/viral vector systems for improved piggyBac DNA transposon in vivo delivery. Mol Ther. 2015;23(4):667–674. doi: 10.1038/mt.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meyerholz DK, et al. Immunohistochemical Detection of Markers for Translational Studies of Lung Disease in Pigs and Humans. Toxicol Pathol. 2016;44(3):434–441. doi: 10.1177/0192623315609691. [DOI] [PMC free article] [PubMed] [Google Scholar]