

Abstract
Globoid cell leukodystrophy (GLD), also known as Krabbe disease, is a fatal demyelinating disease accompanied by the formation of giant, multinucleated cells called globoid cells. Previously believed to be a byproduct of inflammation, these cells can be found early in disease before evidence of any damage. The precise mechanism by which these globoid cells cause oligodendrocyte dysfunction is not completely understood, nor is their cell type defined. In this review we outline the idea that microglial cells are transformed into an unknown, and undefined, novel M3 phenotype in GLD, which is cytotoxic to oligodendrocytes, leading to disease progression.
Keywords: microglia, globoid cell leukodystrophy, globoid cells
1. Globoid Cell Leukodystrophy (GLD)
Globoid cell leukodystrophy (GLD), also known as Krabbe disease, is a genetic demyelinating disease that affects the nervous system. GLD results from loss of function mutations in the gene encoding galactosylceramidase (galc) which results in a pathological accumulation of a cytotoxic lipid intermediate called galactosylsphingosine, or psychosine (Graziano and Cardile 2014). It is thought that the supraphysiological level of psychosine is the root etiology of this disease, yet the process of demyelination is not understood. The ‘psychosine hypothesis’ has been the predominant explanation for GLD pathology for over 40 years, and postulates that the accumulation of psychosine is responsible for the death of myelinating oligodendrocytes, resulting in the profound demyelination observed in GLD (Suzuki 1998). However, presence of multi-nucleated phagocytes called globoid cells, can be identified well before overt demyelination in GLD (Fig. 1A). Globoid cells are present in fetal tissue before the synthesis of myelin occurs (Kobayashi et al. 1988; Suzuki 1998). Studies in our lab have demonstrated that psychosine, when added to mixed glial cultures, consisting of astrocytes and microglia, can activate microglia, leading to the formation of globoid cells, and under conditions which mimic the extracellular environment of the GLD brain, can be cytotoxic to oligodendrocytes (Ijichi et al. 2013). Our study suggests that a pathophysiological effect of psychosine is not limited to oligodendrocyte death, but rather elevated psychosine in GLD may impact many CNS cell types. In particular, our recent studies have shown that one consequence of elevated psychosine in GLD is microglial activation that is mediated, at least in part, by psychosine-mediated activation of astrocytes. The early activation of glia, specifically astrocytes and microglia, observed in GLD neurospecimens at autopsy, may be a key cause contributing to demyelination in this disease. In this review we propose a microglial hypothesis for the pathology of GLD based on recent findings in the field which advances our understanding on the cell biology of this condition and may open new avenues of therapeutic intervention for GLD.
Figure 1. Temporal Comparisons of GLD Abnormalities in Twitcher Mice and the Microglial Hypothesis.
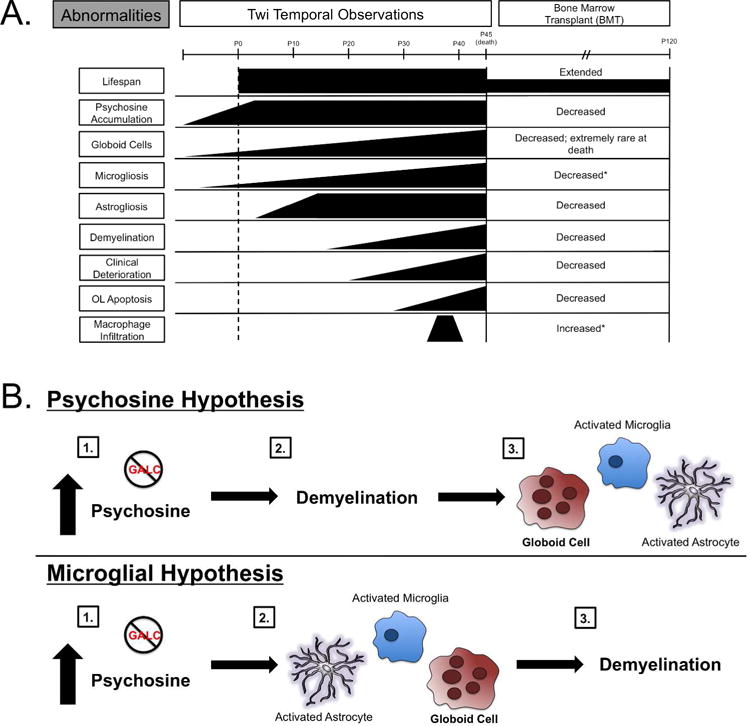
A. Bone marrow transplant of wild type mice into twitcher mice resulted in an extended lifespan to about P120 compared to average twitcher death of P45, a decrease in overall psychosine accumulation, a decrease in globoid cells, a decrease in demyelination, and an increase in macrophage infiltration (Hoogerbrugge et al. 1988a). * indicates unknown activation state. B. The psychosine hypothesis postulates that psychosine accumulation kills oligodendrocytes leading to demyelination and eventual inflammation, such as globoid cell formation, activated microglia, and activated astrocytes. The microglial hypothesis postulates that psychosine accumulation leads to activated astrocytes, microglia, and globoid cell formation, which then induces demyelination.
1.1 Brief Overview of GLD
Globoid cell leukodystrophy is a genetic sphingolipidosis that results in central and peripheral demyelination, and is a classic example of a lysosomal storage disease, as the GALC enzyme is no longer functioning to degrade psychosine, leading to its accumulation (Graziano and Cardile 2014). When expressed, GALC breaks down galactolipids found predominantly in the nervous system and kidneys, where lipids contribute to myelin and membranes, respectively (Suzuki 1998). Natural levels of some galactolipids can function to regulate cycloxygenase inflammatory activity (Hou et al. 2007), whereas loss of GALC function in GLD results in accumulation of psychosine which can be cytotoxic. Using a sensitive assay method, elevated levels of psychosine have been found to accumulate in all tissues in fetal (22–52 weeks old) and infantile (0–2 years old) GLD cases, including the brain, spinal cord, kidney liver, spleen, and lung, concurrently with the characteristic formation of globoid cells present in nervous system tissues (Kobayashi et al. 1988; Svennerholm et al. 1980). The natural function of psychosine in homeostasis and physiology is not known.
GLD is an orphan disease, and it is often grouped together with other infrequent diseases of CNS white matter, referred to as leukodystrophies. In the general population, GLD has a prevalence of 1:250,000 out of live births (Barczykowski et al. 2012). This infantile form of the disease is fatal, with death before the age of 2. In the early onset infantile type of GLD symptoms begin to emerge within the first 6 months of life (Duffner et al. 2009b; Graziano and Cardile 2014). These include irritability, limb stiffness, seizure, vomiting, and quickly progress into rapid and severe motor and mental retardation. Lastly, most patients develop blindness and deafness, and progressively exhibit less responsiveness to their environment (Suzuki 1998). Currently, there are no effective long-term or widely available treatments for this disease. Hematopoietic stem cell transplantation (HSCT) has been found to ameliorate the disease if performed during the neonatal period (Escolar et al. 2005; Peters et al. 2003), but for most, management of symptoms and convalescent treatments are the only options.
1.2 Classification and Therapy for GLD
Based on the age at symptom onset there are four different forms of GLD, the infantile (0–6 months old), late infantile (7 months - 3 years old), juvenile (4–8 years old), and lastly the adult form (over 9 years old) (Kolodny et al. 1991). The infantile form of the disease represents over 90% of all GLD cases, where these patients typically die within the two years of clinical disease onset (Duffner et al. 2009b; Hagberg et al. 1963). The clinical symptoms of later onset GLD are similar to the infantile symptoms, which are mentioned in the above section, yet the disease course and severity does differ between the onset ages and patients (Duffner et al. 2012; Phelps et al. 1991). Typically the progression and severity of the disease is milder in comparison to early-onset infantile GLD (Duffner et al. 2012).
Currently, the most effective diagnostic test for GLD is an assay for GALC enzymatic activity, which is then confirmed by genetic mutation analysis. In a blood sample, for example, if GALC activity is found to be less than 0.5 nmol/h/mg protein, then the patient is considered at risk for GLD (Duffner et al. 2009a). In addition to these diagnostic procedures, a confirmed diagnosis of GLD also requires neurological and non-neurological tests to assess the list of characteristic features and aforementioned symptoms.
The primary approach of current therapies for GLD has focused on restoring functional GALC in patients. The physiological objective of this enzyme replacement therapy is to deplete the accumulated psychosine to mitigate pathology including oligodendrocyte cell loss. Many of these therapies have shown promise using twitcher mice, the authentic enzymatic murine model of GLD discovered in 1976 as a spontaneous mutation in the galc gene in C57BL/6 mice at Jackson Labs (see below). In twitcher (twi) mice, direct administration of the recombinant GALC enzyme was found to be unsuccessful in reversing the twitcher phenotype as the protein did not efficiently incorporate into the CNS (Umezawa et al. 1985). An alternate method of bone marrow transplantation (BMT) was later shown to increase GALC activity by 7–8 fold and also reduce psychosine accumulation in twi mice. In addition, BMT was also found to reduce the presence of globoid cells in the CNS (Hoogerbrugge et al. 1988a). Additional evidence of remyelination was also found in twi mice that received BMT which identified improved CNS myelination in association with an increased lifespan (Fig. 1A) (Hoogerbrugge et al. 1988a). Translation of these findings to the clinical setting has resulted in what is currently the only treatment that increases lifespan for GLD patients: hematopoietic stem cell transplantation (HSCT). This is an aggressive and often risky treatment approach, especially for very young patients. Cells for HSCT are generally sourced from the bone marrow or umbilical cord. Transplanted donor cells may provide a dual benefit: a) quelling inflammation and b) providing a source of functional GALC that can supplant the mutated form in the host CNS (Escolar et al. 2005). Therapeutic correction of GALC deficiency in the brain of HSCT recipients is still questionable. BMT and other virus-based gene therapies are all intended to provide enzyme replacement as a means to remedy the accumulated psychosine in the patient CNS and decrease, or potentially reverse tissue damage. After several years of HSCT some GLD patients have shown improvement, but the effectiveness and widespread availability of HSCT is limited. More importantly, the efficacy of HSCT is improved when performed in the pre-symptomatic period before major damage has occurred but also when the patients are typically very young (Duffner et al. 2012; Krivit et al. 1998), but also less suited to endure the treatment itself.
2. Pathophysiology of GLD
2.1 Mouse Model of GLD: Twitcher Mouse
In 1976 the Jackson Laboratory discovered mice of the inbred C57/BL6 strain that developed a tremor, had a low body weight, and had progressive weakness in the limbs, then died prematurely around postnatal day (P) 45. Post-mortem analyses of these mice revealed significant demyelination both in the CNS and PNS, as well as the presence of multinucleated globoid cells: all features comparable to human GLD pathology (Duchen et al. 1980; Suzuki and Suzuki 1995). Based on their visible tremor and weakness, which starts at about P20, the mice were named “twitchers” (Twi). Twi mice closely resemble the biochemical and neuropathological findings, as well as the clinical course of the human disease. Disease progression in twi mice is rapid and mice rarely survive beyond 45 days of age (Suzuki and Taniike 1995). Genetic analysis of twi mice determined that the basis for this mouse form of GLD was a missense mutation resulting in a guanine to adenine substitution at residue 1017 in the galc gene (Sakai et al. 1996). Twi mice also develop highly elevated levels of psychosine comparable to the accumulation found in humans in the nervous system (Shinoda et al. 1987). This mouse has proved to be an authentic enzymatic recapitulation of GLD and thus emerged as an incredibly useful resource for studying this disease, as pathological features are essentially identical with those of human infantile GLD (Suzuki and Suzuki 1995).
2.2 Galactosylceramidase (GALC) and the Psychosine Hypothesis
GALC is a galactolipid hydrolase that resides in the lysosome, and hydrolyzes galactolipids, breaks large galactolipids including psychosine and galactosylceramides, into constituent components of galactose and their sphingoid bases (Suzuki and Suzuki 1970). In a normal nervous system, substrates of GALC are processed by the lysosome, and the recycled components are able to be reused in lipid synthesis (Kolter and Sandhoff 2006). Due to the mutations in galc, degradation of cerebrosides, the principal glycosphingolipid in brain tissue, is impaired. This results in psychosine accumulation in GLD affected individuals during development (Graziano and Cardile 2014). There are over 110 identified mutations in the human galc gene, but the most common mutation identified in infantile GLD is a 30 Kb deletion at position 502 within intron 10 (Luzi et al. 1995). In addition, there are several other mutations found within various populations, including the point mutation G1582A, which is found in the Israeli population (Rafi et al. 1995; Wenger et al. 2000). Regardless of where the mutation is, all patients have significantly lower levels of functionally active GALC (Jalal et al. 2012).
Psychosine, a galactolipid, is considered a natural byproduct made during membrane lipid synthesis globally. Within the lipid-rich environment of the CNS biosynthesis of galactolipids reaches a peak during the most active period of myelination which occurs during the first 18 months of life in humans and the first 15–25 days in rodents (Costantino-Ceccarini and Morell 1972; Miyatake and Suzuki 1972; Vanier and Svennerholm 1976). This correlation suggests that perturbation in lipid synthesis renders myelination particularly susceptible to disruption in GLD. Psychosine is produced within a cell by the binding of uridine diphosphate (UDP)-galactose to sphingosine, which may be catalyzed by an enzyme, UDP-galactose:ceramide galactosyltransferase (CGT) (Cleland and Kennedy 1960). Expression of CGT has been found to be higher in myelinating cells, when compared to neurons, making them more susceptible to accumulate psychosine (Schaeren-Wiemers et al. 1995). Because lipid synthesis is an ongoing process during development, exceptionally high psychosine levels can accumulate before myelination occurs in GLD (Kobayashi et al. 1988). For instance, elevated psychosine is found in human fetal GLD tissue, at 21 weeks of gestation, when there is neither overt myelination nor demyelination (Ida et al. 1994). Accumulation of psychosine is over 100 fold higher in the GLD nervous system than in other organs, including the kidney, liver, and thymus, underlining the susceptibility of the CNS to psychosine (White et al. 2009; Zhu et al. 2012). Psychosine is believed to accumulate in lipid rafts, which can then alter many signaling pathways (White et al. 2009), and has also been shown to accumulate both intracellular and extracellularly, yet the precise mechanism of how psychosine contributes to GLD pathology is unclear. In general psychosine is cytotoxic to many cell types, although the natural physiological function in homeostasis remains unknown. The accumulation of psychosine in preterm fetal tissues, when overt demyelination is not possible, suggests that 1) the origin of GLD pathogenesis is much more than just a demyelinating disease, 2) that psychosine may have other sources in addition to myelinating cells, and 3) psychosine must have impact on other cell types that contribute to this disease.
2.3 Globoid Cells, Microglia, and GLD Pathology
Globoid cells are one of the hallmarks of GLD pathology. These cells are multinucleated phagocytes, and typically are 10–15 μm in diameter (Duchen et al. 1980; Pollanen and Brody 1990). They have been found to contain fatty filamentous inclusions that stain positive for periodic acid-Schiff (PAS), which may be phagocytized myelin, as these cells are often found in proximity to sites where demyelination is prominent (Itoh et al. 2002; Suzuki 1970). (Fig. 1A). Although globoid cells are characteristic of this disease, and even its namesake, the origins and function of globoid cells have not been addressed until recently.
Globoid cells have been to thought to originate from CNS resident microglia and/or infiltrating peripheral macrophages. The basis for this thinking has been that both populations of CNS resident microglia and peripheral macrophages are immunoreactive to the same markers MAC-1, ferrin, CD68, and Iba1. Previous work by Wenger et al., has reported globoid cells are not found in the PNS, supporting that globoid cells are of microglial origin rather than from macrophages (Wenger et al. 1999). However, others have suggested globoid cells may be found in the PNS, but only at later stages of the disease when the blood nerve barrier loses its sealing properties (Kobayashi et al. 1985). Interestingly, in twi mice the ablation of CXCR2, a key factor for macrophage infiltration, did not stop globoid cell formation, strengthening the argument for microglia as the cell of origin for globoid cells in GLD (Reddy et al. 2013). Based on other diseases where multi-nucleated “giant cells” are observed, including human immunodeficient virus (HIV)-encephalopathy, this specialized and unique phagocytic phenotype is believed to be a secondary response of the phagocytes to prolonged inflammation resulting from preceding and ongoing tissue damage (Masliah et al. 1996; McNally and Anderson 2011). On the basis of this interpretation, until recently, globoid cell formation had been thought to be a result of chronic demyelination and inflammation seen in GLD; however, globoid cells are found earlier than demyelination in GLD and even before damage-associated inflammation is measured (Potter et al. 2013). Together these lines of evidence point to globoid cells being of microglial origin and for microglia having more than a reactionary role in disease pathology in GLD (Claycomb et al. 2014; Ida et al. 1994; Martin et al. 1981).
Our data further support globoid cells as coming exclusively from microglia. This conclusion is based on our work showing that isolated macrophages, when exposed to psychosine do not become globoid cells, while microglia do (Ijichi et al. 2013). In addition, when staining human GLD tissue, and “globoid-like” cells in vitro, these cells were found to be immunoreactive for Iba1, a marker of phagocytes, and CD16/32 a pro-inflammatory microglial and macrophage marker (Claycomb et al. 2014). At present activated microglia are thought to mimic the activation patterns of peripheral macrophages, which are known to have three states: an M0 resting state, an M1 pro-inflammatory state, and an M2 anti-inflammatory state (Michell-Robinson et al. 2015). These states can be identified using antibodies against specific cell surface markers (I). For example, classically activated M1 macrophages, or microglia, can be identified by CD16/32, while alternatively activated M2 macrophages and microglia can be identified by elevated expression of the the mannose receptor. Just like their macrophage counterparts, resting microglia can be activated to an M1 phenotype by exposure to pro-inflammatory cytokines such as TNF-α and IL-6, which are both associated with psychosine-mediated cell death (Formichi et al. 2007). These M1-inducing cytokines in turn trigger release of additional pro-inflammatory cytokines and chemokines: IL-1β, TNF-α, IL-6, IL-23, MIP-1β, and MCP-1 (Boche et al. 2013; Luzi et al. 2009), which can induce further cell death. Activated M1 microglia have been shown to be cytotoxic towards mature oligodendrocytes in vitro, via the secretion of factors such as TNF-α and nitric oxide (Merrill et al. 1993; Zajicek et al. 1992). IL-4, IL-13, and IL-10 are associated with activating microglia and macrophages towards the M2 phenotype, which can then be identified using markers such as arginase-1 (Arg-1) and CD206, also known as the mannose receptor (Michell-Robinson et al. 2015). These activated cells can promote phagocytosis, extracellular matrix construction, and tissue repair via the secretion of factors such as TGFβ, IL-10, and IL-13 (Tang and Le 2015). Twi mice have been found to have increased levels of Arg-1, CD206, and TGFβ, suggesting that the M2 phenotype may also play a role in this disease (Table I) (Kondo et al. 2011). Future studies will be needed to fully characterize the timing, extent, and roles of activation markers as they relate to microglial phenotypes in this disease.
Table I. Microglial Activation in Twitcher Mice.
Proposed nomenclature of psychosine activated microglia, M3.1 and M3.2. In GLD, M3.1 cells are psychosine activated and grown on tenascin-C, become multi nucleated, show the classic M1 markers, and are cytotoxic. M3.2 cells are also psychosine-activated, multi nucleated, but exhibit M2 markers and are non cytotoxic.
| Nomenclature | Activating Molecules | Markers | Secreted Factors | Roles | |
|---|---|---|---|---|---|
|
M1 (classical activation) |
LPS IFN-γ TNF-α*1 |
CD86*1 MHCII*1 CD16/32*1 |
iNOS*1 IL-1β*1 TNF-α*1 IL-6*1 IL-23 |
- Cytotoxic - Astrocyte activation - Pro-inflammatory |
|
|
M2 (alternative activation) |
IL-4 IL-13*2 IL-10 |
Arg1*1 CD206*1 |
TGFβ*1 IL-10 IL-13*2 |
- Phagocytosis - Anti-inflammatory - Tissue Repair - Neurotrophic |
|
| Proposed | |||||
|
M3.1 (multi-nucleated) |
Psychosine* Tenascin-C*3 |
MHCII*1 CD16/32*1 |
TNF-α*1 | - Activation with multinucleation - Cytotoxic phenotype - Phagocytosis |
|
|
M3.2 (multi-nucleated) |
Psychosine* Laminin |
CD206*1 | ? | - Activation with multinucleation - Non-cytotoxic phenotype - Phagocytosis |
|
indicates an increase in twitcher mouse CNS
Interestingly, when twi mice were crossed with a macrophage-deficient osteopetrotic mutant mouse the number of non-myelinated axons was significantly higher in the mutant mouse when compared to a twi mouse, and this appeared to be due to impaired remyelination rather than accelerated demyelination (Kondo et al. 2011). The lack of remyelination in the mutant macrophage-deficient twi mice was due to the lack of phagocytosis of myelin debris, which is carried out by macrophages. Overall, this study demonstrates that macrophages in GLD are beneficial in order to promote myelin repair, which suggests that macrophages do not become cytotoxic globoid cells when exposed to demyelination and psychosine accumulation. Instead, macrophages may physiologically counteract the dysfunction of CNS resident microglia.
3. The Cell Biology of the Psychosine Hypothesis Revisited
The early pathological activation of microglia into inflammatory globoid cells, caused by the accumulation of psychosine in the fetus, may be the primary cause of cell death in this disease, rather then just the accumulation of psychosine. We propose that an earlier pathogenic role for microglia in this disease complements the long-standing psychosine hypothesis of GLD. The psychosine hypothesis was formulated by Miyatake and Suzuki in 1972, and states that the abnormal accumulation of psychosine in GLD causes a direct death of oligodendrocytes (Fig. 1B) (Miyatake and Suzuki 1972). Many studies since have confirmed the cytotoxic potential for psychosine to elicit cytotoxic effects directly on oligodendrocytes in vitro (Cho et al. 1997; Haq et al. 2003; Jatana et al. 2002; Zaka and Wenger 2004), which has lent support to this hypothesis.
3.1 Revisiting the Psychosine Hypothesis
Psychosine is not only cytotoxic to oligodendrocytes, but is also toxic to neurons in vitro (Castelvetri et al. 2011; Sugama et al. 1990). Axonal swelling is observed prior to demyelination in the twi mouse, and even at one week of age there is abnormal axonal transport in twitcher mice (Cantuti Castelvetri et al. 2013; Castelvetri et al. 2011). Research done using the GALCtwi-5J mouse, a spontaneous mutation in GALC that matches the missense mutation in patients with infantile GLD, shows the typical neuropathological hallmarks including globoid cells, gliosis, and psychosine accumulation, but the CNS shows no demyelination or axonal loss (Potter et al. 2013). This suggests that the accumulation of psychosine may not directly lead to myelin loss in this disease, and instead may cause neuroinflammation eventually leading to death. This mouse line also demonstrates that the formation of globoid cells occurs before significant demyelination, and is therefore not a secondary inflammatory reaction.
Psychosine has been found to inhibit cytokinesis in various cell types including U937 monocytes and HeLa cells, leading to the formation of globoid-like cells in vitro, but their cytotoxic properties were not investigated (Kanazawa et al. 2000). The inhibition of cytokinesis by psychosine may account for the formation of globoid cells, but this still does not explain their purpose in the disease, or how exactly accumulated psychosine accounts for all aspects of GLD.
Oligodendrocyte cell death is attributed to the intracellular accumulation of psychosine, which can then induce cellular apoptosis (Giri et al. 2008). A study published by Kondo and colleagues transplanted oligodendrocytes isolated from twi mice into the shiverer mouse, a model of hypomyelination, and demonstrated that the transplanted oligodendrocytes survived and myelinated axons in the shiverer mouse (Kondo et al. 2005). The GLD brain environment may contribute to the damage to oligodendrocytes not limited to the effects of psychosine directly. Most importantly the psychosine hypothesis does not take into account the cytotoxicity of globoid cells, and how these cells are not just a byproduct of damage-induced inflammation since they are found in the fetal spinal cord well before myelin damage is observed (Ida et al. 1994; Martin et al. 1981). Since globoid cells are found before myelin damage occurs there is an inherent, underlying inflammation that can induce the activation of globoid cells to cause oligodendrocyte cell death, and a subsequent increase in overall CNS inflammation. Therefore in light of these initial findings much more focus needs to put into investigating the function of globoid cells and their contribution to GLD pathology.
3.2 The Microglial Hypothesis of GLD
Microglia are CNS-resident macrophages, which originate from hematopoietic stem cells (HSCs) from the yolk sac that enter the developing neuroepithelium around embryonic day (E)8.5–9.5 in mice (Alliot et al. 1999; Alliot et al. 1991), and in human fetuses microglia-like cells can be detected at three weeks of gestational age (Hutchins et al. 1990). The primary role of microglia is to survey the CNS and defend against any sort of invasion and traumatic injuries (Ginhoux et al. 2013). Microglia have many other roles in the CNS, such as maintaining CNS homeostasis (Cunningham et al. 2013), regulating synapse formation (Schafer et al. 2013), and aiding tissue maintenance and repair (Michell-Robinson et al. 2015).
Since the development of the psychosine hypothesis microglia have not been a primary focus in studying GLD pathology, but evidence shows strong implications that microglia play an important role in GLD. Sites of demyelination in twi mice almost always coexist with astrogliosis and highly activated microglia (Ohno et al. 1993). In addition, the CNS of twi mice has been found to contain high levels of proinflammatory cytokines including IL-6, TNFα, iNOS, and RANTES, which are all associated with activated microglia (Table I) (Kondo et al. 2011; Wu et al. 2000). Based on these data, and since globoid cells are found in fetal tissues before the presence of overt demyelination, we propose that globoid cells are an aberrant form of activated microglia.
In the past couple of years activation states of microglia have been studied and further elucidated into two classic categories: the M1, classic activation, and the M2, alternative activation (Gordon 2003). When referring to microglia switching between the activation states the term ‘polarization’ is used, as it refers to a shift in molecular profile of the cell from a basal condition (Michell-Robinson et al. 2015). Typically, in the literature, M1 cells are activated via LPS and/or IFN-γ, while M2 cells are activated by IL-4 or -13 (Table I) (Martinez and Gordon 2014; Michell-Robinson et al. 2015). M1 cells are characterized by their rounded, amoeboid morphology, known to be a hyperactive state, while M2 cells have a more elongated morphology (Mantovani et al. 2004; Van Ginderachter et al. 2006). The M1 activated cells express surface makers such as CD80, CD86, CD16/32, CCR7, and produce reactive nitrogen species and pro-inflammatory cytokines such as IL-12 and TNF-α (Fairweather and Cihakova 2009; Van Ginderachter et al. 2006). In turn, M1 activated microglia can induce increased inflammation onto other cell types, such as astrocytes. By contrast, M2 polarized microglia produce anti-inflammatory cytokines such as IL-10, and show increased phagocytic activity (Biswas and Mantovani 2010). In addition, these cells support tissue remodeling and promote fibrosis through arginase-1 activity, which can also be used as a marker for this microglial phenotype (Hesse et al. 2001; Wynn 2004). Interestingly, in vitro, M1 polarized microglia exert cytotoxic effects on both neurons and oligodendrocytes, while M2 microglia promote neurite outgrowth, phagocytosis, and extracellular matrix (ECM) reconstruction (Hu et al. 2012; Kigerl et al. 2009; Ponomarev et al. 2007). However, these distinctions are not as simple as just M1 and M2; overlapping phenotypes have been observed (Biswas and Mantovani 2010; David and Kroner 2011). For example, there are several distinct subgroups within the M2 phenotype, including M2a, M2b, and M2c. The M2a activated cell is associated with the anti-inflammatory phenotype, and is induced via IL-4 and IL-13 (Leidi et al. 2009; Taylor et al. 2005), M2b is associated with pro-inflammatory cell surface expression, activated by LPS (Edwards et al. 2006), and lastly, M2c is associated with tissue remodeling, and is activated via IL-10 and TGF-β1 (Leidi et al. 2009; Mantovani et al. 2004). Based on the varying states of microglia activation, these likely reflect a spectrum of activity states rather than a set of distinct activated phenotypes.
Damage in the CNS typically entails the recruitment of microglia resulting in an innate immune response, yet the role of microglial polarization in damage and disease is just being elucidated. In experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis, a lack of IL-4 in the CNS impaired M2 polarization of microglia and exacerbated symptoms (Ponomarev et al. 2007). Examination of post-mortem human tissue from patients with multiple sclerosis active lesions showed both M1 and M2 microglia (Vogel et al. 2013). In a murine model of focal demyelination by injection of lysolecithin, microglia polarized to the M2 phenotype, which was shown to promote oligodendrocyte differentiation and remyelination (Miron et al. 2013). Many studies demonstrate the neuroprotection and remodeling properties of M2 polarized microglia in demyelinating diseases, yet globoid cells, a microglial cell, have not been investigated.
3.3 Do Globoid Cells Fit the “Classical” M1 and “Alternatively Activated” M2 Phenotypes?
Globoid cells have yet to be categorized within these macrophage/microglial phenotypes, but understanding the polarization of these cells might help in further elucidating the role they play in this disease. Characterization of the inflammatory profile in sick (P45) twi mouse spinal white matter, Kondo and colleagues found that both M1 and M2 microglia markers are up-regulated in twi mice, including iNOS, CD16/32, TNF-α, and CD86 for the M1 cells, and arginase-1 and CD206 for the M2 cells (Table I) (Gordon 2003; Kigerl et al. 2009; Kondo et al. 2011; Mantovani et al. 2005). TNF-α, generally produced by M1 polarized cells, works as a destructive pro-inflammatory cytokine, but it has also been shown to promote proliferation of oligodendrocyte progenitor cells (Arnett et al. 2001). Arginase-1 is a known M2 polarization marker that counteracts myelin inhibition (Cai et al. 2002). High expressions of IL-6, iNOS, and IGF- 1 were all reported in twi mouse CNS tissue, which all represent the M1 phenotype of inflammation (Kondo et al. 2011; Michell-Robinson et al. 2015). Interestingly, an increase in CD206 (mannose receptor) is seen in the twi mice, underlining an M2 anti-inflammatory phenotype (Kondo et al. 2011). It is important to note that these analyses were done on Iba1 microglia and infiltrating macrophages but not specifically on multinucleated globoid cells, which also express distinct patterns of M1 and/or M2 phenotype markers.
The definitions of M1 and M2 macrophage activation markers emerged from understanding macrophage responses to cytokines and/or pathogens. This classification however, may not appropriately or adequately categorize the unique nature of microglial activation or the phenotype of globoid cells in GLD. We suggest that that the adoption of a new subclass is needed to distinguish instances where M1/M2-like classification is neither appropriate nor adequate due to the “non-traditional” psychosine-activation of microglia in GLD. Hence, we suggest that microglia under circumstances like GLD may be denoted by a new classification: M3. This M3 designation should be used to identify activated microglia and globoid cells that are distinguished from macrophages, because there is increasing evidence microglia can express unique complexity when assessing M1 and M2 markers (Michell-Robinson et al. 2015), including CD16/32 and CD206 markers in GLD (Table I) (Claycomb et al. 2014). Altered microglia have also been found in the mouse model of amyotrophic lateral sclerosis (ALS), where they express both M1 and M2 markers, where both neuroprotective and toxic factors are concurrently upregulated (Chiu et al. 2013). This study, using deep RNA sequencing, suggests that there are unique “mutant” microglia in this mouse model of ALS during the process of neurodegeneration. It may be worth to repeat this type of study on the mutant twi mice to investigate these altered globoid microglia. Another transcriptome-based study showed a spectrum of macrophage activation states, using diverse activation signals, which extend past the M1 and M2 model (Xue et al. 2014). All of these studies demonstrate a uniqueness of microglia in terms of their pathophysiological response to inflammation and injury. We propose that emerging evidence supports a need for a new designation to encompass the activation states of microglia in neurological diseases. This M3 designation, while perhaps highlighting microglia as a distinct cell type from macrophages, also leaves the purpose of these cells and the plastic nature of microglia, particularly from our example of GLD that may extend to other forms of neuropathology in which enlarged, multi-nucleated microglia have been identified.
3.4 How do ECM components regulate the formation of globoid cells?
The ECM is an intricate molecular network that occupies extracellular space in tissues. The ECM can be altered in many CNS diseases, including Alzheimer’s disease (Morawski et al. 2012), stroke (Ji and Tsirka 2012), and MS (Bonneh-Barkay and Wiley 2009) and influences inflammatory responses. We have found that the make up of the ECM within the GLD CNS adds another important aspect of GLD pathology that can influence microglia and globoid cell function as well as the expression of classical polarization markers. In the following segment, we will outline evidence for how M3 activated microglia demonstrate features of both classical M1 (M3.1) and alternate M2 (M3.2) macrophage activation.
Astrocytes play a fundamental role in the homeostasis of the ECM. For example, astrocytes from chronically demyelinated lesions in multiple sclerosis have been shown to produce high levels of tenascin-C (TnC), which negatively influences remyelination (Gutowski et al. 1999). TnC is absent in most healthy tissues, but is specifically expressed in response to injury (Chiquet-Ehrismann and Chiquet 2003). Examination of TnC in the brain of twi mice and from GLD post-mortem tissue led to the discovery of abnormally increased levels of TnC (Claycomb et al. 2014). Furthermore, TnC is a key molecule enhancing cytotoxic properties of globoid cells. For example, we found that TnC but not laminin, which is another important ECM protein (Bandtlow and Zimmermann 2000), facilitated the formation of globoid cells from primary cultures of astrocytes and microglia exposed to psychosine (Claycomb et al. 2014). In these conditions, globoid cells plated on TnC express M1 MHCII, while those plated on laminin express M2 CD206 (Claycomb et al. 2014). In addition, globoid cells stimulated with psychosine were much more cytotoxic to oligodendrocytes when grown on TnC than laminin, and produced much higher levels of TNF-α (Claycomb et al. 2014). These data suggest that TnC can regulate globoid cell phenotype towards a more inflammatory M1-like state. Interestingly, globoid cells still do not fit with the classic M1/M2 phenotypes as they express both M1 and M2 polarization markers and are also multinucleated. These features, which are currently a focus of intense study underlie our position that an M3 classification may be warranted (Table I). For example, when microglia are activated by psychosine and also grown on TnC they tend to polarize toward an M3.1 state. This sub-class of M3 denotes that they are non-traditionally activated (i.e. via psychosine) and yet bear many similarities to the classic M1 phenotype. For example, they are cytotoxic, release pro-inflammatory factors, and even stain for markers such as CD16/32. Instead, globoid cells grown on laminin polarize towards an M3.2 phenotype, where they are activated via a non-pathogen and noncytokine route, and are similar to the classic M2 phenotype. The accumulation of TnC in GLD can contribute to the white matter pathology by altering globoid cell phenotype towards the M3.1, inducing increased inflammation and damage. ECM regulation of microglial activation has also been investigated in rat brain after cryoinjury showing M2 regulation of microglia by increased levels of the ECM protein fibronectin (Kim et al. 2013). Vitronectin has also been shown to play a role by activating microglia towards the M1 pro-inflammatory phenotype (Welser-Alves et al. 2011). Understanding how these phenotypes contribute to GLD pathology, and how the ECM may be playing a role, may allow for therapeutic targeting of these cells in order to prevent cytotoxicity and promote regeneration in GLD patients.
3.5 Are globoid cells a developmental microglial aberration?
Blood monocytes, when isolated from mice, and treated with psychosine do not produce globoid cells, while isolated microglia do (Ijichi et al. 2013). In addition, unlike monocytes, microglia are not derived from the bone marrow but originate from hematopoietic stem cells in the yolk sac during development (Ginhoux et al. 2013; Kierdorf et al. 2013). GLD is considered a neurodevelopmental disorder, as many patients do not survive past two years of age in the infantile type (Duffner et al. 2009b). Since microglia are of a separate origin from macrophages, their development may be altered in GLD which could explain why they tend to phenotypically be M3.1.
The way microglial cells are differentiated from the yolk sac is still unknown, specifically the precise factors that direct this differentiation, which is believed to be regulated at the tissue level (Prinz and Priller 2014). Evidence has shown that microglia develop in an IRF-8 and PU.1 dependent manner, which are both transcription factors necessary for the development of microglia (Rosenbauer and Tenen 2007). Neither of these transcription factors have been investigated in either twi mouse or human GLD patients, but may be worthwhile, as GLD is a developmental disorder.
In human fetuses microglia-like cells have been detected at 3 weeks of gestational age (Hutchins et al. 1990), and at 16 weeks a major influx of microglia starts, and they become widely distributed in the neural tube by 22 weeks (Verney et al. 2010). By 35 weeks differentiated microglia populations can be detected (Esiri et al. 1991). During the critical window of development wherein microglia populate the developing GLD brain the aberrant environment containing psychosine and marked elevation in inflammatory factors may result in errors that impact future phenotype and function of microglial overall. For example, IRF-8, a crucial regulator required for the differentiation of microglia during embryonic development (Kierdorf et al. 2013), is also essential for transforming microglia into the reactive phenotype (Masuda et al. 2012). In GLD and twi mice there may be a dysregulation of this transcription factor leading to dysfunctionally activated microglia: globoid cells. In addition, TGF-β has also been proposed to play a major role in microglial development, inducing a ‘functional molecular signature’ in developing microglia (Butovsky et al. 2014; Gosselin et al. 2014). A dysfunction of TGF-β during embryonic development may also lead to formation of globoid cells. The process of microglial development, as it is separate from macrophage development, is still being understood and fleshed out, the more insight we have may help further understand the origin and purpose of globoid cells in GLD.
5. Therapeutic Opportunities to Target Microglia in GLD
5.1 Current Therapies
All current treatments for GLD are aimed to administer functional GALC in order to reduce psychosine accumulation to prevent oligodendrocyte cell death, and neurodegeneration. Studies using the twi mice reported that intravenous or intraperitoneal administration of GALC was unsuccessful, as it never become incorporated into the CNS (Kobayashi and Suzuki 1982; Umezawa et al. 1985). Bone marrow transplantation (BMT) was found to increase GALC activity, reduce psychosine accumulation, lowered the amount of globoid cells, and showed remyelination in twi mice (Hoogerbrugge et al. 1988a). In addition, an enzyme replacement therapy using an adeno-associated virus (AAV) has increased the lifespan of twi mice by 20–25 days (Lattanzi et al. 2014; Rafi et al. 2015). It has also been shown that treating twi mice with an AAV2 genome construct expressing mouse GALC led to an extended and symptom-free lifespan up to 8 months of age, with a slower symptom progression (Lin et al. 2015; Rafi et al. 2012). Combining HSCT and lentiviral gene-transfer in twi mice extended lifespan and preserved myelin in the sciatic nerve (Galbiati et al. 2009; Hawkins-Salsbury et al. 2015; Rafi et al. 2015).
The current standard of care therapy for GLD is hematopoietic stem cell transplantation. HSCT appears to provide some level of GALC correction via chimerism of bone marrow cells (Hoogerbrugge et al. 1988b). This therapy has shown to extend lifespan, and improve cognition, gait, motor coordination, and verbal learning, if administered in the pre-symptomatic period of disease (Escolar et al. 2005; Krivit et al. 1998). Typically the effectiveness of HSCT after diagnosis is greater in patients with the late-onset type than infantile GLD (Duffner et al. 2012). HSCT is a possible therapeutic option for some GLD patients, but there is a need for a safer, long-term therapy.
5.2 Microglial Targeting
Accumulating evidence indicated a pathogenic potential of globoid cells. Globoid cells may be aberrantly formed cytotoxic microglia, and hence, a potential target for therapy. Pharmacological regulation of TNF-α and IL-1 in mutant microglia may be an interesting alternative treatment. Pentoxifyline or dexamethasone are two compounds that could serve this purpose (Chao et al. 1992). Facilitating an anti-inflammatory M2 microglia polarization maybe also beneficial. Two selective serotonin reuptake inhibitors (SSRIs), fluoxetine and S-citalopram, down-regulate M1 activation and up-regulate M2 activation in mouse primary microglia (Su et al. 2015). Peroxisome proliferator activated receptor y (PPARy) agonists, such as rosiglitazone or pioglatzone, also shift microglia from an M1 phenotype towards an M2 anti-inflammatory phenotype in a mouse model of Parkinson’s disease (Pisanu et al. 2014). Treatments that promote M2 activated phenotypes also increase expression of molecules such as IL-4, IL-13, and IL-10 may also prove helpful to reduce inflammatory factors that ensue damage of oligodendrocytes. Future studies exploring this new role of microglia could yield important new opportunities to address neuropathology in GLD, and to identify compounds that could be combined with current HSCT and gene therapy approaches.
6. Conclusions
Evidence that globoid cells are found in the GLD fetus, before major demyelination and inflammation has had a chance to occur, underlines a key and new pathogenic role for microglia in this disease (Ida et al. 1994). Increasing evidence indicates that in neurological diseases microglia, and not macrophages, can adopt unique profiles of activation markers and become multinucleated (Ijichi et al. 2013). We have shown that isolated microglia when treated with psychosine and exposed to different ECM proteins can develop different activated phenotypes with distinct effects on oligodendrocyte toxicity (Table I) (Claycomb et al. 2014). We propose that activated microglia may warrant their own category (designated ‘M3’) to reflect the distinct differences microglial cells develop in neurological conditions that may not be appropriately ascribed to either the classical (M1) or alternative (M2) activation classes of macrophages. For example, based on our studies, M3.1 psychosine-activated microglia are cytotoxic towards oligodendrocytes when exposed to TnC, while M3.2 microglia are psychosine-activated but noncytotoxic to oligodendrocytes when exposed to laminin, yet both produce elevated expression of TNF and express M1 and M2 markers (Claycomb et al. 2014). Differentiating microglia from macrophages may help future studies define unique aspects of microglial physiology, such as the development of multi-nucleated microglia found in diseases such as GLD, ALS (Fendrick et al. 2007), HIV encephalitis (Budka 1986), Alzheimer’s disease (Hornik et al. 2014), other leukodystrophies (Budka 1986; Elleder 1984), and other conditions. In terms of understanding GLD, future characterization of the globoid cell phenotype may advance our strategies for developing treatments for this disease. Currently, only HSCT has been shown to be beneficial in treating this disease, but it is not always effective and widespread availability is limited. Further studying the role of globoid cells in this disease may help find future targets to slow down and treat the rapid symptoms. For example, inhibiting the M3.1 activation of microglia to becoming globoid cells may prove beneficial. In broader terms, refocusing the definitions of microglial activation may enhance our understanding of these unique cells both in the developing nervous system during disease.
Significance Statement.
Current thinking on the sequence of events underlying the profound demyelination in GLD suggests a cell-autonomous loss of oligodendrocytes with psychosine accumulation as the initiating event of a reactive cellular cascade. Recent findings suggest that this order should be reconsidered, and this review outlines how activation of microglia to an activated multi-nucleated phenotype, which in this disease are called ‘globoid cells’ and can occur in response to psychosine, may begin a cell-mediated process of demyelination. The microglial hypothesis of GLD provides new opportunities for therapeutic intervention.
Acknowledgments
Funding Sources:
The National Multiple Sclerosis Society (RG 5001-A-1 to SJC), Legacy of Angels Foundation (to ERB), and NIH grants (R21NS087578 to SJC; R01NS065808 and R21NS087474 to ERB).
Footnotes
Associate Editor: Ernesto Bongarzone
Conflict of Interest Statement: SJC and AMN declare no conflict of interest. ERB is a consultant for Lysosomal Therapeutics, Inc.
Role of Authors: SJC and AMN drafted the manuscript and ERB provided critical revision of the manuscript.
Literature Cited
- Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res. 1999;117(2):145–152. doi: 10.1016/s0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- Alliot F, Lecain E, Grima B, Pessac B. Microglial progenitors with a high proliferative potential in the embryonic and adult mouse brain. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(4):1541–1545. doi: 10.1073/pnas.88.4.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nature neuroscience. 2001;4(11):1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Bandtlow CE, Zimmermann DR. Proteoglycans in the developing brain: new conceptual insights for old proteins. Physiol Rev. 2000;80(4):1267–1290. doi: 10.1152/physrev.2000.80.4.1267. [DOI] [PubMed] [Google Scholar]
- Barczykowski AL, Foss AH, Duffner PK, Yan L, Carter RL. Death rates in the U.S. due to Krabbe disease and related leukodystrophy and lysosomal storage diseases. American journal of medical genetics Part A. 2012;158a(11):2835–2842. doi: 10.1002/ajmg.a.35624. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nature immunology. 2010;11(10):889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathology and applied neurobiology. 2013;39(1):3–18. doi: 10.1111/nan.12011. [DOI] [PubMed] [Google Scholar]
- Bonneh-Barkay D, Wiley CA. Brain extracellular matrix in neurodegeneration. Brain pathology. 2009;19(4):573–585. doi: 10.1111/j.1750-3639.2008.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budka H. Multinucleated giant cells in brain: a hallmark of the acquired immune deficiency syndrome (AIDS) Acta neuropathologica. 1986;69(3–4):253–258. doi: 10.1007/BF00688301. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17(1):131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Deng K, Mellado W, Lee J, Ratan RR, Filbin MT. Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron. 2002;35(4):711–719. doi: 10.1016/s0896-6273(02)00826-7. [DOI] [PubMed] [Google Scholar]
- Cantuti Castelvetri L, Givogri MI, Hebert A, Smith B, Song Y, Kaminska A, Lopez-Rosas A, Morfini G, Pigino G, Sands M, Brady ST, Bongarzone ER. The sphingolipid psychosine inhibits fast axonal transport in Krabbe disease by activation of GSK3beta and deregulation of molecular motors. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33(24):10048–10056. doi: 10.1523/JNEUROSCI.0217-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelvetri LC, Givogri MI, Zhu H, Smith B, Lopez-Rosas A, Qiu X, van Breemen R, Bongarzone ER. Axonopathy is a compounding factor in the pathogenesis of Krabbe disease. Acta neuropathologica. 2011;122(1):35–48. doi: 10.1007/s00401-011-0814-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Hu S, Close K, Choi CS, Molitor TW, Novick WJ, Peterson PK. Cytokine release from microglia: differential inhibition by pentoxifylline and dexamethasone. The Journal of infectious diseases. 1992;166(4):847–853. doi: 10.1093/infdis/166.4.847. [DOI] [PubMed] [Google Scholar]
- Chiquet-Ehrismann R, Chiquet M. Tenascins: regulation and putative functions during pathological stress. The Journal of pathology. 2003;200(4):488–499. doi: 10.1002/path.1415. [DOI] [PubMed] [Google Scholar]
- Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S, Myers RM, Maniatis T. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell reports. 2013;4(2):385–401. doi: 10.1016/j.celrep.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KH, Kim MW, Kim SU. Tissue culture model of Krabbe’s disease: psychosine cytotoxicity in rat oligodendrocyte culture. Dev Neurosci. 1997;19(4):321–327. doi: 10.1159/000111228. [DOI] [PubMed] [Google Scholar]
- Claycomb KI, Winokur PN, Johnson KM, Nicaise AM, Giampetruzzi AW, Sacino AV, Snyder EY, Barbarese E, Bongarzone ER, Crocker SJ. Aberrant production of tenascin-C in globoid cell leukodystrophy alters psychosine-induced microglial functions. Journal of neuropathology and experimental neurology. 2014;73(10):964–974. doi: 10.1097/NEN.0000000000000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland WW, Kennedy EP. The enzymatic synthesis of psychosine. The Journal of biological chemistry. 1960;235:45–51. [PubMed] [Google Scholar]
- Costantino-Ceccarini E, Morell P. Biosynthesis of brain sphingolipids and myelin accumulation in the mouse. Lipids. 1972;7(10):656–659. doi: 10.1007/BF02533072. [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Martinez-Cerdeno V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33(10):4216–4233. doi: 10.1523/JNEUROSCI.3441-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nature reviews Neuroscience. 2011;12(7):388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- Duchen LW, Eicher EM, Jacobs JM, Scaravilli F, Teixeira F. Hereditary leucodystrophy in the mouse: the new mutant twitcher. Brain: a journal of neurology. 1980;103(3):695–710. doi: 10.1093/brain/103.3.695. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Barczykowski A, Kay DM, Jalal K, Yan L, Abdelhalim A, Gill S, Gill AL, Carter R. Later onset phenotypes of Krabbe disease: results of the world-wide registry. Pediatric neurology. 2012;46(5):298–306. doi: 10.1016/j.pediatrneurol.2012.02.023. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Caggana M, Orsini JJ, Wenger DA, Patterson MC, Crosley CJ, Kurtzberg J, Arnold GL, Escolar ML, Adams DJ, Andriola MR, Aron AM, Ciafaloni E, Djukic A, Erbe RW, Galvin-Parton P, Helton LE, Kolodny EH, Kosofsky BE, Kronn DF, Kwon JM, Levy PA, Miller-Horn J, Naidich TP, Pellegrino JE, Provenzale JM, Rothman SJ, Wasserstein MP. Newborn screening for Krabbe disease: the New York State model. Pediatric neurology. 2009a;40(4):245–252. doi: 10.1016/j.pediatrneurol.2008.11.010. discussion 253–245. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Jalal K, Carter RL. The Hunter’s Hope Krabbe family database. Pediatric neurology. 2009b;40(1):13–18. doi: 10.1016/j.pediatrneurol.2008.08.011. [DOI] [PubMed] [Google Scholar]
- Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80(6):1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elleder M. Ito cells in lysosomal storage disorders. An ultrastructural study. Virchows Arch B Cell Pathol Incl Mol Pathol. 1984;46(1–2):13–19. doi: 10.1007/BF02890291. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, Morse R, Krivit W, Kurtzberg J. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. The New England journal of medicine. 2005;352(20):2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Esiri MM, al Izzi MS, Reading MC. Macrophages, microglial cells, and HLA-DR antigens in fetal and infant brain. J Clin Pathol. 1991;44(2):102–106. doi: 10.1136/jcp.44.2.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairweather D, Cihakova D. Alternatively activated macrophages in infection and autoimmunity. J Autoimmun. 2009;33(3–4):222–230. doi: 10.1016/j.jaut.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendrick SE, Xue QS, Streit WJ. Formation of multinucleated giant cells and microglial degeneration in rats expressing a mutant Cu/Zn superoxide dismutase gene. Journal of neuroinflammation. 2007;4:9. doi: 10.1186/1742-2094-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formichi P, Radi E, Battisti C, Pasqui A, Pompella G, Lazzerini PE, Laghi-Pasini F, Leonini A, Di Stefano A, Federico A. Psychosine-induced apoptosis and cytokine activation in immune peripheral cells of Krabbe patients. Journal of cellular physiology. 2007;212(3):737–743. doi: 10.1002/jcp.21070. [DOI] [PubMed] [Google Scholar]
- Galbiati F, Givogri MI, Cantuti L, Rosas AL, Cao H, van Breemen R, Bongarzone ER. Combined hematopoietic and lentiviral gene-transfer therapies in newborn Twitcher mice reveal contemporaneous neurodegeneration and demyelination in Krabbe disease. J Neurosci Res. 2009;87(8):1748–1759. doi: 10.1002/jnr.22006. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Frontiers in cellular neuroscience. 2013;7:45. doi: 10.3389/fncel.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri S, Khan M, Nath N, Singh I, Singh AK. The role of AMPK in psychosine mediated effects on oligodendrocytes and astrocytes: implication for Krabbe disease. Journal of neurochemistry. 2008;105(5):1820–1833. doi: 10.1111/j.1471-4159.2008.05279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nature reviews Immunology. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, Glass CK. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159(6):1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano AC, Cardile V. History, genetic, and recent advances on Krabbe disease. Gene. 2014 doi: 10.1016/j.gene.2014.09.046. [DOI] [PubMed] [Google Scholar]
- Gutowski NJ, Newcombe J, Cuzner ML. Tenascin-R and C in multiple sclerosis lesions: relevance to extracellular matrix remodelling. Neuropathology and applied neurobiology. 1999;25(3):207–214. doi: 10.1046/j.1365-2990.1999.00176.x. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Sourander P, Svennerholm L. Diagnosis of Krabbe’s infantile leucodystrophy. Journal of neurology, neurosurgery, and psychiatry. 1963;26:195–198. doi: 10.1136/jnnp.26.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq E, Giri S, Singh I, Singh AK. Molecular mechanism of psychosine-induced cell death in human oligodendrocyte cell line. J Neurochem. 2003;86(6):1428–1440. doi: 10.1046/j.1471-4159.2003.01941.x. [DOI] [PubMed] [Google Scholar]
- Hawkins-Salsbury JA, Shea L, Jiang X, Hunter DA, Guzman AM, Reddy AS, Qin EY, Li Y, Gray SJ, Ory DS, Sands MS. Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2015;35(16):6495–6505. doi: 10.1523/JNEUROSCI.4199-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse M, Modolell M, La Flamme AC, Schito M, Fuentes JM, Cheever AW, Pearce EJ, Wynn TA. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. Journal of immunology. 2001;167(11):6533–6544. doi: 10.4049/jimmunol.167.11.6533. [DOI] [PubMed] [Google Scholar]
- Hoogerbrugge PM, Poorthuis BJ, Romme AE, van de Kamp JJ, Wagemaker G, van Bekkum DW. Effect of bone marrow transplantation on enzyme levels and clinical course in the neurologically affected twitcher mouse. The Journal of clinical investigation. 1988a;81(6):1790–1794. doi: 10.1172/JCI113521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogerbrugge PM, Suzuki K, Suzuki K, Poorthuis BJ, Kobayashi T, Wagemaker G, van Bekkum DW. Donor-derived cells in the central nervous system of twitcher mice after bone marrow transplantation. Science. 1988b;239(4843):1035–1038. doi: 10.1126/science.3278379. [DOI] [PubMed] [Google Scholar]
- Hornik TC, Neniskyte U, Brown GC. Inflammation induces multinucleation of Microglia via PKC inhibition of cytokinesis, generating highly phagocytic multinucleated giant cells. J Neurochem. 2014;128(5):650–661. doi: 10.1111/jnc.12477. [DOI] [PubMed] [Google Scholar]
- Hou CC, Chen YP, Wu JH, Huang CC, Wang SY, Yang NS, Shyur LF. A galactolipid possesses novel cancer chemopreventive effects by suppressing inflammatory mediators and mouse B16 melanoma. Cancer research. 2007;67(14):6907–6915. doi: 10.1158/0008-5472.CAN-07-0158. [DOI] [PubMed] [Google Scholar]
- Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43(11):3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- Hutchins KD, Dickson DW, Rashbaum WK, Lyman WD. Localization of morphologically distinct microglial populations in the developing human fetal brain: implications for ontogeny. Brain Res Dev Brain Res. 1990;55(1):95–102. doi: 10.1016/0165-3806(90)90109-c. [DOI] [PubMed] [Google Scholar]
- Ida H, Rennert OM, Watabe K, Eto Y, Maekawa K. Pathological and biochemical studies of fetal Krabbe disease. Brain & development. 1994;16(6):480–484. doi: 10.1016/0387-7604(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Ijichi K, Brown GD, Moore CS, Lee JP, Winokur PN, Pagarigan R, Snyder EY, Bongarzone ER, Crocker SJ. MMP-3 mediates psychosine-induced globoid cell formation: implications for leukodystrophy pathology. Glia. 2013;61(5):765–777. doi: 10.1002/glia.22471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Hayashi M, Fujioka Y, Nagashima K, Morimatsu Y, Matsuyama H. Immunohistological study of globoid cell leukodystrophy. Brain & development. 2002;24(5):284–290. doi: 10.1016/s0387-7604(02)00057-8. [DOI] [PubMed] [Google Scholar]
- Jalal K, Carter R, Yan L, Barczykowski A, Duffner PK. Does galactocerebrosidase activity predict Krabbe phenotype? Pediatric neurology. 2012;47(5):324–329. doi: 10.1016/j.pediatrneurol.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Jatana M, Giri S, Singh AK. Apoptotic positive cells in Krabbe brain and induction of apoptosis in rat C6 glial cells by psychosine. Neurosci Lett. 2002;330(2):183–187. doi: 10.1016/s0304-3940(02)00655-9. [DOI] [PubMed] [Google Scholar]
- Ji K, Tsirka SE. Inflammation modulates expression of laminin in the central nervous system following ischemic injury. Journal of neuroinflammation. 2012;9:159. doi: 10.1186/1742-2094-9-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa T, Nakamura S, Momoi M, Yamaji T, Takematsu H, Yano H, Sabe H, Yamamoto A, Kawasaki T, Kozutsumi Y. Inhibition of cytokinesis by a lipid metabolite, psychosine. The Journal of cell biology. 2000;149(4):943–950. doi: 10.1083/jcb.149.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C, Muller DN, Luckow B, Brocker T, Debowski K, Fritz G, Opdenakker G, Diefenbach A, Biber K, Heikenwalder M, Geissmann F, Rosenbauer F, Prinz M. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nature neuroscience. 2013;16(3):273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29(43):13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Ahn M, Choi S, Kim M, Sim KB, Kim J, Moon C, Shin T. Potential role of fibronectin in microglia/macrophage activation following cryoinjury in the rat brain: an immunohistochemical study. Brain research. 2013;1502:11–19. doi: 10.1016/j.brainres.2013.01.043. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Katayama M, Bourque E, Suzuki K, Suzuki K. The twitcher mouse: positive immunohistochemical staining of globoid cells with monoclonal antibody against Mac-1 antigen. Brain research. 1985;352(1):49–54. doi: 10.1016/0165-3806(85)90086-0. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Goto I, Yamanaka T, Suzuki Y, Nakano T, Suzuki K. Infantile and fetal globoid cell leukodystrophy: analysis of galactosylceramide and galactosylsphingosine. Ann Neurol. 1988;24(4):517–522. doi: 10.1002/ana.410240407. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Suzuki K. The twitcher mouse: fate of exogenously administered [3H]galactosyl-sphingosine. Advances in experimental medicine and biology. 1982;152:253–259. [PubMed] [Google Scholar]
- Kolodny EH, Raghavan S, Krivit W. Late-onset Krabbe disease (globoid cell leukodystrophy): clinical and biochemical features of 15 cases. Dev Neurosci. 1991;13(4–5):232–239. doi: 10.1159/000112166. [DOI] [PubMed] [Google Scholar]
- Kolter T, Sandhoff K. Sphingolipid metabolism diseases. Biochim Biophys Acta. 2006;1758(12):2057–2079. doi: 10.1016/j.bbamem.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Adams JM, Vanier MT, Duncan ID. Macrophages counteract demyelination in a mouse model of globoid cell leukodystrophy. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31(10):3610–3624. doi: 10.1523/JNEUROSCI.6344-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Wenger DA, Gallo V, Duncan ID. Galactocerebrosidase-deficient oligodendrocytes maintain stable central myelin by exogenous replacement of the missing enzyme in mice. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(51):18670–18675. doi: 10.1073/pnas.0506473102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivit W, Shapiro EG, Peters C, Wagner JE, Cornu G, Kurtzberg J, Wenger DA, Kolodny EH, Vanier MT, Loes DJ, Dusenbery K, Lockman LA. Hematopoietic stem-cell transplantation in globoid-cell leukodystrophy. The New England journal of medicine. 1998;338(16):1119–1126. doi: 10.1056/NEJM199804163381605. [DOI] [PubMed] [Google Scholar]
- Lattanzi A, Salvagno C, Maderna C, Benedicenti F, Morena F, Kulik W, Naldini L, Montini E, Martino S, Gritti A. Therapeutic benefit of lentiviral-mediated neonatal intracerebral gene therapy in a mouse model of globoid cell leukodystrophy. Hum Mol Genet. 2014;23(12):3250–3268. doi: 10.1093/hmg/ddu034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidi M, Gotti E, Bologna L, Miranda E, Rimoldi M, Sica A, Roncalli M, Palumbo GA, Introna M, Golay J. M2 macrophages phagocytose rituximab-opsonized leukemic targets more efficiently than m1 cells in vitro. Journal of immunology. 2009;182(7):4415–4422. doi: 10.4049/jimmunol.0713732. [DOI] [PubMed] [Google Scholar]
- Lin DS, Hsiao CD, Lee AY, Ho CS, Liu HL, Wang TJ, Jian YR, Hsu JC, Huang ZD, Lee TH, Chiang MF. Mitigation of cerebellar neuropathy in globoid cell leukodystrophy mice by AAV-mediated gene therapy. Gene. 2015;571(1):81–90. doi: 10.1016/j.gene.2015.06.049. [DOI] [PubMed] [Google Scholar]
- Luzi P, Abraham RM, Rafi MA, Curtis M, Hooper DC, Wenger DA. Effects of treatments on inflammatory and apoptotic markers in the CNS of mice with globoid cell leukodystrophy. Brain research. 2009;1300:146–158. doi: 10.1016/j.brainres.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzi P, Rafi MA, Wenger DA. Characterization of the large deletion in the GALC gene found in patients with Krabbe disease. Human molecular genetics. 1995;4(12):2335–2338. doi: 10.1093/hmg/4.12.2335. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23(4):344–346. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends in immunology. 2004;25(12):677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Martin JJ, Leroy JG, Ceuterick C, Libert J, Dodinval P, Martin L. Fetal Krabbe leukodystrophy. A morphologic study of two cases. Acta neuropathologica. 1981;53(2):87–91. doi: 10.1007/BF00689987. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Ge N, Mucke L. Pathogenesis of HIV-1 associated neurodegeneration. Crit Rev Neurobiol. 1996;10(1):57–67. doi: 10.1615/critrevneurobiol.v10.i1.30. [DOI] [PubMed] [Google Scholar]
- Masuda T, Tsuda M, Yoshinaga R, Tozaki-Saitoh H, Ozato K, Tamura T, Inoue K. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell reports. 2012;1(4):334–340. doi: 10.1016/j.celrep.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally AK, Anderson JM. Foreign body-type multinucleated giant cells induced by interleukin-4 express select lymphocyte co-stimulatory molecules and are phenotypically distinct from osteoclasts and dendritic cells. Exp Mol Pathol. 2011;91(3):673–681. doi: 10.1016/j.yexmp.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. Journal of immunology. 1993;151(4):2132–2141. [PubMed] [Google Scholar]
- Michell-Robinson MA, Touil H, Healy LM, Owen DR, Durafourt BA, Bar-Or A, Antel JP, Moore CS. Roles of microglia in brain development, tissue maintenance and repair. Brain: a journal of neurology. 2015;138(Pt 5):1138–1159. doi: 10.1093/brain/awv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nature neuroscience. 2013;16(9):1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatake T, Suzuki K. Galactosylsphingosine galactosyl hydrolase. Partial purification and properties of the enzyme in rat brain. The Journal of biological chemistry. 1972;247(17):5398–5403. [PubMed] [Google Scholar]
- Morawski M, Bruckner G, Jager C, Seeger G, Matthews RT, Arendt T. Involvement of perineuronal and perisynaptic extracellular matrix in Alzheimer’s disease neuropathology. Brain pathology. 2012;22(4):547–561. doi: 10.1111/j.1750-3639.2011.00557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Komiyama A, Martin PM, Suzuki K. Proliferation of microglia/macrophages in the demyelinating CNS and PNS of twitcher mouse. Brain research. 1993;602(2):268–274. doi: 10.1016/0006-8993(93)90692-g. [DOI] [PubMed] [Google Scholar]
- Peters C, Steward CG, National Marrow Donor P, International Bone Marrow Transplant R, Working Party on Inborn Errors EBMTG Hematopoietic cell transplantation for inherited metabolic diseases: an overview of outcomes and practice guidelines. Bone marrow transplantation. 2003;31(4):229–239. doi: 10.1038/sj.bmt.1703839. [DOI] [PubMed] [Google Scholar]
- Phelps M, Aicardi J, Vanier MT. Late onset Krabbe’s leukodystrophy: a report of four cases. Journal of neurology, neurosurgery, and psychiatry. 1991;54(4):293–296. doi: 10.1136/jnnp.54.4.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisanu A, Lecca D, Mulas G, Wardas J, Simbula G, Spiga S, Carta AR. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiology of disease. 2014;71:280–291. doi: 10.1016/j.nbd.2014.08.011. [DOI] [PubMed] [Google Scholar]
- Pollanen MS, Brody BA. Fetal globoid cell leukodystrophy. Arch Pathol Lab Med. 1990;114(2):213–216. [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27(40):10714–10721. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter GB, Santos M, Davisson MT, Rowitch DH, Marks DL, Bongarzone ER, Petryniak MA. Missense mutation in mouse GALC mimics human gene defect and offers new insights into Krabbe disease. Human molecular genetics. 2013;22(17):3397–3414. doi: 10.1093/hmg/ddt190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nature reviews Neuroscience. 2014;15(5):300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- Rafi MA, Luzi P, Chen YQ, Wenger DA. A large deletion together with a point mutation in the GALC gene is a common mutant allele in patients with infantile Krabbe disease. Hum Mol Genet. 1995;4(8):1285–1289. doi: 10.1093/hmg/4.8.1285. [DOI] [PubMed] [Google Scholar]
- Rafi MA, Rao HZ, Luzi P, Curtis MT, Wenger DA. Extended normal life after AAVrh10- mediated gene therapy in the mouse model of Krabbe disease. Mol Ther. 2012;20(11):2031–2042. doi: 10.1038/mt.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafi MA, Rao HZ, Luzi P, Luddi A, Curtis MT, Wenger DA. Intravenous injection of AAVrh10-GALC after the neonatal period in twitcher mice results in significant expression in the central and peripheral nervous systems and improvement of clinical features. Molecular genetics and metabolism. 2015;114(3):459–466. doi: 10.1016/j.ymgme.2014.12.300. [DOI] [PubMed] [Google Scholar]
- Reddy AS, Patel JR, Vogler C, Klein RS, Sands MS. Central nervous system pathology progresses independently of KC and CXCR2 in globoid-cell leukodystrophy. PloS one. 2013;8(6):e64647. doi: 10.1371/journal.pone.0064647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nature reviews Immunology. 2007;7(2):105–117. doi: 10.1038/nri2024. [DOI] [PubMed] [Google Scholar]
- Sakai N, Inui K, Tatsumi N, Fukushima H, Nishigaki T, Taniike M, Nishimoto J, Tsukamoto H, Yanagihara I, Ozono K, Okada S. Molecular cloning and expression of cDNA for murine galactocerebrosidase and mutation analysis of the twitcher mouse, a model of Krabbe’s disease. Journal of neurochemistry. 1996;66(3):1118–1124. doi: 10.1046/j.1471-4159.1996.66031118.x. [DOI] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, van der Bijl P, Schwab ME. The UDP-galactose:ceramide galactosyltransferase: expression pattern in oligodendrocytes and Schwann cells during myelination and substrate preference for hydroxyceramide. Journal of neurochemistry. 1995;65(5):2267–2278. doi: 10.1046/j.1471-4159.1995.65052267.x. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Stevens B. The “quad-partite” synapse: microglia-synapse interactions in the developing and mature CNS. Glia. 2013;61(1):24–36. doi: 10.1002/glia.22389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda H, Kobayashi T, Katayama M, Goto I, Nagara H. Accumulation of galactosylsphingosine (psychosine) in the twitcher mouse: determination by HPLC. Journal of neurochemistry. 1987;49(1):92–99. doi: 10.1111/j.1471-4159.1987.tb03399.x. [DOI] [PubMed] [Google Scholar]
- Su F, Yi H, Xu L, Zhang Z. Fluoxetine and S-citalopram inhibit M1 activation and promote M2 activation of microglia in vitro. Neuroscience. 2015;294:60–68. doi: 10.1016/j.neuroscience.2015.02.028. [DOI] [PubMed] [Google Scholar]
- Sugama S, Kim SU, Ida H, Eto Y. Psychosine cytotoxicity in rat neural cell cultures and protection by phorbol ester and dimethyl sulfoxide. Pediatr Res. 1990;28(5):473–476. doi: 10.1203/00006450-199011000-00011. [DOI] [PubMed] [Google Scholar]
- Suzuki K. Ultrastructural study of experimental globoid cells. Lab Invest. 1970;23(6):612–619. [PubMed] [Google Scholar]
- Suzuki K. Twenty five years of the “psychosine hypothesis”: a personal perspective of its history and present status. Neurochemical research. 1998;23(3):251–259. doi: 10.1023/a:1022436928925. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Suzuki K. The twitcher mouse: a model for Krabbe disease and for experimental therapies. Brain pathology. 1995;5(3):249–258. doi: 10.1111/j.1750-3639.1995.tb00601.x. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Suzuki Y. Globoid cell leucodystrophy (Krabbe’s disease): deficiency of galactocerebroside beta-galactosidase. Proceedings of the National Academy of Sciences of the United States of America. 1970;66(2):302–309. doi: 10.1073/pnas.66.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Taniike M. Murine model of genetic demyelinating disease: the twitcher mouse. Microscopy research and technique. 1995;32(3):204–214. doi: 10.1002/jemt.1070320304. [DOI] [PubMed] [Google Scholar]
- Svennerholm L, Vanier MT, Mansson JE. Krabbe disease: a galactosylsphingosine (psychosine) lipidosis. J Lipid Res. 1980;21(1):53–64. [PubMed] [Google Scholar]
- Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol. 2015 doi: 10.1007/s12035-014-9070-5. [DOI] [PubMed] [Google Scholar]
- Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol. 2005;23:901–944. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- Umezawa F, Eto Y, Tokoro T, Ito F, Maekawa K. Enzyme replacement with liposomes containing beta-galactosidase from Charonia lumpas in murine globoid cell leukodystrophy (twitcher) Biochemical and biophysical research communications. 1985;127(2):663–667. doi: 10.1016/s0006-291x(85)80212-6. [DOI] [PubMed] [Google Scholar]
- Van Ginderachter JA, Movahedi K, Hassanzadeh Ghassabeh G, Meerschaut S, Beschin A, Raes G, De Baetselier P. Classical and alternative activation of mononuclear phagocytes: picking the best of both worlds for tumor promotion. Immunobiology. 2006;211(6–8):487–501. doi: 10.1016/j.imbio.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Vanier M, Svennerholm L. Chemical pathology of Krabbe disease: the occurrence of psychosine and other neutral sphingoglycolipids. Advances in experimental medicine and biology. 1976;68:115–126. doi: 10.1007/978-1-4684-7735-1_8. [DOI] [PubMed] [Google Scholar]
- Verney C, Monier A, Fallet-Bianco C, Gressens P. Early microglial colonization of the human forebrain and possible involvement in periventricular white-matter injury of preterm infants. J Anat. 2010;217(4):436–448. doi: 10.1111/j.1469-7580.2010.01245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel DY, Vereyken EJ, Glim JE, Heijnen PD, Moeton M, van der Valk P, Amor S, Teunissen CE, van Horssen J, Dijkstra CD. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. Journal of neuroinflammation. 2013;10:35. doi: 10.1186/1742-2094-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welser-Alves JV, Boroujerdi A, Tigges U, Milner R. Microglia use multiple mechanisms to mediate interactions with vitronectin; non-essential roles for the highly-expressed alphavbeta3 and alphavbeta5 integrins. Journal of neuroinflammation. 2011;8:157. doi: 10.1186/1742-2094-8-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger DA, Rafi MA, Luzi P, Datto J, Costantino-Ceccarini E. Krabbe disease: genetic aspects and progress toward therapy. Mol Genet Metab. 2000;70(1):1–9. doi: 10.1006/mgme.2000.2990. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Victoria T, Rafi MA, Luzi P, Vanier MT, Vite C, Patterson DF, Haskins MH. Globoid cell leukodystrophy in cairn and West Highland white terriers. J Hered. 1999;90(1):138–142. doi: 10.1093/jhered/90.1.138. [DOI] [PubMed] [Google Scholar]
- White AB, Givogri MI, Lopez-Rosas A, Cao H, van Breemen R, Thinakaran G, Bongarzone ER. Psychosine accumulates in membrane microdomains in the brain of krabbe patients, disrupting the raft architecture. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29(19):6068–6077. doi: 10.1523/JNEUROSCI.5597-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Combs C, Cannady SB, Geldmacher DS, Herrup K. Beta-amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol Aging. 2000;21(6):797–806. doi: 10.1016/s0197-4580(00)00219-0. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nature reviews Immunology. 2004;4(8):583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, Beyer M, Latz E, Freeman TC, Ulas T, Schultze JL. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajicek JP, Wing M, Scolding NJ, Compston DA. Interactions between oligodendrocytes and microglia. A major role for complement and tumour necrosis factor in oligodendrocyte adherence and killing. Brain: a journal of neurology. 1992;115(Pt 6):1611–1631. [PubMed] [Google Scholar]
- Zaka M, Wenger DA. Psychosine-induced apoptosis in a mouse oligodendrocyte progenitor cell line is mediated by caspase activation. Neurosci Lett. 2004;358(3):205–209. doi: 10.1016/j.neulet.2003.12.126. [DOI] [PubMed] [Google Scholar]
- Zhu H, Lopez-Rosas A, Qiu X, Van Breemen RB, Bongarzone ER. Detection of the neurotoxin psychosine in samples of peripheral blood: application in diagnostics and follow-up of Krabbe disease. Arch Pathol Lab Med. 2012;136(7):709–710. doi: 10.5858/arpa.2011-0667-LE. [DOI] [PMC free article] [PubMed] [Google Scholar]