

Abstract
Krabbe disease (KD) is caused by mutations in the galactosylceramidase (GALC) gene, which encodes a lysosomal enzyme that degrades galactolipids, including galactosylceramide and galactosylsphingosine (psychosine). GALC deficiency results in progressive intracellular accumulation of psychosine, which is believed to be the main cause for the demyelinating neurodegeneration in KD pathology. Umbilical cord blood transplantation slows disease progression if performed presymptomatically, but carries a significant risk of morbidity and mortality. Accurate presymptomatic diagnosis is therefore critical to facilitate the efficacy of existing transplant approaches and avoid unnecessary treatment of children who will not develop KD. Unfortunately current diagnostic criteria, including GALC activity, genetic analysis, and psychosine measurement, are insufficient for secure presymptomatic diagnosis. Herein, we performed a global metabolomic analysis to identify pathogenetic metabolic pathways and novel biomarkers implicated in the authentic mouse model of KD, twitcher. At a time point before onset of signs of disease, twitcher hindbrains had metabolic profiles similar to wild type, with the exception of a decrease in metabolites related to glucose energy metabolism. Instead, many metabolic pathways were altered after early signs of disease in the twitcher, including decreased phospholipid turnover, restricted mitochondrial metabolism of branched-chain amino acids, increased inflammation, neurotransmitter metabolism and osmolytes. Hypoxanthine, a purine derivative, is increased before signs of disease appear, suggesting its potential as a biomarker for early diagnosis of KD. Additionally, given the early changes in glucose metabolism in the pathogenesis of KD, diagnostic modalities that report metabolic function, such as positron emission tomography, may be useful in KD.
Keywords: Krabbe disease, globoid cell leukodystrophy, metabolomics, twitcher, glucose metabolism, mitochondrial dysfunction, oxidative stress, biomarker
Graphical Abstract
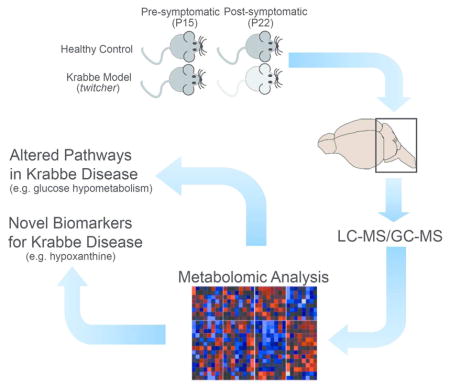
We performed a global metabolomic analysis from the hindbrains of pre-symptomatic and post-symptomatic Krabbe mice. We identified several altered metabolic pathways and a number of novel potential biomarkers that may be beneficial for patients with Krabbe disease.
INTRODUCTION
Krabbe disease (KD) is a fatal neurodegenerative leukodystrophy that typically affects children less than two years of age. Although there is no cure for this disease, umbilical cord transplantation has proven to be beneficial if delivered to pre-symptomatic patients and can extend patient lifespan and improve quality of life (Escolar et al. 2005). Current diagnostic criteria of KD include a reduction in leukocyte or fibroblast GALC activity, mutation analysis, and clinical history. However, recent studies suggest that these parameters may not directly correlate with severity of symptoms, including age of onset, progression of disease and survival, for reasons yet unknown (Wenger 2011b). Recently, we reported that measurement of GALC activity in lysosomal fractions may better discriminate infants at high risk for the infantile phenotype, from children who will develop later-onset phenotypes (Shin et al. 2016), although additional patient samples are required for further validation.
The twitcher mouse is an authentic murine model for KD (Duchen et al. 1980) and carries a spontaneous nonsense mutation in exon 10 of Galc with no residual GALC activity (Sakai et al. 1996). The background strain of the twitcher has been shown to play an important role in determining phenotype. In the congenic C57BL/6 background, the clinical course is severe and signs of central nervous system disease appear by postnatal day 21 (P21) with death as early as P40-45. Alternatively, twitcher mice bred in a mixed background containing C57BL/6 and either FVB or CE/J show a protracted clinical course, with death at P50 or even P90, respectively (Duchen et al. 1980; Santambrogio et al. 2012). Regardless of background, head tremors and decreased body weight are often the earliest clinical findings, while muscle weakness in the hindlimbs is an additional prominent feature. Myelin loss occurs in both the central and peripheral nervous systems, and is accompanied by astrocytic gliosis and the accumulation of multinucleated globoid cells, which are considered pathognomonic for KD (Suzuki 2004b).
Previously, metabolic profiling of the twitcher on the C57BL/6 background using electro spray ionization (ESI) tandem mass spectrometry (MS/MS) showed that the bioactive sphingolipids sphingosine-1-phosphate, C18:0 ceramide, C22:0 ceramide and C24:0 ceramide levels were reduced in the pons/medulla of twitcher mice at P31 and P35-37 span (Esch et al. 2003). Additionally, that study found a trend towards decreased levels of long chain fatty acids and increased levels of shorter chain fatty acids in galactosylceramides and ceramides from twitcher mice compared with WT (Esch et al. 2003). In a more recent study using gas chromatography with electron impact mass spectrometry (GC-EI-MS), the fatty acid (FA) composition of the P25 and P35 twitcher on the C57BL/6 background was further investigated (Zanfini et al. 2014). A number of FAs, including one saturated FA, three monounsaturated FAs, three polyunsaturated FAs (PUFA) and one plasmalogen species were significantly altered in twitcher brain specimens. Three PUFAs were also found to be significantly altered in twitcher serum, though they were dissimilar to the PUFA changes reported in the brain (Zanfini et al. 2014). In a third study, the 1H and 13C magnetic resonance spectra (MRS) technique was applied to measure metabolites of spinal cord, cerebellum, and forebrain with mild signs of disease (P30) and full signs of disease (P40) in twitcher mice on the mixed background of C57BL/6 and FVB, revealing glucose hypometabolism, alterations in neurotransmitter content, N-acetylaspartate, N-acetylaspartylglutamate, and osmolyte levels, in close agreement with the progression of astrogliosis, microglia activation, apoptosis, and neurodegeneration (Meisingset et al. 2013). A major limitation to all of these studies is that profiled metabolites came only from twitcher mice with established disease. To be useful as a biomarker or for diagnostic purposes, it is critically important that these metabolites would be significantly changed at time points prior to the onset of disease.
The primary aim of this study is two-fold: 1) to discover if there are additional biomarkers that could be used for early diagnosis of KD patients, and 2) to further characterize the metabolic pathways influenced by KD pathology. Answers to these two questions are crucially important for accurate diagnosis and for the design of more effective therapies. We analyzed global metabolites of twitcher, both before and after the onset of disease signs, using the recently developed LC/MS and GC/MS metabolomics-profiling techniques (Metabolon, Inc.). Our investigations found several key metabolites that were present at altered concentrations from WT. Many of these metabolites are tightly associated with pathways: namely mitochondrial fuel selection and energy production, inflammation, neurotransmitter metabolism, and osmotic regulation.
MATERIALS AND METHODS
Mice
Experiments were conducted according to the protocols approved by the Institutional Animal Care and Use Committee of University at Buffalo and Roswell Park Cancer Institute. Twitcher mice (RRID:IMSR_JAX:000845) were maintained on the background of C57BL/6N. Breeder C57BL/6N mice were purchased from Charles River. Freshly collected hindbrain regions from twitcher and WT littermate mice at P8, P15, P22, and P34 days were transferred into polypropylene cryovials, snap-frozen in liquid nitrogen and kept at −80°C until the analysis. The time between dissection and freezing of tissues was under one minute for all tissues.
Metabolomic analysis
Tissue samples were prepared for analysis using a proprietary series of organic and aqueous extractions to remove the protein fraction while allowing maximum recovery of small molecules. The resulting extract was divided into two fractions: one for analysis by LC/MS and one for analysis by GC/MS. The LC/MS portion of the platform is consisted of an electrospray ionization source and linear ion-trap mass analyzer. The sample extract was split into two aliquots, dried, then reconstituted in acidic or basic LC-compatible solvents. One aliquot was analyzed using acidic positive ion optimized conditions and the other using basic negative ion optimized conditions in two independent injections using separate dedicated columns. Extracts reconstituted in acidic conditions were gradient eluted using water and methanol both containing formic acid, while the basic extracts contained ammonium bicarbonate. The MS analysis alternated between MS and data-dependent MS2 scans using dynamic exclusion. The samples destined for GC/MS analysis were re-dried under vacuum desiccation for a minimum of 24 hours prior to being derivatized under dried nitrogen using bistrimethyl-silyl-triflouroacetamide. The GC column was 5% phenyl and the temperature ramp is from 40°C to 300°C in a 16 minute period. Samples were analyzed on a Thermo-Finnigan Trace DSQ fast-scanning single-quadrupole mass spectrometer using electron impact ionization.
Data extraction and compound identification
Raw data was extracted, peak-identified and QC processed by Metabolon Inc. Briefly, metabolites were identified by comparison to library entries of purified standards or recurrent unknown entities. Metabolon’s library is based on authenticated standards that contain the retention time/index (RI), mass to charge ratio (m/z), and chromatographic data (including MS/MS spectral data). Biochemical identifications were based on retention index within a narrow RI window of the proposed identification, nominal mass match to the library +/− 0.4 amu, and the MS/MS forward and reverse scores between the experimental data and authentic standards. The MS/MS scores were based on a comparison of the ions present in the experimental spectrum to the ions present in the library spectrum. The raw data of this study is deposited in the freely accessible metabolomicsworkbench.org.
Hypoxanthine/Xanthine, S-adenosylhomocysteine, Branched chain amino acids, Glucose, and Glucose 6-phosphate measurements
Hypoxanthine/Xanthine concentrations were measured using the Hypoxanthine/Xanthine fluorometric assay kit (BioVision; RRID:SCR_005057, Catalog # K685). Hypoxanthine/Xanthine is specifically oxidized by the Xanthine Enzyme Mix to form an intermediate, which reacts with the developer and probe to form a product that can be measured fluorometrically (Ex/Em = 535/587 nm). S-Adenosylhomocysteine (SAH) concentrations in brain extracts were measured using an S-Adenosylhomocysteine ELISA kit (Cell Biolabs, Inc., Catalog #STA-671) following the manufacturer’s protocols. This assay is a competitive ELISA where samples and an anti-SAH antibody are added to a plate coated with an SAH conjugate. The sample and conjugate compete for antibody binding, which generates a reverse curve. Samples with high SAH levels will bind the majority of the antibody, which gets washed away and results in a low absorbance, while samples with low SAH levels will leave more antibody available to bind to the conjugate, producing a high signal. Branched chain amino acid (BCAA) levels were measured by the Branched Chain Amino Acid (Leu/Ile/Val) Colorimetric Assay Kit (BioVision; RRID:SCR_005057, Catalog #K564), which utilizes an enzyme reaction in which BCAA are oxidatively deaminated producing NADH, which reduces the probe and thus generates a colored product measured at 450 nm. Intracellular or serum glucose and glucose 6-phosphate concentrations were measured using the Glucose and Glucose 6-phosphate assay kits (BioVision; RRID:SCR_005057, Catalog #K606 and K657), respectively. The kits provide direct measurement of glucose or glucose 6-phosphate in serum and tissue. The provided enzyme mix specifically oxidizes glucose or glucose 6-phosphate to generate a product that reacts with a dye to generate fluorescence (Ex/Em = 535/587 nm) or color (450 nm), respectively. To measure metabolites in serum-free brain and liver tissues, mouse in anesthesia were cardiac perfused with cold phosphate-buffered saline (PBS). Dissected brains and livers were subsequently snap frozen using liquid nitrogen and kept at −80°C until the analysis. The samples for comparison group were handled by the same way to minimize any variation among them.
Statistical Calculation
Two types of statistical analyses were performed with the program “R” (http://cran.r-project.org; RRID:SCR_000036). (1) ANOVA comparisons were used to identify metabolites that differed significantly between ages or genotypes following log transformation and imputation of missing values, if any, with the minimum observed value for each compound. The q-value describes the false discovery rate; a low q-value (q<0.10) is an indication of high confidence in a result. (2) Random Forest analysis was used for classification, which gives an estimate of how well a metabolite can be classified in a new data set into each group. Random forests create a set of classification trees based on continual sampling of the experimental units and compounds. Then each observation is classified based on the majority votes from all the classification trees. (3) Student t tests were performed using Prism (RRID:SCR_002798) when comparing metabolite concentrations for validation studies in the brain, serum and liver. Statistical significance was defined as p<0.05.
RESULTS
Summary of metabolomic analysis and metabolic pathways that were significantly affected
To find novel biomarkers and uncharacterized biochemical pathways implicated in Krabbe disease pathogenesis, we profiled the metabolome of twitcher mice, and their wildtype (WT) littermates, at P15 and P22. These time points were selected to correspond to before and after onset of signs of disease on the C57BL/6 background (Suzuki 2004b). We analyzed thirty-two specimens in four cohorts, which included eight mice per cohort. The cohorts were defined as follows: 1) twitcher at P15, 2) twitcher at P22, 3) WT at P15 and 4) WT at P22. We specifically chose the hindbrain for each mouse, including the rostral end of spinal cord, as Krabbe pathology is more advanced in the hindbrain compared to the forebrain, which is likely related to the caudal to rostral pattern of myelination in the developing brain (Levine et al. 1994). We identified a total of 314 compounds in each specimen, and conducted a two-way analysis of variance (ANOVA) with post-test comparisons to reveal a number of statistically significant differences (p≤0.05) across both genotype and age (Table 1 and Supplementary Table 1). The false discovery rate (q-value) was also calculated to take into account the multiple comparisons that normally occur in metabolomic-based studies. Psychosine was significantly increased at both P15 and P22 in the hindbrains of twitcher mice compared to WT (Fig. 1). Generally, the WT and twitcher brains showed very similar levels of metabolite change over time. Specifically, comparison of the WT brain profiles between P15 and P22 indicated that almost half of the metabolites, 149 out of 314 total, were significantly changed and the same was true of the twitcher brains between P15 and P22, 151 out of 314. The P15-22 span is a very active time period in mouse brain development and maturation, especially in regards to the myelin sheath, and the mouse metabolome is known to show extreme variation during development (Houtkooper et al. 2011). At P15, before the onset of central nervous system (CNS) pathology in the twitcher mouse, only 12 compounds were found to be statistically significantly changed between WT and twitcher mice (Fig. 2A and C). At P22, at a time point shortly after signs relevant to the CNS have appeared, the number of statistically changed metabolites rose to 52 (Fig. 2B and C).
Table 1.
Statistical summary of metabolomic profile of hindbrains of twitcher and WT control mice from a total of 314 named metabolites
| 2-way ANOVA comparisons |
|
|
|
|
||||
|---|---|---|---|---|---|---|---|---|
| total metabolites (p≤0.05) | 149 | 151 | 12 | 52 | ||||
metabolites (
 ) ) |
|
|
|
|
||||
| total metabolites (0.05<p<0.10) | 17 | 17 | 10 | 15 | ||||
| metabolites (
) |
|
|
|
|
A total of 314 metabolites were identified in the hindbrain specimens, and two-way analysis of variance (ANOVA) with post-test comparisons revealed varying numbers of statistically significant differences across the various comparisons of both genotype and time point (See also Supplementary Table 1), with statistical significance (p≤0.05). Comparison of the WT brain profiles on P15 and P22 indicated that almost half - 149 out of 314 - of the metabolites were significantly altered and the same was true of the twitcher brains, with 151 out of 314 changing significantly between P15 and P22. At P15, only 12 compounds exhibited statistically significant changes between WT and twitcher mice, whereas this number rose to 52 at P22.
Figure 1.

Galactosylsphingosine (psychosine) was highly increased in both pre- and post-symptomatic twitcher mice compared to WT. The top and bottom bars with whiskers represent the entire spread of the data points for the samples. Within the boxplot, the plus (+) indicates the mean value, and the horizontal dividing line is the median value. The top and bottom of the box represent the seventy-fifth and twenty-fifth percentile, with the whiskers indicating the maximum and minimum points.
Figure 2.

Metabolomic profiling of hindbrains of twitcher and WT control mice. A) Non-supervised z-score plot of 314 compounds from P15 WT (n = 8) and P15 twitcher (n = 8) hindbrains, normalized to the mean of the WT samples. Each black or red point represents one metabolite in WT or twitcher sample, respectively. Arrows depict 12 metabolites that were found to be statistically significantly changed (p ≤ 0.05) by ANOVA comparisons. B) The same z-score plot of 314 compounds from P22 wildtype (n = 8) and P22 twitcher (n = 8). Arrows depict 52 metabolites that were found to be statistically significantly changed (p ≤ 0.05) by ANOVA comparisons. C) List of significantly changed metabolites from parts A (top) and B (bottom) listed in blue or red, representing a decrease or increase from WT respectively.
We used Random Forest (RF) analysis as a supervised clustering method (Goldstein et al. 2010) to discover common metabolic pathways that were altered between WT and twitcher hindbrains. The predictive accuracy for RF analysis comparing twitcher mice to WT littermates at P15 was not very accurate (31%) reflecting the overall similarity of the two genotypes before the onset of signs of disease. Nonetheless, the metabolites that were altered were categorized to the glycolysis and pentose phosphate pathways on the Biochemical Importance Plot (Fig. 3A), implicating these pathways as early manifestations of differences between WT and twitcher. On the other hand, classification of the P22 specimens for both WT and twitcher cohorts had an RF predictive analysis of 100%. The Biochemical Importance Plot for the P22 comparison was dominated by markers associated with mitochondrial energy metabolism, inflammation, and oxidative stress (Fig. 3B). A heat map of the metabolic pathways highlighted by RF clustering shows a decrease in some metabolites involved in glucose metabolism in P15 twitcher hindbrains (Fig. 3C–D). In twitcher hindbrains at P22 there was also a decrease in phospholipid turnover and sugar alcohols involved in osmolyte regulation, and an increase in branched chain amino acid (BCAA) metabolism, inflammation and oxidative stress, and neurotransmitter and osmolyte metabolism (Fig. 3C). The heat map also shows that these metabolic pathways that were changed at the P22 time point were largely unchanged before signs of disease at P15. Fourteen other metabolites were also found to change significantly in twitcher hindbrains at one of these two time points, though they were not ordered into specific metabolic pathways by RF clustering (Fig. 3C). A full analysis of all 314 metabolites at both time points and genotypes is available in Supplementary Tables 1 and 2.
Figure 3.

Classification of metabolites by Random Forest statistical analysis. A) Random Forest classification in hindbrains of P15 twitcher compared to P15 WT gave a predictive accuracy of 31%, suggesting key differences in glycolysis and pentose phosphate pathway metabolism. B) Random Forest in P22 twitcher compared to P22 WT gave a predictive accuracy of 100%, with differences in fuel selection and energy metabolism, inflammation and oxidative stress markers. C–D) All 314 analyzed metabolites were subjected to supervised clustering via Random Forest classification to predict metabolic pathways that were globally changed in the twitcher hindbrain, yielding glucose metabolism, phospholipid turnover, branched chain amino acid metabolism, inflammation and oxidative stress and neurotransmitter and osmolyte metabolism. C) Heat map shows the relative concentration of 70 metabolites (rows), grouped by sample type (column). Sample types are grouped by genotype (WT and twitcher) and age (P15 and P22). Colored squares represent the relative concentration of a given metabolite, defined as the number of standard deviations above or below the median value. B.C.A.A. = Branched Chain Amino Acids, NeuroT= neurotransmitters. D) Heat map shows the fold change performed on the 70 metabolites from C. Red and blue shaded cells indicate p≤0.05 (red indicates that the mean values are significantly higher for that comparison; blue values significantly lower). Light red and light blue shaded cells indicate 0.05<p<0.10 (light red indicates that the mean values trend higher for that comparison; light blue values trend lower). Two single ANOVA comparisons (columns) analyzed the mean concentration of each metabolite as follows: (i) P15 twitcher to P15 WT mice and (ii) P22 twitcher to P22 WT mice.
Glucose usage was reduced in twitcher mice before disease onset
Glucose is the primary energy source used by all cells of the brain (Vannucci et al. 1998). Glycolysis and gluconeogenesis are the key regulatory metabolic pathways that control glucose homeostasis. The metabolites glucose 6-phosphate (G6P), fructose 6-phosphate (F6P) and 3-phosphoglycerate, members of both the glycolysis and gluconeogenesis pathways, were found to be significantly lower in P15 twitcher hindbrains compared to WT controls (Figs. 3C–D and 6), indicating glucose hypometabolism. F6P is converted to mannose 6-phosphate (M6P), an additional hexose phosphate, by the M6P isomerase (DeRossi et al. 2006). M6P was also significantly reduced in the P15 twitcher brains and may represent a possible compromise to maintain lysosomal integrity in twitcher mice, since M6P is important to traffic lysosomal proteins, like GALC, to the lysosome via the M6P receptor.
Figure 6.

Illustration of key metabolic pathways that were altered in the hindbrain of twitcher, an authentic mouse model of Krabbe disease. For simplicity, not all biochemical steps or enzymes involved in the metabolic pathways are shown. We found that glycolysis and gluconeogenesis (A), the pentose phosphate pathway (B), membrane lipid homeostasis (C), and branched-chain amino acid metabolism (D) are all involved in the pathogenesis of the twitcher mice.
The pentose phosphate pathway (PPP) is a key metabolic pathway that runs parallel to glycolysis and allows for both the reduction of NADP+ to NADPH and the reversible conversion between glycolytic intermediates and pentose phosphates (Wamelink et al. 2008). Similarly to intermediates of glycolysis/gluconeogenesis, PPP metabolites including sedoheptulose 7-phosphate and ribulose/xylulose 5-phosphate, were also decreased at P15 in the twitcher (Figs. 3C–D and 6). Further evidence of altered glucose homeostasis was suggested by the significant elevation of 1,5-anhydroglucitol (1,5-AG) in the twitcher samples at P22. 1,5-AG is a good postprandial indicator of glycemic control in plasma, where its levels are inversely correlated with glucose (Buse et al. 2004), though it is unknown whether this same relationship holds true in tissues as well. Interestingly, all of these changes are specific to the earlier time point, as all metabolites return to WT levels one week later. Taken together, these changes suggest that alterations in glucose metabolism precede or coincide with the onset of neurological signs in twitcher mice. Glucose hypometabolism seen in the P15 twitcher may be evident, at a presymptomatic time point in Krabbe disease, by using 18F-fluorodeoxyglucose Positron Emission Tomography (FDG-PET) (Marcus et al. 2014). In fact, glucose hypometabolism is a common trait of other disorders involving oligodendrocytes such as multiple sclerosis and various leukodystrophies (Bakshi et al. 1998; Bluml et al. 2001; Meisingset et al. 2013).
Reduced phospholipid and membrane turnover in twitcher brains
Phospholipids are the major component of the cell membrane and are involved in a number of important structural and signaling roles in the eukaryotic cell (Lombard et al. 2012). Glycerophospholipids are synthesized by the addition of a head group to diacylglycerol (DAG) (Lagace and Ridgway 2013), a signaling molecule that is in turn synthesized by glycerol 3-phosphate (G3P) (Fig. 6). We found a significantly reduced level of G3P, and a structurally related metabolite, glycerol 2-phosphate, in the brains of P22 twitcher mice (Fig. 3C–D). The reduction in G3P, the key building block in the synthesis of all glycerophospholipids, suggests an overall reduction in the breakdown and/or synthesis of all membrane glycerophospholipids, including phosphatidylcholine (PC) and phosphatidylglycerol. PC can also be synthesized or degraded to glycerophosphocholine (GPC) by the addition or removal of an acyl chain. Consistent with reductions in G3P, there was a trend towards a decrease in glycerophosphocholine (GPC) in the P22 twitcher. PC levels did not differ between WT and twitcher brains (Supplementary Tables 1 and 2), but its derivative, cytidine 5′-diphosphocholine (CDP-choline), was increased in P15 twitcher brains. Since CDP-choline increases glucose metabolism in the brain and cerebral blood flow (Watanabe et al. 1975), significantly upregulated CDP-choline in the P15 twitcher may indicate a compensatory regulation of glucose hypometabolism at an earlier stage of the disease.
Cholesterol is an essential structural lipid of all membranes including the myelin sheath of oligodendrocytes and Schwann cells. We found that the cholesterol precursors, lanosterol and lathosterol, were also dramatically reduced in P22 twitcher specimens compared to WT, further suggesting disrupted membrane homeostasis and possibly reflective of decreased myelin turnover. In fact, the P22 time point falls within the most active period of myelination for rodents, which typically spans P15 to P25. At this point, galactosylceramide biosynthesis is known to peak and myelin is thought to rapidly turn over (Costantino-Ceccarini and Morell 1972). Generally, the process of myelin turnover requires the break-down of galactosylceramide into galactose and ceramide by GALC (Platt and Walkley 2004) and these products are re-used in a separate remyelination pathway (Suzuki 2004a). In twitcher mice and Krabbe patients, this break-down is blocked by the loss of GALC activity, and may therefore explain why metabolites necessary for membrane synthesis are reduced in the P22 twitcher. Furthermore, the P22 twitcher also has reduced citrulline which is used as a post-translational modification on select proteins, including myelin basic protein (Wood and Moscarello 1989).
N-acetylaspartate (NAA), a derivative of aspartic acid, emits the largest signal in Magnetic Resonance Spectrometry (MRS) of human brain, and are decreased in numerous neuropathological conditions (Deicken et al. 2000). Interestingly, NAA is significantly reduced in P22 twitcher compared to WT (Fig. 3C–D) and may represent a reliable biomarker for KD progression in close agreement with a previous study (Meisingset et al. 2013). Since one of the primary functions served by NAA is a source of acetate for lipid and myelin synthesis in oligodendrocytes (Chakraborty et al. 2001), the reduction of NAA may reflect a decrease in myelin turnover in the P22 twitcher. Taken together, these results suggest that membrane homeostasis, including sphingolipids, glycerophospholipids, cholesterol and myelin, may be disrupted in twitcher brains before the signs of disease begin, and is readily perturbed once early signs are present.
Elevated oxidation of BCAAs in P22 twitcher
In the brain, the branched chain amino acids (BCAAs) valine, isoleucine, and leucine, are important for the synthesis of the excitatory neurotransmitter glutamate and the inhibitory neurotransmitter GABA (Yudkoff 1997). All three BCAAs were found to be significantly increased in the P22 twitcher brain (Fig. 3C–D). Evidence of increased BCAA metabolism in the P22 twitcher hindbrain was also supported by the significant accumulation of a number of BCAA oxidative acyl-carnitine derivatives including acetylcarnitine, propionylcarnitine, isobutyrylcarnitine, 3-dehydrocanitine, 2-methylbutyrylcarnitine (C5), β-hydroisovalerylcarnitine, butyrylcarnitine, and hydroxybutyrylcarnitine (Figs. 3C–D and 6). Carnitine and acyl-carnitines are present in the mitochondria where they play an important role in generating energy from lipids by β-oxidation as well as in maintaining equilibrium between metabolic pathways in the cytoplasm and mitochondria. The exchange of acyl-carnitines for coenzyme A is believed to be an important mechanism to maintain the limited pools of free coenzyme A in cells (Ramsay and Zammit 2004). We found a relative reduction in the levels of free coenzyme A and an increase in free carnitine in twitcher brains at P22. We also found a significant decrease in the biosynthetic precursor for carnitine, deoxycarnitine (Fig. 3C–D). Overall, this pattern suggests that the entire carnitine pathway is altered in twitcher hindbrains after onset of disease. The build-up of acyl-carnitines may specifically reflect the incomplete oxidation of BCAAs or fatty acids, and may therefore be a sign of mitochondrial dysfunction (Wanders et al. 2012). In fact, mitochondrial dysfunction has recently been postulated to represent an underlying role in the pathogenesis of KD, as psychosine is thought to induce apoptotic and necrotic cell death by increasing mitochondrial calcium and reactive oxygen species (ROS) production (Voccoli et al. 2014).
Biomarkers of inflammation and anti-oxidation were elevated in the twitcher at both P15 and P22
Despite the absence of external signs relevant to CNS disease in the P15 twitcher, microglia and astrocytes have already begun to show subtle activation at P15 in some twitcher hindbrain regions (Taniike and Suzuki 1994). Microgliosis and astrocytosis are known to be progressive and eventually span the entire central nervous system of affected twitcher mice (Levine et al. 1994). Not surprisingly, in the P22 twitcher hindbrains, the inflammatory mediators corticosterone and 12-HETE, were significantly elevated as well as the dipeptide proline-hydroxy-proline which reflects the inflammatory matrix metalloproteinase-catalyzed breakdown of collagen (Cechowska-Pasko et al. 2006) (Fig. 3C–D). Similarly, there were also complementary signs of anti-oxidant mediators which could protect against such oxidative stress in the P22 twitcher. For example, there was a significant elevation of the potent endogenous antioxidant uric acid, which can safely react with highly toxic hydroxyl radicals and hypochlorous acid. Uric acid is synthesized in a two-step enzymatic reaction from hypoxanthine, which was also found to be significantly increased in the twitcher hindbrain. Alternatively, uric acid can be synthesized by purine nucleotides, adenosine and guanosine. In P22 twitcher hindbrains we found a significant reduction in these purine nucleotides and some of their derivatives including, adenosine, guanosine, adenosine 5′-diphosphate (ADP), N1-methyladenosine, and guanosine 5′-diphospho-fucose (Fig. 3C–D). Other antioxidant molecules were also found to be increased in the P22 twitcher samples, including α-tocopherol, which protects cell membranes from free radicals and is upregulated with increased microglial activation and neuroinflammation (Khanna et al. 2015).
Neurotransmitters and Neuromodulators
Serotonin’s precursor, tryptophan, and its breakdown product, 5-hydroxyindoleacetate, were elevated in P22 twitcher compared to WT specimens (Fig. 3C–D). Similarly, phenylalanine, the precursor for the catecholamine neurotransmitters (dopamine, norepinephrine and epinephrine) was also significantly increased in P22 twitcher hindbrains. The polyamine putrescine - both a regulator of proliferation/chromatin condensation as well as a precursor for GABA (γ-aminobutyric acid) (Iacomino et al. 2012) - was also increased in the brains of P22 twitcher mice. Furthermore, serine was also highly upregulated in P22 twitcher, which serves as a neuromodulator by coactivating NMDA receptors. In fact, serine has recently been considered as a potential biomarker for early diagnosis of Alzheimer’s disease and has been found to be present at a relatively high concentration in the cerebrospinal fluid of patients that later developed Alzheimer’s disease (Madeira et al. 2015). The changes in these neurotransmitter precursors reflect changes in neurotransmitter metabolism which may be further altered as the disease progresses.
Osmotic Regulation
KD is an inflammatory disease and pathological tissues usually contain obvious levels of edema and swelling. The P22 twitcher had changes in a range of compounds known to serve as osmotic regulators, including a decrease in betaine and increases in urea, threonine and allo-threonine. Homoserine (also called isothreonine), an intermediate in the biosynthesis of threonine, was found to be significantly reduced. Additionally, the polyol arabitol was found to be significantly decreased at P22. A similar trend, albeit non-significant, was found to occur for other polyols including mannitol, sorbitol and ribitol. Polyols are all known to be active osmolytes (Yancey 2005). Taken together, these altered metabolites may suggest a modest sign of altered osmo-regulation in the brains of twitcher at P22 (Fig. 3C–D). However, because these biochemicals have multiple roles in metabolism, it is difficult to discern whether these observed changes are truly linked to osmotic regulation.
Other observations and potential biomarkers for early diagnosis
Disease-specific metabolic profiles have proven to be useful measures for early diagnosis and surveillance of disease progression (Zhang et al. 2015). We hypothesized that metabolites that were significantly altered at the P15 time point before onset of signs of disease could represent potential biomarkers for early diagnosis of KD. In this study, we found that four metabolites: S-adenosylhomocysteine, cytidine 5′-diphosphocholine, 1-palmitoylglycerophosphoserine, and hypoxanthine, were significantly increased in the P15 twitcher as compared to WT (Figs. 2A, 2C, and 4A). Instead, eight metabolites were found to be significantly decreased in the P15 twitcher hindbrain: glucose 6-phosphate, fructose 6-phosphate, 3-phosphoglycerate, sedoheptulose 7-phosphate, ribulose/xylulose 5-phosphate, mannose 6-phosphate, 10-heptadecenoate, and 2-linoleoylglycerol (Figs. 2C and 3C–D).
Figure 4.

Potential biomarkers for early diagnosis of Krabbe disease. (A) Metabolomics analysis revealed that four metabolites: S-adenosylhomocysteine, hypoxanthine, cytidine 5′-diphosphocholine, and 1-palmitoylglycerophosphoserine, were significantly increased in twitcher brains before signs of disease at P15 as compared to WT. Red arrow indicates that the mean values are significantly higher (p ≤ 0.05), and pink arrow indicates the mean values trend higher (0.05<p <0.10) by ANOVA comparisons. (B) ELISA analysis for S-adenosylhomocysteine showed that the concentration of this metabolite was not correlated with progression of Krabbe pathology in twitcher mice. (C) Fluorometric assay for Hypoxanthine/Xanthine showed that concentrations were higher in the twitcher brain as compared to WT at P15, before disease onset. *p < 0.05 (Student’s t-test; n = 3; P = 0.013).
S-adenosylhomocysteine (SAH) is an amino acid derivative and an intermediate, byproduct, or modulator of several metabolic pathways, including the activated methyl cycle and cysteine biosynthesis. It is also a product of S-adenosyl-methionine (SAM)-dependent methylation of biological molecules, including DNA, RNA, and histones and other proteins. Elevated SAH is considered a risk factor for many diseases, including cancer and neurodegenerative diseases (Herrmann and Obeid 2007). We investigated the potential of SAH as a biomarker in KD progression by performing an enzyme-linked immunosorbent assay (ELISA) on three brain specimens from twitcher and WT littermates through various time points. Unfortunately, the level of SAH was not altered much as the disease progressed (Fig. 4B). This represents a major limitation in using SAH as a biomarker for KD, as SAH would not be adequate for the surveillance of disease progression and response to therapy. We next investigated the potential of hypoxanthine as a biomarker in KD progression. Hypoxanthine is the precursor of the antioxidant uric acid and a purine metabolite involved in the purine nucleotide salvage pathway. We performed a fluorometric Xanthine enzyme assay on three brain specimens from twitcher and WT brains at various time points. We validated that hypoxanthine was indeed elevated at the P15 time point in twitcher brains (Fig. 4C). Hypoxanthine levels were also increased at the earlier P8 time point and continued to be upregulated as the disease progressed, though they were not statistically significant except at P15. These data suggest that, if hypoxanthine was also elevated in the blood or cerebrospinal fluid (CSF) of twitcher mice and KD patients, it may be a potential biomarker for both diagnosis and clinical follow up of patients with Krabbe disease.
Since many metabolites are extremely volatile and are known to be degraded or metabolized very quickly, we collected tissues for the metabolomic analysis without perfusion and snap-froze them in liquid nitrogen to minimize the dissection time, as described in the previous brain metabolomics studies (Davidovic et al. 2011; Kopp et al. 2010; Salek et al. 2010). To know if residual serum left in the brain contributed to the observed changes in the metabolomics analysis, we analyzed several metabolites in PBS-perfused hindbrains. Interestingly, measured glucose concentrations in the hindbrains were highly dependent on residual serum and were significantly reduced in both the WT and the twitcher PBS-perfused brain (Fig. 5A). Regardless of perfusion status, however, the glucose concentrations measured in the brain were extremely low and barely detectable. There was no statistical significant difference in glucose concentrations between P22 WT and twitcher hindbrains. Conversely, the metabolites glucose 6-phosphate, hypoxanthine, and BCAAs were not changed with PBS-perfusion. The concentration of these metabolites was over a full order of magnitude higher in the hindbrain than glucose, which may explain why a small amount of residual serum would not significantly alter their measured concentrations (Fig. 5B). To further understand the glucose usage defects in the twitcher mouse, we analyzed glucose concentrations in the serum and in the PBS-perfused liver, a key organ in regulating glucose metabolism (Fig. 5C). We also measured the concentration of glucose 6-phosphate, the first glucose intracellular intermediate in both the serum and the liver. Interestingly, glucose levels in the serum of P15 twitcher mice were significantly reduced compared to wildtype littermates, which may have contributed to the non-significant trend in the metabolomics study. However, we did not observe any changes in glucose concentrations in the liver, or in glucose 6-phosphate concentrations in the serum or liver. These findings suggest that the decrease in glucose metabolism is likely related to altered brain glucose usage, as opposed to metabolic alterations in peripheral glucose homeostasis. The complex alterations in twitcher glucose metabolism and glucose usage may therefore reveal the opportunity to diagnose Krabbe disease by in vivo imaging modalities like FDG-PET.
Figure 5.

Effect of serum contribution on hindbrain metabolite concentration and glucose usage in serum and liver. (A) The concentration of glucose was measured in the hindbrains of PBS-perfused and non-perfused WT and twitcher P22 mice. Glucose, which is detected at low concentrations in the hindbrain, is significantly reduced by PBS-perfusion for both genotypes (Student’s t-test; n = 3; P = 0.002). The concentration of glucose was not significantly altered between the genotypes at P22. (B) The concentrations of other important metabolites in the hindbrains were not significantly altered by PBS perfusion. The metabolites glucose 6-phosphate, hypoxanthine/xanthine and BCAAs were all significantly altered in the metabolomics analysis and were found to occur at hindbrain concentrations of at least one order of magnitude higher than glucose. All three of these metabolites were not found to be significantly altered by PBS perfusion in the P22 twitcher hindbrain. (C) Glucose concentrations in the serum of P15 twitcher mice are significantly reduced (Student’s t-test; n = 3; P = 0.015), but are not significantly changed at P22. Conversely, glucose 6-phosphate concentrations are unchanged at both time points. Neither glucose, nor glucose 6-phosphate were significantly altered at either time point in the twitcher liver.
DISCUSSION
Patients with infantile KD exhibit severe and rapidly progressive neurological deterioration. Treatment with cord blood transplant is effective if initiated pre-symptomatically, but has a significant morbidity and mortality—presymptomatic diagnosis must be accurate. Unfortunately, secure diagnosis is complicated by the broad spectrum of clinical variability seen in KD patients with the same mutation or GALC activity. The identification of reliable biomarkers and underlying biological pathways relevant in Krabbe pathogenesis is therefore necessary to advance diagnosis, prognosis, and timely treatment of KD patients. While galactosylceramide is thought to be the major physiologic substrate for GALC, the enzyme also catabolizes galactosylsphingosine (psychosine), monogalactosyl diglyceride, and under specific assay conditions, lactosylceramide (Wenger et al. 2001).
Psychosine accumulates throughout the nervous system after loss of enzymatic function of GALC and is thought to cause oligodendrocyte apoptosis and degeneration of axons (Wenger 2011a). It has been proposed that psychosine measurement could help monitor disease progression and be used to diagnose and evaluate Krabbe patients (Turgeon et al. 2015). However, a number of animal and human studies suggest that psychosine accumulation may not be as direct an indicator for KD progression as previously thought. Firstly, twitcher mice deficient for the enzyme thought to generate psychosine, UDP-galactose:ceramide galactosyl transferase (CGT), show a similar neuronal degeneration as seen in the classical twitcher mouse, despite undetectable levels of brain psychosine (Ezoe et al. 2000). Secondly, mice deficient for the sphingolipid activator protein A (saposin A), an enzyme required for GALC activation in vivo, are phenotypically similar (though milder) to the twitcher mouse. Interestingly, the accumulation of psychosine in the brains of saposin A deficient mice was only twice normal compared to the 10–20 fold increase in the twitcher mice, even when both mice had progressed to similar clinical phenotypes (Matsuda et al. 2007). Furthermore, in the Galctwi-5j mouse, which has a spontaneous mutation in Galc that matches the Glu130Lys missense mutation found in some KD patients, psychosine levels in Galctwi-5j mice do not correlate with the regions exhibiting the disturbances in myelination/axonopathy (Potter et al. 2013). Lastly, in a study investigating residual GALC activity in cells from KD patients, it was found that some mutations from affected patients had reduced GALC activity for galactosylceramide, while having intact activity for psychosine (Harzer et al. 2002). Taken together, these studies strongly suggest that psychosine levels may not always correlate with clinical symptoms.
Our approach, therefore, was to investigate metabolites other than psychosine that could contribute to disease progression in KD, with the intent of exploiting them as novel biomarkers. Advanced metabolomic analysis provides a detailed and unbiased snapshot of the global metabolic profile that exists within a tissue. By comparing relative changes in the abundance of biochemical species between WT and twitcher tissues, we were able to highlight a number of previously undocumented potential biomarker candidates as well as entire metabolic pathways that were altered in KD and may underlie the disease process. In the hindbrains of twitcher mice, we found four upregulated and eight downregulated metabolites prior to signs of disease, and 31 upregulated and 21 downregulated metabolites immediately after the onset of disease. The role of each of these metabolites and correlation to disease pathogenesis remains unclear but warrants further investigation. For example, a number of our findings were consistent with previous metabolomic studies in other demyelinating diseases including multiple sclerosis (Bhargava et al. 2015; Cocco et al. 2016; Gonzalo et al. 2012; Mangalam et al. 2013; Pieragostino et al.; Tavazzi et al. 2011). In particular, in a metabolomics study from the CSF of a rat model of Experimental Autoimmune Enchephalomyelitis (EAE), BCAAs and putrescine were both found to increase with disease progression (Noga et al. 2012). Similarly, a metabolomic analysis in the serum of a Relapsing Remitting EAE model in SJL mice, found significant increases in alpha-tocopherol and a number of acyl-carnitine species, as well as a reduction in 1,5-anhydroglucitol and 3-phosphoglycerate (Mangalam et al. 2013). Some of our detected changes, therefore, may represent altered metabolic pathways attributed directly to oligodendrocyte dysfunction and demyelination. Alternatively, other changes may represent a global alteration in the metabolism and equilibrium of the tissue, unrelated to demyelination. At P15, before the onset of signs of disease, the twitcher specimens had similar metabolic profiles compared to WT specimens. The only metabolic pathway that was altered at this early time point reflected decreased metabolites associated with glucose energy metabolism (Figs. 3B and 6), indicating an alteration of glucose uptake or usage before the clinical signs of disease. As predicted, the major differences between WT and twitcher hindbrains were observed on postnatal day 22, after signs of disease were apparent, including a decrease in phospholipid turnover, signs of restricted mitochondrial metabolism of branched-chain amino acids, elevated biomarkers of inflammation and oxidative stress and alterations in neurotransmitter and osmolyte metabolism (Fig. 6). Changes in these metabolites and in the overall metabolic pathways may be useful as part of a multimodal approach to correlate and collocate metabolic and structural abnormalities in the brain, which could provide more sensitive detection of early changes in disease. For example, given the early changes in brain glucose metabolism during the transition toward the clinical phenotype, it seems reasonable that the use of FDG-PET may provide useful imaging parameters that could complement the structural information acquired by magnetic resonance imaging. These modalities, used in tandem or even in combination, may be useful for more accurate diagnosis of Krabbe disease by recognizing both structural and metabolic changes in the brain.
Our study confirms and significantly extends upon previous studies that analyze metabolites in twitcher mice (Esch et al. 2003; Meisingset et al. 2013; Zanfini et al. 2014). Firstly, we used a high-throughput analytical metabolomics platform incorporating positive and negative mode LC/MS and GC/MS analyses, which included an extensive biochemical library that contained over 2,300 named and 5,200 unnamed compounds. The previous studies profiled a more limited number of metabolites focusing on sphingolipid species by using ESI tandem mass spectrometry (MS/MS) (Esch et al. 2003) or 17 peak assigned metabolites using 1H and 13C magnetic resonance spectra (Meisingset et al. 2013). Secondly, previous groups profiled only twitcher brains after signs of disease appeared, as opposed to specimens before signs were apparent. Meisingset et al. (2013) examined spinal cord, cerebellum, and forebrain with mild signs (P30) and advanced signs of disease (P40) in twitcher mice on the mixed background of C57BL/6 and FVB. Esch et al. (2003) used the pons/medulla of twitcher mice at P31 and P35-37 span on the background of C57BL/6, which are in the later stages of disease progression. Metabolite analysis at early time points is important for the discovery of biomarkers with diagnostic implications. For example, Meisingset et al. (2013) identified N-acetyl aspartate (NAA) and lactate as potential biomarkers for the primary oligodendrocyte dysfunction that occurs in Krabbe disease, and myo-inositol and taurine as early indicators of disturbed metabolism in the disease, based on the results in the symptomatic twitcher mice. However, our study showed that NAA levels were not changed in the P15 hindbrain and in fact were significantly reduced in the P22 twitcher (0.91 fold) compared to WT littermates. Furthermore, neither lactate, myo-inositol, nor taurine was changed at the P15 and P22 time points in twitcher hindbrains. A recent study analyzed the fatty acid composition in brain and serum of the twitcher mice on the background of C57BL/6 for the purpose of biomarker discovery for Krabbe disease (Zanfini et al. 2014), and found that C16:1n7c monounsaturated fatty acid (palmitoleate) was significantly increased in the twitcher (at P20-40 span) in brain, as in P22 twitcher brain of our study. However, they showed that C14:0 saturated fatty acid (myristate) was not changed in twitcher P20-40 brain compared to controls, which instead was significantly increased in P22 twitcher brain of our analysis (Supplementary Tables 1 and 2). Unfortunately, they did not measure 10-heptadecenoate (17:1n7) and 10-nonadecenoate (19:1n9), which are decreased in our analysis in P15 and P22 twitcher, respectively. These differences may be accounted for by differences in the brain regions and techniques used for the analysis. Nevertheless, we believe that the volatility of these measurements may suggest that these metabolites have limited diagnostic potential.
In conclusion, by using global metabolomic analysis, we found that before the onset of signs of disease there was an alteration of glucose uptake or usage. After the onset of the disease, there were decreases in metabolic markers of phospholipid turnover, signs of restricted mitochondrial metabolism of branched-chain amino acids, and elevations of biomarkers of inflammation and oxidative stress. Furthermore, we identified hypoxanthine as a potential early biomarker for diagnosis, which may reveal brain damage much earlier than histopathology. Many of the identified markers are novel in their association with the disease. In a further study, we will focus on the validation of changes in these potential biomarkers in twitcher brain, extend analysis of their levels to CSF and serum to ask if they could be assessed in patients, and examine their correlation to Krabbe disease progression and severity in patient material.
Supplementary Material
Significance Statement.
Using a global metabolomic analysis in the authentic mouse model of Krabbe disease, twitcher, we identified metabolic pathways that are altered before disease onset, thus potentially pathogenic, and potential biomarkers. Before disease, twitcher hindbrains had a decrease in metabolites related to glucose metabolism and increased hypoxantine, suggesting that glucose metabolism in positron emission tomography and hypoxanthine are potential early biomarker for diagnosis.
Acknowledgments
Grant information: This work was supported by grants from the National Institutes of Health [R03-NS087359 to D. Shin], the Empire State Development Corporation for The Research Foundation—Krabbe Disease Research Working Capital [W753 to L. Wrabetz and ML. Feltri], the Empire State Development Corporation for Krabbe Disease Research Capital Equipment [U446 to L. Wrabetz and ML. Feltri]; and a grant from Hunter’s Hope Foundation.
Footnotes
Conflict of interest
The authors declare they have no conflict of interest.
Role of authors: All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: DS, LW, LF. Acquisition of data: DS, NW. Analysis and interpretation of data: DS, NW. Drafting of the manuscript: DS, NW. Critical revision of the manuscript for important intellectual content: DS, NW, LW, LF. Statistical analysis: DS, NW. Obtained funding: DS, LW, LF.
References
- Bakshi R, Miletich R, Kinkel P, Emmet M, Kinkel W. High-resolution fluorodeoxyglucose positron emission tomography shows both global and regional cerebral hypometabolism in multiple sclerosis. J Neuroimaging. 1998;8(4):228–234. doi: 10.1111/jon199884228. [DOI] [PubMed] [Google Scholar]
- Bhargava P, Mowry E, Calabresi P. Global metabolomics identifies perturbation of multiple metabolic pathways in Multiple Sclerosis (P5.242) Neurology. 2015;84(14) Supplement P5.242 [Google Scholar]
- Bluml S, Moreno A, Hwang J-H, Ross BD. 1-(13)C glucose magnetic resonance spectroscopy of pediatric and adult brain disorders. NMR Biomed. 2001;14(1):19–32. doi: 10.1002/nbm.679. [DOI] [PubMed] [Google Scholar]
- Buse JB, Freeman JLR, Edelman SV, Jovanovic L, McGill JB. Serum 1,5-Anhydroglucitol (GlycoMark™): A Short-Term Glycemic Marker. Diabetes Technol Ther. 2004;5(3):355–363. doi: 10.1089/152091503765691839. [DOI] [PubMed] [Google Scholar]
- Cechowska-Pasko M, Pałka J, Wojtukiewicz MZ. Enhanced prolidase activity and decreased collagen content in breast cancer tissue. Int J Exp Pathol. 2006;87(4):289–296. doi: 10.1111/j.1365-2613.2006.00486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty G, Mekala P, Yahya D, Wu G, Ledeen R. Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: evidence for myelin-associated aspartoacylase. J Neurochem. 2001;78(4):736–745. doi: 10.1046/j.1471-4159.2001.00456.x. [DOI] [PubMed] [Google Scholar]
- Cocco E, Murgia F, Lorefice L, Barberini L, Poddighe S, Frau J, Fenu G, Coghe G, Murru MR, Murru R, Carratore FD, Atzori L, Marrosu MG. 1H-NMR analysis provides a metabolomic profile of patients with multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2016;3(1):e185. doi: 10.1212/NXI.0000000000000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino-Ceccarini E, Morell P. Biosynthesis of brain sphingolipids and myelin accumulation in the mouse. Lipids. 1972;7(10):656–659. doi: 10.1007/BF02533072. [DOI] [PubMed] [Google Scholar]
- Davidovic L, Navratil V, Bonaccorso CM, Catania MV, Bardoni B, Dumas M-E. A Metabolomic and Systems Biology Perspective on the Brain of the Fragile X Syndrome Mouse Model. Genome Res. 2011;21:2190–2202. doi: 10.1101/gr.116764.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deicken R, Johnson C, Pegues M. Proton magnetic resonance spectroscopy of the human brain in schizophrenia. Rev Neurosci. 2000;11(2–3):147–158. doi: 10.1515/revneuro.2000.11.2-3.147. [DOI] [PubMed] [Google Scholar]
- DeRossi C, Bode L, Eklund EA, Zhang F, Davis JA, Westphal V, Wang L, Borowsky AD, Freeze HH. Ablation of Mouse Phosphomannose Isomerase (Mpi) Causes Mannose 6-Phosphate Accumulation, Toxicity, and Embryonic Lethality. J Biol Chem. 2006;281(9):5916–5927. doi: 10.1074/jbc.M511982200. [DOI] [PubMed] [Google Scholar]
- Duchen LW, Eicher EM, Jacobs JM, Scaravilli F, Teixeira F. Hereditary leucodystrophy in the mouse: the new mutant twitcher. Brain. 1980;103(3):695–710. doi: 10.1093/brain/103.3.695. [DOI] [PubMed] [Google Scholar]
- Esch Sw, Williams TD, Biswas S, Chakrabarty A, Levine SM. Sphingolipid profile in the CNS of the twitcher (Globoid cell leukodystrophy) mouse: A lipidomics approach. Cellular and Molecular Biology. 2003;49(5):779–787. [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, Morse R, Krivit W, Kurtzberg J. Transplantation of Umbilical-Cord Blood in Babies with Infantile Krabbe’s Disease. N Engl J Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Ezoe T, Vanier MT, Oya Y, Popko B, Tohyama J, Matsuda J, Suzuki K, Suzuki K. Biochemistry and Neuropathology of Mice Doubly Deficient in Synthesis and Degradation of Galactosylceramide. J Neurosci Res. 2000;59:170–178. doi: 10.1002/(sici)1097-4547(20000115)59:2<170::aid-jnr3>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Goldstein BA, Hubbard AE, Cutler A, Barcellos LF. An application of Random Forests to a genome-wide association dataset: Methodological considerations & new findings. BMC Genetics. 2010;11:49. doi: 10.1186/1471-2156-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo H, Brieva L, Tatzber F, Jové M, Cacabelos D, Cassanyé A, Lanau-Angulo L, Boada J, Serrano JCE, González C, Hernández L, Peralta S, Pamplona R, Portero-Otin M. Lipidome analysis in multiple sclerosis reveals protein lipoxidative damage as a potential pathogenic mechanism. J Neurochem. 2012;123(4):622–634. doi: 10.1111/j.1471-4159.2012.07934.x. [DOI] [PubMed] [Google Scholar]
- Harzer K, Knoblich R, Rolfs A, Bauer P, Eggers J. Residual galactosylsphingosine (psychosine) b-galactosidase activities and associated GALC mutations in late and very late onset Krabbe disease. Clinica Chimica Acta. 2002;317:77–84. doi: 10.1016/s0009-8981(01)00791-4. [DOI] [PubMed] [Google Scholar]
- Herrmann W, Obeid R. Biomarkers of folate and vitamin B12 status in cerebrospinal fluid. Clin Chem Lab Med. 2007;45(12):1614–1620. doi: 10.1515/CCLM.2007.310. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Argmann C, Houten SM, Canto C, Jeninga EH, Andreux PA, Thomas C, Doenlen Rl, Schoonjans K, Auwerx J. The metabolic footprint of aging in mice. Sci Rep. 2011;1(134) doi: 10.1038/srep00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacomino G, Picariello G, D’Agostino L. DNA and nuclear aggregates of polyamines. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2012;1823(10):1745–1755. doi: 10.1016/j.bbamcr.2012.05.033. [DOI] [PubMed] [Google Scholar]
- Khanna S, Heigel M, Weist J, Gnyawali S, Teplitsky S, Roy S, Sen CK, Rink C. Excessive α-tocopherol exacerbates microglial activation and brain injury caused by acute ischemic stroke. FASEB J. 2015;29(3):828–836. doi: 10.1096/fj.14-263723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp F, Komatsu T, Nomura DK, Trauger SA, Thomas JR, Siuzdak G, Simon GM, Cravatt BF. The glycerophospho-metabolome and its influence on amino acid homeostasis revealed by brain metabolomics of GDE1(−/−) mice. Chem Biol. 2010;17(8):831–840. doi: 10.1016/j.chembiol.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagace T, Ridgway N. The role of phospholipids in the biological activity and structure of the endoplasmic reticulum. Biochimica et Biophysica Acta (BBA) 2013;1833(11):2499–2510. doi: 10.1016/j.bbamcr.2013.05.018. [DOI] [PubMed] [Google Scholar]
- Levine SM, Wetzel DL, Eilert AJ. Neuropathology of twitcher mice: examination by histochemistry, immunohistochemistry, lectin histochemistry and fourier transform infrared microspectroscopy. Int J Dev Neurosci. 1994;12(4):275–288. doi: 10.1016/0736-5748(94)90075-2. [DOI] [PubMed] [Google Scholar]
- Lombard J, López-García P, Moreira D. The early evolution of lipid membranes and the three domains of life. Nat Rev Microbiol. 2012;10(7):507–515. doi: 10.1038/nrmicro2815. [DOI] [PubMed] [Google Scholar]
- Madeira C, Lourenco MV, Vargas-Lopes C, Suemoto CK, Brandão CO, Reis T, Leite REP, Laks J, Jacob-Filho W, Pasqualucci CA, Grinberg LT, Ferreira ST, Panizzutti R. D-serine levels in Alzheimer’s disease: implications for novel biomarker development. Translational Psychiatry. 2015;5:e561. doi: 10.1038/tp.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangalam A, Poisson L, Nemutlu E, Datta I, Denic A, Dzeja P, Rodriguez M, Rattan R, Giri S. Profile of Circulatory Metabolites in a Relapsing-remitting Animal Model of Multiple Sclerosis using Global Metabolomics. J Clin Cell Immunol. 2013;30(4) doi: 10.4172/2155-9899.1000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus C, Mena E, Subramaniam RM. Brain PET in the Diagnosis of Alzheimer’s Disease. Clin Nucl Med. 2014;39(10):e413–e426. doi: 10.1097/RLU.0000000000000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda J, Yoneshige A, Suzuki K. The function of sphingolipids in the nervous system: lessons learnt from mouse models of specific sphingolipid activator protein deficiencies. J Neurochem. 2007;103:32–38. doi: 10.1111/j.1471-4159.2007.04709.x. [DOI] [PubMed] [Google Scholar]
- Meisingset TW, Ricca A, Neri M, Sonnewald U, Gritti A. Region- and age-dependent alterations of glial-neuronal metabolic interactions correlate with CNS pathology in a mouse model of globoid cell leukodystrophy. J Cereb Blood Flow Metab. 2013;33(7):1127–1137. doi: 10.1038/jcbfm.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noga MJ, Dane A, Shi S, Attali A, Aken Hv, Suidgeest E, Tuinstra T, Muilwijk B, Coulier L, Luider T, Reijmers TH, Vreeken RJ, Hankemeier T. Metabolomics of cerebrospinal fluid reveals changes in the central nervous system metabolism in a rat model of multiple sclerosis. Metabolomics. 2012;8(2):253–263. doi: 10.1007/s11306-011-0306-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieragostino D, D’Alessandro M, Ioia Md, Rossi C, Zucchelli M, Urbani A, Ilio CD, Lugaresi A, Sacchettaab P, Boccio PD. An integrated metabolomics approach for the research of new cerebrospinal fluid biomarkers of multiple sclerosis. Mol Biosyst. 11(6):1563–1572. doi: 10.1039/c4mb00700j. [DOI] [PubMed] [Google Scholar]
- Platt F, Walkley SU. In: Lysosomal defects and storage. Platt, Walkley, editors. Oxford: Oxford University Press; 2004. pp. 32–49. [Google Scholar]
- Potter GB, Santos M, Davisson MT, Rowitch DH, Marks DL, Bongarzone ER, Petryniak MA. Missense mutation in mouse GALC mimics human gene defect and offers new insights into Krabbe disease. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt190. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay RR, Zammit VA. Carnitine acyltransferases and their influence on CoA pools in health and disease. Mol Aspects Med. 2004;25(5–6):475–493. doi: 10.1016/j.mam.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Sakai N, Inui K, Tatsumi N, Fukushima H, Nishigaki T, Taniike M, Nishimoto J, Tsukamoto H, Yanagihara I, Ozono K, Okada S. Molecular Cloning and Expression of cDNA for Murine Galactocerebrosidase and Mutation Analysis of the Twitcher Mouse, a Model of Krabbe’s Disease. J Neurochem. 1996;66:1118–1124. doi: 10.1046/j.1471-4159.1996.66031118.x. [DOI] [PubMed] [Google Scholar]
- Salek RM, Xia J, Innes A, Sweatman BC, Adalbert R, Randle S, McGowan E, Emson PC, Griffin JL. A metabolomic study of the CRND8 transgenic mouse model of Alzheimer’s disease. Neurochemistry International. 2010;56(8):937–947. doi: 10.1016/j.neuint.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Santambrogio S, Ricca A, Maderna C, Ieraci A, Aureli M, Sonnino S, Kulik W, Aimar P, Bonfanti L, Martino S, Gritti A. The galactocerebrosidase enzyme contributes to maintain a functional neurogenic niche during early post-natal CNS development. Hum Mol Genet. 2012;21(21):4732–4750. doi: 10.1093/hmg/dds313. [DOI] [PubMed] [Google Scholar]
- Shin D, Feltri ML, Wrabetz L. Altered trafficking and processing of GALC mutants correlates with globoid cell leukodystrophy severity. J Neurosci. 2016;36(6):1858–1870. doi: 10.1523/JNEUROSCI.3095-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K. Krabbe Disease: Myelin Biology and Disorders. 2004a. pp. 841–850. [Google Scholar]
- Suzuki K. Models of Krabbe Disease. 2004b. pp. 1101–1113. [Google Scholar]
- Taniike M, Suzuki K. Spacio-temporal progression of demyelination in twitcher mouse: with clinico-pathological correlation. Acta neuropathol. 1994;88:228–236. doi: 10.1007/BF00293398. [DOI] [PubMed] [Google Scholar]
- Tavazzi B, Batocchi AP, Amorini AM, Nociti V, D’Urso S, Longo S, Gullotta S, Picardi M, Lazzarino G. Serum Metabolic Profile in Multiple Sclerosis Patients. Multiple Sclerosis International. 2011;2011 doi: 10.1155/2011/167156. Article ID 167156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgeon CT, Orsini JJ, Sanders KA, Magera MJ, Langan TJ, Escolar ML, Duffner P, Oglesbee D, Gavrilov D, Tortorelli S, Rinaldo P, Raymond K, Matern D. Measurement of psychosine in dried blood spots — a possible improvement to newborn screening programs for Krabbe disease. J Inherit Metab Dis. 2015;38(5):923–929. doi: 10.1007/s10545-015-9822-z. [DOI] [PubMed] [Google Scholar]
- Vannucci S, Clark R, Koehler-Stec E, Li K, Smith C, Davies P, Maher F, Simpson I. Glucose Transporter Expression in Brain: Relationship to Cerebral Glucose Utilization. Dev Neurosci. 1998;20(4–5):369–379. doi: 10.1159/000017333. [DOI] [PubMed] [Google Scholar]
- Voccoli V, Tonazzini I, Signore G, Caleo M, Cecchini M. Role of extracellular calcium and mitochondrial oxygen species in psychosine-induced oligodendrocyte cell death. Cell Death Dis. 2014;5:e1529. doi: 10.1038/cddis.2014.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamelink M, Struys E, Jakobs C. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. J Inherit Metab Dis. 2008;31(6):703–717. doi: 10.1007/s10545-008-1015-6. [DOI] [PubMed] [Google Scholar]
- Wanders RJA, Duran M, Loupatty FJ. Enzymology of the branched-chain amino acid oxidation disorders: the valine pathway. J Inherit Metab Dis. 2012;35(1):5–12. doi: 10.1007/s10545-010-9236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Kono S, Nakashima Y, Mitsunobu K, Otsuki S. Effects of various cerebral metabolic activators on glucose metabolism of brain. Folia Psychiatr Neurol Jpn. 1975;29(1):67–76. doi: 10.1111/j.1440-1819.1975.tb02324.x. [DOI] [PubMed] [Google Scholar]
- Wenger DA. Krabbe Disease. NCBI Bookshelf 2011a [Google Scholar]
- Wenger DA. Leukodystrophies (International Review of Child Neurology Series) 1. 2011b. Krabbe disease (Globoid cell leukodystrophy) pp. 90–105. [Google Scholar]
- Wenger DA, Suzuki K, Suzuki Y, Suzuki K. Galactosylceramide Lipidosis: Globoid Cell Leukodystrophy (Krabbe Disease) 2001. [Google Scholar]
- Wood D, Moscarello M. The isolation, characterization, and lipid-aggregating properties of a citrulline containing myelin basic protein. J Biol Chem. 1989;264(9):5121–5127. [PubMed] [Google Scholar]
- Yancey PH. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J Exp Biol. 2005;208:2819–2830. doi: 10.1242/jeb.01730. [DOI] [PubMed] [Google Scholar]
- Yudkoff M. Brain metabolism of branched-chain amino acids. Glia. 1997;21(1):92–98. doi: 10.1002/(sici)1098-1136(199709)21:1<92::aid-glia10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Zanfini A, Dreassi E, Berardi A, Piomboni P, Costantino-Ceccarini E, Luddi A. GC-EI-MS Analysis of Fatty Acid Composition in Brain and Serum of Twitcher Mouse. Lipids. 2014;49:1115–1125. doi: 10.1007/s11745-014-3945-0. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Li H, Zhang Z, Yang F, Chen J. Serum Metabolites as Potential Biomarkers for Diagnosis of Knee Osteoarthritis. Dis Markers. 2015;2015:684794. doi: 10.1155/2015/684794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.