

Abstract
Globoid cell leukodystrophy (GLD), or Krabbe disease, is a debilitating and always fatal pediatric neurodegenerative disease caused by a mutation in the gene encoding the hydrolytic enzyme galactosylceramidase (GALC). In the absence of GALC, progressive loss of myelin and accumulation of a neurotoxic substrate lead to incapacitating loss of motor and cognitive function and death typically by two years of age. To date there is no cure.
Recent convincing evidence of the therapeutic potential of combining gene and cell therapies in the murine model of GLD has accelerated the need for validated markers of disease in order to evaluate therapeutic efficacy. Herein we demonstrate clinically relevant and quantifiable measures of central and peripheral nervous system (CNS, PNS) disease progression in the naturally-occurring canine model of GLD. As measured by brainstem auditory evoked response (BAER) testing, GLD dogs demonstrated a significant increase in I–V interpeak latency and hearing threshold at all time points. Motor nerve conduction velocities (NCV) were significantly lower than normal by 12–16 weeks of age and sensory NCV by 8–12 weeks of age, serving as a sensitive indicator of peripheral nerve dysfunction. Post mortem histological evaluations confirmed neuroimaging and electrodiagnostic assessments and detailed loss of myelin and accumulation of storage product in the CNS and PNS. Additionally, cerebrospinal fluid (CSF) psychosine concentrations were significantly elevated in GLD dogs, demonstrating potential as a biochemical marker of disease. These data demonstrate that CNS and PNS disease progression can be quantified over time in the canine model of GLD using identical tools as used to assess human patients.
Keywords: Krabbe disease, large animal model, biomarker, electrophysiological testing, neuroimaging, CSF psychosine
Graphical Abstract
Post mortem histological evaluations confirmed neuroimaging and electrodiagnostic assessments and detailed loss of myelin and accumulation of storage product in the CNS and PNS.
Introduction
Globoid cell leukodystrophy (GLD), also known as Krabbe disease, is a progressive and fatal neurodegenerative lysosomal storage disorder. This inherited disease is caused by deficient activity of galactosylceramidase (GALC), which is responsible for degrading myelin, specifically the myelin lipids galactosylceramide (GalCer) and galactosylsphingosine (psychosine). The absence of sufficient GALC activity results in galactolipid accumulation, oligodendrocyte and Schwann cell death, abnormal central and peripheral nervous system (CNS and PNS) myelination, and the infiltration of multinucleated macrophages (globoid cells) (Wenger et al., 2015).
Symptoms in children emerge in the first year of life and include irritability, stiffness, loss of developmental milestones, and seizures. The disease rapidly progresses to loss of all body faculties and death often by 2 years of age (Duffner et al., 2009, Duffner et al., 2011). To date the only available therapeutic intervention for children with GLD is hematopoietic stem cell transplantation (HSCT) from donor umbilical cord blood or bone marrow. When employed early, HSCT has proven capable of slowing disease progression (Escolar et al., 2005); however, the quality of life in children is poor due to neurologic deterioration resulting in loss of cognitive and motor functions (Aldenhoven and Kurtzberg, 2015).
Development of new and effective therapies for Krabbe disease has been slow due to the dearth of experimental therapies found to substantially improve neurological disease in animal models and the absence of validated surrogate markers of disease that can be monitored as secondary clinical endpoints. However, recent studies in the Twitcher mouse, a murine model of Krabbe disease, have shown that gene therapy with an adeno-associated virus (AAV) vector carrying a wild type copy of murine GALC cDNA synergizes with HSCT to significantly improve motor deficits, survival time, and CNS pathology (Lin et al., 2007, Reddy et al., 2011, Hawkins-Salsbury et al., 2015, Rafi et al., 2015). While clearly provocative, differences in disease phenotype between murine and human Krabbe disease and the small size and limited lifespan of the mouse hinder the translational utility of this model. In contrast, naturally-occurring Krabbe disease in dogs recapitulates the clinical, pathological, and biochemical abnormalities of human disease (Cozzi et al., 1998, Wenger et al., 1999, McGowan et al., 2000, Cantuti-Castelvetri et al., 2015). Additionally, the canine brain, similar in size to that of an infant, allows for the implementation of HSCT and CNS gene transfer methods identical to what would occur in infants, and for the evaluation of the efficacy of these therapies using electrodiagnostic, imaging, and biochemical markers identical to those used in children.
We propose that the canine model of GLD is a critical intermediate step in advancing therapies from the Twitcher mouse into the clinic. In this manuscript, histological evaluations corroborate clinical assessments and detail loss of myelin and accumulation of storage product in the CNS and PNS. Next, we demonstrate quantifiable measures of disease progression in canine GLD using identical electrodiagnostic and imaging tools as those used in pediatric patients. Lastly, using HPLC coupled-mass spectrometry, we establish significant elevations of psychosine in cerebrospinal fluid (CSF) of GLD dogs, suggesting this as a sensitive biochemical marker of disease. These complementary clinical and biochemical markers of disease progression will be used in ongoing preclinical studies in the canine model of GLD, and should benefit future investigational new drug applications.
Materials and methods
Animals
Mixed breed dogs were raised in the National Referral Center for Animal Models of Human Genetic Disease of the School of Veterinary Medicine of the University of Pennsylvania (NIH OD P40-10939) under National Institutes of Health and USDA guidelines for the care and use of animals in research. The experimental protocols were approved by the University’s Institutional Animal Care and Use Committee. An equal number of male and female dogs were use. Whole blood from dogs was tested for the GALC missense mutation using a TaqMan® real-time polymerase chain reaction-based DNA test to identify affected, normal, and heterozygote dogs. The custom TaqMan® SNP genotyping assay included forward primer: ACTGGCCTTACGTGAATCTTCAG and reverse primer: GCTTGGCACCCACAATCC with VIC/NFQ as the reporter/quencher for allele 1 and FAM/NFQ for allele 2. Using a TaqMan® Genotyping Master Mix (Life Technologies #4371353, Grand Island, NY), the assay was run on an Applied Biosystems 7500 platform (Applied Biosystems, Foster City, CA). GLD dogs were euthanized at a humane endpoint (15.7 ± 4.8 weeks of age; mean ± standard deviation; n=19) defined by complete pelvic limb paralysis. Euthanasia was performed using an overdose of intravenous barbiturate. After sacrifice, animals were perfused through the left ventricle with 750 mL of 0.9% cold saline and tissues collected.
Dogs were evaluated weekly via clinical neurological evaluations; approximately every 4 weeks (8 weeks to first day of 12 weeks, 12 weeks to first day of 16 weeks, and 16 weeks to 20 weeks) for electrodiagnostic testing; and at endstage disease (pelvic limb paralysis) for psychosine, imaging, and histological evaluations.
MRI
Dogs were anesthetized with propofol, endotracheally intubated, and maintained on isoflurane anesthesia. Imaging was performed at 1.5 Tesla using a dedicated research MRI scanner (Signa; GE Corporation, Milwaukee, WI). The imaging session was performed as previously described (Cozzi et al., 1998). Briefly, an extremity coil was used in transmit-receive mode to acquire the images. Imaging sessions consisted of sagittal spin-echo imaging with long TR/TE; axial spin-echo imaging with short TR/TE and axial fast-spin echo imaging with long TR/TE sequences; and dorsal plane axial conventional spin-echo imaging with short TR/TE. T2-weighted images were generated using TR=6000 ms, TE=88 ms, 192 phase encode steps and 256 data points. MRI analysis was conducted on 4 endstage GLD affected dogs (9.3 – 16.4 weeks of age) and 2 age-matched normal control dogs. Representative images of 3 regions at the level of the, caudate nucleus, midbrain, and cerebellum, from 1 GLD affected and 1 normal control are shown.
Brain stem auditory evoked response (BAER)
BAER was conducted at 8–12 (normal n=5, GLD n=8); 12–16 (normal n=6, GLD n=6), and 16–20 (normal n=5, GLD n=4) weeks of age in left and right ears of normal and GLD dogs. Dogs were anesthetized with propofol, endotracheally intubated, and maintained on isoflurane anesthesia. BAER data was recorded on a Nicolet Viking Quest machine (Nicolet Biomedical, Madison, WI) using 12 mm, 29 gauge subdermal needle recording electrodes. The active electrode was placed in the skin over the osseous bulla of the stimulated ear, the reference electrode in the skin over the vertex of the skull, and the ground electrode in the skin over the contralateral osseous bulla. Alternating rarefaction and condensation clicks (0.1 ms duration) were delivered to the stimulated ear at 11.1 Hz using a 25 cm plastic tube connected to a plastic ear piece placed within the external ear canal. The filter settings for the amplifier were set to 20 Hz and 3 kHz. One thousand evoked responses were averaged for each tracing obtained. An amplifier sensitivity of 1 uV/cm was used to record the responses; the analysis time was 10 ms. Central conduction time was defined as the time between the first and the fifth peak. Hearing threshold was defined as the sound intensity at which an evoked waveform was first visible.
Nerve conduction velocity
NCV was conducted at 8–12 (normal n=5, GLD n=8); 12–16 (normal n=6, GLD n=6), and 16–20 (normal n=5, GLD n=4) weeks of age in normal and GLD dogs. Dogs were anesthetized with propofol, endotracheally intubated, and maintained on isoflurane anesthesia. NCV was conducted using an electrodiagnostics machine (Nicolet Viking Quest, Nicolet Biomedical). Motor NCV in the tibial, sciatic, and ulnar nerves of the left limb were determined using 12 mm, 29 gauge subdermal needle recording electrodes placed in the interosseous muscle. The tibial nerve was electrically stimulated at the tarsus and stifle. The sciatic nerve was electrically stimulated at the stifle and at the level of the femoral head. The ulnar nerve was stimulated at the carpus and elbow. Nerves were stimulated with monopolar stimulating electrodes placed subcutaneously for a duration of 200 μsec. Stimulus intensity was increased until a M wave of maximal amplitude was obtained. Sensory NCV was determined for the superficial radial nerve. Subcutaneous recording electrodes were placed lateral to the superficial radial nerve at the level of the elbow and the skin over the dorsum of the paw and stimulated with subcutaneous monopolar stimulating electrodes for 200 μsec.
CSF psychosine concentrations
CSF psychosine levels were measured in GLD dogs (n=5) at late stage of disease progression (10.9 – 16.4 weeks of age) and compared to age-matched normal control dogs (n=5). CSF was collected from the cerebllomedullary cistern while under anesthesia for NCV or immediately prior to euthanasia. Protein precipitation was performed to extract psychosine from 50 μL of dog CSF. Deuterated galactosyl sphingosine (psychosine d5: 1ng/mL) was used as an internal standard and was added to the samples before extraction. Quality control (QC) samples were prepared by pooling the extracts from study samples. QC was run every five-study sample to make sure that the analytical method and instrument response was constant. Data is reported as peak area ratio of analyte to internal standard.
Sample analysis was performed with a Shimadzu 20AD HPLC system (Shimadzu Scientific, Somerset, NJ), coupled to a triple quadrupole mass spectrometer (API 4000 QTrap) operated in MRM mode. The positive ion ESI mode was used for detection of psychosine and deuterated psychosine −d5. Data processing was conducted with Analyst 1.5.1 (Applied Biosystems, http://sciex.com/products/software/analyst-software).
Histology
Perfused brains were fixed in 4% paraformaldehyde and paraffin-embedded. Sections were cut at 5 microns and slides were deparafinnized and rehydrated in a series of xylenes and ethanols (100%, 95%, 70%). For myelin staining, slides were incubated in an eriochrome cyanine R solution (#32752, Sigma, St. Louis, MO) with ferric chloride and sulphuric acid for 30 minutes at room temperature, rinsed in running water, followed by incubation in an iron (III) nitrate nonahydrate (Sigma, 216828) differentiating solution for 2–5 minutes and rinsed in running water. Slides were counterstained with eosin and dehydrated in a series of ethanols and xylenes and mounted. For Periodic acid-Schiff staining, slides were incubated in periodic acid solution (#3951, Sigma) for 5 minutes at room temperature, rinsed with several changes of water, followed by incubation in Schiff’s reagent (#3952016, Sigma) for 15 minutes. Slides were rinsed in running water, dehydrated in a series of ethanols and xylenes, and mounted. Brain regions analyzed included sections at the level of the frontal lobe, caudate nucleus, thalamus, midbrain, occipital cortex, cerebellum, and brainstem of endstage 3 GLD affected (9.3 – 16.4 weeks of age) and 2 normal control dogs. Representative images of 3 regions at the level of the, caudate nucleus, midbrain, and cerebellum, from 1 GLD affected and 1 normal control are shown.
Electron microscopy
Samples of tibial, common peroneal, sciatic, ulnar, and superficial radial nerves were processed in routine manner for single nerve fiber teasing and semithin/ultrathin sections as previously described (Vite et al., 2001). Nerves were fixed in universal fixative (37% formaldehyde aqueous solution; 100 ml 100% formalin, 880 ml distilled water, 2.7 g NaOH, 11.6 g NaH2PO4-H2O, and 20 ml 50% glutaraldehyde). Electron microscopy was conducted on ultrathin sections prepared from tibial, sciatic, ulnar, radial, and common peroneal, nerves in a GLD dog at endpoint.
Statistics
Two-tailed, unpaired t-tests and figures were conducted using Graph Pad Prism 6 (Graphpad Prism, La Jolla, CA, http://www.graphpad.com, RRID: SCR_002798). * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
Results
CNS and PNS disease progression in the canine model of Krabbe disease closely recapitulate human disease (Table 1). Affected dogs have a consistent age of onset of clinical signs with pelvic limb ataxia, thoracic limb dysmetria, and head tremor beginning by 4–6 weeks of age; inability to walk without falling and urinary incontinence occurring by 12 weeks of age; and complete paralysis of the pelvic limbs warranting humane euthanasia occurring at 15.7 ± 4.8 weeks of age (mean ± standard deviation; n=19). A variable age of onset of increased tone to the pelvic limbs and decreased myotatic and withdrawal reflexes also occurs.
Table 1.
Phenotypic comparison of Krabbe disease
| Phenotype | Human patients | Murine model | Canine model |
|---|---|---|---|
|
| |||
| Irritability | + | − | − |
| Psychomotor regression | + | ? | ? |
| Cerebellar ataxia/intention tremor | + | + | + |
| Postural reaction deficits | + | + | + |
| Stiffness | + | + | + |
| Spastic paresis/paralysis | + | + | + |
| Hearing loss | + | − | + |
| Vision loss | + | − | + |
| Motor and sensory neuropathy | + | + | + |
| Autonomic nervous system dysfunction | + | + | + |
MRI
Magnetic resonance imaging of the brain of a GLD affected dogs shows T2-weighted bilaterally symmetrical increases in signal intensity of the corona radiata (Figure 1D, arrow), corpus callosum, centrum semiovale, internal capsule (Figure 1E, arrow), and cerebellar white matter (Figure 1F, arrow) when compared to a normal, age-matched control dogs (Figure 1 A–C). Cerebral ventricles are dilated (Figure 1E, arrow) and sulci are widened (Figure 1D, arrow) indicating cerebral atrophy in GLD dogs. Gadolinium enhancement of the white matter was not seen in any of the dogs recently imaged, although it was previously described in a West Highland white terrier (Cozzi et al., 1998).
Figure 1.

Magnetic resonance imaging
T2-weighted magnetic resonance imaging of the brain shows bilaterally symmetrical increases in signal intensity of the corona radiata (D, arrow), corpus callosum, centrum semiovale, and internal capsule (E, arrow) in a GLD dog compared to a normal, age-matched control dog (A, B). Similarly, the cerebellar white matter (F, arrow) and optic radiations were found to be hyperintense in the GLD dog when compared to normal (C). Cerebral ventricles of the GLD dog were dilated (E, arrow) and sulci widened indicating cerebral atrophy (D, arrow).
Brain Histopathology
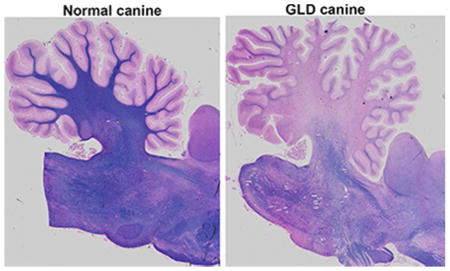
Consistent with MRI findings, GLD affected dogs show severe loss of myelin indicated by iron-eriochrome cyanine R histological stain. Loss of myelin is apparent in all sections of the brain when compared to normal, age-matched, control dogs. Representative images include brain sections at the level of the caudate nucleus, cerebrum at the level of the midbrain, and cerebellum (Figure 2). Numerous white matter tracts including the centrum semiovale, corpus callosum, fimbria hippocampi (Figure 2 inset box), and cerebellar white matter have little to no staining at end stage disease. In contrast, subcortical u-fibers, also known as short association fibers, and the brainstem retained more myelin staining. U-fibers represent a terminal zone of myelination, which are the last areas to myelinate in the normal brain, and experience a slower rate of myelin turnover (Maricich et al., 2007), demonstrating that disparity in normal myelin metabolism may have consequences on disease pathogenesis. Loss of myelin strongly correlated with the presence of periodic acid-Schiff (PAS) positive storage material (Figure 3).
Figure 2.

Myelination in normal and GLD dog brain
Iron-eriochrome cyanine R histological stain shows loss of myelin (purple stain) in a GLD dog at end stage (right panel) compared to a normal age-matched control dog (left panel). Additionally, white mater regions are subjectively small in GLD dogs compared to normal control dogs. Cerebral hemisphere sections are shown at the level of the caudate nucleus (top panel), cerebrum at the level of the midbrain (middle panel), and cerebellum (bottom panel). Insets show 25x magnification.
Figure 3.

Storage material in normal and GLD dog brain
Periodic acid-Schiff histological stain shows accumulation of storage material in GLD affected dog at end stage (right panel) compared to normal age-matched control dog (left panel). Brain sections are shown at the level of the caudate nucleus (top panel), cerebrum at the level of the midbrain (middle panel), and cerebellum (bottom panel). Insets show 25x magnification.
Brainstem auditory evoked response testing
Brainstem auditory evoked response (BAER) testing was conducted in GLD dogs to assess the effect of disease on the auditory pathway over time. In GLD dogs, there was a significant increase (p ≤ 0.001) in central conduction time, measured by the difference in peak I–V latencies, in all age groups analyzed, 8–12 weeks, 12–16 weeks, and 16–20 weeks, when compared to normal, age-matched control dogs (Figure 4 A). A significant increase in hearing threshold over normal was also identified in affected dogs at all time points evaluated (Figure 4 B). The slight reduction in threshold significance at the final time point (p ≤ 0.01) was likely due to the small number of affected animals that survived >16 weeks and still had recordable BAER (n=4). The increase in central conduction time, increase in hearing threshold, and loss of waveform integrity on BAER are all consistent with auditory system demyelination in the GLD dog.
Figure 4.

Brainstem auditory evoked responses (BAERs) in normal and GLD dogs.
BAER was conducted at 8–12 (normal n=5, GLD n=8); 12–16 (normal n=6, GLD n=6), and 16–20 (normal n=5, GLD n=4) weeks of age in left and right ears of normal (solid circle) and GLD dogs (solid square). A) Central conduction time, the time between the first and the fifth peak, was increased in affected dogs. B) Hearing threshold, defined as the sound intensity at which an evoked waveform, was first visible was increased in affected dogs. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
Nerve conduction velocity testing
Nerve conduction velocities were quantified in motor and sensory nerves of GLD dogs and normal, age-matched controls at 3 time points: 8–12, 12–16, and 16–20 weeks of age. In GLD dogs, motor nerve conduction velocities of the tibial (Figure 5 A) and sciatic (Figure 5 B) nerves were significantly decreased by 12–16 weeks of age (p = 0.028, p = 0.004), and were also significantly decreased at 16–20 weeks of age for the sciatic nerve (p = 0.009). NCV of the ulnar nerve (Figure 5 C) was significantly reduced at 8–12 (p = 0.047) and 12–16 (p = 0.005) weeks of age. Significance was not achieved at the latest time point in the tibial and ulnar nerves, likely due to the low number of animals that survived beyond 16 weeks and still had recordable NCV (n=4). Sensory NCV of the radial nerve (Figure 5 D) was the most dramatically decreased with a p value of 0.0001 at both 8–12 and 12–16 weeks of age. Values trended towards significance at endpoint; however, statistics could not be calculated due to insufficient animal numbers (only 2 GLD dogs had recordable sensory NCV 16–20 weeks; in remaining dogs, no response could be evoked). The reduction in conduction velocity of motor and sensory nerves is consistent with severe demyelination of peripheral nerves in GLD dogs.
Figure 5.

Nerve conduction velocity (NCV) in normal and GLD dogs.
NCV was conducted at 8–12 (normal n=5, GLD n=8); 12–16 (normal n=6, GLD n=6), and 16–20 (normal n=5, GLD n=4) weeks of age in normal (solid circle) and GLD dogs (solid square). For pelvic limb motor NCV the A) tibial nerve was stimulated at the tarsus and stifle, the B) sciatic nerve was stimulated at the stifle and at the level of the femoral head, and for the thoracic limb the C) ulnar nerve was stimulated at the carpus and elbow, and for sensory NCV the D) radial nerve at the level of the elbow. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
Electron microscopy of peripheral nerves
Analysis of ultrathin sections of peripheral nerves by electron microscopy revealed that storage inclusions and thinly myelinated fibers were a prominent feature in all nerves analyzed (tibial, sciatic, ulnar, radial, and common peroneal) in one GLD dog at endstage disease. Storage inclusions were present in numerous Schwann cells (Figure 6 A) and in macrophages (Figure 6 B), most commonly located in the endoneurium with occasional presence in the perineurium. The inclusions were highly heterogeneous (arrow in Figure 6 A), with the most prominent and typical morphology consisting of straight, elongated, needle-like, prismatic inclusions. In some cells the prismatic inclusions were voluminous and arranged in different plans of section, giving them a wave-like curvilinear pattern (Figure 6 C, arrow). Other membrane-bound concentric or irregular-shaped inclusions, as well as honeycomb, crystalloid structures frequently accompanied the characteristic prismatic inclusions. Myelin-like lamellar structures occasionally occurred with these inclusions (Figure 6 D, arrow). No inclusions were observed in unmyelinated fibers, which appeared normal. Thinly myelinated fibers were common in all nerves analyzed (Figure 6 E, F, arrows).
Figure 6.

Electron micrographs of peripheral nerve of GLD dog.
Electron micrographs of GLD dog at end stage demonstrating A) storage inclusion in Schwann cells (19,500x, arrow); B) storage inclusions in macrophages (15,700x); C) prismatic inclusions in a ‘wave-like’ curvilinear pattern (41,300x, arrow); D) myelin-like lamellar structures within inclusions (41,300x, arrow); E) thinly myelinated fibers (30,300x, arrow); F) thinly myelinated fibers (19,500x, arrow).
CSF psychosine concentrations
It has previously been shown that psychosine is dramatically increased in the white matter of GLD dog brains (Wenger et al., 1999). For this study, we quantified psychosine concentrations in CSF by HPLC-coupled mass spectrometry. CSF psychosine levels were significantly elevated (p=0.004) in GLD dogs (mean 0.069 ± 0.036, n=5) at late stage of disease progression (10.9–16.4 weeks of age) compared to age-matched normal control dogs (n=5) (Figure 7). The highest CSF psychosine concentration was found in the oldest GLD dog evaluated (16.4 weeks of age). This minimally invasive method of determining CNS-specific disease progression could serve as a valuable biomarker of disease progression; repeated measures over time are now underway.
Figure 7.

CSF psychosine concentration in normal and GLD affected dogs
CSF psychosine concentrations determined by HPLC coupled-mass spectrometry and presented as peak area ratio for GLD affected dogs (solid square; n=5, mean age 13.4 weeks, range 10.9–16.4 weeks), compared to normal control dogs (solid circle; n=5, mean age 18.0 weeks, range 6.1–38.7). ** p ≤ 0.01.
Discussion
Canine GLD was first described in 1966 (Fletcher et al., 1966). In 1996, the disease causing mutation in West Highland white and Cairn terriers was identified as an A to C transversion at cDNA position 473 in the GALC gene, resulting in the replacement of a hydrophobic tyrosine residue with a hydrophilic serine in the protein. Transient transfection of COS-1 cells with the mutant canine GALC cDNA revealed that the mutant protein is not functional (Victoria et al., 1996). Affected dogs, homozygous for the recessive mutation, can be identified at birth from whole blood using real-time PCR amplification of genomic DNA. In 2000, a breeding colony of canine GLD was established and has been maintained at the School of Veterinary Medicine at the University of Pennsylvania. Since then, Krabbe disease has also been characterized in the Irish setter where it occurs due to an insertion mutation (McGraw and Carmichael, 2006); no colony of these dogs is currently established. In light of recent advancements demonstrating unprecedented therapeutic efficacy using a combination of bone marrow transplantation and gene therapy in the Twitcher mouse (Lin et al., 2007, Reddy et al., 2011, Hawkins-Salsbury et al., 2015, Rafi et al., 2015), the canine model of GLD has renewed significance as an intermediate model in translation of therapeutics to the clinic. Here we further characterized the canine model and identified the onset of clinical signs by 4 weeks of age and a survival time to 15.7 ± 4.8 weeks of age (n=19). Electrodiagnostic testing methods, routinely used in affected children, are reported longitudinally in the canine model of GLD, allowing a quantifiable means to track disease progression. Furthermore we have identified that psychosine is elevated in the spinal fluid of affected dogs. Future studies will evaluate its use as a biomarker of disease severity or of therapeutic efficacy.
The clinical data in dogs compares well to what is published in human patients. In children, neurologic evaluations revealed that in 41 patients, abnormal results on motor examination were identified, with 90% demonstrating increased muscle tone, typically in the extremities. Deep tendon reflexes were abnormal in the majority of children (71% decreased reflexes and increased in 19%) and clonus and plantar extensor responses were also common (Duffner et al., 2011). GLD is also naturally occurring in non-human primates. In a natural history study of GLD affected rhesus macaques the onset and progression of symptoms were highly variable and survival widely ranged from 52–642 days (Borda et al., 2008). In this study, the canine model of GLD was found to have repeatable and predictable onset of clinical signs and limited range of survival.
Analysis of MRI scans from 39 early infantile Krabbe patients, those with symptom onset 0–6 months of age, showed abnormalities in 38 of 39 cases. The most common irregularities were T2 signal intensity changes in the deep cerebral white matter periventricular/centrum semiovale (84.6%) and the dentate (84.6%), followed by cerebellar white matter (53.8%), thalamus (41.0%), and parietal-occipital (30.8%) (Abdelhalim et al., 2014). Similar changes were found in our canine model of GLD including T2 signal hyperintensities in numerous white matter tracts. In contrast, there were no remarkable findings on MRI of GLD affected rhesus macaques (Borda et al., 2008).
BAER testing conducted in children with early infantile Krabbe disease revealed prolonged wave I–V interpeak latency in 88% (15/17) of children. Of children that were symptomatic at the time of testing, 100% (13) experienced increased central conduction time while 2 of 4 pre-symptomatic children had similar abnormalities (Aldosari et al., 2004). Similarly, GLD dogs demonstrated a significant increase in I–V interpeak latency at all times analyzed, 8–12, 12–16, and 16–20 weeks of age. As there is overlap in values between normal and GLD affected dogs, BAER is not likely to be a sensitive measure to detect improvements especially in early disease stages following treatment. Instead BAER will be most useful as a repeated measure analysis to study individual animals longitudinally.
Peripheral neuropathy has also been previously evaluated in children affected with early infantile Krabbe disease. Of 24 Krabbe patients assessed, 23 had abnormal nerve conduction velocities, with 82% showing deficient sensory and 82% showing slow motor conduction velocities. Abnormalities in motor conduction velocities were comparable in lower (79%) and upper (80%) extremities. Importantly, the severity of the demyelination on nerve conduction velocity studies correlated well with the clinical severity of the disease (Siddiqi et al., 2006). Similarly, conduction velocities of the median, ulnar, and tibial nerves were significantly slower in GLD-affected rhesus macaques when compared to normal monkeys and also correlated with disease progression (Weimer et al., 2005). Comparably, motor nerve conduction velocities of pelvic (tibial and sciatic nerves) and thoracic limbs (ulnar nerve) were a sensitive indicator of peripheral nerve dysfunction in the canine model of GLD with all conduction velocities significantly lower than normal by 12–16 weeks of age. Interestingly, decreases in sensory (radial) nerve conduction velocities achieved the greatest and earliest significance of 0.0001 by 8–12 weeks of age and mean velocity continued to decrease with age. Unlike BAER, radial NCV resulted in no overlap between normal and GLD affected dogs at any time point and is therefore expected to be a sensitive and robust means to determine therapeutic efficacy even early in disease.
Nerve conduction velocity studies, in combination with neuroimaging studies, have been proposed as the most sensitive measure to assess the severity of Krabbe disease in human patients (Moser, 2006). Herein we demonstrate that we can conduct identical electrodiagnositic and neuroimaging studies in the GLD dog with analogous results as found in human patients. Post mortem histological evaluations verify clinical assessments and detail loss of myelin and accumulation of storage product in the CNS and PNS. Furthermore, we establish the utility of CSF psychosine as a potential biochemical marker of disease. Thus, electrodiagnostic, imaging, and biochemical data may serve as secondary clinical outcome measures in ongoing preclinical studies in the canine model of GLD and could benefit future human clinical trials for Krabbe disease.
Significance statement.
Herein we further characterize the naturally-occurring canine model of globoid cell leukodystrophy (GLD), or Krabbe disease, and demonstrate that we can conduct identical electrodiagnostic and neuroimaging studies in GLD dogs with analogous results as found in human patients. Post-mortem histological evaluations verify clinical assessments and detail loss of myelin and accumulation of storage in the central and peripheral nervous systems. Furthermore, we establish the utility of a minimally-invasive biochemical marker of disease. Thus, electrodiagnostic, imaging, and biochemical data will serve as secondary outcome measures in ongoing preclinical studies in the canine model of GLD and could benefit future clinical trials.
Acknowledgments
Support NIH OD P40-10939 (Vite)
Role of authors
“All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: A.M.B., C.H.V. Acquisition of data: A.M.B., J.H.B., X.J., G.P.S., M.L.P, C.A.F., P.A.O, K.G.B., D.S.O., C.H.V. Analysis and interpretation of data: A.M.B., X.J., K.G.B., D.S.O., C.H.V. Drafting of the manuscript: A.M.B., C.H.V. Names or Initials. Critical revision of the manuscript for important intellectual content: A.M.B., J.H.B., P.A.O, K.G.B., D.S.O., C.H.V. Statistical analysis: A.M.B. Obtained funding: C.H.V. Administrative, technical, and material support: G.P.S., K.G.B., X.J. Study supervision: C.H.V.”
Footnotes
Conflict of interest
There are no identified conflicts of interest
Literature cited
- Abdelhalim AN, Alberico RA, Barczykowski AL, Duffner PK. Patterns of magnetic resonance imaging abnormalities in symptomatic patients with Krabbe disease correspond to phenotype. Pediatric neurology. 2014;50:127–134. doi: 10.1016/j.pediatrneurol.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Aldenhoven M, Kurtzberg J. Cord blood is the optimal graft source for the treatment of pediatric patients with lysosomal storage diseases: clinical outcomes and future directions. Cytotherapy. 2015;17:765–774. doi: 10.1016/j.jcyt.2015.03.609. [DOI] [PubMed] [Google Scholar]
- Aldosari M, Altuwaijri M, Husain AM. Brain-stem auditory and visual evoked potentials in children with Krabbe disease. Clinical neurophysiology: official journal of the International Federation of Clinical Neurophysiology. 2004;115:1653–1656. doi: 10.1016/j.clinph.2004.02.022. [DOI] [PubMed] [Google Scholar]
- Borda JT, Alvarez X, Mohan M, Ratterree MS, Phillippi-Falkenstein K, Lackner AA, Bunnell BA. Clinical and immunopathologic alterations in rhesus macaques affected with globoid cell leukodystrophy. The American journal of pathology. 2008;172:98–111. doi: 10.2353/ajpath.2008.070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L, Maravilla E, Marshall M, Tamayo T, D’Auria L, Monge J, Jeffries J, Sural-Fehr T, Lopez-Rosas A, Li G, Garcia K, van Breemen R, Vite C, Garcia J, Bongarzone ER. Mechanism of neuromuscular dysfunction in Krabbe disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2015;35:1606–1616. doi: 10.1523/JNEUROSCI.2431-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzi F, Vite CH, Wenger DA, Victoria T, Haskins ME. MRI and electrophysiological abnormalities in a case of canine globoid cell leucodystrophy. The Journal of small animal practice. 1998;39:401–405. doi: 10.1111/j.1748-5827.1998.tb03741.x. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Barczykowski A, Jalal K, Yan L, Kay DM, Carter RL. Early infantile Krabbe disease: results of the World-Wide Krabbe Registry. Pediatric neurology. 2011;45:141–148. doi: 10.1016/j.pediatrneurol.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Jalal K, Carter RL. The Hunter’s Hope Krabbe family database. Pediatric neurology. 2009;40:13–18. doi: 10.1016/j.pediatrneurol.2008.08.011. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, Morse R, Krivit W, Kurtzberg J. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. The New England journal of medicine. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Fletcher TF, Kurtz HJ, Low DG. Globoid cell leukodystrophy (Krabbe type) in the dog. Journal of the American Veterinary Medical Association. 1966;149:165–172. [PubMed] [Google Scholar]
- Hawkins-Salsbury JA, Shea L, Jiang X, Hunter DA, Guzman AM, Reddy AS, Qin EY, Li Y, Gray SJ, Ory DS, Sands MS. Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2015;35:6495–6505. doi: 10.1523/JNEUROSCI.4199-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Donsante A, Macauley S, Levy B, Vogler C, Sands MS. Central nervous system-directed AAV2/5-mediated gene therapy synergizes with bone marrow transplantation in the murine model of globoid-cell leukodystrophy. Mol Ther. 2007;15:44–52. doi: 10.1038/sj.mt.6300026. [DOI] [PubMed] [Google Scholar]
- Maricich SM, Azizi P, Jones JY, Morriss MC, Hunter JV, Smith EO, Miller G. Myelination as assessed by conventional MR imaging is normal in young children with idiopathic developmental delay. AJNR American journal of neuroradiology. 2007;28:1602–1605. doi: 10.3174/ajnr.A0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan JC, Haskins M, Wenger DA, Vite C. Investigating demyelination in the brain in a canine model of globoid cell leukodystrophy (Krabbe disease) using magnetization transfer contrast: preliminary results. Journal of computer assisted tomography. 2000;24:316–321. doi: 10.1097/00004728-200003000-00025. [DOI] [PubMed] [Google Scholar]
- McGraw RA, Carmichael KP. Molecular basis of globoid cell leukodystrophy in Irish setters. Veterinary journal. 2006;171:370–372. doi: 10.1016/j.tvjl.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Moser HW. Peripheral nerve involvement in Krabbe disease: a guide to therapy selection and evaluation. Neurology. 2006;67:201–202. doi: 10.1212/01.wnl.0000231531.73713.a9. [DOI] [PubMed] [Google Scholar]
- Rafi MA, Rao HZ, Luzi P, Wenger DA. Long-term Improvements in Lifespan and Pathology in CNS and PNS After BMT Plus One Intravenous Injection of AAVrh10-GALC in Twitcher Mice. Molecular therapy: the journal of the American Society of Gene Therapy. 2015 doi: 10.1038/mt.2015.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy AS, Kim JH, Hawkins-Salsbury JA, Macauley SL, Tracy ET, Vogler CA, Han X, Song S-K, Wozniak DF, Fowler SC, Klein RS, Sands MS. Bone marrow transplantation augments the effect of brain- and spinal cord-directed adeno-associated virus 2/5 gene therapy by altering inflammation in the murine modelof globoid-cell leukodystrophy. J Neurosci. 2011;31:9945–9957. doi: 10.1523/JNEUROSCI.1802-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqi ZA, Sanders DB, Massey JM. Peripheral neuropathy in Krabbe disease: electrodiagnostic findings. Neurology. 2006;67:263–267. doi: 10.1212/01.wnl.0000230153.34613.84. [DOI] [PubMed] [Google Scholar]
- Victoria T, Rafi MA, Wenger DA. Cloning of the canine GALC cDNA and identification of the mutation causing globoid cell leukodystrophy in West Highland White and Cairn terriers. Genomics. 1996;33:457–462. doi: 10.1006/geno.1996.0220. [DOI] [PubMed] [Google Scholar]
- Vite CH, McGowan JC, Braund KG, Drobatz KJ, Glickson JD, Wolfe JH, Haskins ME. Histopathology, electrodiagnostic testing, and magnetic resonance imaging show significant peripheral and central nervous system myelin abnormalities in the cat model of alpha-mannosidosis. Journal of neuropathology and experimental neurology. 2001;60:817–828. doi: 10.1093/jnen/60.8.817. [DOI] [PubMed] [Google Scholar]
- Weimer MB, Gutierrez A, Baskin GB, Borda JT, Veazey RS, Myers L, Phillippi-Falkenstein KM, Bunnell BA, Ratterree MS, England JD. Serial electrophysiologic studies in rhesus monkeys with Krabbe disease. Muscle & nerve. 2005;32:185–190. doi: 10.1002/mus.20350. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Escolar ML, Luzi P, Rafi MA. Krabbe disease (globoid cell leukodystrophy) In: Valle D, et al., editors. The Online Metabolic and Molecular Bases of Inherited Diseases. The McGraw-Hill Medical; 2015. [Google Scholar]
- Wenger DA, Victoria T, Rafi MA, Luzi P, Vanier MT, Vite C, Patterson DF, Haskins MH. Globoid cell leukodystrophy in cairn and West Highland white terriers. The Journal of heredity. 1999;90:138–142. doi: 10.1093/jhered/90.1.138. [DOI] [PubMed] [Google Scholar]