

Abstract
Enzyme replacement therapy and substrate reduction therapy have proved useful in reversing many pathological consequences of many non-neural lysosomal storage diseases but have not yet reversed pathology or influenced disease outcome in Krabbe disease, We will discuss the relative merits of stem cell therapy, molecular chaperone therapy, gene therapy, substrate reduction therapy, enzyme replacement therapy and combination therapy. Given the limitations of these approaches we will introduce the idea of using tiny 6nm intensely fluorescent Quantum dots to deliver a cell-penetrating peptide and His6-tagged beta-D-galactosidase across the blood-brain barrier. We can therefore follow the fate of injected material and ensure that all targets are reached and accumulated material is degraded. Uptake of lysosomal hydrolases is a complex process and our cell-penetrating peptide JB577 is uniquely able to promote endosomal egress of the QD-cargo. We further show that uptake may depend on the charge of the coating of the Quantum dot, specifically that negative charge directs the cargo to neurons. Since Krabbe disease primarily involves glia, specifically oligodendroglia we experimented with many coatings and discovered a positive charged coating (Polyethyleneglycol 600-amino) which had a positive charge and targeted oligodendrocytes. A similar effect was achieved by treating with chondroitinase ABC to degrade the extracellular matrix, indicating that enzyme replacement has several hurdles to overcome before it can become a routine CNS therapy.
Enzyme replacement therapy and substrate reduction therapy have proved useful in reversing many pathological consequences of lysosomal storage disease (LSD) in extraneural tissue of storage disease patients but have not been found to reverse pathology or influence disease outcome in a human neurological lysosomal storage disease such as Krabbe disease. Thus there is an unmet need to come up with a new approach to the delivery of either beta-D-galactocerebrosidase enzyme protein or low molecular weight inhibitors of Ceramide: galactosyltransferase (substrate reduction therapy) to patients with Krabbe disease.
Protein delivery to the CNS
Delivering a protein across a cell membrane and having it retain its biological function inside the cell is a major challenge in biology and medicine because of the blood-brain barrier. Many cells have developed a type of receptor (a “coated pit”) which facilitates the uptake of nutritive proteins such as lipoproteins (the Apo-E LDL receptor) and transferrin but for the most part they have not evolved to take up proteins from the extracellular space and proteins are endocytosed at a very low rate (Masliah and Spencer 2015). Notable exceptions to this rule are the lysosomal hydrolases, some of which use a phosphorylated sugar (Mannose-6-phosphate) and a specific receptor to exchange enzymes between cells. This specialized system has been taken advantage of to carry out enzyme replacement therapy in non-neural sphingolipid storage diseases such as Gaucher and Fabry (Sleat and Wiseman et al. 2013). However, it is not clear that such exchange occurs in the central nervous system (CNS) and it took over 20 years to perfect the cellular uptake of these lysosomal hydrolases by non-CNS tissue. This involved modification of carbohydrate chains to elude mannose receptors in the liver and the precise details are as closely guarded as the formula for Coca–Cola. Further, despite this success, enzyme replacement for other CNS lysosomal storage diseases has been less successful (San Filippo (MPSIII), Pompe etc) and the enzyme is not delivered to human brain cells. In this Review we discuss the current approaches to therapy and the prospects for developing a delivery system to replace defective enzyme in a CNS disease such as Krabbe disease. This would involve using fluorescent membrane-penetrating Quantum dots as the base, solubilizing the QDs with specific charged coatings (Susumu and Uyeda 2007) and attaching both a cell-penetrating peptide and a His6-tagged GalCer:-beta-galactocerebrosidase to Zn on the surface of the QD (Delehanty et al. 2010; Susumu et al. 2011; Walters et al 2012; Boeneman et al. 2013; Agarwal et al. 2015; Walters (2015).
Lysosomal hydrolases can resist extracellular degradation but then must traverse through non-fenestrated capillaries, pass through pericyte and astrocyte end-foot processes and finally diffuse through a highly negatively charged extracellular matrix (proteoglycans) before reaching the target, be it neurons, astrocytes or oligodendrocytes (Schwartz and Domowicz 2014; Masliah and Spencer 2015). Fortunately there are clues as to how one might achieve this from the many viruses (such as HIV) which have evolved mechanisms of cell penetration by using positively charged peptide sequences within their coat proteins, such as the lysine-rich TAT sequence in HIV (Meng et al. 2014). Thus the combination of a polylysine sequence and an LDL receptor-binding sequence from Apo-E in a 39 amino acid peptide facilitated CNS uptake of a lysosomal enzyme (Tripeptidylpeptidase-1) following IV injection into a mouse (Meng et al. 2014). In this review we propose that one can take advantage of unique cell-penetrating peptides coupled to Quantum dots (QDs), and by modifying the charge of the coating charge, deliver enzymes such as GalCer:β-D-galactosidase to its oligodendrocyte target cell in the CNS, and how this might repair pathological processes. Several studies have shown exogenous enzyme to increase the lifespan of enzyme-deficient mice or dogs (Meng et al, 2014, Vuillemenot et al. 2011) so the challenge is to translate this to humans. The roadblocks to replacing aberrant, toxic, proteins in the CNS are huge so this is merely a start down a long road, but the current lack of success in healing a damaged CNS in Krabbe disease (KD) requires such new ideas and new reagents.
Current status of therapy for Krabbe disease
1. Hematopoietic stem cell transplantation (HSCT) in Krabbe disease
Krabbe disease (globoid cell leukodystrophy) (KD) is an autosomal recessive disorder resulting from mutations in the gene for the lysosomal enzyme GalCer:beta-D-galactocerebrosidase (GALC) (Suzuki and Suzuki 2001). The enzyme deficiency leads to the accumulation of GalCer and a cytotoxic metabolite psychosine (galactosylsphingosine) and rapid degeneration of myelinating cells (Wenger et al. 2001). The only approved treatment for presymptomatic infantile patients and later-onset patients is hematopoietic stem cell transplantation (HSCT) but this treatment is less than ideal with most patients still developing progressive problems with gait and language. Several naturally occurring animal models are available for therapeutic efforts to treat Krabbe disease, the most studied being the twitcher (twi) mouse, and these mice have been shown to respond favorably to a range of therapies (Suzuki and Suzuki 2001; Wenger et al. 2001). These are summarized in Fig. 1.
Fig 1. Summary of approaches to KD therapy.

- Blue: Adenoviral infection introduces gene for GALC into the CNS genome producing GALC in all cells infected.
- Green: GALC Enzyme with M-6-P binds receptor, is endocytosed and (hopefully) egresses from the endosome and progresses to the lysosome.
- Red: Chaperone (eg NOEV) is endocytosed and (may) escape from the endosome and reach the ER (where it can promote proper folding of misfolded GALC or turn off the unfolded protein response) resulting from point mutations in the GALC gene.
- Orange: Quantum dots (QDs) with PEG600NH2 positively charged coat and surface Zn bind HIS6-GALC enzyme and JB577 cell-penetrating peptide and gain entry to lysosomes, where QDs are detached and enzyme degrades storage material. This construct should theoretically be able to cross the blood-brain barrier by virtue of its lipophilic character and the vKIKK sequence in JB577.
- Black: Substrate-reduction therapy (SRT). The Ceramide:Galactosyltransferase inhibitor ( a theoretical derivative of PDMP or NOEV) gains entry to ER and inhibits de novo synthesis of GALC, resulting in less storage of GalCer and more importantly less GalSph (Psychosine). Since there are currently no useful inhibitors of the Galactosyltransferase and no easy way of achieving reduced synthesis of psychosine, another approach to SRT would be to inhibit the Galactosyltransferase enzyme by RNAi-based technology, using the VKIKK sequence in JB577 to bind the siRNA for the Cer:galactosyltransferase.
2. Embryonic Stem cells (ESCs) for Krabbe therapy
Stem cells (Olig2 ESCs) can be induced into oligodendrocyte precursor cells (OPCs) by using cytokines and a multi-step differentiation procedure. Target oligodendrocyte markers can be monitored by reverse transcription-polymerase chain reaction (RT-PCR) and immunocytochemistry and the toxicity of the stored psychosine to OPCs is readily determined by a cell proliferation assay. The OPCs are labeled with the lipophilic long-chain dialkylcarbocyanine DiR (D12731), which has excitation and emission maxima in the near infrared region, (where many tissues are optically transparent) and transplanted into the brains of twitcher mice (Kuai et al. 2015). Transplanted cells are typically detected by in-vivo Multispectral Imaging Systems and real-time PCR. In one such study 76% of the OPCs were found to be positive for enhanced green fluorescent protein (eGFP-positive), the eGFP expression being driven by the Olig2 promoter. Such OPCs were more resistant to psychosine toxicity and the Galactosylceramide (GalCer) level in OPCs was also significantly higher than other cells, suggesting that enzyme was being expressed in the correct (lysosomal) subcellular compartment and that some remyelination might be taking place.
In other studies, when Dir-labeled OPCs were injected into the forebrain of post-natal day 10 twitcher mice the transplanted OPCs were myelin basic protein (MBP)-positive and remained along the injection tract as observed by fluorescent microscopy (Hawkins-Salsbury et al. 2015). The level of the Dir fluorescent signal and eGFP mRNA significantly decreased at days 10 and 20 after injection, (as indicated by an in-vivo Multispectral Imaging System and real-time PCR. Unfortunately, because of poor cell survival and limited migration ability, there was no significant improvement in brain GALC activity, MBP level, life span, body weight, and behavioral deficits of the twitcher mice.
3. Molecular chaperone therapy
The beta-D-galactosidase (GALC) inhibitor N-octyl-4-epi-β-valienamine (NOEV) acts as a chaperone to promote refolding of mutant protein in Krabbe (twitcher) mice as well as a murine GM1-gangliosidosis model (Hossain et al. 2015). NOEV strongly inhibited GALC activity in cell lysates of GALC-transfected COS1 cells but in vitro NOEV treatment was able to stabilize GALC activity under heat denaturation conditions and significantly increase the enzyme activity of mutants with late-onset forms. A similar chaperone effect for other lysosomal hydrolases has been reported, for example in infantile Batten disease where the deficient enzyme is palmitoyl:protein thioesterase deficiency (PPT1) (Dawson et al. 2010). One chaperone (Migalastat) has been approved for human use as a chaperone in alpha-galactosidase-deficient Fabry patients but NOEV (Warnock et al. 2010) has not been tested in Krabbe patients as yet. When NOEV was added to cultured COS1 cells expressing mutant GALC activity and human skin fibroblasts from Krabbe disease patients it increased GALC by enhancing the maturation of GALC precursor to its mature active form. NOEV most likely binds to the active site of human GALC protein and has promising potential for patients with Krabbe disease resulting from the late-onset mutations. However its use in humans has been hindered by the problems of delivering the chaperone into human brain without toxicity.
4. Gene therapy
The absence of GALC activity leads to the accumulation of the toxic psychosine and the preferential loss of myelinating cells, leading to astrogliosis and axonopathy. Thus cerebellar ataxia is one of the dominant manifestations in adolescents and adults affected with Krabbe disease (KD). In order to study the efficacy of cerebellum-targeted gene therapy on the cerebellar neuropathology in twitcher mice therapy, Rafi et al. (2015). attempted to reverse the degeneration of Purkinje cells, Bergmann glia, and granule cells by gene Intracerebellar-mediated gene therapy. This efficiently corrected the enzymatic deficiency in the cerebellum, leading to reduced neuroinflammation and demyelination. However, the number, dendritic territory, and axonal processes of Purkinje cells remained normal in the cerebellum of treated twitcher mice, where radial fibers of Bergmann glia spanned the molecular layer and collateral branches ensheathed the dendritic processes of Purkinje cells. Encouragingly, the aberrant expression of neurotrophic factors was reversed in the cerebellum of treated twitcher mice, indicating that the preservation of cellular function and neuronal architecture was being achieved. The life span of the treated twitcher mice was significantly prolonged and their neurobehavioral performance was improved. These findings underscore the complexity of cerebellar neurodegeneration in KD but emphasize the potential effectiveness of gene therapy in reducing neuropathological deficits in KD (Lin et al, 2015). However, there are significant current problems with the safety of such therapies for humans and gene therapy is still a work in progress.
5. Substrate reduction therapy (SRT)
The theoretical basis for SRT is that if you can reduce the synthesis of the storage material (in this case GalCer) with a low-molecular weight inhibitor you can reduce the storage burden. It works in Gaucher disease (“Cerdelga”, at $300,000-plus/year per patient) to inhibit GlcCer synthesis and SRT works to some extent in Fabry disease but not in Niemann-Pick type C and other LSDs (Andersson et al. 2004: Shayman 2014; Marshall et al. 2015). It is now being used in combination with other therapies.
6. Combined Gene therapy, substrate reduction therapy and HSCT
Since KD has proved highly refractory to virtually every experimental therapy attempted, Twitcher mice have been simultaneously treated with CNS-directed gene therapy, substrate reduction therapy, and bone marrow transplantation. This approach tried to simultaneously target the primary pathogenic mechanism (GALC deficiency) and two secondary consequences of GALC namely psychosine accumulation and neuroinflammation. The SRT part used cycloserine, a general inhibitor of sphingolipid synthesis, so the positive results were surprising. The simultaneous treatment of multiple pathogenic targets resulted in an unprecedented increase in life span with improved motor function, persistent GALC expression, nearly normal psychosine levels, and decreased neuroinflammation in the treated Twitcher mice (Hawkins-Salisbury et al. 2015). Thus treating both the primary pathogenic mechanism and several secondary targets could be the way to treat humans with KD. Since cycloserine is not FDA approved it might be possible to get the same inhibition of psychosine synthesis with with low doses of Myriocin.
7. Neonatal lentiviral vector-mediated intracerebral gene therapy (ICGT) combined with transplantation of GALC-overexpressing neural stem cells (NSC) to synergize with bone marrow transplant (BMT)
This combination also provided a dramatic extension of lifespan and extensive clinical-pathological rescue in the KD murine model (Ungari et al. 2015). It could be demonstrated that timely and long-lasting delivery of functional GALC to affected tissues resulted from the complementary mode of action of the treatments. In particular, the contribution of neural stem cell transplantation and ICGT during the early asymptomatic stage of the disease was thought to be instrumental to enhancing long-term therapeutic improvement compared to BMT alone. The study also clarified the relative inputs of the central nervous system, peripheral nervous system and periphery to Krabbe disease, and the relative contribution of treatments to the final therapeutic outcome. This has important implications for treatment strategies proposed for humans and the study gives proof-of-concept for efficacy, tolerability and clinical relevance of the combined gene/cell therapies proposed. This combined approach may eventually constitute a feasible and effective therapeutic opportunity for children affected by Krabbe disease if a suitable viral vector such as AAV9 can be found (Lin et al. 2015).
8. Enzyme replacement therapy
Following the success of enzyme replacement therapy for non-CNS lysosomal storage diseases such as Gaucher disease, (Shayman 2014; Marshal et al. 2015) a significant advance has been the development of a "humanized" mouse model of KD by inserting a human GALC cDNA containing an adult-onset patient mutation into the murine GALC gene. Such mice were found to exhibit all the pathological hallmarks of KD including psychosine accumulation, neuroinflammation, CNS infiltration of macrophages, astrogliosis and demyelination (Matthes et al. 2015). Residual GALC activities in mouse tissues were low and the mice had a reduced lifespan of 46 days. The usefulness of this model for enzyme replacement is that by expressing the human transgene, the mice did not develop an immune response against rhGALC, making it suitable for therapies based on human enzyme. Intravenously injected rhGALC was believed to cross the blood-brain barrier and be targeted to lysosomes of brain macrophages, astrocytes and neurons, but this was not experimentally verified. High-dose enzyme replacement therapy was started at postnatal day 21 and reduced the elevated psychosine levels in the peripheral and central nervous system by 15% However, it did not reduce neuroinflammation, demyelination and the bbb still remains a barrier to treatment of Krabbe disease. A summary of commercial ERT efforts is shown in Table 1.
Table 1.
| Some commercial attempts to correct lysosomal storage diseases in | |||
|---|---|---|---|
| Company | humans Disease | Approach | Effective in CNS |
| Genzyme | Gaucher Hunter/Hurler Marateaux- Lamy |
Purified enzyme Substrate reduction (Cerdelga) | NO |
| Shire | Gaucher Fabry |
Enzyme | NO |
| Bio-Marin | Batten CLN2 (TPP1) | Enzyme | NO |
| Amicus | Fabry Pompe |
Chaperone (Migalastat) | NO |
| Lysogene | MPS III α GMI- gangliosidosis |
Gene therapy | NO |
| Alcyone Life Sci | Microfabrication technology for targeted drug therapy (Human stem cells) | NO | |
| Stem-Cell Inc | Batten CLN1 | HSCs | ? |
Because of all the problems associated with these approaches to therapy we have tried to develop a new approach to the treatment of diseases such as Krabbe disease by using nanoparticles to stabilize and deliver enzyme to specific brain cell types. Several nanoparticles have been used as delivery systems but CdSeZnS Quantum dots have many advantages over other types of nanoparticle (Table 2).
Table 2.
Dawson/Krabbe
| Nanoparticles: comparison of Quantum dots and endosomal escpe peptides (such as JB577) with other particles | ||
|---|---|---|
| Type | Size (nM) | Peptide origin |
| Quantum Dot | 10 | JB577 Modular Peptide |
| Gold | 20 | Prion |
| Gold | 25 | Nuclear entry |
| Superparamagnetic iron oxide | 45 | HIV TAT |
| Liposomes | <100 | Influenza fusogenic peptide |
| Polyglycolic acid | <100 | ER insertion sequence |
| Silica | 25–105 | HIV TAT |
9. Development of a Nanoparticle delivery system to oligodendrocytes
i) Developing a delivery system that would permit us to attach a protein to a fluorescent entity so that we could follow its fate in brain tissue
The rabies virus is able to infect the CNS through receptor-mediated anterograde transport and by using the rabies virus 29 amino acid sequence to facilitate transport across the blood-brain barrier and a rhodamine tag, we could show some neuronal labeling in rat hippocampal slices. However, by using a lysine-rich cell-penetrating lipopepetide (similar in theory to lysine/arginine-rich HIV-TAT which penetrates the CNS to induce “Neuro-AIDS”), Santra et al, (2005) were able to get better neuronal delivery and we have since built on this idea. The lipopeptide we used (JB577 ((AcW•G• (Palmitoyl) Diaminopropyl •VKIKK•P9•G2•H6) works on a similar principle to the lysine-rich peptide found in the HIV GP120 glycoprotein coat of HIV (GRKKRRQRRRGYKCNH2) but has an additional palmitate (linked by an amide bond to a cysteine substitute Dap) (DAP1) which seems to improve its membrane-penetrating properties.. DAP1 was originally synthesized as an inhibitor of palmitoyl:protein thioesterase (PPT1) (Dawson et al. 2010) which could act as a chaperone to refold misfolded PPT1 protein. In order to couple the lipopeptide to the QD we solubilized the QDs (Fig. 2) and then attached a His6 sequence to DAP1 in order to bind to the Zn on the surface of a CdSe/ZNS Quantum Dot (QD) (Fig. 3). These CdSeZnS (6–9 mm) QDs are solubilized in the US Naval Laboratories by coating with dihydrolipoic acid-polyethyleneglycol-based polymers or Zwitterionic ligands such a compact ligand 4 (Fig. 2) which allows up to 50 His6-peptides to bind to one QD. Our lead cell-penetrating peptide JB577 has the unique property (amongst >50 related peptides tested) of penetrating cell membranes to promote endosomal egress (Delehanty et al., 2010, Boeneman et al, 2013). This is a critical requirement for delivery to the brain.
Fig 2.

Structure of compact ligands CL4, CL2 and CL1 used to solubilize CdSe/ZnS Quantum dots and provide a charged surface for targeting the QDs plus TPP1 cargo to neurons (CL4) or Glia (CL1). More conventional positively charged coats would use PEG600 with primary amino groups (Li et al (2012, Walters et al, 2015) to give a net positive charge.
Fig 3.
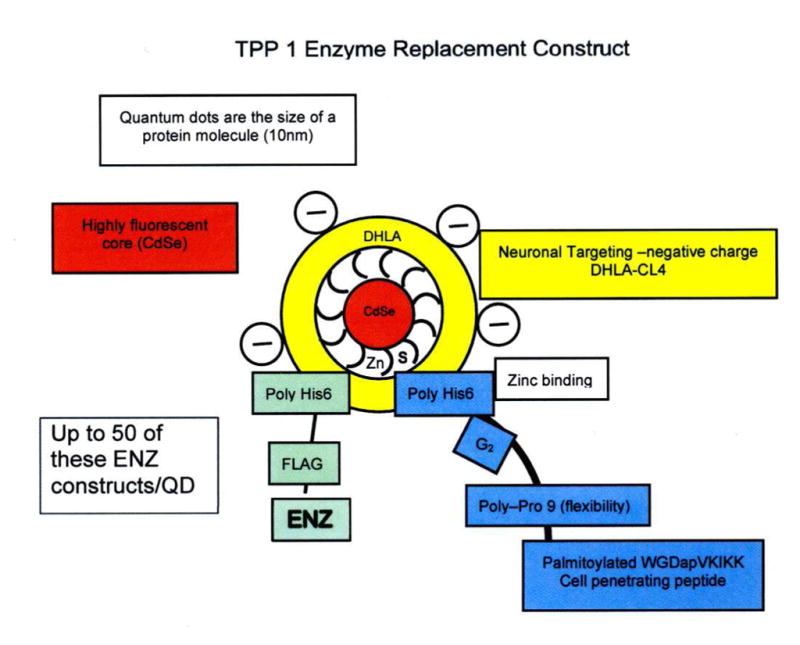

Fig 3A. Schematic representation of Quantum Dot (QD) with attached peptide/GALC-ENZ
- Red: Crystalline semiconductor 6nm core is Cd/Se
- White: Surface coat is Zn.
- Yellow: The solubilizing coat is negatively charged Compact Ligand.4 (Fig. 2) (neuron targeting) or Polyethylene glycol polymer 600 with NH2 positive charge for oligodendrocytes (Walters et al, 2015..
- Blue: Cell penetrating palmitoylated VKIKK peptide (Dap1) has a HIS6 attachment sequence to bind it to Zn and and a peptide spacer (G2P9 to promote attachment.
- Green: Enzyme (eg:GALC) has a C-terminal FLAG (to show lysosomal uptake) HIS6 and a spacer to to attach 20 –35 molecules to the QD.
Fig. 3B. Space-filling model of a Quantum dot bound to JB577 through a Zn-His6 association and showing the site of attachment of RNAi sequences which could be used to supress synthesis of specific enzymes (for example Cer –Glucosyltransferase) and effect substrate-reduction therapy (SRT).
Thus the secret to success was that the Zn coat, which encloses the semiconductor crystalline CdSe core, binds tightly to a sequence of 6 Histidine residues (His6). Using Zn or Ni columns is a common way of purifying His-tagged recombinant proteins and we were able to test a range of proteins (from His6-green fluorescent protein to lysosomal hydrolases) for preservation of activity following attachment to the modified QDs (Walters et al. 2015). We have attached other proteins to QD using this approach, but some require a proline spacer (as used in JB577) and all probably have unique half lives in vivo. We attach the His6 and a detector sequence such as FLAG to the C-terminal end (Fig. 3) since lysosomal hydrolases such as GAL-C are N-terminally processed proteolytically and then become resistant to degradation by proteases and glycosidases. In order to get the QD endocytosed by the cell and released from the endosome into the cytosol or the lysosome we screened dozens of potential cell-penetrating peptides but only lipopeptide JB577, designed by us to be an inhibitor of the lysosomal enzyme palmitoyl: protein thioesterase (PPT1) (Dawson et al, 2010) and based on the Ras4A sequence, was able to achieve this goal (Santra 2005; Delehanty et al. 2010; Susumu et al. 2011; Walters et al 2012; Boeneman et al. 2013; Agarwal et al. 2015; Walters et al.(2015). Omitting the lysine-rich VKIKK sequence resulted in loss of endosomal egress. This approach has been patented by us (US#8,835172). Thus by using the JB577 lipopeptide (WG(N-palmitoyl)DapVKIKKP9G2H6 we have been able to achieve the goal of endosomal uptake, release into the cytosol and delivery to the lysosome. See Fig. 3 for details of a construct which could be used to deliver enzyme to brain. When such a construct was injected into 6-cell thick rat hippocampal slices (Walters et al., 2012, 2015) the QDs appeared to be delivered specifically to neurons, with little delivery to glial cells (Fig. 4).
Fig 4.

Preferential uptake of Quantum dots (QD-CL4-JB577 (red)) by pyramidal neurons in rat hippocampal slices, showing very little uptake by microglia (stained green with Alexa Fluor isolectin GSlB4 from griffonia simplicifolia) or glia (GFAP-data not shown. See Walters et al, 2012 for more details).
ii) Effect of surface charge on uptake in a multicellular, myelinating rat hippocampal slice preparation
As part of our initial studies on rat neonatal hippocampal slices we made the serendipitous discovery that the solubilizing coating on the Quantum dots was a major factor in deciding the cellular targeting of the QD complex, namely that a negatively charged compact ligand (CL) coat (CL4) heavily favored neurons (Walters et al, 2015) and a positively charged coat (PEG600NH2) favored glial cells, especially oligodendrocytes. This opened up the possibility for GALC protein delivery to vulnerable oligodendrocytes in Krabbe disease (Walters et al, 2015) and was attributed to the fact that glial cells secrete a much more complex and extensive extracellular matrix than most neurons, as summarized in Fig. 5. In addition to proteoglycans (chondroitin sulfates, hyaluronic acids, and heparin sulfates) there are negatively-charged, sialic acid-rich PI-linked, O-linked and N-linked glycoproteins in the extracellular matrix and the glymphatic system (Iliff et al, 2012). Finally there are membrane-associated glycosphingolipids associated with extracellular membranous sheets as well as the cell body(gangliosides and sulfatides) and in KD the abnormal GalCer and Psychosine components play a critical role in the pathogenesis of the disease. All these molecules must play a role in modulating the fate of exogenous enzyme en route to the lysosome.
Fig. 5. Representation of the extracellular matrix surrounding a neural cell.

The quantitative and qualitative composition is variable (eg: in the perineural net) but neurons tend to have less negatively charged extracellular matrix than glial cells (especially reactive glial cells following injury) and this should be considered when designating a vehicle to deliver enzyme, chaperones or substrate-reducing drugs to cells. Major proteoglycans are Chondroitin sulfates, heparan sulfates (which are linked to proteins through Gal-Gal-Xylosyl-Serine and hyaluronic acid. Sialic-acid-containing glycoproteins can be GlcNAc-Asn-linked (N), GalNAc-Ser (O-linked) or phosphatidylinositol (PI)-linked. Negatively-charged glycosphingolipids include gangliosides and sulfatides. Sulfatide is 3-O-galactosylceramide and therefore metabolically related to the galactosylsphingosine(psychosine) storage material in KD The ECM can be digested with bacterial chondroitinase ABC, heparanase and sialidase (neuraminidase).
We then discovered that an enzymic digestion of the extracellular matrix with chondrotinase ABC in these 5-cell thick hippocampal slices could greatly facilitate uptake of peptide/protein by oligodendrocytes (Fig. 6A–C) (Walters et al, 2015). A similar approach of chondroitin sulfate digestion has been used to break up reactive astrocytes and glial scars in spinal cord injury and sometimes this increases the chances of recovery in mouse models of spinal cord injury (Galtery et al, 2007, Mountney et al, 2013). Chondroitinase ABC cleaves a particularly broad range of galactosaminoglycans (GalAG) substrates, including CS (chondroitin sulfate), DS (dermatan sulfate) and, to a much lesser extent, hyaluronic acid, (a large polymer of repeating β-D-glucuronic acid)] (1→3) linked to GlcNAc (N-acetyl-D-glucosamine) disaccharides (Schwartz and Domowicz 2014). Sulfated GalAGs are composed of disaccharide repeat units of sulfated uronic acid [IduA (α-L-iduronic acid) or GlcA. These basic disaccharide units are linearly associated via β-(1→4) linkages to form polymers of CS attached at multiple points along a core protein. Biosynthesis of CS involves sulfation of the sugar backbone at various positions, which generates further diversity in their oligosaccharide sequences (Fig. 5). CS is commonly O-sulfated at the C-4 of the N-acetylgalactosamine [C4S (chondroitin-4-sulphate) or CSA], the C-6 of the N-acetylgalactosamine [C6S (chondroitin-6-sulfate) or CSC], or Iduronic acid and C4 of GalNAc (CSB). Other rare modifications in CS, such as 2-O-or 3-O-sulfation of the GlcA moieties, have also been reported (Schwartz and Domowicz, 2013) and digestion of the ECM prior to therapy should be given serious consideration in the future since the extracellular matrix is a major factor to be considered when designing ERT.
Fig 6. Chondrotinase digestion increases uptake of QDs by oligodendrocytes.

Panel A: Rat hippocampal fate of QD-CL4-JB577 showing endosomal uptake and selective entry into neuronal cells at the CA1/CA3 border of the hippocampus but not into cells of the astrocyte-rich layer (bottom left). (Walters et al., 2012). Panel B. Treatment with chondroitinase ABC has little effect on neuronal uptake. Panel C. Treatment with chondroitinase ABC increases uptake into Myelin Basic Protein positive (Green) cells (oligodendrocytes) by 50% (see merged color).. Panel D: Similar treatment does not increase uptake of QDs (Red) by microglia (Green) stained with isolectin antibody by >10%. (See Walters et al, 2015) for more details of quantification.
Future directions
Toxicity with the Cd core may be an issue but we have not observed any toxicity with our QDs. Once we have established where and when the QDs go we can replace the metal core with biodegradeable QDs. To be successful, the enzyme needs to be stable in the specific pathological environment of the KD brain and by recombinantly engineering lysosomal enzymes such as GALC to have a C-terminal His6 tag (they are typically N-terminally processed) and a FLAG tag we can bind the His6protein to the Zn on the surface of the QD and make sure the GALC reaches the damaged parts of the brain.
We need to study GALC fate inside tissues by fluorescence microscopy and electron microscopy (Fig. 8).(Figs. 4, 6, 7 and 8) to make sure it reaches the majority of cells, since all cells express the defect. We can achieve this by addition of a C-terminal FLAG tag so that we can assess with a highly specific antibody, just how long the protein stays attached to the QD. An especially useful system for screening newly developed enzyme-QD constructs is the embryonic chick (Fig. 7A, B), Agarwal et al, 2015). By injecting the QD-peptide/enzyme construct into the spinal canal of Day 4 embryos we can readily see that by Day 6, extensive penetration of the spinal cord can be observed (Fig. 7A). The QDs spread to all parts of the brain and developmental milestones are achieved on time (Agarwal et al, 2015). QDs then spread to all parts of the developing brain and by day 15 the chorionic villus has acquired most of the QDs, which are then most likely excreted, leaving the enzyme in the brain.
We need a mechanism for directing GALC enzyme to oligodendrocytes to reverse their degeneration and this enzyme must be able to traverse the extracellular matrix (Fig. 5). We then need to show that the GALC ends up in lysosomes. Here we can take advantage of the electron-dense nature of the QD to use EM to show where QDs go subcellularly in the target cell (Fig. 7) (Walters et al. 2012) and match this with lysosomal fluorescence such as Lysotracker or anti-FLAG antibody (since only the GALC has this epitope). As we have discussed, injection of QD-CL4-JB577-Enzyme labels mainly neurons (Fig. 6A). Many cells, especially astrocytes, do not take up negatively charged QDs. However, we can treat with chondroitinase ABC and use a positively charged coating for the QD to increase oligodendrocyte uptake to >50% (Fig. 6B) whereas microglia are essentially unaffected (Fig. 6C). Surprisingly, neuraminidase treatment to remove negatively charged sialic acids has thus far had no effect on uptake by neurons. This evidence of lack of uptake by microglia is especially important since we do not want the microglia to remove enzyme that should be targeted to lysosomes. We believe that this QD approach is uniquely capable of delivering enzymes such as GALC to cells in the CNS such as oligodendrocytes. This opens up all kinds of future possibilities including not only proteins but small RNAs and other cell-modifying molecules.
The major test will be to show that JB577-coated QDs can deliver enzyme across capillaries, pass through pericytes and astrocyte end-foot processes and finally diffuse to the target through a highly negatively charged extracellular matrix consisting of proteoglycans, glycoproteins and gangliosides. To therapeutically treat Krabbe disease we need a means of targeting the lysosomal enzyme to oligodendrocytes, which we believe we have achieved by coating the QDs with positively charged compact ligand PEG600NH2 (Walters et al, 2015). We have been able to achieve a similar result in actively myelinating rat hippocampal brain slices by pre-treating with chondoitinase ABC to digest much of the extracellular matrix which surrounds the oligodendrocyte (Walters et al., 2015). The role of cell surface charge in cell uptake (endocytosis) has not been previously studied and represents a new concept in facilitating translational efforts in the CNS such as enzyme replacement therapy. Our novel results suggest a method for directing the targeting and fate of clinically relevant cargo in the brain through the manipulation of both the vehicle’s charge as well as the charge of the target location.
-
The last problem to overcome is how to measure success in ERT of Twitcher mice/KD model systems) and eventually in humans.
-
i)
Improved function, and increased life-span has been used in many studies and is a good measure of success (or lack of it).
-
ii)
Measuring the unique storage product psychosine (detected by MS/MS)(Dawson, 2015), is a powerful technique and using a MALDI approach one can quantify lipids in tiny areas of tissue, getting down to the unicellular level. Although these approaches are not generally available they can be accessed via Core facilities. GalCer is often reduced rather than stored because of hypomyelination but this can be quantified by HPTLC (or MS/MS). GalCer is an important marker for KD since it only synthesized by oligodendrocytes and kidney cells.
-
iii)
Reduced myelin is the hallmark of all forms of KD so microscopy or MBP western blot can be used to assess the efficacy of therapy.
-
iii)
GALC Enzyme assay requires a [3H]-labeled substrate but increased activity can be followed. However, the main caveat here is that the enzyme detected may not be in the cell or in the lysosomal compartment.
-
iv)
Following treatment with Quantum Dots +JB+HIS6/FLAG tagged GALC we can show, by single cell confocal imaging, that GALC is inside the cell (anti- HIS6) and then released from endosomes (FLAG staining of lysosomes). We have extensive evidence that there is endosomal egress for up to 72h (Boeneman et al, 2013.
-
i)
Fig 7. QDs are taken up extensively by neuroblasts in early chick embryo development.

Panel A shows ubiquitous distribution of Quantum dots throughout spinal cord of a Day 6 embryonic chick into which we had injected QD-CL4-JB577 into the spinal canal at Day 4 (see Agarwal et al., 2015 for more details). Panel B shows the extensively QD-labeled choroid plexus at day 15 following injection at day 4.
Conclusions
Most therapeutic molecules fail to cross the bbb – a neurovascular unit composed of endothelial cells, astrocytes, pericytes, and neurons. Physical attempts to open the bbb, for example with osmotic gents such as mannitol, can damage the brain so one emphasis in treating Krabbe and related lysosomal storage diseases has been on specific solutions such as incorporating sequences from the brain LDL transporter together with a polylysine peptide sequence (eg: Meng et al, 2014) or by taking advantage of drugs which modify adenosine receptor signaling (Carman et al, 2013). A key element of bbb function is the expression of ATP binding (ABC) drug efflux transporters which remove drugs which might cross the bbb. To overcome this the drug should have a certain amount of lipofilicity (a fatty acid) and regions of positive charge (lysine rich) all of which are possessed by JB577 – and only by JB577 in our studies. There is increasing evidence that ceramide has a role in multi-drug resistance (MDR) and bbb penetration and unlike SM and GlcCer which project outwards from the cell surface, Ceramide readily translocates across the membrane. Highly malignant tumor cells have high activity of Cer:glucosyltransferase and conversion of Cer to GlcCer is an escape system from Ceramide-induced apoptosis (Pamitoylated peptides appear to mimic ceramide in cell-penetrating ability and It was recognized a number of years ago that some CdSe/ZnS Quantum dots could cross the bbb (Santra et al, 2005). The addition of JB577 has further enhanced this capability.
The general consensus is that psychosine is the toxic agent in KD (Suzuki and Suzuki, 1970, If combination therapy is critical, a future possible approach, since we do not have a really specific inhibitor of GalCer or psychosine synthesis, is to use RNAi techniques to knockdown the Cer: galactosyltransferase. RNA interference is more potent than antisense alone but has been restricted by RNA instability. (Haussecker, and Kay 2015). Double stranded RNA sequence (~70bp) are exported to the cytoplasm where DICER cleaves them into SiRNA of ~21 nucleotides. By embedding the RNA in nanoparticles, companies like Silenseed (targeting mutant K-Ras) and Arrowhead (DPC liposomes) have been able to protect the RNA sufficiently to achieve target-specific RNA knockdown. Li et al (2012) have shown that you can deliver RNAi directly bound to the QD coat and QD-JB is a development of this approach to protect and deliver to the CNS. When coupled with VKIKK as the RNA attachment site, this may help Krabbe therapy by inhibiting synthesis of GalCer through knockdown of GalC synthase.
Significance Statement.
Lysosomal storage diseases such as Krabbe are devastating to families and any therapy requires knowledge of what is stored and where. Thus an understanding of Krabbe disease will increase our understanding of the CNS as well as ultimately providing a means of therapy
Acknowledgments
I would like to thank the National Institutes of Health , NINDS (NS-36866) and NICHD (HD09402) and the Childrens Brain Diseases Foundation for support of this work, None of this would have been possible but for the work of John P. Kilkus, Sylvia A Dawson, Miriam Domowicz, Rishabh Agarwal, Connor Lynch and Ted Getz. We also acknowledge the role of Igor Medintz and colleagues at the US Navy Labs and Philip E. Dawson at Scripps research in La Jolla, CA. There are no conflicts of interest.
Literature cited
- Agarwal R, Domowicz MS, Schwartz NB, Henry J, Medintz I, Delehantyk JB, Stewart MJ, Susumu K, Huston AL, Deschamps JR, Dawson PE, Palomo V, Dawson G. Delivery and Tracking of Quantum Dot Peptide Bioconjugates in an Intact Developing Avian Brain. ACS Chemical Neurosci. 2015;6(3):494–504. doi: 10.1021/acschemneuro.5b00022. Online March. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U, Smith D, Jeyakumar M, Butters TD, Borja MC, Dwek RA, Platt FM. Improved outcome of N-butyldeoxygalactonojirimycin-mediated substrate reduction therapy in a mouse model of Sandhoff disease. Neurobiol Dis. 2004;16(3):506–15. doi: 10.1016/j.nbd.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Boeneman K, Delehanty JB, Blanco-Canosa JB, Susumu K, Stewart M, Huston AL, Dawson G, Ingale S, Walters R, Domowicz M, Dawson PE, Medintz I. Selecting Improved Peptidyl Motifs for Cytosolic Delivery of Disparate Protein and Nanoparticle Materials. ACS Nano. 2013 May 28;7(5):3778–96. doi: 10.1021/nn400702r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson G, Schroeder C, Dawson PE. Palmitoyl:protein thioesterase (PPT1) inhibitors can act as pharmacological chaperones in infantile Batten Disease. Biochem Biophys Res Commun. 2010;395(1):66–69. doi: 10.1016/j.bbrc.2010.03.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson G. Glycosignaling: a general review. In: Yu RK, Schengrund, editors. Glycobiology of the Nervous System. Vol. 17. Springer; 2014. pp. 293–306. PMC Journal. [DOI] [PubMed] [Google Scholar]
- Dawson G. Measuring Brain Lipids. Biochim Biophys Acta 2015. 2015 Aug;1851(8):1026–1039. doi: 10.1016/j.bbalip.2015.02.007. Epub 2015 Feb 18. (special issue on brain lipids) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delehanty JB, Bradburne CE, Boeneman K, Susumu K, Farrell D, Mei BC, Blanco-Canosa JB, Dawson G, Dawson PE, Mattoussi H, Medintz IL. Delivering quantum dot-peptide bioconjugates to the cellular cytosol: escaping from the endolysosomal system. Integr Biol. 2010;2:265–277. doi: 10.1039/c0ib00002g. [DOI] [PubMed] [Google Scholar]
- Hawkins-Salsbury JA, Shea L, Jiang X, Hunter DA, Guzman AM, Reddy AS, Qin EY, Li Y, Gray SJ, Ory DS, Sands MS. Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. J Neurosci. 2015;35:6495–505. doi: 10.1523/JNEUROSCI.4199-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert JA, Dawson G. Neuronal-ceroid Lipofuscinoses:therapeutic strategies, past, present and future. Biochim Biophys Acta. 2006;1762:945–53. doi: 10.1016/j.bbadis.2006.08.004. Review. [DOI] [PubMed] [Google Scholar]
- Hossain MA, Higaki K, Saito S, Ohno K, Sakuraba H, Nanba E, Suzuki Y, Ozono K, Sakai N. Chaperone therapy for Krabbe disease: potential for late-onset GALC mutations. J Hum Genet. 2015 Sep;60(9):539–45. doi: 10.1038/jhg.2015.61. Epub 2015 Jun 25. [DOI] [PubMed] [Google Scholar]
- Kuai XL, Ni RZ, Zhou GX, Zheng BM, Zhang JF, Yi N, Zhao XL, Shao N, Ni WK, Wang ZW. Transplantation of mouse embryonic stem cell-derived oligodendrocytes in the murine model of globoid cell leukodystrophy *. Stem Cell Research & Therapy. 2015;6:30. doi: 10.1186/s13287-015-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Li Z, Ji F, Xiao Z, Wang M, Peng Y, Zhang Y, Liu L, Liang Z, Li F. Delivery of Quantum Dot-siRNA Nanoplexes in SK-N-SH cells for BACE1 gene silencing and intracellular imaging. Molecular Therapy-Nucleic Acids1. 2012;E20:1–10. doi: 10.1038/mtna.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DS, Hsiao CD, Lee AY, Ho CS, Liu HL, Wang TJ, Jian YR, Hsu JC, Huang ZD, Lee TH, Chiang MF. Mitigation of cerebellar neuropathy in globoid cell leukodystrophy mice by AAV-mediated gene therapy. Gene. 2015 Oct 15;571(1):81–90. doi: 10.1016/j.gene.2015.06.049. Epub 2015 Jun 23. [DOI] [PubMed] [Google Scholar]
- Marshall J, McEachern KA, Chuang WL, Hutto E, Siegel CS, Shayman JA, Grabowski GA, Scheule RK, Copeland DP, Cheng SH. Improved management of lysosomal glucosylceramide levels in a mouse model of type 1 Gaucher disease using enzyme and substrate reduction therapy. J Inherit Metab Dis. 2015;33:281–289. doi: 10.1007/s10545-010-9072-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E1, Spencer B. Applications of ApoB LDLR-Binding Domain Approach for the Development of CNS-Penetrating Peptides for Alzheimers’ Disease. Methods Mol Biol. 2015;1324:331–7. doi: 10.1007/978-1-4939-2806-4_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes F, Andersson C, Stein A, Eistrup C, Fogh J, Gieselmann V, Wenger DA, Matzner U. Enzyme replacement therapy of a novel humanized mouse model of globoid cell leukodystrophy. Exp Neurol. 2015;271:36–45. doi: 10.1016/j.expneurol.2015.04.020. Epub 2015 May 6. [DOI] [PubMed] [Google Scholar]
- Meng Y, Sohar I, Sleat DE, Richardson JR, Reuhl KR, Jenkins RB, Sarkar G, Lobel Effective Intravenous Therapy for Neurodegenerative Disease With a Therapeutic Enzyme and a Peptide That Mediates Delivery to the Brain. Mol Ther. 2014;22(3):547–553. doi: 10.1038/mt.2013.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morad SA, Bridges LC, Almeida Larrea AD, Mayen AL, MacDougall MR, Davis TS, Kester M, Cabot MC. Short-chain ceramides depress integrin cell surface expression and function in colorectal cancer cells. Cancer Lett. 2016 Apr 1; doi: 10.1016/j.canlet.2016.03.049. pii: S0304-3835(16)30210–5. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Mountney A, Zahner MA, Sturgill ER, Riley CJ, Aston Martin JW, Oudega A, Schramm LP, Hurtado A, Schnaar RL. Sialidase, Chondroitinase ABC, and Combination Therapy after Spinal Cord Contusion Injury. J Neurotrauma. 2013;30(3):181–190. doi: 10.1089/neu.2012.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafi MA, Rao HZ, Luzi P, Wenger DA. Long-term Improvements in Lifespan and Pathology in CNS and PNS After BMT Plus One Intravenous Injection of AAVrh10-GALC in Twitcher Mice. Mol Ther. 2015 Nov;23(11):1681–90. doi: 10.1038/mt.2015.145. Epub 2015 Sep 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra S, Yang H, Stanley JT, Holloway PH, Moudgil BM, Walter G, Mericle RA. Rapid and effective labeling of brain tissue using TAT-conjugated CdS:Mn/ZnS quantum dots. Chem Commun. 2005:3144–3146. doi: 10.1039/b503234b. [DOI] [PubMed] [Google Scholar]
- Schwartz NB, Domowicz MS. Chemistry and function of glycosaminoglycans in the nervous system. Adv Neurobiol. 2014;9:89–115. doi: 10.1007/978-1-4939-1154-7_5. PMC Journal. [DOI] [PubMed] [Google Scholar]
- Shayman J. Thematic Review Series: recent advances in the treatment of lysosomal storage diseases. J Lipid res. 2014;55:993–994. doi: 10.1194/jlr.E049817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE1, Sun P, Wiseman JA, Huang L, El-Banna M, Zheng H, Moore DF, Lobel P. Extending the mannose 6-phosphate glycoproteome by high resolution/accuracy mass spectrometry analysis of control and acid phosphatase 5-deficient mice. Mol Cell Proteomics. 2013;2(7):1806–17. doi: 10.1074/mcp.M112.026179. Epub 2013 Mar 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susumu K, Oh E, Delehanty JB, Blanco-Canosa JB, Johnson BJ, Jain V, Hervey WJ, Algar WR, Boeneman K, Dawson PE, Medintz IL. Multifunctional compact zwitterionic ligands for preparing robust biocompatible semiconductor quantum dots and gold nanoparticles. J Am Chem Soc. 2011;133:9480–9496. doi: 10.1021/ja201919s. [DOI] [PubMed] [Google Scholar]
- Susumu K, Uyeda HT, Medintz IL, Pons T, Delehanty JB, Mattoussi H. Enhancing the stability and biological functionalities of quantum dots via compact multifunctional ligands. J Am Chem Soc. 2007;129:13987–13996. doi: 10.1021/ja0749744. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Suzuki Y. Globoid cell leukodystrophy (Krabbe’s disease): Deficiency of galactocerebrosidase β-galactosidase. Proc Natl Acad Sci U S A. 1970;66:302–9. doi: 10.1073/pnas.66.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungari S, Montepeloso A, Morena F, Cocchiarella F, Recchia A, Martino S, Gentner B, Naldini L, Biffi A. Design of a regulated lentiviral vector for hematopoietic stem cell gene therapy of globoid cell leukodystrophy. Mol Ther Methods Clin Dev. 2015 Oct 14;2:15038. doi: 10.1038/mtm.2015.38. eCollection 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillemenot BR, Katz ML, Lobel P, O’Neill CA. Intrathecal tripeptidyl-peptidase1 reduces lysosomal storage in a canine model of late infantile neuronal ceroid lipofuscinosis. Mol Genet Metab. 2011;104:325–337. doi: 10.1016/j.ymgme.2011.06.018. [DOI] [PubMed] [Google Scholar]
- Walters R, Kraig RP, Medintz I, Delehanty JB, Stewart MH, Susumu K, Huston AL, Dawson PE, Dawson G. Nanoparticle Targeting to Neurons in a Rat Hippocampal Slice Culture Model. ASN Neuro. 2012;4(6):383–392. doi: 10.1042/AN20120042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters R, Medintz IL, Delehanty JB, Stewart MH, Susumu K, Huston AL, Dawson PE, Dawson G. The Role of Negative Charge in the Delivery of Quantum Dots to Neurons ASN Neuro. 2015 Jul-Aug;:1–12. doi: 10.1177/1759091415592389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnock DG, Bichet DG, Holida M, Goker-Alpan O, Nicholls K, Thomas M, Eyskens F, Shankar S, Adera M, Sitaraman S, Khanna R, Flanagan JJ, Wustman BA, Barth J, Barlow C, Valenzano KJ, Lockhart DJ, Boudes P, Johnson FK. Oral Migalastat HCl Leads to Greater Systemic Exposure and Tissue Levels of Active α-Galactosidase A in Fabry Patients when Co-Administered with Infused Agalsidase. PLoS One. 2015 Aug 7;10(8):e0134341. doi: 10.1371/journal.pone.0134341. eCollection 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger DA, Suzuki K, Suzuki Y, Suzuki K. Galactosylceramide lipidosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. Globoid cell leukodystrophy (Krabbe disease) 8. New York: McGraw-Hill; 2001. pp. 3669–87. The metabolic and molecular bases of inherited disease. [Google Scholar]
- Won JS, Kim J, Paintlia MK, Singh I, Singh AK. Role of endogenous psychosine accumulation in oligodendrocyte differentiation and survival: implication for Krabbe disease. Brain Res. 2013 May 1;1508:44–52. doi: 10.1016/j.brainres.2013.02.024. Epub 2013 Feb 21. [DOI] [PMC free article] [PubMed] [Google Scholar]