

Abstract
Currently, presymtomatic hematopoietic stem and progenitor cell transplantation (HSPCT) is the only therapeutic modality that alleviates Krabbe's disease (KD)‐induced central nervous system damage. However, all HSPCT‐treated patients exhibit severe deterioration in peripheral nervous system function characterized by major motor and expressive language pathologies. We hypothesize that a combination of several mechanisms contribute to this phenomenon, including 1) nonoptimal conditioning protocols with consequent inefficient engraftment and biodistribution of donor‐derived cells and 2) insufficient uptake of donor cell‐secreted galactocerebrosidease (GALC) secondary to a naturally low expression level of the cation‐independent mannose 6‐phosphate‐receptor (CI‐MPR). We have characterized the effects of a busulfan (Bu) based conditioning regimen on the efficacy of HSPCT in prolonging twi mouse average life span. There was no correlation between the efficiency of bone marrow engraftment of donor cells and twi mouse average life span. HSPCT prolonged the average life span of twi mice, which directly correlated with the aggressiveness of the Bu‐mediated conditioning protocols. HSPC transduced with lentiviral vectors carrying the GALC cDNA under control of cell‐specific promoters were efficiently engrafted in twi mouse bone marrow. To facilitate HSPCT‐mediated correction of GALC deficiency in target cells expressing low levels of CI‐MPR, a novel GALC fusion protein including the ApoE1 receptor was developed. Efficient cellular uptake of the novel fusion protein was mediated by a mannose‐6‐phosphate‐independent mechanism. The novel findings described here elucidate some of the cellular mechanisms that impede the cure of KD patients by HSPCT and concomitantly open new directions to enhance the therapeutic efficacy of HSPCT protocols for KD. © 2016 The Authors. Journal of Neuroscience Research Published by Wiley Periodicals, Inc.
Keywords: busulfan conditioning, Galc‐AErbd (ApoE receptor binding domain), myeloid‐specific promoter, erythroid‐specific promoter, AB_313497, AB_10894189, AB_398535, AB_2034021, AB_10563203, AB_10611731, IMSR_JAX:002014, IMSR_JAX:000845
Neonatal bone marrow transplantation (BMT) provides significant (although incomplete) protection for early infantile Krabbe's dieaese (KD) patients’ central nervous system (CNS), preserves their cognitive function, and appears to lengthen their survival (mean of 23–25 months without treatment) up to the second decade of life (Escolar et al., 2005; Duffner et al., 2011, 2009; Wasserstein et al., 2016). However, BMT cannot effectively alleviate the motor and sensory deficiencies caused by peripheral demyelination. This therapeutic approach is premised on the ability of the donor's hematopoietic stem and progenitor cells (HSPC) and their progenies to colonize the host CNS and to secrete functional GALC, which upon mannose‐6‐phosphate (M6P)‐mediated uptake compensates for the lack of host GALC function. Failure to efficiently complete any of these processes potentially contributes to the mechanism that renders KD incurable by HPCT. Host conditioning is aimed at generating space for donor HSPC in host target niches and is considered critical for efficient engraftment. Although not applicable for KD patients, total body irradiation (TBI) is the conditioning method employed in most HSPCT‐based preclinical studies of KD (Lin et al., 2007; Gentner et al., 2010). Notwithstanding the major impact of the conditioning protocol on the clinical outcome of HSPCT in lysosomal storage disorder (LSD) patients, the effects of the aggressiveness of the conditioning protocol on the pathologic course of KD have not been evaluated in a preclinical model. The ability of GALC‐deficient host cells to take up extracellular GALC efficiently is the premise of the HSPCT‐based therapy for KD. Both the cation‐dependent and the cation‐independent M6P receptors (CD‐MPR and CI‐MPR) mediate intracellular trafficking of lysosomal enzymes. However, only the CI‐MPR (also known as IGF2R) facilitates uptake of extracellular proteins via the M6P receptor (M6PR) pathway (Munier‐Lehmann et al., 1996; Dahms et al., 2008; Stein et al., 2010). Several research groups have demonstrated that expression of IGF2R in rodent and human CNS is cell type specific (Nissley et al., 1993; Gonzalez‐Parra et al., 2001; Hawkes and Kar, 2003, 2004; Jofre et al., 2009). Thus, naturally low levels of IGF2R expression in various cell populations could potentially reduce the therapeutic efficacy of HSPCT for KD patients. The ability to mediate cellular uptake of GALC via an M6P‐independent pathway should address this potential weakness. Recent publications described the development of a fusion protein comprising the ApoE1‐receptor binding domain (AErbd) and the lysosomal protein α‐L‐iduronidase (IDUA; Wang et al., 2013; Dai et al., 2014). This facilitated cellular uptake and transcytosis of the novel fusion protein to the CNS via the low‐density lipoprotein (LDL) receptor‐related protein 1 (LRP‐1) pathway, independently of the M6P pathway. Premised on this technology, a novel GALC‐AErbd fusion protein can potentially be used to treat cell populations that naturally do not express sufficient IGF2R.
To date, preclinical trials employing viral vectors for GALC delivery failed to significantly alter the pathologic course of KD in the twi mouse model (Lin et al., 2007, 2005; Gentner et al., 2010; Ungari et al., 2015). Notwithstanding the ongoing growth in the pool of potential bone marrow (BM) donors, it is still possible that the lack of a matched allogeneic donor will render KD patients unsuitable for HSPCT. The ability of lentiviral vectors to efficiently transduce and maintain long‐term transgene expression in patients’ HSPC and their differentiated progenies has been employed in human clinical trials to establish therapeutic autologous HSPC‐based gene replacement protocols for adrenoleukodystrophy (Cartier et al., 2009), metachromatic leukodystrophy (Biffi et al., 2013) and Wiscott‐Aldrich syndrome (Aiuti et al., 2013) patients for whom a matched donor could not be identified. However, cytotoxicity associated with lentiviral vector‐mediated GALC expression in HSPC poses a potential hurdle in employing the aforementioned therapeutic approach for KD patients (Gentner et al., 2010; Visigalli et al., 2010). By incorporating a tandem of target sequences of the HSPC‐specific micro‐RNA mir126 investigators (Gentner et al., 2010; Ungari et al., 2015) minimized GALC expression in HSPC and efficiently engrafted human and mouse HSPC that had been transduced with lentiviral vectors from which the human PGK promoter regulated expression of functional GALC mRNA. We assert that overexpression of target sequence of host miRNAs may alter the natural miRNA system (Ebert and Sharp, 2010). On the other hand, lentiviral vectors that express functional GALC under the control of cell‐specific promoters would further enhance expression of GALC in the relevant HSPC progenies (e.g., microglia), with minimal GALC expression in HSPC. This strategy should facilitate therapeutic autologous HSPCT in KD patients without posing risk/benefit concerns associated with the overexpression of decoy micro‐RNA targets.
MATERIALS AND METHODS
Cells
THP‐1 cells were cultured in RPMI1640 (Hyclone, Logan, UT) with 10% FBS (Atlantic Biologicals, Miami, FL), 2 mM glutamine (Corning Cellgro, Manassas, VA), 100 U/ml penicillin, 100 µg/ml streptomycin, and 250 ng/ml amphotericin B (Corning Cellgro). Murine erythroleukemia (MEL) and 293T cells were maintained in DMEM High‐Glucose (Hyclone) supplemented with 10% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, and 250 ng/ml amphotericin B. During the differentiation, MEL cells were induced by 5 mM hexamethylene bisacetamide (HMBA) and supplemented with 20% FBS. Lineage‐negative cells purified using a mouse lineage cell depletion kit (Miltenyi Biotechnology, San Diego, CA) were cultured in Stem Span SFEM medium (Stemcell, Vancouver, British Columbia, Canada) with 100 ng/ml mouse stem cell factor (SCF), 100 ng/ml Flt3 ligand, 100 ng/ml IL3, and 100 ng/ml TPO (Sigma, St. Louis, MO).
Plasmids
The lentiviral vector packaging cassette ΔNRF and the VSV‐G envelope plasmid were described previously (Kafri et al., 1999). Promoter sequences and codon‐optimized GALC cDNA's were synthesized by GeneArt (Thermo Fisher). The relevant DNA fragments were cloned into lentiviral vectors (pTK134 or pTK208). Vector structures were verified by restriction enzyme analysis and/or sequencing.
Luciferase Assay
Luciferase expression was measured by the Luciferase Assay system from Promega (Madison, WI). Briefly, virus‐transduced cells were lysed with lysis buffer and analyzed on 1420 Multilabel Counter Victor 3 (PerkinElmer, Waltham, MA). Activities were further normalized with protein concentration and viral copy number (VCN) per cell. Protein concentration was determined by the Bradford method.
VCN Quantification
VCN was quantified by multiplex PCR (Suwanmanee et al., 2013) on an ABI7300 real‐time PCR system. NotI794 primer/prober set (left primer 5′‐taagaccaccgcacagca‐3′, right primer 5′‐cacttctccaattgtccctca‐3′; No. 25; Roche Universal Probe Library [UPL]) was used for vectors detection, and paired with two different reference genes, mouse GAPDH primer/probe set or human GUSB primer/probe set (Roche, Indianapolis, IN). DNA samples were treated with dpn I to minimize plasmid contamination before PCR analysis.
293T Uptake and Galc Activity Assay
Cells were incubated with medium containing different GALC variants at 37 °C for 3 hr. After three PBS washes, cells were lysed with RIPA buffer on ice for 30 min. Cell lysates were cleared by centrifugation at 12,000 rpm for 5 min at 4 °C and assayed for GALC activity. For M6P inhibition, 293T cells were pretreated with or without 1 mM M6P for 30 min, followed by incubation of conditioned media with different GALC proteins.
GALC activity assay was performed as described previously (Martino et al., 2009). Briefly, cells were lysed in RIPA buffer supplemented with protease inhibitors (Sigma). Proteins (10 µl, ∼5–10 µg) were incubated with the artificial fluorogenic substrate 4‐methylumbelliferone‐galactopyranoside (1.5 mmol/liter) resuspended in 100 µl 0.1/0.2 mol/liter citrate/phosphate buffer, pH 4.0, in the presence of 11 µmol/liter AgNO3 at 37 °C for 30 min, followed by treatment with 0.2 M sodium carbonate buffer. Fluorescence of liberated 4‐MU was measured on the 1420 Multilabel Counter Victor 3. Free 4‐methylumbelliferone (4‐MU; Sigma) was used as a standard to calibrate β‐galactosidase activity. Results were normalized with protein concentration.
Primary Fibroblast Culture and GALC Activity Assay
Human fibroblasts derived from two patients and two unaffected healthy donors (GM06806, GM04913, GM00041, GM08333; Coriell Institute) were seeded at a density of 10,000 cells/cm2 in growth medium (DMEM, 15% FBS, 2 mM L‐glutamine, nonessential amino acids, penicillin/streptomycin 100 U/ml; Thermo Scientific, Pleasanton, CA). After 2 days, the medium was replaced and changed daily with growth medium supplemented with supernatant derived from cells overexpressing GALC or GALC‐AErdb and from cells transfected with the sole vector as a control. Sister cultures were also treated with 2.5 mM M6P. This treatment was carried out in duplicate for 3 days, after which the cells were washed twice with PBS, collected, pelleted, and resuspended in distilled H2O for GALC activity analysis. Cell suspensions were sonicated (three pulses, 3 sec each, 30% intensity) and used to perform the GALC activity assay, as described by Wiederschain et al. (1992). Briefly, 10 μl lysate was added to 20 μl of a substrate solution containing 6‐hexadecanoylamino‐4‐methylumbelliferyl‐β‐D‐galactoside (HMU‐β‐GAL), mixed, and incubated for 17 hr at 37 °C. After incubation, the reaction was terminated with a solution containing 0.2% SDS and Triton X‐100, pH 10.7, and the fluorescence measured (ex. 370 nm, em. 535 nm) by fluorometry. Results were normalized for protein content.
Animals
Female BoyJ mice (B6.SJL‐Ptprca Pepcb/BoyJ; RRID:IMSR_JAX:002014) at age ∼6–8 weeks were purchased from the Jackson Laboratory. Heterozygous twitcher (GALC+/−) mice on a congenic C57BL/6 background (RRID:IMSR_JAX:000845) were kindly provided by Dr. Steven J. Gray in Gene Therapy Center, University of North Carolina at Chapel Hill (UNC). The mouse colony was maintained under the supervision of T.K., and all procedures were approved by the Institutional animal care and use committee of UNC (IACUC 13‐195.0). Genotyping was carried out by PCR with clipped toe DNA's before postnatal day 8 (date of birth counted as day 0). Briefly, the toes were lyzed in 25 mM NaOH/0.2 mM EDTA at 98 °C for 90 min, followed by neutralization with same volume of 40 mM Tris (pH 5.5). PCR (98 °C 3 min, followed by 40 repeated cycles of 98 °C 10 sec, 62 °C 15 sec, 72 °C 20 sec) was performed with toe DNA and primer pair (left primer 5′‐CACACAACCCAGTTTACTCAACC‐3′, right primer 5′‐GATGGCCCACTGTCTTCAGG‐3′; Precision Melt Supermix; Bio‐Rad, Hercules, CA). Melting curve of knockouts, wild type, and heterozygotes was determined by using a Roche light Cycle480. (The method was developed by Steven J. Gray in the Gene Therapy Center at UNC.) Endpoint reaching animals were euthanized by CO2 asphyxiation in accordance with UNC IACUC protocol (13‐195.0). Endpoint criteria included: weight loss of more than 25% of body weight, difficulties in drinking, respiratory distress and severe hind leg paralysis.
Brain Immunohistochemistry of L‐Cycloserine‐Treated twi Mice
Animals received 25 mg/kg L‐cycloserine (Sigma Aldrich) subcutaneously three times per week starting at postnatal day 6. At postnatal day 21 mice were sacrificed and perfused with PBS. Brains were isolated, fixed for 24 hr in 10% neutral buffered formalin, then transferred to 70% ethanol. Further processes, including tissue embedding, sectioning, HE staining, immunohistochemistry staining, imaging, and analysis, were performed in the UNC Translational Pathology Laboratory. CI‐M6PR rabbit monoclonal Ab (1:100; Cell Signaling Technology, Danvers, MA) was used for immunostaining. Data were analyzed using the eSlide Manager and Image Scope from Leica Biosystems (Buffalo Grove, IL).
Bone Marrow Transplantation
Animals in experimental groups were conditioned with busulfan (Bu; Sigma; injected intraperitoneally [i.p.] (25 mg/kg) at postnatal day 8. On the next day, fresh donor bone marrow (BM) cells from BoyJ mouse tibia, femur, and pelvis were isolated, counted and resuspended in PBS. Each conditioned animal received approximately 5 × 107 total BM cells via i.p. injection.
For transduced lineage‐negative BM cells, total BoyJ bone marrow were isolated on the same day of mice conditioning. Isolated Lin– cells were transduced with lentiviral vectors (vTK1667 or vTK 1784) at m.o.i. 50 (based on 293T cells) in culture medium with an additional 10 µg/ml rapamycin (Sigma) for ∼14–16 hr. Transduced Lin– cells were washed with PBS and mixed with fresh isolated total BM cells for transplantation. Each Bu‐conditioned mouse received 1 x 106 Lin– cells transduced with vTK1667, 1 x 106 Lin– cells transduced with vTK1784, and 1 x 107 fresh isolated total BM cells from BoyJ mice.
Viral Vector Production, Concentration, and Titration
Lentiviral vector production transient three‐plasmid transfection into 293T cells, and viral concentration were described by Kafri et al. (Kafri et al., 1999).The following plasmid amounts were used: 15 µg transfer cassette, 10 µg ΔNRF packaging construct, and 5 µg of the VSV‐G envelope plasmid pMD.G. Viral vectors were concentrated by sucrose gradient ultracentrifugation. The emergence of replication competent retroviruses (RCRs) was ruled out by three independent safety assays (GFP rescue assay, Tat transfer assay, and Gag transfer assay) as described earlier. Titers of physical vector particles were determined by p24gag ELISA using the National Institutes of Health p24 Antigen Capture Assay kit, as previously documented (Kantor et al., 2009). Infectious unit titers were determined by measuring VCN following infection on 293T cell.
Flow Cytometry Analysis
To measure BM engraftment, mouse BM cells from femur/tibia were flushed by PBS and labeled for flow cytometry analysis. After Fc receptor block (BD Biosciences, San Jose, CA), bone marrow cells were stained at 4 °C for 15 min with the following antibodies (BD Biosciences): V450 mouse‐lineage antibody cocktail, PE‐Cy7 rat anti‐mouse Sca1, APC rat anti‐mouse cKit, PE mouse anti‐mouse CD45.1, and FITC mouse anti‐mouse CD45.2. After fixation with 2% PFA at 4 °C for 15 min, samples were ready for flow analysis.
To quantify the hematopoietic‐derived cells in the CNS, mice were perfused with 25 ml cold PBS before brain dissection. CNS‐infiltrated lymphocyte isolation was performed as described by Reddy et al. (2011). Briefly, isolated brains were treated with collagenase buffer at room temperature for 30 min after homogenization and passed through a 70‐μm cell strainer. The hematopoietic‐derived cells were separated on a Percoll (Sigma; GE17‐0891) gradient. Cells were then blocked with Fc receptor and stained with the following fluorophore‐conjugated antibodies: PE mouse anti‐mouse CD45.1 (BioLegend), FITC mouse anti‐mouse CD45.2 (BD Biosciences; 561874), and rat APC anti‐mouse CD11b (BD Biosciences; 553312). Fixed brain samples were used for flow analysis. All the BM and brain data were acquired using CyAn ADP (Beckman Coulter) and analyzed with Summit5.2.
Antibody Characterization
See Table 1 for a list of all antibodies used. The IGF2R (CD222) antibody detected only the expected protein (∼280 kD) on Western blot of mouse brain and liver. The staining pattern of mouse and human tissues (APT84G1 and APT84E1) and their cellular distribution were identical to those described in the human protein atlas (www.proteinatlas.org). The CD45.1 antibody did not react with mouse cells expressing the CD45.2 alloantigen (manufacturer's data sheet). The CD45.2 antibody exclusively detected mouse cells expressing the CD45.2 alloantigen (manufacturer's data sheet). FACscan analysis of BM cells obtained from either C57B6 or Boy.J exhibited specific staining with CD45.2‐ and CD45.1‐directed antibodies, respectively, without detectable cross‐reactivity. Mac1, Sca‐1, cKit (CD117), and lineage antibody cocktails were routinely tested by FACscan analysis and demonstrated population‐specific staining (manufacturer's data sheet).
Table 1.
Primary Antibodies Used
| Antigen | Description of Immunogen | Source, host species, catalog No., clone or lot No., RRID | Concentration used |
|---|---|---|---|
| Insulin‐like growth factor 2 Receptor (IGF2R/CI‐M6PR/CD222) | Synthetic peptide corresponding to residues surrounding Phe1379 of human IGF‐II receptor | Cell Signaling Technology, rabbit monoclonal, 15128 | 1:100 (immunostaining) 1:1,000 (Western blotting) |
| Leukocyte common antigen Ly5.1 (CD45.1) | SJL mouse thymocytes and splenocytes | Bio Legend, mouse monoclonal, 110708 RRID:AB_313497 | 2 µg/ml/106 cell (flow cytometry) |
| Leukocyte common antigen Ly5.2 (CD45.2) | B10.S mouse thymocytes and splenocytes | BD Biosciences, mouse monoclonal, 561874, RRID:AB_10894189 | 2.5 µg/ml/106 cell (flow cytometry) |
| CD11b/Mac‐1a | Mouse splenic cells CD11b | BD Biosciences, rat monoclonal, 553312, clone M1/70, RRID:AB_398535 | 2 µg/ml/106 cell (flow cytometry) |
| Lymphocyte antigen Ly‐6A/E (Sca‐1) | IL‐2‐dependent mouse T cell line CTL‐L | BD Biosciences, rat monoclonal, 561021, RRID:AB_2034021 | 2 µg/ml/106 cell (flow cytometry) |
| SCFR; stem cell factor receptor CD117 (cKit) | Mouse bone marrow mast cells | BD Biosciences, rat monoclonal, 561074, RRID:AB_10563203 | 2 µg/ml/106 cell (flow cytometry) |
| Lineage antibody cocktail, with isotype control | mouse T‐cell receptor (CD3e), mouse splenic cells (target CD11b); mouse abelson leukemia virus‐induced pre‐B tumor cells (target B222); mouse fetal liver (target Ter119, erythroids marker); Ly6G/6C | BD Biosciences, hamster antibody cocktail, 561301, RRID:AB_10611731 | 20 µl/100 µl/106 cell (flow cytometry) |
Statistical Analysis
All statistical analysis was performed via Student's t‐test in Microsoft Excel.
RESULTS
Novel lentiviral vectors carrying myeloid‐ and erythroid/megakaryocytic‐specific promoters facilitate efficient GALC delivery to and engraftment of hematopoietic stem cells in the twitcher (twi) mouse model of KD.
More than 2,000 therapeutic BMTs in human patients suffering from rare genetic disorders (Boelens et al., 2014) attest to BMT as the cornerstone of all therapeutic regimens for LSD. This reality renders KD patients for whom an HSPC donor could not be identified practically untreatable. The ability of lentiviral vectors to efficiently deliver functional cDNAs to patients’ HSPC and to maintain therapeutic levels of transgene products in their progenitors opens a promising therapeutic avenue that can circumvent the need for a matching donor. However, recent studies focusing on lentiviral vector‐mediated GALC delivery to HSPC indicated that GALC overexpression is toxic to HSPC and inhibits the engraftment in hematopoietic tissues (Gentner et al., 2010; Visigalli et al., 2010). We hypothesized that novel lentiviral vectors from which functional GALC cDNA is expressed under control of either myeloid or erythroid/megakaryocytic promoters would minimize toxic GALC expression in HSPC and would facilitate efficient engraftment of vector‐transduced HSPC in hematopoietic tissues. To test this hypothesis, we developed a series of novel vectors from which the GFP and the firefly luciferase reporter genes, as well a codon‐optimized (CO) human GALC cDNA, were expressed under control of either a myeloid‐ or erythroid/megakaryocyte‐specific promoters. The myeloid promoter was synthesized based on the nucleotide sequence of the 146gp91 promoter described earlier (He et al., 2006; Barde et al., 2011). The erythroid/megakaryocyte promoter IHK was developed and characterized earlier (Moreau‐Gaudry et al., 2001). Lentiviral vectors carrying these cDNAs under control of a CMV promoter served as controls (Fig. 1A). Cell‐specific gene expression from the aforementioned vectors was characterized in relevant cell lines. Specifically, THP‐1 and hexamethylene bisacetamide (HMBA)‐induced mouse erythroleukemia cells (MELs) served as myeloid and erythroid reporter cell lines, respectively. Vector‐transduced 293T cells served as controls. As shown in Figure 1B–D and Table 2, highly cell‐specific gene expression was exhibited for the 146gp91 and the IHK promoters in the relevant THP‐1 and HMBA‐induced MEL cells, respectively. Furthermore, GFP expression under control of the 146gp91 promoter in BM cells expressing the myeloid marker Mac1+ was significantly higher than the expression levels detected in vector‐transduced Lin–SCA1+Kit+ (LSK) HSPC (Fig. 1C). No GFP expression was detected in either total cultured lineage‐negative (Lin–) cells or LSK cells following transduction with lentiviral vectors carrying the IHK promoter. Furthermore, a remarkable increase in GFP and GALC expression from the IHK promoter was observed following HMBA‐mediated differentiation of MEL cells (Fig. 1B). On the other hand, efficient GFP expression from lentiviral vectors carrying the CMV promoter was detected in total cultured Lin– and LSK cells. In addition, CMV containing vectors exhibited significantly higher levels of transgene expression in 293T and LSK cells compared with the expression levels detected in THP‐1 and MAC1+ cells, respectively. Premised on the cell specificity of transgene expression exhibited by these vectors in vitro, we sought to employ lentiviral vectors carrying the cell‐specific promoters as a means to deliver GALC expression cassettes to donor HSPCs. As described below and in Figure 3B and Table 3, vector‐transduced HSPC efficiently engrafted the host BM following conditioning. Thus, BM chimerism of donor cells containing one or more vector genomes expressing the GALC under these cell‐specific promoters was not lower than the chimerism of nontransduced HSPC. These findings indicate that efficient lentiviral vector‐mediated delivery of GALC expression cassettes comprising cell specific promoters can be achieved without posing biosafety concerns associated with altering the host miRNA system.
Figure 1.
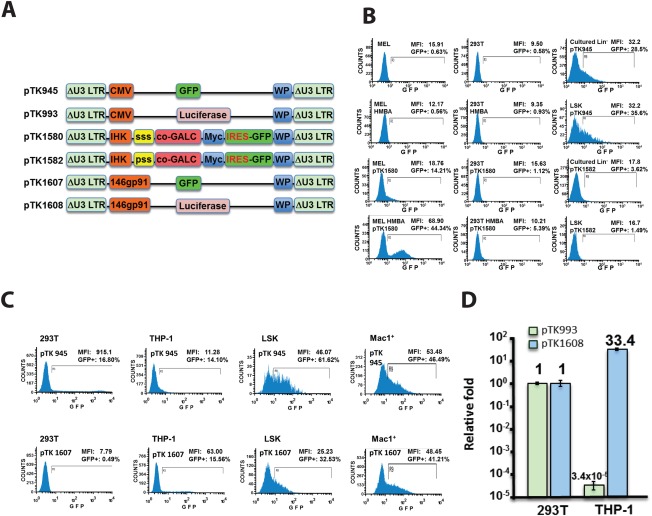
Novel lentiviral vectors support erythroid‐ and myeloid‐specific transgene expression. A: Novel self‐inactivating (SIN) vectors carrying erythroid (IHK)‐ and myeloid (146gp91)‐specific promoters. ssp, Synthetic secretory signal peptide; psp, parental secretory signal peptide; co‐GALC, codon optimized mouse GALC cDNA; myc, myc tag; 146gp91, myeloid‐specific promoter; IHK, erythroid/megakaryocyte‐specific promoter; IRES, internal ribosome entry site; WP, woodchuck hepatitis virus posttranscriptional regulatory element; ΔU3 LTR, Self‐inactivating (SIN) LTR deleted of the parental enhancer promoter. B: Lentiviral vectors carrying the IHK promoter support erythroid‐specific transgene expression in vitro. FACscan analysis of GFP expression in mouse erythroleukemia (MEL) cells, human 293T cells, mouse Lin– cells, and mouse Lin–Sca1+Kit+ (LSK) cells following transduction with lentiviral vectors carrying either the IHK promoter (pTK1580 and pTK1582) or the CMV promoter (pTK945). MEL and 293T cells were analyzed either before or after HMBA‐induced erythroid differentiation. Untransduced MEL and 293T cells served as controls. Percentage of GFP‐positive cells is shown. Mean fluorescence intensity (MFI) presents levels of GFP expression. Note that GFP expression from IHK‐containing lentiviral vectors was detected only in HMBA‐induced MEL cells. C,D: Lentiviral vectors carrying the 146gp91 promoter support myeloid‐specific transgene expression in vitro. C: Lentiviral vectors carrying the GFP reporter gene under control of either the myeloid 146gp91 promoter (pTK1607) or the CMV promoter (pTK945) were employed to transduce human 293T cells, cells of the human THP‐1 monocyte cell line, the above‐mentioned mouse LSK cells, and mouse BM cells expressing the macrophage surface marker Mac1+. FACScan analysis of GFP expression was employed as described for B. Note that high levels GFP expression from the CMV promoter were detected in all target cells excluding the Thp1 cells. CMV‐regulated expression was higher in LSK than in Mac1+ cells. On the other hand, GFP expression driven by146gp91 was high in THP1 and very low in 293T cells. Furthermore GFP expression level in Mac1+ cells was significantly higher than the level of expression detected in LSK cells. D: Lentiviral vectors carrying the firefly luciferase under control of either a myeloid promoter (pTK1607) or a CMV promoter (pTK993) transduced 293T and THP‐1 cells. Luciferase activity in relative light unit (RLU) was normalized per milligram protein and VCN per cell. Luciferase activity generated by each vector in 293T served as a reference baseline. The ratio or fold expression of luciferase activity from these vectors in THP1 cells relative to luciferase expression in 293T cells was calculated. Note that CMV expression in THP‐1 cell was dramatically lower than its expression in 293T cells. On the other hand, luciferase expression per vector genome from the myeloid promoter increased 33‐fold relative to its expression in 293T cells.
Table 2.
Novel Lentiviral Vectors Carrying the IHK Promoter Demonstrate Erythroid Specific Expression of GALC in Differentiated MEL Cellsa
| Galc activity (nmol/hr/mg protein) | |||
|---|---|---|---|
| Cont | pTK1580 | pTK1582 | |
| Naïve MEL | ND | 6.46 | 8.47 |
| Differentiated MEL (HMBA) | ND | 30.70 | 34.03 |
MEL cells were transduced with lentiviral vectors carrying codon‐optimized murine GALC cDNA under control of the erythroid/megakaryocyte‐specific promoter IHK (pTK1580, pTK1582). Vector‐transduced MEL were induced to undergo erythroid differentiated by 5 mM HMBA. GALC activity in vector‐transduced MEL cells prior to and after differentiation was determined and served as a surrogate marker for IHK promoter activity. Note the robust increase in IHK activity following HMBA‐induced differentiation.
Figure 3.
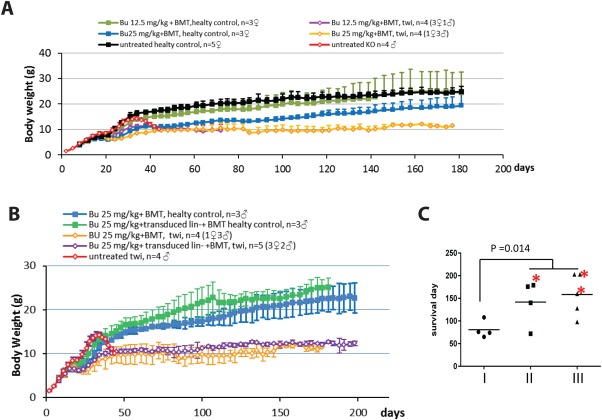
Effects of Bu‐based conditioning protocols on clinical parameters following HSPCT in the twi mouse model of KD. A: Weight gain curves of healthy littermates (control) and twi mice following conditioning protocols that were premised on i.p. injection of either 12.5 mg/kg or 25 mg/kg Bu at postnatal day 8. At postnatal day 9, all conditioned mice were given 5–6 × 107 total BM cells. Mouse weight was measured every 2–3 days. Untreated healthy and twi mice served as controls. B: Weight gain curves of healthy littermates (control) and twi mice following a conditioning protocol in which 25 mg/kg Bu was administered by i.p injection at postnatal day 8. At postnatal day 9, one group of either conditioned healthy mice (controls) or conditioned twi mice was given via i.p. injection 1 × 106 pTK1667‐transduced Lin– cells, 1 × 106 pTK1784‐transduced Lin– cells, and 1 × 107 total bone marrow cells. In addition, one group of either conditioned healthy mice or conditioned twi mice received 5–6 × 107 total BM cells. A group of untreated twi mice served as a control. Mouse weight was measured every 2–3 days. C: Life span of three groups of twi mice (groups I–III) following different conditioning and HSPCT protocols. Mice in group I were conditioned with a low dose (12.5 mg/kg) of Bu and received 5–6 × 107 total BM cells. Mice in group II were conditioned with a high dose (25 mg/kg) of Bu and received 5–6 × 107 total BM cells. Mice in group III were conditioned with a high dose (25 mg/kg) of Bu and received 1 × 106 pTK1667‐transduced Lin– cells, 1 × 106 pTK1784‐transduced Lin– cells, and 1 × 107 total BM cells. Student's t‐test demonstrated a significant increase (n = 4 and 9, P = 0.014) in mouse life span following conditioning with high‐dose Bu compared with the life span of mice conditioned with low‐dose (12.5 mg/kg) Bu. Asterisk indicates that the mice were sacrificed for data collection.
Table 3.
Different Conditioning and HSPCT Protocols and Their Effects on Twi Mouse Life Span, Donor/Host Chimerism in BM and CNS, and VCN in Mouse BMa
| Bu (mg/kg) | Survival study included | Age (days) | LSK | CNS enriched microglial and leukocytes | CNS CD11b | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD45.1 (%) | Ave ± SD | CD45.2 (%) | Ave ± SD | CD45.1 (%) | Ave ± SD | CD45.2 (%) | Ave ± SD | CD45.1 (%) | Ave ± SD | CD45.2 (%) | Ave ± SD | VCN | ||||||
| A | K67L2 | 12.5 | KO, ♀ | 89 | 93.5 | 0.7 | 28.5 | 35.4 | 64.0 | 25.7 | ||||||||
| K68L1 | 12.5 | KO, ♀ | 80 | 94.2 | 1.0 | 38.3 | 26.0 | 58.0 | 26.2 | |||||||||
| K68L4 | 12.5 | KO, ♀ | 77 | 93.6 | 1.2 | 36.5 | 32.3 | 58.6 | 28.2 | |||||||||
| K69L3 | 12.5 | KO, ♂ | 86 | 94.6 | 94.0 ± 0.5 | 0.9 | 1.0 ± 0.2 | 25.8 | 32.3 ± 6.1 | 43.6 | 34.3 ± 7.3 | 57.8 | 59.6 ± 2.9 | 29.2 | 27.3 ± 1.7 | |||
| B | K51R2 | 12.5 | KO, ♀ | Y | 74 | 90.0 | 2.1 | |||||||||||
| K55R3 | 12.5 | KO, ♀ | Y | 65 | 89.3 | 3.9 | ||||||||||||
| K55R1 | 12.5 | KO, ♀ | Y | 76 | 91.6 | 4.1 | ||||||||||||
| K65R2 | 12.5 | KO, ♂ | Y | 108 | 98.9 | 92.5 ± 4.4 | 0.7 | 2.7 ± 1.6 | ||||||||||
| C | K65R4 | 12.5 | He, ♂ | 202 | 97.5 | 0.2 | 14.5 | 71.2 | 7.6 | 88.5 | ||||||||
| K65L1 | 12.5 | He, ♂ | 202 | 97.6 | 0.5 | 12.3 | 76.2 | 6.2 | 89.8 | |||||||||
| K67L1 | 12.5 | WT, ♀ | 103 | 41.3 | 54.8 | 4.8 | 85.6 | 2.0 | 96.6 | |||||||||
| K69L2 | 12.5 | WT, ♂ | 87 | 93.7 | 82.5 ± 27.5 | 0.4 | 13.9 ± 27.2 | 16.6 | 12.0 ± 5.1 | 62.9 | 74.0 ± 9.5 | 7.9 | 5.8 ± 2.6 | 84.0 | 89.8 ± 5.1 | |||
| D | K67R4L4 | 25 | KO, ♀ | 90 | 96.2 | 0.5 | 40.8 | 24.9 | 53.5 | 28.8 | ||||||||
| K68R1 | 25 | KO, ♂ | 78 | 95.1 | 0.6 | 40.5 | 25.3 | 46.7 | 40.1 | |||||||||
| K68L3 | 25 | KO, ♀ | 80 | 96.8 | 1.1 | 37.9 | 21.9 | 57.3 | 23.9 | |||||||||
| K69R4 | 25 | KO, ♀ | 80 | 92.7 | 0.2 | 50.9 | 19.5 | 62.2 | 26.2 | |||||||||
| K69L1 | 25 | KO, ♂ | 87 | 96.5 | 95.4 ± 1.7 | 0.0 | 0.5 ± 0.4 | 27.2 | 39.5 ± 8.5 | 41.9 | 26.7 ± 8.8 | 54.8 | 54.9 ± 5.7 | 24.1 | 28.6 ± 6.7 | |||
| E | K48L2 | 25 | KO, ♂ | Y | 179* | 96.8 | 0.1 | |||||||||||
| K48L3 | 25 | KO, ♂ | Y | 176 (dead) | 97.7 | 0.8 | ||||||||||||
| K50R2 | 25 | KO, ♂ | Y | 140 | 95.0 | 1.5 | ||||||||||||
| K58R3 | 25 | KO, ♀ | Y | 72 | 91.3 | 95.2 ± 2.8 | 1.0 | 0.9 ± 0.6 | ||||||||||
| F | K58R2 | 25 | WT, ♀ | 203 | 97.1 | 0.4 | 23.9 | 56.8 | 7.8 | 83.2 | ||||||||
| K50R1 | 25 | He, ♂ | 264 | 98.9 | 0.5 | 17.6 | 62.9 | 7.1 | 85.6 | |||||||||
| K59R1 | 25 | WT, ♂ | 221 | 98.0 | 0.4 | 10.4 | 73.9 | 4.4 | 92.3 | |||||||||
| K67R1L4 | 25 | WT, ♀ | 103 | 94.3 | 97.1 ± 2.0 | 0.1 | 0.3 ± 0.1 | 11.3 | 15.8 ± 6.3 | 78.2 | 67.9 ± 9.8 | 5.6 | 6.2 ± 1.6 | 90.7 | 87.9 ± 4.2 | |||
| G | K53R3 | 25 | KO, ♂ | Y | 160* (vet) | |||||||||||||
| K59R4L2 | 25 | KO, ♂ | Y | 203* | 96.5 | 0.0 | 37.4 | 17.0 | 64.4 | 19.6 | 0.46 | |||||||
| K53L1 | 25 | KO, ♀ | Y | 98 (dead) | 0.93 | |||||||||||||
| K59L1 | 25 | KO, ♀ | Y | 128 | 96.6 | 0.2 | 1.11 | |||||||||||
| K59L2 | 25 | KO, ♀ | Y | 203 (dead) | 94.6 | 95.9 ± 1.1 | 0.0 | 0.1 ± 0.1 | 1.24 | |||||||||
| H | K59R4L1 | 25 | WT, ♂ | 201 | 95.8 | 0.4 | 19.9 | 64.7 | 10.4 | 81.9 | 0.63 | |||||||
| K64R4 | 25 | WT, ♂ | 182 | 96.1 | 0.0 | 18.3 | 51.7 | 6.1 | 84.1 | 0.02 | ||||||||
| K64L3 | 25 | WT, ♂ | 182 | 96.8 | 0.2 | 30.4 | 33.5 | 18.6 | 64.2 | 0.35 | ||||||||
| K53L3 | 25 | WT, ♀ | 241 | 91.6 | 95.1 ± 2.3 | 0.0 | 0.2 ± 0.2 | 11.0 | 19.9 ± 8.0 | 62.0 | 53.0 ± 14.1 | 5.0 | 10.0 ± 6.2 | 87.7 | 79.5 ± 10.5 | 0.82 | ||
| BoyJ | — | WT, ♀ | ∼150 | 96.8 | 0.1 | 80.7 | 0.6 | 93.3 | 0.2 | |||||||||
| K53R4L1 | — | WT, ♀ | 256 | 0.2 | 94.8 | 0.1 | 84.0 | 0.1 | 96.2 | |||||||||
The dose of Bu employed in the conditioning protocol is indicated. Mice used for the survival study are marked. Mice in groups A–F received 5–6 × 107 total donor BM cells. Mice in groups G and H received 1 × 106 pTK1784‐transduced Lin– cells, 1 × 106 pTK1667‐transduced Lin– cells, and 1 × 107 donor total BM cells. “Age” denotes the age of the mouse at the time of death or when sacrificed experimentally. Only mice involved in the “survival study” died from natural causes or were euthanized in accordance with UNC IACUC protocol. “Dead” indicates that the specific mouse was found dead in its cage; “vet”: denotes that tissues of euthanized mouse were not available; asterisk indicates the mice sacrificed for data collection. LSK, bone marrow Lin–Sca1+ckit+ cells; CNS enriched microglial and leukocytes were isolated by percoll gradient from perfused brain.
L‐Cycloserine Enhances IGF2R Expression in Twi Mouse CNS
The cornerstone of cell‐ and gene therapy‐based therapeutic protocols for LSDs is the phenomenon of “cross‐correction,” in which donor cell‐secreted, M6P‐comprising enzymes are taken up by host enzyme‐deficient cells (either in the periphery or in the CNS). Sufficient expression of IGF2R, the CI‐M6PR in treated cells, is essential to facilitate this therapeutic pathway. However, supraphysiological levels of functional GALC following viral vector‐mediated gene delivery (either alone or in combination with BMT) to twi mouse CNS and PNS resulted in only moderate prolongation of average mouse life span (Lin et al., 2007). Similarly, a substrate reduction approach using L‐cycloserine, an inhibitor of sphingolipids synthesis (which should also decrease accumulation of toxic undegraded metabolites such as psychosine), failed to prolong twi mouse life span (Hawkins‐Salsbury et al., 2015). However, by combining L‐cycloserine treatment with BMT and AAV vector‐based GALC delivery, Hawkins‐Salsbury et al. dramatically prolonged the life span of treated twi mice. Premised on these studies, we theorized that an L‐cycloserine‐mediated increase in IGF2R expression facilitated uptake of functional GALC secreted from donor HSPC and from AAV vector‐transduced cells by twi cell populations that naturally express insufficient levels of the M6P receptor. To test this hypothesis, we employed a semiquantitative immunohistochemistry analysis to characterize IGF2R levels in either L‐cycloserine treated or naïve twi mice. As shown in Figure 2A,B, L‐cycloserine treated‐mice exhibited significantly higher levels of cellular IGF2R(Student's t test, n = 3, P = 0.006). These data further support the notion that insufficient IGF2R expression limits the efficacy of BMT and gene therapy‐based therapeutic protocols to cure KD.
Figure 2.
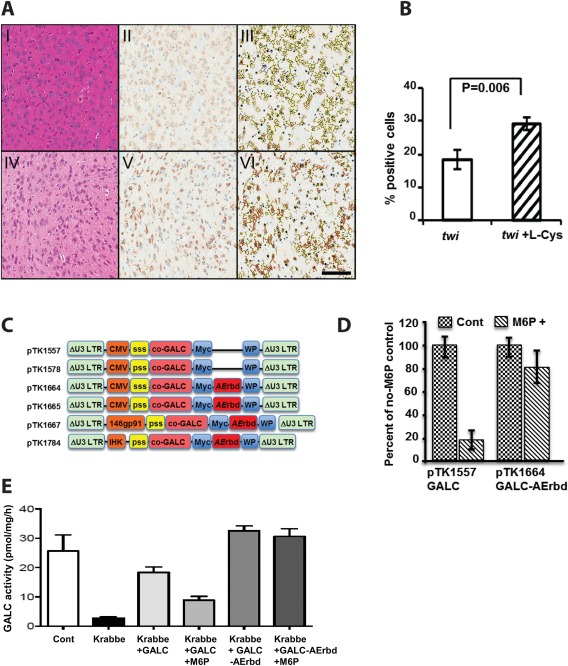
Induction of IGF2R expression in mouse CNS by L‐cycloserine and efficient uptake of a novel GALC‐AErbd fusion protein via an M6P‐independent pathway. A,B: Analysis of IFG2R expression in L‐cycloserine (L‐cys)‐treated and untreated twi mouse CNS. A: Representative sections from untreated (I–III) and L‐cys‐treated (IV–VI) twi mouse diencephalon. Hematoxylin and eosin (HE) staining (I, IV) shows no significant morphological differences between L‐cys‐treated and untreated mice. Immunohistochemical (IHC) staining (II, V) shows enhanced expression of IGF2R in L‐cys treated mouse diencephalon. To semiquantify expression of IGF2R in treated and untreated twi brain, the IHC‐stained sections were masked by red (3 + positive), orange (2 + positive), yellow (1 + positive), and blue (no signal; III, VI). All panels are X20. Scale bar = 100 µm. B: Semiquantification of IGF2R expression using IHC‐stained sagittal brain sections of L‐cys treated and untreated twi mice. Red and orange signals were quantified as positive cells. The percentage of positive cells between two groups were compared by two‐tailed Student's t‐test, N = 3 in each group, P = 0.006. C: Depiction of the lentiviral vectors used to characterize uptake of the novel GALC‐AErdb and its usage. ssp, Synthetic secretory signal peptide; psp, parental secretory signal peptide; co‐GALC, codon optimized mouse GALC cDNA; myc, myc tag; AErbd, ApoE receptor binding domain; 146gp91, myeloid‐specific promoter; IHK, erythroid/megakaryocyte‐specific promoter; WP, woodchuck hepatitis virus posttranscriptional regulatory element; ΔU3 LTR, SIN LTR deleted of the parental enhancer promoter. D: Uptake of GALC and GALC‐AErbd by 293T cells via the IGF2R and the LRP‐1 pathways. 293T cells were cultured either in the presence or in the absence of 1mM M6P (a competitive inhibitor of protein uptake via the IGF2R pathway) in conditioned media containing either GALC (generated by pTK1557‐transduced cells) or GALC‐AErbd (generated by pTK1664‐transduced cells). The GALC activity in these 293Tcells served as a surrogate marker for GALC uptake. Cellular GALC activity in the absence of M6P served as a reference value, considered to be 100%. The ratio of cellular GALC activity in the presence of M6P relative to GALC activity of the same protein without M6P was calculated. Uptake of GALC (pTK1557) was efficiently inhibited by the presence of 1 mM M6P, whereas uptake of the GALC‐AErbd fusion protein (pTK1664) was only slightly affected. E: Uptake of GALC and GALC‐AErbd by healthy and KD human fibroblasts cells via the IGF2R and the LRP‐1 pathways. Human fibroblasts isolated from healthy donors (Cont) and KD patients (Krabbe) were cultured either in the presence or absence of M6P (a competitive inhibitor of protein uptake via the IGF2R pathway) in conditioned media containing either GALC (generated by pTK1578‐transduced cells) or the GALC‐AErbd (generated by pTK1665‐transduced cells). The levels of GALC activity in naïve KD and healthy human fibroblasts are shown. Note that the presence of 2.5 mM M6P efficiently inhibited GALC uptake, whereas GALC‐AErbd uptake was not significantly affected in the presence of M6P.
Efficient M6P‐Independent Uptake of a Novel GALC‐ApoE1 Receptor Binding Domain (GALC‐AErbd) Fusion Protein
Intrigued by the notion that low levels of IGF2R in various CNS and PNS cell populations potentially limit the efficacy of HSPC transplantation and gene therapy protocols for treating KD, we sought to facilitate cellular uptake of secreted GALC independently of the M6P pathway. To this end, we developed a novel fusion protein comprising the GALC and the AErbd.
This approach was premised on earlier studies in which fusion of the AErbd to the lysosomal protein α‐L‐iduronidase (IDUA) facilitated uptake of the novel protein by blood–brain barrier (BBB) endothelial cells, transcytosis to the CNS, and delivery to astrocytes and neurons throughout the cerebral cortex via the LRP‐1 pathway (independently of the M6P pathway) (Wang et al., 2013; Dai et al., 2014; El‐Amouri et al., 2014). The novel GALC‐AErbd cDNA was cloned into lentiviral vectors under control of a CMV, an IHK, or a 146gp91 promoter (Fig. 2C). To characterize the efficiency of LRP‐1‐mediated GALC uptake, 293T cells were transduced with lentiviral vectors expressing either GALC or GALC‐AErbd cDNAs. Conditioned media were collected from vector‐transduced cells and applied to 293T cells and human fibroblasts derived from KD patients, in either the absence or the presence of M6P. Cellular activity of GALC (Wiederschain et al., 1992) in conditioned media‐treated cells served as a surrogate marker for cellular uptake of GALC and GALC‐AErbd. Activity of GALC in untreated healthy and KD human fibroblasts served as reference controls. As shown in Figure 2D,E the presence of M6P, a competitive inhibitor of the IGF2R‐mediated protein uptake pathway, efficiently inhibited uptake of conditioned media GALC by both 293T cells and human KD fibroblasts. On the other hand, M6P treatment had a minimal yet measurable effect on cellular uptake of GALC‐AErbd. These data indicate that cellular uptake of the novel GALC‐AErbd fusion protein can be mediated independently by the IGF2R‐ and the LRP‐1‐specific pathways. This attribute of GALC‐AErbd broadens the spectrum of target cells that can benefit from the “cross‐correction” phenomenon in current and future therapeutic regimens for KD (Polavarapu et al., 2007; Lillis et al., 2008).
Twi mouse life span following HSPCT correlates with the aggressiveness of the preconditioning protocol.
Earlier studies suggested that both host conditioning with either irradiation or chemotherapy and damage to the CNS are required for efficient donor HSPC engraftment in rodent brains (Priller et al., 2001; Mildner et al., 2007; Davoust et al., 2008).
Note that Bu was the major chemotherapy agent employed in earlier lentiviral/HPCT‐based human clinical trials of neurodegenerative diseases (Cartier et al., 2009; Aiuti et al., 2013) as well as in allogeneic HPCT for KD (Escolar et al., 2005), yet irradiation‐conditioning protocols have been employed in most HSPCT‐based preclinical trials investigating KD (Lin et al., 2007; Gentner et al., 2010; Hawkins‐Salsbury et al., 2015). This limits our ability to accurately interpret the therapeutic outcomes described in the above‐mentioned preclinical studies. Furthermore, the correlation between the levels of donor/host chimerism in the bone marrow and the therapeutic benefits of HPCT in KD has not been established. Thus, it is imperative that we further characterize the effects of the conditioning protocol and host health status on the therapeutic outcome of HSPCT in the twi mouse model of KD. To this end, as shown in Figure 4A and Table 3, healthy donor total BM cells (BoyJ expressing CD45.1) were transplanted into host mice (C57B6 expressing CD45.2) including either twi (GALC–/–; groups A, B, D, and E) or healthy hosts (groups C and F). Note that the healthy host population comprised heterozygous twi (GALC+/–) and wild type mice (GALC+/+). Specifically, at postnatal day 8, host mice were conditioned by i.p. injection of either 12.5 (groups A–C) or 25 mg/kg Bu (groups D–H) and 1 day later were given naïve donor total bone marrow cells (5–6 × 107 cells/mouse, groups A–F) i.p. Mouse body weight was measured every 2–3 days. To facilitate characterization of donor cells engraftment in twi host CNS, mice were sacrificed at postnatal days 77–90 (groups A and D). All other twi mice died naturally or were euthanized in accordance with UNC Institutional Animal Care and Use Committee (IACUC) protocol. As shown in Figure 3A,B, untreated twi mice maintained close to normal body weight until week 5, after which dramatic weight loss followed by death occurred in all mice before postnatal day 46. Healthy BMT‐treated mice, which were conditioned with Bu 12.5 mg/kg demonstrated a body weight increase curve that was not significantly different from the curve of untreated healthy mice. However, conditioning of healthy mice with 25 mg/kg Bu inhibited gain in mouse body weight for approximately 2 weeks, after which a continuous increase in body weight was observed. However, at postnatal day 180, the average body weight of this group of mice was still significantly lower than the body weight of untreated or 12.5 mg/kg Bu‐treated mice. For the first 7 weeks of life, the body weight curve of twi mice treated with 25 mg/kg Bu was not significantly different from the body weight curve of their healthy counterparts, which received the same treatment. Interestingly, this group of mice failed to gain weight after postnatal week 7. The maximal average body weight of these mice was 12.5 g; however, in contrast to untreated twi mice, death in this group was not preceded by body weight loss. Because of the milder conditioning protocol, twi mice treated with 12.5 mg/kg of Bu demonstrated early gains in body weight, which started to decline at postnatal week 5 and stabilized within 1 week. The average body weight at death of this group of mice was not significantly different from the average body weight of their twi counterparts that were conditioned with 25 mg/kg of Bu. On the other hand, a highly significant difference was observed in the effects of the two conditioning protocols on the life span of HSPCT‐treated twi mice. As shown in Figure 3C, the average life span of twi mice conditioned with 25 mg/kg Bu was significantly longer than the average life span of those conditioned with 12.5 mg/kg Bu (Student's t‐test, n = 4 and 9, P = 0.014).
Figure 4.
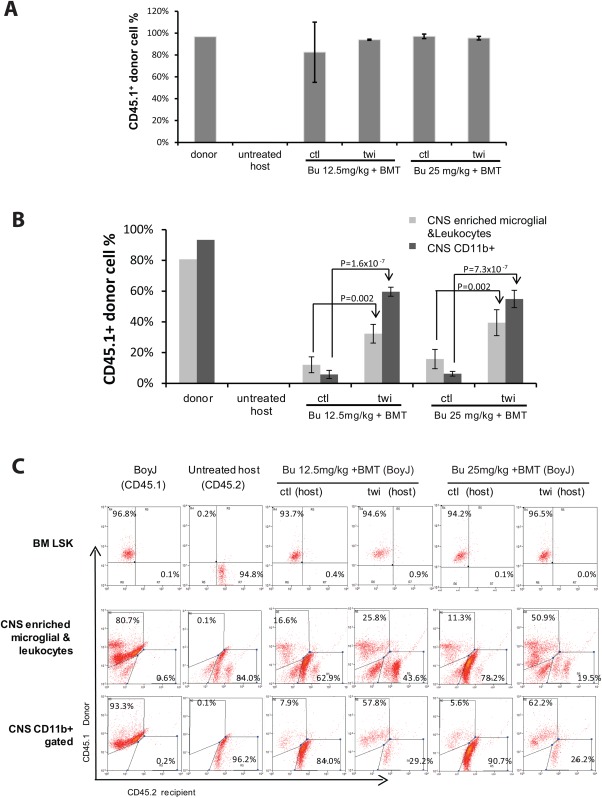
Engraftment efficiency in control and twi mouse BM and CNS following BMT with Bu‐based conditioning protocols. A: Engraftment efficiency of BoyJ donor cells (expressing the CD45.1 marker) in healthy and twi mouse BM following BMT and conditioning protocols, using either 12.5 or 25 mg/kg Bu. Percentage of donor cells in LSK cell populations isolated from these treated mice was determined by FACscan analysis with antibody staining directed to the donor‐specific CD45.1 marker. Note that no significant differences were observed among the different treatment groups. LSK cells isolated from either untreated BoyJ donor (CD45.1) and untreated healthy host (CD45.2) mice served as positive and negative controls. B: Engraftment efficiency of BoyJ donor cells (expressing the CD45.1 marker) in healthy and twi mouse CNS following BMT and conditioning protocols with either 12.5 or 25 mg/kg Bu. Percentage of donor cells in either microglia‐ or leukocyte‐enriched cell populations or CD11b+ cells from treated mouse CNS was determined by FACscan analysis with antibody staining directed to the donor‐specific CD45.1 marker. Note that significant differences in engraftment of donor cells (CD45.1) in treated mouse CNS were observed among the different treatment groups (in contrast to the lack of differences in engraftment efficiency of donor cells in host BM). CNS cells isolated from either untreated BoyJ donors (CD45.1) or untreated healthy host (CD45.2) mice served as positive and negative controls. Student's tests were performed between indicated groups, n = 4 for all groups, P values are listed in the figure. C: FACscan analysis of donor/host chimerism in host BM and CNS following low‐ and high‐dose Bu conditioning and HSPCT in either healthy control (ctl) or twi mice. All host mice (healthy control and twi) expressed the CD45.2 marker. All donor cells derived from BoyJ mice expressd the CD45.1 marker. Top row shows FACscan analysis of CD45.1 (donor) and CD45.2 (host) protein expression in Lin–Sca1+Kit+ (LSK) cells isolated from either healthy (ctl) or twi host mice following conditioning with either low‐dose (12.5 mg/kg) or high‐dose (25 mg/kg) Bu. Untreated BoyJ CD45.1 and the host control (CD45.2) served as references to CD45.1 and CD45.2 expressing cells, respectively. Note highly efficient engraftment in healthy and twi hosts receiving either low‐ or high‐dose Bu‐based conditioning. Middle row shows FACscan analysis of CD45.1 (donor) and CD45.2 (host) protein expression in CNS enriched with microglia and leukocytes isolated from either healthy (ctl) or twi host mice following conditioning with either low‐dose (12.5 mg/kg) or high‐dose (25 mg/kg) Bu. Untreated BoyJ CD45.1 and the host control (CD45.2) served as references to CD45.1 and CD45.2 expressing cells, respectively. Note that a higher level of chimerism was obtained in twi host mice. High‐dose Bu‐based conditioning appears to increase chimerism in twi mouse. Bottom row shows FACscan analysis of CD45.1 (donor) and CD45.2 (host) protein expression in CD11b+ expressing cells isolated from either healthy (ctl) or twi host mice following conditioning with either low‐dose (12.5 mg/kg) or high‐dose (25 mg/kg) Bu. Untreated BoyJ CD45.1 and the host control (CD45.2) served as references to CD11b+ gated cells expressing only CD45.1 and CD45.2, respectively.
The average life span of twi mice following HSPCT did not correlate with the level of donor/host chimerism in the BM.
Next we sought to investigate whether the longer life span of twi mice following high‐dosage of Bu (25 mg/kg) conditioning protocol was secondary to a more efficient engraftment of donor HSPC in the host BM compartment. To this end mouse pluripotent hematopoietic Lin–Sca1+Kit+ (LSK) cells isolated from bone marrow of all mouse groups A–H were FACscan analyzed for CD45.1 (donor)‐ and CD45.2 (host)‐expressing cells. As shown in Figure 4A,C and Table 3, with the exception of a single mouse (group C mouse K67L1) all mice in all treatment groups exhibited high levels of chimerism (>90%). These data strongly suggest that the levels of BM chimerism cannot explain the difference in average twi mouse life span following conditioning with high and low Bu doses. No difference in chimerism was observed between wild‐type and twi mouse hosts.
The levels of donor/host cell chimerism in twi mouse hosts CNS were significantly higher than those found in wild‐type hosts’ CNS.
To understand better the mechanism by which the conditioning protocol affects the life span of BMT‐treated twi mice, we characterized donor/host chimerism in the CNS of treated mouse groups A, C, D, and F. To this end, either CNS enriched microglia and leukcocytes or CD11b‐gated cells were analyzed by FACscan for CD45.1(donor) or CD45.2 (host) expression. As shown in Figure 4B,C and Table 3, no significant differences in the levels of donor/host chimerism in the CNS were found between low (12.5 mg/kg, group A)‐ and high (25 mg/kg, group D)‐dose Bu‐treated twi mice. On the other hand, the level of chimerism in twi mouse CNS was always significantly higher than the chimerism level in the CNS of their healthy counterparts (Stuendent's t test, n = 4, P = 0.002 for CNS enriched microglia and leukocytes and P = 1.6 × 10‐7 for CD11b‐gated cells in low‐dose groups; P = 0.002 for CNS enriched microglia and leukocytes and P = 7.3 x 10‐7 for CD11b‐gated cells in high‐dose groups). These data suggest that KD‐induced pathology may enhance the efficiency of engraftment and colonization of hematopoietic‐derived donor cells in the host CNS.
Transduction of HSPC with lentiviral vectors carrying the GALC‐AErbd cDNA, under the control of either a myeloid‐ or an erythroid/megakaryocyte‐specific promoter, does not reduce engraftment efficiency in host BM.
To increase the efficacy of BMT for KD, we sought to overexpress the novel GALC‐AErbd protein in HSPC‐derived cells. We expected that high levels of donor cell‐secreted GALC‐AErbd, whose uptake by GALC‐deficient host cells is mediated by two independent pathways (M6P and LRP‐1 pathways), would further extend the life span of BMT‐treated twi mice. To this end, Lin– cells isolated from donor BoyJ (CD45.1) mice were transduced at an m.o.i. of 50 with lentiviral vectors from which the GALC‐AErbd cDNA is expressed under control of either the myeloid‐specific promoter 146gp91 (pTK1667) or the erythroid/megakaryocyte‐specific promoter IHK (pTK1784). At postnatal day 8, twi and wild‐type control mice were conditioned by i.p. injection of Bu (25 mg/kg). At postnatal day 9 mice were injected i.p. with the these vector‐transduced cells (1 × 106 cells/mouse of pTK1667‐transduced Lin– cells and 1 × 106 cells/mouse of pTK1784 vector‐transduced Lin– cells) along with 107 naïve total BM cells (Table 3, groups G and H). Wild‐type and twi mice receiving naïve total BM cells (5–6 × 107 cells/mouse) served as controls. Mouse weight was determined every 2–3 days. Mice were euthanized according to UNC IACUC protocol. VCN in mouse total BM cells was determined by qPCR. In addition donor/host chimerism in isolated LSK cells and in treated mouse CNS were determined by FACScan analysis as described above. As shown in Figure 3C, HSPC transduced with lentiviral vectors carrying the GALC‐Arbd did not extend the life span of BMT‐treated twi mice beyond the average life span of these mice following BMT with nontransduced donor HSPC. Similarly, the weight gain curve of either twi or wild‐type mice treated with vector‐transduced HSPC was not significantly different from the weight gain curve of their counterparts, which received donor nontransduced total BM cells (Fig. 3B). These data indicated that, under the conditions described above, lentiviral vector delivery of GALC‐AErbd to donor HSPC did not increase the therapeutic benefits of BMT to twi mice. To evaluate the efficiency of lentiviral vector transduction and engraftment of vector‐transduced HSPC, we characterized donor/host chimerism and VCN in the BM of wild‐type and twi mice. As shown in Table 3, the efficiency of engraftment and colonization of HSPC transduced with lentiviral vectors from which the GALC‐AErbd is expressed under the control of erythroid‐ and myeloid‐specific promoters is similar to that of BM engraftment of nontransduced HSPC. VCN in BM of twi mice (ranging between 0.46 and 1.24) was higher than the VCN in BM of wild‐type mice (ranging between 0.02 and 0.82). Altogether these data indicated that HSPC transduced with lentiviral vectors carrying GALC under control of cell‐specific promoters could colonize the BM compartment in wild‐type and twi mice. Note that in earlier studies significantly higher VCN (five to eight copies) in host mouse BM was required to moderately prolong the life span of GALC deficient mice (Gentner et al., 2010; Ungari et al., 2015). Clearly, additional studies using higher m.o.i. to achieve higher VCN in host BM are required to accurately evaluate the therapeutic potential of the GALC‐AErbd fusion protein.
DISCUSSION
Allogeneic HSPCT with HLA‐matched healthy donor HSPC has been the standard therapeutic approach for a plethora of rare metabolic genetic disorders (such as LSDs, including KD). Since the early 1980s more than 2,000 HSPCTs were performed to treat genetic disorders (Boelens et al., 2014). The degree of success of this approach is mainly disease specific. With regard to LSDs, the spectrum of endpoint therapeutic benefits ranges between largely inefficient even in animal models (Heldermon et al., 2013) to highly beneficial in human patients (Boelens et al., 2013). However, even in the most successful scenario—HSPCT applied to MPS‐IH (Hurler disease) patients—a complete cure is far from being achieved (Aldenhoven et al., 2015). Treating presymptomatic KD patients with HSPCT preserves most of their cognitive function. However, progressive deterioration of the patients’ PNS severely affects gross motor function and expressive language (Escolar et al., 2005; Duffner et al., 2009). We assert that the limited efficacy of the current HSPCT protocols fior curing KD is linked to the basic biological processes on which the HSPCT therapeutic approach for KD is premised. These include the ability of donor HSPC to efficiently colonize the host in proximity to cell populations that are vulnerable to GALC deficiency. Furthermore, the therapeutic effects of HSPCT are heavily dependent on the “cross‐correction” phenomenon, so it is imperative that these host target populations express sufficient levels of IGF2R to facilitate efficient uptake of donor‐derived GALC. Overall, this study was premised on the notion that two independent mechanisms potentially limit the efficacy of current HSPCT protocols at curing KD. Specifically, we hypothesized that both inefficient conditioning protocols (which limit the number and biodistribution of donor‐derived microglia in host CNS) and inefficient host cell uptake of donor cell‐secreted GALC (resulting from lack of sufficient IGF2R) should be addressed to maximize the therapeutic benefits of HSPCT protocols for KD patients.
In addition, we have characterized some of the obstacles and potential solutions associated with the strategy of combining lentiviral vector delivery of GALC with HSPCT‐based therapy for KD. The ability of lentiviral vectors to deliver and maintain long‐term transgene expression in HSPC and their progenies opens new directions in HSPCT‐based therapy for genetic disorders in general and for KD in particular (Miyoshi et al., 1999; Cartier et al., 2009; Aiuti et al., 2013; Biffi et al., 2013). Specifically, lentiviral vectors carrying functional GALC to autologous HSPC circumvent the need to identify an HLA‐matched donor. In addition, overexpression of GALC in vector‐transduced cells should in theory broaden the spectrum of target cells that can benefit from the phenomenon of cross‐correction. However, in recent preclinical studies using mouse models of KD, lentiviral vector‐mediated GALC delivery to HSPC revealed potential limitations in this approach. Overall only a very mild effect on GALC‐deficient mice life span could be achieved, and only in mice showing very high VCN in engrafted donor cells (Gentner et al., 2010; Ungari et al., 2015). Furthermore, GALC cytotoxicity, which inhibited engraftment of vector‐transduced HSPC, necessitated incorporation of an HSPC‐specific miRNA target sequence to minimize GALC expression in and to facilitate efficient engraftment of vector‐transduced HSPC. Notably, a heterogeneous cell population with an average VCN of 8 usually contains cells with VCN higher than 25. The combination of very high VCN and vector design comprising the target sequence to host miRNA raises biosafety concerns. These include the possibility of inadvertently altering the host miRNA system via the “sponge effect” described earlier in which high levels of exogenous miRNA target sequence served as a competitive inhibitor decoy that suppressed specific miRNA function (Ebert and Sharp, 2010). We assert that weaknesses in the lentiviral vector system should be addressed by further vector development rather than by manipulating host biological pathways. To this end, we developed novel lentiviral vectors from which the reporter genes GFP and firefly luciferase (as well as the GALC and the GALC‐AErbd fusion protein) were expressed under control of either an erythroid/megakaryocyte (IHK)‐ or a myeloid (146gp91)‐specific promoter (Moreau‐Gaudry et al., 2001; He et al., 2006; Barde et al., 2011). The novel vectors demonstrated cell‐specific expression in vitro and facilitated efficient GALC‐AErbd delivery to and engraftment of vector‐transduced HSPC. Donor/host chimerism in host BM following transplantation with vector‐transduced cells was equivalent to the chimerism achieved following engraftment of untransduced HSPC. VCN of the novel vectors in twi mouse BM ranged from 0.46 to 1.24. Because, in addition to the vector‐transduced Lin– cells, the transplanted donor cell population included 107 total BM cells, we speculate that VCN in engrafted transduced cells was higher than that described above.
The mild effect of high lentiviral VCN on the average life span of HSPCT‐treated GALC‐deficient mice (Gentner et al., 2010; Ungari et al., 2015) suggested that high levels of vector/donor cell‐derived functional GALC would not cure KD patients. This phenomenon was in line with similar studies in which supraphysiological levels of AAV vector‐delivered GALC failed to significantly alter the life span of twi mice (Lin et al., 2007). We theorized that lack or insufficient levels of IGF2R in GALC‐deficient host cells rendered the cross‐correction phenomenon inefficient. This notion is supported by several studies showing that expression of IGF2R is developmental age and gender dependent (Nissley et al., 1993; Gonzalez‐Parra et al., 2001; Hawkes and Kar, 2003, 2004; Romano et al., 2005; Jofre et al., 2009). Furthermore, analysis of IGF2R expression in healthy and Alzheimer's disease human brains demonstrated regional and cell type specificity (Kar et al., 2006). GALC was shown to express primarily in neurons and not on other cell types in the CNS (astrocytes, oligodendrocytes) that can benefit from the cross‐correction phenomenon. The fact that IGF2R is biallelically expressed in human tissues (Kalscheuer et al., 1993) and is genomically imprinted in mice (Barlow et al., 1991) limits the ability to interpret some of the results obtained in mouse models. Here we demonstrated for the first time an L‐cycloserine‐mediated increase of IGF2R. These findings support our notion that the dramatic prolongation in twi mouse life span upon combining L‐cycloserine treatment with HSPCT and AAV vector‐mediated GALC delivery is secondary to an L‐cycloserine‐induced increase in IGF2R (Hawkins‐Salsbury et al., 2015). Additional characterization of this phenomenon with identification of novel small molecules that enhance IGF2R expression may improve current therapeutic protocols for KD. Intrigued by these findings, we sought to circumvent the need to increase IGF2R expression by directing GALC uptake via an IGF2R‐independent pathway. To this end, we developed the GALC‐AErbd fusion protein, whose uptake by GALC‐deficient cells is mediated by two independent pathways, namely, the IGF2R and the LRP‐1 pathways. Our approach was premised on an earlier study demonstrating efficient LRP‐1‐mediated uptake of erythrocyte/megakaryocyte‐secreted IDUA‐AErbd by neurons and astrocytes throughout the cerebral cortex (Wang et al., 2013). Indeed, here we demonstrate the ability of 293T and human GALC‐deficient fibroblasts to take up the GALC‐AErbd independently of the IGF2R pathway efficiently. We expected that the novel GALC‐AErbd would broaden the spectrum of host cells that benefit from the phenomenon of cross‐correction. The fact that the life span of twi mice engrafted with HSPC transduced with GALC‐AErbd‐expressing vectors was not significantly different from the life span of their counterparts, which received nontransduced total BM cells, was not highly surprising, especially because VCN in these treated mice BM was not higher than 1.24. Note that, in earlier studies, in which lentiviral vectors were employed to deliver GALC to HSPC, relatively high VCN (6–10) in host BM was required to achieve merely mild prolongation in the life span of GALC‐deficient mice (Gentner et al., 2010; Ungari et al., 2015). Facilitating cellular uptake of donor‐cell secreted GALC‐AErbd via an M6PR‐independent pathway and inducing overexpression of IGF2R by L‐cycloserine are two independent strategies to allow efficient HPCT‐mediated cross‐correction of GALC deficiency in twi mouse CNS cells expressing low levels of IGF2R. The GALC‐AErbd approach does not require significant modifications to current HPCT‐based protocols and if successful provides major long‐term therapeutic benefits to HPCT‐treated patients. However, the efficacy of this approach, which is dependent on the level of LRP‐1 in KD patients’ CNS, has not been established. Furthermore, employing this approach to treat other LSDs requires the development of different AErbd‐fusion proteins. The ability of an L‐cycloserine/gene therapy combination to treat twi mice efficiently has opened a new direction in KD therapy. However, the broad spectrum of L‐cycloserine activity and its inherent cytotoxicity render this small molecule unsuitable for human clinical trials. Clearly, less toxic small molecules that upregulate IGF2R expression in vivo have to be identified, and their ability to synergize with gene and cell therapy protocols in curing KD has to be established. Furthermore, this approach necessitates life‐long treatment. Thus, we assert that additional studies are needed to characterize the therapeutic mechanism of this approach and to establish its efficacy (either separately or in combination) for treating LSDs prior to considering their usage in clinical applications.
However, we also realize that a suboptimal conditioning protocol might negatively affect the efficacy of HSPCT for correcting GALC deficiency in preclinical models. Because Bu is the major chemotherapy agent employed in human clinical trials of neurodegenerative diseases (Cartier et al., 2009; Aiuti et al., 2013) as well as in allogeneic HPCT for KD (Escolar et al., 2005), we sought to characterize the dose effects of Bu on the efficacy of HSPCT in the twi mouse model. Overall, the average life span of HSPCT treated twi mice in this study was considerably longer than the average life span of twi mice conditioned by irradiation (Lin et al., 2007, 2005; Gentner et al., 2010; Ungari et al., 2015). This limits our ability to evaluate the efficacy of different therapeutic measures that were tested in irradiated twi mice. The results of this study clearly demonstrate direct correlation between the dose of Bu and the capacity of HSPCT protocols to prolong the average life span of twi mice. Furthermore, colonization of donor cells in twi mouse CNS was significantly more efficient than the engraftment of the same donor cells in healthy mouse CNS. These finding are in line with several earlier studies concluding that both effective conditioning protocols and host CNS pathology are required for optimal colonization of the host CNS with donor hematopoietic progenies (Priller et al., 2001; Mildner et al., 2007; Davoust et al., 2008). This notion raises the possibility that various regions in twi mouse CNS that are relatively healthy at the time of HSPCT are not efficiently colonized with donor cells.
Clearly, further optimization of the conditioning/HSPCT protocols is needed to maximize the clinical outcome of HSPCT in the twi model of KD. We anticipate that the results of this study will pave the way for the development of new GALC fusion proteins with a broadened spectrum of target cells in the CNS.
CONFLICT OF INTEREST STATEMENT
ERB is a consultant for Lysosomal Therapeutics, Inc.
ROLE OF AUTHORS
All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: TK. Acquisition of data: PH, YL, NN‐F, GS, YB. Analysis, interpretation of data, and drafting of the manuscript: TK, RM, PH. Statistical analysis consulted with DM. Critical revision of the manuscript for important intellectual content: DP, MSS, ERB, TK. Obtained funding: TK, MSS, ERB.
ACKNOWLEDGMENTS
We thank the staffs of the UNC Translational Pathology Laboratory (TPL) for expert technical assistance. The UNC TPL is supported in part by grants from the National Cancer Institute (2‐P30‐CA016086‐40) and the UNC University Cancer Research Fund (UCRF). We thank Dr. Dvir Kafri for editing the manuscript. This article is dedicated to the U.S. Marine Corps and the Gold Star families and in memory of the MS St. Louis.
SIGNIFICANCE Although presymtomatic hematopoietic stem and progenitor cell transplantation (HSPCT) is the standard therapeutic modality for Krabbe's disease, it fails to provide a curative solution. Newly developed lentiviral vectors carrying myeloid‐ and erythroid‐specific promoters improve the therapeutic potential of HSPCT by facilitating GALC delivery to host HSPC without compromising their ability to engraft the host bone marrow. Low levels of CI‐MPR (also known as IGF2R) may limit the efficacy of HSPCT for curing KD. A novel GALC‐AErdb fusion whose uptake by host cells is mediated by an IGF2R‐independent pathway may resolve this limitation. It appears that an aggressive busulfan conditioning protocol significantly extends the life span of twi mice.
REFERENCES
- Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, Bosticardo M, Evangelio C, Assanelli A, Casiraghi M, Di Nunzio S, Callegaro L, Benati C, Rizzardi P, Pellin D, Di Serio C, Schmidt M, Von Kalle C, Gardner J, Mehta N, Neduva V, Dow DJ, Galy A, Miniero R, Finocchi A, Metin A, Banerjee PP, Orange JS, Galimberti S, Valsecchi MG, Biffi A, Montini E, Villa A, Ciceri F, Roncarolo MG, Naldini L. 2013. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott‐Aldrich syndrome. Science 341:1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldenhoven M, Jones SA, Bonney D, Borrill RE, Coussons M, Mercer J, Bierings MB, Versluys B, van Hasselt PM, Wijburg FA, van der Ploeg AT, Wynn RF, Boelens JJ. 2015. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: results after implementation of international guidelines. Biol Blood Marrow Transplant 21:1106–1109. [DOI] [PubMed] [Google Scholar]
- Barde I, Laurenti E, Verp S, Wiznerowicz M, Offner S, Viornery A, Galy A, Trumpp A, Trono D. 2011. Lineage‐ and stage‐restricted lentiviral vectors for the gene therapy of chronic granulomatous disease. Gene Ther 18:1087–1097. [DOI] [PubMed] [Google Scholar]
- Barlow DP, Stoger R, Herrmann BG, Saito K, Schweifer N. 1991. The mouse insulin‐like growth factor type‐2 receptor is imprinted and closely linked to the Tme locus. Nature 349:84–87. [DOI] [PubMed] [Google Scholar]
- Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, Baldoli C, Martino S, Calabria A, Canale S, Benedicenti F, Vallanti G, Biasco L, Leo S, Kabbara N, Zanetti G, Rizzo WB, Mehta NA, Cicalese MP, Casiraghi M, Boelens JJ, Del Carro U, Dow DJ, Schmidt M, Assanelli A, Neduva V, Di Serio C, Stupka E, Gardner J, von Kalle C, Bordignon C, Ciceri F, Rovelli A, Roncarolo MG, Aiuti A, Sessa M, Naldini L. 2013. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341:1233158. [DOI] [PubMed] [Google Scholar]
- Boelens JJ, Aldenhoven M, Purtill D, Ruggeri A, Defor T, Wynn R, Wraith E, Cavazzana‐Calvo M, Rovelli A, Fischer A, Tolar J, Prasad VK, Escolar M, Gluckman E, O'Meara A, Orchard PJ, Veys P, Eapen M, Kurtzberg J, Rocha V, Eurocord, Inborn Errors Working Party of European Bone Marrow Transplant Group . 2013. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood 121:3981–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boelens JJ, Orchard PJ, Wynn RF. 2014. Transplantation in inborn errors of metabolism: current considerations and future perspectives. Br J Haematol 167:293–303. [DOI] [PubMed] [Google Scholar]
- Cartier N, Hacein‐Bey‐Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal‐Cortivo L, Caccavelli L, Mahlaoui N, Kiermer V, Mittelstaedt D, Bellesme C, Lahlou N, Lefrere F, Blanche S, Audit M, Payen E, Leboulch P, l'Homme B, Bougneres P, Von Kalle C, Fischer A, Cavazzana‐Calvo M, Aubourg P. 2009. Hematopoietic stem cell gene therapy with a lentiviral vector in X‐linked adrenoleukodystrophy. Science 326:818–823. [DOI] [PubMed] [Google Scholar]
- Dahms NM, Olson LJ, Kim JJ. 2008. Strategies for carbohydrate recognition by the mannose 6‐phosphate receptors. Glycobiology 18:664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M, Han J, El‐Amouri SS, Brady RO, Pan D. 2014. Platelets are efficient and protective depots for storage, distribution, and delivery of lysosomal enzyme in mice with Hurler syndrome. Proc Natl Acad Sci U S A 111:2680–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoust N, Vuaillat C, Androdias G, Nataf S. 2008. From bone marrow to microglia: barriers and avenues. Trends Immunol 29:227–234. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Barczykowski A, Jalal K, Yan L, Kay DM, Carter RL. 2011. Early infantile Krabbe disease: results of the World‐Wide Krabbe Registry. Pediatr Neurol 45:141–148. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Caviness VS Jr., Erbe RW, Patterson MC, Schultz KR, Wenger DA, Whitley C. 2009. The long‐term outcomes of presymptomatic infants transplanted for Krabbe disease: report of the workshop held on July 11 and 12, 2008, Holiday Valley, New York. Genet Med 11:450–454. [DOI] [PubMed] [Google Scholar]
- Ebert MS, Sharp PA. 2010. MicroRNA sponges: progress and possibilities. RNA 16:2043–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Amouri SS, Dai M, Han JF, Brady RO, Pan D. 2014. Normalization and improvement of CNS deficits in mice with Hurler syndrome after long‐term peripheral delivery of BBB‐targeted iduronidase. Mol Ther 22:2028–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, Morse R, Krivit W, Kurtzberg J. 2005. Transplantation of umbilical‐cord blood in babies with infantile Krabbe's disease. N Engl J Med 352:2069–2081. [DOI] [PubMed] [Google Scholar]
- Gentner B, Visigalli I, Hiramatsu H, Lechman E, Ungari S, Giustacchini A, Schira G, Amendola M, Quattrini A, Martino S, Orlacchio A, Dick JE, Biffi A, Naldini L. 2010. Identification of hematopoietic stem cell‐specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci Translational Med 2:58ra84. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Parra S, Argente J, Chowen JA, van Kleffens M, van Neck JW, Lindenbeigh‐Kortleve DJ, Drop SL. 2001. Gene expression of the insulin‐like growth factor system during postnatal development of the rat pituitary gland. J Neuroendocrinol 13:86–93. [DOI] [PubMed] [Google Scholar]
- Hawkes C, Kar S. 2003. Insulin‐like growth factor‐II/mannose‐6‐phosphate receptor: widespread distribution in neurons of the central nervous system including those expressing cholinergic phenotype. J Comp Neurol 458:113–127. [DOI] [PubMed] [Google Scholar]
- Hawkes C, Kar S. 2004. The insulin‐like growth factor‐II/mannose‐6‐phosphate receptor: structure, distribution and function in the central nervous system. Brain Res Rev 44:117–140. [DOI] [PubMed] [Google Scholar]
- Hawkins‐Salsbury JA, Shea L, Jiang X, Hunter DA, Guzman AM, Reddy AS, Qin EY, Li Y, Gray SJ, Ory DS, Sands MS. 2015. Mechanism‐based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. J Neurosci 35:6495–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Qiang M, Ma W, Valente AJ, Quinones MP, Wang W, Reddick RL, Xiao Q, Ahuja SS, Clark RA, Freeman GL, Li S. 2006. Development of a synthetic promoter for macrophage gene therapy. Hum Gene Ther 17:949–959. [DOI] [PubMed] [Google Scholar]
- Heldermon CD, Qin EY, Ohlemiller KK, Herzog ED, Brown JR, Vogler C, Hou W, Orrock JL, Crawford BE, Sands MS. 2013. Disease correction by combined neonatal intracranial AAV and systemic lentiviral gene therapy in Sanfilippo syndrome type B mice. Gene Ther 20:913–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jofre GF, Balmaceda V, Sartor T, Carvelli L, Barrera P, Sosa MA. 2009. Organ‐specific changes in the expression of mannose‐6‐phosphate receptors during postnatal development in rats. Cells Tissues Organs 190:27–33. [DOI] [PubMed] [Google Scholar]
- Kafri T, van Praag H, Ouyang L, Gage FH, Verma IM. 1999. A packaging cell line for lentivirus vectors. J Virol 73:576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalscheuer VM, Mariman EC, Schepens MT, Rehder H, Ropers HH. 1993. The insulin‐like growth factor type‐2 receptor gene is imprinted in the mouse but not in humans. Nat Genet 5:74–78. [DOI] [PubMed] [Google Scholar]
- Kantor B, Ma H, Webster‐Cyriaque J, Monahan PE, Kafri T. 2009. Epigenetic activation of unintegrated HIV‐1 genomes by gut‐associated short chain fatty acids and its implications for HIV infection. Proc Natl Acad Sci U S A 106:18786–18791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S, Poirier J, Guevara J, Dea D, Hawkes C, Robitaille Y, Quirion R. 2006. Cellular distribution of insulin‐like growth factor‐II/mannose‐6‐phosphate receptor in normal human brain and its alteration in Alzheimer's disease pathology. Neurobiol Aging 27:199–210. [DOI] [PubMed] [Google Scholar]
- Lillis AP, Van Duyn LB, Murphy‐Ullrich JE, Strickland DK. 2008. LDL receptor‐related protein 1: unique tissue‐specific functions revealed by selective gene knockout studies. Physiol Rev 88:887–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Donsante A, Macauley S, Levy B, Vogler C, Sands MS. 2007. Central nervous system‐directed AAV2/5‐mediated gene therapy synergizes with bone marrow transplantation in the murine model of globoid‐cell leukodystrophy. Mol Ther 15:44–52. [DOI] [PubMed] [Google Scholar]
- Lin D, Fantz CR, Levy B, Rafi MA, Vogler C, Wenger DA, Sands MS. 2005. AAV2/5 vector expressing galactocerebrosidase ameliorates CNS disease in the murine model of globoid‐cell leukodystrophy more efficiently than AAV2. Mol Ther 12:422–430. [DOI] [PubMed] [Google Scholar]
- Martino S, Tiribuzi R, Tortori A, Conti D, Visigalli I, Lattanzi A, Biffi A, Gritti A, Orlacchio A. 2009. Specific determination of beta‐galactocerebrosidase activity via competitive inhibition of beta‐galactosidase. Clin Chem 55:541–548. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Bruck W, Priller J, Prinz M. 2007. Microglia in the adult brain arise from Ly‐6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci 10:1544–1553. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Smith KA, Mosier DE, Verma IM, Torbett BE. 1999. Transduction of human CD34+ cells that mediate long‐term engraftment of NOD/SCID mice by HIV vectors. Science 283:682–686. [DOI] [PubMed] [Google Scholar]
- Moreau‐Gaudry F, Xia P, Jiang G, Perelman NP, Bauer G, Ellis J, Surinya KH, Mavilio F, Shen CK, Malik P. 2001. High‐level erythroid‐specific gene expression in primary human and murine hematopoietic cells with self‐inactivating lentiviral vectors. Blood 98:2664–2672. [DOI] [PubMed] [Google Scholar]
- Munier‐Lehmann H, Mauxion F, Bauer U, Lobel P, Hoflack B. 1996. Re‐expression of the mannose 6‐phosphate receptors in receptor‐deficient fibroblasts. Complementary function of the two mannose 6‐phosphate receptors in lysosomal enzyme targeting. J Biol Chem 271:15166–15174. [DOI] [PubMed] [Google Scholar]
- Nissley P, Kiess W, Sklar M. 1993. Developmental expression of the IGF‐II/mannose 6‐phosphate receptor. Mol Reprod Dev 35:408–413. [DOI] [PubMed] [Google Scholar]
- Polavarapu R, Gongora MC, Yi H, Ranganthan S, Lawrence DA, Strickland D, Yepes M. 2007. Tissue‐type plasminogen activator‐mediated shedding of astrocytic low‐density lipoprotein receptor‐related protein increases the permeability of the neurovascular unit. Blood 109:3270–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priller J, Flugel A, Wehner T, Boentert M, Haas CA, Prinz M, Fernandez‐Klett F, Prass K, Bechmann I, de Boer BA, Frotscher M, Kreutzberg GW, Persons DA, Dirnagl U. 2001. Targeting gene‐modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment. Nat Med 7:1356–1361. [DOI] [PubMed] [Google Scholar]
- Reddy AS, Kim JH, Hawkins‐Salsbury JA, Macauley SL, Tracy ET, Vogler CA, Han X, Song SK, Wozniak DF, Fowler SC, Klein RS, Sands MS. 2011. Bone marrow transplantation augments the effect of brain‐ and spinal cord‐directed adeno‐associated virus 2/5 gene therapy by altering inflammation in the murine model of globoid‐cell leukodystrophy. J Neurosci 31:9945–9957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano PS, Carvelli L, Lopez AC, Jofre G, Sartor T, Sosa MA. 2005. Developmental differences between cation‐independent and cation‐dependent mannose‐6‐phosphate receptors in rat brain at perinatal stages. Brain Res Dev Brain Res 158:23–30. [DOI] [PubMed] [Google Scholar]
- Stein S, Ott MG, Schultze‐Strasser S, Jauch A, Burwinkel B, Kinner A, Schmidt M, Kramer A, Schwable J, Glimm H, Koehl U, Preiss C, Ball C, Martin H, Gohring G, Schwarzwaelder K, Hofmann WK, Karakaya K, Tchatchou S, Yang R, Reinecke P, Kuhlcke K, Schlegelberger B, Thrasher AJ, Hoelzer D, Seger R, von Kalle C, Grez M. 2010. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med 16:198–204. [DOI] [PubMed] [Google Scholar]
- Suwanmanee T, Hu G, Gui T, Bartholomae CC, Kutschera I, von Kalle C, Schmidt M, Monahan PE, Kafri T. 2013. Integration‐deficient lentiviral vectors expressing codon‐optimized R338LhFIX restore normal hemostasis in hemophilia B mice. Mol Ther 22:567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungari S, Montepeloso A, Morena F, Cocchiarella F, Recchia A, Martino S, Gentner B, Naldini L, Biffi A. 2015. Design of a regulated lentiviral vector for hematopoietic stem cell gene therapy of globoid cell leukodystrophy. Mol Ther Methods Clin Dev 2:15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visigalli I, Ungari S, Martino S, Park H, Cesani M, Gentner B, Sergi Sergi L, Orlacchio A, Naldini L, Biffi A. 2010. The galactocerebrosidase enzyme contributes to the maintenance of a functional hematopoietic stem cell niche. Blood 116:1857–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, El‐Amouri SS, Dai M, Kuan CY, Hui DY, Brady RO, Pan D. 2013. Engineering a lysosomal enzyme with a derivative of receptor‐binding domain of apoE enables delivery across the blood–brain barrier. Proc Natl Acad Sci U S A 110:2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserstein MP, Andriola M, Arnold G, Aron A, Duffner P, Erbe RW, Escolar ML, Estrella L, Galvin‐Parton P, Iglesias A, Kay DM, Kronn DF, Kurtzberg J, Kwon JM, Langan TJ, Levy PA, Naidich TP, Orsini JJ, Pellegrino JE, Provenzale JM, Wenger DA, Caggana M. 2016. Clinical outcomes of children with abnormal newborn screening results for Krabbe disease in New York State. Genet Med (in press). [DOI] [PubMed] [Google Scholar]
- Wiederschain G, Raghavan S, Kolodny E. 1992. Characterization of 6‐hexadecanoylamino‐4‐methylumbelliferyl‐beta‐D‐galactopyranoside as fluorogenic substrate of galactocerebrosidase for the diagnosis of Krabbe disease. Clin Chim Acta 205:87–96. [DOI] [PubMed] [Google Scholar]