

Abstract
The formation and maintenance of an organism is highly dependent on the orderly control of cell growth, differentiation, death, and migration. These processes are tightly regulated by signaling cascades in which a limited number of molecules dictate these cellular events. While these signaling pathways are highly conserved across species and cell types, the functional outcomes that result from their engagement are specified by the context in which they are activated. Using the Neurofibromatosis type-1 (NF1) cancer predisposition syndrome as an illustrative platform, we discuss how NF1/RAS signaling can create functional diversity at multiple levels (molecular, cellular, tissue, and genetic/genomic). As such, the ability of related molecules (e.g., K-RAS, H-RAS) to activate distinct effectors, as well as cell type- and tissue-specific differences in molecular composition and effector engagement, generate numerous unique functional effects. These variations, coupled with a multitude of extracellular cues and genomic/genetic changes that each modify the innate signaling properties of the cell, enable precise control of cellular physiology in both health and disease. Understanding these contextual influences is important when trying to dissect the underlying pathogenic mechanisms of cancer relevant to molecularly-targeted therapeutics.
Keywords: glioma, RAS, mTOR, NF1, astrocyte, neuron, nervous system
Graphical abstract
1. Introduction
“The only man I know who behaves sensibly is my tailor; he takes my measurements anew each time he sees me. The rest go on with their old measurements and expect me to fit them.”
- George Bernard Shaw
Precise control of cell behavior involves continual sensing of a variety of physical and chemical cues from the external milieu, and interpreting those signals to respond in an appropriate manner. These extracellular cues take the form of cell-bound and diffusible ligands that bind specific receptors on cells to initiate a cascade of signaling events that culminate in changes in protein function through post-translational (e.g., phosphorylation) or transcriptional (e.g., mRNA or miRNA) alterations. In this regard, signaling represents the language of the cell, where molecules (words) and cellular context (syntax) serve as units of informational content. Creating meaningful sentences and paragraphs requires that a limited number of molecules can be used in combinatorial and situation-specific manners to produce a diversity of outputs and responses relevant to the specific environment at that particular time. Unfortunately, when we study signaling pathways in normal cells or in the setting of cancer, we often fail to consider how the cellular language conferred by these pathways is influenced by context, that is, the different extracellular signals present in the immediate milieu, the various adaptive responses that limit and promote intracellular signal transduction, the innate properties of distinct cell types responding to these cues, and the impact of epigenetic/genomic changes on the ultimate consequence of these informational signals. In this review, we use the Neurofibromatosis type 1 (NF1) predisposition syndrome as an illustrative platform to discuss how heterogeneity can be generated at the molecular, cellular, tissue, and genomic/genetic levels. Moreover, we suggest that the precision created by this “contextual signaling” enables diverse outcomes to arise from engagement of a limited number of key signaling pathways.
1.1 Neurofibromatosis type 1
Neurofibromatosis type 1 (NF1) is a monogenic syndrome affecting 1 in every 2,500 individuals worldwide. Caused by a germline mutation in the NF1 gene, affected children and adults are prone to the development of benign and malignant tumors. In addition, numerous other organ systems are affected, leading to skeletal, cardiovascular, dermatologic, ophthalmologic, endocrinologic, and neurological abnormalities. This latter group of clinical features is the most common, and includes peripheral nerve tumors (neurofibromas and malignant peripheral nerve sheath tumors) and brain tumors (optic pathway and brainstem gliomas, malignant glioma).
Neurofibromas and optic pathway gliomas (OPGs) form following bi-allelic NF1 loss, which is the consequence of somatic inactivation of the remaining normal NF1 gene and total loss of NF1 protein (neurofibromin) expression. Individuals with NF1 start life with a germline NF1 gene mutation, which increases their chance of cancer, since only one additional genetic event is required (somatic NF1 gene loss). As neurofibromin functions as a GTPase-activating protein (GAP) to accelerate the conversion of active GTP-bound rat sarcoma (RAS) to inactive RAS-GDP, neurofibromin loss in neoplastic cells leads to RAS hyperactivation and engagement of multiple RAS downstream effectors, culminating in increased cell growth. In addition, tumorigenesis is further controlled by non-neoplastic cells in the local microenvironment that elaborate key stromal growth factors and chemokines. Moreover, the impact of NF1 loss is highly dependent on the specific cell type and tissue, as well as by modifying genomic loci and cooperating genetic mutations. The influence of all of these factors may partly explain why individuals with NF1 exhibit such a wide range of clinical variability. In this regard, children and adults from the same family with an identical germline NF1 gene mutation can manifest different medical features of NF1 and have markedly distinct clinical courses and severity. Understanding these factors is vitally important for providing risk assessments to affected individuals, as well as for designing personalized therapies for these clinical abnormalities when they develop.
2. Molecular Level
While neurofibromin functions as a RAS-GAP, the consequence of bi-allelic NF1 loss on signaling pathway activation and cell growth is not identical in all cell types. This reflects the fact that a signaling molecule (e.g., RAS) is not a single molecule, but rather comprises a family of highly homologous proteins with slightly different functions. Moreover, RAS can signal by engaging a variety of downstream effector proteins to control cell biology. Lastly, some signaling effector proteins function as part of a multi-molecular complex whose composition determines which signaling intermediates are activated and what cellular output is controlled (Figure 1).
Figure 1.

Neurofibromin loss activates RAS signaling to potentially activate a large number of downstream effector proteins.
2.1 Signaling molecules comprise a group of functionally-distinct proteins
One mechanism by which RAS pathway activation can generate distinct cellular responses is through differential engagement of RAS/RAS effector proteins. In this regard, RAS, rapidly accelerated fibrosarcoma (RAF) and protein kinase B (AKT) each comprise families of molecularly-similar, but functionally-distinct, proteins.
RAS exists as at least four highly homologous RAS proteins (H-RAS, N-RAS, K-RAS4A, K-RAS4B), which differ in their potency to activate downstream effectors [1]. While these molecules share 85% amino acid sequence similarity, the specific functions of each RAS protein are dictated by distinct 25-residue hypervariable regions (HVR) at the carboxyl terminus. Differential lipid modifications within this HVR direct H-RAS and N-RAS to the plasma membrane, while K-RAS is trafficked to different domains within the plasma membrane [2, 3]. In addition, oncogenic K-RAS activation (Noonan syndrome) increases cellular proliferation and differentiation, while hyperactive N-RAS (Noonan syndrome) promotes cell survival [4]. Similarly, mutational H-RAS activation (Costello syndrome) stimulates tumor angiogenesis [5] and DNA synthesis [6], while mutational K-RAS activation has no effect [7]. Moreover, homozygous inactivation of K- RAS is embryonically lethal, whereas N-RAS or H-RAS knockout mice are viable [8, 9]. Lastly, only mutationally-activated K-RAS, but not H-RAS or N-RAS, increased neural stem cell proliferation in vitro and astroglial differentiation in vitro and in vivo [10].
One of the direct targets of RAS is the RAF serine/threonine-specific protein kinase, which, like RAS, comprises three distinct molecules (A-RAF, B-RAF, C-RAF [RAF-1]). These proteins are differentially expressed and regulated, and are non-redundant in their ability to activate their downstream effectors. RAF molecules, though structurally similar, regulate MEK1/2 with varied affinity through the formation of multiple RAF heterodimers. In this regard, the B-RAF/C-RAF heterodimer stimulates MEK much more efficiently than B-RAF or C-RAF activation alone [11]. Additionally, B-RAF and C-RAF require additional post-translational modifications for full activity [12, 13]. Moreover, this family of protein kinases interacts with numerous adapters, kinases, G-proteins, and chaperones to create signaling diversity [14].
Similarly, AKT is encoded by three separate genes, PKBα (AKT1) PKBβ (AKT2) and PKBγ (AKT3), whose activation also has different consequences. As such, Akt1 knockout mice display defects in overall growth, while Akt2 null mice are insulin intolerant and demonstrate a diabetes-like syndrome [15]. In sharp contrast, Akt3 knockout mice have a selective reduction in brain size, reflecting its robust expression in brain tissues [16].
2.2 Individual signaling molecules activate different effectors
GTP-bound RAS can directly activate at least three different proteins (RAF, phosphoinositide 3-kinase [PI3K], and protein kinase C-zeta [PKCζ]), which transduce growth-promoting messages through distinct signaling pathways [17–19].
RAS-dependent RAF activation leads to the sequential phosphorylation of mitogen-activated protein kinase kinase (MEK) and p44/p42 extracellular signal-related kinase (ERK) [20]. While neurofibromin loss and activation of MEK/ERK can lead to increased proliferation through ERK activation of transcriptional factors [21], it can also increase cell proliferation through a 90 kDa ribosomal S6 kinase (p90-RSK)/mechanistic target of rapamycin (mTOR)-dependent manner [22]. In addition, neurofibromin controls glial and neuronal differentiation in a RAF/MEK-dependent, but mTOR-independent, manner in brain neural stem cells (NSCs) by activating the Jagged1/Notch pathway [23]. Finally, oncogenic KRAS increases NSC growth by negatively regulating the retinoblastoma protein in a RAF-dependent, but MEK-independent, fashion [10].
As another RAS effector, PI3K signaling is required for a wide variety of critical cell processes [19, 24]. RAS activity increases PI3K activation, which allows phosphatidylinositol (4,5)-biphosphate (PIP2) to convert into phosphatidylinositol (3,4,5)-triphosphate (PIP3). PIP3 then recruits protein kinase B (AKT) to the plasma membrane, promoting PI3K-mediated phosphoinositide dependent protein kinase-1 (PDPK1/PDK1) phosphorylation and activation of AKT. However, AKT-mediated mTOR activation can occur through a variety of mechanisms, including direct mTOR activation [25], phosphorylation of the proline-rich in AKT substrate of 40 kDa (PRAS40) [26], or through inactivation of the tuberous sclerosis complex (TSC) [27].
Lastly, RAS can transmit its growth promoting signaling through atypical Protein Kinase C molecules, like PKCζ, either involving PI3K [28] or independent of PI3K [29]. In neurons, reduced neurofibromin function leads to RAS-mediated PKCζ activation, resulting in G protein-coupled receptor kinase 2 (GRK2) suppression of G protein-coupled receptor function and reduced cyclic adenosine monophosphate (cAMP) production [17]. These distinct neurofibromin/RAS downstream pathways (RAF/MEK, PI3K/AKT, and PKCζ) create a diversity of signaling pathway activation following NF1 loss in specific cell types.
2.3 Individual molecules form protein complexes necessary for signaling transduction
Beyond RAS/RAS effector families and differential engagement of RAS downstream effectors, another mechanism for generating functional diversity is through the formation of multi-protein signaling complexes. Critical for neurofibromin/RAS growth control is the mTOR complex. mTOR operates in as at least two molecularly- and functionally-distinct protein complexes. mTOR complex 1 (mTORC1) consists of regulatory-associated protein of mTOR (Raptor) and PRAS40. Raptor is required for mTOR kinase activity by directly binding and activating mTORC1 effectors, such as the translational regulators p70 S6 kinase (S6K) and 4E (elF4E) binding protein (4EBP1) [30]. In contrast, mTOR complex 2 (mTORC2) contains rapamycin-insensitive companion of mTOR (Rictor), protein observed with rictor (Protor), and mammalian stress-activated MAP kinase interacting protein-1 (mSIN1), which are all required for the activation of mTORC2-specific effectors, AKT [31], protein kinase Cα,β, γ (PKCα, β, γ) [32], and serum- and glucocorticoid-induced protein kinase 1 (SGK1) [33]. While mTORC1 is critical for mediating ribosomal biogenesis and translational control, mTORC2 is essential for controlling cell survival, migration, and cytoskeletal dynamics [34].
3. Cellular Level
In addition to signaling diversity generated at the molecular level, there are cell type-specific constraints that operate to alter the way neurofibromin regulates RAS pathway activation and cell function. These include differences between cell types, as well as differences between the same cell type within a given tissue (Figure 2).
Figure 2.

The consequence of neurofibromin loss and RAS activation is dictated by cell type- and brain region-specific differences.
3.1 Cell type-specific control of RAS effector engagement
While neurofibromin suppresses RAS activity to control cell growth and survival in all cell types examined to date, how neurofibromin signals to its downstream effectors to mediate this effect depends entirely on the individual cell type. As such, central nervous system (CNS) neuronal growth and survival relies on RAS/PKCζ regulation of cAMP [17], rather than through MEK/ERK or PI3K [22]. In contrast, brain astroglial and NSCs use neurofibromin to control cell growth through the RAS/mTOR pathway [22, 35, 36]. However, Nf1-deficient astrocyte proliferation is dependent on mTOR/Rac1 activation [37], whereas NSCs use RAS/mTORC2/AKT/p27 signaling [36]. Additionally, neurofibromin control of mast cell function operates through RAS/PI3K/Rac1 [38], whereas Nf1-deficient osteoblast growth is dependent on RAS/ERK/RSK/activating transcription factor 4 (ATF4) signaling [39]. In microglia, neurofibromin controls cell proliferation and activation through the Rac1/mixed-lineage protein kinase 3 (MLK3)/mitogen-activated protein kinase kinase 4 (MKK4)/c-Jun N-terminal kinase (JNK) pathway [40]. In addition, the signature genetic alteration in low-grade gliomas involves the fusion of the BRAF gene to the amino terminal of the KIAA1549 protein product (f-BRAF). Similar to neurofibromin, ectopic f-BRAF expression increases RAS/MEK signaling in both astrocytes and NSCs, but only increases proliferation in NSCs and eventual tumor formation as a result of ERK-mediated mTOR activation [36, 41].
3.2 Regional constraints dictate RAS pathway function
Another contextual determinant that impacts RAS pathway signaling is the regional identity of the cell. In this respect, isolated astroglial cells have different levels of neurofibromin expression, with significantly higher Nf1 protein expression in the optic nerve, brainstem and cerebellar astrocytes relative to those from the neocortex [42]. For example, reduced neurofibromin expression in CNS neurons results in attenuated survival and neurite outgrowth, which depends on neurofibromin/RAS-controlled cAMP production [43], whereas, Nf1-deficient peripheral nervous system (PNS) neurons have increased survival and longer neuritic processes, reflecting RAS-mediated AKT hyperactivation [44]. Finally, Nf1 loss in NSCs has differential effects depending on the region of the brain in which they reside. For example, Nf1-deficient NSCs from the third ventricular and brainstem exhibit increased proliferation and gliogenesis, whereas those from the lateral ventricle or neocortex of the same mouse do not [36].
4. Tissue Level
While it is clear that cell type and tissue location can differentially influence RAS signaling and functional outcome, additional factors operate at the tissue level to affect RAS pathway signaling (Figure 3). In the case of low-grade tumors arising in children and adults with NF1 (neurofibromas, OPGs), evidence exists for an obligate role for cellular and acellular determinants in tumor formation and maintenance. While less is known about the role of acellular factors in NF1-associated tumors (extracellular matrix [ECM] components), expression of the ECM components, laminin-2α and integrin α6β1, potentiates high-grade glioma cell growth [45, 46].
Figure 3.

Non-neoplastic cells and acellular signaling in the local microenvironment control RAS-dependent neoplastic cell growth.
In experimental mouse models of plexiform neurofibroma, both macrophages and bone marrow-derived mast cells control tumor formation and growth through the activation of RAS signaling. Using a combination of bone marrow transplantation and pharmacological inhibitor approaches, mast cells are required for murine plexiform neurofibroma formation and continued growth [47]. In these experiments, mast cells recruited by Nf1-deficient Schwann cell-produced stem cell factor (SCF or KIT-ligand) secrete cytokines that increase neurofibroma growth [48]. Moreover, SCF can also stimulate Nf1+/− mast cells to increase fibroblast proliferation and collagen deposition, thereby forming a permissive tumor microenvironment [49]. Interruption of this paracrine circuit with the c-KIT inhibitor, Imatinib, reduced plexiform neurofibroma growth [47].
In human low-grade gliomas, resident brain tissue macrophages (microglia) comprise 30–50% of the cells [50], where they are critical for murine Nf1 OPG formation and progression [51]. As such, pharmacologic or genetic reduction of microglial function results in reduced tumor proliferation in vivo [40, 50, 52]. Since these tumor-associated microglia are the likely source of stromal chemokines and growth factors, one large-scale RNA-sequencing effort identified several potential candidates. The most promising molecule, the (C-C motif) ligand 5 (CCL5) chemokine, increased Nf1-deficient optic nerve astrocyte proliferation in vitro. Importantly, inhibition of CCL5 using a neutralizing antibody resulted in reduced tumor growth in vivo [53], which operates in a RAS/AKT-dependent fashion [54].
NF1-associated CNS/PNS tumors arise in close proximity to central and peripheral nerves, raising the intriguing possibility that neuronal activity might influence tumorigenesis or tumor growth. Elegant studies in high-grade glioma (HGG) model systems have shown that neuronal activity increases HGG growth and proliferation in a PI3K/mTOR-dependent manner [55]. As such, increased cortical neuronal activity, controlled optogenetically in a mouse xenograft model, resulted in greater glioma growth and proliferation in vivo. Using a proteomic strategy, several secreted factors were identified from stimulated cortical brain slides, culminating in the discovery of neuroligin-3 (NLGN3) as the responsible neuron-derived mitogen [55].
5. Genomic/Genetic Level
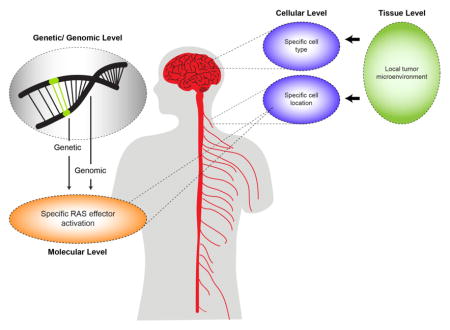
Beyond the influences of the individual signaling molecules in different cell types and the impact of factors from the local tissue microenvironment, another mechanism by which diversity can be created is at the level of genetic and genomic changes. In the context of NF1, these include the specific germline NF1 gene mutation, cooperating genetic mutations, and genomic modifier loci (Figure 4).
Figure 4.

Genetic and genomic factors differentially impact on neurofibromin/RAS signaling and tumor formation.
5.1. The Nf1 germline mutation
Every individual with NF1 is born with a germline mutation in the NF1 gene; however, >98% of individuals harbor a unique mutation. While there are few clear relationships between the specific germline NF1 gene mutation (genotype) and the clinical features of the disease (phenotype), converging evidence from epidemiologic, human NF1-patient iPSCs, and genetically-engineered mouse studies suggest that the specific germline NF1 gene mutation may be one predictive risk factor. As such, germline NF1 gene mutations have been reported in families who exhibit other hallmarks of the disease, but do not develop cutaneous neurofibromas (c2979-2972 delAAT and Arg1809 missense mutations) [56–58]. These early-phase observations are bolstered by studies employing NF1-patient derived iPSCs and derivative neural progenitor cells. In these experiments, different NF1 germline mutations have differential effects on neurofibromin expression and function, with some mutations leading to minor reductions in neurofibromin levels and others with >70% decreases [59]. Using genetically-engineered mice designed to harbor the R681X mutation observed in a patient with an OPG or a missense mutation (G848R) found in individuals with spinal neurofibromatosis, differential effects of these mutations were found. Mice with the R681X mutation had in >70% reductions in neurofibromin levels, whereas those with the G848R mutation had <25% reductions [54]. Importantly, only those mice with the R681X mutation developed OPGs.
5.2. Cooperating genetic mutations
In addition to NF1 gene inactivation, additional molecular changes have been identified in human NF1-associated OPG that converge on the same signaling pathway regulated by neurofibromin [60, 61]. For example, a heterozygous PTEN deletion was identified in one NF1-OPG, raising the possibility that these coincident genetic changes cooperate with NF1 loss to increase glioma growth and lead to clinically more aggressive neoplasms. Consistent with this idea, Nf1 genetically-engineered mice that also harbored a heterozygous PTEN mutation exhibited larger tumors with greater proliferation as a result of increased AKT activation [62].
5.3. Genomic modifier loci
Another way neurofibromin/RAS function can be modulated involves genomic modifier genes. While there is evidence for differences in racial and ethnic group risks for brain tumors [63], the most compelling data to support the existence of modifier loci derives from Nf1 genetically-engineered mouse models [64]. In these studies, NPcis mice, which carry mutations in the Nf1 and Trp53 genes on the same chromosome, develop HGG. However, NPcis mice on a C57BL/6 background developed brain tumors with high penetrance, whereas those on other genetic backgrounds do not. Further analyses of NPcis mice on different genetic backgrounds revealed the presence of modifier loci that influence spinal tumor development and brain astrocytoma formation [65], and high-grade peripheral nerve and brain tumor resistance in a sex-specific manner [66]. While it is not known how these modifiers operate to control tumorigenesis, one mechanism might involve differential Nf1 gene expression [67].
6. Conclusions
Creating functional diversity from one protein and/or pathway through contextual signaling allows for a great deal of outcome specificity without having to increase the amount of genetic material, and thus is evolutionarily efficient. Using NF1 as a model, we have outlined how RAS signaling can be contextually modified at the molecular, cellular, tissue, and genomic/genetic levels (Figure 5). In this regard, each specific genetic mutation, signaling effector family member, cell type, and tissue work in a combination with one another, as well as with genomic constraints, to encode a curated milieu that determines an explicit functional outcome and tumor pathology. As such, each RAS downstream effector can function in several states depending on the context in which it is activated.
Figure 5.

Contextual signaling in cancer operates at the genetic/genomic, molecular, cellular and tissue levels.
The assumption that canonical signaling pathways function in a linear and static manner across all cell types and tissues may not fully represent the manner in which they truly operate. As detailed in this review, converging evidence from several types of experiments suggest that understanding the most accurate context in which mitogenic signaling drives cell growth yields a clearer picture of the mechanisms underlying NF1 heterogeneity at the cellular, tissue, and organismal levels. A deeper appreciation of contextual signaling may improve our understanding of the basic principles that govern development and is also likely to lead to the design of more effective therapies for diseases characterized by inappropriate RAS/RAS pathway activation, such as seen in NF1-related tumors. Defining and encoding these variables relative to disease pathogenesis will hopefully result in better risk assessment strategies and the individualized therapies.
Highlights.
Cellular responses are dictated by context-dependent signaling pathway engagement
A limited number of molecules can activate multiple downstream effectors
Specific effector pathways are activated in a cell type and tissue-specific manner
Extracellular cues and genomic/genetic factors modify signaling pathway output
Acknowledgments
We thank all of the members of the Gutmann laboratory for their insightful comments during the preparation of this manuscript. This work was partially funded by grants from the National Cancer Institute (R01-CA195692-01) and Children’s Tumor Foundation (Synodos NF1 Low-Grade Glioma Initiative) to D.H.G. S.B.W. is supported by the Neurology T32 Training Grant (NS007205).
Abbreviations
- AKT
protein kinase B
- ATF4
activating transcription factor 4
- cAMP
cyclic adenosine monophosphate
- CCL5
(C-C motif) ligand 5
- CNS
central nervous system
- 4EBP1
4E (elF4E) binding protein
- ECM
extracellular matrix
- ERK
p44/p42 extracellular signal-related kinase
- GAP
GTPase-activating protein
- GRK2
G protein-coupled receptor kinase 2
- HGG
high grade glioma
- HVR
hypervariable region
- iPSCs
induced pluripotent stem cells
- JNK
c-Jun N-terminal kinases
- MEK
mitogen-activated protein kinase kinase
- MKK4
mitogen-activated protein kinase kinase 4
- MLK3
mixed-lineage protein kinase 3
- mSIN1
mammalian stress-activated MAP kinase interacting protein-1
- mTOR
mechanistic target of rapamycin
- mTORC1
mTOR complex 1
- mTORC2
mTOR complex 2
- NF1
Neurofibromatosis type 1
- NLGN3
neuroligin-3
- NSCs
neural stem cells
- OPG
optic pathway glioma
- PDPK1/PDK1
phosphoinositide dependent protein kinase-1
- PI3K
phosphoinositide 3-kinase
- PIP2
phosphatidylinositol (4,5)-biphosphate
- PIP3
phosphatidylinositol (3,4,5)-triphosphate
- PKCα,β, γ
protein kinase Cα,β, γ
- PKCζ
protein kinase C-zeta
- PNS
peripheral nervous system
- PRAS40
proline-rich in AKT substrate of 40 kDa
- Protor
protein observed with rictor
- PTEN
phosphate and tensin homolog
- Rac1
ras-related C3 botulinum toxin substrate 1
- RAF
rapidly accelerated fibrosarcoma
- Raptor
regulatory-associated protein of mTOR
- RAS
rat sarcoma
- Rictor
rapamycin-insensitive companion of mTOR
- p90-RSK
ribosomal S6 kinase
- SCF
stem cell factor
- S6K
p70 S6 kinase
- SGK1
serum- and glucocorticoid-induced protein kinase 1
- TSC
tuberous sclerosis complex
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yan J, Roy S, Apolloni A, Lane A, Hancock JF. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem. 1998;273(37):24052–6. doi: 10.1074/jbc.273.37.24052. [DOI] [PubMed] [Google Scholar]
- 2.Jang H, Abraham SJ, Chavan TS, Hitchinson B, Khavrutskii L, Tarasova NI, Nussinov R, Gaponenko V. Mechanisms of membrane binding of small GTPase K-Ras4B farnesylated hypervariable region. J Biol Chem. 2015;290(15):9465–77. doi: 10.1074/jbc.M114.620724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu S, Banerjee A, Jang H, Zhang J, Gaponenko V, Nussinov R. GTP Binding and Oncogenic Mutations May Attenuate Hypervariable Region (HVR)-Catalytic Domain Interactions in Small GTPase K-Ras4B, Exposing the Effector Binding Site. J Biol Chem. 2015;290(48):28887–900. doi: 10.1074/jbc.M115.664755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, Settleman J, Giovannini M, Jacks T. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40(5):600–8. doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arbiser JL, Moses MA, Fernandez CA, Ghiso N, Cao Y, Klauber N, Frank D, Brownlee M, Flynn E, Parangi S, Byers HR, Folkman J. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc Natl Acad Sci U S A. 1997;94(3):861–6. doi: 10.1073/pnas.94.3.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson MF, Paterson HF, Marshall CJ. Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature. 1998;394(6690):295–9. doi: 10.1038/28425. [DOI] [PubMed] [Google Scholar]
- 7.Okada F, Rak JW, Croix BS, Lieubeau B, Kaya M, Roncari L, Shirasawa S, Sasazuki T, Kerbel RS. Impact of oncogenes in tumor angiogenesis: mutant K-ras up-regulation of vascular endothelial growth factor/vascular permeability factor is necessary, but not sufficient for tumorigenicity of human colorectal carcinoma cells. Proc Natl Acad Sci U S A. 1998;95(7):3609–14. doi: 10.1073/pnas.95.7.3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esteban LM, Vicario-Abejon C, Fernandez-Salguero P, Fernandez-Medarde A, Swaminathan N, Yienger K, Lopez E, Malumbres M, McKay R, Ward JM, Pellicer A, Santos E. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol Cell Biol. 2001;21(5):1444–52. doi: 10.1128/MCB.21.5.1444-1452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koera K, Nakamura K, Nakao K, Miyoshi J, Toyoshima K, Hatta T, Otani H, Aiba A, Katsuki M. K-ras is essential for the development of the mouse embryo. Oncogene. 1997;15(10):1151–9. doi: 10.1038/sj.onc.1201284. [DOI] [PubMed] [Google Scholar]
- 10.Bender RH, Haigis KM, Gutmann DH. Activated k-ras, but not h-ras or N-ras, regulates brain neural stem cell proliferation in a raf/rb-dependent manner. Stem Cells. 2015;33(6):1998–2010. doi: 10.1002/stem.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rushworth LK, Hindley AD, O’Neill E, Kolch W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol. 2006;26(6):2262–72. doi: 10.1128/MCB.26.6.2262-2272.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 1999;18(8):2137–48. doi: 10.1093/emboj/18.8.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marais R, Light Y, Paterson HF, Mason CS, Marshall CJ. Differential regulation of Raf-1, A-Raf and B-Raf by oncogenic ras and tyrosine kinases. J Biol Chem. 1997;272(7):4378–83. doi: 10.1074/jbc.272.7.4378. [DOI] [PubMed] [Google Scholar]
- 14.Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351(Pt 2):289–305. [PMC free article] [PubMed] [Google Scholar]
- 15.Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, Sundararajan D, Chen WS, Crawford SE, Coleman KG, Hay N. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17(11):1352–65. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25(5):1869–78. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anastasaki C, Gutmann DH. Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum Mol Genet. 2014;23(25):6712–21. doi: 10.1093/hmg/ddu389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74(1):205–14. doi: 10.1016/0092-8674(93)90307-c. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370(6490):527–32. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 20.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–67. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 21.Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, Franklin RA, McCubrey JA. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17(7):1263–93. doi: 10.1038/sj.leu.2402945. [DOI] [PubMed] [Google Scholar]
- 22.Kaul A, Toonen JA, Cimino PJ, Gianino SM, Gutmann DH. Akt- or MEK-mediated mTOR inhibition suppresses Nf1 optic glioma growth. Neuro Oncol. 2015;17(6):843–53. doi: 10.1093/neuonc/nou329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YH, Gianino SM, Gutmann DH. Neurofibromatosis-1 regulation of neural stem cell proliferation and multilineage differentiation operates through distinct RAS effector pathways. Genes Dev. 2015;29(16):1677–82. doi: 10.1101/gad.261677.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2(4):339–45. [PubMed] [Google Scholar]
- 25.Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344(Pt 2):427–31. [PMC free article] [PubMed] [Google Scholar]
- 26.Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ, Roth RA. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem. 2003;278(12):10189–94. doi: 10.1074/jbc.M210837200. [DOI] [PubMed] [Google Scholar]
- 27.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4(9):658–65. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 28.Mendez R, Kollmorgen G, White MF, Rhoads RE. Requirement of protein kinase C zeta for stimulation of protein synthesis by insulin. Mol Cell Biol. 1997;17(9):5184–92. doi: 10.1128/mcb.17.9.5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diaz-Meco MT, Lozano J, Municio MM, Berra E, Frutos S, Sanz L, Moscat J. Evidence for the in vitro and in vivo interaction of Ras with protein kinase C zeta. J Biol Chem. 1994;269(50):31706–10. [PubMed] [Google Scholar]
- 30.Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278(18):15461–4. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 31.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 32.Gan X, Wang J, Wang C, Sommer E, Kozasa T, Srinivasula S, Alessi D, Offermanns S, Simon MI, Wu D. PRR5L degradation promotes mTORC2-mediated PKC-delta phosphorylation and cell migration downstream of Galpha12. Nat Cell Biol. 2012;14(7):686–96. doi: 10.1038/ncb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pearce LR, Sommer EM, Sakamoto K, Wullschleger S, Alessi DR. Protor-1 is required for efficient mTORC2-mediated activation of SGK1 in the kidney. Biochem J. 2011;436(1):169–79. doi: 10.1042/BJ20102103. [DOI] [PubMed] [Google Scholar]
- 34.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122–8. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 35.Banerjee S, Crouse NR, Emnett RJ, Gianino SM, Gutmann DH. Neurofibromatosis-1 regulates mTOR-mediated astrocyte growth and glioma formation in a TSC/Rheb-independent manner. Proc Natl Acad Sci U S A. 2011;108(38):15996–6001. doi: 10.1073/pnas.1019012108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee da Y, Yeh TH, Emnett RJ, White CR, Gutmann DH. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev. 2010;24(20):2317–29. doi: 10.1101/gad.1957110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banerjee S, Byrd JN, Gianino SM, Harpstrite SE, Rodriguez FJ, Tuskan RG, Reilly KM, Piwnica-Worms DR, Gutmann DH. The neurofibromatosis type 1 tumor suppressor controls cell growth by regulating signal transducer and activator of transcription-3 activity in vitro and in vivo. Cancer Res. 2010;70(4):1356–66. doi: 10.1158/0008-5472.CAN-09-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDaniel AS, Allen JD, Park SJ, Jaffer ZM, Michels EG, Burgin SJ, Chen S, Bessler WK, Hofmann C, Ingram DA, Chernoff J, Clapp DW. Pak1 regulates multiple c-Kit mediated Ras-MAPK gain-in-function phenotypes in Nf1+/− mast cells. Blood. 2008;112(12):4646–54. doi: 10.1182/blood-2008-04-155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elefteriou F, Benson MD, Sowa H, Starbuck M, Liu X, Ron D, Parada LF, Karsenty G. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 2006;4(6):441–51. doi: 10.1016/j.cmet.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daginakatte GC, Gianino SM, Zhao NW, Parsadanian AS, Gutmann DH. Increased c-Jun-NH2-kinase signaling in neurofibromatosis-1 heterozygous microglia drives microglia activation and promotes optic glioma proliferation. Cancer Res. 2008;68(24):10358–66. doi: 10.1158/0008-5472.CAN-08-2506. [DOI] [PubMed] [Google Scholar]
- 41.Kaul A, Chen YH, Emnett RJ, Dahiya S, Gutmann DH. Pediatric glioma-associated KIAA1549:BRAF expression regulates neuroglial cell growth in a cell type-specific and mTOR-dependent manner. Genes Dev. 2012;26(23):2561–6. doi: 10.1101/gad.200907.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yeh TH, Lee da Y, Gianino SM, Gutmann DH. Microarray analyses reveal regional astrocyte heterogeneity with implications for neurofibromatosis type 1 (NF1)-regulated glial proliferation. Glia. 2009;57(11):1239–49. doi: 10.1002/glia.20845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown JA, Gianino SM, Gutmann DH. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J Neurosci. 2010;30(16):5579–89. doi: 10.1523/JNEUROSCI.3994-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vogel KS, Brannan CI, Jenkins NA, Copeland NG, Parada LF. Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell. 1995;82(5):733–42. doi: 10.1016/0092-8674(95)90470-0. [DOI] [PubMed] [Google Scholar]
- 45.Lathia JD, Li M, Hall PE, Gallagher J, Hale JS, Wu Q, Venere M, Levy E, Rani MR, Huang P, Bae E, Selfridge J, Cheng L, Guvenc H, McLendon RE, Nakano I, Sloan AE, Phillips HS, Lai A, Gladson CL, Bredel M, Bao S, Hjelmeland AB, Rich JN. Laminin alpha 2 enables glioblastoma stem cell growth. Ann Neurol. 2012;72(5):766–78. doi: 10.1002/ana.23674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang P, Rani MR, Ahluwalia MS, Bae E, Prayson RA, Weil RJ, Nowacki AS, Hedayat H, Sloan AE, Lathia JD, Rich JN, Tipps R, Gladson CL. Endothelial expression of TNF receptor-1 generates a proapoptotic signal inhibited by integrin alpha6beta1 in glioblastoma. Cancer Res. 2012;72(6):1428–37. doi: 10.1158/0008-5472.CAN-11-2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, Yang X, Knowles S, Horn W, Li Y, Zhang S, Yang Y, Vakili ST, Yu M, Burns D, Robertson K, Hutchins G, Parada LF, Clapp DW. Nf1-dependent tumors require a microenvironment containing Nf1+/−- and c-kit-dependent bone marrow. Cell. 2008;135(3):437–48. doi: 10.1016/j.cell.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen S, Burgin S, McDaniel A, Li X, Yuan J, Chen M, Khalaf W, Clapp DW, Yang FC. Nf1−/− Schwann cell-conditioned medium modulates mast cell degranulation by c-Kit-mediated hyperactivation of phosphatidylinositol 3-kinase. Am J Pathol. 2010;177(6):3125–32. doi: 10.2353/ajpath.2010.100369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang FC, Ingram DA, Chen S, Hingtgen CM, Ratner N, Monk KR, Clegg T, White H, Mead L, Wenning MJ, Williams DA, Kapur R, Atkinson SJ, Clapp DW. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/− mast cells. J Clin Invest. 2003;112(12):1851–61. doi: 10.1172/JCI19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simmons GW, Pong WW, Emnett RJ, White CR, Gianino SM, Rodriguez FJ, Gutmann DH. Neurofibromatosis-1 heterozygosity increases microglia in a spatially and temporally restricted pattern relevant to mouse optic glioma formation and growth. J Neuropathol Exp Neurol. 2011;70(1):51–62. doi: 10.1097/NEN.0b013e3182032d37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet. 2007;16(9):1098–112. doi: 10.1093/hmg/ddm059. [DOI] [PubMed] [Google Scholar]
- 52.Pong WW, Higer SB, Gianino SM, Emnett RJ, Gutmann DH. Reduced microglial CX3CR1 expression delays neurofibromatosis-1 glioma formation. Ann Neurol. 2013;73(2):303–8. doi: 10.1002/ana.23813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Solga AC, Pong WW, Kim KY, Cimino PJ, Toonen JA, Walker J, Wylie T, Magrini V, Griffith M, Griffith OL, Ly A, Ellisman MH, Mardis ER, Gutmann DH. RNA Sequencing of Tumor-Associated Microglia Reveals Ccl5 as a Stromal Chemokine Critical for Neurofibromatosis-1 Glioma Growth. Neoplasia. 2015;17(10):776–88. doi: 10.1016/j.neo.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toonen JA, Anastasaki C, Smithson LJ, Gianino SM, Li K, Kesterson RA, Gutmann DH. NF1 germline mutation differentially dictates optic glioma formation and growth in neurofibromatosis-1. Hum Mol Genet. 2016 doi: 10.1093/hmg/ddw039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, Gibson EM, Mount CW, Polepalli J, Mitra SS, Woo PJ, Malenka RC, Vogel H, Bredel M, Mallick P, Monje M. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell. 2015;161(4):803–16. doi: 10.1016/j.cell.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pinna V, Lanari V, Daniele P, Consoli F, Agolini E, Margiotti K, Bottillo I, Torrente I, Bruselles A, Fusilli C, Ficcadenti A, Bargiacchi S, Trevisson E, Forzan M, Giustini S, Leoni C, Zampino G, Digilio MC, Dallapiccola B, Clementi M, Tartaglia M, De Luca A. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur J Hum Genet. 2015;23(8):1068–71. doi: 10.1038/ejhg.2014.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rojnueangnit K, Xie J, Gomes A, Sharp A, Callens T, Chen Y, Liu Y, Cochran M, Abbott MA, Atkin J, Babovic-Vuksanovic D, Barnett CP, Crenshaw M, Bartholomew DW, Basel L, Bellus G, Ben-Shachar S, Bialer MG, Bick D, Blumberg B, Cortes F, David KL, Destree A, Duat-Rodriguez A, Earl D, Escobar L, Eswara M, Ezquieta B, Frayling IM, Frydman M, Gardner K, Gripp KW, Hernandez-Chico C, Heyrman K, Ibrahim J, Janssens S, Keena BA, Llano-Rivas I, Leppig K, McDonald M, Misra VK, Mulbury J, Narayanan V, Orenstein N, Galvin-Parton P, Pedro H, Pivnick EK, Powell CM, Randolph L, Raskin S, Rosell J, Rubin K, Seashore M, Schaaf CP, Scheuerle A, Schultz M, Schorry E, Schnur R, Siqveland E, Tkachuk A, Tonsgard J, Upadhyaya M, Verma IC, Wallace S, Williams C, Zackai E, Zonana J, Lazaro C, Claes K, Korf B, Martin Y, Legius E, Messiaen L. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation. Hum Mutat. 2015;36(11):1052–63. doi: 10.1002/humu.22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Upadhyaya M, Huson SM, Davies M, Thomas N, Chuzhanova N, Giovannini S, Evans DG, Howard E, Kerr B, Griffiths S, Consoli C, Side L, Adams D, Pierpont M, Hachen R, Barnicoat A, Li H, Wallace P, Van Biervliet JP, Stevenson D, Viskochil D, Baralle D, Haan E, Riccardi V, Turnpenny P, Lazaro C, Messiaen L. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet. 2007;80(1):140–51. doi: 10.1086/510781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anastasaki C, Woo AS, Messiaen LM, Gutmann DH. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum Mol Genet. 2015;24(12):3518–28. doi: 10.1093/hmg/ddv103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodriguez EF, Scheithauer BW, Giannini C, Rynearson A, Cen L, Hoesley B, Gilmer-Flynn H, Sarkaria JN, Jenkins S, Long J, Rodriguez FJ. PI3K/AKT pathway alterations are associated with clinically aggressive and histologically anaplastic subsets of pilocytic astrocytoma. Acta Neuropathol. 2011;121(3):407–20. doi: 10.1007/s00401-010-0784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodriguez FJ, Ligon AH, Horkayne-Szakaly I, Rushing EJ, Ligon KL, Vena N, Garcia DI, Cameron JD, Eberhart CG. BRAF duplications and MAPK pathway activation are frequent in gliomas of the optic nerve proper. J Neuropathol Exp Neurol. 2012;71(9):789–94. doi: 10.1097/NEN.0b013e3182656ef8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaul A, Toonen JA, Gianino SM, Gutmann DH. The impact of coexisting genetic mutations on murine optic glioma biology. Neuro Oncol. 2015;17(5):670–7. doi: 10.1093/neuonc/nou287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abadin SS, Zoellner NL, Schaeffer M, Porcelli B, Gutmann DH, Johnson KJ. Racial/Ethnic Differences in Pediatric Brain Tumor Diagnoses in Patients with Neurofibromatosis Type 1. J Pediatr. 2015;167(3):613–20. e1–2. doi: 10.1016/j.jpeds.2015.04.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26(1):109–13. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- 65.Amlin-Van Schaick J, Kim S, Broman KW, Reilly KM. Scram1 is a modifier of spinal cord resistance for astrocytoma on mouse Chr 5. Mamm Genome. 2012;23(3–4):277–85. doi: 10.1007/s00335-011-9380-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walrath JC, Fox K, Truffer E, Gregory Alvord W, Quinones OA, Reilly KM. Chr 19(A/J) modifies tumor resistance in a sex- and parent-of-origin-specific manner. Mamm Genome. 2009;20(4):214–23. doi: 10.1007/s00335-009-9179-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hawes JJ, Tuskan RG, Reilly KM. Nf1 expression is dependent on strain background: implications for tumor suppressor haploinsufficiency studies. Neurogenetics. 2007;8(2):121–30. doi: 10.1007/s10048-006-0078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]