

Summary
Abnormal levels of reactive oxygen species (ROS) and inflammatory cytokines have been observed in the skeletal muscle during muscle wasting including sarcopenia. However, the mechanisms that signal ROS production and prolonged maintenance of ROS levels during muscle wasting are not fully understood. Here, we show that myostatin (Mstn) is a pro‐oxidant and signals the generation of ROS in muscle cells. Myostatin, a transforming growth factor‐β (TGF‐β) family member, has been shown to play an important role in skeletal muscle wasting by increasing protein degradation. Our results here show that Mstn induces oxidative stress by producing ROS in skeletal muscle cells through tumor necrosis factor‐α (TNF‐α) signaling via NF‐κB and NADPH oxidase. Aged Mstn null (Mstn−/−) muscles, which display reduced sarcopenia, also show an increased basal antioxidant enzyme (AOE) levels and lower NF‐κB levels indicating efficient scavenging of excess ROS. Additionally, our results indicate that both TNF‐α and hydrogen peroxide (H2O2) are potent inducers of Mstn and require NF‐κB signaling for Mstn induction. These results demonstrate that Mstn and TNF‐α are components of a feed forward loop in which Mstn triggers the generation of second messenger ROS, mediated by TNF‐α and NADPH oxidase, and the elevated TNF‐α in turn stimulates Mstn expression. Higher levels of Mstn in turn induce muscle wasting by activating proteasomal‐mediated catabolism of intracellular proteins. Thus, we propose that inhibition of ROS induced by Mstn could lead to reduced muscle wasting during sarcopenia.
Keywords: antioxidant enzyme, myostatin, reactive oxygen species, sarcopenia, skeletal muscle, tumor necrosis factor‐α
Introduction
Skeletal muscle is a metabolically active tissue that is highly susceptible to wasting during various disease conditions like cancer, chronic obstructive pulmonary disease, sepsis and also with aging (sarcopenia). Several studies have reported an increase in reactive oxygen species (ROS) in muscle in response to infection, inflammatory stimulus and/or aging (Sohal & Weindruch, 1996; Powers et al., 2005). Reactive oxygen species are molecules formed by incomplete reduction of oxygen, and under normal physiological conditions, ROS get detoxified in the cell with the help of the intracellular AOEs and non‐enzymatic antioxidants. Physiological levels of ROS play an important role in myoblast proliferation by regulating G1 to S phase transition (Jahnke et al., 2009). Reduced mitochondrial biogenesis, reduced H2O2 in the mitochondria and decreased release of Ca2+ are shown to be associated with the initiation of differentiation of myoblasts (Jahnke et al., 2009). Furthermore, in a recent study, Handayaningsih et al. (2011) have demonstrated that IGF‐I induced myotubes hypertrophy by signaling via ROS in vitro. However, excessive generation of ROS causes extensive oxidative damage to nucleic acids and proteins. Two main sources of ROS in the skeletal muscle are mitochondria and peroxisomes. During aging, mitochondrial dysfunction occurs owing to several causes including dysregulated expression of mitochondrial genes and mutations in mitochondrial DNA leading to oxidative stress (Aiken et al., 2002; Zahn et al., 2006; Herbst et al., 2007). In addition, lipid peroxidation is significantly higher in aged human skeletal muscle, which is indicative of oxidative stress (Mecocci et al., 1999; Pansarasa et al., 1999; Fanòet al., 2001).
In the aging muscle, higher levels of ROS have been linked to the occurrence of chronic inflammation (Peake et al., 2010). The combination of increased oxidative stress and inflammation during muscle wasting seems to be causal to the induction of cytokines such as TNF‐α and its downstream target NF‐κB and NAD(P)H oxidase (Kim et al., 2007). NF‐κB, a transcription factor, is generally sequestered in the cytoplasm bound to IκB in an inactive complex. Once activated by a stimulus including ROS, IκB gets phosphorylated by IKK and NF‐κB p65 subunit translocates from the cytoplasm into the nucleus and regulates the expression of its target genes. Some of the known downstream targets that NF‐κB up‐regulates include muscle‐specific E3 ligases such as MuRF1 involved in muscle wasting (Cai et al., 2004). NF‐κB is also implicated in the loss of myogenic regulatory factor myoD messenger RNA in skeletal muscle, thus enhancing the muscle degeneration (Guttridge et al., 2000).
In recent years, a growth and differentiation factor, myostatin (Mstn) has emerged as an inducer of muscle wasting. Myostatin is an inhibitor of myogenesis regulating both the proliferation and the differentiation of myoblasts (Thomas et al., 2000; Langley et al., 2002) and overexpression of Mstn in vivo results in cachexia in mice (Zimmers et al., 2002; Reisz‐Porszasz et al., 2003). One of the mechanisms by which Mstn has been shown to induce cachexia is by activating the ubiquitin proteolytic system through FoxO1 (McFarlane et al., 2006). In recent past, several studies have reported an increase in Mstn mRNA and/or protein levels during aging in humans and rodents (Yarasheski et al., 2002; Baumann et al., 2003; Raue et al., 2006; Léger et al., 2008), while Welle et al. (2002) reported that Mstn mRNA levels were unchanged during aging. Using Mstn−/− mice, it was found that lack of Mstn helped muscle regenerate better in the old mice and recently it was reported that Mstn inactivation enhanced bone density, insulin sensitivity, and heart function in aged mice (Siriett et al., 2007; Morissette et al., 2009). Overall, Mstn seems to be playing a role in muscle function during aging also.
It is well established now that skeletal muscle is prone to oxidative stress during aging, which augments chronic low‐level inflammation in the muscle by regulating factors like TNF‐α, IL6, IL‐1, and NF‐κB, which in turn can cause oxidative stress. The combined impact of the oxidative stress and inflammation is thought to be responsible for perturbation of the anabolic signaling proteins such as insulin‐like growth factor 1 and activating catabolic signals such as proteasomal‐mediated degradation of proteins. However, what has not been very well understood so far are the mechanisms that initiate TNF‐α induction and thereby activate ROS production. Furthermore, the molecular mechanisms that maintain the chronic elevated levels of ROS during aging have also not been thoroughly understood.
Here, in this manuscript we present evidence that Mstn is a pro‐oxidant capable of signaling ROS production via NF‐κB and TNF‐α in muscle cells. Furthermore, we show that Mstn feeds forward the expression of redox‐sensitive transcription factor, NF‐κB, through induction of pro‐inflammatory cytokine TNF‐α. Lack of functional Mstn offers resistance to oxidative stress in the aging muscle by enhancing the antioxidant response and reducing NF‐κB levels. Based on our results, we speculate that Mstn plays a critical role in integrating redox signaling, inflammatory and catabolic signaling pathways during ROS‐mediated muscle wasting.
Results
Mstn induces ROS in differentiating C2C12 myoblasts
Increased levels of Mstn are associated with cachexia, sarcopenia, and disuse atrophy (Yarasheski et al., 2002; McFarlane et al., 2006). Similarly, ROS or oxidative stress plays an important role in inducing muscle atrophy (Powers et al., 2005), sarcopenia, and cachexia (Li & Reid, 2000; Gomes‐Marcondes & Tisdale, 2002; Mantovani et al., 2004; Moylan & Reid, 2007). Hence, we asked whether Mstn could induce ROS production by treating differentiating C2C12 cells with Mstn and estimating the levels of ROS. The results show that ROS production was significantly increased upon Mstn treatment (Fig. 1A,B).
Figure 1.
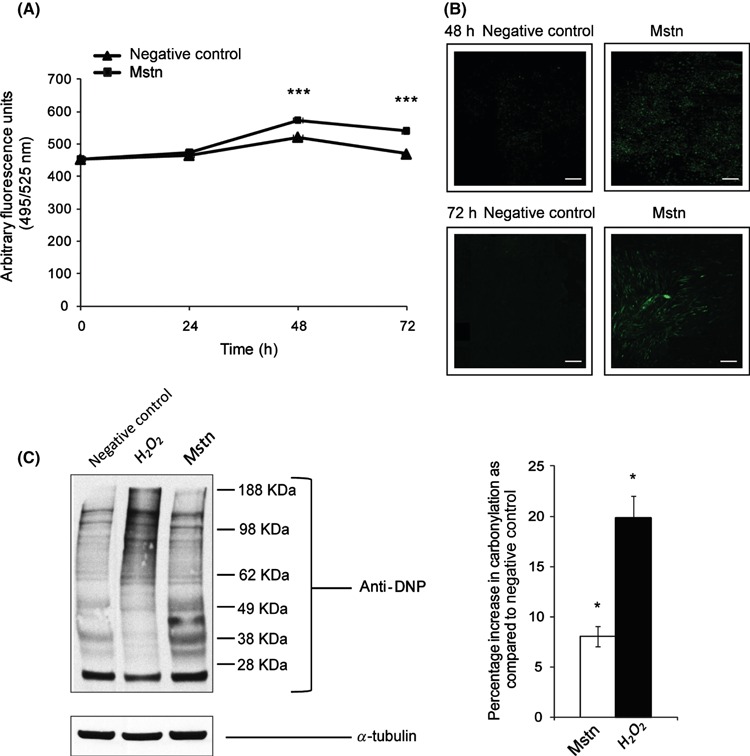
Myostatin (Mstn) induces reactive oxygen species (ROS) in C2C12 myoblasts. (A) ROS production was measured using a fluorescent multilabel plate reader in differentiating C2C12 cells treated with Mstn (3.5 ng mL−1) using a fluorescent probe, CM‐H2DCFDA (Molecular Probes). The graph shows relative intensities expressed as arbitrary fluorescent units. ROS production is significantly increased (***P < 0.001) upon Mstn treatment during differentiation when compared to the untreated cells (n = 4). (B) ROS production was also measured in Mstn‐treated C2C12 cells in Permanox chamber slides. The fluorescence was viewed under the Leica upright microscope, and images were taken at 10× magnification. Increased fluorescence (green) intensity is directly proportional to increased ROS production in cells (n = 4). (C) Effect of Mstn and H2O2 on protein carbonylation in C2C12 myoblasts. Left panel – representative gel showing the increased protein carbonylation in H2O2 (0.05 mm) or Mstn (3.5 ng mL−1) treated C2C12 cells as detected by Oxyblot assay kit (Millipore Corp.). Right panel – corresponding densitometry analysis of the blot showing the percentage increase in carbonylation as compared to the untreated cells (*P < 0.05) (n = 2). α‐tubulin was used as an internal control for equal protein loading on the gel.
Mstn increases protein carbonylation in C2C12 myoblasts
Reactive oxygen species are molecules that cause extensive oxidative damage to nucleic acids, proteins, and various components of the cell. The protein oxidation by ROS leads to various protein modifications including protein carbonylation (Stadtman, 2001). To find out whether ROS induced by Mstn results in oxidation and subsequent carbonylation, protein lysates of C2C12 cells treated with H2O2 (potent pro‐oxidant) or Mstn were subjected to immunoblotting with anti‐DNP antibody. Densitometry analysis of the oxyblot clearly indicates an increased protein carbonylation in response to H2O2 and Mstn treatment of C2C12 cells (Fig. 1C). Hence, the results indicate that ROS produced by Mstn causes protein carbonylation in C2C12 myoblasts.
Mstn up‐regulates the activity of AOEs and their gene expression in differentiating C2C12 myoblasts
Under normal physiological conditions, cells respond to the changes in ROS levels by the production of non‐enzymatic antioxidants and AOEs. To investigate whether ROS induced by Mstn also elicit changes in antioxidant enzyme levels, enzyme activities of superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) were determined in protein extracts from C2C12 cells treated with Mstn. Increase in the activity of SOD (Fig. 2A), CAT (Fig. 2B), and GPx (Fig. 2C) was observed in differentiating C2C12 cells treated with Mstn. To determine whether Mstn increases the transcription of the genes encoding AOE, RT–qPCR analysis was performed. The results show that mRNA expression of Sod1 (Fig. 2E) and Cat (Fig. 2F) was up‐regulated upon Mstn treatment during C2C12 myoblast differentiation. To determine whether Mstn treatment also regulates the levels of non‐enzymatic antioxidant‐reduced glutathione (GSR), the activity of GSR produced in differentiating C2C12 cells was assayed. Results show that GSR levels are significantly elevated on treatment with Mstn (Fig. 2D). Together, these results indicate that Mstn increases the levels of AOEs and GSR in differentiating C2C12 myotubes. Similarly, Mstn also increased the AOE and GSR enzyme activities in primary myoblasts isolated from 8‐week‐old wild‐type (WT) mice (data not shown). However, in Mstn−/− primary myoblasts isolated from 8‐week‐old Mstn−/− mice, Mstn failed to increase the expression and activity of AOEs (data not shown). These results indicate that Mstn induces AOEs in WT primary myoblasts, whereas Mstn−/− primary myoblasts are resistant to oxidative stress generated by Mstn, and thus, the AOE activity remained unchanged on Mstn treatment.
Figure 2.
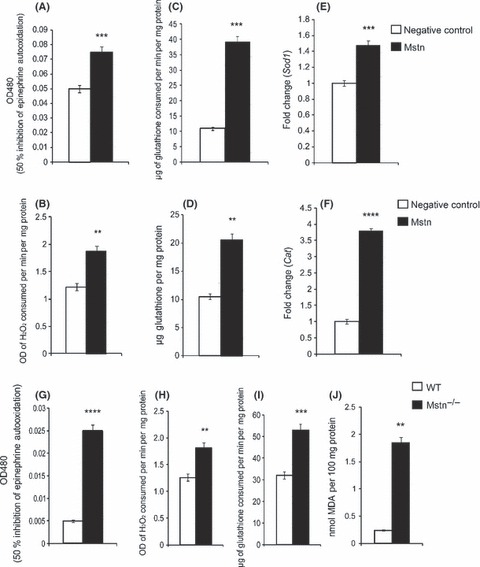
Myostatin (Mstn) up‐regulates the activity and mRNA expression of AOEs in differentiating C2C12 myoblasts. C2C12 cells were treated with Mstn (3 μg mL−1) in the differentiation media, and total cell lysates were made at indicated time points. The enzyme assays were performed for SOD (A), CAT (B), GPx (C), and GSR (D). The values are mean ± SE of four independent experiments: **P < 0.01 and ***P < 0.001 when compared to the negative control (n = 4). C2C12 cells were treated with Mstn (3.5 ng mL−1) and gene expression analysis was performed for Sod1 and Cat. The values are mean ± SE of four independent experiments. An increase in gene expression of Sod1 (E, ***P < 0.001) and Cat (F, ****P < 0.0001) was observed upon Mstn treatment during differentiation (n = 4). Mstn−/− primary myoblasts have elevated basal AOE levels and increased lipid peroxidation. Enzyme activities of SOD (G), CAT (H), and GPx (I) were analyzed in the total cell lysates of differentiating primary myoblasts from WT and Mstn−/− mice (8‐week old) at indicated time points. The values are given as mean ± SE of four independent experiments: **P < 0.05, ***P < 0.001, and ****P < 0.0001 denote significant increase compared to the WT primary myoblasts. (J) The lipid peroxidation was determined in the differentiating primary myoblasts at the indicated time point. The values are given as nmoles of malonaldehyde produced and are mean ± SE of four independent experiments; **P < 0.01 denotes significant increase relative to the WT primary myoblasts.
Basal AOE activity in Mstn−/− mice is higher than the WT mice
To further understand the regulation of antioxidant enzyme gene expression by Mstn, the basal levels of the AOE activity during differentiation were compared between the WT and Mstn−/− primary myoblasts. Our results show that Mstn−/− myoblasts have higher basal levels of the antioxidant enzymes (SOD, CAT and GPx) when compared to the WT myoblasts (Fig. 2G,H,I, respectively).
NADH/NAD(P)H oxidase, enzymes of mitochondrial ETC (mETC) and TNF‐α are involved in ROS production by Mstn
To investigate the mechanism involved in Mstn‐induced ROS generation, cell signaling inhibitors, which include Indomethacin, L‐NAME, Oxypurinol, Quinacrine, Diphenylene iodonium chloride, 4,4′‐Diisothiocyanatostilbene‐2,2′‐disulfonic acid disodium salt (DIDS), Carbonyl cyanide 3‐chlorophenylhydrazone (CCCP), and 2‐Thenoyltrifluoroacetone (TTFA) (Table 1), that inhibit various pathways and enzymes involved in ROS production were used (Iuchi et al., 2003). Reactive oxygen species production by Mstn was significantly inhibited in cells treated with Quinacrine, DIDS, CCCP, and TTFA inhibitors (Fig. 3A). These inhibitors block free radical generation by NADH/NAD(P)H oxidase, the various enzymes and complexes of mitochondria and mETC. Thus, the inhibitor studies show that NADH/NAD(P)H oxidase and mETC are involved in the production of ROS by Mstn in muscle cells. To further establish that NADPH oxidase is involved in Mstn signaling, the mRNA expression of NADPH oxidase 1 (Nox1) in differentiating C2C12 myoblasts treated with Mstn was analyzed. The results show an up‐regulation of Nox1 (Fig. 3B) upon treatment with Mstn.
Table 1.
The reactive oxygen species inhibitors, the respective enzymes/pathways inhibited, and the concentration used
| Inhibitor | Enzymes/pathways inhibited | Concentration used (μm) | Duration of treatment (min) |
|---|---|---|---|
| Indomethacin | Cyclooxygenase | 100 | 15 |
| L‐NAME (Nω‐nitro‐L‐arginine methyl ester) | NOS | 1 | 15 |
| Oxypurinol | Xanthine oxidase | 100 | 15 |
| Quinacrine | NADH oxidase | 6 | 5 |
| Diphenylene iodonium chloride | NAD(P)H oxidase, NOS and Complex I of mETC | 50 | 5 |
| 4,4′‐Diisothiocyanatostilbene‐2,2′‐disulfonic acid disodium salt (DIDS) | Superoxide release from mitochondria | 100 | 10 |
| Carbonyl cyanide 3‐chlorophenylhydrazone (CCCP) | Mitochondrial membrane proton gradient | 0.5 | Along with Mstn in differentiation media |
| 2‐Thenoyltrifluoroacetone (TTFA) | Complex II of mETC | 100 | Along with Mstn in differentiation media |
Figure 3.
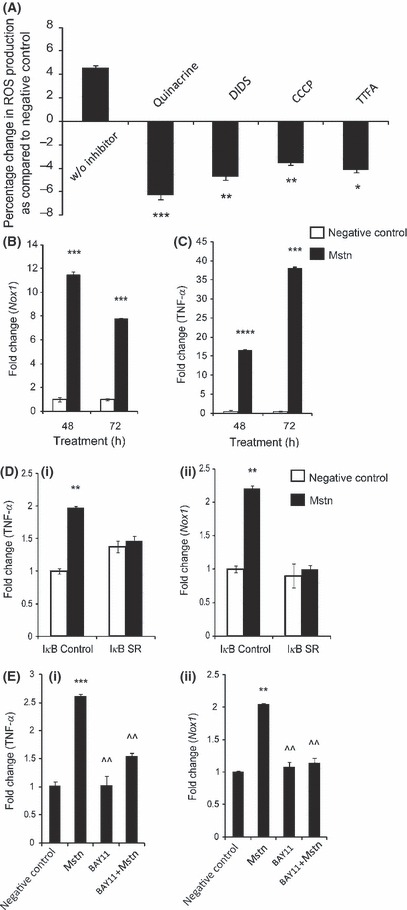
Effect of the inhibitors of NADH/NAD(P)H oxidase and enzymes of mitochondrial ETC (mETC) on myostatin (Mstn)‐induced reactive oxygen species (ROS) generation. (A) C2C12 cells were treated for 48 h with Mstn (3.5 ng mL−1) in the presence of ROS cell signaling inhibitors, and ROS content was analyzed using the fluorescent probe, CM‐H2DCFDA (Molecular Probes). Reactive oxygen species production was significantly decreased (*P < 0.05, **P < 0.01 and ***P < 0.001) in cells treated with Quinacrine, DIDS, CCCP, and TTFA when compared to the cells treated with Mstn alone (no inhibitor). The data are expressed as percentage increase or decrease in ROS production (n = 4). mRNA expression for Nox1 and tumor necrosis factor‐α (TNF‐α ) was determined in C2C12 cells treated with Mstn (3.5 ng mL−1) in differentiation media at indicated time points. The values are mean ± SE of four independent experiments. ***P < 0.001 and ****P < 0.0001 denote significant fold increase in Nox1 (B) and TNF‐α (C) mRNA expression relative to the negative control. Mstn induces ROS through TNF‐α via NF‐κB signaling. (D) Representative graph showing mRNA expression of TNF‐α (i) and Nox1 (ii) in IκB‐α control and IκB‐α‐SR‐expressing C2C12 cells which lack NF‐κB activity treated for 48 h with Mstn (3.5 ng mL−1) in proliferation media. The values are mean ± SE of two independent experiments. **P < 0.01 denotes significant increase when compared to untreated cells (n = 2). (E) Representative graph showing mRNA expression of TNF‐α (i) and Nox1 (ii) in C2C12 cells treated for 1 h with BAY11‐7085 (20 μm mL−1) and for 48 h with Mstn (3.5 ng mL−1) in proliferation media. The values are mean ± SE of two independent experiments. **P < 0.01 and ***P < 0.001 denote significant increase when compared to untreated cells; ^^P < 0.01 denotes significant decrease when compared to Mstn‐treated cells (n = 2).
Lipid peroxidation is also known to give rise to ROS; therefore, we treated WT myoblasts with Mstn and analyzed the lipid peroxidation by measuring thiobarbituric acid–reactive substances (TBARS) expressed in terms of malonaldehyde (MDA). Thiobarbituric acid–reactive substances estimation has been commonly used previously but its drawbacks are overestimation of the product and inconsistency (Garcia et al., 2005). Nevertheless, Mstn treatment of WT myoblasts decreased TBARS levels (Fig. S1A, Supporting information), suggesting that Mstn induced the generation of ROS independent of lipid peroxidation. An increase in lipid peroxidation during differentiation in Mstn−/− myotubes resulting in increased production of TBARS was observed (Fig. 2J). This increase in lipid peroxidation could be attributed to the increased number of mitochondria in the Mstn−/− muscles (unpublished results from our laboratory).
Mstn induces ROS through TNF‐α and NF‐κB signaling
As TNF‐α is known to regulate NADPH oxidase (Kim et al., 2007), we next asked whether Mstn induces TNF‐α gene expression, which in turn up‐regulates Nox1 gene. The RT–qPCR results confirmed that Mstn treatment potently induces TNF‐α expression in differentiating C2C12 cells (Fig. 3C). To ascertain whether Mstn requires NF‐κB activity to induce TNF‐α and Nox1, we used a stable C2C12 cell line that expresses the IκB‐α SR protein. The IκB‐α SR protein acts as a potent and specific inhibitor of NF‐κB activity (Guttridge et al., 1999). As shown in Fig. 3D, treatment of control cells with Mstn leads to an increase in TNF‐α (Fig. 3D (i)) and Nox1 (Fig. 3D (ii)) expression. However, in IκB‐α‐SR‐expressing C2C12 cells, Mstn fails to induce the expression of TNF‐α (Fig. 3D (i)) and Nox1 (Fig. 3D (ii)). In another experiment, NF‐κB activity was inhibited by the compound BAY 11‐7085, which prevents NF‐κB activation by blocking IκB‐α phosphorylation (Ladner et al., 2003). When C2C12 cells were first treated with BAY 11‐7085 compound, Mstn failed to up‐regulate TNF‐α and Nox1 mRNA expression (Fig. 3E (i) and (ii), respectively). Hence, these results confirm that Mstn requires NF‐κB activity to induce TNF‐α and Nox1 expression.
Normally, NF‐κB exists in the cytoplasm in an inactive form bound to IκB‐α. Various stimuli, for example, TNF‐α are known to transduce the signal starting at proximal kinase IKK, which is known to phosphorylate IκB‐α on Serine32/36. The phosphorylation of IκB‐α is necessary for its degradation and release of NF‐κB from the inactive complex. Concomitantly, the phosphorylation of NF‐κB and its translocation from the cytoplasm to the nucleus renders NF‐κB to bind to the regulatory sequences of the target genes. Therefore, we next studied the signaling events leading to the activation of NF‐κB upon Mstn treatment. Whole cell lysates, nuclear and cytoplasmic extracts of C2C12 cells treated with Mstn during proliferation were analyzed by Western blotting using NF‐κB p65, p‐NF‐κB p65, IκB‐α, p‐IκB‐α, and IKKα antibodies (Fig. 4A). Whole cell protein lysates did not show a significant difference in NF‐κB p65 level on Mstn treatment (Lanes 2 and 4); however, nuclear extracts showed increased NF‐κB p65 level upon 48 and 72 h of Mstn treatment (Lanes 2 and 4) compared to the untreated cells (Lanes 1 and 3). A decrease in NF‐κB p65 content was observed in the cytoplasmic extract at 48 h on Mstn treatment (Lane 2), at the same time a decrease in p‐NF‐κB p65 level (Fig. 4A; Right panel (i)) was observed in the nuclear extract (Lane 2). An increase in NF‐κB p65 level in cytoplasmic extract and p‐NF‐κB p65 level in nuclear extract was observed on 72 h of Mstn treatment (Lane 4). After 48 h of treatment with Mstn, IκB‐α content was decreased (Lane 2) and p‐IκB‐α level (Fig. 4A; Right panel (ii)) remained unchanged (Lane 2), whereas 72 h of Mstn treatment increased IκB‐α level (Lane 4) and p‐IκB‐α level (Lane 4). IKKα level is increased in C2C12 cells treated for 72 h with Mstn (Lane 4). Thus, these results indicate that Mstn is able to stimulate degradation of IκB‐α by increasing IKKα and activating NF‐κB transcription factor.
Figure 4.
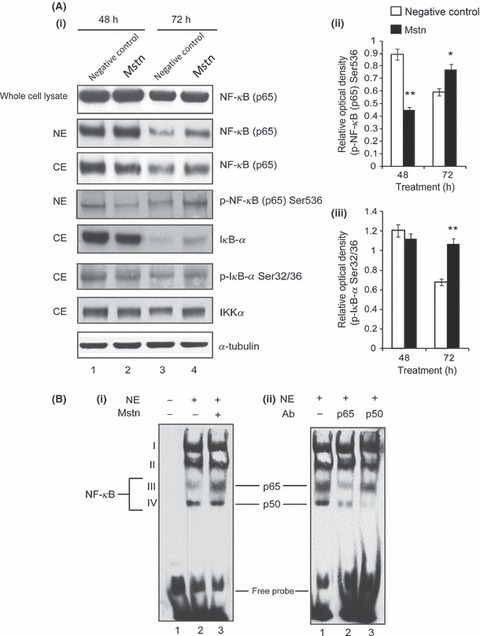
Myostatin (Mstn) induces reactive oxygen species (ROS) through tumor necrosis factor‐α (TNF‐α) via NF‐κB signaling. (A) Effect of Mstn on NF‐κB and IκB‐α in proliferating C2C12 myoblasts. Western blotting analysis was performed on whole cell lysates, nuclear and cytoplasmic extracts obtained from C2C12 cells treated with Mstn (3.5 ng mL−1) for indicated time points. (i) Left panel – representative immunoblot showing protein levels of NF‐κB (p65), p‐NF‐κB (p65), IκB‐α, p‐IκB‐α, and IKKα in negative control (Lanes 1 and 3) and Mstn (Lanes 2 and 4)‐treated whole protein lysates, nuclear and cytoplasmic extracts at indicated time points. α‐tubulin was used as an internal control for equal protein loading on the gel. Right panel – corresponding densitometry analysis of (ii) p‐NF‐κB (p65) and (iii) p‐IκB‐α showing significant increase or decrease in protein content upon Mstn treatment (*P < 0.05, **P < 0.01) (n = 2). (B) Mstn‐mediated enhancement of NF‐κB binding activity on mouse TNF‐α promoter. Electrophoretic mobility shift assay was performed using nuclear extracts from proliferating C2C12 cells treated with Mstn (3.5 ng mL−1) for 48 h. (i) Left panel – representative gel showing the increased activity of NF‐κB upon Mstn treatment. Lane 1, oligo only; Lane 2, untreated; Lane 3, Mstn treated. Electrophoretic mobility shift assay was also performed with nuclear extracts pre‐incubated with the specific antibody. (ii) Right panel – representative gel showing NF‐κB complexes containing p65 or p50 subunits. Lane 1, no antibody added; Lane 2, p65‐specific antibody added; Lane 3, p50‐specific antibody added (n = 2).
As Mstn treatment results in increased translocation of NF‐κB into the nucleus, we next performed experiments to investigate whether Mstn treatment results in enhanced binding of NF‐κB to the NF‐κB binding sites on TNF‐α promoter. Electrophoretic mobility shift assay (EMSA) was performed using nuclear extracts made from C2C12 cells treated with Mstn and the oligonucleotides bearing NF‐κB binding site of the mouse TNF‐α promoter. Four shifted bands (complex I, II, III, and IV) were observed on EMSA, and addition of Mstn to C2C12 cells led to increased NF‐κB DNA binding activity as shown by an increased level of complex III (Fig. 4B (i); Lanes 2 and 3). To prove that complex III and IV contains p65 and p50, respectively, we performed a super shift analysis with specific antibodies. Our results revealed that complex III could be super shifted (and hence diminished band intensity) with p65‐specific antibodies, whereas and complex IV was super shifted with p50 subunit‐specific antibodies (Fig. 4B (ii); Lanes 2 and 3). These observations further indicate that increased availability of NF‐κB in nucleus because of Mstn signaling results in enhanced binding to the NF‐κB binding sites in TNF‐α promoter/enhancer sequences.
5‐Aminosalicylic acid (5‐ASA) inhibits NF‐κB activation and IκB degradation; therefore, we next used 5‐ASA to further confirm that NF‐κB activity is required for Mstn‐mediated up‐regulation of TNF‐α and Nox1. Our results show that TNF‐α (Fig. 5A) and Nox1 (Fig. 5B) expression was significantly down‐regulated with 5‐ASA treatment, along with Mstn in comparison with cells treated with Mstn only. Western blotting analysis for NF‐κB p65 was performed on whole cell lysates, nuclear and cytoplasmic extracts of proliferating C2C12 cells treated for 48 h with Mstn along with 5‐ASA. Whole cell protein lysates showed significant decrease in NF‐κB p65 level in cells treated with 5‐ASA (Fig. 5C; Lanes 3 and 4) when compared to untreated (Lane 1) and Mstn‐treated cells (Lane 2); nuclear extracts show significant decrease in NF‐κB p65 levels (Fig. 5C) on treatment with 5‐ASA (Lane 3) along with Mstn (Lane 4) when compared to cells treated with Mstn only (Lane 2); cytoplasmic extracts show corresponding increase in NF‐κB p65 levels (Fig. 5C; Lanes 1 and 3). These results indicate that Mstn‐mediated activation and translocation of NF‐κB is required for TNF‐α and Nox1 expression.
Figure 5.
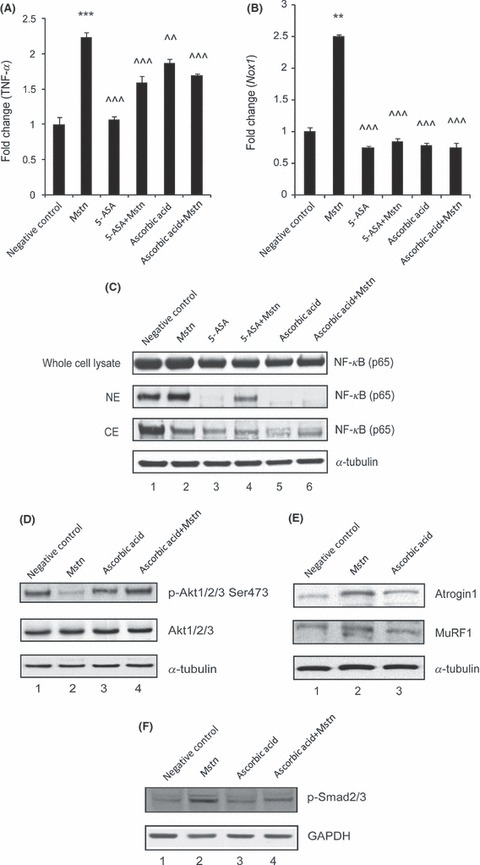
5‐ASA and ascorbic acid inhibit Mstn‐induced ROS via tumor necrosis factor‐α (TNF‐α) and NF‐κB signaling. mRNA expression analysis was performed on C2C12 cells treated with 5‐ASA (10 μm mL−1) and ascorbic acid (100 μm mL−1) along with Mstn (3 μg mL−1). Representative graphs show fold change in the mRNA expression of TNF‐α (A) and Nox1 (B). The values are mean ± SE of two independent experiments. **P < 0.01 and ***P < 0.001 denote significant increase when compared to untreated cells; ^^P < 0.01 and ^^^P < 0.001 denote significant decrease when compared to Mstn‐treated cells (n = 2). (C) Western blotting analysis showing the level of NF‐κB‐p65 at 48 h time point in whole cell lysates, nuclear and cytoplasmic extracts of proliferating C2C12 cells, untreated (Lane 1), treated with Mstn (Lane 2), 5‐ASA (Lane 3), 5‐ASA and Mstn (Lane 4), ascorbic acid (Lane 5), and ascorbic acid and Mstn (Lane 6). α‐tubulin was used as an internal control for equal protein loading on the gel. Mstn induces the muscle‐specific E3 ligase expression via ROS‐dependent pathway. Western blotting analysis showing the levels of p‐Akt and Akt (48 h) (D), Atrogin1 and MuRF1 (72 h) (E) in the C2C12 cells, untreated (Lane 1), treated with Mstn (3 μg mL−1) (Lane 2), ascorbic acid (100 μm mL−1) (Lane 3), and ascorbic acid and Mstn (Lane 4,D) (n = 2). α‐tubulin was used as an internal control for equal protein loading on the gels. (F) Representative gel showing p‐Smad2/3 protein levels in C2C12 cells, untreated (Lane 1), treated with Mstn (3 μg mL−1) (Lane 2), ascorbic acid (100 μm mL−1) (Lane 3), and ascorbic acid and Mstn (Lane 4) for 48 h during proliferation. GAPDH was used as an internal control for equal protein loading on the gel.
Antioxidants are known to detoxify free radicals and protect against ROS in skeletal muscles. Hence, we treated proliferating C2C12 cells with ascorbic acid, an antioxidant, along with Mstn to investigate whether antioxidants are able to inhibit Mstn‐induced ROS signaling. Our results demonstrate that ascorbic acid significantly down‐regulated TNF‐α (Fig. 5A) and Nox1 (Fig. 5B) mRNA expression mediated by Mstn. Western blotting analysis for NF‐κB p65 on whole cell protein lysates showed significant decrease in NF‐κB p65 level in cells treated with ascorbic acid (Fig. 5C; Lanes 5 and 6) when compared to untreated (Lane 1) and Mstn‐treated cells (Lane 2); nuclear extracts show significant decrease in NF‐κB p65 levels (Fig. 5C) on treatment with ascorbic acid (Lane 5) along with Mstn (Lane 6) when compared to cells treated with Mstn only (Lane 2); cytoplasmic extracts show corresponding increase in NF‐κB p65 levels (Fig. 5C; Lanes 5 and 6).
Mstn induces the muscle‐specific E3 ligase expression via ROS‐dependent pathway
We previously showed that Mstn inhibits Akt phosphorylation and is a potent inducer of muscle‐specific E3 ligases such as Atrogin1 and MuRF1 (McFarlane et al., 2006). Given that these two E3 ligases can be activated through increased ROS levels (Li et al. 2003; Cai et al., 2004), we further investigated whether Mstn induces E3 ligases in a ROS‐dependent pathway. Western blotting analysis confirmed that treatment with Mstn does indeed inhibit Akt phosphorylation (Fig. 5D; Lane 2) and up‐regulate Atrogin1 and MuRF1 level (Fig. 5E; Lane 2) in C2C12 cells in the absence of ascorbic acid, a potent antioxidant. Ascorbic acid failed to induce the expression of Atrogin1 and MuRF1 (Fig. 5E; Lane 3). Furthermore, ascorbic acid was also able to rescue Mstn‐mediated inhibition of Akt phosphorylation (Fig. 5D; Lanes 3 and 4). It is known that Mstn signals by activating receptor Smads, Smad2 and Smad3 (Zhu et al., 2004). Western blotting analysis for p‐Smad2/3 (Fig. 5F) revealed that ascorbic acid (Lane 3) treatment along with Mstn (Lane 4) reduced p‐Smad2/3 level when compared to cells treated with Mstn only (Lane 2). These results thus indicate that Mstn signals the up‐regulation of E3 ligases through ROS.
TNF‐α in turn induces Mstn expression, secretion, and signaling in a feed forward manner
The above‐mentioned results clearly indicate that Mstn activates TNF‐α expression and thereby induces ROS production. To see whether elevated TNF‐α in turn increases Mstn for a sustained ‘feed forward’ mechanism, we next investigated Mstn mRNA levels in C2C12 cells treated with TNF‐α. Our results show that Mstn mRNA expression was indeed significantly elevated (Fig. 6A). Consistent with mRNA expression, Mstn protein content was up‐regulated with TNF‐α (Fig. 6B; Lane 2) treatment. Consistent with an increase in intracellular Mstn levels, TNF‐α treatment also resulted in elevated Mstn secretion (Fig. 6C). These results confirm that TNF‐α induces Mstn at the transcriptional, translational, and secretion level. To further investigate whether elevated Mstn levels, as a result of TNF‐α treatment, result in increased Smad2/3 phosphorylation, C2C12 cells were treated with TNF‐α and protein extract was analyzed for p‐Smad2/3 protein content. Western blotting analysis revealed that TNF‐α (Fig. 6D; Lane 3) significantly increased p‐Smad2/3 level when compared to the untreated cells (Fig. 6D; Lane 1).
Figure 6.
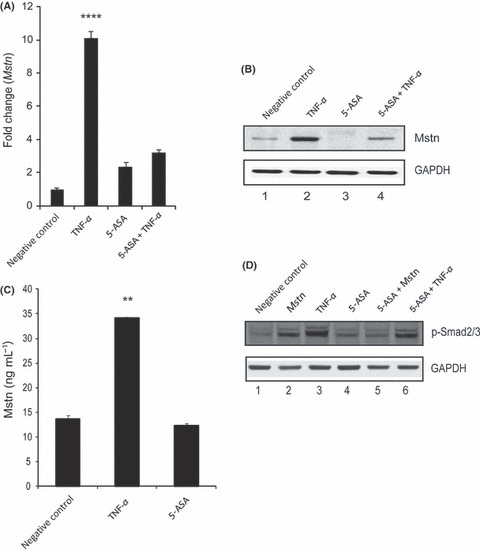
Tumor necrosis factor‐α (TNF‐α) induces myostatin (Mstn) expression, secretion, and signaling in a feed forward mechanism (A) Representative graph showing mRNA expression of Mstn and (B) representative gel showing protein levels of Mstn (∼56 KDa‐full length) in C2C12 cells, untreated (Lane 1), treated with TNF‐α (10 ng mL−1) (Lane 2), 5‐ASA (10 μm mL−1) (Lane 3), and 5‐ASA and TNF‐α (Lane 4) for 72 h in proliferation media. The values are mean ± SE of two independent experiments. Level of significance was compared to the untreated control (****P < 0.0001) (n = 2). GAPDH was used as an internal control for equal protein loading on the gel. (C) Level of secreted Mstn in the cell culture media from C2C12 cells treated with TNF‐α (10 ng mL−1) and 5‐ASA (10 μm mL−1) was determined by EIA. The values are mean ± SE of two independent experiments. The concentration of Mstn is given as ng mL−1, and the level of significance was determined with respect to the negative control (**P < 0.01) (n = 2). (D) Representative gel showing p‐Smad2/3 protein levels in C2C12 cells, untreated (Lane 1), treated with Mstn (3 μg mL−1) (Lane 2), TNF‐α (10 ng mL−1) (Lane 3), 5‐ASA (10 μm mL−1) (Lane 4), 5‐ASA and Mstn (Lane 5), and 5‐ASA and TNF‐α (Lane 6) for 72 h during proliferation. GAPDH was used as an internal control for equal protein loading on the gel.
TNF‐α induces Mstn expression via NF‐κB signaling
Tumor necrosis factor‐α has been shown to induce gene expression via NF‐κB pathway. Therefore, using 5‐ASA we wanted to ascertain whether NF‐κB is involved in TNF‐α‐mediated Mstn induction. Tumor necrosis factor‐α signaling was inhibited with 5‐ASA treatment, hence there was no significant increase in Mstn mRNA (Fig. 6A). We also observed that 5‐ASA treatment reduced basal levels and TNF‐α‐mediated up‐regulation of Mstn protein (Fig. 6B; Lanes 3 and 4) and Mstn secretion (Fig. 6C). Consistent with reduced levels of Mstn, 5‐ASA treatment interfered with phosphorylation of Smad2/3 by Mstn (Fig. 6D; Lanes 4, 5, and 6).
Myostatin regulatory region contains more than one NF‐κB binding sites. As TNF‐α induces Mstn expression, secretion, and signaling, we wanted to investigate whether this induction was by NF‐κB binding to the Mstn enhancer region to regulate its gene expression. Electrophoretic mobility shift assay was performed using nuclear extracts of C2C12 cells treated with TNF‐α and the oligonucleotides containing NF‐κB binding site from mouse Mstn enhancer region. As shown in Fig. 7A, treatment of C2C12 cells with TNF‐α led to significantly increased NF‐κB DNA binding activity. To further confirm that TNF‐α regulates Mstn via NF‐κB, the ability of TNF‐α to up‐regulate Mstn mRNA expression and protein was tested in the C2C12 cell line expressing the IκB‐α SR protein which inhibits NF‐κB activity. As shown in Fig. 7B (i), TNF‐α is unable to increase the mRNA expression of Mstn in the absence of NF‐κB activity (IκB‐α SR) when compared to the control cells (IκB‐α control). Western blotting analysis also revealed that TNF‐α does not up‐regulate Mstn level when NF‐κB was inhibited by the IκB‐α SR protein (Fig. 7B (ii); Lanes 3 and 4). In an independent study, we used the blocker of NF‐κB, BAY 11‐7085, and the results show that upon inhibiting NF‐κB activity, TNF‐α does not up‐regulate Mstn mRNA expression (Fig. 7C (i)) and protein level (Fig. 7C (ii); Lanes 3 and 4). Hence, these results confirm that TNF‐α regulates Mstn via NF‐κB in C2C12 myoblasts.
Figure 7.
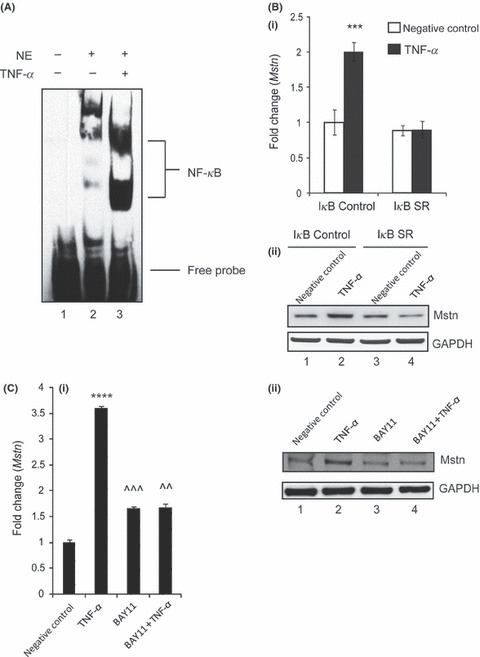
Tumor necrosis factor‐α (TNF‐α) induces myostatin (Mstn) expression via NF‐κB signaling. (A) TNF‐α‐mediated enhancement of NF‐κB binding on mouse Mstn promoter. Electrophoretic mobility shift assay was performed using nuclear extracts from proliferating C2C12 cells treated with TNF‐α (10 ng mL−1) for 72 h. Representative gel showing the significant increase in the binding activity of NF‐κB upon TNF‐α treatment. Lane 1, oligo only; Lane 2, untreated; Lane 3, TNF‐α treated. (B) Representative graph showing mRNA expression of Mstn (i) and representative gel showing protein levels of Mstn (ii) in IκB‐α‐SR‐expressing C2C12 cells treated for 72 h with TNF‐α (10 ng mL−1) in proliferation media; IκB‐α C (control cells) untreated (Lane 1), TNF‐α treated (Lane 2), IκB‐α SR cells untreated (Lane 3), and TNF‐α treated (Lane 4). The values are mean ± SE of two independent experiments. ***P < 0.001 denotes significant increase in mRNA expression when compared to untreated cells (n = 2). GAPDH was used as an internal control for equal protein loading on the gel. (C) Representative graph showing mRNA expression of Mstn (i) and representative gel showing protein levels of Mstn (ii) in C2C12 cells treated for 1 h with BAY11‐7085 (20 μm mL−1) and for 72 h with TNF‐α (10 ng mL−1) in proliferation media; Lane 1, untreated; Lane 2, TNF‐α treated; Lane 3, BAY11‐7085 treated; and Lane 4, BAY11‐7085 and TNF‐α‐treated cells. The values are mean ± SE of two independent experiments. ****P < 0.0001 denotes significant increase in gene expression when compared to untreated cells; ^^P < 0.01 and ^^^P < 0.001 denote significant decrease in mRNA expression when compared to Mstn‐treated cells (n = 2). GAPDH was used as an internal control for equal protein loading on the gel.
H2O2 also induces Mstn expression, secretion, and signaling
In addition to TNF‐α, we next tested whether pro‐oxidants like H2O2 could also induce Mstn levels in C2C12 cells. When C2C12 cells were treated with H2O2, Mstn mRNA expression was significantly elevated (Fig. 8A), whereas a significant down‐regulation of Mstn expression was observed when ROS was inhibited by ascorbic acid treatment (Fig. 8A). Consistent with mRNA levels, Mstn protein content was also up‐regulated with H2O2 (Fig. 8B; Lane 2) treatment, which was down‐regulated with ascorbic acid (Fig. 8B; Lane 3) treatment when compared to untreated cells (Fig. 8B; Lane 1). In addition, we also performed EIA and our results showed that Mstn secretion into media was significantly up‐regulated on treatment with H2O2 while ascorbic acid treatment did not result in any change (Fig. 8C). Furthermore, treatment of C2C12 with H2O2 led to inhibition of proliferation (Fig. S1B, Supporting information) and differentiation (Fig. S1C, Supporting information) of myoblasts similar to that seen in case of Mstn. These results confirm that pro‐oxidants increase Mstn expression, secretion, and signaling.
Figure 8.
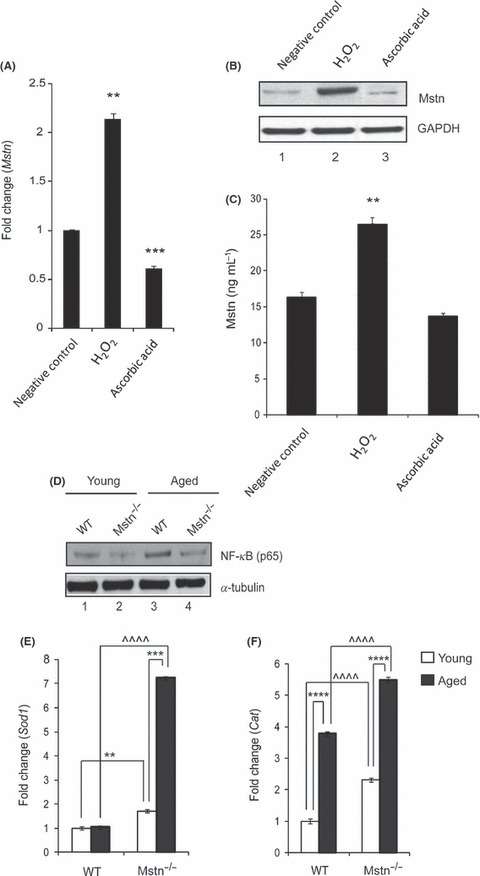
Hydrogen peroxide (H2O2) induces myostatin (Mstn) expression, secretion, and signaling (A) Representative graph showing mRNA expression of Mstn and (B) representative gel showing protein levels of Mstn in C2C12 cells, untreated (Lane 1), treated with H2O2 (0.05 mm) (Lane 2), and ascorbic acid (100 μm mL−1) (Lane 3) for 72 h in proliferation media. The values are mean ± SE of two independent experiments. Significant increase (**P < 0.01) or decrease (***P < 0.001) in mRNA expression was compared to the untreated control (n = 2). GAPDH was used as an internal control for equal protein loading on the gel. (C) Level of secreted Mstn in the cell culture media from C2C12 cells treated with H2O2 (0.05 mm) and ascorbic acid (100 μm mL−1) was determined by EIA. The values are mean ± SE of two independent experiments. The concentration of Mstn is given as ng mL−1, and the level of significance was determined with respect to the negative control (**P < 0.01) (n = 2). Absence of Mstn during aging decreases NF‐κB levels and increases AOE gene expression in skeletal muscle. Western blotting analysis showing the level of NF‐κB‐p65 in protein lysates from Gas muscle of young (6‐week old) and aged (2‐year old) WT and Mstn−/− mice; young WT (Lane 1), young Mstn−/− (Lane 2), aged WT (Lane 3), and aged Mstn−/− (Lane 4). α‐tubulin was used as an internal control for equal protein loading on the gel. mRNA expression analysis in Gas muscle from young and aged WT and Mstn−/− mice was performed. Representative graphs show fold change in the mRNA expression of Sod1 (E) and Cat (F). The values are mean ± SE of three independent experiments. **P < 0.01, ***P < 0.001, and ****P < 0.0001 denote significant increase when compared to young and aged mice, and ^^^^P < 0.0001 denotes significant increase when compared to WT and Mstn−/− mice (n = 3).
Absence of Mstn helps to maintain AOE levels in skeletal muscle during aging
Elevated ROS levels in skeletal muscle have been shown to be one of the hallmarks of sarcopenia. Given that Mstn protein levels have been shown to be increased during aging (Baumann et al., 2003) and that Mstn−/− mice have reduced sarcopenia, we decided to study the role of Mstn‐induced ROS during aging. Western blotting analysis for NF‐κB p65 was performed on protein lysates from gastrocnemius (Gas) muscle from young (6‐week old) and aged (2 years) WT and Mstn−/− mice (Fig. 8D). When compared to the young WT mice (Lane 1), the aged WT mice showed significant increase in NF‐κB p65 level (Lane 3). However, both young (Lane 2) and aged (Lane 4) mice of Mstn−/− genotype expressed significantly lower levels of NF‐κB p65. Similarly, TNF‐α and Nox1 mRNA expression was also down‐regulated in Mstn−/− young and aged mice (data not shown).
To determine the antioxidant status of aging mice, basal AOE levels were measured in Gas muscle isolated from young and aged WT and Mstn−/− mice. A significant increase in the mRNA expression of AOEs, Sod1 (Fig. 8E) and Cat (Fig. 8F) was noted in young and aged Mstn−/− mice when compared to WT mice of similar age. In fact, AOE expression was up‐regulated in aged Mstn−/− mice in comparison with young Mstn−/− mice. With aging, Sod1 expression was not increased in WT mice, whereas in the absence of Mstn, there was an increase in Sod1 expression. These results support the notion that the absence of Mstn helps in maintaining the antioxidant status in skeletal muscle during aging.
Discussion
A major cause of aging and several diseases is thought to be the cumulative oxidative stress, resulting from the production of ROS because of mitochondrial dysfunction (Aiken et al., 2002; Zahn et al., 2006; Herbst et al., 2007). However, the underlying signaling mechanisms that induce ROS during aging are not well understood. Here, we show that Mstn is a potent inducer of ROS in myotubes and that lack of Mstn enhances the antioxidant response. Furthermore, we also show that the increased ROS levels in turn induce Mstn. Our studies therefore identify a novel “feed forward” mechanism for a sustained ROS production in Mstn‐elevated situations such as aging and other muscle wasting conditions.
Myostatin, a member of the TGF‐β super‐family, is now widely accepted as an inhibitor of skeletal muscle growth. Overexpression of Mstn in mice has been shown to cause cachexia (Zimmers et al., 2002; Reisz‐Porszasz et al., 2003), and increased levels of Mstn are also observed during muscle wasting seen in human immunodeficiency virus (HIV) infection, cancer, and chronic kidney diseases. Elevated Mstn in these conditions is believed to increase protein degradation, decrease myogenesis, and reduce protein synthesis. Muscle wasting in the above‐mentioned conditions also involves ROS production, which has been found to be associated with protein catabolism and ubiquitin‐proteasome pathway (Li & Reid, 2000; Gomes‐Marcondes & Tisdale, 2002). In addition, oxidative stress during aging is considered to be a major contributor of muscle atrophy (Sohal & Weindruch, 1996; Powers et al., 2005).
Several lines of evidence presented in this study suggest that Mstn is an inducer of ROS in the muscle cells. Our results demonstrate that Mstn induces ROS production in myoblasts during proliferation as well as during differentiation. Increase in ROS levels was evident from the incorporation of the fluorescent dye in the Mstn‐treated C2C12 cells coinciding with increased carbonylation of the cellular proteins. Even though carbonylation of proteins is considered to be a standard marker for oxidative stress, there are limitations of Oxyblot assay for determination of carbonylated proteins in terms of sensitivity (Nystrom, 2005). In addition to carbonylation of proteins, previous studies have also reported an increase in AOE levels in C2C12 cells when exposed to pro‐oxidants like paraquat (Franco et al., 1999). Indeed, in Mstn‐treated muscle cells we detected an increase not only in carbonylation of proteins but also in the levels of AOE. These results thus confirm that increased Mstn levels lead to the induction of oxidative stress in skeletal muscle cells. Considering that Mstn is a pro‐oxidant, we argued that in the absence of Mstn, ROS levels and thus AOE levels should be lower in Mstn−/− primary myoblasts. However, what we observed was that differentiating primary myoblasts isolated from 8‐week‐old Mstn−/− mice have higher basal levels of AOEs compared to WT mice. This observation could be accounted for by increased muscle mass in Mstn−/− mice as compared to the WT mice. Nevertheless, the higher basal levels of AOEs appear to confer resistance to oxidative stress in Mstn−/− mice. This finding is supported by the result that when differentiating Mstn−/− primary myoblasts were treated with Mstn, no significant increase in AOE activity was observed implying that Mstn−/− primary myoblasts already had sufficient levels of AOE to combat additional oxidative stress (data not shown). Similarly, in the 2‐year‐old Mstn−/− mice the AOE activity remains up‐regulated as compared to the old wild‐type mice. In line with these observations, it has been suggested that AOEs and non‐enzyme scavengers of ROS like GSR are extremely important to maintain the balance of redox system (Valko et al., 2007), thus accentuated AOE activity in Mstn−/− muscle would make it less susceptible to oxidative damage. Our results show that with aging, there are changes in the basal activity of different AOEs. Aged Mstn−/− mice are found to have higher basal levels of AOEs compared to aged wild‐type mice; this might be one of the reasons for aged Mstn−/− mice to maintain the muscle mass and function.
To delineate the mechanism(s) through which ROS maybe induced by Mstn, we used cell signaling inhibitors to block the pathways involved in the generation of ROS. The results show that NADH/NAD(P)H oxidase and mitochondrial ETC are involved in ROS induction by Mstn because blocking Nox and mitochondrial ETC reduces ROS production by Mstn. Based on AOE analysis in the Mstn‐treated myoblasts, NADPH oxidase‐derived ROS are most likely to be superoxide radicals, hydroxyl radicals, and H2O2. In addition, the contribution of lipid peroxidation toward ROS was discounted by the observation that TBARS levels were down‐regulated by Mstn. Previously, it has been reported that TNF‐α induces oxidative stress in skeletal muscles (Buck & Chojkier, 1996) and its expression is regulated through NF‐κB transcription factor. We also clearly show by molecular analysis that treatment of myoblasts with Mstn increases NF‐κB translocation into nucleus, which induced the transcription of TNF‐α (Fig. 3C,D (i)). Induction of ROS by Mstn via TNF‐α was further confirmed by the fact that in the absence of Mstn, there is a reduction in TNF‐α and Nox1 mRNA levels in young and aged Mstn−/− mice when compared to WT mice (data not shown). These data are also supported by an earlier report where low circulatory levels of TNF‐α were reported in Mstn−/− mice (Wilkes et al., 2009). Previously, it has been reported that TNF‐α induces oxidative stress by activating Nox1 expression (Kim et al., 2007). Indeed, in this study also, we clearly show that the increased TNF‐α is responsible for the increased expression of NAD(P)H oxidase. Hence, we propose that Mstn increases the levels of ROS by Nox1 pathway through NF‐κB. Several lines of evidence from our data support this hypothesis. Firstly, the oxidative stress induced by Mstn was alleviated upon treatment with NF‐κB inhibitors (5‐ASA and BAY 11‐7085). Furthermore, loss of Mstn leads to decrease in NF‐κB levels in the Gas muscles of Mstn−/− mice (Fig. 8D). It is also noteworthy that aging leads to an increase in the levels of NF‐κB in the Gas muscles of 2‐year‐old WT mice. However, such an increase was not observed in the NF‐κB levels in the aged Mstn−/− muscle. Taken together, these results confirm a hypothesis that Mstn signals via NF‐κB, TNF‐α, and Nox1 to induce ROS and are also exemplified in vivo. Furthermore, the oxidative stress induced by Mstn was alleviated upon treatment with the antioxidant, ascorbic acid, which supports evidence indicating that the administration of antioxidants is effective to protect against ROS in chronically loaded muscles of aged rats (Ryan et al., 2008).
A higher level of ROS has previously been shown to inhibit Akt by activating protein phosphatase (PP2A) that dephosphorylates Ser473 on Akt. Mstn has been shown to inhibit Akt phosphorylation (McFarlane et al., 2006), and the molecular mechanism for the dephosphorylation has not been identified so far. Current results suggest that induced ROS by Mstn could be one of the possible molecular mechanisms behind Mstn‐induced inhibition of Akt phosphorylation. Hypophosphorylation of Akt by either blocking IGF or increasing Mstn levels leads to skeletal muscle wasting by inducing muscle‐specific E3 ligases, Atrogin1 and MuRF1. In this study, ascorbic acid and 5‐ASA treatment of Mstn‐treated myoblasts were able to rescue p‐Akt level and down‐regulate the protein content of E3 ligases Atrogin1 and MuRF1 by Smad2/3‐dependent mechanism. These results therefore further suggest that increased ROS produced by Mstn could potentially induce muscle wasting by activating ubiquitin proteolytic pathway.
Another important discovery that is described here is that ROS itself is a potent inducer of Mstn. Furthermore, we show that TNF‐α and NF‐κB are required for this ‘feed forward’ induction of Mstn by ROS (Fig. 9). Inhibition of either TNF‐α induction by ROS or NF‐κB activity and binding to Mstn enhancer results in reduced Mstn levels in myoblasts and myotubes, which further confirms that ROS induce Mstn via TNF‐α‐ and NF‐κB‐dependent pathway. It is noteworthy that in a related context of muscle wasting associated with renal failure, recently, Zhang et al. (2011) reported that Mstn is responsible for this muscle wasting and that TNF‐α stimulates Mstn mRNA and protein by a NF‐κB‐dependent pathway in C2C12 myotubes.
Figure 9.
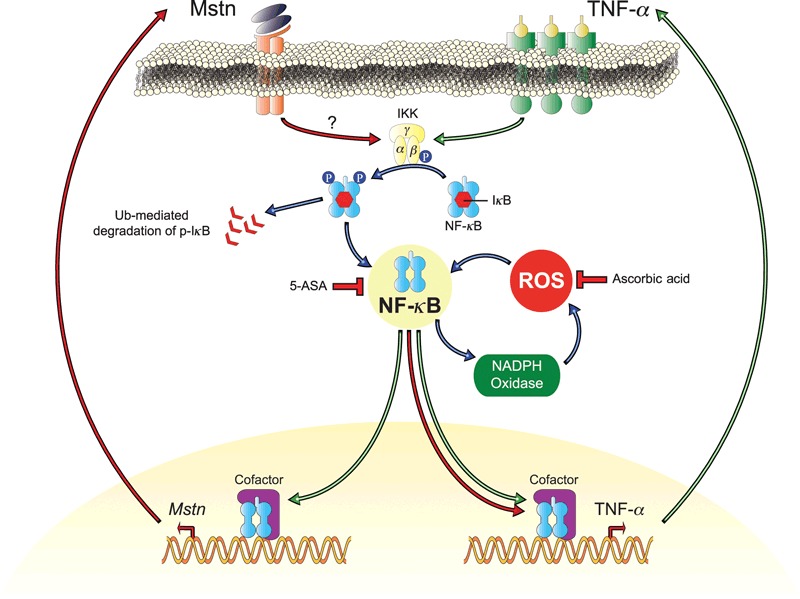
Proposed mechanism by which myostatin (Mstn) induces reactive oxygen species (ROS) in skeletal muscle. Increased Mstn induces tumor necrosis factor‐α (TNF‐α) production via NF‐κB signaling, increasing the production of ROS by NADPH oxidase. The induced ROS results in a feed forward loop further increasing Mstn levels via NF‐κB signaling of TNF‐α. The increased ROS levels result in a decrease in muscle protein synthesis and an increase in protein degradation leading to skeletal muscle wasting. 5‐ASA and ascorbic acid block these pathways to reduce ROS, Mstn, and TNF‐α.
Therefore, in the present study, using an integrative approach from cell cultures to mice, we demonstrate that Mstn and TNF‐α are components of a feed forward loop in which Mstn triggers the generation of second messenger ROS, mediated by TNF‐α and NADPH oxidase. We speculate that higher levels of ROS induce muscle wasting via proteasomal‐mediated degradation of proteins. Furthermore, we also propose that increased ROS in turn induces TNF‐α which stimulates Mstn expression via pro‐inflammatory transcription factor NF‐κB thus enabling a ‘feed forward’ and sustained induction of ROS during muscle wasting. Myostatin protein levels in skeletal muscle and circulation is reported to increase during aging and cachexia (Yarasheski et al., 2002; Baumann et al., 2003; Raue et al., 2006; Léger et al., 2008), which would overlap with the increase in pro‐inflammatory and redox‐sensitive NF‐κB. As stated earlier, Mstn−/− aged mice have lower levels of NF‐κB in the muscle, which makes Mstn a perfect candidate to integrate inflammatory and ROS signaling during aging‐related muscle wasting.
Experimental procedures
Animals
Young, 5‐ to 8‐week‐old C57Bl/6J male mice (WT) were obtained from National University of Singapore‐Centre for Animal Resources, Singapore, Republic of Singapore. Young, 5‐ to 8‐week‐old Mstn−/− mice were obtained from Mstn−/− heterozygous mice breeding (gifted by Prof S.J Lee (Johns Hopkins University, Baltimore, MD, USA)) at Nanyang Technological University Animal house, Singapore, Republic of Singapore. Wild‐type and Mstn−/− mice (2‐year old) were aged at AgResearch, Hamilton, NZ. All animals had free access to chow diet and water. All experimental procedures were approved by the Institutional Animal Care and Use Committee, Singapore.
Reagents and proteins
Recombinant human Mstn protein used in the experiments was produced and purified in the laboratory according to established protocol (Sharma et al., 1999). Conditioned media containing Mstn was obtained from CHO cell line that expresses Mstn (Zimmers et al., 2002). The concentration of Mstn in the conditioned media was determined by EIA (Immundiagnostik AG, Bensheim, Germany). Recombinant human TNF‐α was purchased from Miltenyi Biotec., Bergisch Gladbach, Germany. For other reagents and chemicals, please refer to Data S1, Supporting information.
Cell culture
The established ATCC mammalian (murine) cell line C2C12 myoblasts were used (Yaffe & Saxel, 1977). The cell stocks were stored in C2C12 proliferation media: Dulbecco’s modified Eagle’s medium (DMEM) (PAA Laboratories, Inc., Pasching, Austria), 10% fetal bovine serum (HyClone Laboratories, Inc., Logan, Utah, USA), 1% penicillin/streptomycin (P/S) (Gibco, Carlsbad, CA, USA) media with 5% dimethyl sulfoxide (Sigma‐Aldrich, St. Louis, MO, USA) in liquid nitrogen. The cells were passaged in proliferation media and differentiated in C2C12 differentiation media: DMEM, 2% Horse serum (Gibco), and 1% P/S were used to differentiate the myoblasts to myotubes.
The NF‐κB inhibitor cell line, which is the IκB‐α super‐repressor (IκB‐α SR) C2C12 cells, was gifted by Guttridge et al. (1999). The IκB‐α control and IκB‐α‐SR‐expressing C2C12 cells were cultured in proliferation media containing 0.5 mg mL−1 geneticin (Invitrogen, Carlsbad, CA, USA).
Myoblast proliferation assay
C2C12 cells were seeded in 96‐well plates in proliferation media at a cell density of 1000 cells per well and incubated at 37 °C and 5% CO2. After overnight attachment, the cells were treated with H2O2 (0.05 and 0.1 mm), and the negative control was treated with autoclaved Milli Q water in fresh proliferation media. Proliferating cells were fixed at 24‐h intervals with 100 μL of formal saline fixative (10% formaldehyde (Merck & Co., Inc., NJ, USA) and 0.9% saline). The proliferation was assessed by methylene blue photometric end point assay (Oliver et al., 1989).
Myoblast differentiation assay
C2C12 cells were seeded on Thermanox plastic cover slips (Nalge Nunc International, Rochester, NY, USA) in 24‐well plates in proliferation media at a cell density of 25 000 cells per cm2. After overnight incubation of cells at 37 °C and 5% CO2, the cells were treated with various concentrations of H2O2 (0.05 mm) and the negative control was treated with autoclaved Milli Q water in differentiation media. The differentiating cells and myotubes were fixed at 24‐h intervals. The myotubes were stained with hematoxylin and eosin. Images of non‐overlapping areas from two cover slips were taken in a bright field microscope at 10× magnification.
Isolation of primary myoblasts (satellite cells)
The hind limb skeletal muscles from mice were dissected and used for isolation of primary myoblasts. Primary myoblasts were isolated according to the established protocol (Partridge, 1997; McCroskery et al., 2003). Isolated satellite cells were trypsinized and seeded for various assays.
Treatment of C2C12 cells and primary myoblasts
Based on the results obtained from the proliferation and differentiation assays on C2C12 cells treated with H2O2, 0.05 mm was chosen as concentration of H2O2 to cause mild oxidative stress, and hereby, this concentration was used for the treatment of cells. Recombinant Mstn, 3 μg mL−1 concentrations used in this study inhibited 30–40% proliferation of myoblasts. Thus, this concentration of Mstn was used to cause mild oxidative stress in the cells. The conditioned media from CHO cells was found to have Mstn at a concentration of 3.5 ng mL−1. Based on earlier studies (Yamashita et al., 2008; Grzelkowska‐Kowalczyk & Wieteska‐Skrzeczyńska, 2010), 10 ng mL−1 TNF‐α was used to treat C2C12 cells. Also, 10 μm mL−1 of 5‐ASA (Yan & Polk, 1999) and 100 μm mL−1 ascorbic acid (Ciafrèet al., 2007) was used to treat C2C12 cells during proliferation and differentiation. BAY 11‐7085 at the concentration of 20 μm mL−1 (Ladner et al., 2003) was used to inhibit NF‐κB activity in proliferating C2C12 cells. C2C12 cells and primary myoblasts following various treatments for time indicated were collected in TRIZOL (Invitrogen) for RNA isolation.
Determination of ROS production
Real‐time intracellular measurement of ROS
Reactive oxygen species was determined using a fluorescent probe, 5‐(and‐6)‐chloromethyl‐2′, 7′‐dichlorodihydrofluorescein diacetate acetyl ester (CM‐H2DCFDA) (Molecular Probes, Carlsbad, CA, USA). C2C12 cells were seeded in 96‐well plates in proliferation media at a cell density of 6000 cells per well and incubated at 37 °C and 5% CO2. After overnight attachment of cells, they were treated with Mstn in fresh differentiation media. Subsequently, at 24‐h time point intervals, the media were removed and the cells were incubated with 5 μm of CM‐H2DCFDA (Molecular Probes) in phosphate‐buffered saline (PBS) for 30 min at 37 °C and 5% CO2. Upon incubation, the dye was removed and the 200 μL of differentiation media was added in each well. Fluorescence was determined at various time points using a fluorescent multilabel plate reader at excitation/emission wavelengths of 495 and 525 nm, respectively.
Visualization of Mstn‐induced ROS
C2C12 cells were seeded onto Permanox chamber slides (Nalge Nunc International) in proliferation media at a cell density of 25 000 cells per cm2 and incubated at 37 °C and 5% CO2. After overnight attachment, the cells were treated with Mstn in fresh differentiation media. Subsequently, at 24‐h time intervals, the media were removed and the cells were incubated with 5 μm of CM‐H2DCFDA (Molecular Probes) in PBS for 30 min at 37 °C and 5% CO2. Upon incubation, the dye was removed and 0.5 mL of differentiation media was added in each well. The fluorescence was viewed under the Leica upright microscope, and images were taken at 10× magnification.
Protein isolation
C2C12 cells and primary myoblasts following various treatments for the indicated time were collected in protein lysis buffer [50 mm Tris pH 7.6, 250 mm NaCl, 5 mm EDTA, 0.1% Igepal‐Nonidet P‐40 (NP‐40) (Fluka, St. Louis, MO, USA), and complete protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA)], and protein lysates were made as previously described (McFarlane et al., 2006). Gas muscle from young and aged WT and Mstn−/− mice were homogenized in the protein lysis buffer, and the clear supernatants were collected. These protein lysates were used for Oxyblot assay (Millipore Corp., Billerica, MA, USA) and Western blotting analysis. Cytoplasmic and nuclear fractions from proliferating C2C12 cells following Mstn treatment were isolated as previously described (Ye et al., 1996).
Oxyblot assay
To detect carbonyl groups introduced into proteins by oxidative reactions because of ROS, Oxyblot assay using Oxyblot Protein Oxidation Detection Kit (Millipore Corp.) was performed. Briefly, 15 μg of the protein lysate collected from C2C12 cells upon various treatments was derivatized with 1× 2,4‐dinitrophenylhydrazine solution. The derivatized samples were loaded onto a 4–12% Bis‐Tris polyacrylamide gel (Invitrogen) and electroblotted onto a nitrocellulose membrane. After blocking with 1× PBS‐T, BSA, pH 7.2, the membrane was incubated with 1:200 diluted rabbit anti‐DNP primary antibody followed by incubation with 1:300 diluted goat anti‐rabbit IgG (HRP conjugated) secondary antibody. After subsequent washes, the membrane was developed using chemiluminescent reagents (Western Lightning Plus ECL, PerkinElmer, Waltham, MA, USA) and was exposed to X‐ray film, and the signal intensity was calculated using a densitometer to determine the extent of carbonylation of the proteins.
Cell and muscle homogenates for AOE assays
To obtain cell homogenates for antioxidant enzyme assays, the C2C12 cells and primary myoblasts were seeded in 6‐well plates at a cell density of 25 000 cells per cm2. After overnight incubation at 37 °C and 5% CO2, the cells were treated with the above‐mentioned concentration of recombinant Mstn or conditioned media Mstn – the negative control being the buffer or control conditioned CHO media, respectively, in differentiation media. The 10% cell homogenate (1 mL of cell lysis buffer was added to the cells and scraped off the plate and resuspended in 9 mL of cell lysis buffer) was collected at 24‐h time points in cell lysis buffer (0.02 m Tris‐HCl (pH 7.0), 150 mm NaCl, and 1 mm EDTA). Similarly, for AOE assays in muscle samples (Quadriceps), the muscles were weighed and a 10% homogenate was made in lysis buffer (0.02 m Tris‐HCl, pH 7.0) using a Teflon homogenizer in ice‐cold condition. The homogenates were centrifuged at 2750 g for 10 min at 4 °C. The supernatants were used for determination of various non‐enzymatic and enzymatic antioxidants.
Estimation of protein concentrations using Bradford’s (Bio‐Rad) assay
The protein concentrations of the cell and muscle homogenates, cell protein lysates, nuclear and cytoplasmic extracts were determined using Bradford’s assay (Bradford, 1976) (Bio‐Rad Laboratories Inc., Hercules, CA, USA).
Estimation of antioxidant enzymes
Superoxide dismutase
About 0.1 mL of cell homogenate was diluted by adding 0.9 mL of distilled water, and 1.5 mL of 0.1 m carbonate buffer, pH 10.2 (containing 57 mg EDTA per 100 mL), was added to it. The reaction was initiated by the addition of 0.4 mL of 3 mm epinephrine. The change in absorbance per minute was measured at 480 nm for 3 min. Activity is expressed as the optical density (at 480 nm) of the enzyme required to exhibit 50% inhibition of epinephrine auto‐oxidation.
Catalase
The activity of catalase was determined by the method of Beers & Sizer (1952). The activity was expressed as optical density of H2O2 consumed per minute per milligram protein.
Glutathione peroxidase
Activity of glutathione peroxidase was determined using the method of Rotruck et al. (1973) with some modifications. Briefly, 0.4 m sodium phosphate buffer (pH 7.0), 10 mm sodium azide, 4 mm reduced glutathione, the cell lysate or muscle homogenate, and 2.5 mm hydrogen peroxide were mixed and incubated at 37 °C for 3 min. After terminating the reaction with 10% TCA, the residual glutathione content was determined by the addition of 0.3 m disodium hydrogen phosphate and 0.6 mm DTNB reagent. The activity of GPx was expressed as μg of glutathione consumed per minute per milligram protein.
Estimation of antioxidant‐reduced glutathione
The reduced glutathione was determined by the method of Moron et al. (1979). The amount of glutathione was expressed as μg mg−1 protein.
Estimation of lipid peroxidation product
The lipid peroxidation product (MDA) was determined by thiobarbituric acid reaction as described by Ohkawa et al. (1979). The lipid peroxide content was expressed as nmoles MDA per 100 mg protein.
Identification of signaling pathway and enzymes involved through which Mstn induces ROS
To determine the enzymes involved and the cell signaling pathway involved in Mstn‐induced ROS, various cell signaling inhibitors were used (Iuchi et al., 2003). The inhibitors used are as mentioned in Table 1. C2C12 cells were seeded in 96‐well plates in proliferation media at a cell density of 6000 cells per well and incubated at 37 °C and 5% CO2. After overnight attachment, the cells were treated with Mstn in fresh differentiation media for wells assigned to inhibitors 1–6. For inhibitor 7, the cells in the assigned wells were treated with Mstn and 0.5 μm CCCP and for inhibitor 8, Mstn and 100 μm TTFA in fresh differentiation media. After 48 h of Mstn treatment, inhibitors 1–6 (Table 1) in differentiation media were added to the wells. After the stipulated time (Table 1), the cells were incubated with 5 μm of CM‐H2DCFDA (Molecular Probes) in PBS for 30 min at 37 °C and 5% CO2. Upon incubation, the dye was removed and 200 μL of differentiation media per well was added. Fluorescence was determined using a fluorescent multilabel plate reader at excitation/emission wavelengths of 495 and 525 nm, respectively.
RT–qPCR (reverse transcriptase–quantitative polymerase chain reaction)
RNA was isolated from C2C12 cells that were subjected to various treatments during proliferation and differentiation using TRIZOL protocol (Invitrogen) and also from muscles (Gas), and cDNA was synthesized using Superscript First‐Strand Synthesis System for RT–PCR according to the manufacturer’s (Bio‐Rad Laboratories Inc.) instructions. Quantitative analysis of the gene expression by real‐time PCR was carried out using CFX96 (Bio‐Rad Laboratories Inc.) system. Each real‐time PCR (10 μL) contained 2 μL of cDNA, 5 μL of SsoFast Evagreen (Bio‐Rad Laboratories Inc.) and forward and reverse primers (Table S2, Supporting information) at a final concentration of 2.5 μm. All reactions were performed using the following thermal cycler conditions: 98 °C for 3 min, followed by 45 cycles of a three‐step reaction, denaturation at 98 °C for 3 s, annealing at 60 °C for 10 s and extension at 72 °C for 10 s, and final extension at 95 °C for 10 s. The reaction was followed by a melt curve from 65 °C to 95 °C in 5 s increments of 0.5 °C to ensure amplification specificity. Transcript levels were normalized to GAPDH transcript levels. Relative fold change in expression was calculated using the ΔΔCT method.
Western blot analysis
Total protein (15–40 μg) was separated on 4–12% Bis‐Tris polyacrylamide gels (Invitrogen) and transferred to nitrocellulose membrane by electroblotting. The membranes for probing with primary antibodies NF‐κB‐p65, Mstn, p‐Akt1/2/3 (Ser473), Akt1/2/3, Atrogin1, and MuRF1 (Table S1, Supporting information) were blocked for 1 h at room temperature in 0.3% BSA, 1% polyethylene glycol (Sigma‐Aldrich) and 1% polyvinylpyrrolidone (Sigma‐Aldrich) in 1× TBS‐T and then incubated overnight at 4 °C with the above‐mentioned primary antibodies. The membranes for probing with p‐Smad2/3, IKKα, α‐tubulin and GAPDH (Table S1) were blocked in 5% non‐fat milk in 1× TBS‐T overnight at 4 °C and then incubated for 3 h at room temperature with the above‐mentioned primary antibodies. The membranes for detecting p‐NF‐κB‐p65 (Ser536), IKB‐α, and p‐IKB‐α (Ser32/36) (Table S1) were blocked for 1 h at room temperature in 5% BSA TBS‐T and then incubated overnight at 4 °C with the specific primary antibodies. After subsequent washes and incubation with respective secondary antibodies (Table S1) for 1 h at room temperature, the HRP activity was detected using chemiluminescent reagents (Western Lightning Plus ECL).
Electrophoretic mobility shift assay
The oligonucleotides containing the NF‐κB binding site on mouse Mstn promoter (5′‐CTCAAACAGGGGGCTTTCCCTCCTCAATATC‐3′) and mouse TNF‐α promoter (5′‐GATCCAGCTCAGGGGGAGGCCCCAAGGCCTGAC‐3′) were hybridized to their respective complementary strands and labeled at the 3′ end with biotin tetra‐ethyleneglycol (Sigma‐Aldrich). The EMSAs were performed using the Lightshift Chemiluminescent EMSA kit (Thermo Scientific Pierce Biotechnology, Rockford, IL, USA). Nuclear extracts obtained from Mstn‐treated proliferating C2C12 cells were incubated with the biotin labeled probe (final concentration 20 fmol) in a final volume of 20 μL containing binding buffer (100 mm Tris, 500 mm KCl, 10 mm DTT, pH 7.5), glycerol, MgCl2, poly(dI‐dC), and NP‐40. After 20 min of incubation at room temperature, samples were subjected to electrophoresis on a 5% acrylamide gel containing tris‐borate‐EDTA (Promega Corporation, Madison, WI, USA) at 80 V at 4 °C. The samples were then transferred to a membrane at 200 mA for 2 h at 4 °C and UV‐crosslinked after transfer. The membrane was probed with stabilized streptavidin–horseradish peroxidase conjugate and developed using the Lightshift Chemiluminescent EMSA kit (Thermo Scientific Pierce Biotechnology).
To confirm the presence of NF‐κB p65 and NF‐κB p50 in the retarded complex, nuclear extracts from C2C12 cells were pre‐incubated for 20 min at room temperature with 1 μL of 1:1 diluted (in PBS) NF‐κB p65 (sc‐372) and NF‐κB p50 (sc‐7178) antibody before incubation with the labeled probe for another 20 min at room temperature. The samples were then subjected to EMSA as mentioned earlier.
Mstn Enzyme immunoassay (EIA)
To estimate the Mstn secreted into the media, Mstn EIA kit (Immundiagnostik AG) was used and the competitive immunoassay was performed according to the manufacturer’s instructions. Briefly, standards with defined Mstn concentrations and media that were to be assayed for Mstn were incubated with polyclonal antibodies against human recombinant Mstn in glass vials. During the incubation, free Mstn would be bound by the Mstn antibodies. The pre‐incubates were then transferred into the microtiter wells coated with recombinant Mstn. The unbound antibodies in the pre‐incubates then bind to the immobilized antigen and are detected using a peroxidase‐conjugated secondary antibody and TMB as a substrate. The absorbance of the samples is read using an ELISA microtiter plate reader at 450 nm. The concentration of Mstn secreted into the media is expressed as ng mL−1.
Statistical analysis
Statistical analysis was performed using anova, and the results were considered significant at P < 0.05. Results were expressed as SEM (Standard Error of Mean).
Author contributions
Mridula Sharma and Ravi Kambadur planned the project, analyzed and interpreted the data and prepared the manuscript. Sandhya Sriram performed majority of the experiments and analyzed the data. Subha Subramanian, Durga Sathiakumar, and Rithika Venkatesh helped in performing some of the experiments. Monica Senna Salerno and Craig Desmond McFarlane helped in planning experiments on aging animals.
Supporting information
Data S1 Reagents and chemicals.
Table S1 List of Antibodies used.
Table S2 List of qPCR primers used.
Fig. S1 Mstn down regulates lipid peroxidation in differentiating WT primary myoblasts (A) WT primary myoblasts were treated with Mstn (3 μg/ml) in the differentiation media and total cell lysates were made at indicated time points.
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer‐reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting info item
Supporting info item
Acknowledgments
We thank Prof S J Lee for providing Mstn−/− heterozygous mice, Dr. Denis C Guttridge for IκB‐SR‐expressing C2C12 cells and Dr. Esther Latres for the primary antibodies to Atrogin1 and MuRF1. We are also thankful to Sabeera Bonala and Kelvin, Tan Suan Liang, for their help. We are grateful to Academic Research Council (Ministry of Education, Singapore, Republic of Singapore) and National Research Foundation, Singapore, Republic of Singapore for funding.
References
- Aiken J, Bua E, Cao Z, Lopez M, Wanagat JON, McKenzie D, McKiernan S (2002) Mitochondrial DNA deletion mutations and sarcopenia. Ann. N Y Acad. Sci. 959, 412–423. [DOI] [PubMed] [Google Scholar]
- Baumann AP, Ibebunjo C, Grasser WA, Paralkar VM (2003) Myostatin expression in age and denervation‐induced skeletal muscle atrophy. J. Musculoskelet. Neuronal Interact. 3, 8–16. [PubMed] [Google Scholar]
- Beers RF, Sizer IW (1952) A Spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 195, 133–140. [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Buck M, Chojkier M (1996) Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. EMBO J. 15, 1753–1765. [PMC free article] [PubMed] [Google Scholar]
- Cai D, Frantz JD, Tawa NE, Melendez PA, Oh B‐C, Lidov HGW, Hasselgren P‐O, Frontera WR, Lee J, Glass DJ, Shoelson SE (2004) IKKβ/NF‐κB Activation Causes Severe Muscle Wasting in Mice. Cell 119, 285–298. [DOI] [PubMed] [Google Scholar]
- Ciafrè SA, Niola F, Giorda E, Farace MG, Caporossi D (2007) CoCl2‐simulated hypoxia in skeletal muscle cell lines: role of free radicals in gene up‐regulation and induction of apoptosis. Free Radic. Res. 41, 391–401. [DOI] [PubMed] [Google Scholar]
- Fanò G, Mecocci P, Vecchiet J, Belia S, Fulle S, Polidori M, Felzani G, Senin U, Vecchiet L, Beal M (2001) Age and sex influence on oxidative damage and functional status in human skeletal muscle. J. Muscle Res. Cell Motil. 22, 345–351. [DOI] [PubMed] [Google Scholar]
- Franco AA, Odom RS, Rando TA (1999) Regulation of antioxidant enzyme gene expression in response to oxidative stress and during differentiation of mouse skeletal muscle. Free Radic. Biol. Med. 27, 1122–1132. [DOI] [PubMed] [Google Scholar]
- Garcia YJ, Rodríguez‐Malaver AJ, Peñaloza N (2005) Lipid peroxidation measurement by thiobarbituric acid assay in rat cerebellar slices. J. Neurosci. Methods 144, 127–135. [DOI] [PubMed] [Google Scholar]
- Gomes‐Marcondes MCC, Tisdale MJ (2002) Induction of protein catabolism and the ubiquitin‐proteasome pathway by mild oxidative stress. Cancer Lett. 180, 69–74. [DOI] [PubMed] [Google Scholar]
- Grzelkowska‐Kowalczyk K, Wieteska‐Skrzeczyńska W (2010) Treatment with TNF‐α and IFN‐γ alters the activation of SER/THR protein kinases and the metabolic response to IGF‐I in mouse C2C12 myogenic cells. Cell. Mol. Biol. Lett. 15, 13–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr (1999) NF‐kappa B controls cell growth and differentiation through transcriptional regulation of Cyclin D1. Mol. Cell. Biol. 19, 5785–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttridge DC, Mayo MW, Madrid LV, Wang C‐Y, Baldwin AS Jr (2000) NF‐κB‐induced loss of myod messenger rna: possible role in muscle decay and cachexia. Science 289, 2363–2366. [DOI] [PubMed] [Google Scholar]
- Handayaningsih A‐E, Iguchi G, Fukuoka H, Nishizawa H, Takahashi M, Yamamoto M, Herningtyas E‐H, Okimura Y, Kaji H, Chihara K, Seino S, Takahashi Y (2011) Reactive oxygen species play an essential role in IGF‐I signaling and IGF‐I‐induced myocyte hypertrophy in C2C12 myocytes. Endocrinology 152, 912–921. [DOI] [PubMed] [Google Scholar]
- Herbst A, Pak JW, McKenzie D, Bua E, Bassiouni M, Aiken JM (2007) Accumulation of Mitochondrial DNA deletion mutations in aged muscle fibers: evidence for a causal role in muscle fiber loss. J. Gerontol. A Biol. Sci. Med. Sci. 62, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iuchi T, Akaike M, Mitsui T, Ohshima Y, Shintani Y, Azuma H, Matsumoto T (2003) Glucocorticoid excess induces superoxide production in vascular endothelial cells and elicits vascular endothelial dysfunction. Circ. Res. 92, 81–87. [DOI] [PubMed] [Google Scholar]
- Jahnke VE, Sabido O, Freyssenet D (2009) Control of mitochondrial biogenesis, ROS level, and cytosolic Ca2 + concentration during the cell cycle and the onset of differentiation in L6E9 myoblasts. Am. J. Physiol. Cell. Physiol. 296, C1185–C1194. [DOI] [PubMed] [Google Scholar]
- Kim Y‐S, Morgan MJ, Choksi S, Liu Z‐g (2007) TNF‐induced activation of the Nox1 NADPH oxidase and its role in the induction of Necrotic Cell Death. Mol. Cell 26, 675–687. [DOI] [PubMed] [Google Scholar]
- Ladner KJ, Caligiuri MA, Guttridge DC (2003) Tumor necrosis factor‐regulated biphasic activation of NF‐κB is required for cytokine‐induced loss of skeletal muscle gene products. J. Biol. Chem. 278, 2294–2303. [DOI] [PubMed] [Google Scholar]
- Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R (2002) Myostatin inhibits myoblast differentiation by down‐regulating MyoD expression. J. Biol. Chem. 277, 49831–49840. [DOI] [PubMed] [Google Scholar]
- Léger B, Derave W, De Bock K, Hespel P, Russell AP (2008) Human sarcopenia reveals an increase in SOCS‐3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res. 11, 163–175B. [DOI] [PubMed] [Google Scholar]
- Li Y‐P, Chen Y, Li AS, Reid MB (2003) Hydrogen peroxide stimulates ubiquitin‐conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am. J. Physiol. Cell. Physiol. 285, C806–C812. [DOI] [PubMed] [Google Scholar]
- Li Y‐P, Reid MB (2000) NF‐κB mediates the protein loss induced by TNF‐α in differentiated skeletal muscle myotubes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1165–R1170. [DOI] [PubMed] [Google Scholar]
- Mantovani F, Piazza S, Gostissa M, Strano S, Zacchi P, Mantovani R, Blandino G, Del Sal G (2004) Pin1 links the activities of c‐Abl and p300 in regulating p73 function. Mol. Cell 14, 625–636. [DOI] [PubMed] [Google Scholar]
- McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R (2003) Myostatin negatively regulates satellite cell activation and self‐renewal. J. Cell Biol. 162, 1135–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane C, Plummer E, Thomas M, Hennebry A, Ashby M, Ling N, Smith H, Sharma M, Kambadur R (2006) Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF‐κB‐independent, FoxO1‐dependent mechanism. J. Cell. Physiol. 209, 501–514. [DOI] [PubMed] [Google Scholar]
- Mecocci P, Fanó G, Fulle S, MacGarvey U, Shinobu L, Polidori MC, Cherubini A, Vecchiet J, Senin U, Beal MF (1999) Age‐dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Radic. Biol. Med. 26, 303–308. [DOI] [PubMed] [Google Scholar]
- Morissette MR, Stricker JC, Rosenberg MA, Buranasombati C, Levitan EB, Mittleman MA, Rosenzweig A (2009) Effects of myostatin deletion in aging mice. Aging Cell. 8, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moron MS, Depierre JW, Mannervik B (1979) Levels of glutathione, glutathione reductase and glutathione S‐transferase activities in rat lung and liver. Biochim. Biophys. Acta 582, 67–78. [DOI] [PubMed] [Google Scholar]
- Moylan JS, Reid MB (2007) Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 35, 411–429. [DOI] [PubMed] [Google Scholar]
- Nystrom T (2005) Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 24, 1311–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 95, 351–358. [DOI] [PubMed] [Google Scholar]
- Oliver M, Harrison N, Bishop J, Cole P, Laurent G (1989) A rapid and convenient assay for counting cells cultured in microwell plates: application for assessment of growth factors. J. Cell Sci. 92, 513–518. [DOI] [PubMed] [Google Scholar]
- Pansarasa O, Bertorelli L, Vecchiet J, Felzani G, Marzatico F (1999) Age‐dependent changes of antioxidant activities and markers of free radical damage in human skeletal muscle. Free Radic. Biol. Med. 27, 617–622. [DOI] [PubMed] [Google Scholar]
- Partridge TA (1997) Tissue culture of skeletal muscle. Methods Mol. Biol. 75, 131–144. [DOI] [PubMed] [Google Scholar]
- Peake JM, Della Gatta P, Cameron‐Smith D (2010) Aging and its effects on inflammation in skeletal muscle at rest and following exercise‐induced muscle injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, 1485–1495. [DOI] [PubMed] [Google Scholar]
- Powers SK, Kavazis AN, DeRuisseau KC (2005) Mechanisms of disuse muscle atrophy: role of oxidative stress. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R337–R344. [DOI] [PubMed] [Google Scholar]
- Raue U, Slivka D, Jemiolo B, Hollon C, Trappe S (2006) Myogenic gene expression at rest and after a bout of resistance exercise in young (18–30 yr) and old (80–89 yr) women. J. Appl. Physiol. 101, 53–59. [DOI] [PubMed] [Google Scholar]
- Reisz‐Porszasz S, Bhasin S, Artaza JN, Shen R, Sinha‐Hikim I, Hogue A, Fielder TJ, Gonzalez‐Cadavid NF (2003) Lower skeletal muscle mass in male transgenic mice with muscle‐specific overexpression of myostatin. Am. J. Physiol. Endocrinol. Metab. 285, E876–E888. [DOI] [PubMed] [Google Scholar]
- Rotruck JT, Pope AL, Ganther HE, Swanson AB, Hafeman DG, Hoekstra WG (1973) Selenium: biochemical role as a component of glutathione peroxidase. Science 179, 588–590. [DOI] [PubMed] [Google Scholar]
- Ryan MJ, Dudash HJ, Docherty M, Geronilla KB, Baker BA, Haff GG, Cutlip RG, Away SE (2008) Aging‐dependent regulation of antioxidant enzymes and redox status in chronically loaded rat dorsiflexor muscles. J. Gerontol. A Biol. Sci. Med. Sci. 63, 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Kambadur R, Matthews KG, Somers WG, Devlin GP, Conaglen JV, Fowke PJ, Bass JJ (1999) Myostatin, a transforming growth factor‐β superfamily member, is expressed in heart muscle and is upregulated in cardiomyocytes after infarct. J. Cell. Physiol. 180, 1–9. [DOI] [PubMed] [Google Scholar]
- Siriett V, Salerno MS, Berry C, Nicholas G, Bower R, Kambadur R, Sharma M (2007) Antagonism of myostatin enhances muscle regeneration during sarcopenia. Mol. Ther. 15, 1463–1470. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R (1996) Oxidative stress, caloric restriction, and aging. Science 273, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman ER (2001) Protein oxidation in aging and age‐related diseases. Ann N Y Acad. Sci. 928, 22–38. [DOI] [PubMed] [Google Scholar]
- Thomas M, Langley B, Berry C, Sharma M, Kirk S, Bass J, Kambadur R (2000) Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J. Biol. Chem. 275, 40235–40243. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84. [DOI] [PubMed] [Google Scholar]
- Welle S, Bhatt K, Shah B, Thornton CA (2002) Insulin‐like growth factor‐1 and myostatin mRNA expression in muscle: comparison between 62‐77 and 21‐31 yr old men. Exp. Gerontol. 37, 833–839. [DOI] [PubMed] [Google Scholar]
- Wilkes JJ, Lloyd DJ, Gekakis N (2009) Loss‐of‐function mutation in myostatin reduces tumor necrosis factor α production and protects liver against obesity‐induced insulin resistance. Diabetes 58, 1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D, Saxel O (1977) Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270, 725–727. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Otsuka F, Mukai T, Otani H, Inagaki K, Miyoshi T, Goto J, Yamamura M, Makino H (2008) Simvastatin antagonizes tumor necrosis factor‐{alpha} inhibition of bone morphogenetic proteins‐2‐induced osteoblast differentiation by regulating Smad signaling and Ras/Rho‐mitogen‐activated protein kinase pathway. J. Endocrinol. 196, 601–613. [DOI] [PubMed] [Google Scholar]
- Yan F, Polk DB (1999) Aminosalicylic acid inhibits IκB kinase α phosphorylation of IκBα in mouse intestinal epithelial cells. J. Biol. Chem. 274, 36631–36636. [DOI] [PubMed] [Google Scholar]
- Yarasheski KE, Bhasin S, Sinha‐Hikim I, Pak‐Loduca J, Gonzalez‐Cadavid NF (2002) Serum myostatin‐immunoreactive protein is increased in 60‐92 year old women and men with muscle wasting. J. Nutr. Health Aging 6, 343–348. [PubMed] [Google Scholar]
- Ye J, Cippitelli M, Dorman L, Ortaldo J, Young H (1996) The nuclear factor YY1 suppresses the human gamma interferon promoter through two mechanisms: inhibition of AP1 binding and activation of a silencer element. Mol. Cell. Biol. 16, 4744–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn JM, Sonu R, Vogel H, Crane E, Mazan‐Mamczarz K, Rabkin R, Davis RW, Becker KG, Owen AB, Kim SK (2006) Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2, 1058–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Rajan V, Lin E, Hu Z, Han HQ, Zhou X, Song Y, Min H, Wang X, Du J, Mitch WE (2011) Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 25, 1653–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Topouzis S, Liang L‐f, Stotish RL (2004) Myostatin signaling through Smad2, Smad3 and Smad4 is regulated by the inhibitory Smad7 by a negative feedback mechanism. Cytokine 26, 262–272. [DOI] [PubMed] [Google Scholar]
- Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Se‐Jin L (2002) Induction of cachexia in mice by systemically administered myostatin. Science 296, 1486–1488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Reagents and chemicals.
Table S1 List of Antibodies used.
Table S2 List of qPCR primers used.
Fig. S1 Mstn down regulates lipid peroxidation in differentiating WT primary myoblasts (A) WT primary myoblasts were treated with Mstn (3 μg/ml) in the differentiation media and total cell lysates were made at indicated time points.
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer‐reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting info item
Supporting info item