

Abstract
Idiopathic achalasia is an archetype esophageal motor disorder, causing significant impairment of eating ability and reducing quality of life. The pathophysiological underpinnings of this condition are loss of esophageal peristalsis and insufficient relaxation of the lower esophageal sphincter (LES). The clinical manifestations include dysphagia for both solids and liquids, regurgitation of esophageal contents, retrosternal chest pain, cough, aspiration, weight loss and heartburn. Even though idiopathic achalasia was first described more than 300 years ago, researchers are only now beginning to unravel its complex etiology and molecular pathology. The most recent findings indicate an autoimmune component, as suggested by the presence of circulating anti-myenteric plexus autoantibodies, and a genetic predisposition, as suggested by observed correlations with other well-defined genetic syndromes such as Allgrove syndrome and multiple endocrine neoplasia type 2 B syndrome. Viral agents (herpes, varicella zoster) have also been proposed as causative and promoting factors. Unfortunately, the therapeutic approaches available today do not resolve the causes of the disease, and only target the consequential changes to the involved tissues, such as destruction of the LES, rather than restoring or modifying the underlying pathology. New therapies should aim to stop the disease at early stages, thereby preventing the consequential changes from developing and inhibiting permanent damage. This review focuses on the known characteristics of idiopathic achalasia that will help promote understanding its pathogenesis and improve therapeutic management to positively impact the patient’s quality of life.
Keywords: Achalasia, Treatment, Autoimmune disease, Pathophysiology
Core tip: Primary achalasia is associated with loss of ganglion cells in the esophagus and in the lower esophageal sphincter. In the last decade, achalasia pathophysiology has been widely studied, and investigations have aimed to expand clinical management beyond mere treatment of symptoms and towards attacking the specific cause of the disease. While the etiologies remain unclear and no cure exists, the most recent findings suggest an interaction between autoimmune and inflammatory responses, possibly triggered by viral infection, in genetically susceptible individuals. This article reviews the recent advances in understanding the disease pathogenesis with implications for improving therapeutic management.
INTRODUCTION
Idiopathic achalasia is a primary motility disease of the esophagus that has a worldwide prevalence of 10 individuals in every 100000 and 1 new case reported per year for every 100000[1,2]. Clinically, it is characterized by two principal findings from esophageal manometry: (1) loss of esophageal peristalsis; and (2) incomplete relaxation of the lower esophageal sphincter (LES)[3,4]. Although its etiology has not yet been completely elucidated, autoimmune, viral and neurodegenerative factors have been proposed as potentially causative or promoting[3,5]. What has been studied and established is that there is a degenerative process, of unknown origin, that involves loss of ganglion cells in the myenteric (Auerbach’s) plexus of the esophagus and is accompanied by local and systemic inflammation[6] and subsequent loss of important neurotransmitters such as the vasoactive intestinal peptide (VIP) and nitric oxide (NO)[7].
The principal (presenting) symptoms of the disease arise secondary to the partial and progressive obstruction of the gastroesophageal junction (GEJ) and aperistalsis, and include dysphagia, regurgitation, weight loss, retrosternal chest pain and heartburn, all of which represent the current therapeutic targets[8]. However, the low prevalence of idiopathic achalasia can delay diagnosis and the similarity of its presenting symptomology to that of gastroesophageal reflux disease (GERD) can lead to misdiagnosis[9]. In general, patients presenting with progressive dysphagia and regurgitation are treated with proton pump inhibitors, the usual treatment for GERD, and non-response to treatment is considered an indicator for idiopathic achalasia suspicion.
The routine work-up for clinical suspicion of idiopathic achalasia includes upper endoscopy, which helps to exclude other differential diagnosis such as tumors and will show a dilated esophagus retaining undigested food and saliva, a barium esophagram, which will show esophageal dilatation, poor emptying, aperistalsis and a narrowed GEJ “bird-beak sign”, and lastly a high-resolution manometry (HRM) that will confirm the diagnosis and facilitate disease subtype classification[3,8]. The following manometrical characteristics correspond to the 3 different subtypes, as outlined by the Chicago Classification criteria: achalasia with absence of peristalsis (“classic”), type I; classic achalasia with esophageal compression, type II; classic achalasia with spastic contractions, type III[10]. This classification system also represents an important tool regarding outcomes after treatments for the disease[11].
In the last decade, researchers have turned their attention towards determining the pathophysiological underpinnings of achalasia in order to not just treat the symptoms but to target the specific cause(s) of the disease that remain unclear and currently do not have a cure.
ETIOLOGY AND PHYSIOPATHOLOGY
Primary achalasia is associated with loss of ganglion cells in the esophagus and in the LES. The loss of ganglion cells has been associated with both inflammation and deposition of collagen. Findings from the collective research suggest that the disease process involves an interaction between autoimmune and inflammatory responses, possibly triggered by viral infection, in genetically susceptible individuals (Figure 1).
Figure 1.
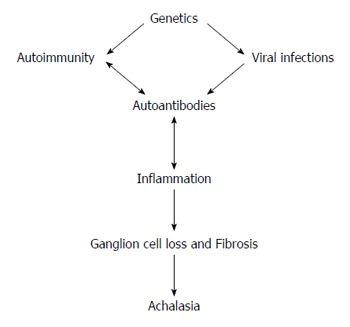
Main mechanisms in the etiopathogenesis of achalasia.
Ganglion cell loss
The enteric nervous system is distributed along the gastrointestinal tract, including the esophagus. The myenteric plexus is situated between the circular and longitudinal smooth muscle layers of the gut and consists of postganglionic neurons that differentiate into excitatory cholinergic neurons and inhibitory nitrergic neurons. While the excitatory neurons release acetylcholine, the inhibitory neurons release the free radical NO and the neurotransmitter/anti-inflammatory cytokine VIP; the coordinated release creates the balance of relaxation and contraction that is vital for normal esophageal peristalsis[12].
Achalasia is known to be caused by the reduction of interstitial cells of Cajal, but most importantly to a selective loss of inhibitory ganglion in the myenteric plexus of the esophagus, which is associated with a decrease of NO and VIP. These pathophysiological and morphologic features have been demonstrated in a benzyldimethyltetradecylammonium chloride-induced amyenteric rat model and in human tissues[5,13-20]. In addition, the ability of nitrergic nerves to mediate esophageal neuromuscular functions, such as LES relaxation and normal peristalsis, was demonstrated in experimental animal systems.
Animal studies have also provided the first clues about the genetic underpinnings of achalasia. Mice with neuronal NO synthase 1 gene disruption (nNOS-/-) presented with significantly increased hypertensive LES, and consequently markedly impaired relaxation that developed into an achalasia-like LES dysfunction and gastroparesis; these findings contrast with the hypotensive LES present in the W/Wv mice that lack the interstitial cells of Cajal as intermediary of LES relaxation[21,22].
Human studies have also suggested a significantly decreased or absent NO innervation in the myenteric plexus of patients with achalasia[23]. Immunohistochemical studies of biopsies from patients with achalasia who underwent surgical treatment showed that levels of the VIP, nNOS, neural proteins, S-100, substance P and protein gene product 9.5 (PGP9.5) were significantly lower than in healthy individuals[5,13,24]. Thus, impaired production of NO and VIP are involved in the pathophysiology of achalasia, and - as discussed later in this review - may be genetic components and therapeutic targets.
An underlying pathogenic mechanism in the early stage of achalasia is the esophageal myenteric immune-mediated response and inflammatory state accompanied by T cell infiltration, triggered by a yet unknown etiologic factor. This pathologic condition may cause neuritis and ganglionitis, with no initial ganglion cell loss or with only mild to severe fibrosis in the smooth muscle layer. A proteomic analysis of serum from patients with achalasia and from healthy individuals showed disease-related up-regulation of transthyretin (TTR); TTR is a serum and cerebrospinal fluid carrier of the thyroid hormone thyroxine (T4) and a retinol-binding protein that is associated with familial amyloid polyneuropathy, and its observed up-regulation corroborates with the consequent neural degeneration observed in patients[25]. Moreover, other studies of achalasia patients have shown increased deposits of the complement complex C5b-C9 and of IgM within or proximal to ganglion cells of myenteric plexus[26]. The imbalance produced by loss of VIP- and NO-secreting neurons can lead to irreversible esophageal motor dysfunction. Furthermore, the progressive destruction of myenteric ganglion cells and the occurrence of neural fibrosis would lead to the classic subtype of achalasia[19,27].
In addition to the destruction of inhibitory ganglionic cells (due to the abundant inflammatory infiltrates and autoimmunity), hypertrophy and neuronal fibrosis accompany the disease progression[28]. Most reports of pathological findings for human cases show the presence of neuronal autoantibodies[29,30]. These autoimmune constituents have been shown to directly contribute to the destruction of the myenteric plexus[31,32]. In particular, Bruley des Varannes et al[23] showed that serum from patients with achalasia, and not from patients with GERD, can induce phenotypic and functional changes in myenteric neurons that reproduce the disease characteristics, thereby refuting the theory that these autoantibodies could be an epiphenomenon.
Infections
In order to explain the loss of ganglion cells in achalasia, several studies have explored the potential of pathogenic factors as the disease-initiating agents, in particular those related to chronic latent or active neurotrophic viral or bacterial infections. The proposed virus candidates have included herpes simplex virus (HSV) (a neurotropic virus with predilection for squamous epithelium), John Cunningham (commonly known as “JC”) virus, bornavirus, varicella zoster, measles, and human papilloma virus[33-36]. In fact, Boeckxstaens[35] proposed that during latent HSV-1 infection, the virus persists in the neurons of the LES and esophagus, giving rise to a persistent immune activation that in genetically predisposed individuals can elicit neuronal destruction. Moreover, an epidemiological study and genotype-phenotype analysis carried out by Becker et al[37] demonstrated that patients were frequently affected by viral infections, especially varicella zoster virus infections, before achalasia onset, and that pregnancy may stimulate the disease in females who are carriers of the HLA-DQβ1 insertion[37].
Several studies of esophageal biopsy specimens from patients with idiopathic achalasia have used the polymerase chain reaction to investigate the presence of various virus strains. Infection with and active replication of HSV-1, but not cytomegalovirus or Epstein-Barr virus, were detected along the myenteric plexus. The preferential ports of entry for HSV-1 are the perioral and esophageal mucosa. Furthermore, herpes viruses exhibit a strong tropism for nerve fibers and following the primary exposure the viruses can remain in a latent form in neuronal nuclei[6,34]. T lymphocytes are known to specifically respond to HSV-1 antigens, and Facco et al[34] demonstrated that achalasia patients have a significantly higher rate of oligoclonal CD3+/CD8+ lymphocytic infiltrates in LES, as compared with healthy controls.
Since not all patients with viral infections develop achalasia, it has been proposed that specific genetic changes affecting the immune system may create susceptibility to this disease. Chronic viral infection could trigger an aberrant immune response that under an appropriate genetic and environmental background would facilitate the loss of esophageal neurons. In support of this theory, detailed examinations of the myenteric plexus of patients with achalasia have shown infiltration of CD3+/CD45RO+ T cells, predominantly CD8+ T cytotoxic lymphocytes expressing activation markers[5,17,18,38].
Autoimmunity
The proposed autoimmune etiology of achalasia is supported by the presence of anti-myenteric antibodies in the circulation and inflammatory T cell infiltrates in the myenteric plexus, as well as demonstrated statistical correlations between the disease and particular HLA class II antigens. Autoimmune diseases often occur in association with one another, either involving a single individual or within a family. It has been proposed that the etiology of achalasia includes an autoimmune component. Findings from a recent study, and numerous case reports, have characterized patients with achalasia as being 3.6-times more likely to have autoimmune diseases, including uveitis (RR = 259), Sjögren’s syndrome (RR = 37), systemic lupus erythematosus (RR = 43), type I diabetes (RR = 5.4), hypothyroidism (RR = 8.5), rheumatoid arthritis (RR = 2.4), scleroderma, ankylosing spondylitis, myasthenia gravis, Guillain-Barre syndrome, autoimmune acquired hemophilia A, polyglandular autoimmune syndrome type II, psoriasis and asthma[29,39-45]. Intriguingly, the younger population of patients with achalasia was shown to have a higher prevalence of autoimmune comorbidities (RR = 3.3)[43], compared with an older population of patients with achalasia. Finally, there are some reports of patients with achalasia responding to immunosuppressive drugs, lending further credence to the notion that this disease has an autoimmune component[42].
It is most likely that the etiology of achalasia is multifactorial, involving genetic and immune-related factors, possibly both pathogen-/environmental- and host-derived[15]. Such an etiological profile may trigger damaging autoimmune mechanisms or chronic inflammation.
Inflammation
Histological and immunohistochemical studies of the esophageal tissues of patients with achalasia have also found inflammatory infiltrates of varying intensity around myenteric neurons, contrasting with the non-infiltrate findings of control groups with normal myenteric plexus[6,18,38,46]. CD3+, CD4+, CD25+ and CD8+ T lymphocytes predominated in all the diseased tissues, as well as CD20+ B lymphocytes and eosinophilic granulocytes but to a lesser degree, with detection of occasional plasma and mast cells along the nerve fascicles and around the ganglion cells[5,6,18,46]. Moreover, T and B inflammatory infiltrates predominated in tissues with advanced stage disease (> 10-year symptom history)[5].
A serum proteomic analysis has demonstrated that C4B5, C3, cyclin-dependent kinase 5, and α2-macroglobulin are up-regulated in achalasia patients, as compared with controls, corroborating the theory of immune-mediated response and/or neural degeneration components of the disease pathogenesis[25]. Moreover, another study of achalasia patients had shown that the complement complex C5b-C9 (membrane attack complex) and IgM are deposited within or at the ganglion cells of the myenteric plexus[26].
Turnover of extracellular matrix and some CD4+ T cell subsets and cytokines have been also described in achalasia patients. In particular, a significant increase has been observed in the expression of matrix metalloproteinase-9 (MMP-9, also known as 92 kDa gelatinase) and its tissue inhibitor, TIMP1, in LES from patients with achalasia, as compared to controls[6]. Some tissue and circulating CD4+ T cell subsets have been characterized in patients with achalasia as well (Figure 2).
Figure 2.

Hypothetical interplay of cytokines in the pathophysiology of achalasia. IL: Interleukin; IFN: Interferon.
Up-regulated expression of interleukin-22 (IL-22) has been detected in tissue (especially in myenteric plexus)[6] and circulating cells of patients with achalasia (Figure 3). IL-22 belongs to the IL-10 superfamily, and acts as an initiator of the innate immune response against pathogens in gut epithelial and respiratory cells, as a modulator of tissue repair/regeneration processes, and as a regulator of antibody production. IL-22 is synthesized by the T helper (Th) cell subsets of Th22 and Th17. The Th22 cells differentiate from naïve T cells in response to TNF-α and IL-6 signals, and subsequently synthesize and secrete IL-26, IL-13 and IL-22; IL-26 plays important roles in cellular proliferation and survival, antimicrobial peptide production, epithelial renewal and immunity[47].
Figure 3.
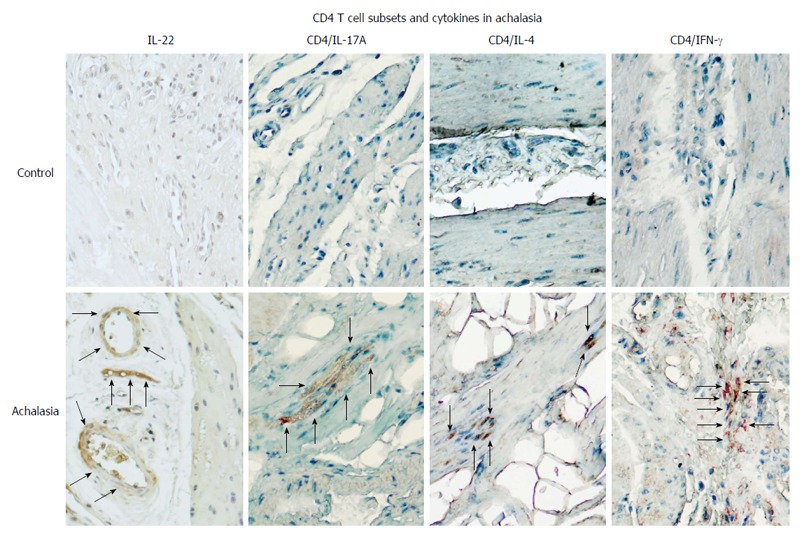
CD4+ T cell subsets and their expression of pro-inflammatory/anti-fibrogenic cytokines in achalasia. Immunohistochemical analysis of IL-22, CD4/IL-17 (Th17 cells), CD4/IL-4 (Th2 cells), and CD4/IFN-γ (Th1 cells) in specimens of esophagus from healthy control donors and achalasia patients. Arrows denote the immunoreactive cells. Original magnification × 320.
The cytokine IL-17A is produced by the Th17 subset and is a key mediator of auto-inflammatory diseases. Under both physiological and pathological conditions, IL-17A acts to stimulate T cells, increase the production of autoantibodies and inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-8, IL-17, IL-22, etc) and chemokines (CCL2, CCL7, CCL20, CXCL1, CXCL5), induce neutrophil recruitment through chemokine regulation, activate innate immune cells and enhance B cell functions[48]. The frequency of IL-17A-secreting cells is reportedly higher in the peripheral cells and myenteric plexus of esophageal tissue of patients with achalasia, as compared to controls (Figure 3)[6].
Another important cytokine, interferon-gamma (IFN-γ), may play a role in achalasia pathogenesis. Produced by activated T cells and natural killer cells (NKs), IFN-γ regulates various immune and inflammatory responses. Specifically, this cytokine potentiates the effects of the type I IFNs, recruits leukocytes to infected tissue in order to potentiate beneficial inflammation, and stimulates macrophages to engulf and kill bacteria. IFN-γ released by Th1 cells is also important in regulating the Th2 response. As IFN-γ is vitally implicated in the regulation of immune response, aberrant regulation of its production can lead to autoimmune diseases. Moreover, IFN-γ inhibits collagen synthesis and induces the production of chemokines including CXCR3, CXCL9, CXCL10 and CXCL11[48]. Studies have found that patients with achalasia have a significantly higher percentage of circulating and tissue IFN-γ+/CD4+ T cells, as compared to controls (Figure 3)[6].
Increased expression of IL-1β, IL-2 and TNF-β has also been detected in tissue from achalasia patients, as compared with controls[34]. Other cytokines with dual anti-inflammatory/pro-fibrogenic functions have been evaluated in patients with achalasia. Under normal physiologic conditions, TGF-β1 controls cellular growth, proliferation, differentiation, negative regulation of inflammation, collagen synthesis and apoptosis; patients with achalasia have shown significantly increased TGF-β1+ cell expression in the myenteric plexus of esophageal biopsies, as compared to tissues from controls[6].
IL-4 is an anti-inflammatory cytokine that inhibits the synthesis of several important cytokines (IL-1β, TNF-α, IL-6, IL-17A, etc), regulates B cell proliferation and differentiation, and functions as a potent inhibitor of apoptosis. IL-4 is synthesized primarily by Th2 cells and is required for the initiation and maintenance of fibrosis. The IL-4-expressing CD4+ Th2 subset is defined by production of IL-4, IL-5, IL-9 and IL-13 and functions in type 2 immunity for fighting off infectious disease, the process of which involves neutralization of toxins, maintenance of metabolic homeostasis, regulation of wound healing, regeneration of tissue and production and deposition of fibrosis-enhancing collagen; in addition, these cells suppress autoimmune disease mediated by Th1 cells[49]. Studies have shown that patients with achalasia have significantly higher circulating and tissue IL-4+ cell percentage, as compared to controls (Figure 3)[6].
IL-13 has functions similar to IL-4; in contrast, however, it regulates the type I collagen gene, thereby participating in fibrosis. Its expression pattern in patients with achalasia is also similar to that of IL-4[6].
Immune “regulatory cells” (such as the T regulatory cells commonly known as Tregs) include a variety of cell subpopulations with specialized functions that allow them to exert cell extrinsic immunosuppression and tolerance to self as well to foreign antigens. Tregs modulate the natural course of protective immune responses in order to limit tissue damage and autoimmunity. In addition, they suppress immunologic responses by producing granzymes and perforins, depleting IL-2, secreting suppressor molecules such as IL-19 and TGF-β1, and diminishing the functions of antigen presenting cells (APCs) that otherwise promote anergy or apoptosis of effector T cells. As such, the Tregs play important roles in tissue repair and homeostasis[50]. Patients with achalasia show a higher Treg frequency in the myenteric plexus of esophageal tissue, as compared to controls[6]. Nonetheless, Sodikoff et al[46] reported that Foxp3 (the distinguishing marker of Tregs) was not detectable in achalasia biopsies of their cohort, contrasting our findings in which a higher percentage of CD25+/Foxp3+ cells were detected in esophageal smooth muscle tissue from patients with type III achalasia, followed by types I and II, as compared to controls. These discrepant findings may be explained by different techniques used to conserve the samples prior to examination or to perform the immunohistochemical evaluation, or the findings may reflect important differences in disease evolution among the two studies’ cohorts (Figure 4)[46].
Figure 4.
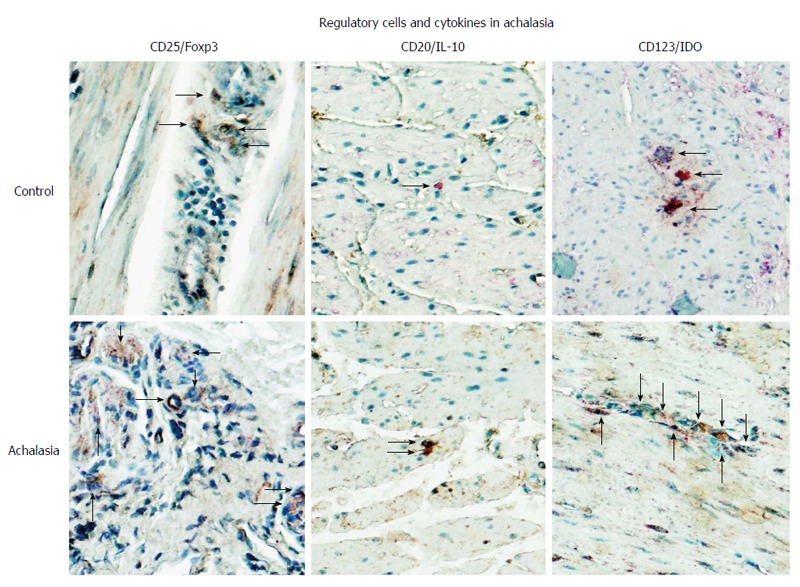
Regulatory cells in achalasia. Immunohistochemical analysis of CD25/Foxp3 (T regulatory cells), CD20/IL-10 (B regulatory cells) and CD123/IDO (plasmacytoid dendritic regulatory cells) in specimens of esophagus from healthy control donors and achalasia patients. Arrows denote immunoreactive cells. Original magnification × 320.
In addition to Tregs, a newly described population of regulatory B cells (termed Bregs) has also been shown to contribute to immunosuppression, not only in autoimmune diseases but also in inflammatory and organ/tissue transplant conditions; regardless, however, the effects are direct and occur via enhancement of Treg function. This CD19+CD24hiCD38hi immature/transitional T1 B cell subset suppresses the differentiation of Th1 cells in an IL-10-dependent manner[51]. Intriguingly, biopsies of myenteric plexus obtained from patients with achalasia showed a higher relative IL-10-producing B cell percentage than tissues from a control group (Figure 4)[6].
Lastly, it is known that dendritic plasmacytoid regulatory cells (termed pDCregs) are a sub-population of immune cells that express the indoleamine 2,3-dioxygenase (IDO) enzyme that is responsible for mediating tryptophan metabolism, which suppresses T effector cell activity and induces CD4+/CD25hi regulatory T cell polarization. IDO-mediated deprivation of tryptophan halts the proliferation of T cells at mid-G1 phase, which in concert with the pro-apoptotic activity of kynurenine leads to immune tolerance. IDO has a selective role in Th2 differentiation and is regulated positively during antigenic presentation and the functional complexing of CTLA-4/B7-1/B7-2 in lymphocytes and dendritic cells. In addition IDO contributes to the immune responses to pathogens, being up-regulated by circulating nucleic acids (from host and non-host genomes) through the activation of TLR4 and TLR9, and it contributes to adaptive immunity processes that subsequently modulate the inflammatory process[52]. Patients with achalasia have shown a higher frequency of pDCregs in the myenteric plexus of esophageal tissue, as compared to control tissues (Figure 4).
Autoantibodies
The observation of increased prevalence of circulating IgG antibodies against myenteric plexus in most patients with achalasia has led to the suggestion of a role for autoantibodies in the pathogenesis of this disease. Studies have also demonstrated a notable absence of anti-myenteric autoantibody in achalasia-free controls, patients with Hirschsprung’s disease, esophageal cancer, peptic esophagitis, gastroesophageal reflux or myasthenia gravis[53-55]. Nonetheless, a study by Moses et al[32] suggested that these circulatory antibodies are more likely the result of a non-specific reaction to the disease process, rather than being the cause of the disease; this idea was supported by detection of similar antibodies in patients without achalasia. In accordance with the hypotheses, evidence of autoantibodies against myenteric neurons were detected in serum samples from patients with achalasia, especially in carriers of HLA DQA1*0103 and DQB1*0603 alleles[55]. Recently, Kallel-Sellami et al[30], as well as our group[6], determined the levels of circulating anti-myenteric antibodies in serum from patients with achalasia; the measurements in both studies were carried out with the commercially available kit Neurology Mosaic 1 (Euroimmun, Leubeck, Germany) that involves a standard indirect immunofluorescence screening assay using frozen monkey nerves, cerebellum and intestinal tissue as antigenic substrates. The prevalence of nuclear or cytoplasmic circulating antibodies against myenteric plexus in the sera from idiopathic achalasia patients was 63% and 100% vs 12% and 0% in the sera from healthy donors, respectively; moreover, most antibodies showed positive reaction in the nuclear and nucleolar compartments of cells in the myenteric plexus[6,30]. These two studies also analyzed the target antigens of circulating anti-myenteric autoantibodies by testing sera with the Neuronal Antigens Profile Plus RST kit (Euroimmun) that involves immunoblotting for a panel of individual neuronal antigens, including amphiphysin, CV2, PNMA2 (Ma-2/Ta), onconeuronal antigens (Ri, Yo, Hu), recoverin, SOX-1 and titin. A majority (69%) of the sera samples from the idiopathic achalasia patients reacted with PNMA2 (Ma2/Ta), and a few (8%) reacted with the recoverin antigen that is related to Sjögren’s syndrome[6,30].
Other autoantibodies have been detected in serum from non-diabetic patients with achalasia, including glutamic acid descarboxylase-65 (GAD65) antibody, which showed a remarkably higher frequency compared to the control subjects (21% vs 2.5%)[29]. The enzyme GAD65 converts glutamic acid to gamma-aminobutyric acid, and is expressed in GABAergic nerve terminals in the enteric nervous system. GAD65 antibodies are reportedly present in approximately 80% of patients with type I diabetes mellitus and in approximately 20% of patients with various organ-specific neurological disorders, including myasthenia gravis, Lambert-Eaton syndrome, autoimmune dysautonomias and encephalopathies[29]. Approximately 40% of the achalasia patients evaluated in similar studies have shown at least one other organ-specific autoantibody, namely thyroid or gastric parietal cell antibodies, and > 40% of patients have shown anti-nuclear antibodies[23,29].
Genetics
Genetic factors may play an important role in the development and progression of achalasia. The existence of familial cases suggests that achalasia may be inherited and thus have a genetic component. Furthermore, statistical correlations of achalasia with well-defined genetic syndromes have been found. For example, mutation in the ALADIN 12q13 gene, which is associated with Allgrove syndrome, specifically (Triple-A) in exons 1, 2, 7, 8, 10-14 and 16, and a poly(A) tract, shows clinical manifestations of achalasia, alacrima, and adrenocorticotrophic hormone-resistant adrenal insufficiency[56-58]. In addition, achalasia has been associated with the multiple endocrine neoplasia type 2 (MEN 2) B syndrome, specifically a germline mutation in exon 16 (M918T) of the RET proto-oncogene on chromosome 10q11 that leads to a methionine-threonine amino acid substitution in the tyrosine kinase domain and thus constitutive activation of the oncogene. The MEN 2 mutation can manifest as one of three types of cancer predisposition, all with an autosomal dominant mode of inheritance; these include the familial medullary thyroid carcinoma, MEN2A and MEN 2B forms[27]. Riley-Day syndrome (familial dysautonomia) and Smith-Lemli-Optiz syndrome (elevated concentrations of the 7- and 8-dehydrocholesterol due to reductase deficiency) have also been linked to achalasia[27]. The combination of achalasia with Hirschsprung’s disease or aganglionic megacolon has been also described[59]. Up to 2% of children with Down’s syndrome have achalasia, presumably due to a significant reduction in the number of neurons present in the esophageal plexus of this population[56,60]. Finally, congenital central hypoventilation syndrome has been also associated with achalasia in children[56].
As idiopathic achalasia is a relatively rare disorder, another approach to genetic analysis of these cases has been to identify candidate genes via identification of single nucleotide polymorphisms (SNPs) and classification of their clinical phenotype. The neuronal nitric oxide synthase (NOS1) gene is located on human chromosome 12q24.2, and a disease-related microsatellite (CA repeat) polymorphism has been found within the 3’-untranslated region (UTR) of exon 29. Additionally, an exome analysis of two siblings with infant-onset achalasia revealed homozygosity for a premature stop codon in the gene encoding NOS1 (at residue Tyr1202, instead of at residue 1435). Kinetic analyses and molecular modeling indicated that the truncated protein product has defective folding capacity, as well as defective capabilities for NO production and binding of co-factors[15,61]. Other genetic polymorphisms of NOS gene isoforms that have been discovered involve the endothelial NOS4a4a, inducible NOS22GA and neuronal NOS29TT[62]. The receptor of vasoactive intestinal polypeptide (VIPR1) gene is located on chromosome 3p22. VIPR1 belongs to the secretin receptor family, a group of G-protein coupled receptors expressed by immune cells (T cells, macrophages and dendritic cells) and myenteric neurons of the distal esophagus and LES. It is highly polymorphic, and five SNPs have been reported in patients with late achalasia, including (rs421558) intron-1, (rs437876) intron-4, (rs417387) intron-6, and (rs896 and rs9677) 3’-UTR[63]. The IL-23 receptor (IL-23R) gene is located on chromosome 1p31 and its encoded protein, IL-23R, is expressed by Th17 cells and has been associated with chronic autoimmune disorders. One study showed an IL-23R gene polymorphism, wherein arginine replaces glutamine at codon 381, as significantly more common in patients with achalasia than in healthy controls[15,64]. The protein tyrosine phosphatase non-receptor 22 (PTPN22) gene is located on chromosome 1p13.3-p13, within a region known to be associated with autoimmune disease; the encoded protein, an intracellular lymphoid-specific tyrosine phosphatase (Lyp), is a down-regulator of T cell activation. A SNP in the PTPN22 gene at position 1858C/T, which leads to a replacement of arginine with tryptophan in codon 620, has been shown to increase risk of achalasia in females of Spanish descent[15,65]. Polymorphisms in the IL10 gene have been associated with different autoimmune conditions, such as systemic lupus erythematosus, type 1 diabetes, ulcerative colitis and asthma; for achalasia, however, the GCC haplotype of the IL10 promoter has been associated with a lower risk of the disease[15,66]. Finally, polymorphisms in the IL-33 gene, which encodes the IL-1 cytokine family member IL-33 and is known to play a critical role in chronic inflammatory autoimmune diseases, have been reported as more frequent in females with achalasia, as compared to controls; specifically, the polymorphisms are the rs3939286 SNP and the rs7044343T/rs3939286A risk haplotype[67].
Human leukocyte antigen (HLA) class II antigens have also been associated with autoimmune diseases such as systemic lupus erythematosus, Sjögren’s syndrome, and other connective tissue diseases. In addition, the myenteric infiltrates are predominantly T cell lymphocytes than can recognize certain class II antigens. Associations between achalasia and HLA-DQβ1 (HLA-DQB1*05:03 and HLA-DQB1*06:01), HLA-DQα1 (HLA-DQA1*01:03), and HLA-DQβ1 (HLA-DQB1*03:01 and HLA-DQB1*03:04) have been determined[15,45]. In fact, many studies have shown a positive association with this disease and the various class II HLA antigens, including DQw1, DQA1*0103, DQB1*0601, DQB1*0602, DQB1*0603, DQB1*0601, DQB1*0502, and DQB1*0503 alleles, in Caucasians. Moreover, patients with the DQA1*0103 and DQB1*0603 alleles have been shown to have a significantly higher prevalence of anti-myenteric antibody[37,54,55,67-70]. In a study of 1068 cases of achalasia from central Europe, Spain and Italy, an 8-residue insertion at position 227-234 in the cytoplasmic tail of HLA-DQβ1 (encoded by HLA-DQB1*05:03 and HLA-DQB1*06:01) was characterized as conferring the strongest risk for achalasia. Two amino acid substitutions in the extracellular domain of HLA-DQα1 at position 41 (lysine encoded by HLA-DQA1*01:03) and of HLA-DQβ1 at position 45 (glutamic acid encoded by HLA-DQB1*03:01 and HLA-DQB1*03:04) were characterized as independently conferring achalasia risk[70]. Moreover, the HLA-DQβ1 insertion was characterized as a strong risk factor for achalasia and showed a particular geospatial north-south gradient among Europeans. The finding of this insertion being less common in northern European populations, as compared with those from the southern regions mirrored the differential prevalence of the disease between populations[4]. This geographic profile may reflect a genetic predisposition, putting certain individuals at increased risk of developing achalasia, possibly after exposure to some particular environmental conditions. It is important to note, here, that not all patients with achalasia carry the putative “predisposing” HLA and not all people with the HLA have the disease[37,71].
Thus, the initial event that triggers achalasia may be the result of a repetitive insult produced by neurotropic virus infection, likely HSV-1[6,34-36,72,73], which induces a conspicuous and persistent inflammation at the perineural level, in the myenteric plexus. Not all infected patients will develop achalasia, on account of a genetic predisposition to develop a chronic auto-inflammatory response that has the potential to progress to the disease[35] (Figure 5).
Figure 5.
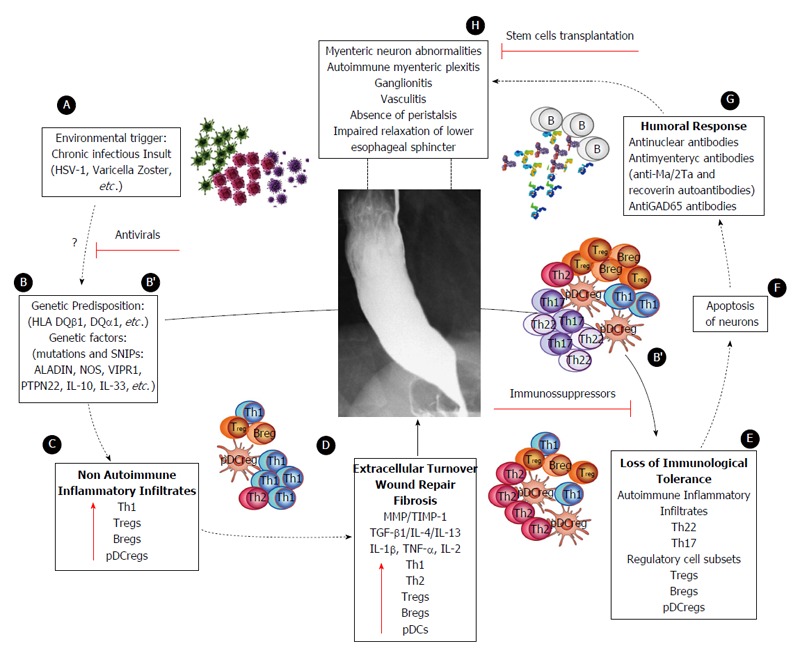
Proposed model of achalasia pathophysiology (modified from Furuzawa-Carballeda et al[6]). A: An initial active or latent infectious insult, likely involving a neurotropic virus such as the herpes family of viruses or varicella zoster, which have predilection for squamous epithelium and neurons and may cause ganglion cell damage which would be limited to the esophagus; B: Some individuals with genetic predisposition will progress with an aggressive inflammatory response; C: At a very early stage of the disease, the inflammatory infiltrates may be predominantly composed of Th1, Th2 and regulatory cell subsets [T regulatory cells (Tregs), B regulatory cells (Bregs) and plasmacytoid dendritic regulatory cells (pDCregs)]; D: Repair of tissue after injury would require orchestrated coordination of several cell types and biosynthetic processes and would be coordinated by an interacting group of pro- and anti-inflammatory cytokines, fibrous extracellular matrix (ECM) proteins to replace lost or damaged tissue, and products of metabolism such as oxygen radicals. The most prominent pro-fibrogenic cytokines are TGFβ, IL-4 and IL-13. ECM also mediates cellular crosstalk, and does so in two ways. Newly deposited ECM is then rebuilt over time to emulate normal tissue. Matrix proteinases and their inhibitors (TIMPs) also are important, during wound repair, tissue remodeling and fibrosis. B’; E: If steps A-D occur repeatedly, such as in a chronic infection condition, only those individuals with genetic predisposition to developing a long-lasting autoinflammatory response will progress to development of the disease (loss of peripheral tolerance). Thus, autoinflammatory infiltrates would be predominantly composed of Th22, Th17 and regulatory subpopulations; F: Degeneration and significant loss of nerve fibers, associated with autoinflammatory infiltrates of the myenteric plexus, provide evidence of an immune-mediated destruction of the inhibitory neurons, not only by necrosis but also apoptosis (Fas/FasL overexpression); G: Autoimmune etiology of achalasia is further supported by the presence of anti-myenteric autoantibodies in sera; H: Pathophysiologically, achalasia is caused by autoinflammation, degeneration of nerves in the esophagus, plexitis, abnormalities in microvasculature, ganglionitis, and finally by the loss of inhibitory ganglion in the myenteric plexus. Red lines: Potential therapeutic targets.
THERAPEUTIC APPROACHES
Currently, there is no cure for achalasia. Treatment goals are to ameliorate the patient’s symptoms, improve esophageal body emptying and limit esophageal dilation, all of which can be accomplished by resolving the esophageal outflow obstruction (Table 1). The different therapeutic approaches available today, including pharmacotherapy, botulinum toxin injections, endoscopical dilatations, esophageal stents, peroral endoscopy myotomy and surgical treatment for achalasia (Figure 6), all aim to treat the symptoms but are not capable of use as preventives or address the underlying pathology of the disease[8,74,75].
Table 1.
Current treatment options in achalasia
| Treatment option | Pros | Cons | Success rate |
| Oral agents | Non operative patients, on demand, dose adjustment | Adverse events, low duration, not a definitive method | 28%-66% reduction of LES pressure |
| Pneumatic dilation | Short recovery, low procedure time, best non-surgical method | Perforation, multiple procedures needed, post procedure reflux | 66%-90% 1 yr and 48% 10 yr |
| Heller myotomy | Most durable effect | Not applicable for high risk surgical patients, post-surgical reflux, anesthesia required | 93% 1 yr |
| 69%-80% 10 yr | |||
| Self-expanding metal stent | Good palliative option, high risk surgical patients | Expensive, stent migration, reflux (single center experience) | 100% 1 mo, |
| 83% 10 yr | |||
| POEM | Non-surgical, -low and -high risk patients | Complications (pneumothorax, reflux), not widely available, expertise | 5%-62% reduction of LES pressure |
Figure 6.
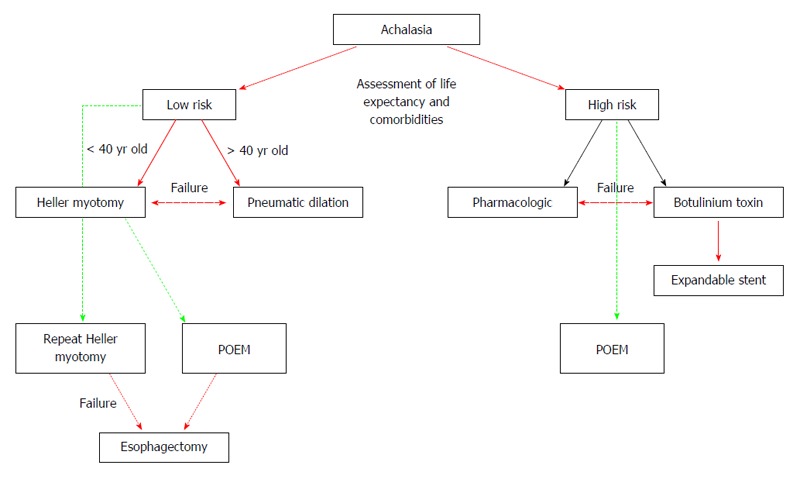
Proposed algorithm for the treatment of Achalasia patients (modified from Sioulas et al[95]). Red single arrow: recommended strategy; Red bold arrow: treatment for low-risk; Black single arrow: treatment for high-risk; Green dashed line: potential, low evidence treatment; Red dashed double arrow: alternative for treatment failure.
Pharmacologic therapy
Calcium channel blockers, long-acting nitrates and phosphodiesterase type 5 inhibitors, to name a few, have been used for achalasia; however, none has proven an effective therapy. Use of these drugs is only recommended in the early stages of the disease, as a temporary regimen immediately prior to another definitive treatment, or in patients who are unable or unwilling to undergo any other of the therapeutic alternatives[3,8]. Nevertheless, these drugs are capable of providing a clinical response, albeit an incomplete and short-acting one. Another important feature of these drugs are the side effects, which are generally poorly tolerated and span the spectrum from inconvenient (headache) to severe (hypotension and edema).
Endoscopic therapies
Botulinum toxin is a presynaptic inhibitor of acetylcholine release from motor neurons. Endoscopic intrasphinteric injection of botulinum toxin A has been demonstrated as a safe approach to achieve short-term improvement of symptoms (85% of the patients experience improvement with a single injection)[76]; however, this therapy loses efficacy over the long-term, resulting in patients frequently requiring increasingly repeated injections (60% of patients starting at 6 mo after the first injection, and 30% at 1 year after). Although some authors advocate for its use as a bridge therapy, to be administered while another definite treatment option is actively sought for the patient under care, they note that it should be applied with caution as it is not free of complications[77]. Perhaps botulinum toxin has a role in treating patients in whom dilation or myotomy are contraindicated[3,8,78].
Endoscopical dilatation (ED) has been demonstrated as the most effective nonsurgical treatment of achalasia, but it has the highest rates of complications. For this procedure, pneumatic dilators are preferred over rigid and using a greater diameter of the dilator produces better long-term results (i.e., change from 3.0 to 4.0 mm diameter increased the 4-year follow-up success rate to 93%). Dysphagia success rates at the 5-year and 15-year follow-ups were reported as 40%-78% and 12%-58%, respectively[8]. The complications of ED are various and include esophageal perforation (0%-16%; as low as 1.9% in experienced hands), GERD (15%-33%) and intramural hematomas. Due to the risk of esophageal perforation, all patients must first be confirmed as candidates for subsequent surgical interventions that will repair the damage, and in cases of GERD post-dilation proton pump inhibitors are recommended[3,8].
Another treatment alternative for the treatment of achalasia includes self-expanded metal stents (SEMS). Experience is limited using this treatment option and includes a study with 75 achalasia patients in which a 30 mm SEMS was temporally placed under fluoroscopic guidance. After 4-5 d, SEMS where endoscopically removed and patients followed for up to 10 years. Success rate at 1 mo was 100% and 83% at 10 years[79]. Another study compared the efficacy of different SEMS diameters (20, 25 and 30 mm) in achalasia patients. A total of 90 patients were included and followed at 10 years and showed better success rate (83.3%) with a 30 mm SEMS compared to lower diameters[80]. SEMS have shown promising results but experience is limited to a single institution and cannot be widely recommended and more studies are needed in order to include this in the treatment algorithm for the treatment of achalasia.
Surgical therapies
The surgical approach that involves disruption of the muscle fibers of the LES (known as a Heller myotomy) has been demonstrated as very effective in treating patients with achalasia. Since its first description in 1913, this procedure has undergone multiple modifications that have optimized its application and outcome[8,74]. The current laparoscopic Heller myotomy (LHM) has reported success rates of 95% and 75% at 5-year and 15.8-year follow-ups, respectively. Comparison of the LHM approach against a combination approach of LHM + fundoplication showed that the latter reduced GERD rates substantially (48% vs 9%)[81]. Of note, it is recommended to perform a partial fundoplication after the Heller myotomy[82-84].
Peroral endoscopic myotomy
Peroral endoscopic myotomy (POEM) is a newly developed technique that involves creation of a submucosal tunnel to disrupt the muscular layers of the esophagus, and it is considered a promising approach for treating achalasia with minimal complications. It was developed by Inoue et al[85] in 2010 as a method that combines the benefits of myotomy with those of an endoscopic application; since then, this less invasive procedure has proven to be feasible, safe and effective. A meta-analysis found that POEM efficacy for dysphagia may be similar to that of laparoscopic myotomy, but with the added benefit of a lower hospital stay[86]. Like myotomy, POEM modifies the LES pressure and esophageal body motility. The rates of short-term clinical success (at 1-year follow-up) have been high, from 82% to 100%. Although long-term outcomes have not been reported yet, the 2-mo, 2-year and 3-year success rates are high, at 91.3%, 91.0% and 88.5%, respectively[86].
NEW PERSPECTIVES AND FUTURE TREATMENTS
The therapeutic approaches available today for the treatment of achalasia implies the destruction of the LES, rather than restoring or modifying the underlying pathology of the disease. In the last 10 years the immune-mediated hypothesis to explain the pathophysiology of achalasia has gained strong support based on objective evidence[6]. Considering an autoimmune etiology, it is theoretically reasonable to treat patients with immune modulatory drugs in the early stages of the disease, when there is presumably still a number of functional neurons that can be protected[69].
To our knowledge, there are only three case reports of corticosteroid use (specifically of prednisolone, methylprednisolone or beclomethasone) alone or combined with other immunosuppressive therapy (specifically of methotrexate, azathioprine or cyclophosphamide) for achalasia and the results show dramatic improvement of the clinical picture, with complete recovery of peristalsis corroborated by HRM[39,42,87,88]. This feature reinforces the concept of a cause-effect relationship of the immune-mediated insult damaging the enteric innervation. Although the studies show an important improvement, until today there is no definitive evidence that has been reported to support the widespread application of this therapeutic approach. Thus, future studies to estimate the benefit of immunosuppressive therapy are still required.
A possible alternative therapeutic approach, which still lacks strong evidential support, is transplantation of neural progenitor cells. Recently, investigators have demonstrated that stem cells with neurogenic potential can successfully engraft, survive, migrate and differentiate into neurons and glia within the aganglionic intestine. Moreover, preliminary evidence has indicated that transplanted cell-based therapies can lead to a functional recovery of aganglionic gastrointestinal diseases, including achalasia[69,89]. This finding opens a promising avenue of scientific investigation for future studies to improve screening for early diagnosis or genetic predisposition[37,45,54,55,67-70] for idiopathic achalasia.
Last but not least, the use of antiviral therapy in patients with recent-onset achalasia is promising. By this therapeutic approach, the antigenic challenge would be eliminated and the immune response might be controlled in these patients[6,34-36,72,73] (Figure 5 and Table 2).
Table 2.
Pathophysiology mechanisms and potential therapeutic targets in achalasia
| Physiopathology mechanism | Therapeutic target |
| Incomplete relaxation of lower esophageal sphincter | 3Botulinum toxin injection[3,8,74-77] |
| 3Pneumatic dilatation[3,8] | |
| 3Heller myotomy[8,74,81-84] | |
| 3Self-Expanding metal stent[79,80] | |
| 3POEM[85,86,91] | |
| Loss of inhibitory ganglion neurons and decrease of NO and VIP | 3Nitrates administration (isosorbide dinitrate and nitroglycerin)[3,7,8] |
| 1Treatment with immunosuppressive drugs (prednisolone, methylprednisolone, beclomethasone, methotrexate, azathioprine or cyclophosphamide) in the early stage of the disease[39,42,87,88] | |
| 2Transplantation of neural progenitor cells[69,89] | |
| Immune-mediate response, inflammation, organ-specific autoimmune disease, and autoantibodies that causes neuritis and glanglionitis | 1Treatment with immunosuppressive drugs (prednisolone, methylprednisolone, beclomethasone, methotrexate, azathioprine or cyclophosphamide) to avoid immune-mediate insult damaging the enteric innervation. Suppress the whole activated immune system, exerts antiproliferative and pro-apoptotic effects, particularly on activated T cells and suppress antibody formation by B cells[39,42,87,88] |
| 2Biologic therapy (monoclonal antibodies: TNF-α inhibitors, IL-6 inhibitor, anti-CD20 antibody, CTLA-4 antibody, IFN-g antibody, etc) in the early stage of the disease[48,93] | |
| 2Administration of regulatory cells to downregulate inflammation and to induce immunological tolerance[50-52] | |
| Fibrosis | 2Extracellular matrix remodeling (Polymerized type I collagen)[94] |
| Neurotrophic viral infection (Herpes simplex virus, varicella zoster, measles, etc.), molecular mimicry, bystander activation, viral persistence and polyclonal activation | 2Antiviral therapy in patients with recent-onset achalasia[6,34-36,72,73] |
| Genetics | 2Genomic medicine for decrease the effect of certain environmental factors, such as infectious agents, on the burden of disease[61-66] |
Drugs or experimental procedures with therapeutic benefit;
Drugs or experimental procedures with potential therapeutic;
Therapies with marginal benefit.
CONCLUSION
New research has introduced different perspectives regarding the possible etiology of achalasia. In the last 10 years, the immune-mediated hypothesis (as the primary pathophysiologic abnormality) has gained strong objective support. Yet, the therapeutic approaches available today for the treatment of achalasia still do not resolve the cause of the disease; instead, they target the consequences, focusing on the destruction of the LES rather than on restoring or modifying the underlying pathology. With better understanding of the pathophysiology of achalasia, new therapies may prevent permanent damage and stop the disease at early stages.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Mexico
Peer-review report classification
Grade A (Excellent): A, A
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: The authors report no conflicts of interest.
Peer-review started: April 6, 2016
First decision: June 20, 2016
Article in press: August 5, 2016
P- Reviewer: Garcia-Olmo D, Macedo G, Mestieri LHM, Wang HT S- Editor: Yu J L- Editor: A E- Editor: Ma S
References
- 1.Sadowski DC, Ackah F, Jiang B, Svenson LW. Achalasia: incidence, prevalence and survival. A population-based study. Neurogastroenterol Motil. 2010;22:e256–e261. doi: 10.1111/j.1365-2982.2010.01511.x. [DOI] [PubMed] [Google Scholar]
- 2.Enestvedt BK, Williams JL, Sonnenberg A. Epidemiology and practice patterns of achalasia in a large multi-centre database. Aliment Pharmacol Ther. 2011;33:1209–1214. doi: 10.1111/j.1365-2036.2011.04655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaezi MF, Pandolfino JE, Vela MF. ACG clinical guideline: diagnosis and management of achalasia. Am J Gastroenterol. 2013;108:1238–149; quiz 1250. doi: 10.1038/ajg.2013.196. [DOI] [PubMed] [Google Scholar]
- 4.Pandolfino JE, Kwiatek MA, Nealis T, Bulsiewicz W, Post J, Kahrilas PJ. Achalasia: a new clinically relevant classification by high-resolution manometry. Gastroenterology. 2008;135:1526–1533. doi: 10.1053/j.gastro.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gockel I, Bohl JR, Doostkam S, Eckardt VF, Junginger T. Spectrum of histopathologic findings in patients with achalasia reflects different etiologies. J Gastroenterol Hepatol. 2006;21:727–733. doi: 10.1111/j.1440-1746.2006.04250.x. [DOI] [PubMed] [Google Scholar]
- 6.Furuzawa-Carballeda J, Aguilar-León D, Gamboa-Domínguez A, Valdovinos MA, Nuñez-Álvarez C, Martín-del-Campo LA, Enríquez AB, Coss-Adame E, Svarch AE, Flores-Nájera A, et al. Achalasia--An Autoimmune Inflammatory Disease: A Cross-Sectional Study. J Immunol Res. 2015;2015:729217. doi: 10.1155/2015/729217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boeckxstaens GE. Novel mechanism for impaired nitrergic relaxation in achalasia. Gut. 2006;55:304–305. doi: 10.1136/gut.2005.078402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hunter D, Osborn W, Wang K, Kazantseva N, Hattrick-Simpers J, Suchoski R, Takahashi R, Young ML, Mehta A, Bendersky LA, et al. Giant magnetostriction in annealed Co(1-x)Fe(x) thin-films. Nat Commun. 2011;2:518. doi: 10.1038/ncomms1529. [DOI] [PubMed] [Google Scholar]
- 9.Ates F, Vaezi MF. The Pathogenesis and Management of Achalasia: Current Status and Future Directions. Gut Liver. 2015;9:449–463. doi: 10.5009/gnl14446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kahrilas PJ, Bredenoord AJ, Fox M, Gyawali CP, Roman S, Smout AJ, Pandolfino JE. The Chicago Classification of esophageal motility disorders, v3.0. Neurogastroenterol Motil. 2015;27:160–174. doi: 10.1111/nmo.12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torresan F, Ioannou A, Azzaroli F, Bazzoli F. Treatment of achalasia in the era of high-resolution manometry. Ann Gastroenterol. 2015;28:301–308. [PMC free article] [PubMed] [Google Scholar]
- 12.Patel DA, Kim HP, Zifodya JS, Vaezi MF. Idiopathic (primary) achalasia: a review. Orphanet J Rare Dis. 2015;10:89. doi: 10.1186/s13023-015-0302-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gockel I, Bohl JR, Eckardt VF, Junginger T. Reduction of interstitial cells of Cajal (ICC) associated with neuronal nitric oxide synthase (n-NOS) in patients with achalasia. Am J Gastroenterol. 2008;103:856–864. doi: 10.1111/j.1572-0241.2007.01667.x. [DOI] [PubMed] [Google Scholar]
- 14.Herbella FA, Oliveira DR, Del Grande JC. Are idiopathic and Chagasic achalasia two different diseases? Dig Dis Sci. 2004;49:353–360. doi: 10.1023/b:ddas.0000020486.71719.62. [DOI] [PubMed] [Google Scholar]
- 15.Ghoshal UC, Daschakraborty SB, Singh R. Pathogenesis of achalasia cardia. World J Gastroenterol. 2012;18:3050–3057. doi: 10.3748/wjg.v18.i24.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petersen RP, Martin AV, Pellegrini CA, Oelschlager BK. Synopsis of investigations into proposed theories on the etiology of achalasia. Dis Esophagus. 2012;25:305–310. doi: 10.1111/j.1442-2050.2009.01030.x. [DOI] [PubMed] [Google Scholar]
- 17.Kilic A, Krasinskas AM, Owens SR, Luketich JD, Landreneau RJ, Schuchert MJ. Variations in inflammation and nerve fiber loss reflect different subsets of achalasia patients. J Surg Res. 2007;143:177–182. doi: 10.1016/j.jss.2007.03.050. [DOI] [PubMed] [Google Scholar]
- 18.Raymond L, Lach B, Shamji FM. Inflammatory aetiology of primary oesophageal achalasia: an immunohistochemical and ultrastructural study of Auerbach’s plexus. Histopathology. 1999;35:445–453. doi: 10.1046/j.1365-2559.1999.035005445.x. [DOI] [PubMed] [Google Scholar]
- 19.Park W, Vaezi MF. Etiology and pathogenesis of achalasia: the current understanding. Am J Gastroenterol. 2005;100:1404–1414. doi: 10.1111/j.1572-0241.2005.41775.x. [DOI] [PubMed] [Google Scholar]
- 20.Singaram C, Sweet MA, Gaumnitz EA, Bass P, Snipes RL. Evaluation of early events in the creation of amyenteric opossum model of achalasia. Neurogastroenterol Motil. 1996;8:351–361. doi: 10.1111/j.1365-2982.1996.tb00273.x. [DOI] [PubMed] [Google Scholar]
- 21.Sivarao DV, Mashimo HL, Thatte HS, Goyal RK. Lower esophageal sphincter is achalasic in nNOS(-/-) and hypotensive in W/W(v) mutant mice. Gastroenterology. 2001;121:34–42. doi: 10.1053/gast.2001.25541. [DOI] [PubMed] [Google Scholar]
- 22.Müller M, Colcuc S, Drescher DG, Eckardt AJ, von Pein H, Taube C, Schumacher J, Gockel HR, Schimanski CC, Lang H, et al. Murine genetic deficiency of neuronal nitric oxide synthase (nNOS(-/-) ) and interstitial cells of Cajal (W/W(v) ): Implications for achalasia? J Gastroenterol Hepatol. 2014;29:1800–1807. doi: 10.1111/jgh.12600. [DOI] [PubMed] [Google Scholar]
- 23.Bruley des Varannes S, Chevalier J, Pimont S, Le Neel JC, Klotz M, Schafer KH, Galmiche JP, Neunlist M. Serum from achalasia patients alters neurochemical coding in the myenteric plexus and nitric oxide mediated motor response in normal human fundus. Gut. 2006;55:319–326. doi: 10.1136/gut.2005.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoshino M, Omura N, Yano F, Tsuboi K, Kashiwagi H, Yanaga K. Immunohistochemical study of the muscularis externa of the esophagus in achalasia patients. Dis Esophagus. 2013;26:14–21. doi: 10.1111/j.1442-2050.2011.01318.x. [DOI] [PubMed] [Google Scholar]
- 25.Im SK, Yeo M, Lee KJ. Proteomic identification of proteins suggestive of immune-mediated response or neuronal degeneration in serum of achalasia patients. Gut Liver. 2013;7:411–416. doi: 10.5009/gnl.2013.7.4.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Storch WB, Eckardt VF, Junginger T. Complement components and terminal complement complex in oesophageal smooth muscle of patients with achalasia. Cell Mol Biol (Noisy-le-grand) 2002;48:247–252. [PubMed] [Google Scholar]
- 27.Gockel HR, Gockel I, Schimanski CC, Schier F, Schumacher J, Nöthen MM, Lang H, Müller M, Eckardt AJ, Eckardt VF. Etiopathological aspects of achalasia: lessons learned with Hirschsprung’s disease. Dis Esophagus. 2012;25:566–572. doi: 10.1111/j.1442-2050.2011.01277.x. [DOI] [PubMed] [Google Scholar]
- 28.Akiho H, Ihara E, Motomura Y, Nakamura K. Cytokine-induced alterations of gastrointestinal motility in gastrointestinal disorders. World J Gastrointest Pathophysiol. 2011;2:72–81. doi: 10.4291/wjgp.v2.i5.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraichely RE, Farrugia G, Pittock SJ, Castell DO, Lennon VA. Neural autoantibody profile of primary achalasia. Dig Dis Sci. 2010;55:307–311. doi: 10.1007/s10620-009-0838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kallel-Sellami M, Karoui S, Romdhane H, Laadhar L, Serghini M, Boubaker J, Lahmar H, Filali A, Makni S. Circulating antimyenteric autoantibodies in Tunisian patients with idiopathic achalasia. Dis Esophagus. 2013;26:782–787. doi: 10.1111/j.1442-2050.2012.01398.x. [DOI] [PubMed] [Google Scholar]
- 31.Dhamija R, Tan KM, Pittock SJ, Foxx-Orenstein A, Benarroch E, Lennon VA. Serologic profiles aiding the diagnosis of autoimmune gastrointestinal dysmotility. Clin Gastroenterol Hepatol. 2008;6:988–992. doi: 10.1016/j.cgh.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moses PL, Ellis LM, Anees MR, Ho W, Rothstein RI, Meddings JB, Sharkey KA, Mawe GM. Antineuronal antibodies in idiopathic achalasia and gastro-oesophageal reflux disease. Gut. 2003;52:629–636. doi: 10.1136/gut.52.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robertson CS, Martin BA, Atkinson M. Varicella-zoster virus DNA in the oesophageal myenteric plexus in achalasia. Gut. 1993;34:299–302. doi: 10.1136/gut.34.3.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Facco M, Brun P, Baesso I, Costantini M, Rizzetto C, Berto A, Baldan N, Palù G, Semenzato G, Castagliuolo I, et al. T cells in the myenteric plexus of achalasia patients show a skewed TCR repertoire and react to HSV-1 antigens. Am J Gastroenterol. 2008;103:1598–1609. doi: 10.1111/j.1572-0241.2008.01956.x. [DOI] [PubMed] [Google Scholar]
- 35.Boeckxstaens GE. Achalasia: virus-induced euthanasia of neurons? Am J Gastroenterol. 2008;103:1610–1612. doi: 10.1111/j.1572-0241.2008.01967.x. [DOI] [PubMed] [Google Scholar]
- 36.Sinagra E, Gallo E, Mocciaro F, Stella M, Malizia G, Montalbano LM, Orlando A, D’Amico G, Cottone M, Rizzo AG. JC Virus, Helicobacter pylori, and oesophageal achalasia: preliminary results from a retrospective case-control study. Dig Dis Sci. 2013;58:1433–1434. doi: 10.1007/s10620-012-2485-9. [DOI] [PubMed] [Google Scholar]
- 37.Becker J, Niebisch S, Ricchiuto A, Schaich EJ, Lehmann G, Waltgenbach T, Schafft A, Hess T, Lenze F, Venerito M, et al. Comprehensive epidemiological and genotype-phenotype analyses in a large European sample with idiopathic achalasia. Eur J Gastroenterol Hepatol. 2016;28:689–695. doi: 10.1097/MEG.0000000000000602. [DOI] [PubMed] [Google Scholar]
- 38.Clark SB, Rice TW, Tubbs RR, Richter JE, Goldblum JR. The nature of the myenteric infiltrate in achalasia: an immunohistochemical analysis. Am J Surg Pathol. 2000;24:1153–1158. doi: 10.1097/00000478-200008000-00014. [DOI] [PubMed] [Google Scholar]
- 39.Kornizky Y, Heller I, Isakov A, Shapira I, Topilsky M. Dysphagia with multiple autoimmune disease. Clin Rheumatol. 2000;19:321–323. doi: 10.1007/s100670070055. [DOI] [PubMed] [Google Scholar]
- 40.Emami MH, Raisi M, Amini J, Daghaghzadeh H. Achalasia and thyroid disease. World J Gastroenterol. 2007;13:594–599. doi: 10.3748/wjg.v13.i4.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quidute AR, Freitas EV, Lima TG, Feitosa AM, Santos JP, Correia JW. Achalasia and thyroid disease: possible autoimmune connection? Arq Bras Endocrinol Metabol. 2012;56:677–682. doi: 10.1590/s0004-27302012000900013. [DOI] [PubMed] [Google Scholar]
- 42.Al-Jafar H, Laffan M, Al-Sabah S, Elmorsi M, Habeeb M, Alnajar F. Severe recurrent achalasia cardia responding to treatment of severe autoimmune acquired haemophilia. Case Rep Gastroenterol. 2012;6:618–623. doi: 10.1159/000343435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Booy JD, Takata J, Tomlinson G, Urbach DR. The prevalence of autoimmune disease in patients with esophageal achalasia. Dis Esophagus. 2012;25:209–213. doi: 10.1111/j.1442-2050.2011.01249.x. [DOI] [PubMed] [Google Scholar]
- 44.Amr BS, Mamillapalli C. Achalasia in a Patient with Polyglandular Autoimmune Syndrome Type II. Case Rep Gastroenterol. 2015;9:160–164. doi: 10.1159/000430493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Becker J, Haas SL, Mokrowiecka A, Wasielica-Berger J, Ateeb Z, Bister J, Elbe P, Kowalski M, Gawron-Kiszka M, Majewski M, et al. The HLA-DQβ1 insertion is a strong achalasia risk factor and displays a geospatial north-south gradient among Europeans. Eur J Hum Genet. 2016;24:1228–1231. doi: 10.1038/ejhg.2015.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sodikoff JB, Lo AA, Shetuni BB, Kahrilas PJ, Yang GY, Pandolfino JE. Histopathologic patterns among achalasia subtypes. Neurogastroenterol Motil. 2016;28:139–145. doi: 10.1111/nmo.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xin N, Namaka MP, Dou C, Zhang Y. Exploring the role of interleukin-22 in neurological and autoimmune disorders. Int Immunopharmacol. 2015;28:1076–1083. doi: 10.1016/j.intimp.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 48.Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun. 2008;31:252–256. doi: 10.1016/j.jaut.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. 2015;15:271–282. doi: 10.1038/nri3831. [DOI] [PubMed] [Google Scholar]
- 50.Perdigoto AL, Chatenoud L, Bluestone JA, Herold KC. Inducing and Administering Tregs to Treat Human Disease. Front Immunol. 2015;6:654. doi: 10.3389/fimmu.2015.00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding T, Yan F, Cao S, Ren X. Regulatory B cell: New member of immunosuppressive cell club. Hum Immunol. 2015;76:615–621. doi: 10.1016/j.humimm.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 52.Vorobjova T, Uibo O, Heilman K, Uibo R. Increased density of tolerogenic dendritic cells in the small bowel mucosa of celiac patients. World J Gastroenterol. 2015;21:439–452. doi: 10.3748/wjg.v21.i2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Storch WB, Eckardt VF, Wienbeck M, Eberl T, Auer PG, Hecker A, Junginger T, Bosseckert H. Autoantibodies to Auerbach’s plexus in achalasia. Cell Mol Biol (Noisy-le-grand) 1995;41:1033–1038. [PubMed] [Google Scholar]
- 54.Verne GN, Sallustio JE, Eaker EY. Anti-myenteric neuronal antibodies in patients with achalasia. A prospective study. Dig Dis Sci. 1997;42:307–313. doi: 10.1023/a:1018857617115. [DOI] [PubMed] [Google Scholar]
- 55.Ruiz-de-León A, Mendoza J, Sevilla-Mantilla C, Fernández AM, Pérez-de-la-Serna J, Gónzalez VA, Rey E, Figueredo A, Díaz-Rubio M, De-la-Concha EG. Myenteric antiplexus antibodies and class II HLA in achalasia. Dig Dis Sci. 2002;47:15–19. doi: 10.1023/a:1013242831900. [DOI] [PubMed] [Google Scholar]
- 56.Hallal C, Kieling CO, Nunes DL, Ferreira CT, Peterson G, Barros SG, Arruda CA, Fraga JC, Goldani HA. Diagnosis, misdiagnosis, and associated diseases of achalasia in children and adolescents: a twelve-year single center experience. Pediatr Surg Int. 2012;28:1211–1217. doi: 10.1007/s00383-012-3214-3. [DOI] [PubMed] [Google Scholar]
- 57.Zimmer V, Vanderwinden JM, Zimmer A, Ostertag D, Strittmatter M, Koehler K, Huebner A, Lammert F. Organ-specific Neurodegeneration in Triple A syndrome-related Achalasia. Am J Med. 2015;128:e9–12. doi: 10.1016/j.amjmed.2015.04.025. [DOI] [PubMed] [Google Scholar]
- 58.Thomas J, Subramanyam S, Vijayaraghavan S, Bhaskar E. Late onset adrenal insufficiency and achalasia in Allgrove syndrome. BMJ Case Rep. 2015;2015:pii bcr2014208900. doi: 10.1136/bcr-2014-208900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taguchi T, Ieiri S, Miyoshi K, Kohashi K, Oda Y, Kubota A, Watanabe Y, Matsufuji H, Fukuzawa M, Tomomasa T. The incidence and outcome of allied disorders of Hirschsprung’s disease in Japan: Results from a nationwide survey. Asian J Surg. 2015 doi: 10.1016/j.asjsur.2015.04.004. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 60.Sosa-Stanley J, Vandendool K, Kiev J. Redo Heller Myotomy for Achalasia in a Patient with Down Syndrome: a Case Report. W V Med J. 2015;111:16–18. [PubMed] [Google Scholar]
- 61.Shteyer E, Edvardson S, Wynia-Smith SL, Pierri CL, Zangen T, Hashavya S, Begin M, Yaacov B, Cinamon Y, Koplewitz BZ, et al. Truncating mutation in the nitric oxide synthase 1 gene is associated with infantile achalasia. Gastroenterology. 2015;148:533–536.e4. doi: 10.1053/j.gastro.2014.11.044. [DOI] [PubMed] [Google Scholar]
- 62.Singh R, Ghoshal UC, Misra A, Mittal B. Achalasia Is Associated With eNOS4a4a, iNOS22GA, and nNOS29TT Genotypes: A Case-control Study. J Neurogastroenterol Motil. 2015;21:380–389. doi: 10.5056/jnm14123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paladini F, Cocco E, Cascino I, Belfiore F, Badiali D, Piretta L, Alghisi F, Anzini F, Fiorillo MT, Corazziari E, et al. Age-dependent association of idiopathic achalasia with vasoactive intestinal peptide receptor 1 gene. Neurogastroenterol Motil. 2009;21:597–602. doi: 10.1111/j.1365-2982.2009.01284.x. [DOI] [PubMed] [Google Scholar]
- 64.de León AR, de la Serna JP, Santiago JL, Sevilla C, Fernández-Arquero M, de la Concha EG, Nuñez C, Urcelay E, Vigo AG. Association between idiopathic achalasia and IL23R gene. Neurogastroenterol Motil. 2010;22:734–78, e218. doi: 10.1111/j.1365-2982.2010.01497.x. [DOI] [PubMed] [Google Scholar]
- 65.Santiago JL, Martínez A, Benito MS, Ruiz de León A, Mendoza JL, Fernández-Arquero M, Figueredo MA, de la Concha EG, Urcelay E. Gender-specific association of the PTPN22 C1858T polymorphism with achalasia. Hum Immunol. 2007;68:867–870. doi: 10.1016/j.humimm.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 66.Nuñez C, García-González MA, Santiago JL, Benito MS, Mearín F, de la Concha EG, de la Serna JP, de León AR, Urcelay E, Vigo AG. Association of IL10 promoter polymorphisms with idiopathic achalasia. Hum Immunol. 2011;72:749–752. doi: 10.1016/j.humimm.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 67.Latiano A, De Giorgio R, Volta U, Palmieri O, Zagaria C, Stanghellini V, Barbara G, Mangia A, Andriulli A, Corinaldesi R, et al. HLA and enteric antineuronal antibodies in patients with achalasia. Neurogastroenterol Motil. 2006;18:520–525. doi: 10.1111/j.1365-2982.2006.00772.x. [DOI] [PubMed] [Google Scholar]
- 68.De la Concha EG, Fernandez-Arquero M, Mendoza JL, Conejero L, Figueredo MA, Perez de la Serna J, Diaz-Rubio M, Ruiz de Leon A. Contribution of HLA class II genes to susceptibility in achalasia. Tissue Antigens. 1998;52:381–384. doi: 10.1111/j.1399-0039.1998.tb03059.x. [DOI] [PubMed] [Google Scholar]
- 69.Boeckxstaens GE, Zaninotto G, Richter JE. Achalasia. Lancet. 2014;383:83–93. doi: 10.1016/S0140-6736(13)60651-0. [DOI] [PubMed] [Google Scholar]
- 70.Gockel I, Becker J, Wouters MM, Niebisch S, Gockel HR, Hess T, Ramonet D, Zimmermann J, Vigo AG, Trynka G, et al. Common variants in the HLA-DQ region confer susceptibility to idiopathic achalasia. Nat Genet. 2014;46:901–904. doi: 10.1038/ng.3029. [DOI] [PubMed] [Google Scholar]
- 71.Lenz TL, Deutsch AJ, Han B, Hu X, Okada Y, Eyre S, Knapp M, Zhernakova A, Huizinga TW, Abecasis G, et al. Widespread non-additive and interaction effects within HLA loci modulate the risk of autoimmune diseases. Nat Genet. 2015;47:1085–1090. doi: 10.1038/ng.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Niwamoto H, Okamoto E, Fujimoto J, Takeuchi M, Furuyama J, Yamamoto Y. Are human herpes viruses or measles virus associated with esophageal achalasia? Dig Dis Sci. 1995;40:859–864. doi: 10.1007/BF02064992. [DOI] [PubMed] [Google Scholar]
- 73.Ganem D, Kistler A, DeRisi J. Achalasia and viral infection: new insights from veterinary medicine. Sci Transl Med. 2010;2:33ps24. doi: 10.1126/scitranslmed.3000986. [DOI] [PubMed] [Google Scholar]
- 74.Torres-Villalobos G, Martin-Del-Campo LA. Surgical treatment for achalasia of the esophagus: laparoscopic heller myotomy. Gastroenterol Res Pract. 2013;2013:708327. doi: 10.1155/2013/708327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gyawali CP. Achalasia: new perspectives on an old disease. Neurogastroenterol Motil. 2016;28:4–11. doi: 10.1111/nmo.12750. [DOI] [PubMed] [Google Scholar]
- 76.Annese V, Bassotti G, Coccia G, Dinelli M, D’Onofrio V, Gatto G, Leandro G, Repici A, Testoni PA, Andriulli A. A multicentre randomised study of intrasphincteric botulinum toxin in patients with oesophageal achalasia. GISMAD Achalasia Study Group. Gut. 2000;46:597–600. doi: 10.1136/gut.46.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chao CY, Raj A, Saad N, Hourigan L, Holtmann G. Esophageal perforation, inflammatory mediastinitis and pseudoaneurysm of the thoracic aorta as potential complications of botulinum toxin injection for achalasia. Dig Endosc. 2015;27:618–621. doi: 10.1111/den.12392. [DOI] [PubMed] [Google Scholar]
- 78.Pasricha PJ, Ravich WJ, Hendrix TR, Sostre S, Jones B, Kalloo AN. Intrasphincteric botulinum toxin for the treatment of achalasia. N Engl J Med. 1995;332:774–778. doi: 10.1056/NEJM199503233321203. [DOI] [PubMed] [Google Scholar]
- 79.Zhao JG, Li YD, Cheng YS, Li MH, Chen NW, Chen WX, Shang KZ. Long-term safety and outcome of a temporary self-expanding metallic stent for achalasia: a prospective study with a 13-year single-center experience. Eur Radiol. 2009;19:1973–1980. doi: 10.1007/s00330-009-1373-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li YD, Cheng YS, Li MH, Chen NW, Chen WX, Zhao JG. Temporary self-expanding metallic stents and pneumatic dilation for the treatment of achalasia: a prospective study with a long-term follow-up. Dis Esophagus. 2010;23:361–367. doi: 10.1111/j.1442-2050.2010.01048.x. [DOI] [PubMed] [Google Scholar]
- 81.Richards WO, Torquati A, Holzman MD, Khaitan L, Byrne D, Lutfi R, Sharp KW. Heller myotomy versus Heller myotomy with Dor fundoplication for achalasia: a prospective randomized double-blind clinical trial. Ann Surg. 2004;240:405–12; discussion 412-5. doi: 10.1097/01.sla.0000136940.32255.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rawlings A, Soper NJ, Oelschlager B, Swanstrom L, Matthews BD, Pellegrini C, Pierce RA, Pryor A, Martin V, Frisella MM, et al. Laparoscopic Dor versus Toupet fundoplication following Heller myotomy for achalasia: results of a multicenter, prospective, randomized-controlled trial. Surg Endosc. 2012;26:18–26. doi: 10.1007/s00464-011-1822-y. [DOI] [PubMed] [Google Scholar]
- 83.Kumagai K, Kjellin A, Tsai JA, Thorell A, Granqvist S, Lundell L, Håkanson B. Toupet versus Dor as a procedure to prevent reflux after cardiomyotomy for achalasia: results of a randomised clinical trial. Int J Surg. 2014;12:673–680. doi: 10.1016/j.ijsu.2014.05.077. [DOI] [PubMed] [Google Scholar]
- 84.Rebecchi F, Giaccone C, Farinella E, Campaci R, Morino M. Randomized controlled trial of laparoscopic Heller myotomy plus Dor fundoplication versus Nissen fundoplication for achalasia: long-term results. Ann Surg. 2008;248:1023–1030. doi: 10.1097/SLA.0b013e318190a776. [DOI] [PubMed] [Google Scholar]
- 85.Inoue H, Minami H, Kobayashi Y, Sato Y, Kaga M, Suzuki M, Satodate H, Odaka N, Itoh H, Kudo S. Peroral endoscopic myotomy (POEM) for esophageal achalasia. Endoscopy. 2010;42:265–271. doi: 10.1055/s-0029-1244080. [DOI] [PubMed] [Google Scholar]
- 86.Youn YH, Minami H, Chiu PW, Park H. Peroral Endoscopic Myotomy for Treating Achalasia and Esophageal Motility Disorders. J Neurogastroenterol Motil. 2016;22:14–24. doi: 10.5056/jnm15191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.De Giorgio R, Guerrini S, Barbara G, Stanghellini V, De Ponti F, Corinaldesi R, Moses PL, Sharkey KA, Mawe GM. Inflammatory neuropathies of the enteric nervous system. Gastroenterology. 2004;126:1872–1883. doi: 10.1053/j.gastro.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 88.Savarino E, Gemignani L, Zentilin P, De Bortoli N, Malesci A, Mastracci L, Fiocca R, Savarino V. Achalasia with dense eosinophilic infiltrate responds to steroid therapy. Clin Gastroenterol Hepatol. 2011;9:1104–1106. doi: 10.1016/j.cgh.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 89.Wagner JP, Sullins VF, Dunn JC. A novel in vivo model of permanent intestinal aganglionosis. J Surg Res. 2014;192:27–33. doi: 10.1016/j.jss.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Krill JT, Naik RD, Vaezi MF. Clinical management of achalasia: current state of the art. Clin Exp Gastroenterol. 2016;9:71–82. doi: 10.2147/CEG.S84019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dobrowolsky A, Fisichella PM. The management of esophageal achalasia: from diagnosis to surgical treatment. Updates Surg. 2014;66:23–29. doi: 10.1007/s13304-013-0224-1. [DOI] [PubMed] [Google Scholar]
- 92.Marano L, Pallabazzer G, Solito B, Santi S, Pigazzi A, De Luca R, Biondo FG, Spaziani A, Longaroni M, Di Martino N, et al. Surgery or Peroral Esophageal Myotomy for Achalasia: A Systematic Review and Meta-Analysis. Medicine (Baltimore) 2016;95:e3001. doi: 10.1097/MD.0000000000003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rosman Z, Shoenfeld Y, Zandman-Goddard G. Biologic therapy for autoimmune diseases: an update. BMC Med. 2013;11:88. doi: 10.1186/1741-7015-11-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Furuzawa-Carballeda J, Ortíz-Ávalos M, Lima G, Jurado-Santa Cruz F, Llorente L. Subcutaneous administration of polymerized type I collagen downregulates interleukin (IL)-17A, IL-22 and transforming growth factor-β1 expression, and increases Foxp3-expressing cells in localized scleroderma. Clin Exp Dermatol. 2012;37:599–609. doi: 10.1111/j.1365-2230.2012.04385.x. [DOI] [PubMed] [Google Scholar]
- 95.Sioulas AD, Malli C, Dimitriadis GD, Triantafyllou K. Self-expandable metal stents for achalasia: Thinking out of the box! World J Gastrointest Endosc. 2015;7:45–52. doi: 10.4253/wjge.v7.i1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]