

Abstract
Colonic inflammation is required to heal infections, wounds, and maintain tissue homeostasis. As the seventh hallmark of cancer, however, it may affect all phases of tumor development, including tumor initiation, promotion, invasion and metastatic dissemination, and also evasion immune surveillance. Inflammation acts as a cellular stressor and may trigger DNA damage or genetic instability, and, further, chronic inflammation can provoke genetic mutations and epigenetic mechanisms that promote malignant cell transformation. Both sporadical and colitis-associated colorectal carcinogenesis are multi-step, complex processes arising from the uncontrolled proliferation and spreading of malignantly transformed cell clones with the obvious ability to evade the host’s protective immunity. In cells upon DNA damage several proto-oncogenes, including c-MYC are activated in parelell with the inactivation of tumor suppressor genes. The target genes of the c-MYC protein participate in different cellular functions, including cell cycle, survival, protein synthesis, cell adhesion, and micro-RNA expression. The transcriptional program regulated by c-MYC is context dependent, therefore the final cellular response to elevated c-MYC levels may range from increased proliferation to augmented apoptosis. Considering physiological intestinal homeostasis, c-MYC displays a fundamental role in the regulation of cell proliferation and crypt cell number. However, c-MYC gene is frequently deregulated in inflammation, and overexpressed in both sporadic and colitis-associated colon adenocarcinomas. Recent results demonstrated that endogenous c-MYC is essential for efficient induction of p53-dependent apoptosis following DNA damage, but c-MYC function is also involved in and regulated by autophagy-related mechanisms, while its expression is affected by DNA-methylation, or histone acetylation. Molecules directly targeting c-MYC, or agents acting on other genes involved in the c-MYC pathway could be selected for combined regiments. However, due to its context-dependent cellular function, it is clinically essential to consider which cytotoxic drugs are used in combination with c-MYC targeted agents in various tissues. Increasing our knowledge about MYC-dependent pathways might provide direction to novel anti-inflammatory and colorectal cancer therapies.
Keywords: c-MYC, Therapy, Apoptosis, Autophagy, Colon, Inflammation, Colorectal cancer
Core tip: The c-MYC gene is frequently deregulated in colonic inflammation, and overexpressed in both sporadic and colitis-associated colon adenocarcinomas. Endogenous c-MYC is essential for efficient induction of p53-dependent apoptosis following DNA damage, moreover its function is also involved in and regulated by autophagy-related mechanisms, and its expression is affected by DNA-methylation, or histone acetylation. Increasing our knowledge about MYC-dependent pathways might provide direction to novel colonic anti-inflammatory and anti-cancer strategies.
INTRODUCTION
Chronic, non-infectious inflammatory and cancerous colonic diseases currently represent a major threat to human health worldwide. Inflammation is required to fight microbial infections, heal wounds, and maintain tissue homeostasis, however, it could lead to cancer. As the seventh hallmark of cancer it may affect all phases of tumor development, including tumor initiation, promotion, invasion and metastatic dissemination, and also evasion immune surveillance[1]. Inflammation acts as a cellular stressor and may trigger DNA damage or genetic instability, and, further, chronic inflammation can provoke genetic mutations and epigenetic mechanisms that promote malignant cell transformation[1,2]. Both sporadical and colitis-associated colorectal carcinogenesis are multi-step, complex processes arising from the uncontrolled proliferation and spreading of malignantly transformed cell clones with the obvious ability to evade the host’s protective immunity[3,4]. Therefore to develop more effective therapeutic strategies for colorectal cancer (CRC) it is quite challenging due to its heterogeneity and phenotypic diversity.
The MYC-family of cellular proto-oncogenes encodes three highly related nuclear phosphoproteins, namely c-MYC, N-MYC, and L-MYC[5]. c-MYC is a basic-helix-loop-helix-leucine zipper protein with a proto-oncogene function, being involved in cell proliferation, transformation, and death[6]. Data from chromatin immunoprecipitation studies demonstrate that c-MYC protein occupies regulatory regions of up to 15% of all genes, and can both activate or repress the expression of several target genes[7,8] (Figure 1A). The target genes of c-MYC participate in different cellular functions, including cell cycle, survival, protein synthesis, cell adhesion, and microRNA (miRNA) expression[7] (Figure 1B). The transcriptional program regulated by c-MYC is context dependent, therefore the final cellular response to elevated c-MYC levels may range from raised proliferation to augmented apoptosis[7]. In the absence of c-MYC cell cycle kinetics is strongly reduced[9].
Figure 1.
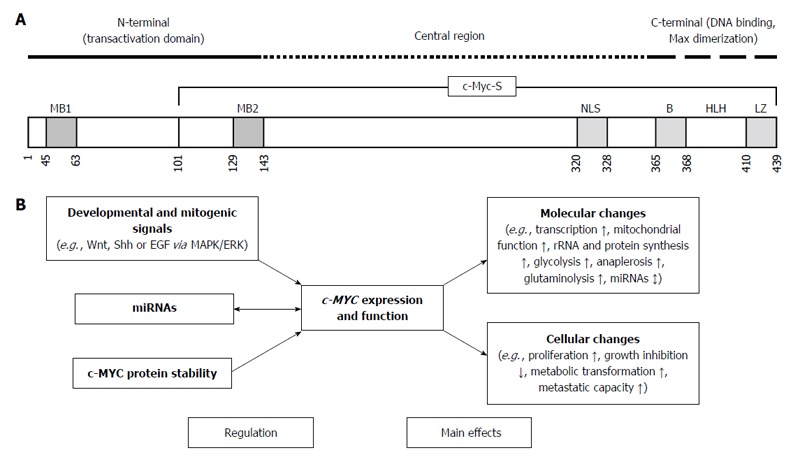
Schematic representation of the structure, regulation and main effects of c-MYC. A: The c-MYC protein consists of three domains: N-terminal, Central region, and C-terminal. The Central region and the C-terminal domain of c-MYC are responsible for protein-protein interactions that result in transcriptional repression by c-MYC. The C-terminal domain contains a basic (B) helix-loop-helix (HLH)-leucine zipper (LZ) motif that is necessary for interaction with different proteins (such as Max), and physiological recognition of DNA target sequences[7,58]; B: Developmental and mitogenic signals tightly regulate c-MYC gene expression both in normal (nontransformed) and in transformed cells via the MAPK/ERK pathway. MicroRNAs display a dual-faced role in the c-MYC regulatory network; both as regulators and as targets of c-MYC. The stability of the c-MYC protein also represents a particularly effective mechanism of gene regulation. c-Myc-S: Truncated c-Myc protein; MB1 and MB2: Evolutionarily conserved Myc Box sequences; NLS: Nuclear localization signal; Shh: Sonic hedgehog; EGF: Epidermal growth factor; MAPK: Mitogen-activated protein kinase; ERK: Extracellular signal-regulated kinase.
As a result of synergistic or sequential damage of DNA in normal colonic epithelial cells, several proto-oncogenes, including c-MYC are activated in parallel with the inactivation of tumor suppressor genes, leading finally to the alteration of DNA repair systems and apoptosis regulation. Accumulation of the damaged DNA may ultimately cause cellular transformation. In this article we try to summarize the complex interactions of c-MYC-signaling within physiological intestinal epithelial homeostasis, inflammatory and cancerous colonic diseases, and the related therapeutic aspects.
CONTROL AND EFFECTS OF MYC GENE EXPRESSION
During recent years, several basic cellular functions of MYC have been established[10]. MYC plays a master regulator role of cell growth and proliferation, and it also controls stemness by maintaining pluripotency and self-renewal. On the other hand, MYC can sensitize cells to apoptosis, regulate cellular senescence, and is involved in DNA damage responses[10].
As a central, dual-faced regulator gene, MYC is controlled by several different mechanisms. Growth factor-dependent signals have been identified to control MYC expression. Growth factors like Ets-1 or E2F1 enhance transcription from the MYC promoter[11]. The β-catenin/TCF site also mediates the induction of the MYC promoter in regards to the Wingless type (Wnt)-signaling pathway[12]. Additionally, growth factor-dependent pulse of phosphoinositol (PI)3-kinase protects c-MYC protein from proteosomal degradation[13]. In contrast, the Smad and E2F4 containing repressor complex which forms on the MYC promoter after transforming growth factor (TGF)-β stimulus suppresses MYC expression and enhances the anti-proliferative effects of TGF-β[14,15].
Elevated levels of c-MYC protein strongly sensitize epithelial cells toward proapoptotic stimuli like DNA damage[16]. As a result, downregulation of c-MYC is necessary for cell cycle arrest, and survival of cells in response to DNA damage[17]. Since in the presence of strong mitogenic signals the downregulation of MYC-expression is required for proto-oncogene-induced cellular senescence[18], c-MYC may be involved in tumor-suppressive mechanisms as well. In case of epithelial and mesenchymal stem cells, however, MYC-expression can be restricted even in the presence of several growth factors and cytokines[19]. These observations indicate that MYC-expression plays a dual-faced role regarding cellular survival and tissue homeostasis.
In physiological circumstances, negative feedback regulatory loops also play an important role in decreasing cellular c-MYC levels[20]. Negative feedback regulation is frequently disturbed in the course of tumorigenic transformation, permitting transformed cells to overexpress MYC[20]. Epigenetic factors, such as miRNAs, are also involved in downregulation of MYC in response to DNA damaging agents[17].
Regarding the colon, the protein kinase MK5 have been also identified as a negative regulator of MYC expression[15]. Expression of MK5 itself is regulated by MYC, since MYC binds to the promoter of the MK5 gene, therefore activates its expression. As a result, MYC and MK5 form a negative feedback loop, in which FoxO proteins have been identified as key mediators[15].
c-MYC IN PHYSIOLOGICAL INTESTINAL HOMEOSTASIS
Considering intestinal homeostasis, c-MYC expressed in the entire intestinal tract displays a fundamental role[21]. In the small intestine c-MYC regulates the appropriate number of epithelial cells within the crypts[9]. Muncan et al[9] reported that upon conditional deletion of c-MYC gene crypt epithelial cells become smaller as compared to normal ones. Moreover, in the absence of c-MYC protein epithelial cell proliferation became reduced[9]. On the other hand, it was unexpectedly found in mice that conditional deletion of c-Myc in adult intestinal epithelium by utilizing a Cre-estrogen receptor fusion transgene driven by the intestine-specific villin promoter did not induce an overt phenotype[22]. According to this result the proliferation and expansion of intestinal epithelial progenitors can occur in a Myc-independent manner, as well. The difference between the studies of Muncan et al[9] and Bettess et al[22] most likely relates to deletion efficiency accomplished with the different Cre transgenes in the earliest crypt progenitors. Regarding apoptotic cell death, c-MYC does not influence epithelial apoptotic rate in the small intestine, it induces apoptosis only in the colon[9,23].
In the intestine cell proliferation and differentiation are under the tight control of the Wnt/β-catenin signaling[24]. In mice c-MYC is a critical downstream effector of cellular proliferation induced by the Wnt/β-catenin pathway[25,26]. Following epithelial injury, the c-Myc 3’Wnt responsive DNA elements (WRE)-dependent regulation of the expression of the c-Myc gene seems to be essential for maintaining intestinal homeostasis and regeneration[27].
In colonic epithelial cells, c-MYC-induced apoptosis can be either p53-dependent or independent[28-30]. Basically, in cells the level of p53 expression is low, but its expression is elevated upon stress responses[31,32]. By promoting proteosomic degradation mouse double minute (Mdm)-2 is a negative regulator of the p53 protein[33]. As a regulatory loop, p53 transcriptionally upregulates Mdm2[33]. Alternative reading frame (Arf) also has a role in this regulatory mechanism, since it inhibits the function of Mdm2 and c-MYC[33,34]. By increasing Arf expression, c-MYC protein displays a prominent role in p53 regulation leading finally to p53-dependent apoptosis[35]. The crosstalk between c-MYC and p53 is essential in inducing pro-survival or pro-death responses to apoptotic stimuli.
Upon modulating apoptotic signals c-MYC is able to regulate intrinsic apoptosis independently from p53, as well[36,37]. c-MYC can also alter the balance between the pro- and antiapoptotic members of the Bcl2 (B-cell/lymphoma 2)-family[16,38]. Bcl2 can inhibit c-MYC mediated apoptosis, however, on the other hand, c-MYC overexpression suppresses the antiapoptotic Bcl2 protein and mRNA levels[39]. To suppress the antiapoptotic Bcl2 expression the DNA-binding activity of c-MYC is required[40]. In mice, c-MYC may induce apoptosis via the activation of the proapoptotic protein, Bax[41]. c-MYC also participates in the extrinsic apoptotic pathways[38,42,43]. Therefore, it is difficult to predict which c-MYC target genes are responsible for the final biological effects. It is likely that the current status of cell physiology ultimately influences the outcome of c-MYC overexpression, and affects c-MYC regulating the apoptotic process in colonic epithelial cells.
c-MYC IN COLONIC INFLAMMATION
As a hallmark of cancer, inflammation may lead to tumor formation. Acute and chronic colonic inflammation disrupts the integrity of the epithelial layer, moreover can lead to regenerative cell proliferation, and even fibrosis. In animal colitis models the use of glycogen synthase kinase (GSK)3β inhibitors mitigated disease symptoms by reducing pro-inflammatory immune response[44]. It has been shown, that during the recovery phase of dextran sulfate sodium (DSS)-induced colitis GSK3β inhibition by lithum chloride promotes colonic regeneration. The explanation of this effect is that lithium treatment increased the expression of Myc transcripts, MYC proteins, and the expression of several Wnt/MYC target genes in the colonic epithelium[45].
Additionally, in humans the steady-state levels of several nuclear proto-oncogenes including c-MYC and N-MYC were demonstrated to be lower in epithelial cells from involved or uninvolved inflammatory disease bowel (IBD) samples than in normal epithelial cells from either sporadic colon cancer or diverticulitis patients[46]. In active inflammation the downexpression of c-MYC in IBD epithelium may result in attenuated cell proliferation, therefore may contribute to mucosal ulceration. On the other hand, c-MYC may also be involved in epithelial regeneration after inflammatory damage by altering apoptotic cell death.
It is a known fact, that patients with chronic, longstanding IBD have an increased risk for developing colitis-associated cancer (CAC). By using whole-exome sequencing analysis it has been recently demonstrated that -among others- the MYC genomic locus is more frequently amplified in CAC than sporadic colorectal cancers[47]. Moreover, genomic alterations observed in CAC are distinct form those found in sporadic CRCs, and vary by type of IBD[48]. Proteomic network analyses have identified proteins related to mitochondria, oxidative activity, calcium-binding proteins, and c-MYC that play roles in early and late stage colitis-associated neoplastic progression, respectively[49]. c-MYC is often overexpressed in dysplastic cells in chronic longstanding ulcerative colitis, the precursor to CAC[50,51]. Taking together these data, it seems that the complex role and final effects of c-MYC in inflammatory colon mucosa are context- and microenvironment dependent.
c-MYC IN COLORECTAL CANCER
The c-MYC oncogene is frequently deregulated in human cancers and is overexpressed in up to 70%-80% of colon adenocarcinomas[52]. Since c-MYC is a downstream target of the APC (adenomatous polyposis coli) gene, and APC itself is inactivated in most colorectal cancers[53], it is not surprising that in early and advanced stages of colorectal carcinogenesis c-MYC is overexpressed at both the mRNA and protein levels[54,55].
The imbalance of cell proliferation and apoptosis is a key component in initiation of colorectal tumorigenesis. Basically, overexpression of c-MYC could lead to apoptosis[38], indicating its crucial role for determining cell survival and/or apoptotic pathways[36]. Under pathological conditions deregulated Wnt/β-catenin signaling promotes CRC by activating the expression of c-MYC[56]. Moreover, c-MYC-triggered apoptosis provides an inherent “fail-safe” program to check unlimited cell growth. The extent of apoptotic cell death is in correlation with the level of c-MYC expression[57].
In case of early to late colorectal adenomas significant correlation of nuclear β-catenin and c-MYC nuclear expression was found with the size of colon adenomas, but not with their cellular proliferative activity[58]. This phenomenon implies a dose-dependent function of β-catenin. Without nuclear β-catenin, T-cell factor family (TCF) proteins are bound by a co-repressor, and this complex acts as transcriptional repressor of the target genes[59,60]. Nuclear β-catenin competes with the co-repressor for TCF binding in a dose-dependent manner. In colorectal cancer cells the disruption of β-catenin/TCF-4 activity induces a rapid G1 arrest and blocks the physiologically active genetic program in the proliferative compartment of colonic crypts. Simultaneously, an intestinal differentiation program is induced, in which c-MYC plays a switch role by direct repression of the p21CIP1/WAF1 promoter. Following disruption of β-catenin/TCF-4 activity, the decreased expression of c-MYC results in p21CIP1/WAF1 transcription, which in turn mediates G1 arrest and differentiation[12].
Though several in vitro studies proved that c-MYC has the ability to sensitize or induce apoptosis[61,62], its role in apoptotic cell-death is not well established and unclear in vivo. In a recent article, Phesse et al[63] demonstrated for the first time in an in vivo model that endogenous c-MYC is essential for efficient induction of p53-dependent apoptosis following DNA damage. It has been long known that p53 serves a key element in the development of sporadic colorectal cancer[64], and further, it is also involved in colitis-associated carcinogenesis[65]. Until now, in the gut c-MYC was considered as a fundamentally expressed gene responsible for epithelial regeneration and the regulation of the number of crypt cells. Phesse et al[63] concluded that c-MYC serves as a universal regulator of apoptosis in in vivo systems suggesting an important and new aspect of colorectal carcinogenesis (Figure 2). On the other hand, the exact mechanisms linking c-MYC levels to Mdm2 expression still remain unclear. In accordance with recent results[63,66,67], one can speculate that c-MYC may directly inhibit Mdm2 transcription. The induction of the c-MYC-dependent apoptosis program requires c-MYC expression to exceed a threshold, which is defined by Bcl2 family proteins in a cell-, tissue type and milieu-specific fashion[23]. In the colon, however, the different behaviour of the apoptosis regulator Bax, controlled by c-MYC may suggest the existence of a different apoptotic program of epithelial cells.
Figure 2.

Schematic illustration of the relation of c-MYC to p53 following DNA damage in the intestine.
A recent report has demonstrated a role of AMBRA1 (activating molecule in Beclin-1 regulated autophagy) in both the autophagic pro-survival response and Beclin-1-dependent autophagy in embryonic stem cells[68]. AMBRA1 has been shown to be a crucial regulator of autophagy and apoptosis in colorectal cancer cells that maintains the balance between these cellular mechanisms[69]. AMBRA1 promoted dephosphorylation and degradation of c-MYC, and favors the interaction between c-MYC and PP2A (a c-MYC phosphatase), leading finally reduced cell divison rate[70]. AMBRA1 has been recently characterized as a target of mTOR (mammalian target of rapamycin) in the autophagy process[71]. Furthermore, the AMBRA1/PP2A-mediated regulation of c-MYC is also under mTOR control[70], indicating the key role of mTOR in regulating cellular fate by interfering with its metabolic status (Figure 3).
Figure 3.
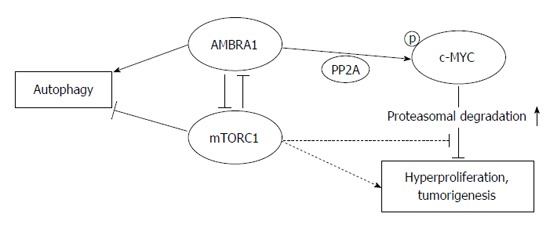
Interplay of AMBRA1, c-MYC and mTOR in colorectal cancer cells. AMBRA1 links autophagy to cell proliferation by facilitating c-MYC demolition. AMBRA1 promotes c-MYC phosphorilation and proteasomal degradation, therefore prevents hyperprolifearion and tumorigenesis. mTORC1 negatively controls the function of AMBRA1, thus finally supporting c-MYC-driven cell proliferation. Arrows represent stimulation or increase; blocked arrows represent inhibition; broken lines represent indirect effects. AMBRA1: Activating molecule in Beclin-1 regulated autophagy; mTORC1: Mammalian target of rapamycin complex 1; PP2A: Protein phosphatase 2A.
The MK5 kinase regulates the translation of c-MYC, since it is required for the expression of miR-34b/c that bind to the 3’UTR of MYC. The MK5-MYC negative regulatory feedback loop has been found to be disrupted during colorectal tumorigenesis[15]. Two changes may explain the disruption of this regulatory circuit. First, silencing of the miR-34B/c gene promoters by DNA methylation[72]. Second, the expression of MK5 is downregulated in colorectal tumors by a currently unknown mechanism[15]. Depletion of MK5 regulates Ephrin B1, a MYC-repressed gene that is involved in the progression of p53-deficient colorectal tumors[73].
ANTI-INFLAMMATORY THERAPEUTIC ASPECTS OF c-MYC IN THE COLON
Mesenchymal stem cell transplantation (MSCT) has been reported effective in the treatment of IBD as it can restore epithelial barrier integrity, induce immune suppression, and stimulate regeneration of endogenous host progenitor cells[74-78]. Mesenchymal stem cells can be engrafted into the damaged mucosa and even differentiated into colonic interstitial cells[79]. The pathobiologic background of this reparative process, however, is not well known. In an IBD-MSCT rat model, when intestinal epithelium was inflamed, the canonical Wnt signaling was found to be activated by Wnt3a and inhibited by GSK-3β and APC[78]. Shortly after MSCT, the elevated c-Myc and downregulated Apc gene expressions facilitate mesenchymal stem cell proliferation, and then differentiation into intestinal epithelial cells in the anaphase, by reducing the expression of c-Myc. These changes promoted intestinal stem cell proliferation and repaired the intestinal mucosa. Though, MSCT is a useful therapeutic possibility in IBD models, the parallel use of GSK3β inhibitors after MSCT may be therapeutically useful to enhance MYC-signaling, hence promoting reparative cell proliferation[45].
Traditionally, the pathomechanism of Crohn’s disease has been associated with Th1 cytokine profile. In an experimental autoimmune inflammation model it was demonstrated that inhibiting the functions of BET (bromo and extra-terminal domain)-family proteins during early T-cell differentiation resulted in long-lasting suppression of the pro-inflammatory functions of Th1 cells[80]. These effects were mimicked by an inhibitor of c-MYC function, as well, implicating reduced expression of c-Myc as one avenue by which BET-inhibitors suppressed the inflammatory functions of T-cells. hypothetically, BET and c-MYC inhibition may have therapeutic potential in Crohn’s disease (Table 1).
Table 1.
Therapeutic options based on c-MYC targeting are represented by various strategies in inflammatory and cancerous colonic disorders
| Main mode of action | Potential pathways/agents |
| Inflammatory colonic disorders | |
| Upregulation of c-MYC expression | GSK inhibitors ± MSCT |
| Inhibition of c-MYC signaling | BET inhibitors ± c-MYC inhibitors |
| (suppression of Th1 function) | |
| Cancerous colonic diseases | |
| Downregulation of c-MYC expression | dose-dependent gene and protein expression suppression; PPAR-γ (5-ASA, mesalazine) |
| suppressing protein expression by UDCA | |
| crosstalk with integrins | |
| E2F1 inhibition (downregulatin GCN5 expression) | |
| FGFR kinase inhibition | |
| epigenetic regulation by miR-320b | |
| siRNA blocking of ABC-transporters | |
| lncRNAs (blocking of PARROT or CCAT1-L) | |
| siRNAs using PEI-PGMA platform | |
| modified ODC promoter | |
| Promoting c-MYC degradation | 26S proteosomal pathway (aspirin) |
| SIRT2 inhibition | |
| Inhibition of c-MYC signaling | Omomyc |
| BET (+ Wnt/MAPK) inhibitors | |
GSK: Glycogen synthase kinnase; MSCT: Mesenchymal stem cell transplantation; PPAR-γ: Peroxisome proliferator-activated receptor-γ; 5-ASA: 5-aminosalicylate; UDCA: Ursodeoxycholic acid; E2F1: E2F Transcription factor 1; GCN5: Histone acetyltransferase; FGFR: Fibroblast growth factor receptor; miR-320b: Micro-ribonucleic acid-320b; siRNA: Small interfering ribonucleic acid; ABC: Adenosine triphosphate-binding casette; lncRNA: Long noncoding ribonucleic acid; PARROT: Proliferation associated RNA and regulator of translation; CCAT1-L: Longer isoform of colon cancer associated transcript 1; PEI-PGMA: Polyethileneimine-polyglycidal methacrylate; ODC: Ornithine decarboxylase; SIRT2: Sirtuine2; BET: Bromo- and extra-terminal domain; MAPK: Mitogen-activated protein kinase.
ANTI-CANCER THERAPEUTIC ASPECTS OF c-MYC IN THE COLON
Cancer cells have been reported to display cell cycle arrest, differentiation, senescence or cell death after MYC inhibition via different molecular mechanisms (Table 1).
5-aminosalicylic acid has been identified as an agonist of peroxisome proliferator-activated receptor (PPAR)-γ[81]. Activation of PPAR-γ induces apoptosis by downregulating c-MYC[82,83]. Regrading aspirin the involvement of the 26S proteasomal pathway has been found in decreasing c-MYC expression in a concentration-dependent fashion[84]. Due to its oncogenic activities including cell growth, proliferation, angiogenesis, genomic instability and blocking differentiation, the downregulation of c-MYC would be expected to have important clinical implications[85].
Ursodeoxycholic acid (UDCA) displays chemopreventive action against chemical and colitis-associated colonic carcinogenesis[86]. One possible explanation of this effect is the inhibition of cell proliferation by suppressing c-MYC protein expression and, as a consequence, cell cycle regulatory molecules including cyclin-dependent kinase-4 and -6[86]. According to this result, c-MYC is a target molecule of UDCA in colon carcinoma cells. However, mapping the benefits of UDCA administration for CRC chemoprevention at population level needs further studies.
Integrins, containing noncovalently associated α/β heterodimers provide dynamic cell to cell linkage and cell attachment to matrix molecules. While in normal human intestinal epithelium α1β1 integrins are usually expressed in the lower third of crypts[87], in colorectal cancers and colon cancer cell lines integrin-α1 is expressed up to 65% of cases[88]. c-MYC regulates several integrin subunits, thus influences various functions of integrins regarding colon cancer cell proliferation, migration, and survival[89-92]. A combination of anti-MYC and -integrin targeted therapies hence may represent novel aspects of anti-tumor strategies in colon cancers.
Aberrant kinase activation originated from mutation, amplification, or translocation can drive growth and survival in several human cancers[93,94]. In gastric cancer, the crosstalk between fibroblast growth factor receptor (FGFR)2 and CD44 has been found to maintain cancer stemness by reciprocically regulating their expression via differentially regulating c-MYC transcription[95]. Since FGFR2 has been found to be amplified in the NCI-H716 colorectal cancer cell line[96], this result suggests that emerging FGFR inhibitor therapeutics may have efficacy in a subset of colon cancer driven by FGFR2 amplification.
It has been shown that the inhibition of BET protein family impairs the proliferation of several cancer cell lines[97-99]. These effects are partly mediated by c-MYC repression[98]. In a recent study of Tögel et al[100] the authors investigated the effect of BET inhibitors on proliferation and c-MYC expression within 20 CRC cell lines. They have found that JQ1, a BET inhibitor, administered together with Wnt or MAPK inhibitors sufficiently downregulates the expression of c-MYC, thus inhibits CRC cell proliferation. Based on these results, this kind of combined therapy seems to be effective in CRC treatment.
Upon recent results, targeting c-MYC can also be considered as a promising anti-cancer therapeutic strategy[23,101-104]. c-MYC inhibition with a protein fragment called Omomyc has been shown to be very effective to regress epithelial cell-derived tumors in mice models[23,105]. Omomyc has been found to be a pan-MYC family (c-, N- and L-MYC) inhibitor, potentially useful for cancers carrying any MYC family member amplification[106].
In case of cancers in which cell growth is not dependent on amplified MYC family genes, MYC suppression alone is not enough for a sufficient therapeutic effect. In animal models of Myc-driven cancers, reversion of the tumor by Myc suppression has been impeded by the parallel repression of TP53 or retinoblastoma-1 proteins underlining the relevance of these pathways to be intact for the treatment of cancers by MYC targeting[107-109].
Using a focused RNA interference library for genes involved in epigenetic regulation, sirtuin2 (SIRT2), an NAD(+)-dependent deacetylase, has been identified as a modulator of the therapy response to EGFR inhibitors in colon and lung cancers[110]. Thiomyristoyl lysine compound (TM), a SIRT2 inhibitor with high potency and specificity, has broad anti-cancer activity. SIRT2 inhibition was found to promote c-MYC ubiquitination and degradation, hence it may be a potential target for c-MYC-driven cancers including colorectal carcinoma[111].
Recent studies have suggested that the elevated expression of general control nonrepressed protein 5 (GCN5), a histone acetyltransferase can often be detected in human cancers[112]. GCN5 expression is elevated in colon cancer, and its overexpression is regulated by c-MYC[113]. By suppressing GCN5 human colon cancer cell growth can be inhibited. Furthermore, the suppression of the proapoptotic transcription factor E2F1-induced GCN5 transcription facilitates E2F1-induced cell death, implying a negative feedback in apoptosis regulation[113]. According to these results, GCN5 seems to be a potential therapeutic target for human colon cancers.
Regarding transcription factor-based therapies of tumorous diseases inhibition of c-MYC may also represent a promising option[101,114]. Numerous cytotoxic agents, and ionizing radiation have been shown to induce apoptosis following DNA damage. Since most of the anti-cancer drugs are used in combination with the potential of genotoxicity, it is of importance to further assess the role of c-MYC in response to DNA damage.
Therapeutic approaches that would allow the reprogramming and returning of altered c-MYC activity within tumor cells are also promising therapeutical strategies. RNA interference technology is one of these modalities. MiRNAs are key post-transcriptional regulators of genetic networks. Single-stranded mature miRNAs associated with Argonaute proteins form the core of a gene regulatory complex [i.e. RNA-induced silencing complex (RISC)]. MiRNA-RISC-mediated gene inhibition can be materialized by three processes: (1) site-specific cleavage; (2) enhanced mRNA degradation; and (3) translational inhibition[115]. Evidences indicate that post-transcriptional miRNA-mediated gene expression regulation can act as tumor suppressor or onogene in CRC[116]. Currently, miR-320b has been found to be significantly downregulated in CRC tumor tissues. In addition, miR-320b overexpression has been found to correlate with decreased cell growth both in vitro and in vivo. Moreover, it has been also demonstrated that miR320b directly targets c-MYC, and its overexpression in SW-480, SW-620, HCT-116, LoVo, and HK293 CRC cell lines decreases c-MYC expression at gene and protein level as well[117]. According to these results, increasing miR-320b gene expression may represent a potential therapeutic approach in CRC.
Colorectal cancer stem cells (CSCs) has an important role in tumor initiation, progression, and recurrence. c-MYC was found to be highly expressed in CD133+ colon CSCs[118]. The overexpression of ATP-binding casette (ABC) transporters in cancer cells can result in therapy resistance by exporting anti-tumor drugs[119]. Recently, c-MYC expression has been effectively blocked on mRNA and protein level by c-MYC small interfering RNA (siRNA), moreover c-MYC silencing sensitized CD133+ CSCs to chemotherapy-induced cytotoxicity by downregulating the expression of ABC transporter proteins[120].
In eukaryotic cells a vast number of noncoding RNA species are transcribed. Among them, long noncoding RNAs (lncRNAs) have been widely implicated in post-transcriptional gene expression regulation. The expression level of lncRNAs is usually very low and tissue-specific[121]. c-MYC can regulate the expression of lncRNAs, some of these may also contribute to the transcription of c-MYC target genes[122]. It has been reported that proliferation associated RNA and regulator of translation (PARROT), an lncRNA dynamically expressed in both transformed and normal cells contributes to proliferation in senescence and cancer. PARROT has been also identified as an upstream regulator of c-MYC. Its depletion results in the depletion of c-MYC mRNA and protein expression, subsequently altering cell growth and proliferation[121]. In gastric cancer c-MYC activates the expression of colon cancer associated transcript 1 (CCAT1) lncRNA, leading to an increased proliferation and migration of cancer cells[123]. CCAT1-L, a longer isoform of CCAT1, has been reported to regulate MYC expression in colon cancer. It is supposed that CCAT1-L allows the interaction between the enhancer and the c-MYC promoter thus promotes tumorigenesis[124].
Achieving effective intracellular delivery of therapeutic RNA interfering molecules such as siRNAs or short hairpin RNAs (shRNAs) is quite challenging. In a recent study, spherical nucleic acid-gold nanoparticle conjugates have been shown to selectively induce apoptosis in glioma cells in vivo[125]. However, the used 21 base siRNA duplexes were quite unstable. ShRNAs with a transient period of expression are better suited for long-term effectiveness, due to their ability to produce siRNAs continuously within cancer cells, thus resulting in prolonged suppression of target genes[126]. Until today, shRNAs have been delivered effectively in vivo using viral vectors. Among nonviral vectors, polyethileneimine (PEI) is the most widely used, gold-standard agent[127]. However, the major disadvantage of PEI is its cytotoxicity[128]. On the other hand, it has been demonstrated that anchoring multiple PEI chains to macromolecule polyglycidal methacrylate (PGMA) nanoparticles dramatically reduces their cytotoxicity, while achieving efficient nanoparticle endocytosis[129]. Using the PGMA platform effective delivery of small oligos (anti-miRs and mimics) and larger encoding shRNAs were performed in a wide variety of cancer cell lines including colorectal ones. Furthermore, the effectiveness of this therapy was validated for in vivo tumor suppression using transgenic mouse models. It was found that oral delivery of the c-Myc-conjugated nanoparticles to an Apc-deficient crypt progenitor colon cancer model resulted in an increased host survival and re-entered intestinal tissue to a non-Wnt-deregulated state[126]. According to these results, it seems that careful design of nonviral nanoparticles may help to made RNA interference technology an affordable and amenable therapy for CRC.
Regarding tumor-specific cytotoxicity, viral-directed enzyme prodrug therapy may also represent an ideal alternative[130]. However, the viruses used to deliver cDNAs encoding prodrug-activating enzymes can transduce normal cells, not just tumor cells. To achieve tumor-specific expression of the delivered cDNAs is to regulate transcription of the prodrug-activating enzyme with a promoter that is preferentially activated by tumor cells. MYC-responsive, modified ornithine decarboxylase (ODC) promoter/enhancer sequences have been identified that upregulate target protein expression in SW480 an HT29 colon cancer cells overexpressing the c-MYC protein. The modified ODC promoter may be useful in achieving tissue-specific expression of target proteins in cancers overexpress c-MYC[131].
CONCLUSION
The incidence of inflammatory colonic disorders is increasing worldwide. Though inflammation is required to heal infections, wounds, and maintain tissue homeostasis, as the seventh hallmark of cancer, however, it may affect all stages of tumor development. c-MYC, with its dual-faced role in cell proliferation and death, is implicated in several aspects of inflammatory tissue damage and repair. Since the therapeutic potential of c-MYC influencing therapies has not studied yet in the clinic, additional studies are needed to determine whether long-term treatment with c-MYC targeting agents can therapeutically suppress ongoing inflammation.
Colorectal carcinogenesis is a complex, multistep process that is driven by the accumulation of multiple genetic alterations. c-MYC is overexpressed in several types of malignant tumors including colorectal cancer, and is necessary for the uncontrolled proliferation of cancer cells. Single or combined therapies based on c-MYC targeting are represented by various strategies. Molecules directly targeting c-MYC, or agents acting on other genes involved in the c-MYC pathway could be selected for combined regiments. However, due to its context-dependent cellular function, it is clinically essential to consider which cytotoxic drugs are used in combination with c-MYC targeted agents in various tissues. Noncoding small RNAs have been recently implicated in anti-cancer therapies[132]. Regardless of the therapy applied, it is important to first determine the molecular pathways underlying the agents to inform the therapy design. Combining c-MYC-targeting agents with specific noncoding RNAs may lead to the development of novel colorectal cancer therapies.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Hungary
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: All authors declare no conflict of interest.
Peer-review started: July 11, 2016
First decision: July 29, 2016
Article in press: August 23, 2016
P- Reviewer: Lakatos PL, Kadiyska TK, Shimoyama S, Yochum GS S- Editor: Gong ZM L- Editor: A E- Editor: Ma S
References
- 1.Lazebnik Y. What are the hallmarks of cancer? Nat Rev Cancer. 2010;10:232–233. doi: 10.1038/nrc2827. [DOI] [PubMed] [Google Scholar]
- 2.Basith S, Manavalan B, Yoo TH, Kim SG, Choi S. Roles of toll-like receptors in cancer: a double-edged sword for defense and offense. Arch Pharm Res. 2012;35:1297–1316. doi: 10.1007/s12272-012-0802-7. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Axelrad JE, Lichtiger S, Yajnik V. Inflammatory bowel disease and cancer: The role of inflammation, immunosuppression, and cancer treatment. World J Gastroenterol. 2016;22:4794–4801. doi: 10.3748/wjg.v22.i20.4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mukherjee B, Morgenbesser SD, DePinho RA. Myc family oncoproteins function through a common pathway to transform normal cells in culture: cross-interference by Max and trans-acting dominant mutants. Genes Dev. 1992;6:1480–1492. doi: 10.1101/gad.6.8.1480. [DOI] [PubMed] [Google Scholar]
- 6.Hermeking H, Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- 7.Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 8.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 9.Muncan V, Sansom OJ, Tertoolen L, Phesse TJ, Begthel H, Sancho E, Cole AM, Gregorieff A, de Alboran IM, Clevers H, et al. Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Mol Cell Biol. 2006;26:8418–8426. doi: 10.1128/MCB.00821-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsson LG, Henriksson MA. The Yin and Yang functions of the Myc oncoprotein in cancer development and as targets for therapy. Exp Cell Res. 2010;316:1429–1437. doi: 10.1016/j.yexcr.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 11.Roussel MF, Davis JN, Cleveland JL, Ghysdael J, Hiebert SW. Dual control of myc expression through a single DNA binding site targeted by ets family proteins and E2F-1. Oncogene. 1994;9:405–415. [PubMed] [Google Scholar]
- 12.van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 13.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen CR, Kang Y, Siegel PM, Massagué J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell. 2002;110:19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- 15.Kress TR, Cannell IG, Brenkman AB, Samans B, Gaestel M, Roepman P, Burgering BM, Bushell M, Rosenwald A, Eilers M. The MK5/PRAK kinase and Myc form a negative feedback loop that is disrupted during colorectal tumorigenesis. Mol Cell. 2011;41:445–457. doi: 10.1016/j.molcel.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 16.Maclean KH, Keller UB, Rodriguez-Galindo C, Nilsson JA, Cleveland JL. c-Myc augments gamma irradiation-induced apoptosis by suppressing Bcl-XL. Mol Cell Biol. 2003;23:7256–7270. doi: 10.1128/MCB.23.20.7256-7270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cannell IG, Kong YW, Johnston SJ, Chen ML, Collins HM, Dobbyn HC, Elia A, Kress TR, Dickens M, Clemens MJ, et al. p38 MAPK/MK2-mediated induction of miR-34c following DNA damage prevents Myc-dependent DNA replication. Proc Natl Acad Sci USA. 2010;107:5375–5380. doi: 10.1073/pnas.0910015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hydbring P, Bahram F, Su Y, Tronnersjö S, Högstrand K, von der Lehr N, Sharifi HR, Lilischkis R, Hein N, Wu S, et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc Natl Acad Sci USA. 2010;107:58–63. doi: 10.1073/pnas.0900121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laurenti E, Wilson A, Trumpp A. Myc’s other life: stem cells and beyond. Curr Opin Cell Biol. 2009;21:844–854. doi: 10.1016/j.ceb.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Penn LJ, Brooks MW, Laufer EM, Land H. Negative autoregulation of c-myc transcription. EMBO J. 1990;9:1113–1121. doi: 10.1002/j.1460-2075.1990.tb08217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679–683. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bettess MD, Dubois N, Murphy MJ, Dubey C, Roger C, Robine S, Trumpp A. c-Myc is required for the formation of intestinal crypts but dispensable for homeostasis of the adult intestinal epithelium. Mol Cell Biol. 2005;25:7868–7878. doi: 10.1128/MCB.25.17.7868-7878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, Evan GI. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell. 2008;14:447–457. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, Vass JK, Athineos D, Clevers H, Clarke AR. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–679. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- 26.Yekkala K, Baudino TA. Inhibition of intestinal polyposis with reduced angiogenesis in ApcMin/+ mice due to decreases in c-Myc expression. Mol Cancer Res. 2007;5:1296–1303. doi: 10.1158/1541-7786.MCR-07-0232. [DOI] [PubMed] [Google Scholar]
- 27.Konsavage WM, Jin G, Yochum GS. The Myc 3’ Wnt-responsive element regulates homeostasis and regeneration in the mouse intestinal tract. Mol Cell Biol. 2012;32:3891–3902. doi: 10.1128/MCB.00548-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyer N, Kim SS, Penn LZ. The Oscar-worthy role of Myc in apoptosis. Semin Cancer Biol. 2006;16:275–287. doi: 10.1016/j.semcancer.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 29.Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 30.Sakamuro D, Eviner V, Elliott KJ, Showe L, White E, Prendergast GC. c-Myc induces apoptosis in epithelial cells by both p53-dependent and p53-independent mechanisms. Oncogene. 1995;11:2411–2418. [PubMed] [Google Scholar]
- 31.Hock A, Vousden KH. Regulation of the p53 pathway by ubiquitin and related proteins. Int J Biochem Cell Biol. 2010;42:1618–1621. doi: 10.1016/j.biocel.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 32.Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- 33.Shi D, Gu W. Dual Roles of MDM2 in the Regulation of p53: Ubiquitination Dependent and Ubiquitination Independent Mechanisms of MDM2 Repression of p53 Activity. Genes Cancer. 2012;3:240–248. doi: 10.1177/1947601912455199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 35.Bouchard C, Lee S, Paulus-Hock V, Loddenkemper C, Eilers M, Schmitt CA. FoxO transcription factors suppress Myc-driven lymphomagenesis via direct activation of Arf. Genes Dev. 2007;21:2775–2787. doi: 10.1101/gad.453107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoffman B, Liebermann DA. The proto-oncogene c-myc and apoptosis. Oncogene. 1998;17:3351–3357. doi: 10.1038/sj.onc.1202592. [DOI] [PubMed] [Google Scholar]
- 37.Vadde R, Radhakrishnan S, Reddivari L, Vanamala JK. Triphala Extract Suppresses Proliferation and Induces Apoptosis in Human Colon Cancer Stem Cells via Suppressing c-Myc/Cyclin D1 and Elevation of Bax/Bcl-2 Ratio. Biomed Res Int. 2015;2015:649263. doi: 10.1155/2015/649263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene. 2008;27:6462–6472. doi: 10.1038/onc.2008.312. [DOI] [PubMed] [Google Scholar]
- 39.Eischen CM, Packham G, Nip J, Fee BE, Hiebert SW, Zambetti GP, Cleveland JL. Bcl-2 is an apoptotic target suppressed by both c-Myc and E2F-1. Oncogene. 2001;20:6983–6993. doi: 10.1038/sj.onc.1204892. [DOI] [PubMed] [Google Scholar]
- 40.Eischen CM, Woo D, Roussel MF, Cleveland JL. Apoptosis triggered by Myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol Cell Biol. 2001;21:5063–5070. doi: 10.1128/MCB.21.15.5063-5070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dansen TB, Whitfield J, Rostker F, Brown-Swigart L, Evan GI. Specific requirement for Bax, not Bak, in Myc-induced apoptosis and tumor suppression in vivo. J Biol Chem. 2006;281:10890–10895. doi: 10.1074/jbc.M513655200. [DOI] [PubMed] [Google Scholar]
- 42.Hueber AO, Zörnig M, Lyon D, Suda T, Nagata S, Evan GI. Requirement for the CD95 receptor-ligand pathway in c-Myc-induced apoptosis. Science. 1997;278:1305–1309. doi: 10.1126/science.278.5341.1305. [DOI] [PubMed] [Google Scholar]
- 43.Klefstrom J, Arighi E, Littlewood T, Jäättelä M, Saksela E, Evan GI, Alitalo K. Induction of TNF-sensitive cellular phenotype by c-Myc involves p53 and impaired NF-kappaB activation. EMBO J. 1997;16:7382–7392. doi: 10.1093/emboj/16.24.7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uddin MJ, Jeong SO, Zheng M, Chen Y, Cho GJ, Chung HT, Joe Y. Carbon monoxide attenuates dextran sulfate sodium-induced colitis via inhibition of GSK-3β signaling. Oxid Med Cell Longev. 2013;2013:210563. doi: 10.1155/2013/210563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raup-Konsavage WM, Cooper TK, Yochum GS. A Role for MYC in Lithium-Stimulated Repair of the Colonic Epithelium After DSS-Induced Damage in Mice. Dig Dis Sci. 2016;61:410–422. doi: 10.1007/s10620-015-3852-0. [DOI] [PubMed] [Google Scholar]
- 46.Alexander RJ, Panja A, Kaplan-Liss E, Mayer L, Raicht RF. Expression of protooncogene-encoded mRNA by colonic epithelial cells in inflammatory bowel disease. Dig Dis Sci. 1996;41:660–669. doi: 10.1007/BF02213120. [DOI] [PubMed] [Google Scholar]
- 47.Robles AI, Traverso G, Zhang M, Roberts NJ, Khan MA, Joseph C, Lauwers GY, Selaru FM, Popoli M, Pittman ME, et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology. 2016;150:931–943. doi: 10.1053/j.gastro.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yaeger R, Shah MA, Miller VA, Kelsen JR, Wang K, Heins ZJ, Ross JS, He Y, Sanford E, Yantiss RK, et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct From Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology. 2016;151:278–287.e6. doi: 10.1053/j.gastro.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brentnall TA, Pan S, Bronner MP, Crispin DA, Mirzaei H, Cooke K, Tamura Y, Nikolskaya T, Jebailey L, Goodlett DR, et al. Proteins That Underlie Neoplastic Progression of Ulcerative Colitis. Proteomics Clin Appl. 2009;3:1326. doi: 10.1002/prca.200900061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ciclitira PJ, Macartney JC, Evan G. Expression of c-myc in non-malignant and pre-malignant gastrointestinal disorders. J Pathol. 1987;151:293–296. doi: 10.1002/path.1711510409. [DOI] [PubMed] [Google Scholar]
- 51.Pavelic ZP, Pavelic L, Kuvelkar R, Gapany SR. High c-myc protein expression in benign colorectal lesions correlates with the degree of dysplasia. Anticancer Res. 1992;12:171–175. [PubMed] [Google Scholar]
- 52.Erisman MD, Rothberg PG, Diehl RE, Morse CC, Spandorfer JM, Astrin SM. Deregulation of c-myc gene expression in human colon carcinoma is not accompanied by amplification or rearrangement of the gene. Mol Cell Biol. 1985;5:1969–1976. doi: 10.1128/mcb.5.8.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 54.Imaseki H, Hayashi H, Taira M, Ito Y, Tabata Y, Onoda S, Isono K, Tatibana M. Expression of c-myc oncogene in colorectal polyps as a biological marker for monitoring malignant potential. Cancer. 1989;64:704–709. doi: 10.1002/1097-0142(19890801)64:3<704::aid-cncr2820640323>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 55.Erisman MD, Scott JK, Watt RA, Astrin SM. The c-myc protein is constitutively expressed at elevated levels in colorectal carcinoma cell lines. Oncogene. 1988;2:367–378. [PubMed] [Google Scholar]
- 56.Shah M, Rennoll SA, Raup-Konsavage WM, Yochum GS. A dynamic exchange of TCF3 and TCF4 transcription factors controls MYC expression in colorectal cancer cells. Cell Cycle. 2015;14:323–332. doi: 10.4161/15384101.2014.980643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 58.Brabletz T, Herrmann K, Jung A, Faller G, Kirchner T. Expression of nuclear beta-catenin and c-myc is correlated with tumor size but not with proliferative activity of colorectal adenomas. Am J Pathol. 2000;156:865–870. doi: 10.1016/s0002-9440(10)64955-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cavallo RA, Cox RT, Moline MM, Roose J, Polevoy GA, Clevers H, Peifer M, Bejsovec A. Drosophila Tcf and Groucho interact to repress Wingless signalling activity. Nature. 1998;395:604–608. doi: 10.1038/26982. [DOI] [PubMed] [Google Scholar]
- 60.Roose J, Molenaar M, Peterson J, Hurenkamp J, Brantjes H, Moerer P, van de Wetering M, Destrée O, Clevers H. The Xenopus Wnt effector XTcf-3 interacts with Groucho-related transcriptional repressors. Nature. 1998;395:608–612. doi: 10.1038/26989. [DOI] [PubMed] [Google Scholar]
- 61.Juin P, Hueber AO, Littlewood T, Evan G. c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 1999;13:1367–1381. doi: 10.1101/gad.13.11.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.You Z, Madrid LV, Saims D, Sedivy J, Wang CY. c-Myc sensitizes cells to tumor necrosis factor-mediated apoptosis by inhibiting nuclear factor kappa B transactivation. J Biol Chem. 2002;277:36671–36677. doi: 10.1074/jbc.M203213200. [DOI] [PubMed] [Google Scholar]
- 63.Phesse TJ, Myant KB, Cole AM, Ridgway RA, Pearson H, Muncan V, van den Brink GR, Vousden KH, Sears R, Vassilev LT, et al. Endogenous c-Myc is essential for p53-induced apoptosis in response to DNA damage in vivo. Cell Death Differ. 2014;21:956–966. doi: 10.1038/cdd.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 65.Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G7–17. doi: 10.1152/ajpgi.00079.2004. [DOI] [PubMed] [Google Scholar]
- 66.Slack A, Chen Z, Tonelli R, Pule M, Hunt L, Pession A, Shohet JM. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc Natl Acad Sci USA. 2005;102:731–736. doi: 10.1073/pnas.0405495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saxena A, Rorie CJ, Dimitrova D, Daniely Y, Borowiec JA. Nucleolin inhibits Hdm2 by multiple pathways leading to p53 stabilization. Oncogene. 2006;25:7274–7288. doi: 10.1038/sj.onc.1209714. [DOI] [PubMed] [Google Scholar]
- 68.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 69.Gu W, Wan D, Qian Q, Yi B, He Z, Gu Y, Wang L, He S. Ambra1 is an essential regulator of autophagy and apoptosis in SW620 cells: pro-survival role of Ambra1. PLoS One. 2014;9:e90151. doi: 10.1371/journal.pone.0090151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, De Zio D, Nazio F, Antonioli M, D’Orazio M, et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol. 2015;17:706. doi: 10.1038/ncb3171. [DOI] [PubMed] [Google Scholar]
- 71.Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15:406–416. doi: 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- 72.Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y, Tokino T. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 2008;68:4123–4132. doi: 10.1158/0008-5472.CAN-08-0325. [DOI] [PubMed] [Google Scholar]
- 73.Batlle E, Henderson JT, Beghtel H, van den Born MM, Sancho E, Huls G, Meeldijk J, Robertson J, van de Wetering M, Pawson T, et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell. 2002;111:251–263. doi: 10.1016/s0092-8674(02)01015-2. [DOI] [PubMed] [Google Scholar]
- 74.Yabana T, Arimura Y, Tanaka H, Goto A, Hosokawa M, Nagaishi K, Yamashita K, Yamamoto H, Adachi Y, Sasaki Y, et al. Enhancing epithelial engraftment of rat mesenchymal stem cells restores epithelial barrier integrity. J Pathol. 2009;218:350–359. doi: 10.1002/path.2535. [DOI] [PubMed] [Google Scholar]
- 75.Lazebnik LB, Konopliannikov AG, Kniazev OV, Parfenov AI, Tsaregorodtseva TM, Ruchkina IN, Khomeriki SG, Rogozina VA, Konopliannikova OA. [Use of allogeneic mesenchymal stem cells in the treatment of intestinal inflammatory diseases] Ter Arkh. 2010;82:38–43. [PubMed] [Google Scholar]
- 76.Nemoto Y, Kanai T, Takahara M, Oshima S, Nakamura T, Okamoto R, Tsuchiya K, Watanabe M. Bone marrow-mesenchymal stem cells are a major source of interleukin-7 and sustain colitis by forming the niche for colitogenic CD4 memory T cells. Gut. 2013;62:1142–1152. doi: 10.1136/gutjnl-2012-302029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sémont A, Demarquay C, Bessout R, Durand C, Benderitter M, Mathieu N. Mesenchymal stem cell therapy stimulates endogenous host progenitor cells to improve colonic epithelial regeneration. PLoS One. 2013;8:e70170. doi: 10.1371/journal.pone.0070170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xing Y, Chen X, Cao Y, Huang J, Xie X, Wei Y. Expression of Wnt and Notch signaling pathways in inflammatory bowel disease treated with mesenchymal stem cell transplantation: evaluation in a rat model. Stem Cell Res Ther. 2015;6:101. doi: 10.1186/s13287-015-0092-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hayashi Y, Tsuji S, Tsujii M, Nishida T, Ishii S, Iijima H, Nakamura T, Eguchi H, Miyoshi E, Hayashi N, et al. Topical implantation of mesenchymal stem cells has beneficial effects on healing of experimental colitis in rats. J Pharmacol Exp Ther. 2008;326:523–531. doi: 10.1124/jpet.108.137083. [DOI] [PubMed] [Google Scholar]
- 80.Bandukwala HS, Gagnon J, Togher S, Greenbaum JA, Lamperti ED, Parr NJ, Molesworth AM, Smithers N, Lee K, Witherington J, et al. Selective inhibition of CD4+ T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors. Proc Natl Acad Sci USA. 2012;109:14532–14537. doi: 10.1073/pnas.1212264109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rousseaux C, Lefebvre B, Dubuquoy L, Lefebvre P, Romano O, Auwerx J, Metzger D, Wahli W, Desvergne B, Naccari GC, et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med. 2005;201:1205–1215. doi: 10.1084/jem.20041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yamakawa-Karakida N, Sugita K, Inukai T, Goi K, Nakamura M, Uno K, Sato H, Kagami K, Barker N, Nakazawa S. Ligand activation of peroxisome proliferator-activated receptor gamma induces apoptosis of leukemia cells by down-regulating the c-myc gene expression via blockade of the Tcf-4 activity. Cell Death Differ. 2002;9:513–526. doi: 10.1038/sj.cdd.4401000. [DOI] [PubMed] [Google Scholar]
- 83.Shimada T, Kojima K, Yoshiura K, Hiraishi H, Terano A. Characteristics of the peroxisome proliferator activated receptor gamma (PPARgamma) ligand induced apoptosis in colon cancer cells. Gut. 2002;50:658–664. doi: 10.1136/gut.50.5.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ai G, Dachineni R, Muley P, Tummala H, Bhat GJ. Aspirin and salicylic acid decrease c-Myc expression in cancer cells: a potential role in chemoprevention. Tumour Biol. 2016;37:1727–1738. doi: 10.1007/s13277-015-3959-0. [DOI] [PubMed] [Google Scholar]
- 85.Chu EC, Chai J, Ahluwalia A, Tarnawski AS. Mesalazine downregulates c-Myc in human colon cancer cells. A key to its chemopreventive action? Aliment Pharmacol Ther. 2007;25:1443–1449. doi: 10.1111/j.1365-2036.2007.03336.x. [DOI] [PubMed] [Google Scholar]
- 86.Peiró-Jordán R, Krishna-Subramanian S, Hanski ML, Lüscher-Firzlaff J, Zeitz M, Hanski C. The chemopreventive agent ursodeoxycholic acid inhibits proliferation of colon carcinoma cells by suppressing c-Myc expression. Eur J Cancer Prev. 2012;21:413–422. doi: 10.1097/CEJ.0b013e32834ef16f. [DOI] [PubMed] [Google Scholar]
- 87.Beaulieu JF. Differential expression of the VLA family of integrins along the crypt-villus axis in the human small intestine. J Cell Sci. 1992;102(Pt 3):427–436. doi: 10.1242/jcs.102.3.427. [DOI] [PubMed] [Google Scholar]
- 88.Boudjadi S, Carrier JC, Beaulieu JF. Integrin α1 subunit is up-regulated in colorectal cancer. Biomark Res. 2013;1:16. doi: 10.1186/2050-7771-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ni H, Dydensborg AB, Herring FE, Basora N, Gagné D, Vachon PH, Beaulieu JF. Upregulation of a functional form of the beta4 integrin subunit in colorectal cancers correlates with c-Myc expression. Oncogene. 2005;24:6820–6829. doi: 10.1038/sj.onc.1208848. [DOI] [PubMed] [Google Scholar]
- 90.Groulx JF, Giroux V, Beauséjour M, Boudjadi S, Basora N, Carrier JC, Beaulieu JF. Integrin α6A splice variant regulates proliferation and the Wnt/β-catenin pathway in human colorectal cancer cells. Carcinogenesis. 2014;35:1217–1227. doi: 10.1093/carcin/bgu006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dydensborg AB, Teller IC, Groulx JF, Basora N, Paré F, Herring E, Gauthier R, Jean D, Beaulieu JF. Integrin alpha6Bbeta4 inhibits colon cancer cell proliferation and c-Myc activity. BMC Cancer. 2009;9:223. doi: 10.1186/1471-2407-9-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boudjadi S, Carrier JC, Groulx JF, Beaulieu JF. Integrin α1β1 expression is controlled by c-MYC in colorectal cancer cells. Oncogene. 2016;35:1671–1678. doi: 10.1038/onc.2015.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Untawale S, Zorbas MA, Hodgson CP, Coffey RJ, Gallick GE, North SM, Wildrick DM, Olive M, Blick M, Yeoman LC. Transforming growth factor-alpha production and autoinduction in a colorectal carcinoma cell line (DiFi) with an amplified epidermal growth factor receptor gene. Cancer Res. 1993;53:1630–1636. [PubMed] [Google Scholar]
- 94.Ooi A, Takehana T, Li X, Suzuki S, Kunitomo K, Iino H, Fujii H, Takeda Y, Dobashi Y. Protein overexpression and gene amplification of HER-2 and EGFR in colorectal cancers: an immunohistochemical and fluorescent in situ hybridization study. Mod Pathol. 2004;17:895–904. doi: 10.1038/modpathol.3800137. [DOI] [PubMed] [Google Scholar]
- 95.Park J, Kim SY, Kim HJ, Kim KM, Choi EY, Kang MS. A reciprocal regulatory circuit between CD44 and FGFR2 via c-myc controls gastric cancer cell growth. Oncotarget. 2016;7:28670–28683. doi: 10.18632/oncotarget.8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mathur A, Ware C, Davis L, Gazdar A, Pan BS, Lutterbach B. FGFR2 is amplified in the NCI-H716 colorectal cancer cell line and is required for growth and survival. PLoS One. 2014;9:e98515. doi: 10.1371/journal.pone.0098515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3:308–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tögel L, Nightingale R, Chueh AC, Jayachandran A, Tran H, Phesse T, Wu R, Sieber OM, Arango D, Dhillon AS, et al. Dual Targeting of Bromodomain and Extraterminal Domain Proteins, and WNT or MAPK Signaling, Inhibits c-MYC Expression and Proliferation of Colorectal Cancer Cells. Mol Cancer Ther. 2016;15:1217–1226. doi: 10.1158/1535-7163.MCT-15-0724. [DOI] [PubMed] [Google Scholar]
- 101.Chen BJ, Wu YL, Tanaka Y, Zhang W. Small molecules targeting c-Myc oncogene: promising anti-cancer therapeutics. Int J Biol Sci. 2014;10:1084–1096. doi: 10.7150/ijbs.10190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Poortinga G, Quinn LM, Hannan RD. Targeting RNA polymerase I to treat MYC-driven cancer. Oncogene. 2015;34:403–412. doi: 10.1038/onc.2014.13. [DOI] [PubMed] [Google Scholar]
- 103.Wu X, Cai ZD, Lou LM, Zhu YB. Expressions of p53, c-MYC, BCL-2 and apoptotic index in human osteosarcoma and their correlations with prognosis of patients. Cancer Epidemiol. 2012;36:212–216. doi: 10.1016/j.canep.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 104.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sodir NM, Swigart LB, Karnezis AN, Hanahan D, Evan GI, Soucek L. Endogenous Myc maintains the tumor microenvironment. Genes Dev. 2011;25:907–916. doi: 10.1101/gad.2038411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fiorentino FP, Tokgün E, Solé-Sánchez S, Giampaolo S, Tokgün O, Jauset T, Kohno T, Perucho M, Soucek L, Yokota J. Growth suppression by MYC inhibition in small cell lung cancer cells with TP53 and RB1 inactivation. Oncotarget. 2016;7:31014–31028. doi: 10.18632/oncotarget.8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci USA. 2007;104:13028–13033. doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Giuriato S, Ryeom S, Fan AC, Bachireddy P, Lynch RC, Rioth MJ, van Riggelen J, Kopelman AM, Passegué E, Tang F, et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc Natl Acad Sci USA. 2006;103:16266–16271. doi: 10.1073/pnas.0608017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Felsher DW. MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes Cancer. 2010;1:597–604. doi: 10.1177/1947601910377798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bajpe PK, Prahallad A, Horlings H, Nagtegaal I, Beijersbergen R, Bernards R. A chromatin modifier genetic screen identifies SIRT2 as a modulator of response to targeted therapies through the regulation of MEK kinase activity. Oncogene. 2015;34:531–536. doi: 10.1038/onc.2013.588. [DOI] [PubMed] [Google Scholar]
- 111.Jing H, Hu J, He B, Negrón Abril YL, Stupinski J, Weiser K, Carbonaro M, Chiang YL, Southard T, Giannakakou P, et al. A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity. Cancer Cell. 2016;29:607. doi: 10.1016/j.ccell.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 112.Chen L, Wei T, Si X, Wang Q, Li Y, Leng Y, Deng A, Chen J, Wang G, Zhu S, et al. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. J Biol Chem. 2013;288:14510–14521. doi: 10.1074/jbc.M113.458737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yin YW, Jin HJ, Zhao W, Gao B, Fang J, Wei J, Zhang DD, Zhang J, Fang D. The Histone Acetyltransferase GCN5 Expression Is Elevated and Regulated by c-Myc and E2F1 Transcription Factors in Human Colon Cancer. Gene Expr. 2015;16:187–196. doi: 10.3727/105221615X14399878166230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hurley LH, Von Hoff DD, Siddiqui-Jain A, Yang D. Drug targeting of the c-MYC promoter to repress gene expression via a G-quadruplex silencer element. Semin Oncol. 2006;33:498–512. doi: 10.1053/j.seminoncol.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 115.Gu S, Kay MA. How do miRNAs mediate translational repression? Silence. 2010;1:11. doi: 10.1186/1758-907X-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liu M, Chen H. The role of microRNAs in colorectal cancer. J Genet Genomics. 2010;37:347–358. doi: 10.1016/S1673-8527(09)60053-9. [DOI] [PubMed] [Google Scholar]
- 117.Wang H, Cao F, Li X, Miao H, E J, Xing J, Fu CG. miR-320b suppresses cell proliferation by targeting c-Myc in human colorectal cancer cells. BMC Cancer. 2015;15:748. doi: 10.1186/s12885-015-1728-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Grunt TW, Hebar A, Laffer S, Wagner R, Peter B, Herrmann H, Graf A, Bilban M, Posch M, Hoermann G, et al. Prominin-1 (CD133, AC133) and dipeptidyl-peptidase IV (CD26) are indicators of infinitive growth in colon cancer cells. Am J Cancer Res. 2015;5:560–574. [PMC free article] [PubMed] [Google Scholar]
- 119.Suresh PK, Varadharaj VA, Mathiyazhagan J. ABC transporters in anticancer drug transport - Lessons for Therapy, Drug Development and Delivery Systems. Int J Drug Dev Res. 2015;7:127–136. [Google Scholar]
- 120.Zhang HL, Wang P, Lu MZ, Zhang SD. [c-Myc regulation of ATP-binding cassette transporter reverses chemoresistance in CD133(+) colon cancer stem cells] Sheng Li Xue Bao. 2016;68:171–178. [PubMed] [Google Scholar]
- 121.Vučićević D, Gehre M, Dhamija S, Friis-Hansen L, Meierhofer D, Sauer S, Ørom UA. The long non-coding RNA PARROT is an upstream regulator of c-Myc and affects proliferation and translation. Oncotarget. 2016 doi: 10.18632/oncotarget.8985. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Winkle M, van den Berg A, Tayari M, Sietzema J, Terpstra M, Kortman G, de Jong D, Visser L, Diepstra A, Kok K, et al. Long noncoding RNAs as a novel component of the Myc transcriptional network. FASEB J. 2015;29:2338–2346. doi: 10.1096/fj.14-263889. [DOI] [PubMed] [Google Scholar]
- 123.Yang F, Xue X, Bi J, Zheng L, Zhi K, Gu Y, Fang G. Long noncoding RNA CCAT1, which could be activated by c-Myc, promotes the progression of gastric carcinoma. J Cancer Res Clin Oncol. 2013;139:437–445. doi: 10.1007/s00432-012-1324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xiang JF, Yin QF, Chen T, Zhang Y, Zhang XO, Wu Z, Zhang S, Wang HB, Ge J, Lu X, et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014;24:513–531. doi: 10.1038/cr.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jensen SA, Day ES, Ko CH, Hurley LA, Luciano JP, Kouri FM, Merkel TJ, Luthi AJ, Patel PC, Cutler JI, et al. Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci Transl Med. 2013;5:209ra152. doi: 10.1126/scitranslmed.3006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tangudu NK, Verma VK, Clemons TD, Beevi SS, Hay T, Mahidhara G, Raja M, Nair RA, Alexander LE, Patel AB, et al. RNA Interference Using c-Myc-Conjugated Nanoparticles Suppresses Breast and Colorectal Cancer Models. Mol Cancer Ther. 2015;14:1259–1269. doi: 10.1158/1535-7163.MCT-14-0970. [DOI] [PubMed] [Google Scholar]
- 127.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A. A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol Ther. 2005;11:990–995. doi: 10.1016/j.ymthe.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 129.Iyer KS, Zdyrko B, Malz H, Pionteck J, Luzinov I. Polystyrene Layers Grafted to Macromolecular Anchoring Layer. Macromolecules. 2003;36:6519–6526. [Google Scholar]
- 130.Meck MM, Wierdl M, Wagner LM, Burger RA, Guichard SM, Krull EJ, Harris LC, Potter PM, Danks MK. A virus-directed enzyme prodrug therapy approach to purging neuroblastoma cells from hematopoietic cells using adenovirus encoding rabbit carboxylesterase and CPT-11. Cancer Res. 2001;61:5083–5089. [PubMed] [Google Scholar]
- 131.Iyengar RV, Pawlik CA, Krull EJ, Phelps DA, Burger RA, Harris LC, Potter PM, Danks MK. Use of a modified ornithine decarboxylase promoter to achieve efficient c-MYC- or N-MYC-regulated protein expression. Cancer Res. 2001;61:3045–3052. [PubMed] [Google Scholar]
- 132.Huang H, Weng H, Zhou H, Qu L. Attacking c-Myc: targeted and combined therapies for cancer. Curr Pharm Des. 2014;20:6543–6554. doi: 10.2174/1381612820666140826153203. [DOI] [PubMed] [Google Scholar]