

Abstract
Histoplasma capsulatum is a dimorphic fungus that develops a yeast-like morphology in host's tissue, responsible for the pulmonary disease histoplasmosis. The recent increase in the incidence of histoplasmosis in immunocompromised patients highlights the need of understanding immunological controls of fungal infections. Here, we describe our discovery of the role of endogenous galectin-1 (Gal-1) in the immune pathophysiology of experimental histoplasmosis. All infected wild-type (WT) mice survived while only 1/3 of Lgals1−/− mice genetically deficient in Gal-1 survived 30 days after infection. Although infected Lgals1−/− mice had increased proinflammatory cytokines, nitric oxide (NO), and elevations in neutrophil pulmonary infiltration, they presented higher fungal load in lungs and spleen. Infected lung and infected macrophages from Lgals1−/− mice exhibited elevated levels of prostaglandin E2 (PGE2, a prostanoid regulator of macrophage activation) and prostaglandin E synthase 2 (Ptgs2) mRNA. Gal-1 did not bind to cell surface of yeast phase of H. capsulatum, in vitro, suggesting that Gal-1 contributed to phagocytes response to infection rather than directly killing the yeast. The data provides the first demonstration of endogenous Gal-1 in the protective immune response against H. capsulatum associated with NO and PGE2 as an important lipid mediator in the pathogenesis of histoplasmosis.
1. Introduction
Histoplasmosis is a worldwide known disease caused by the fungus Histoplasma capsulatum. The real geographic distribution of this mycosis could be more widespread than what was previously thought [1, 2]. The incidence of this fungal disease is higher in the Mid- and Southeast USA, Latin America, China, and other world areas [2]. Additionally, asymptomatic cases are escalating and are reported to predominately affect immunocompromised individuals as an acute pulmonary infection similar to mild flu-like symptoms [1, 3, 4]. Likewise, the most severe symptomatic form of the disease, referred to as disseminated histoplasmosis, develops most commonly in immunosuppressed patients. However, unlike the mild form, disseminated histoplasmosis can lead to death [4]. Although antifungal therapies have been used against the fungus, there are no current alternative therapies to treat or protect against H. capsulatum infection.
H. capsulatum is a dimorphic, facultative, intracellular pathogen found as a yeast phase when in host tissue [5]. In the early stages of infection, the fungus is phagocytosed by resident alveolar macrophages, dendritic cells, and neutrophils [6]. Once phagocytosed, the fungus survives in the phagosome and consequently transforms into a yeast. In immunocompromised individuals or when left untreated, the reservoir phagocytes can travel to lymphatic tissue and spread infection. However, induction of a strong cellular immune response can contain or clear the infected phagocytes, therefore preventing the spread of the infection. An effective host defense to H. capsulatum infection is dependent on adequate activation of T cells and phagocytes [6, 7]. Appropriately, the balance between the Th1 and Th2 response is fundamental for solving H. capsulatum infection [6, 7], with Th1 proinflammatory cytokines IFN-γ (interferon-γ), interleukin-12 (IL-12), TNF-α (tumor necrosis factor-α), and GM-CSF (granulocyte macrophage colony-stimulating) being essential to elicit macrophage activation and clearance of H. capsulatum. In addition, a balanced production of lipid mediators, such as prostaglandin E2 (PGE2) and leukotriene B4 (LTB4), is critical for the host defense in histoplasmosis, since high levels of PGE2 and low levels of LTB4 impair the yeast clearance and increase the severity of this fungal disease [8, 9]. Nitric oxide also participates in the host defense against H. capsulatum [10, 11]; however, overproduction of this mediator increases the susceptibility of the host to yeast infection [8, 12].
In addition to cytokines and lipid mediators, a member of the galectin family, known as galectin-3 (Gal-3), has been suggested to be involved in the immune response against H. capsulatum infection [13]. Galectins belong to an endogenous lectin family that recognizes glycans present in microorganisms and participates in the pathophysiology of inflammatory responses, infectious diseases, autoimmunity, and cancer [14–18].
Interestingly, galectin-1 (Gal-1) has been shown to participate in an innate and adaptive immune response to different models of experimental infections such as in Trypanosoma cruzi (T. cruzi) [19], situation in which a dual role for this lectin was described. These authors showed that, in a low concentration, Gal-1 was able to decrease proinflammatory interleukin-12 (IL-12) and nitric oxide (NO), while in a high concentration, it has induced infected macrophage apoptosis. Gal-1 was also found to promote Human Immunodeficiency Virus-1 (HIV-1) infectivity [20]; in dengue virus infection, it could cause an inhibitory effect on virus replication [21]. Thus, several Gal-1 exogenous properties have been related to CRD binding to cell surface receptors, modulating immune cell functions, migration, differentiation, activation, and cell survival [22–27]. Nevertheless, the interactions of this lectin with the intracellular ligands can also occur independently to carbohydrates [28, 29].
Although Gal-1 can participate in various pathophysiological processes, there is little information about the role of Gal-1 in fungal infections. Therefore, the present study evaluated the biological impact of the absence of Gal-1 on a murine model of histoplasmosis. While mice genetically deficient in Gal-3 (Lgals3−/−) were able to clear H. capsulatum infection more efficiently than wild-type (WT) mice [13], it was reported for the first time that Gal-1 (Lgals1−/−) mice are more susceptible to H. capsulatum infection compared to WT group. This unique immune phenotype suppresses the host response against the fungus and is followed by high levels of neutrophil infiltration and proinflammatory cytokines in the lungs which causes a strong anti-inflammatory response with high levels of PGE2 and NO. These findings indicate a novel contribution of endogenous Gal-1 to the development of a protective immune response to H. capsulatum.
2. Results
2.1. Lgals1−/−-Infected Mice Fail to Control H. capsulatum Infection
WT and Lgals1−/− mice were injected with 5 × 105 H. capsulatum yeasts cells directly into the lungs and survival was monitored up to 30 days. 14 days after infection, Lgals1−/−-infected mice began to die. 33% of Lgals1−/−-infected mice survived 30 days after infection, whereas 100% of the infected WT mice survived over that period (Figure 1).
Figure 1.
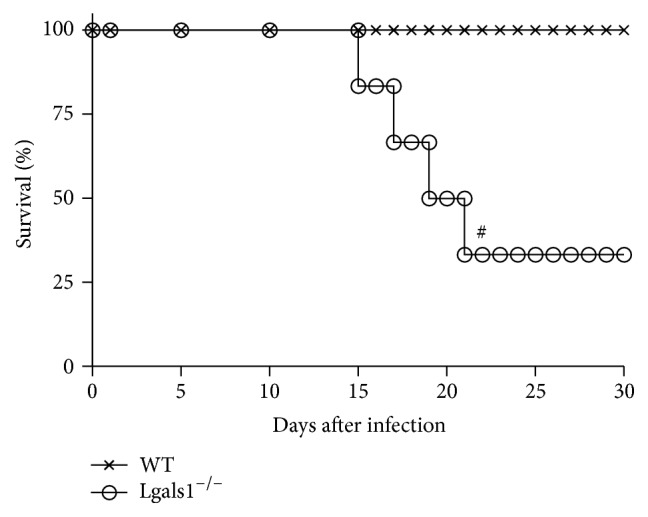
Gal-1 knockout mice are sensitive to H. capsulatum infection. WT and Lgals1−/− mice were infected i.t. with 5 × 105 viable H. capsulatum yeast (sublethal inoculum) and survival was monitored for 30 days. Control mice (uninfected) received i.t. 100 μL PBS (data not shown). Data are representative of one of the two experiments performed independently (n = 10 per group) and Mantel-Cox log-rank (χ 2 “chi-squared”) was used. # p < 0.05 WT versus Lgals1−/−, both infected.
2.2. Lgals1−/−-Infected Mice Have a Higher Yeast Load and Infiltration of Neutrophils in the Lung
To determine if the high mortality rate in Lgals1−/− mice is correlated with impaired fungal clearance, the H. capsulatum load was quantified in the lung and spleen. Considering that Lgals1−/− mice began to die day 14 after infection and that day 15 after infection is a critical point on the evolution of experimental histoplasmosis using mutant mice and a sublethal fungus dose [9], on day 15 after infection, lung parenchymal histopathological analysis and quantification of fungal burden in the lung and spleen were performed. Lgals1−/−-infected mice presented a higher number of yeast cells in pulmonary parenchyma (Figures 2(a) and 2(b)) and higher fungal load in lung (Figure 2(c)) and spleen (Figure 2(d)). Even though infected Lgals1−/− mice presented a higher fungal burden in the lung, an increased neutrophil influx was detected in their pulmonary tissue (Figures 3(a) and 3(b)). It is known that an efficient immune response against H. capsulatum is associated with fungicidal/fungistatic effects of pulmonary infiltrated phagocytes [6, 30]. Thus, these findings suggest that endogenous Gal-1 is required to develop a protective immune response against H. capsulatum and that Gal-1 could be associated with the control of fungal replication as an efficient anti-H. capsulatum activity along with effectors functions and regulation of tissue accumulation of neutrophils.
Figure 2.

Lgals1−/− mice have a higher yeast burden in the lung and spleen day 15 after infection. On day 15 after infection with H. capsulatum (5 × 105 viable yeasts), animals were sacrificed and tissue samples were harvested. (a) Lung sections (5 μm) from WT and Lgals1−/− mice were stained with silver (Grocott's methanamine silver (GMS); bar: 50 μm; insert bar: 25 μm) and (b) yeast cells were quantified (yeast/mm2 lung) using magnifications ×400. Fungal burden was quantified from tissue homogenates and expressed as the number of colony-forming units (CFU) per gram of tissue CFU/g in lung (c) and CFU per spleen (d). Data are representative of one of the two experiments performed independently (n = 10 per group). Values are mean ± SEM. # p < 0.05 WT versus Lgals1−/−, both infected.
Figure 3.

Lgals1−/− mice have increased neutrophil infiltration in the lung parenchyma. H. capsulatum-infected mice were euthanized on day 15 after infection and lung sections (5 μm) were embedded in paraffin blocks. Lung sections from WT + H. capsulatum (I, bar: 100 μm; III, bar: 25 μm) and Lgals1−/− + H. capsulatum (II, bar: 100 μm; IV, bar: 25 μm) were stained with hematoxylin (a) and neutrophils were quantified (neutrophils/mm2) using magnifications ×400 (b). Data are representative of one of the two experiments performed independently (n = 10 per group). Values are mean ± SEM. # p < 0.05 WT versus Lgals1−/−, both infected.
2.3. Lgals1−/−-Infected Mice Show Increased Proinflammatory Cytokines in the Lung
It is well known that increased expression of inflammatory cytokines, including IL-12, IFN-γ, and TNF-α, is critical for the immune-protective response in H. capsulatum-infected mice [31–34]. Thus, to analyze the pattern of inflammatory cytokines in WT and Lgals1−/− mice day 15 after H. capsulatum infection, the levels of IL-12p40, TNF-α, IL-1α, IL-10, IL-4, and IL-6 in the pulmonary homogenates were measured. There were higher levels of IL-12p40 (Figure 4(a)) and IL-1α (Figure 4(c)) and similar concentrations of TNF-α (Figure 4(b)) in homogenized lungs of Lgals1−/−-infected mice, when compared to WT infected mice. Furthermore, no statistically significant differences in TNF-α (Figure 4(b)) were observed, and IL-10, IL-4, and IL-6 (data not shown) were not produced in detectable levels.
Figure 4.

Lgals1−/− mice exhibit increased inflammatory response day 15 after infection with H. capsulatum. Cytokines IL-12p40 (a), TNF-α (b), IL-1-α (c), and NO2 − (d) were quantified from homogenized lungs on day 15 after infection with H. capsulatum. Cytokines levels (pg/mL) were determined in the supernatants by ELISA and NO2 − (μM) by using a Griess reaction. Data are representative of one of the two experiments performed independently (n = 10 per group). Values are mean ± SEM. ∗ p < 0.05 infected mice versus control (uninfected), # p < 0.05 WT versus Lgals1−/−, both infected.
2.4. Lgals1−/−-Infected Mice Demonstrate Prostaglandin E2 and Nitric Oxide Overproduction
Based on the aforementioned results, it was also analyzed whether inflammatory mediators, such as NO and PGE2, are associated with increased levels of proinflammatory cytokines and consequently immunosuppression in the absence of endogenous Gal-1 in experimental histoplasmosis. It has been reported that the inhibition of COX-2 improves the host defense against H. capsulatum [8]. Therefore, PGE2 was quantified from homogenized lungs derived from infected WT and Lgals1−/− mice on day 15 after H. capsulatum infection. The lungs of infected Lgals1−/− mice exhibited higher levels of PGE2 (Figure 5(a)) when compared to infected WT mice. Thus, consistent with other published results [8, 35], these findings suggest that higher levels of PGE2 may contribute to susceptibility of infected Lgals1−/− mice. Interestingly, not only PGE2 but also NO levels in the lung of this group were increased when compared to WT (Figure 4(d)).
Figure 5.

Absence of endogenous Gal-1 increases prostaglandin PGE2 production and Ptges2 expression in peritoneal macrophages. (a) In vivo prostaglandin E2 was quantified in supernatants from homogenized lungs on day 15 after infection with H. capsulatum (5 × 105 yeasts/mice) by ELISA. (b) 5 × 105 peritoneal macrophages were incubated in vitro with H. capsulatum (MOI 1 : 1) during 2 and 24 hours and mRNA levels for Ptges2 were quantified and plotted as Fold Regulation by Log2. In addition, PGE2 was assessed in vitro in the supernatants by ELISA 24 hours after infection (c). In vivo data are representative of one of the two experiments performed independently (n = 10 per group). Values are mean ± SEM. ∗ p < 0.05 infected mice versus control (uninfected), # p < 0.05 WT versus Lgals1−/−, both infected.
2.5. Uninfected Lgals1−/− Macrophages Express High Levels of Prostaglandin E Synthase 2 after Fungal Infection
The immune response against H. capsulatum is mediated by Th1 cells, which requires macrophages activation [6, 7]. The pathogenic yeast fungus replicates inside these cells and results in metabolites of arachidonic acids production, such as prostaglandins and leukotrienes [35]. To assess the role of endogenous Gal-1 in PGE2 production, prostaglandin E synthase 2 (Ptges2) mRNA expression in peritoneal macrophages from Lgals1−/− and WT mice infected or not with H. capsulatum in vitro was evaluated. Interestingly, 24 hours after H. capsulatum infection, Lgals1−/− macrophages had increased Ptges2 mRNA expression when compared to infected WT macrophages. In addition, higher levels of PGE2 were detected in the supernatants 24 hours after the infection of Lgals1−/− macrophages when compared to WT macrophages (Figure 5(c)). Thus, the in vitro results correlate with overproduction of prostaglandins in vivo (Figure 5(a)).
2.6. Galectin-1 Does Not Bind to and Kill the Yeast Form of H. capsulatum
Recently, it was reported that galectins can bind glycans not only on the host cell surface, but also on molecules on pathogens, which has been found to result in pathogen killing and modulation of immune responses against bacterial infections [36, 37]. To assess the binding capacity of Gal-1 on H. capsulatum surface, biotinylated-human recombinant Gal-1 (hrGal-1 : 1 μM and 4 μM) was incubated with the yeast form of H. capsulatum. Gal-1 did not bind to the yeast form of this fungus (Figure 6(a)) although the hrGal-1 was active, since it did bind to glycans on HL-60 cells (Figure 6(b)). As expected, different concentrations of hrGal-1 (0.5, 1.0, 2.5, 4.0, and 10.0 μM) did not alter the viability of H. capsulatum after 24 and 48 h of in vitro incubation (Figure 6(c)). This result suggests that the binding effect can be related to killing activity as Stowell et al. [36] described for E. coli strains in the presence of Gal-4 and Gal-8. Thus, the yeast form of H. capsulatum seems not to express ligands for Gal-1 and indicates that the protective mechanistic effects of Gal-1 to H. capsulatum infection do not involve Gal-1 binding to the yeast.
Figure 6.

Gal-1 does not bind and kill the yeast form of H. capsulatum. (a) Yeasts were incubated for 1 hour at 4°C with 1.0 μM and 4.0 μM biotinylated-hrGal-1, in the presence or absence of 20 mM lactose (Gal-1 inhibitor) or sucrose (control, noninhibitor). After that, yeasts were incubated with streptavidin-FITC and labeled cells were acquired on a FACS Canto (Becton Dickinson, Mountain View, CA, USA) and analyzed in the DIVA software (Becton Dickinson). (b) As a control, HL-60 cells (1 × 106) were incubated with 1 μM biotinylated-hrGal-1 for 1 hour at 4°C, in presence or absence of 20 mM lactose or sucrose. (c) Several hrGal-1 concentrations (0.5, 1.0, 2.5, 4.0, and 10 μM) were incubated with 1 × 106 H. capsulatum cells during 24 and 48 h. After each time, relative fluorescent units (RFU) (560–590 nm) were measured and represent yeast cells metabolically active through the dye resazurin reagent. Data are representative of two independent experiments and expressed as the mean ± SEM.
3. Discussion
Galectins have been described as regulators of immune response in models of inflammatory and infectious diseases and host pathogen recognition [14, 25, 27, 36, 38–41]. Gal-1 and Gal-3 are the best studied members of the galectin family and the expression of these proteins is increased or decreased in distinct cell types following infections caused by different pathogens [42, 43]. Previous reports demonstrate that Gal-3 participates in yeast infections [13, 39, 44]; however, the role of Gal-1 in fungal diseases has not yet been explored. Although the expression of Gal-3 in dendritic cells is not upregulated in WT mice infected with H. capsulatum, mice genetically deficient in Gal-3 clear this fungal infection more efficiently than WT mice [13], showing that high Gal-3 expression in WT mice is not required for the participation in the immune response against H. capsulatum and may actually contribute to pathogenesis [13].
Unexpectedly, Gal-3 knockout mice are more susceptible to Candida albicans infection than WT mice and the susceptibility is associated with high fungal burden in the brain. Additionally, Gal-3, but not Gal-1, can induce yeast cell death upon binding to β-1,2-linked oligomannosides on the surface of pathogenic fungus Candida albicans [44]. Thus, Gal-3 and Gal-1 appear to be differentially involved in host defense mechanisms against fungal infections, and this feature may arise from the specific pathogen. In disseminated candidiasis model, the absence of Gal-3 is responsible for increased susceptibility [39]. In the present study, in contrast to Gal-3-deficient mice [13], the novel observation that the absence of endogenous Gal-1 increased susceptibility to H. capsulatum accompanied by higher fungal loads in the lung and spleen was made. Recently, it was reported that Lgals1−/− mice infected intradermally with T. cruzi are resistant to this parasitic infection compared to their WT counterparts and this resistant phenotype could be associated with a dysfunction in the regulatory properties of Gal-1 followed by high production of Th1 proinflammatory cytokines and improvement of Th1 and CD8+ T cells responses [25]. However, another report from the same group described that Lgals1−/− mice infected intraperitoneally with T. cruzi showed elevated parasitemia, less tissue inflammation, and higher mortality rates as compared to infected WT mice [45]. These authors suggest that this discrepancy could be associated with the presence of different phagocytes at sites of infection and distinct local immune response induced by T. cruzi. Based on these reports and the present data, it is suggested that the infection of Lgals1−/− mice, intratracheally, with H. capsulatum promotes a unique immunophenotype that suppresses the host response against the fungus. This special immunological scenario is characterized by an imbalanced inflammation associated with high levels of neutrophil infiltration and proinflammatory cytokines in the lungs that causes a strong anti-inflammatory response induced by high levels of PGE2 and nitric oxide that could modulate phagocyte and T cell functions.
Based on evidence that Gal-4 and Gal-8 can bind and kill bacteria that express a human blood group B-like antigen and a common mammalian antigen α-Gal [36, 46], it is hypothesized that Gal-1 might have the same effect on the yeast form of H. capsulatum. However, in contrast to Gal-4 and Gal-8 killing activities toward bacteria, Gal-1 neither bound to nor killed the yeast form (Figure 6). This data suggests that the ability of Gal-1 to contribute to proper control of the fungal infection arises from an indirect contribution, since Gal-1 is clearly involved in the modulation of immune response against H. capsulatum.
Next, it was evaluated whether the absence of Gal-1 could interfere with the recruitment of neutrophils to the lungs during the infection, since this lectin could modulate the inflammatory response [24, 47]. It is known that neutrophil migration to sites of infection helps the clearance of pathogens [48]. Human neutrophils are able to impair the growth of H. capsulatum yeast form, and this microbiostatic effect is mediated mostly by compounds present in the azurophil granules [49]. Moreover, in experimental histoplasmosis, depletion of GR-1+ cells, primarily neutrophils, promotes the increase in fungal load in the lungs and spleens and decreases the survival of animals even in the presence of high levels of TNF-α and NO [50]. Previous reports demonstrate that mice genetically deficient in Gal-1 have enhanced neutrophil emigration in response to IL-1β compared to their wild-type counterparts [51]. Furthermore, in an animal model of zymosan-induced peritonitis, exogenous Gal-1 was shown to cause decreased production of proinflammatory cytokines and expression of adhesion molecules on the surface of neutrophils, thus diminishing their rates of migration [47]. The present results are consistent with those of others, indicating that H. capsulatum promotes intense neutrophil recruitment in the lung of Lgals1−/− mice (Figure 3); however, these phagocytes were not able to clear the fungus in the lung pulmonary parenchyma. Other authors have demonstrated that upregulation of proinflammatory cytokines/chemokines resulted in higher numbers of lung neutrophils and also reduced the capacity of the host defense to eliminate the fungus [9]. Since Gal-1 can modulate the adhesion molecules expression as well as releasing mediators of immune response [22–24, 47], it was evaluated whether the increase of neutrophil infiltration into the lung was associated with exacerbation of cytokines during the inflammatory response against H. capsulatum infection. The intense neutrophil accumulation in the lung of Lgals1−/− mice could be explained by high levels of IL-1α (Figure 4(c)), since this cytokine is a chemoattractant for neutrophils [52, 53]. Moreover, the presence of high number of neutrophils may be a major source of IL-12 detected in the lung from infected-Lgals1−/− mice, as neutrophils have been reported to produce IL-12 [54]. Curiously, inhibition of dectin-1 expression, a host receptor for fungal beta-glucan, reduces the severity of fungus infection and its effect was associated with decrease of proinflammatory cytokines, including IL-12, and neutrophil infiltration [55]. Furthermore, it is known that proinflammatory cytokines, including IL-12 [32, 34, 56, 57], are essential for host defense against H. capsulatum. Conversely, on the present model, the increase of IL-12 did not promote fungal clearance in the lungs of Lgals1−/− mice. Based on these results, it may be hypothesized that the excessive production of IL-12p40 and IL-1α in Lgals1−/−-infected mice is deleterious to the animals. Interestingly, Lgals1−/− mice are more resistant to Trypanosoma cruzi infection than wild-type mice and this phenotype is associated with upregulation of IFN-γ and no significant production of IL17A [25]. However, the HSV-1 infection in Lgals1−/− mice promotes a severe disease, compare to wild-type, that is correlated with the elevated number of neutrophil infiltrations and IFN-γ-producing CD4 T cells and no significant change of IL-17-producing T cell in the ocular [58]. Then, considering that (i) immunoregulatory properties of Gal-1 are associated with regulation of TH 1 and TH 17 responses [59], (ii) IL-12 and IL-23 share p40 subunit [60], and (iii) IL-17/IL-23-axis cytokines participate in immune response against H. capsulatum infection [33], further investigation should be done in order to elucidate the impact of IL17/IL23 in experimental histoplasmosis in the absence of endogenous Gal-1. In addition to cytokine production, it was analyzed whether microbicidal factors, such as NO, could be modulated by the deficiency of Gal-1, which could underlie the suppression of host defense against H. capsulatum. It was found that the deficiency of Gal-1 promotes the increase of NO concentration in the lung of infected mice when compared with infected WT mice. These results are in concordance with other studies that show that Gal-1 negatively modulates the NO production by activating macrophage or microglia-like cells [23, 61] and activated microglia from Lgals1−/− mice produce high concentration of NO [62]. Moreover, the high levels of NO produced (Figure 4(d)) in the lung have no microbicidal effect on H. capsulatum, since lungs from Gal-1 Lgals1 −/− mice had higher CFU (Figure 2). Thus, NO appears to be important for the host defense against primary infection by H. capsulatum [11]; nonetheless, the overproduction of NO has also been shown to suppress phagocytic activities of macrophage in H. capsulatum infection and inhibit the CD4 T cells proliferation response to T. cruzi infection [12, 34, 50].
Alveolar macrophages are the first line of host defense in the lung against respiratory pathogens, and this phagocyte is an important source of lipid mediators, such as PGE2 in infected lung [63]. PGE2 has an important role in suppression of host defense involved modulation of alveolar macrophages functions in different pulmonary infection models, such as Streptococcus pneumoniae [64], Klebsiella pneumoniae [65], Pseudomonas aeruginosa [66], and recently H. capsulatum [8]. Lung and macrophages from Lgals1−/−-infected mice produced higher levels of PGE2 when compared to WT mice. Then, it is hypothesized that high levels of NO and PGE2 in the lungs of Lgals1−/−-infected mice inhibit the effector functions of macrophages and neutrophils against H. capsulatum. Whether the absence of endogenous Gal-1 can inhibit the effector functions of neutrophils against H. capsulatum remains unknown, though.
PGE2 is able to inhibit IL-12 production by macrophage and dendritic cells [67], although lung parenchyma from infected Lgals1−/− mice contained higher levels of IL-12 than those from infected WT mice even in the presence of high levels of PGE2. This finding is in agreement with other studies reporting that the inhibition of prostaglandin has no effects on the production of IL-12 in H. capsulatum-infected mice [8]. In addition, the present data is similar to others, demonstrating that the immunoregulatory effects of Gal-1 (endogenous or exogenous) are associated with suppression of Th1 cytokines, including the negative modulation of IL-12 production by activated macrophage or tolerogenic dendritic cells [19, 68–71].
Because of the low yield of murine alveolar macrophages, peritoneal macrophages from Lgals1−/− mice were used to examine the ability of endogenous Gal-1 to modulate the expression of mRNA Ptges2 and PGE2 after H. capsulatum infection (Figure 5(b)). The high fungus burden in lungs and spleen in infected Lgals1−/− mice could be associated with the downregulation effects of PGE2 in antimicrobial functions of phagocytes [8, 72].
Exaggerated inflammatory response could be responsible for higher production of PGE2 in lungs of H. capsulatum-infected Lgals1−/− mice that inhibited fungal clearance, since the PGE2 biosynthesis is increased under inflammatory conditions, and this prostanoid has been described to impair phagocytosis and kill by alveolar macrophages [73]. In addition, the effector functions of phagocytes from Lgals1−/− could be altered, since Gal-1 is a multifunctional molecule with intra- and extracellular effects [28, 29]. This immune suppressive effect is in line with current results, demonstrating the positive impact on mRNA Ptgs2 expression and PGE2 secretion of the Gal-1 deficiency in macrophages from Lgals1−/− mice after fungal infection. This data is in agreement with the results described by Rabinovich and colleagues, since this lectin can reduce arachidonic acid release and PGE2 secretion from activated macrophage [27]. Besides that, celecoxib treatment, a selective cyclooxygenase 2 inhibitor, improved the immune response against H. capsulatum infection through the inhibition of prostaglandin production [8]. Curiously, celecoxib induces expression of Gal-1 in activated macrophage and Gal-1 could be involved in the anti-inflammatory mechanisms of this drug [74]. Furthermore, Gal-1 inhibited the expression of activating transcription factor 3, a negative regulator of mRNA Ptgs2 in macrophage [75]. Despite that, further investigations are needed to elucidate the mechanism by which Gal-1 inhibits Ptgs2 expression. It has now been shown that PGE2 is a DAMP (damage-associated molecular patterns) and is induced and released by dying cells, which leads to suppressed expression of genes associated with inflammation and thereby limits immunostimulatory activities [76]. Also, PGE2 is downregulated in human systemic inflammatory diseases and mice with reduced PGE2 exhibit systemic inflammation [77]. In summary, the present results demonstrate that the endogenous Gal-1 plays an important role in host defense against Histoplasma capsulatum modulating of PGE2, IL-12, and NO production, as well as pulmonary neutrophil accumulation. Future studies are needed to better understand the cellular and molecular mechanisms in which endogenous galectin-1 could participate in host defense against fungus infection.
4. Materials and Methods
4.1. Animals
Six-to-eight-week-old wild-type (WT) male mice and mice genetically deficient in Gal-1 (Lgals1−/−), both in a C57BL/6J background, were housed and bred at the animal facility of the School of Pharmaceutical Sciences of Ribeirão Preto (University of São Paulo, Brazil). Wild-type mice were originally purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and Lgals1−/− mice were provided by Dr. Richard D. Cummings (Department of Surgery, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA, USA). The experimental protocol was approved and conducted in accordance with guidelines of the Institutional Animal Care Committee. To check the depletion of Lgals1, Gal-1 expression (mRNA and protein) analysis on WT and Lgals1−/− cells were performed as previously described [21] using conventional RT-PCR and western blot, respectively (data not shown). We used C57BL/6J mice as wild-type counterparts in our experiments.
4.2. H. capsulatum Strain and Infection of Mice
H. capsulatum strain was isolated from a patient at the Clinical Hospital, School of Medicine of Ribeirão Preto, University of São Paulo, and the characterization and preparation of H. capsulatum yeast cells were performed as previously described [9, 78, 79]. The yeast cultures were used at ≥90% viability according to fluorescein diacetate (Sigma-Aldrich, St. Louis, MO) and ethidium bromide (Sigma-Aldrich) staining [80]. Mice were given intratracheally (i.t.) dispersion containing 100 μL phosphate buffered saline (PBS, vehicle control) or a sublethal dose in PBS (5 × 105 yeasts/animal). The appropriate inoculum size was chosen based on procedure described by Sá-Nunes and colleagues [78]. On day 15 after infection, both uninfected and infected mice were euthanized in a CO2 chamber, and lungs and spleens were collected for analyses.
4.3. Fungal Load and Histopathology
H. capsulatum-infected mice were euthanized on day 15 after infection and tissue samples were harvested. Lung sections (5 μM) were embedded in paraffin blocks and stained with Grocott's methanamine silver (GMS) and quantification of yeasts was expressed as yeast/mm2 (original magnification: 400x). Also, fungal burden was determined from homogenized lung and spleen (Mixer Homogenizer; Labortechnik, Staufen, Germany) as previously described [7, 9]. Serial dilutions of these tissue homogenates were plated onto BHI blood agar and incubated at 37°C for 21 days. The results were expressed as mean colony-forming units (CFU) per gram of lung ± SEM (CFU/g) or CFU per whole spleen ± SEM (CFU/spleen). Lungs were collected, fixed in 10% formaldehyde, and embedded in paraffin blocks. For neutrophils analyses, lung sections (5 μm) were stained with hematoxylin and eosin (H&E) and the cells were quantified in the ocular lens containing 10 × 10 graticules (0.0624 mm2 each in magnifications: 400x). The results are expressed as neutrophils/mm2.
4.4. Measurement of Cytokines, PGE2, and Nitric Oxide
Lungs were collected 15 days after infection, weighed and homogenized (Mixer Homogenizer; Labortechnik, Staufen, Germany) in 2 mL of RPMI1640 (Sigma) and the supernatants were stored at −70°C until being assayed. Commercially available ELISA antibodies were used to measure TNF-α, IL-1α, IL-12p40, IL-10, IL-4, and IL-6 (BD OptEIA ELISA sets; BD Pharmingen) according to the instructions of the manufacturer. PGE2 from lung homogenate and from in vitro assay (in vitro assay is described below) were purified by Sep-Pak C18 cartridges according to the manufacturer's instructions (Waters Corp., Milford, MA). Quantification of PGE2 was assessed also by ELISA (Cayman Chemical, Ann Arbor, MI) and the results for cytokines and PGE2 are expressed in ng/mL. The sensitivity of the assay was <10 pg/mL. Nitrite (NO2 −) concentrations (μM) in lung homogenates was measured by Griess reaction using a standard curve with serial dilutions of NaNO2 (Sigma-Aldrich). Griess reagent was used in order to measure NO levels indirectly from nitrite as described previously [10].
4.5. Gene Expression by Real-Time Polymerase Chain Reaction (qRT-PCR)
4.5.1. In Vitro Assay
WT or Lgals1−/− peritoneal macrophages (5 × 105 cells/well) were incubated with H. capsulatum (MOI 1 : 1) during 2 and 24 hours. PGE2 was assessed in the supernatants 24 hours after infection and expression of mRNA was performed in plated macrophages 2 and 24 hours after H. capsulatum exposure.
4.5.2. Gene Expression
Total mRNA was isolated using the RNeasy Mini kit (Qiagen Inc., Valencia, CA), according to the manufacturer's instructions. cDNA (complementary DNA) was synthesized from 600 ng of total RNA using random primers (High Capacity cDNA Reverse Transcription Kit, Applied Biosystems, Temecula, CA). Aliquots of 2 μL of the total cDNA were amplified by qRT-PCR (StepOne Plus, Applied Biosystems, Singapore) using the primers (IDT®, Integrated DNA Technologies, California, USA) for Ptges2 (the gene encoding prostaglandin E synthase 2, Mm.PT. 58.7480753) and probe (TaqMan® Gene Expression Assay, Applied Biosystems, Foster City, USA). Actb (Mm00607939) was used as reference gene. Amplification was performed in duplicate under the following conditions: denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 minute. Relative quantification was performed using the ΔΔCt method and plotted as Fold Increase or Fold Regulation by Log2.
4.6. Human Recombinant Galectin-1 (hrGal-1) Purification
hrGal-1 was prepared as previously described [26, 81]. Briefly, purified hrGal-1 was treated with 100 mM iodoacetamide (Sigma-Aldrich) in 100 mM lactose/PBS overnight at 4°C [82]. To ensure that hrGal-1 samples were endotoxin-free, Detoxi-Gel Endotoxin removing gel (Pierce Biotechnology, Rockford, IL) was used and hrGal-1 activity was assessed by haemagglutination (data not shown).
4.7. Binding by Flow Cytometry and Resazurin Cell Viability Assays
To measure the capacity of Gal-1 to bind on yeast form of H. capsulatum, 1 μM and 4 μM biotinylated-hrGal-1 were incubated for 1 hour at 4°C, in presence or absence of 20 mM lactose or sucrose (Sigma-Aldrich). After washing, yeasts were incubated with streptavidin-FITC (Jackson IR) for 30 minutes at 4°C, washed, and formalin-fixed (1% in PBS). Labeled cells were acquired on a FACS Canto (Becton Dickinson, Mountain View, CA, USA) and analyzed in the DIVA software (Becton Dickinson). As a control, we used HL-60 cells obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in RPMI medium supplemented with 10% fetal bovine serum. To test H. capsulatum viability in a presence of Gal-1, we incubated, in vitro, several hrGal-1 concentrations (10, 4, 2.4, 1, and 0.5 μM) with 1 × 106 yeast cells during 24 and 48 h. The relative fluorescent units (RFU) using a plate reader were detected (560–590 nm) in order to analyze the number of yeast cells metabolically active using the dye resazurin reagent (Sigma-Aldrich).
4.8. Statistical Analysis
The data are presented as the mean ± SEM. Comparisons were performed using an ANOVA followed by a Bonferroni posttest by the Prism 4.0 statistical program (GraphPad Software, San Diego, CA). Survival analyses were performed using the Mantel-Cox log-rank (χ 2 “chi-squared”) test. Differences in survival were analyzed by the log-rank test. Values of p < 0.05 were considered statistically significant.
Acknowledgments
The authors thank Dr. Seema R. Patel for the critical review of the manuscript, Dr. Connie M. Arthur for helpful discussions, and Rubens Eduardo da Silva for the excellent assistance with animal handling and technical support. This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, Grant nos. 2007/02487-3, 2007/00840-8, and 2011/17611-7), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Grant nos. 557403/2008-1, 467646/2014-7), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Grant no. BEX 9320/13-0), and National Institutes of Health (NIH, AL101982, to Richard D. Cummings). Also, the research leading to these results has received support and funding from the Núcleo de Apoio à Pesquisa em Doenças Inflamatórias (NAPDIN, Grant no. 11.1.21625.01.0).
Abbreviations
- Gal-1:
Galectin-1
- Lgals1−/−:
Galectin-1 deficient mice
- WT:
Wild-type mice
- Th1 and Th2:
Helper T cell responses
- IFN:
Interferon
- IL:
Interleukin
- TNF-α:
Tumor necrosis factor-α
- GM-CSF:
Granulocyte macrophage colony-stimulating
- PGE2:
Prostaglandin E2
- Ptgs2:
Prostaglandin E synthase 2
- NO2−:
Nitrite
- NO:
Nitric oxide
- HIV:
Human Immunodeficiency Virus-1
- HTLV-1:
Human T Lymphotropic Virus-1
- BALF:
Bronchoalveolar Lavage Fluid.
Competing Interests
The authors declare no competing interests.
References
- 1.Allendoerfer R., Deepe G. S., Jr. Infection with Histoplasma capsulatum: host-fungus interface. Revista Iberoamericana de Micologia. 1998;15(4):256–260. [PubMed] [Google Scholar]
- 2.Antinori S. Histoplasma capsulatum: more widespread than previously thought. The American Journal of Tropical Medicine and Hygiene. 2014;90(6):982–983. doi: 10.4269/ajtmh.14-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown G. D., Denning D. W., Gow N. A. R., Levitz S. M., Netea M. G., White T. C. Hidden killers: human fungal infections. Science Translational Medicine. 2012;4(165) doi: 10.1126/scitranslmed.3004404.165rv13 [DOI] [PubMed] [Google Scholar]
- 4.Adenis A. A., Aznar C., Couppié P. Histoplasmosis in HIV-infected patients: a review of new developments and remaining gaps. Current Tropical Medicine Reports. 2014;1:119–128. doi: 10.1007/s40475-014-0017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deepe G. S., Jr., Gibbons R. S., Smulian A. G. Histoplasma capsulatum manifests preferential invasion of phagocytic subpopulations in murine lungs. Journal of Leukocyte Biology. 2008;84(3):669–678. doi: 10.1189/jlb.0308154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroetz D. N., Deepe G. S. The role of cytokines and chemokines in Histoplasma capsulatum infection. Cytokine. 2012;58(1):112–117. doi: 10.1016/j.cyto.2011.07.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allendoerfer R., Boivin G. P., Deepe G. S., Jr. Modulation of immune responses in murine pulmonary histoplasmosis. Journal of Infectious Diseases. 1997;175(4):905–914. doi: 10.1086/513989. [DOI] [PubMed] [Google Scholar]
- 8.Pereira P. A. T., Trindade B. C., Secatto A., et al. Celecoxib improves host defense through prostaglandin inhibition during histoplasma capsulatum infection. Mediators of Inflammation. 2013;2013:11. doi: 10.1155/2013/950981.950981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medeiros A. I., Sá-Nunes A., Soares E. G., Peres C. M., Silva C. L., Faccioli L. H. Blockade of endogenous leukotrienes exacerbates pulmonary histoplasmosis. Infection and Immunity. 2004;72(3):1637–1644. doi: 10.1128/iai.72.3.1637-1644.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lane T. E., Wu-Hsieh B. A., Howard D. H. Antihistoplasma effect of activated mouse splenic macrophages involves production of reactive nitrogen intermediates. Infection and Immunity. 1994;62(5):1940–1945. doi: 10.1128/iai.62.5.1940-1945.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lane T. E., Otero G. C., Wu-Hsieh B. A., Howard D. H. Expression of inducible nitric oxide synthase by stimulated macrophages correlates with their antihistoplasma activity. Infection and Immunity. 1994;62(4):1478–1479. doi: 10.1128/iai.62.4.1478-1479.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu-Hsieh B. A., Chen W., Lee H.-J. Nitric oxide synthase expression in macrophages of Histoplasma capsulatum-infected mice is associated with splenocyte apoptosis and unresponsiveness. Infection and Immunity. 1998;66(11):5520–5526. doi: 10.1128/iai.66.11.5520-5526.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu S.-Y., Yu J.-S., Liu F.-T., Miaw S.-C., Wu-Hsieh B. A. Galectin-3 negatively regulates dendritic cell production of IL-23/IL-17-axis cytokines in infection by Histoplasma capsulatum . The Journal of Immunology. 2013;190(7):3427–3437. doi: 10.4049/jimmunol.1202122. [DOI] [PubMed] [Google Scholar]
- 14.Rabinovich G. A., Gruppi A. Galectins as immunoregulators during infectious processes: from microbial invasion to the resolution of the disease. Parasite Immunology. 2005;27(4):103–114. doi: 10.1111/j.1365-3024.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- 15.Laderach D. J., Compagno D., Toscano M. A., et al. Dissecting the signal transduction pathways triggered by galectin-glycan interactions in physiological and pathological settings. IUBMB Life. 2010;62(1):1–13. doi: 10.1002/iub.281. [DOI] [PubMed] [Google Scholar]
- 16.Liu F.-T., Yang R.-Y., Hsu D. K. Galectins in acute and chronic inflammation. Annals of the New York Academy of Sciences. 2012;1253(1):80–91. doi: 10.1111/j.1749-6632.2011.06386.x. [DOI] [PubMed] [Google Scholar]
- 17.Thiemann S., Baum L. G. Galectins and immune responses—just how do they do those things they do? Annual Review of Immunology. 2016;34(1):243–264. doi: 10.1146/annurev-immunol-041015-055402. [DOI] [PubMed] [Google Scholar]
- 18.Wdowiak K., Spychalowicz W., Fajkis M., Wojnar J. Galectins in hematological malignancies-role, functions and potential therapeutic targets. Postępy Higieny i Medycyny Doświadczalnej. 2016;70:95–103. doi: 10.5604/17322693.1194808. [DOI] [PubMed] [Google Scholar]
- 19.Zúñiga E., Gruppi A., Hirabayashi J., Kasai K. I., Rabinovich G. A. Regulated expression and effect of galectin-1 on Trypanosoma cruzi-infected macrophages: modulation of microbicidal activity and survival. Infection and Immunity. 2001;69(11):6804–6812. doi: 10.1128/iai.69.11.6804-6812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ouellet M., Mercier S., Pelletier I., et al. Galectin-1 acts as a soluble host factor that promotes HIV-1 infectivity through stabilization of virus attachment to host cells. The Journal of Immunology. 2005;174(7):4120–4126. doi: 10.4049/jimmunol.174.7.4120. [DOI] [PubMed] [Google Scholar]
- 21.Toledo K. A., Fermino M. L., Del Cistia Andrade C., et al. Galectin-1 exerts inhibitory effects during DENV-1 infection. PLoS ONE. 2014;9(11, article e112474) doi: 10.1371/journal.pone.0112474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stowell S. R., Qian Y., Karmakar S., et al. Differential roles of galectin-1 and galectin-3 in regulating leukocyte viability and cytokine secretion. Journal of Immunology. 2008;180(5):3091–3102. doi: 10.4049/jimmunol.180.5.3091. [DOI] [PubMed] [Google Scholar]
- 23.Correa S. G., Sotomayor C. E., Aoki M. P., Maldonado C. A., Rabinovich G. A. Opposite effects of galectin-1 on alternative metabolic pathways of L-arginine in resident, inflammatory, and activated macrophages. Glycobiology. 2003;13(2):119–128. doi: 10.1093/glycob/cwg010. [DOI] [PubMed] [Google Scholar]
- 24.Norling L. V., Sampaio A. L. F., Cooper D., Perretti M. Inhibitory control of endothelial galectin-1 on in vitro and in vivo lymphocyte trafficking. The FASEB Journal. 2008;22(3):682–690. doi: 10.1096/fj.07-9268com. [DOI] [PubMed] [Google Scholar]
- 25.Poncini C. V., Ilarregui J. M., Batalla E. I., et al. Trypanosoma cruzi infection imparts a regulatory program in dendritic cells and T cells via galectin-1-dependent mechanisms. Journal of Immunology. 2015;195(7):3311–3324. doi: 10.4049/jimmunol.1403019. [DOI] [PubMed] [Google Scholar]
- 26.Dias-Baruffi M., Zhu H., Cho M., Karmakar S., McEver R. P., Cummings R. D. Dimeric galectin-1 induces surface exposure of phosphatidylserine and phagocytic recognition of leukocytes without inducing apoptosis. Journal of Biological Chemistry. 2003;278(42):41282–41293. doi: 10.1074/jbc.m306624200. [DOI] [PubMed] [Google Scholar]
- 27.Rabinovich G. A., Sotomayor C. E., Riera C. M., Bianco I., Correa S. G. Evidence of a role for galectin-1 in acute inflammation. European Journal of Immunology. 2000;30(5):1331–1339. doi: 10.1002/(sici)1521-4141(200005)30:5<1331::aid-immu1331>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 28.Liu F.-T., Patterson R. J., Wang J. L. Intracellular functions of galectins. Biochimica et Biophysica Acta (BBA)—General Subjects. 2002;1572(2-3):263–273. doi: 10.1016/s0304-4165(02)00313-6. [DOI] [PubMed] [Google Scholar]
- 29.Vladoiu M. C., Labrie M., St-Pierre Y. Intracellular galectins in cancer cells: potential new targets for therapy (review) International Journal of Oncology. 2014;44(4):1001–1014. doi: 10.3892/ijo.2014.2267. [DOI] [PubMed] [Google Scholar]
- 30.Kurita N., Brummer E., Yoshida S., Nishimura K., Miyaji M. Antifungal activity of murine polymorphonuclear neutrophils against histoplasma capsulatum. Medical Mycology. 1991;29(3):133–143. doi: 10.1080/02681219180000241. [DOI] [PubMed] [Google Scholar]
- 31.Allendoerfer R., Deepe G. S., Jr. Intrapulmonary response to Histoplasma capsulatum in gamma interferon knockout mice. Infection and Immunity. 1997;65(7):2564–2569. doi: 10.1128/iai.65.7.2564-2569.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deepe G. S., Jr., Gibbons R., Woodward E. Neutralization of endogenous granulocyte-macrophage colony-stimulating factor subverts the protective immune response to Histoplasma capsulatum . The Journal of Immunology. 1999;163(9):4985–4993. [PubMed] [Google Scholar]
- 33.Deepe G. S., Jr., Gibbons R. S. Interleukins 17 and 23 influence the host response to Histoplasma capsulatum. Journal of Infectious Diseases. 2009;200(1):142–151. doi: 10.1086/599333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou P., Sieve M. C., Bennett J., et al. IL-12 prevents mortality in mice infected with Histoplasma capsulatum through induction of IFN-γ . Journal of Immunology. 1995;155(2):785–795. [PubMed] [Google Scholar]
- 35.Wolf J. E., Massof S. E., Peters S. P. Alterations in murine macrophage arachidonic acid metabolism following ingestion of nonviable Histoplasma capsulatum . Infection and Immunity. 1992;60(7):2559–2564. doi: 10.1128/iai.60.7.2559-2564.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stowell S. R., Arthur C. M., Dias-Baruffi M., et al. Innate immune lectins kill bacteria expressing blood group antigen. Nature Medicine. 2010;16(3):295–301. doi: 10.1038/nm.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerliani J. P., Stowell S. R., Mascanfroni I. D., Arthur C. M., Cummings R. D., Rabinovich G. A. Expanding the universe of cytokines and pattern recognition receptors: galectins and glycans in innate immunity. Journal of Clinical Immunology. 2011;31(1):10–21. doi: 10.1007/s10875-010-9494-2. [DOI] [PubMed] [Google Scholar]
- 38.Sato S., St-Pierre C., Bhaumik P., Nieminen J. Galectins in innate immunity: dual functions of host soluble β-galactoside-binding lectins as damage-associated molecular patterns (DAMPs) and as receptors for pathogen-associated molecular patterns (PAMPs) Immunological Reviews. 2009;230(1):172–187. doi: 10.1111/j.1600-065x.2009.00790.x. [DOI] [PubMed] [Google Scholar]
- 39.Linden J. R., De Paepe M. E., Laforce-Nesbitt S. S., Bliss J. M. Galectin-3 plays an important role in protection against disseminated candidiasis. Medical Mycology. 2013;51(6):641–651. doi: 10.3109/13693786.2013.770607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fichorova R. N., Yamamoto H. S., Fashemi T., et al. Trichomonas vaginalis lipophosphoglycan exploits binding to galectin-1 and -3 to modulate epithelial immunity. Journal of Biological Chemistry. 2016;291(2):998–1013. doi: 10.1074/jbc.M115.651497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muglia C. I., Gobbi R. P., Smaldini P., et al. Inflammation controls sensitivity of human and mouse intestinal epithelial cells to galectin-1. Journal of Cellular Physiology. 2016;231(7):1575–1585. doi: 10.1002/jcp.25249. [DOI] [PubMed] [Google Scholar]
- 42.Camby I., Le Mercier M., Lefranc F., Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006;16(11):137R–157R. doi: 10.1093/glycob/cwl025. [DOI] [PubMed] [Google Scholar]
- 43.Vasta G. R. Roles of galectins in infection. Nature Reviews Microbiology. 2009;7(6):424–438. doi: 10.1038/nrmicro2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kohatsu L., Hsu D. K., Jegalian A. G., Liu F.-T., Baum L. G. Galectin-3 induces death of Candida species expressing specific β-1,2-linked mannans. Journal of Immunology. 2006;177(7):4718–4726. doi: 10.4049/jimmunol.177.7.4718. [DOI] [PubMed] [Google Scholar]
- 45.Benatar A. F., García G. A., Bua J., et al. Galectin-1 prevents infection and damage induced by Trypanosoma cruzi on cardiac cells. PLOS Neglected Tropical Diseases. 2015;9(10) doi: 10.1371/journal.pntd.0004148.e0004148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stowell S. R., Arthur C. M., McBride R., et al. Microbial glycan microarrays define key features of host-microbial interactions. Nature Chemical Biology. 2014;10(6):470–476. doi: 10.1038/nchembio.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gil C. D., Gullo C. E., Oliani S. M. Effect of exogenous galectin-1 on leukocyte migration: modulation of cytokine levels and adhesion molecules. International Journal of Clinical and Experimental Pathology. 2010;4(1):74–84. [PMC free article] [PubMed] [Google Scholar]
- 48.Kolaczkowska E., Kubes P. Neutrophil recruitment and function in health and inflammation. Nature Reviews Immunology. 2013;13(3):159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 49.Newman S. L., Gootee L., Gabay J. E. Human neutrophil-mediated fungistasis against Histoplasma capsulatum: localization of fungistatic activity to the azurophil granules. The Journal of Clinical Investigation. 1993;92(2):624–631. doi: 10.1172/jci116630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sá-Nunes A., Medeiros A. I., Sorgi C. A., et al. Gr-1+ cells play an essential role in an experimental model of disseminated histoplasmosis. Microbes and Infection. 2007;9(12-13):1393–1401. doi: 10.1016/j.micinf.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 51.Cooper D., Norling L. V., Perretti M. Novel insights into the inhibitory effects of Galectin-1 on neutrophil recruitment under flow. Journal of Leukocyte Biology. 2008;83(6):1459–1466. doi: 10.1189/jlb.1207831. [DOI] [PubMed] [Google Scholar]
- 52.Schmitz N., Kurrer M., Bachmann M. F., Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. Journal of Virology. 2005;79(10):6441–6448. doi: 10.1128/jvi.79.10.6441-6448.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faccioli L. H., Souza G. E. P., Cunha F. Q., Poole S., Ferreira S. H. Recombinant interleukin-1 and tumor necrosis factor induce neutrophil migration ‘in vivo’ by indirect mechanisms. Agents and Actions. 1990;30(3-4):344–349. doi: 10.1007/bf01966298. [DOI] [PubMed] [Google Scholar]
- 54.Romani L., Mencacci A., Cenci E., et al. Neutrophil production of IL-12 and IL-10 in candidiasis and efficacy of IL-12 therapy in neutropenic mice. The Journal of Immunology. 1997;158(11):5349–5356. [PubMed] [Google Scholar]
- 55.Zhong J., Huang W., Deng Q., et al. Inhibition of TREM-1 and Dectin-1 alleviates the severity of fungal keratitis by modulating innate immune responses. PLoS ONE. 2016;11(3) doi: 10.1371/journal.pone.0150114.e0150114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allendoerfer R., Deepe G. S., Jr. Regulation of infection with Histoplasma capsulatum by TNFR1 and -2. The Journal of Immunology. 2000;165(5):2657–2664. doi: 10.4049/jimmunol.165.5.2657. [DOI] [PubMed] [Google Scholar]
- 57.Deepe G. S., Jr. Immune response to early and late Histoplasma capsulatum infections. Current Opinion in Microbiology. 2000;3(4):359–362. doi: 10.1016/s1369-5274(00)00104-1. [DOI] [PubMed] [Google Scholar]
- 58.Rajasagi N. K., Suryawanshi A., Sehrawat S., et al. Galectin-1 reduces the severity of herpes simplex virus-induced ocular immunopathological lesions. Journal of Immunology. 2012;188(9):4631–4643. doi: 10.4049/jimmunol.1103063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Toscano M. A., Bianco G. A., Ilarregui J. M., et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nature Immunology. 2007;8(8):825–834. doi: 10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- 60.Floss D. M., Schröder J., Franke M., Scheller J. Insights into IL-23 biology: from structure to function. Cytokine & Growth Factor Reviews. 2015;26(5):569–578. doi: 10.1016/j.cytogfr.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 61.Wang J., Xia J., Zhang F., et al. Galectin-1-secreting neural stem cells elicit long-term neuroprotection against ischemic brain injury. Scientific Reports. 2015;5, article 9621 doi: 10.1038/srep09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pineda D., Ampurdanés C., Medina M. G., et al. Tissue plasminogen activator induces microglial inflammation via a noncatalytic molecular mechanism involving activation of mitogen-activated protein kinases and Akt signaling pathways and AnnexinA2 and Galectin-1 receptors. Glia. 2012;60(4):526–540. doi: 10.1002/glia.22284. [DOI] [PubMed] [Google Scholar]
- 63.Hempel S. L., Monick M. M., Hunninghake G. W. Lipopolysaccharide induces prostaglandin H synthase-2 protein and mRNA in human alveolar macrophages and blood monocytes. The Journal of Clinical Investigation. 1994;93(1):391–396. doi: 10.1172/jci116971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aronoff D. M., Lewis C., Serezani C. H., et al. E-prostanoid 3 receptor deletion improves pulmonary host defense and protects mice from death in severe Streptococcus pneumoniae infection. Journal of Immunology. 2009;183(4):2642–2649. doi: 10.4049/jimmunol.0900129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Serezani C. H., Chung J., Ballinger M. N., Moore B. B., Aronoff D. M., Peters-Golden M. Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. American Journal of Respiratory Cell and Molecular Biology. 2007;37(5):562–570. doi: 10.1165/rcmb.2007-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Domingo-Gonzalez R., Katz S., Serezani C. H., Moore T. A., LeVine A. M., Moore B. B. Prostaglandin E2-induced changes in alveolar macrophage scavenger receptor profiles differentially alter phagocytosis of Pseudomonas aeruginosa and Staphylococcus aureus post-bone marrow transplant. Journal of Immunology. 2013;190(11):5809–5817. doi: 10.4049/jimmunol.1203274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rodríguez M., Domingo E., Municio C., et al. Polarization of the innate immune response by prostaglandin E2: a puzzle of receptors and signals. Molecular Pharmacology. 2014;85(1):187–197. doi: 10.1124/mol.113.089573. [DOI] [PubMed] [Google Scholar]
- 68.Santucci L., Fiorucci S., Rubinstein N., et al. Galectin-1 suppresses experimental colitis in mice. Gastroenterology. 2003;124(5):1381–1394. doi: 10.1016/S0016-5085(03)00267-1. [DOI] [PubMed] [Google Scholar]
- 69.Ilarregui J. M., Croci D. O., Bianco G. A., et al. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nature Immunology. 2009;10(9):981–991. doi: 10.1038/ni.1772. [DOI] [PubMed] [Google Scholar]
- 70.Kuo P.-L., Hung J.-Y., Huang S.-K., et al. Lung cancer-derived galectin-1 mediates dendritic cell anergy through inhibitor of DNA binding 3/IL-10 signaling pathway. Journal of Immunology. 2011;186(3):1521–1530. doi: 10.4049/jimmunol.1002940. [DOI] [PubMed] [Google Scholar]
- 71.Fulcher J. A., Hashimi S. T., Levroney E. L., et al. Galectin-1-matured human monocyte-derived dendritic cells have enhanced migration through extracellular matrix. The Journal of Immunology. 2006;177(1):216–226. doi: 10.4049/jimmunol.177.1.216. [DOI] [PubMed] [Google Scholar]
- 72.Medeiros A., Peres-Buzalaf C., Fortino Verdan F., Serezani C. H. Prostaglandin E2 and the suppression of phagocyte innate immune responses in different organs. Mediators of Inflammation. 2012;2012:13. doi: 10.1155/2012/327568.327568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Serezani C. H., Perrela J. H., Russo M., Peters-Golden M., Jancar S. Leukotrienes are essential for the control of Leishmania amazonensis infection and contribute to strain variation in susceptibility. Journal of Immunology. 2006;177(5):3201–3208. doi: 10.4049/jimmunol.177.5.3201. [DOI] [PubMed] [Google Scholar]
- 74.Rezaie F., Salimi M., Ghahremani M. H., Vaziri B. Potential molecular targets in chemopreventative action of celecoxib: a proteomics analysis of J774.A1 macrophage-like cell line. Molecular BioSystems. 2011;7(4):1306–1311. doi: 10.1039/c0mb00201a. [DOI] [PubMed] [Google Scholar]
- 75.Hellmann J., Tang Y., Zhang M. J., et al. Atf3 negatively regulates Ptgs2/Cox2 expression during acute inflammation. Prostaglandins & Other Lipid Mediators. 2015;116-117:49–56. doi: 10.1016/j.prostaglandins.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hangai S., Ao T., Kimura Y., et al. PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(14):3844–3849. doi: 10.1073/pnas.1602023113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Duffin R., O'Connor R. A., Crittenden S., et al. Prostaglandin E2 constrains systemic inflammation through an innate lymphoid cell-IL-22 axis. Science. 2016;351(6279):1333–1338. doi: 10.1126/science.aad9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sá-Nunes A., Medeiros A. I., Nicolete R., et al. Efficacy of cell-free antigens in evaluating cell immunity and inducing protection in a murine model of histoplasmosis. Microbes and Infection. 2005;7(4):584–592. doi: 10.1016/j.micinf.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 79.Medeiros A. I., Silva C. L., Malheiro A., Maffei C. M. L., Faccioli L. H. Leukotrienes are involved in leukocyte recruitment induced by live Histoplasma capsulatum or by the β-glucan present in their cell wall. British Journal of Pharmacology. 1999;128(7):1529–1537. doi: 10.1038/sj.bjp.0702912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Corrêa B., Purchio A., Paula C. R., Gambale W., Shikanai-Yasuda M. A. Fluorescent method (fluorescein diacetate and ethidium bromide) to study the viability of Cryptococcus neoformans in liquor. Revista do Instituto de Medicina Tropical de Sao Paulo. 1990;32(1):46–50. [PubMed] [Google Scholar]
- 81.Cho M., Cummings R. D. Galectin-1, a β-galactoside-binding lectin in Chinese hamster ovary cells. II. Localization and biosynthesis. The Journal of Biological Chemistry. 1995;270(10):5207–5212. doi: 10.1074/jbc.270.10.5207. [DOI] [PubMed] [Google Scholar]
- 82.Whitney P. L., Powell J. T., Sanford G. L. Oxidation and chemical modification of lung β-galactoside-specific lectin. Biochemical Journal. 1986;238(3):683–689. doi: 10.1042/bj2380683. [DOI] [PMC free article] [PubMed] [Google Scholar]