

Abstract
Background
Gestational diabetes mellitus (GDM) is one of the most common medical complications of pregnancy, and has important health implications for mother and child. Changes in the fetoplacental vessels may predict those in the vasculature of the developing fetus, as these have been implicated in the pathogenesis of human GDM. This study aimed to determine the differences in the localization and expression level of VEGFA and VEGFR2 between placentas of women with GDM and placentas of normal pregnancies, which is the first step in elucidating the possible roles of VEGFA and VEGFR2 in the altered uteroplacental function resulting from maternal hyperglycaemia and ultimately in the manifestation of GDM.
Methods
The expressions of VEGFA and VEGFR2 mRNA and protein in 20 samples from each group were analyzed by real-time PCR, immunohistochemistry and Western blot. The placental blood barrier and angiogenesis were observed by the transmission electron microscopy (TEM) in10 GDM samples and ten controls.
Results
The expression levels of VEGFA and VEGFR2 mRNA and protein were significantly decreased in the GDM group (P < 0.05 or 0.01). Immunohistochemical analysis showed the reduced expression of VEGFA and VEGFR2 protein in GDM-affected placental tissues, and the degenerative alterations of the terminal villi vascular.
Conclusion
The expressions of VEGFA and VEGFR-2 mRNAs and protein were reduced in GDM-affected placental tissues, suggesting that maternal GDM affects the pathophysiological function of placentas.
Electronic supplementary material
The online version of this article (doi:10.1186/s12958-016-0191-8) contains supplementary material, which is available to authorized users.
Keywords: GDM, Placenta, Vascular endothelial growth factor, Receptors, Fetal development
Background
Gestational diabetes mellitus (GDM) is defined as any degree of glucose intolerance with onset or first recognition during pregnancy, regardless of whether or not diabetes persists after pregnancy [1–3]. Pregnancy is a diabetogenic state characterized by impaired insulin sensitivity, especially in the second trimester. The major factors contributing to GDM are the placental hormones, such as human placental lactogen, progesterone, cortisol, growth hormone and prolactin. These hormones cause the decreased phosphorylation of insulin receptor substrate-1 (IRS-1), resulting in profound insulin resistance [4–6]. As reported previously, IRS-1 decreases 50–60 % of insulin sensitivity and 50 % of beta cell function in a normal pregnancy [7], and to maintain euglycemia, the pancreas should compensate by increasing insulin secretion by 2–2.5 times. However, beta cell function deteriorates in GDM, particularly during the first phase of insulin secretion.
Gestational diabetes mellitus (GDM) is one of the most common medical complications of pregnancy, and has important health implications for mother and child. Mothers with GDM have an excess of hypertensive disorders during pregnancy, recurrence of gestational diabetes [8], post-partum diabetes and cardiovascular disease thereafter [9–11]. Pregnancies complicated by GDM are associated with several adverse outcomes in the offspring including macrosomia and a longer-term risk of the development of obesity and type 2 diabetes [12].
Fetoplacental vessels are found in chorionic villi bathed in maternal blood, and this close proximity permits efficient exchange of solutes and gases between the maternal and fetal circulations without intermingling of the two. The arrangement allows the development, growth, and remodeling of fetoplacental vessels to be matched to the fetal need while rendering them vulnerable to changes on both the maternal and fetal sides of the placenta. Any pathological alterations in maternal hemodynamics; maternal blood properties (such as hypoxia or hyperglycemia); or growth factors such as vascular endothelial growth factors (VEGFs), soluble fms-like tyrosine kinase-1 (sFlt1), soluble endoglin (sEng) and inflammatory mediators may directly influence the growth, maintenance, and function of fetoplacental vessels. Furthermore, changes in the fetoplacental vessels may predict those in the vasculature of the developing fetus, as these have been implicated in the pathogenesis of human GDM.
VEGF is a homodimeric disulfide-linked glycoprotein involved in both angiogenesis (growth of new blood vessels from existing ones) and vasculogenesis (de novo formation of blood vessels) [13, 14]. VEGF family members include VEGFA, (PlGF, VEGFB, VEGFC, and VEGFD) and two members that are not expressed in mammals (VEGFE, which is expressed in viruses, and VEGFF, which is found in snake venom) [15, 16]. VEGFs initiate cellular responses by interacting with tyrosine kinase receptors on the cell surface. Tyrosine kinase receptors are a specific type of protein kinase receptor that functions by phosphorylating the substrate to stimulate cellular responses. The VEGF receptor (VEGFR) consists of an extracellular domain, a single transmembrane spanning region, and an intracellular component containing a tyrosine kinase domain. There are three main VEGF-R subtypes: VEGFR1 (Flt-1), VEGFR2 (KDR/Flk-1), and VEGFR3 (Flt-4). VEGFA binds to VEGFR1 and VEGFR2; VEGFB, to VEGFR1; and VEGFC and VEGFD, to VEGFR2 and VEGFR3 [17]. The soluble form of VEGFR1 (sFlt1 or sVEGFR1) is a splice variant lacking the transmembrane and cytoplasmic domains. Therefore, it circulates and acts as a potent VEGF and PlGF antagonist by preventing them from reacting with their endogenous receptors [18, 19].
This study aimed to determine the differences in the localization and expression level of VEGFA and VEGFR2 between placentas of women with GDM and placentas of normal pregnancies, which is the first step in elucidating the possible roles of VEGFA and VEGFR2 in the altered uteroplacental function resulting from maternal hyperglycaemia and ultimately in the manifestation of GDM.
Methods
Subjects
Women with a history of pregestational diabetes and those with a non-singleton index pregnancy were excluded. A 75-g oral glucose tolerance test (OGTT) was performed with plasma glucose measurement fasting and at 1 and 2 h for women at 24–28 weeks of gestation who were not previously diagnosed with overt diabetes. The OGTT should be performed in the morning after an overnight fast of at least 8 h. The diagnosis of GDM is made when any of the following plasma glucose values are exceeded, based on the American Diabetes Association [20]: (1) fasting ≥ 5.1 mmol/l (92 mg/dl), but <7.0 mmol(126 mg/dl); (2) 1 h ≥ 10.0 mmol/l (180 mg/dl); (3) 2 h ≥ 8.5 mmol/l (153 mg/dl). After GDM was diagnosed, those GDM women were asked diet control to meet the satisfying range of fasting blood glucose (3.3 to 5.6 mmol/L). These patients who were consistently not following the strict management of GDM, without medical therapy initiated. Fourteen patients from this group (n = 20) were obese [BMI > 28, BMI = Body weight (kg)/Body surface area2 (m2)] and four were hypertensive. The clinical data on maternal age and weight, number of gestational weeks, mode of delivery, BMI and weight of placenta were summarized in Table 1. Twenty women with normal pregnancies matched with GDM women for number of gestational weeks, maternal age and mode of delivery were recruited as the control group (n = 20). Women in each group provided their placental samples at delivery with the signed informed consent. The study procedure was approved by the ethics committee of the First Affiliated Hospital of Nanjing Medical University (2013-SR-018, 2013) Additional file 1: Table S1.
Table 1.
Clinical characteristics of GDM women and the control group. Results were expressed as the mean ± SD or number (percentage)
| Characteristics | Control group (n = 20) | GDM group (n = 20) | P value |
|---|---|---|---|
| Gestation, weeks | 39.41 ± 0.96 | 39.00 ± 0.90 | 0.34a |
| Maternal age, years | 28.4 ± 5.5 | 31.4 ± 6.7 | 0.13a |
| Maternal BMI, kg/m2 | 24.27 ± 2.18 | 28.23 ± 2.94 | 0.02a* |
| Mode of delivery | Cesarean | Cesarean | - |
| Sex of newborn | 0.53b | ||
| Male | 9(45) | 12(60) | |
| Female | 11(55) | 8(40) | |
| Birth weight of newborn, g | 3621 ± 380 | 3744 ± 537 | 0.29a |
| Placental weights, g | 734 ± 31.5 | 752 ± 33.2 | 0.11a |
BMI body weight (kg)/body surface area2 (m2). aStudent’s t test.; b x 2 test. *P < 0.05
Placental sample collection
In this study, all the subjects underwent delivery by cesarean section due to various reasons such as anxiety and tension, a burning desire of cesarean section and excessive afraid of pain,, other than those related to pathological procedures, to avoid the potential affects of the delivery procedure or other pathological factors on the expression of VEGF system. The placental specimens were weighed and performed on the basis of obstetric indications. The tissue specimen was dissected from the placental subchorial zone corresponding to the umbilical cord insertion (approximately 5 cm away from the site of cord insertion), while avoiding areas of infarction and hematomas. Placentas with abnormal umbilical cord insertions such as velamentous cord insertion were excluded from the analysis. Tissue fragments from the placenta were cut longitudinally from the maternal side to the fetal side. Placental tissues were divided into three parts, maternal, middle, and fetal, as described by Sood et al. [21]. The middle part, consisting of homogeneous villous tissues, was collected and placed in ice-cold phosphate-buffered saline (PBS), cleaned of blood, and immediately cut into four 1 cm × 1 cm × 1 cm fragments, which were frozen and stored at –80 °C for RNA analysis.
TEM examination
For the ultrastructural examination, ten women in each group were randomly selected to provide their placental samples at delivery (Additional file 1: Table S1). Until delivery, eight GDM women (8/10) kept in the A1 class (fasting glucose less than 5.8 mmol/L, postprandial blood glucose less than 6.7 mmol/L) after good diet control. Two GDM women (2/10) were classed as A2 (fasting blood glucose higher than or equal to 5.8 mmol/L, postprandial blood glucose higher than or equal to 6.7 mmol/L) because of their poor diet control. The levels of fasting blood glucose and HbA1c in those GDM women with good diet control in the last weeks of gestation were kept in the range of 5.1 to 5.6 mmol/L. However, the level of fasting blood glucose (6.1 and 11.3 mmol/L) and the level of HbA1c (10.2 and 10.3 mmol/L) in two GDM women with poor diet control were higher than the satisfactory criteria. Women with normal pregnancies matched with GDM women for number of gestational weeks, maternal age and mode of delivery were recruited as control (Additional file 1: Table S1). Three villous tissue blocks per placental sample were prepared for the ultrastructural examination using TEM. The samples were fixed in 2.5 % glutaraldehyde in cacodylate buffer, and stored in cacodylate buffer with 0.05 M saccharose (pH 7.2) at 4 °C until processing. The villous tissues were then post-fixed in 1 % OsO4 for 2 h at 4 °C, routinely processed in a graded series of acetone, then infiltrated with acetone-araldite and embedded in araldite. For orientation, semithin sections (thickness, 1 μm) were stained with thionine. Ultrathin sections (thickness, 80 nm) were treated (double contrast) with uranyl acetate (25 min) and 8 % lead nitrate (5 min) and then systematically examined in a JEM-1010 electron microscope (JEOL Ltd).
Terminal villi were evaluated with respect to the placental blood barrier (thickness of the vasculo-syncytial membrane, thickness of the syncytiotrophoblast (ST) basal membrane (BM), and thickness of the endothelial BM). For each tissue block, five microscopic fields were randomly selected and systematically investigated at 5000×, 12,000×, and 25,000× magnification for quantitative analysis. Thus, a total of 15 random fields were recorded and analyzed per placenta to minimize individual differences. The following three measurements were performed for each field: thickness of the vasculo-syncytial membrane from the intervillous space to the fetal vessels, perpendicular to the BM, at 5000× magnification; microvillous density per 10 μm of length at 12 000× magnification; and thickness of the ST or endothelial BM was measured at 25,000× magnification. Images were analyzed using the TEM Image Platform (Olympus) to perform random measurements. The two operators who carried out the microscopic analyses were blinded to the placental group until the end of the study.
Immunohistochemistry
Twenty tissue blocks in each group were sectioned and fixed to slides with the placenta and stained together. VEGF-A immunostaining: Slides were de-waxed, rehydrated, antigen retrieved and bathed in hydrogen peroxide for 30 mins. Blocking was done with Normal Donkey Serum (NDS). Rabbit anti-VEGFA antibody (VEGF (ab105219) (1:50) (500 μg/ml) (Abcam) and DAR-555 (1:1000) were used as primary and secondary antibody respectively.
VEGFR2 Immunostaining: Slides were de-waxed, rehydrated, antigen retrieved and bathed in hydrogen peroxide for 30 mins. NDS was employed to block non-specific antibody binding. Rabbit anti-VEGFR2 antibody (55B11) (1:200) (100 μg/ml) (Cell Signaling) and DAR-555 (1:1000) were used as primary and secondary antibody respectively.
RNA extraction and cDNA preparation
Total RNA was extracted from twenty placenta tissues from each group using Trizol Reagent (Invitrogen, Carlsbad, CA). RNA was treated with DNase using TURBO DNA-free kit (Ambion, Austin, TX), and purified with an RNeasy mini kit (Qiagen Inc., Valencia, CA). Spectrophotometric analysis and gel electrophoresis were used to determine the yield, purity, and integrity, and to ensure the lack of genomic DNA contamination in the samples. cDNA was prepared from total RNA using a high-capacity cDNA reverse transcriptase kit (Applied Biosystems, Foster City, CA).
First-strand synthesis was performed on 2 μg of total RNA using Superscript II/III ribonuclease H-reverse transcriptase (Invitrogen, Australia).
Real-time PCR
The mRNA expression of VEGF-A and VEGFR-2 in the placental samples obtained from the GDM and control pregnancies were quantified in an ABI Prism 7700 (Perkin-Elmer/Applied Biosystems) using prevalidated Assays on Demand (consisting of a 20× mix of unlabeled HLX1-PCR primers and FAM-dye labeled TaqMan MGB probe; HLX1 Assays on Demand, catalog no. 4331182; Applied Biosystems). Gene expression was quantified as the second step in a two-step RT-PCR protocol according to the manufacturer’s instructions. In brief, the 20-μL PCR reaction mix contained TaqMan Universal PCR master mix, 1× Assays on Demand gene expression assay mix, and 1 μL of placental cDNA. The sample was amplified for 40 cycles, including a denaturation step at 95 °C for 15 s and an annealing/extension step at 60 °C for 30 s. The oligonucleotide primers used for the amplification of the gene VEGF-A and VEGFR-2 were as follows: VEGF-A forward primer, 5′-TGCGGATCAAACCTCACCAA-3′ and VEGF-A reverse primer, 5′-TGTCACATACGC TCCAGGACTT-3′; VEGFR-2 forward primer, 5′-GTGTCAGAATCCCTGCGAAGTA-3′ and VEGFR-2 reverse primer, 5′-GAAATGGGATTGGTAAGGATGA-3′. Actin was used as a housekeeping gene. The primers were designed using Primer Express 1.5 software (Applied Biosystems). A mixed sample was used as the sample of quality control (QC). In every assay, the QC sample was added to two wells in the same 96-well plate. The average Ct of actin was calculated and normalized to test the variation within a assay. The relative value of target gene in the QC sample was 2(Ct2-Ct1), which was used to correct the variation between assays. The relative quantitation of VEGF-A and VEGFR-2 expression normalized to actin was calculated according to the 2–ΔΔT method of Livak and Schmittgen [22] using a term control as a calibrator (ABI Prism 7700 sequence detection system, User Bulletin no. 2, 2001).
Western immunoblotting
Twenty samples from each group were tested as following protocol. Total protein was extracted from 20 mg of snap-frozen placental tissue in 200 μL of RIPA lysis buffer containing 50 mmol/L of Tris-HCl (pH 7.4), 1 % Triton X-100, 1 % sodium deoxycholate, 150 mmol/L NaCl, 0.1 % sodium dodecyl sulfate (SDS), sodium orthovanadate, sodium fluoride, ethylenediaminetetraacetic acid (EDTA), and leupeptin using an Ultra-Turrax (Ika-Labortechnik). The homogenized samples were centrifuged at 10,000 g for 5 min at 4 °C to sediment any insoluble material. The protein concentration of the supernatant was determined using an Enhanced BCA protein assay kit (Beyotime, China) with bovine serum albumin (Sigma Aldrich, Australia) as the standard. Approximately 25 μg of protein per lane was fractionated using 10 % SDS-polyacrylamide gel electrophoresis (PAGE). The proteins were electrophoretically transferred to nitrocellulose membranes and blocked with 5 % nonfat milk in Tris-buffered saline (pH 7.4). Purified rabbit polyclonal VEGFA (1/200; ab105219; VEGFA-121, 165, and 189; Abcam) and rabbit monoclonal VEGFR2 (1/1000; 55B11; Cell Signaling) were used as the primary antibodies. Antibody binding was visualized using peroxidase-conjugated rabbit anti-mouse (Zymed, Mulgrave, Australia) secondary antibody, and analyzed by autoradiography using Fluorchem™ 5500 (Amersham Biotech, Shanghai, China).
The total protein in each well was detected by Coomassie blue staining to ensure constant protein load. A mixed sample as the QC sample was added to two wells in every plate from different assay. Tublin was used as a housekeeping protein. The densitometry of tublin was determined, and then the relative value of target protein in the QC sample was normalized. The variation within a assay was evaluated by two relative values, while the variation between assays was also corrected by the mean relative value. The expressions of VEGFA and VEGFR2 protein were semiquantitatively determined using Scanning Densitometry (Image Quant, Australia).
Statistical analysis
Data were expressed as the mean and standard deviation (SD). Statistical analyses were performed using SPSS software (Statistical Analysis System, version 17.0 for Windows). Statistical differences between the two groups were analyzed using Student’s t test for those data in the normal distribution. Statistical significance was set at p < 0.05.
Results
Summary of the clinical features
As shown in Table 1, the GDM group (n = 20) had a significantly higher mean BMI than the control group (28.23 ± 2.94 kg/m2 versus 24.27 ± 2.18 kg/m2, P = 0.02). Other parameters such as gestation, maternal age, mode of delivery, sex and birth weight of the newborn, and placental weight did not significantly differ between the groups.
TEM analysis
The placenta barrier was composed of ST, endothelium and the space between them. ST was a continuous syncytial layer with multiple nucleus and numerous apical microvilli, while CT had a single large nucleu and scattered distributed beneath the syncytium so that could not be seen in all the visual fields, which is analogous in both groups. BM of ST was a continuous, uniform, and thin basal lamina where collagen fibrils occasionally deposited, which was same to the BM of fetal endothelium. The stroma was the connective tissue core of the chorionic villous space separating ST from the capillary endothelium BM, and contained different kinds of stromal cells and bundles of collagen fibrils.
The placental blood barrier and angiogenesis were specially observed by TEM analysis. The vasculo-syncytial membrane (VSM) and BM of ST were significantly higher (P < 0.05) in the GDM group (6746.15 ± 1270.22 nm and 1077.49 ± 194.39 nm, respectively; Fig. 1a and d; n = 10) than those in the control group (4591.34 ± 1178.60 nm and 707.54 ± 256.56 nm, respectively; Fig. 1b and e; n = 10). The density of ST apical microvilli per unit surface area in the GDM group (44.36 ± 21.95 per 10 μm) was significantly lower than that in the control group (77.13 ± 20.82 per 10 μm; P < 0.05), as shown in Fig. 1g–i.
Fig. 1.
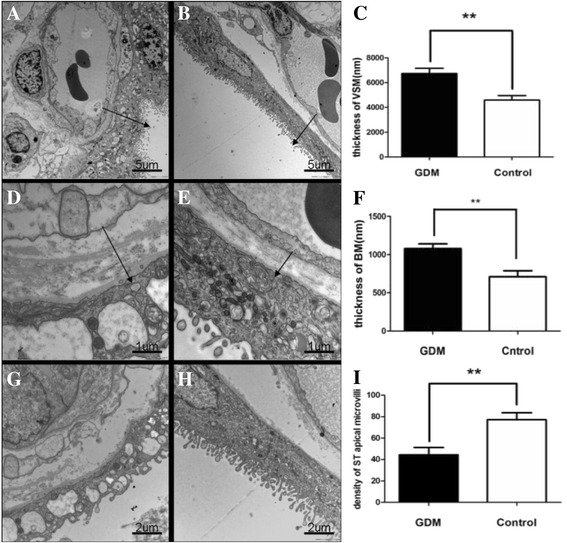
Comparison of semiquantitative parameters of placental ultrastructures between the GDM group (n = 20) and the control group (n = 20). a-b. The terminal villi of GDM (a) and control (b) placenta. c Placental barrier thickness (arrow) of the GDM group (6746.15 ± 1270.22 nm, n =10) was significantly thicker than that of the control group (4591.34 ± 1178.60 nm, n = 10; P < 0.05), Bar = 5um. d-e The terminal villi of GDM (d) and control (e) placenta. f. The BM of ST in the GDM group (1077.49 ± 194.39 nm nm, n = 10) was thicker than that in the control group (707.54 ± 256.56 nm, n = 10; P < 0.05), Bar = 1um. g-h The density of ST apical microvilli in GDM (g) and control (h) placenta. i The density of ST apical microvilli in the GDM group (44.36 ± 21.95 per 10um, n = 10) was significantly lower than that in the control group (77.13 ± 20.82 per 10um, n =10; P < 0.05), and even microvilli-free in some areas, Bar =2um
Localization and distribution of VEGFA and VEGFR2
IHC was used to investigate the cellular distribution of VEGFA and VEGFR2 proteins in GDM-affected (n = 20) and control (n = 20) placental tissues. Figure 2a and c showed the representative images of VEGFA immunoreactivity in the ST layer of term control tissues (Fig. 2a) and term GDM tissues (Fig. 2c). Qualitative assessment of the immunoreactivity revealed that the VEGFA protein level was lower in the placentas of the GDM group than in those of the control group but with no discernable difference in protein localization. Figure 2b and d showed the localization and distribution of the VEGFR2 protein in the placental tissues. Qualitative analysis revealed weaker staining in the endothelial cells of fetal vessels in the placentas of the GDM group (Fig. 2d) than that in the control group (Fig. 2b), but no discernable difference in the localization of VEGFR2 was noted.
Fig. 2.
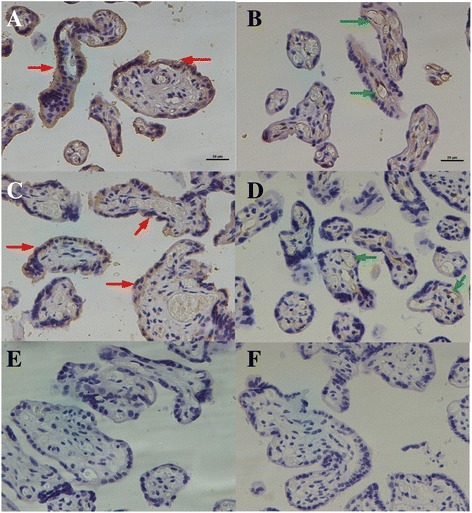
Immunohistochemical staining to study the cellular localization of VEGFA and VEGFR2 proteins in the GDM and control placentas. Representative controls (a, c and e) and GDM placentas (b, d and f) were stained with rabbit monoclonal antibodies to VEGFA (a and b) and VEGFR2(c and d). e and f Omission of the primary antibody as control. Syncytiotrophoblasts (red arrows) and fetal vessels (green arrows). Scale bars: 20 μm
VEGFA and VEGFR2 mRNA expression
Figure 3 illustrated the expressions of VEGFA and VEGFR2 mRNAs. Relative to the level of the Actin housekeeping gene, the VEGFA mRNA level was significantly decreased in the GDM group (n = 20) when compared with the control group (n = 20) (1.23 ± 0.53 versus 4.19 ± 2.61, P < 0.05; Fig. 3a). Similarly, the relative expression of VEGFR2 mRNA was significantly reduced in the GDM group when compared with control group (1.66 ± 0.33 versus 3.64 ± 0.95, P < 0.01; Fig. 3b).
Fig. 3.
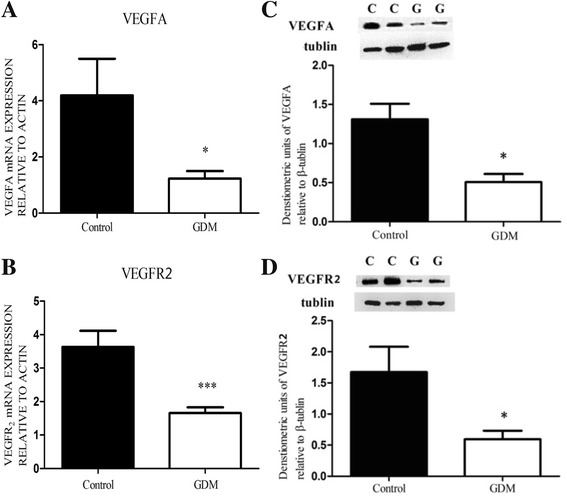
Expressions of VEGFA and VEGFR2 mRNAs and proteins in GDM (n = 20) and control (n = 20) placental tissues by real-time PCR and Western immunoblotting. Relative quantification of VEGFA (a) and VEGFR2 (b) mRNA expression normalized to the expression of ACTIN in all the samples. Data were analyzed according to the 2–ΔΔCT method. A representative immunoblots for VEGFA (c) and VEGFR2 (d) were shown. Immumoblots representing β-tubulin protein (middle panels) showed the loading equally total protein (25 μg). Semi-quantitative analyses of VEGFA and VEGFR2 immunoreactive proteins were performed relative to β-tubulin (bottom panels). The Y-axis represents the expression levels of VEGFA and VEGFR2 related to actin or tubulin. *P < 0.05, **P < 0.01, Student’s t test
Protein expression of VEGFA and VEGFR2
Figure 3 also showed the representative immunoblots of VEGFA and VEGFR2 proteins in the GDM-affected (n = 20) and control placentas (n = 20). Representative immunoblots of the 51-kDa housekeeping protein β-tubulin was used to illustrate the protein load of all samples. The 27-kDa immunoreactive VEGFA protein was observed in both groups. Semi-quantitative densitometry showed the decreased expression of VEGFA protein in the GDM group when compared with the control group (0.51 ± 0.21 versus 1.31 ± 0.40, P < 0.05; Fig. 3c). Figure 3d showed a 210-kDa protein of VEGFR2 in the GDM and control samples. Densitometry also showed the decreased VEGFR2 protein expression in the GDM group when compared with the control group (0.59 ± 0.49 versus 1.60 ± 1.47, P < 0.05).
Discussion
The primary origin of fetal derangements in gestational diabetic mothers is fetal hyperglycaemia resulting from maternal hyperglycaemia. This hyperglycaemia may result in the metabolic and hormonal change in the fetus. Once the fetal pancreas commences to produce and secrete insulin in the second trimester [23], fetal hyperglycaemia will result in fetal hyperinsulinemia and stimulation of fetal metabolism. Lower fetal oxygen content associated by higher lactate concentrations may certainly reflect enhanced fetal metabolism as a result of hyperglycaemia and hyperinsulinemia [24]. Consequently, the improvement of fetal oxygen demands will aggravate the chronic fetal hypoxia [25]. The hyperglycemia of GDM develops during pregnancy and clinically manifests only in the late second trimester and thus may have an impact on placental processes occurring in later stages of pregnancy, such as angiogenesis and microvascular remodeling. VEGF/VEGFR-2, which was selected as representative factors related to the placental blood barrier and angiogenesis in this study, is important in promoting vascular endothelial cell growth and in increasing the number of vessels and capillaries [22], thereby ensuring the adequate supply of nutrients to the fetus, through classical feedback mechanism. We found that the expressions of VEGFA and VEGFR2 was significantly lower in the placentas of women with GDM although we did not show the relationship between above expressions and the increased glucose levels or abnormal levels in their OGTTs due to the limited sample size. There is consistent evidence that the concentration of VEGF in GDM maternal and cord plasma was decreased [26]. VEGF mRNA expression and protein production are oxygen-dependent, which are known to be up-regulated by hypoxia [27]. Just because of the strong proangiogenic potency of hypoxia through regulating multiple steps of vascular growth [28], chronic fetal hypoxia as the consequence of maternal diabetes may thus stimulate placental vasculogenesis and angiogenesis by increasing the growth factors expression in the placenta and fetus. Parallel to its regulation by oxygen, placental VEGF is at a high level in the first trimester when oxygen levels are low and decline thereafter towards term of gestation [29–31], moreover the extent of hyperglycemia other than hypoxia may also contribute and modify its effect [32]. Thus, the condition of hyperglycemia induces a state of mild and persistent ischemia and hypoxia with subsequent increase of hypercapillarization. In contrast, high blood glucose levels especially poorly controlled in GDM trigger severe hypoxia/ischemia, with inhibition of binding VEGF/VEGFR-2 and consequent reduction of hypercapillarization, and therefore fetal hypoxia as a result of maternal diabetes does not stimulate the expression of VEGF in the third trimester [33]. The complex process of villous development and maturity might be also influenced by the maternal and fetal oxidative and other angiogenetic milieu, which will result in endothelial dysfunction and oxidative stress [34]. As the placenta is one of major sources of VEGF during pregnancy, our findings suggested that the reduced expression of VEGF in placenta in the GDM group may contribute to the pathogenic vascular defects which could be observed on the histological examination. Two other studies have also reported that the placental expression and fetal cord levels in GDM are significantly lower than normal [32,35].
These data were confirmed by immunolocalization of VEGF and VEGFR-2. The distribution of VEGF was observed in the placentas of women with GDM, and no VEGF was detected in the placental cellular compartments, which suggested a decrease in VEGF production. VEGF was always detected in the in the ST layer of term control tissues and term GDM tissues [32], in women with gestational diabetes there was a significant decrease in VEGF expression profile when compared to normoglycemic women. The target cells for VEGF in the chorionic villi were determined based on the immunolocalization of VEGFR-2, which expression was particularly low in the placental capillary endothelial cells of gestational diabetes, in contrast to normoglycemic women. The binding between VEGF and VEGFR-2 triggers a signaling pathway that negatively regulates angiogenesis.
Our finding showed that distinct alternations of ultrastructure in GDM placenta comparing with control placenta. The significant differences in ultrastructure may be summarized as the thickening of the vasculo-syncytial membrane (VSM) and BM of ST, a decreased number of ST apical microvilli per unit surface area in the GDM group. These changes related to the reduced VEGFA and VEGFR2 expressions may have adversely affected the transport efficiency of the placental vasculature by the decreased transport of oxygen, nutrients, and waste across the placenta [36]. To explore the comparable and inchoate changes in the VEGFA and VEGFR2 expression and placental ultrastructure, we excluded those cases with adverse pregnancy outcomes or pathological placental tissuess in this study. Our results suggested that this effect may be a response to the placental hypoxia induced by hyperglycemia, which might have potential affect on the placental and fetal development and growth during pregnancy.
Conclusion
This observational study revealed the reduced expressions of VEGFA and VEGFR2 in the placental tissues obtained from women with GDM. It should be noticed for both clinicians and GDM women that the reduced VEGF/ VEGFR2 may affect the placental blood barrier and angiogenesis, and consequent placental function, in the pregnancy with GDM.
Acknowledgements
We thank Mr. Gao Chao who helped us in molecular experiments, and thank Liling Zhou and Qin Lu for their help in TEM work and statistics.
Funding
This work was supported by grants from the China 973 Program (2012CB944703), the project of State Key Laboratory of Reproductive Medicine, Nanjing Medical University (SKLRM-KF-1110), and the China Jiangsu Science & Technology Department Program (BM2013058, BL2012009) and PAPD project of Jiangsu Higher Education Institutions.
Availability of data and materials
Not applicable.
Authors’ contributions
MQ and SL finished the experiments, wrote the first draft of manuscript. LX, MY and XW collected samples, participated discussion. CY and LJ are teachers of MQ and SL. CY and LJ designed the project, reviewed experiment and data, revised the manuscript. XH is a teacher of SL. XH participated in design, sample collection and statistics. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors have reviewed the final version of manuscript, and approved it for publication.
Ethics approval and consent to participate
The study procedure was approved by the ethics committee of the First Affiliated Hospital of Nanjing Medical University (2013-SR-018, 2013). Women in each group provided their placental samples at delivery with the signed informed consent. See the supplemental data.
Abbreviations
- BM
Basal membrane
- BMI
Body mass index
- GDM
Gestational diabetes mellitus
- HbA1c
Glycosylated hemoglobin A1c
- IRS-1
Insulin receptor substrate-1
- NDS
Normal donkey serum
- OGTT
Oral glucose tolerance test
- PBS
Phosphate-buffered saline
- sEng
soluble endoglin
- sFlt1
soluble fms-like tyrosine kinase-1
- ST
Syncytiotrophoblast
- TEM
Transmission electron microscopy
- VEGF
Vascular endothelial growth factor
- VEGFA
Vascular endothelial growth factor A
- VEGFR
Vascular endothelial growth factor receptor
- VEGFR2
Vascular endothelial growth factor receptor-2
- VSM
Vasculo-syncytial membrane
Additional file
The detailed clinical information of 10 cases for TEM exam from each group. (DOCX 15 kb)
Contributor Information
Qian Meng, Email: lygmq6326@163.com.
Li Shao, Email: shao-li-28@163.com.
Xiucui Luo, Email: lxc8816@sina.com.
Yingping Mu, Email: lygmyp@sina.com.
Wen Xu, Email: xuwenyankao2010518@126.com.
Li Gao, Email: gaoli_gao@126.com.
Haoqin Xu, Phone: 86-25-68302608, Email: 1325825613@qq.com.
Yugui Cui, Email: cuiygnj@njmu.edu.cn.
References
- 1.Berger H, Crane J, Farine D. SOGC clinical practice guidelines. Screening for gestational diabetes mellitus. J Obstet Gynaecol Can. 2002;24:894–912. doi: 10.1016/S1701-2163(16)31047-7. [DOI] [PubMed] [Google Scholar]
- 2.Brody SC, Harris R, Lohr K. Screening for gestational diabetes mellitus: a summary of the evidence for the US preventive services task force. Obstet Gynecol. 2003;101:380–92. doi: 10.1016/s0029-7844(02)03057-0. [DOI] [PubMed] [Google Scholar]
- 3.American Diabetes Association Gestational diabetes mellitus. Diabetes Care. 2003;27:S88–90. doi: 10.2337/diacare.27.2007.s88. [DOI] [PubMed] [Google Scholar]
- 4.FriedmanJE IT, Shao J, Huston L, Highman T, Catalano P. Impaired glucose transport and insulin receptor tyrosine phosphorylation in skeletal muscle from obese women with gestational diabetes. Diabetes. 1999;48:1807–14. doi: 10.2337/diabetes.48.9.1807. [DOI] [PubMed] [Google Scholar]
- 5.Ryan EA, Enns L. Role of gestational hormones in the induction of insulin resistance. J Clin Endocrinol Metab. 1988;67:341–7. doi: 10.1210/jcem-67-2-341. [DOI] [PubMed] [Google Scholar]
- 6.Kuhl C. Etiology and pathogenesis of gestational diabetes. Diabetes Care. 1998;21:B19–26. doi: 10.2337/diacare.21.3.C19. [DOI] [PubMed] [Google Scholar]
- 7.Cianni GD, Miccoli R, Volpe L, Lencioni C, Del Prato S. Intermediate metabolism in normal pregnancy and in gestational diabetes. Diabetes Metab Res Rev. 2003;19:259–70. doi: 10.1002/dmrr.390. [DOI] [PubMed] [Google Scholar]
- 8.Getahun D, Nath C, Ananth C, Chavez M, Smulian J. Gestational diabetes in the United States: temporal trends 1989 through. Am J Obstet Gynecol. 2004;2008:198–525. doi: 10.1016/j.ajog.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 9.Bellamy L, Casas J, Hingorani A, Williams D. Type 2 diabetes mellitus after gestational diabetes: a systematic review and meta-analysis. Lancet. 2009;373:1773–9. doi: 10.1016/S0140-6736(09)60731-5. [DOI] [PubMed] [Google Scholar]
- 10.Rivero K, Portal V, Vieria M, Behle I. Prevalence of the impaired glucose metabolism and its association with risk factors for coronary artery disease in women with gestational diabetes. Diabetes Res Clin Pract. 2008;79:433–7. doi: 10.1016/j.diabres.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 11.Heitritter S, Solomon C, Mitchell G, Skali-Ounis N, Seely E. Subclinical inflammation and vascular dysfunction in women with previous gestational diabetes mellitus. J Clin Endocrinol Metab. 2005;90:3983–8. doi: 10.1210/jc.2004-2494. [DOI] [PubMed] [Google Scholar]
- 12.Reece EA. The fetal and maternal consequences of gestational diabetes mellitus. J Matern Fetal Neonatal Med. 2010;23:199–203. doi: 10.3109/14767050903550659. [DOI] [PubMed] [Google Scholar]
- 13.Connolly DT. Vascular permeability factor: a unique regulator of blood vessel function. J Cell Biochem. 1991;47(3):219–23. doi: 10.1002/jcb.240470306. [DOI] [PubMed] [Google Scholar]
- 14.Nagy JA, Dvorak AM, Dvorak HF. VEGF-A(164/165) and PlGF: roles in angiogenesis and arteriogenesis. Trends Cardiovasc Med. 2003;13(5):169–75. doi: 10.1016/S1050-1738(03)00056-2. [DOI] [PubMed] [Google Scholar]
- 15.Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312(5):549–60. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin Sci (Lond) 2005;109(3):227–41. doi: 10.1042/CS20040370. [DOI] [PubMed] [Google Scholar]
- 17.Yamazaki Y, Morita T. Molecular and functional diversity of vascular endothelial growth factors. Mol Divers. 2006;10:515–27. doi: 10.1007/s11030-006-9027-3. [DOI] [PubMed] [Google Scholar]
- 18.Kendall RL, Wang G, Thomas KA. Identification of a natural soluble form of the vascular endothelial growth factor receptor, FLT-1, and its heterodimerization with KDR. Biochem Biophys Res Commun. 1996;226(2):324–8. doi: 10.1006/bbrc.1996.1355. [DOI] [PubMed] [Google Scholar]
- 19.Shibuya M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct Funct. 2001;26(1):25–35. doi: 10.1247/csf.26.25. [DOI] [PubMed] [Google Scholar]
- 20.American Diabetes Association. Standards of Medical Care in Diabetes-2011. Diab Care. 2010;34(1):S11-S61. [DOI] [PMC free article] [PubMed]
- 21.Sood R, Zehnder JL, Druzin ML, Brown PO. Gene expression patterns in human placenta. Proc Natl Acad Sci U S A. 2006;103:5478–83. doi: 10.1073/pnas.0508035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrara N, Gerber HP, Lecouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 23.Jeon J, Correa-Medina M, Ricordi C, Edlund H, Diez JA. Endocrine cell clustering during human pancreas development. J Histochem Cytochem. 2009;57(9):811–24. doi: 10.1369/jhc.2009.953307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taricco E, Radaelli T, Rossi G, de Santis Nobile MS, Bulfamante GP, Avagliano L, Cetin I. Effects of gestational diabetes on fetal oxygen and glucose levels in vivo. BJOG. 2009;116(13):1729–35. doi: 10.1111/j.1471-0528.2009.02341.x. [DOI] [PubMed] [Google Scholar]
- 25.Escobar J, Teramo K, Stefanovic V, Andersson S, Asensi MA, Arduini A, Cubells E, Sastre J, Vento M. Amniotic fluid oxidative and nitrosative stress biomarkers correlate with fetal chronic hypoxia in diabetic pregnancies. Neonatology. 2013;103(3):193–8. doi: 10.1159/000345194. [DOI] [PubMed] [Google Scholar]
- 26.Madazli R, Tuten A, Calay Z, Uzun H, Uludag S, Ocak V. The incidence of placental abnormalities, maternal and cord plasma malondialdehyde and vascular endothelial growth factor levels in women with gestational diabetes mellitus and nondiabetic controls. Gynecol Obstet Investig. 2008;65(4):227–32. doi: 10.1159/000113045. [DOI] [PubMed] [Google Scholar]
- 27.Helske S, Vuorella P, Carpen O, Horning C, Weich H, Halmesmaki E. Expression of vascular endothelial growth factor receptors 1, 2 and 3 in placentas from normal and complicated pregnancies. Mol Hum Reprod. 2001;7:205–10. doi: 10.1093/molehr/7.2.205. [DOI] [PubMed] [Google Scholar]
- 28.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9(6):677–84. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 29.Ahmed A, Li XF, Dunk C, Whittle MJ, Rushton DI, Rollason T. Colocalisation of vascular endothelial growth factor and its flt-1 receptor in human placenta. Growth Factors. 1995;12(3):235–43. doi: 10.3109/08977199509036883. [DOI] [PubMed] [Google Scholar]
- 30.Sharkey AM, Charnock-Jones DS, Boocock CA, Brown KD, Smith SK. Expression of mRNA for vascular endothelial growth factor in human placenta. J Reprod Fertil. 1993;99(2):609–15. doi: 10.1530/jrf.0.0990609. [DOI] [PubMed] [Google Scholar]
- 31.Vuorela P, Hatva E, Lymboussaki A, Kaipainen A, Joukov V, Persico MG, Alitalo K, Halmesmäki E. Expression of vascular endothelial growth factor and placenta growth factor in human placenta. Biol Reprod. 1997;56(2):489–94. doi: 10.1095/biolreprod56.2.489. [DOI] [PubMed] [Google Scholar]
- 32.Pietro L, Daher S, Rudge MV, Calderon IM, Damasceno DC, Sinzato YK, Bandeira C, Bevilacqua E. Vascular endothelial growth factor (VEGF) and VEGF-receptor expression in placenta of hyperglycemic pregnant women. Placenta. 2010;31(9):770–80. doi: 10.1016/j.placenta.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 33.Chang SC, Yang WCV. Hyperglycemia induces altered expressions of angiogenesis associated molecules in the trophoblast. Evid Based Complement Alternat Med. 2013;2013:457971. doi: 10.1155/2013/457971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mordwinkin NM, Ouzounian JG, Yedigarova L, Montoro MN, Louie SG, Rodgers KE. Alteration of endothelial function markers in women with gestational diabetes and their fetuses. J Matern Fetal Neonatal Med. 2013;26(5):507–12. doi: 10.3109/14767058.2012.736564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lassus P, Teramo K, Nupponen I, Markkanen H, Cederqvist K, Andersson S. Vascular endothelial growth factor and angiogenin levels during fetal development and in maternal diabetes. Biol Neonate. 2003;84(4):287–92. doi: 10.1159/000073636. [DOI] [PubMed] [Google Scholar]
- 36.Maid S, Okail AL, Omar S, Attas AL. Histological changes in placental syncytiotrophoblasts of poorly controlled gestational diabetic patients. Endocr J. 1994;41(4):355–60. doi: 10.1507/endocrj.41.355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.