

Abstract
Research and development of new drugs requires both long time and high costs, whereas safety and tolerability profiles make the success rate of approval very low. Drug repurposing, applying known drugs and compounds to new indications, has been noted recently as a cost-effective and time-unconsuming way in developing new drugs, because they have already been proven safe in humans. In this review, we discuss drug repurposing of approved cardiovascular drugs, such as aspirin, beta-blockers, angiotensin converting enzyme inhibitors, angiotensin II receptor blockers, cardiac glycosides and statins. Regarding anti-tumor activities of these agents, a number of experimental studies have demonstrated promising pleiotropic properties, whereas all clinical trials have not shown expected results. In pathological conditions other than cancer, repurposing of cardiovascular drugs is also expanding. Numerous experimental studies have reported possibilities of drug repurposing in this field and some of them have been tried for new indications (‘bench to bedside’), while unexpected results of clinical studies have given hints for drug repurposing and some unknown mechanisms of action have been demonstrated by experimental studies (‘bedside to bench’). The future perspective of experimental and clinical studies using cardiovascular drugs are also discussed.
Keywords: Drug repurposing, Drug repositioning, Cardiovascular drugs, Second label indication, Pleiotropic properties
Background
Drug repurposing is a way to identify a new indication for existing drugs and compounds and is also called as drug repositioning, drug rescue or drug re-profiling. Drug repurposing generates lower costs and shorter time until approval than developing a drug de novo, because all phases of clinical trials have already performed for approved drugs and the information regarding side effects, pharmacokinetics and interaction with other drugs has been collected. Drug repurposing could also be useful for the treatment of rare or orphan diseases without any proven treatments [1].
In this review, we discuss drug repurposing of approved cardiovascular drugs, such as aspirin, beta-blockers, angiotensin converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), cardiac glycosides and statins, which are commonly prescribed for the treatment and/or prevention of cardiovascular diseases. We focus on pleiotropic properties and action mechanisms of each agent in tumor progression and metastasis. Aspirin not only prevents platelet function but also suppresses tumor cell proliferation directly. Beta-blockers inhibit tumor angiogenesis and invasiveness and induce apoptosis. ACE inhibitors and ARBs suppress tumor growth, angiogenesis and invasiveness. Pro-apoptotic activity of cardiac glycosides and anti-proliferative activity of statins are also described. Subsequently, we summarize the findings of clinical studies investigating the relationship between cardiovascular drugs and cancer. Cancer-related events have not been included in the primary endpoints in most of the large-scale cardiovascular clinical trials, which makes it difficult to evaluate whether cardiovascular drugs really exhibit anti-tumor effects identified in experimental research. Nevertheless, aspirin and beta-blockers have advanced to randomized clinical trials (RCTs) to confirm their anti-tumor effects, while findings have been inconsistent with regard to cancer and ACE inhibitors, ARBs, cardiac glycosides or statins.
Repurposing of cardiovascular drugs is also expanding in pathological conditions other than cancer.
Numerous experimental studies have reported possibilities of drug repurposing in the cardiovascular field and some of them have been tried for new indications (‘bench to bedside’), whereas unexpected results of clinical studies have given hints for drug repurposing and the unknown mechanisms of action have been demonstrated by experimental studies (‘bedside to bench’). The excellent examples of drug repurposing are propranolol for infantile hemangioma, beta-blockers for migraine, preoperative statins for perioperative risk reduction and minoxidil for androgenic alopecia, which are prescribed in daily clinical practice. Beta-blockers might be effective for the treatment of cirrhosis and osteoporosis. RCTs are examining whether losartan and statins are effective for Marfan’s syndrome and contrast-induced nephropathy, respectively. In addition to the available knowledge, the future perspective of experimental and clinical studies using cardiovascular drugs are also discussed.
Repurposing of cardiovascular drugs in cancer
Aspirin and cancer
Aspirin is commonly used for the treatment and prevention of atherosclerotic diseases, and pharmacological targets of aspirin are two isoforms of cyclooxygenase (COX) enzyme, COX-1 and COX-2 [2]. COX-1 is a constitutive enzyme expressed in most mammalian tissues and produces thromboxane A2 (TXA2) in platelets, which promotes platelets aggregation and adherence of platelets to tumor cells and thus prevents immune cells from recognizing and eliminating them, resulting in increased distant metastasis. On the other hand, COX-2 is a rapidly inducible enzyme during inflammation and dominantly produces prostaglandin E2 (PGE2) in tumor cells compared with COX-1, and PGE2 is thought to play an important role in accelerating cell proliferation and tumor growth. Aspirin administered at low doses (50–100 mg daily) and high doses (>325 mg daily) selectively blocks COX-1 and COX-2 in an irreversible manner, respectively. The putative action mechanisms of aspirin in tumor progression and metastasis are displayed in Fig. 1. Since the anti-tumor effect of aspirin was first reported in tumor-bearing mice in 1972 [3, 4], a number of subsequent experimental studies have supported this evidence [5]. As most clinical trials have shown a significant reduction in cancer risk and cancer-associated death in patients taking aspirin at a low dose, but not a high dose [6, 7], one important mechanism of tumor suppression by aspirin is proposed as inhibition of COX-1. With this activity, low-dose aspirin could prevent platelets from binding to tumor cells, resulting in suppression of distant metastasis and improved survival. In addition, PGE2 was upregulated in colon cancer [8] and administration of PGE2 enhanced tumor growth and angiogenesis [9]. As PGE2 was significantly suppressed in human colons when aspirin was administered even at a low dose (81 mg daily) [10], Suppression of PGE2 might be also important in anti-tumor activity of aspirin.
Fig. 1.
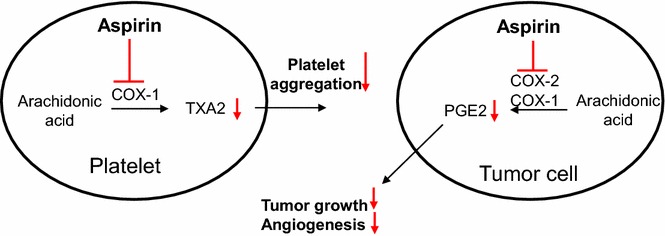
Putative mechanisms of action of low-dose aspirin in platelets and tumor cells in suppressing tumor growth. Low-dose aspirin exerts an inhibitory effect on platelet aggregation by suppressing production of TXA2 through inhibition of COX-1 in platelets. Thus, low-dose aspirin prevents platelets from binding to tumor cells, resulting in suppression of distant metastasis. On the other hand, PGE2, which is upregulated in colon cancer cells, is suppressed by low-dose aspirin, leading to inhibition of tumor growth and angiogenesis. COX-1 cyclooxygenase-1, COX-2 cyclooxygenase-2, PGE2 prostaglandin E2, TXA2 thromboxane A2
In clinical fields, a case–control study first demonstrated that aspirin use was associated with reduced risk of CRC (colorectal cancer) (risk ratio (RR) 0.53, 95 % confidence interval [CI] 0.40–0.71, p < 0.001) in 1988 [11]. Since then, a number of observational studies have shown that regular aspirin use significantly reduced risk of several cancers including CRC [12], esophageal cancer [13], gastric cancer [13], breast cancer [13] and prostate cancer [14–16]. In addition, Rothwell et al. reported that regular aspirin use reduced not only risk of distant metastasis [Hazard ratio (HR) 0.64, 95 % CI 0.48–0.84, p = 0.001] [17], but also cancer-related death [Odds ratio (OR) 0.79, 95 % CI 0.68–0.92, p = 0.003] [7]. Regarding the dose and the duration of aspirin, a meta-analysis of the five RCTs showed that aspirin at low dose (75–300 mg daily) reduced the 20-year incidence and mortality of CRC (incidence HR 0.75, 95 % CI 0.56–0.97, p = 0.02; mortality HR 0.61, 95 % CI 0.43–0.87, p = 0.005) and that the effects of aspirin increased with the duration of the treatment [6]. The results of recent meta-analysis are summarized in Table 1. Thus, aspirin could be effective for the prevention and/or the treatment of cancers. However, these findings are based on the results of observational studies and RCTs to evaluate the effects of aspirin on cardiovascular events. In addition, bleeding and gastrointestinal complications should be taken into consideration in the use of aspirin. To investigate the efficacy and safety of aspirin, the Aspirin in Reducing Events in the Elderly (ASPREE; NCT01038583) study, a RCT, is ongoing. Currently aspirin should be administered only for patients with cardiovascular diseases, not for the prevention of cancer.
Table 1.
Anti-tumor effects of aspirin in recent meta-analyses
| Author (year) [reference] | Number of studies (number of patients) | Dose of aspirin (mg) | Type of cancer | Main findings |
|---|---|---|---|---|
| González-Pérez et al. (2003) [13] | 4, 5 and 11 | Any | Esophageal, gastric and breast cancer | Aspirin reduced the incidence of esophageal cancer (RR 0·51, 95 % CI 0.38–0.69), gastric cancer (RR 0.73, 0.63–0.84) and breast cancer (RR 0.77, 0.69–0.86), derived from four, five and eleven studies respectively |
| Flossmann et al. (2007) [12] | 2 (5061) | 300< | CRC | Aspirin reduced the incidence of CRC (HR 0.74, 95 % CI 0.56–0.97, p = 0.02) |
| Rothwell et al. (2010) [6] | 5 | 75–300 | CRC | Low-dose aspirin reduced the 20-year incidence and mortality of CRC (incidence HR 0.75, 95 % CI 0.56–0.97, p = 0.02; mortality HR 0.61, 95 % CI 0.43–0.87, p = 0.005) |
| Rothwell et al. (2011) [7] | 8 | 75< | Any | Regular aspirin use reduced cancer-related death (OR 0.79, 95 % CI 0.68–0.92, p = 0.003). Therapeutic effects increased with duration of aspirin use |
| Rothwell et al. (2012) [17] | 5 | 75< | Any | Regular aspirin use reduced the risk of distant metastasis (HR 0.64, 95 % CI 0.48–0.84, p = 0.001) |
Beta-blockers and cancer
Previous experimental studies have demonstrated that chronic stress, depression and social isolation are associated with tumor progression [18–21]. As catecholamines such as norepinephrine and epinephrine are elevated under chronic stress and their effects are mainly mediated through beta-adrenoreceptors, activation of beta-adrenoreceptors by catecholamines is believed to play an important role in tumor progression. Indeed, the presence of beta-adrenoreceptors has been shown in the cell lines of breast cancer [22], pancreatic cancer [23], nasopharyngeal cancer [24] and ovarian cancer [25], and catecholamines significantly increased cell proliferation as well as cell migration in human cancer cell lines [26, 27]. Furthermore, in a mouse model of ovarian cancer, beta-adrenergic stimulation not only increased angiogenesis and tumor invasion through the cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) pathway [25], but also prevented the cancer cells from apoptosis by activating focal adhesion kinase (FAK) [28]. Additionally, a recent study showed that beta-adrenergic activation increased distant metastasis by 30 times through M2 macrophage infiltration in a mouse model of breast cancer [29].
On the other hand, beta-blockers mainly block beta-adrenoreceptors, and have been investigated as treatment for malignancies as well as cardiovascular diseases. Generally prescribed beta-blockers are classified into three types depending on selectivity of receptor subtypes, beta-1 selective beta-blockers such as bisoprolol, non-selective beta-blockers (NSBB) such as propranolol and nadolol, and alpha- and beta-blockers such as carvedilol, which block beta-1, all types of, and alpha- and beta-adrenoreceptors respectively. All of these beta-blockers are widely used for the treatment of heart failure, hypertension, ischemic heart disease and arrhythmias in the cardiovascular field. A number of experimental studies have also shown the anti-tumor effects of beta-blockers. In human cancer cell lines, catecholamine-induced proliferation and migration were inhibited by NSBB [26, 27], and enhanced invasiveness [30] and activated FAK by catecholamine were completely blocked by propranolol in a mouse model of ovarian cancer [28]. The action mechanisms of beta-blockers in tumor cells described above are currently proposed, as shown in Fig. 2. Moreover, in a mouse model of breast cancer, propranolol counteracted catecholamine-induced metastasis to distant tissues through M2 macrophage infiltration [29], which indicates the importance of beta-adrenergic signaling in M2 macrophage. Interestingly, a recent publication demonstrated that bisoprolol improved cardiac function and survival in a dose-dependent manner in rats with cancer cachexia, suggesting the favorable effects of beta-blockers in the terminal stage of cancer [31].
Fig. 2.
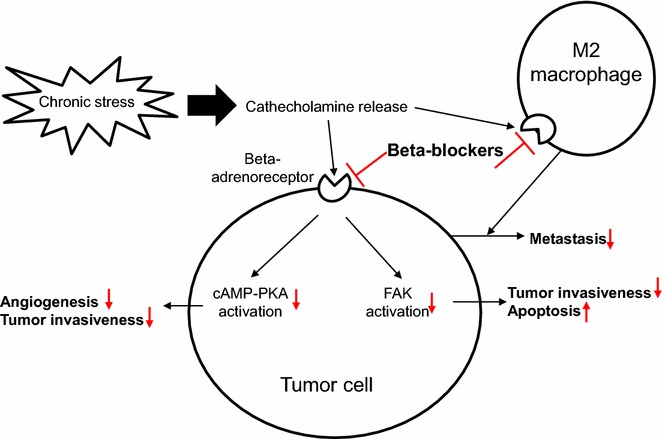
Putative mechanisms of action of beta-blockers in preventing tumor progression. Catecholamines are elevated under chronic stress and bind to beta-adrenoreceptors, resulting in activation of cAMP-PKA pathway and FAK, which accelerates tumor angiogenesis and invasion, and prevents cancer cells from apoptosis respectively. Beta-blockers blocks beta-adrenoreceptors, so that they are believed to suppress tumor growth and invasion. cAMP cyclic AMP, FAK protein kinase A, FAK focal adhesion kinase
In clinical settings, several epidemiological studies have examined the potential effect of beta-blockers on the incidence and the outcome of cancer. The results have been inconsistent [32–37], as shown in Table 2, but some of them demonstrated that the use of beta-blockers was associated with improved overall survival in patients with certain types of cancer such as breast cancer (HR 0.19, 95 % CI 0.06–0.60) [32], ovarian cancer (HR 0.54, 95 % CI 0.31–0.94, p = 0.02) [33] and non-small cell lung carcinoma (HR 0.78, 95 % CI 0.63–0.97, p = 0.02) [34]. In addition, a recent meta-analysis of 12 clinical studies have shown that beta-blocker usage was associated with significantly improved overall survival (HR 0.79, 95 % CI 0.67–0.93, p = 0.004) [38]. Beta-blockers appeared to have a greater effect in patients with early-stage cancer or cancer treated with primary surgery than those with late-stage cancer or cancer treated without primary surgery [38].
Table 2.
Anti-tumor effects of beta-blockers in recent clinical studies
| Authors (year), reference | Number of patients taking beta-blockers | Type of cancer | Main findings |
|---|---|---|---|
| Fryzek et al. (2006) [155] | NA | Breast cancer | The use of beta-blockers was not associated the risk of breast cancer (RR 1.07, 95 % CI 074–1.56) |
| Assimes et al. (2008) [156] | 1788 | Any | Beta-blockers significantly reduced the risk of cancer (OR 0.9, 95 % CI 0.85–0.96) |
| Powe et al. (2010) [157] | 43 | Breast cancer | Patients taking beta-blockers had a 57 % reduced risk of metastasis (Hazard ratio 0.43, 95 % CI 0.20–0.93) |
| Barron et al. (2011) [32] | 70 | Breast cancer | Propranolol reduced cancer-related mortality (HR 0.19, 95 % CI 0.06–0.60) |
| Ganz et al. (2011) [36] | 204 | Breast cancer | Beta-blocker usage was not associated with improved overall survival (HR 1.04, 95 % CI 0.72–1.51) |
| Lemeshow et al. (2011) [37] | 275 | Melanoma | Beta-blockers reduced all-cause mortality (HR 0.81, 95 % CI 0.67–0.97) |
| Diaz et al. (2012) [33] | 23 | Ovarian cancer | Beta-blockers improved overall survival (HR 0.54, 95 % CI 0.31–0.94, p = 0.02) |
| Wang et al. (2013) [34] | 155 | Non-small cell lung carcinoma | Beta-blockers improved overall survival (HR 0.78, 95 % CI 0.63–0.97, p = 0.02) |
| Grytli et al. (2014) [35] | 1115 | Prostate carcinoma | The use of beta-blockers was not associated with reduced all-cause mortality (HR 0.92, 95 % CI 0.83–1.02) |
| Choi et al. (2014) [38] | 6717 | Any | Beta-blocker usage was associated with significantly improved overall survival (HR 0.79, 95 % CI 0.67–0.93, p = 0.004) |
Thus, experimental and clinical evidences have indicated the efficacy of beta-blockers for the treatment of cancer. Although there are no clinical trials to clarify it prospectively, to confirm these promising effects of beta-blockers on cancer, two RCTs are ongoing to investigate the preventive role of beta-blockers in patients with breast cancer (NCT 00502684) and CRC (NCT 00888797) undergoing surgery with curative intent.
ACE inhibitors, ARBs and cancer
Angiotensin II promotes vasoconstriction and sodium reabsorption via angiotensin II type 1 (AT1) receptors in the renin-angiotensin system (RAS), resulting in increased blood pressure. Both of ACE inhibitors and ARBs, agents blocking RAS, are widely used in the cardiovascular fields for the treatment of heart failure, hypertension and old myocardial infarction. In addition to the systemic RAS, recently much attention has been paid to the existence of the local RAS in tumor cells. Indeed, AT1 receptors have been identified in various types of human cancer, such as renal cell carcinoma [39], laryngeal carcinoma [40], pancreatic cancer [41], ovarian cancer [42], breast cancer [43], melanoma [44], and intrahepatic cholangiocarcinoma [45]. Moreover, immunohistochemical analysis showed co-localization of AT1 receptor and vascular endothelial growth factor (VEGF), a major angiogenic protein, in pancreatic cancer cells [46], and expression of AT1 receptor was detected more frequently in high-grade invasive ovarian cancer than in benign ovarian tumor, and was positively correlated with expression of VEGF [42]. Administration of angiotensin II upregulated VEGF [42, 43, 46] and increased tumor angiogenesis [42], tumor growth [45], and tumor invasiveness [42]. Thus, these findings suggested that the local RAS controlled VEGF and tumor progression, and then blockade of the local RAS has been noted as a promising strategy for the treatment of cancer. Indeed, ACE inhibitors suppressed VEGF expression, VEGF-induced angiogenesis and tumor growth [47, 48] and ARBs also showed similar effects in certain cancer cell lines and animal cancer models [41–43, 45, 46]. Proposed action mechanisms are presented in Fig. 3.
Fig. 3.
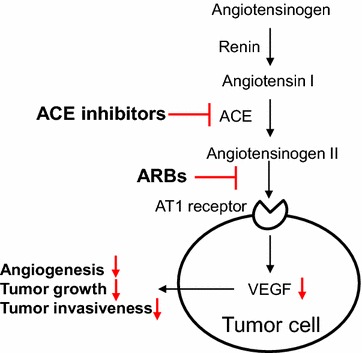
Local RAS, ACE inhibitors and ARBS in tumor cells. In tumor cells, angiotensin II promotes VEGF production via AT1 receptor, resulting in increased angiogenesis. ACE inhibitors and ARBs attenuate local RAS and reduce VEGF-dependent angiogenic signals in cancer
In 1998, an observational study first demonstrated that hypertensive patients taking ACE inhibitors had a reduced cancer risk compared with patients in the control group (RR 0.72, 95 % CI 0.55–0.92) [49]. However, following clinical studies failed to show the favorable effects of ACE inhibitors on cancer risk or outcome [50–54]. For example, Lindholm et al. also investigated a protective role of ACE inhibitors for cancer in elderly patients with hypertension. In this study, ACE inhibitor usage was not associated with decreased risk of new cancer occurrence (standardized incidence ratio 0.99, 95 % CI 0.86–1.13) [52]. Furthermore, in 2010 a meta-analysis of five RCTs revealed that the use of ARBs increased cancer risk (RR 1.08, 95 % CI 1.01–1.16) [55]. However, subsequent several meta-analyses showed no significant association between the use of ARBs and new cancer risk [56, 57] (Table 3). Judging from the results of clinical studies, there is no reason to administer ACE inhibitors or ARBs for the prevention and/or treatment of cancer despite the favorable findings in experimental research. Further long-term prospective trials are needed.
Table 3.
Anti-tumor effects of ACE inhibitors or ARBs in recent clinical studies
| Authors (year), reference | Number of patients taking ACE inhibitors or ARBs | Medication | Type of cancer | Main findings |
|---|---|---|---|---|
| Ronquist et al. (2004) [54] | 100 | ACE inhibitors | Prostate cancer | Current use of ACE inhibitors was not associated with decreased risk of prostate cancer (OR 0.9, 95 % CI 0.7–1.1) |
| Sjoberg et al. (2007) [53] | 62 and 101 | ACE inhibitors | Esophageal and gastric cancer | Current use of ACE inhibitors did not decrease the risk of esophageal and gastric cancer (OR 0.79, 95 % CI 0.60–1.05 for esophageal cancer; OR 1.11, 95 % CI 0.88–1.39 for gastric cancer) |
| Sipahi et al. (2010) [55] | 2510 | ARB | Any | Patients taking ARBs had a significantly increased risk of new cancer development (RR 1.08, 95 % CI 1.01–1.15; p = 0.016) |
| Pasternak et al. (2011) [56] | 3954 | ARB | Any | ARB did not increase the risk of cancer (RR 0.99, 95 % CI 0.95–1.03) |
| The ARB Trialists Collaboration (2011) [57] | 4549 | ARB | Any | There was no association between ARB usage and cancer incidence (OR 1.00, 95 % CI 0.95–1.04) |
Cardiac glycosides and cancer
Cardiac glycosides were originally derived from the foxglove. Two types of cardiac glycosides, digoxin and digitoxin, have been currently prescribed to treat heart failure or to reduce heart rate in the cardiovascular field, whereas the relationship between cardiac glycosides and cancer have been noted since Shiratori et al. [58] reported the antiproliferative effect of cardiac glycosides in cancer cells in1967. Sodium- and potassium-activated adenosine triphosphatase (Na+-K+-ATPase), the primary target of cardiac glycosides, exports three sodium ions in exchange for two potassium ions using ATP as an energy source, thereby maintaining the cell membrane potential. Indeed, increased expression of Na+-K+-ATPase was found in gastric [59] and bladder cancer cells [60], and elevated Na+-K+-ATPase activity was observed in highly invasive human renal carcinoma cells [61]. Cardiac glycosides bind to Na+-K+-ATPase and disrupt its abilities in tumor cells. Namely, cardiac glycosides decreased the membrane potential and increase intracellular Na+, which caused the induction of apoptosis in human cancer cells [62], and also increased intracellular Ca2+ in human prostate adenocarcinoma cells, resulting in activation of calcineurin and transcriptional upregulation of Fas ligand, which can induce apoptosis [63]. Some other mechanisms of cardiac glycosides such as suppression nuclear factor-kappaB [64] and inhibition of DNA topoisomerase II [65] were also shown to induce apoptosis in tumor cells (Fig. 4). The complex mechanism in cardiac glycoside-induced apoptosis has been already well documented [66–68].
Fig. 4.
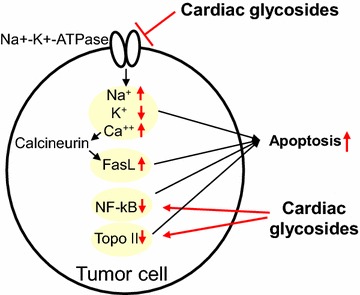
Cardiac glycoside-induced apoptosis in tumor cells. Cardiac glycosides bind to Na+-K+-ATPase and decrease the membrane potential and increase intracellular Na+ and Ca++ in certain human cancer cell lines, resulting in activation of calcineurin and transcriptional upregulation of Fas ligand. Cardiac glycosides also suppressthe expression of nuclear factor-kappaB and inhibit DNA topoisomerase II. All of these activities induce apoptosis in human cancer cells
In clinical settings, there were several studies evaluating the effects of cardiac glycosides on the development and progression of cancer. Early observational studies showed that the use of cardiac glycosides reduced the rate of recurrence and the malignant grade of breast cancer [69, 70] and a subsequent 20-year follow-up revealed that patients with cardiac glycosides had a significantly lower mortality rate (6 %) than those without cardiac glycosides (34 %) [71]. These findings seemed to indicate the anti-tumor effect of cardiac glycosides, but the results from recent reports have been inconsistent with them. In 2001, a large cohort study (n = 9271) revealed that patients taking cardiac glycosides had a higher incidence of cancer compared with controls [72]. Moreover, the use of cardiac glycosides was associated with an increased risk of invasive breast cancer (RR 1.30, 95 % CI 1.14–1.48) among post-menopausal women [73] and the incidence of estrogen receptor-positive breast cancer was significantly higher than that of estrogen receptor-negative breast cancer in women taking cardiac glycosides [74]. Cardiac glycosides also increased the risk of corpus uteri cancer (RR 1.48, 95 % CI 1.32–1.65), whereas did not affect the incidence of ovary cancer (RR 1.06, 95 % CI 0.92–1.22) and cervix cancer (RR 1.00, 95 % CI 0.79–1.25) [75] and reduced the risk of prostate cancer (RR 0.76, 95 % CI 0.61–0.95) [76] (Table 4). These findings suggest that the estrogenic effect of cardiac glycosides might play an important role in inhibiting cancer, but further experimental studies and RCTs setting cancer-related events as primary endpoints should be performed to confirm this hypothesis.
Table 4.
Relationship between cardiac glycosides and the incidence of cancer in recent clinical studies
| Authors (year), reference | Number of patients taking cardiac glycosides | Medication | Type of cancer | Main findings |
|---|---|---|---|---|
| Haux et al. (2001) [72] | 9271 | Digitoxin | Any | Digitoxin use increased the risk of cancer (SIR 1.27, 95 % CI 1.18–1.37). Plasma digitoxin levels were negatively correlated with the risk of cancer |
| Ahern et al. (2008) [73] | 2890 | Digoxin | Breast cancer | Digoxin use was associated with the increased risk of breast cancer (OR 1.30, 95 % CI 1.14–1.48). The risk was positively correlated with the duration of digoxin exposure (OR for 7–18 years of digoxin use 1.39, 95 % CI 1.10–1.74) |
| Biggar et al. (2011) [74] | 104,648 | Digoxin | Breast cancer | Current digoxin use increased the risk of breast cancer (RR, 1.39; 95 % CI, 1.32–1.46). In digoxin users, the risk was higher for ER-positive breast cancers (RR, 1.35; 95 % CI, 1.26–1.45) than for ER-negative breast cancers (RR, 1.20; 95 % CI, 1.03–1.40) |
| Biggar et al. (2012) [75] | 104,648 | Digoxin | Corpus uteri cancer | Current digoxin use increased the risk of corpus uteri cancer (RR 1.48, 95 % CI 1.32–1.65). (RR 1.06, 95 % CI 0.92–1.22) (RR 1.00, 95 % CI 0.79–1.25) |
| Biggar et al. (2012) [75] | 104,648 | Digoxin | Ovary cancer | Current digoxin use increased the risk of ovary cancer (RR 1.06, 95 % CI 0.92–1.22) |
| Biggar et al. (2012) [75] | 104,648 | Digoxin | Cervix cancer | Current digoxin use increased the risk of cervix cancer (RR 1.00, 95 % CI 0.79–1.25) |
| Platz et al. (2011) [76] | 936 | Digoxin | Prostate cancer | Current digoxin use decreased the risk of prostate cancer (RR 0.76, 95 % CI 0.61–0.95) |
Statins and cancer
Statins, competitive inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase, block the formation of mevalonate in the mevalonate pathway to synthesize cholesterol in liver and are commonly administered for the treatment of hyperlipidemia and the secondary prevention of atherosclerotic diseases such as myocardial infarction and ischemic stroke. Geranylgeranyl pyrophosphate (GGPP) and farnesyl pyrophosphate (FPP) are downstream products of the mevalonate pathway and used as substrates in protein prenylation, which is essential for localization of proteins in cell membranes [77]. As GGPP and FPP are also inhibited by statins, it has been proposed that statins could cause apoptosis in tumor cells (Fig. 5). Indeed, experimental studies have shown that statins suppressed proliferation in multiple human cancer cell lines through this inhibitory activity [78–80]. Despite this promising anti-tumor effect of statins, the results of clinical studies are controversial. Initial clinical trials showed that statins were associated with a significant reduction in overall cancer incidence [81, 82]. Some subsequent meta-analyses have demonstrated that statins significantly reduced the risk of prostate [83], esophageal [84] and gastric cancer [85], while other recent meta-analyses failed to show benefits of statins in cancer-associated mortality [86], and in the risk of lung [87] and skin cancer [88]. The effects of statins on cancer in recently published meta-analyses are presented in Table 5. On the other hand, statins enhanced the efficacy of treatment in acute myeloid leukemia [89] and hepatocellular carcinoma [90], when combined with chemotherapeutic agents. Further clinical investigation should be performed, since in most of clinical studies to investigate the role of statins, the primary endpoint was not set on cancer-related events and the observation period was not long enough to observe the development and/or prevention of cancer.
Fig. 5.
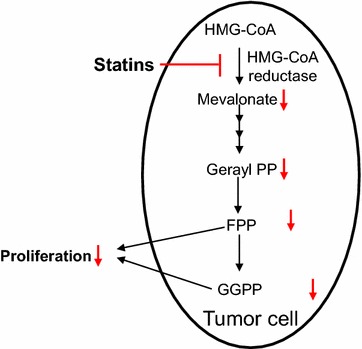
Effects of statins on mevalonate pathway in tumor cells. Downstream products in the mevalonate pathway such as geranylgeranyl pyrophosphate (GGPP) and farnesyl pyrophosphate (FPP) are substrates in protein prenylation. By inhibiting the formation of GGPP and FPP, statins exert anti-proliferatic activity in tumor cells
Table 5.
Effect of statins on cancer in recent meta-analyses
| Authors (year), reference | Number of studies | Type of cancer | Main findings |
|---|---|---|---|
| Bansal et al. (2012) [83] | 15 cohort and 12 case–control studies | Prostate cancer | Statins decreased the risk of prostate cancer (RR 0.93, 95 % CI 0.30–0.86) and advanced prostate cancer (RR 0.80, 95 % CI 0.70–0.90) |
| Singh et al. (2013) [84] | 13 studies (including a post hoc analysis of 22 RCTs) | Esophageal cancer | Statins reduced the risk of esophageal cancer (OR 0.72 95 % CI 0.60–0.86) |
| Wu et al. (2013) [85] | 3 post hoc analyses of 26 RCTs and 8 observational studies | Gastric cancer | Statin use was associated with a decreased risk of gastric cancer (RR 0.73, 95 % CI 0.58–0.93) |
| Emberson et al. (2012) [86] | 27 RCTs | Any | Statins did not reduce the incidence of, or mortality from, any type of cancer (RR 1.00, 95 % CI 0.96–1.05 for incidence; RR 1.00, 95 % CI 0.93–1.08) |
| Tan et al. (2013) [87] | 5 RCTs, 7 cohort and 7 case–control studies | Lung cancer | Statin did not decrease the risk of lung cancer either among RCTs (RR 0.91, 95 % CI 0.76–1.09), cohort studies (RR 0.94, 95 % CI 0.82–1.07) and case–control studies (RR 0.82, 95 % CI 0.57–1.16) |
| Zhang et al. (2014) [88] | 29 studies (including a post hoc analysis of 8 RCTs) | Skin cancer | Statins did not reduce the risk of skin cancer among melanoma (RR 0.94, 95 % CI 0.85–1.04) or non-melanoma skin cancer (RR 1.03, 95 % CI 0.90–1.19) |
Repurposing of cardiovascular drugs in pathological conditions other tan cancer
Propranolol and infantile hemangioma
The efficacy of oral propranolol for infantile hemangioma was first reported in 2008 [91]. Several clinical studies [92–94], including two small RCTs [95, 96], also revealed that infantile hemangioma regressed with the treatment of propranolol. Furthermore, in 2015, a large-scale RCT showed that propranolol at a dose of 3 mg per kg for 6 months was well-tolerated and effective in the treatment of infantile hemangioma [97]. At present, propranolol is regarded as a first-line therapy for infantile hemangioma, though there is room for further research into duration or regimen of propranolol.
Beta-blockers and migraine
The efficacy of beta-blockers for the prevention of migraine has been evaluated in RCTs. A controlled double-blind trial showed that propranolol was more efficacious than placebo and as efficacious as cyproheptadine in reducing frequency, duration and severity of migraine attacks [98]. Moreover, the efficacy of combination of propranolol and cyproheptadine was greater than that of propranolol or cyproheptadine alone. Subsequently, another controlled double blind trial revealed the more potent effect of metoprolol for the prevention of migraine attacks than aspirin [99]. In this study, treatment effectiveness was defined as a 50 % decrease in the rate of migraine attacks and the response rate was 45.2 % in metoprolol group and 29.6 % in aspirin group respectively. In the current guidelines, propranolol and metoprolol are listed up as effective medications for the treatment of migraine [100].
Beta-blockers and cirrhosis
In patients with cirrhosis, portal hypertension gradually develops, leading to variceal bleeding, hepatic encephalopathy, ascites, spontaneous bacterial translocation, hepatorenal syndrome. They have hyperdynamic circulatory abnormalities, such as an increased cardiac output and a decreased peripheral vascular resistance. As a result, sympathetic nervous system is activated in this condition. To counteract these abnormalities and reduce portal pressure, NSBB is regarded as effective [101]. Selective beta-1 antagonists were less beneficial for the treatment of cirrhosis [102, 103], which indicates the importance of both beta-1 and beta-2 adrenergic pathways. Recently, carvedilol has been noted as a promising medication for portal hypertension due to its anti-alpha-1 adrenergic activity, which decreases the hepatic vascular resistance, in addition to the beta-blocking activities [104].
In 1981, Lebre et al. first reported the effects of NSBB on the secondary prevention of variceal bleeding in patients with cirrhosis. Since then, many clinical studies have been performed to examine the role of NSBB in patients with chronic liver disease, cirrhosis and portal hypertension. A meta-analysis of 12 RCTs (n = 769) demonstrated that the beta-blocker use reduced the rate of recurrent bleeding and mortality compared to placebo [105]. There is no significant difference between beta-blocker alone and the combination of beta-blocker and nitrate, despite the previous findings indicating the effectiveness of nitrate for the treatment of cirrhosis [106]. In a 2012 meta-analysis, combining NSBB and endoscopic variceal ligation resulted in less recurrent bleeding than either treatment alone [107].
In addition, the use of NSBBs has been considered to be effective for the primary prophylaxis as well as the secondary prophylaxis. A meta-analysis demonstrated that NSBB decreased variceal bleeding and mortality in patients with cirrhosis without previous gastrointestinal bleeding [108]. The current guidelines for the management of portal hypertension, Baveno VI, also recommend that patients with high-risk small varices or large/medium varices should receive NSBB, if not contradictory, or endoscopic variceal ligation for the primary prevention of variceal bleeding [109].
On the other hand, there are few studies to investigate the role of NSBB to prevent the formation of varices. Groszmann et al. [110] demonstrated that NSBB did not prevent the new development of varices in patients with cirrhosis and portal hypertension without varices. The use of NSBB for the pre-primary prophylaxis is not recommended in the current guidelines [109].
In addition, it would be noted that an observational study showed the harmful effect of NSBB in patients with advanced cirrhosis and refractory ascites. In this study, the use of NSBB was associated with significantly poor 1-year survival compared with control (19 vs 64 %, p < 0.0001) [111]. However, further studies should be performed to evaluate the efficacy and safety of NSBB in this population.
Beta-blockers and osteoporosis
Previous experimental studies revealed that beta-adrenoreceptors are expressed on osteoblastic and osteoclastic cells and that a beta agonist stimulates osteoblasts resulting in bone resorption [112]. These findings supported that a beta agonist decreased bone mass, while a beta antagonist increased bone mass in mice [113].
Recently the importance of sympathetic nervous system in bone remodeling has been focused on. Ducy et al. [114] demonstrated that intracerebroventricular infusion of leptin inhibited bone formation and decreased bone mass in mice, which indicated the central role of leptin in bone remodeling. Furthermore, Takeda et al. [113] showed that the effect of leptin was mediated by beta2-adrenoreceptors on osteoblasts. Namely, blockade of beta2-adrenoreceptors caused bone formation [113], while stimulation of beta2-adrenoreceptors resulted in bone resorption [115]. These findings suggest that beta-blockers could be a therapeutic option for osteoporosis.
There are numerous clinical studies which evaluated the effects of beta-blockers on the risk of osteoporotic fractures in humans. The results are inconsistent, however recent meta-analyses have shown that beta-blockers reduced the risk of fracture by approximately 15 %, independent of gender, fracture site and dose [116, 117]. The most recent meta-analysis (n = 1,644,570) has demonstrated that beta-blocker use was associated with a significantly lower risk of fractures (16 studies, RE pooled ES = 0.86, 95 % CI 0.78–0.93) [118]. Intriguingly, beta1-selective beta-blockers significantly reduced the risk of any fracture compared with NSBB (6 studies, RE pooled ES = 0.82, 95 % CI 0.69–0.97) [118]. These findings do not support the proposed mechanism in which bone formation is promoted by the blockade of beta2-adrenoreceptor.
Additionally, hemodynamic alteration with beta-blockers might affect the incidence of falls and fractures. Namely, anti-arrhythmic actions could prevent subjects, especially elderly people, from a fall, while (orthostatic) hypotension and/or bradycardia could make them injured in a fall, sometimes a hip-fracture. Although findings of clinical studies have been inconsistent with regard to beta-blockers and risk of falls, there are no evidence that beta-blockers increased the incidence of falls [119–121].
Collectively, the roles of beta-adrenoreceptors in bone remodeling are complex and further studies are needed to clarify them. Asbeta-blockers appear to reduce the risk of osteoporotic fracture in clinical settings, RCTs should be performed to evaluate the efficacy and safety of beta-blockers in this condition.
Losartan and Marfan’s syndrome
Marfan’s syndrome is caused by mutations in the gene encoding fibrillin-1 [122], which regulates the transforming growth factor beta (TGF-beta) signaling pathway. In a mouse model of Marfan’s syndrome, fibrillin-1 deficiency was associated with increased TGF-beta signaling [123, 124], which is thought to contributes to the development of aortic aneurysm. In 2006, Habashi et al. [125] showed that losartan, one of the ARBs, suppressed the excessive TGF-beta signaling and prevented the formation of aortic aneurysm in fibrillin-1 deficient mice. In this study, the losartan treatment was significantly efficacious for the prevention of aortic aneurysm compared with the propranolol treatment [125]. Subsequently, the effect of losartan was examined in patients with Marfan’s syndrome. Two small cohort studies demonstrated that losartan significantly slowed the rate of aortic growth in pediatric patients with Marfan’s syndrome [126, 127] and in a RCT, aortic dilation rate was reduced by losartan in adult patients with Marfan’s syndrome [128]. However, a recent RCT showed that there was no significant difference in the progression of aortic diameter between losartan-treated and atenolol-treated patients with Marfan’s syndrome [129]. Further experimental and clinical investigation should be performed, though the suppression of enhanced TGF-beta signaling seems important in this condition.
Preoperative statins and perioperative risk
Experimental research has demonstrated that statins improve endothelial function, attenuate vascular and myocardial remodeling, inhibit vascular inflammation and oxidation, and stabilize atherosclerotic plaques as pleiotropic effects beyond cholesterol lowering [130], which are expected to prevent plaque rupture and subsequent cardiovascular events in the perioperative period. Indeed, several observational studies have suggested that statin use decreased short-term mortality and myocardial infarction [131–134]. In addition, a RCT showed that atorvastatin was associated a significant reduction in the incidence of cardiac events at 6 months follow-up after vascular surgery [135]. In the light of these findings, both the current ESC/ESA and ACC/AHA guidelines give a class I recommendation for continuing perioperative statins [136, 137]. In statin-naïve patients, two meta-analyses showed the preventive effect of statins on perioperative myocardial infarction and death, which allows a class IIa recommendation in the ESC/ESA and ACC/AHA guidelines for pre-operative initiation of statins for patients undergoing vascular surgery [136, 137]. In non-cardiac surgery, evidence is insufficient, but statins appear more beneficial in patients with accumulated cardiovascular risk and statins are often prescribed in such case in clinical practice.
Statin and contrast-induced nephropathy
Contrast-induced nephropathy (CIN) is characterized by acute kidney injury, caused by contrast medium. To prevent CIN, there are limited strategies such as volume expansion, use of iso-osmolar contrast and less amount of contrast media. The pathophysiological mechanisms of CIN are not fully understood, but contrast media might cause renal vasoconstriction, oxidative stress, inflammation and direct tubular necrosis [138]. As statins improve endothelial function and exert anti-inflammatory and antioxidative properties as well as lower cholesterol [139–141], statins have been regarded as promising therapeutic approach. Experimental research suggested that statins ameliorated acute ischemic renal failure and prevented tubular necrosis in rats [142]. In clinical setting, a recent meta-analysis seven RCTs showed that the use of high-dose statins significantly reduced the risk of CIN compared to that of low-dose statins or placebo (RR 0.51, 95 % CI 0.34–0.76) [143]. In addition, a large-scale RCT, Novel Approaches for Preventing or Limiting Events (NAPLES) II trial, showed that a single high-dose of atorvastatin significantly reduced the incidence of CIN compared with placebo in patients with chronic kidney disease undergoing elective coronary angiography (OR 0.22, 95 % CI 0.07–0.22) [144]. Another large-scale RCT, protective effect of rosuvastatin and antiplatelet therapy on contrast-induced acute kidney injury and myocardial damage in patients with acute coronary syndrome (PRATO-ACS) trial, randomly assigned patients with acute coronary syndrome undergoing an early coronary angiography to rosuvastatin or placebo, and revealed that high-dose rosuvastatin prevented CIN (OR 0.38, 95 % CI 0.20–0.71) [145]. These findings support the use of statins for the prevention of CIN in coronary angiography, while future research should investigate effect of statins in other examinations or treatments such as computed tomography, as well as timing of initiation and duration of statin treatment.
Minoxidil and androgenic alopecia
Oral minoxidil had been originally developed as an antihypertensive drug in 1960s and was reported to cause hypertrichosis as well as an antihypertensive effect in 1970s/80s [146, 147], which led to the research of topical minoxidil for the treatment of alopecia [148]. Several mechanisms how minoxidil improves alopecia have been proposed. Minoxidil stimulates cutaneous blood flow through local vasodilatory effect in human scalps [149], up-regulates the expression of VEGF in human hair dermal papilla cells [150], and prolongs the anagen period of hair follicle through the opening of potassium channels [151]. All of these actions could help to promote or maintain hair growth.
In male alopecia, 2 and 5 % minoxidil solutions have been approved and the 5 % solution has shown higher efficacy than the 2 % solution [152]. Recently, the foam type has been newly developed, and the effect of 5 % minoxidil foam has been comparable to the 5 % minoxidil solution. The minoxidil treatment during 5 years has shown the continuous efficacy [153], while the therapeutic effect has disappeared in 24 weeks after discontinuation [154]. In female alopecia, only the 2 % minoxidil solution or foam are currently used. As higher dose seems to show higher efficacy both in males and females, further clinical studies might allow higher dose to be approved.
Conclusions
A number of studies have demonstrated anti-tumor activities of cardiovascular drugs in tumor cells and animal models, while findings of clinical trials, including large-scale RCTs, have often been inconsistent with those of preclinical studies or other clinical trials. Meta-analysis might some contributions to this ‘dissociation’. Meta-analysis is convenient and widely used, while we should pay appropriate attention to the fact that meta-analysis is hard to gather detailed data such as patient characteristics and easily biased. It could be important to consider cancer type, cancer stage and patient characteristics such as age, sex, body mass index in evaluating cancer. Regarding aspirin and beta-blockers, RCTs are ongoing and the results are anticipated with great interest.
In the pathophysiological conditions other than cancer, some cardiovascular drugs have already obtained new indications, such as propranolol for infantile hemangioma, beta-blockers for migraine, and minoxidil for androgenic alopecia. Preoperative use of statins for perioperative risk reduction may be used for patients undergoing non-cardiovascular surgery.
Thus, based on the available knowledge and information, it would be expected that unknown mechanisms of action of drugs are investigated by experimental studies and that clinical evidences are established by well-organized RCTs. Close link between experimental and clinical studies is essential.
Authors’ contributions
JI wrote the manuscript. MK, NE and JS revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We acknowledge support by the German Research Foundation and the Open Access Publication Funds of the Gottingen University.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- ACE
angiotensin converting enzyme
- ARBs
angiotensin II receptor blockers
- AT1
angiotensin II type 1
- cAMP
cyclic adenosine monophosphate
- CI
confidence interval
- CIN
contrast-induced nephropathy
- COX
cyclooxygenase
- CRC
colorectal cancer
- FAK
focal adhesion kinase
- FPP
farnesyl pyrophosphate
- GGPP
geranylgeranyl pyrophosphate
- HR
hazard ratio
- Na+-K+-ATPase
sodium- and potassium-activated adenosine triphosphatase
- NSBB
non-selective beta-blockers
- OR
odds ratio
- PGE2
prostaglandin E2
- PKA
protein kinase A
- RAS
renin-angiotensin system
- RCTs
randomized controlled trials
- RR
risk ratio
- TGF-beta
transforming growth factor beta
- TXA2
thromboxane A2
- VEGF
vascular endothelial growth factor
Contributor Information
Junichi Ishida, Phone: +49 (0) 551 39-66380, Email: juishida-circ@umin.ac.jp.
Masaaki Konishi, Email: m_koni524@hotmail.com.
Nicole Ebner, Email: nicole.ebner@med.uni-goettingen.de.
Jochen Springer, Email: jochen.springer@med.uni-goettingen.de.
References
- 1.Sardana D, Zhu C, Zhang M, Gudivada RC, Yang L, Jegga AG. Drug repositioning for orphan diseases. Brief Bioinform. 2011;12:346–356. doi: 10.1093/bib/bbr021. [DOI] [PubMed] [Google Scholar]
- 2.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 3.Gasic GJ, Gasic TB, Murphy S. Anti-metastatic effect of aspirin. Lancet. 1972;2:932–933. doi: 10.1016/S0140-6736(72)92581-0. [DOI] [PubMed] [Google Scholar]
- 4.Kolenich JJ, Mansour EG, Flynn A. Haematological effects of aspirin. Lancet. 1972;2:714. doi: 10.1016/S0140-6736(72)92124-1. [DOI] [PubMed] [Google Scholar]
- 5.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 6.Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, Meade TW. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376:1741–1750. doi: 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
- 7.Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011;377:31–41. doi: 10.1016/S0140-6736(10)62110-1. [DOI] [PubMed] [Google Scholar]
- 8.Pugh S, Thomas GA. Patients with adenomatous polyps and carcinomas have increased colonic mucosal prostaglandin E2. Gut. 1994;35:675–678. doi: 10.1136/gut.35.5.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shao J, Jung C, Liu C, Sheng H. Prostaglandin E2 Stimulates the beta-catenin/T cell factor-dependent transcription in colon cancer. J Biol Chem. 2005;280:26565–26572. doi: 10.1074/jbc.M413056200. [DOI] [PubMed] [Google Scholar]
- 10.Ruffin MT, Krishnan K, Rock CL, Normolle D, Vaerten MA, PetersGolden M, Crowell J, Kelloff G, Boland CR, Brenner DE. Suppression of human colorectal mucosal prostaglandins: determining the lowest effective aspirin dose. J Natl Cancer Inst. 1997;89:1152–1160. doi: 10.1093/jnci/89.15.1152. [DOI] [PubMed] [Google Scholar]
- 11.Kune GA, Kune S, Watson LF. Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res. 1988;48:4399–4404. [PubMed] [Google Scholar]
- 12.Flossmann E, Rothwell PM. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet. 2007;369:1603–1613. doi: 10.1016/S0140-6736(07)60747-8. [DOI] [PubMed] [Google Scholar]
- 13.González-Pérez A, Rodríguez LAG, López-Ridaura R. Effects of non-steroidal anti-inflammatory drugs on cancer sites other than the colon and rectum: a meta-analysis. BMC Cancer. 2003;3:28. doi: 10.1186/1471-2407-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elwood PC, Gallagher AM, Duthie GG, Mur LA, Morgan G. Aspirin, salicylates, and cancer. Lancet. 2009;373:1301–1309. doi: 10.1016/S0140-6736(09)60243-9. [DOI] [PubMed] [Google Scholar]
- 15.Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012;13:518–527. doi: 10.1016/S1470-2045(12)70112-2. [DOI] [PubMed] [Google Scholar]
- 16.Bosetti C, Rosato V, Gallus S, Cuzick J, La Vecchia C. Aspirin and cancer risk: a quantitative review to 2011. Ann Oncol. 2012;23:1403–1415. doi: 10.1093/annonc/mds113. [DOI] [PubMed] [Google Scholar]
- 17.Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379:1591–1601. doi: 10.1016/S0140-6736(12)60209-8. [DOI] [PubMed] [Google Scholar]
- 18.Bukberg J, Penman D, Holland JC. Depression in hospitalized cancer patients. Psychosom Med. 1984;46:199–212. doi: 10.1097/00006842-198405000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Spiegel D, Giese-Davis J. Depression and cancer: mechanisms and disease progression. Biol Psychiatry. 2003;54:269–282. doi: 10.1016/S0006-3223(03)00566-3. [DOI] [PubMed] [Google Scholar]
- 20.Spiegel D. Health caring. psychosocial support for patients with cancer. Cancer. 1994;74:1453–1457. doi: 10.1002/1097-0142(19940815)74:4+<1453::AID-CNCR2820741609>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 21.Chida Y, Hamer M, Wardle J, Steptoe A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat Clin Pract Oncol. 2008;5:466–475. doi: 10.1038/ncponc1134. [DOI] [PubMed] [Google Scholar]
- 22.Vandewalle B, Revillion F, Lefebvre J. Functional beta-adrenergic receptors in breast cancer cells. J Cancer Res Clin Oncol. 1990;116:303–306. doi: 10.1007/BF01612908. [DOI] [PubMed] [Google Scholar]
- 23.Weddle DL, Tithoff P, Williams M, Schuller HM. Beta-adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis. 2001;22:473–479. doi: 10.1093/carcin/22.3.473. [DOI] [PubMed] [Google Scholar]
- 24.Yang EV, Sood AK, Chen M, Li Y, Eubank TD, Marsh CB, Jewell S, Flavahan NA, Morrison C, Yeh PE, et al. Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 2006;66:10357–10364. doi: 10.1158/0008-5472.CAN-06-2496. [DOI] [PubMed] [Google Scholar]
- 25.Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori M, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12:939–944. doi: 10.1038/nm1447. [DOI] [PubMed] [Google Scholar]
- 26.Wu WK, Wong HP, Luo SW, Chan K, Huang FY, Hui MK, Lam EK, Shin VY, Yi NY, Yang YH. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone from cigarette smoke stimulates colon cancer growth via β-adrenoceptors. Cancer Res. 2005;65:5272–5277. doi: 10.1158/0008-5472.CAN-05-0205. [DOI] [PubMed] [Google Scholar]
- 27.Masur K, Niggemann B, Zanker KS, Entschladen F. Norepinephrine-induced migration of SW 480 colon carcinoma cells is inhibited by β-blockers. Cancer Res. 2001;61:2866–2869. [PubMed] [Google Scholar]
- 28.Sood AK, Armaiz-Pena GN, Halder J, Nick AM, Stone RL, Hu W, Carroll AR, Spannuth WA, Deavers MT, Allen JK, et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J Clin Invest. 2010;120:1515–1523. doi: 10.1172/JCI40802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sloan EK, Priceman SJ, Cox BF, Yu S, Pimentel MA, Tangkanangnukul V, Arevalo JM, Morizono K, Karanikolas BD, Wu L, et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010;70:7042–7052. doi: 10.1158/0008-5472.CAN-10-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sood AK, Bhatty R, Kamat AA, Landen CN, Han L, Thaker PH, Li Y, Gershenson DM, Lutgendorf S, Cole SW. Stress hormone-mediated invasion of ovarian cancer cells. Clin Cancer Res. 2006;12:369–375. doi: 10.1158/1078-0432.CCR-05-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Springer J, Tschirner A, Haghikia A, von Haehling S, Lal H, Grzesiak A, Kaschina E, Palus S, Potsch M, von Websky K, et al. Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. Eur Heart J. 2014;35:932–941. doi: 10.1093/eurheartj/eht302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K. Beta blockers and breast cancer mortality: a population- based study. J Clin Oncol. 2011;29:2635–2644. doi: 10.1200/JCO.2010.33.5422. [DOI] [PubMed] [Google Scholar]
- 33.Diaz ES, Karlan BY, Li AJ. Impact of beta blockers on epithelial ovarian cancer survival. Gynecol Oncol. 2012;127:375–378. doi: 10.1016/j.ygyno.2012.07.102. [DOI] [PubMed] [Google Scholar]
- 34.Wang HM, Liao ZX, Komaki R, Welsh JW, O’Reilly MS, Chang JY, Zhuang Y, Levy LB, Lu C, Gomez DR. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann Oncol. 2013;24:1312–1319. doi: 10.1093/annonc/mds616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grytli HH, Fagerland MW, Fosså SD, Taskén KA. Association between use of β-blockers and prostate cancer–specific survival: a cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur Urol. 2014;65:635–641. doi: 10.1016/j.eururo.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 36.Ganz PA, Habel LA, Weltzien EK, Caan BJ, Cole SW. Examining the influence of beta blockers and ACE inhibitors on the risk for breast cancer recurrence: results from the LACE cohort. Breast Cancer Res Treat. 2011;129:549–556. doi: 10.1007/s10549-011-1505-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lemeshow S, Sørensen HT, Phillips G, Yang EV, Antonsen S, Riis AH, Lesinski GB, Jackson R, Glaser R. β-Blockers and survival among Danish patients with malignant melanoma: a population-based cohort study. Cancer Epidemiol Biomark Prev. 2011;20:2273–2279. doi: 10.1158/1055-9965.EPI-11-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi CH, Song T, Kim TH, Choi JK, Park JY, Yoon A, Lee YY, Kim TJ, Bae DS, Lee JW, Kim BG. Meta-analysis of the effects of beta blocker on survival time in cancer patients. J Cancer Res Clin Oncol. 2014;140:1179–1188. doi: 10.1007/s00432-014-1658-7. [DOI] [PubMed] [Google Scholar]
- 39.Goldfarb DA, Diz DI, Tubbs RR, Ferrario CM, Novick AC. Angiotensin II receptor subtypes in the human renal cortex and renal cell carcinoma. J Urol. 1994;151:208–213. doi: 10.1016/s0022-5347(17)34918-2. [DOI] [PubMed] [Google Scholar]
- 40.Marsigliante S, Resta L, Muscella A, Vinson GP, Marzullo A, Storelli C. AT1 angiotensin II receptor subtype in the human larynx and squamous laryngeal carcinoma. Cancer Lett. 1996;110:19–27. doi: 10.1016/S0304-3835(96)04449-7. [DOI] [PubMed] [Google Scholar]
- 41.Fujimoto Y, Sasaki T, Tsuchida A, Chayama K. Angiotensin II type 1 receptor expression in human pancreatic cancer and growth inhibition by angiotensin II type 1 receptor antagonist. FEBS Lett. 2001;495:197–200. doi: 10.1016/S0014-5793(01)02377-8. [DOI] [PubMed] [Google Scholar]
- 42.Suganuma T, Ino K, Shibata K, Kajiyama H, Nagasaka T, Mizutani S, Kikkawa F. Functional expression of the angiotensin II type 1 receptor in human ovarian carcinoma cells and its blockade therapy resulting in suppression of tumor invasion, angiogenesis, and peritoneal dissemination. Clin Cancer Res. 2005;11:2686–2694. doi: 10.1158/1078-0432.CCR-04-1946. [DOI] [PubMed] [Google Scholar]
- 43.Herr D, Rodewald M, Fraser HM, Hack G, Konrad R, Kreienberg R, Wulff C. Potential role of renin-angiotensin-system for tumor angiogenesis in receptor negative breast cancer. Gynecol Oncol. 2008;109:418–425. doi: 10.1016/j.ygyno.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 44.Otake AH, Mattar AL, Freitas HC, Machado CM, Nonogaki S, Fujihara CK, Zatz R, Chammas R. Inhibition of angiotensin II receptor 1 limits tumor-associated angiogenesis and attenuates growth of murine melanoma. Cancer Chemother Pharmacol. 2010;66:79–87. doi: 10.1007/s00280-009-1136-0. [DOI] [PubMed] [Google Scholar]
- 45.Okamoto K, Tajima H, Ohta T, Nakanuma S, Hayashi H, Nakagawara H, Onishi I, Takamura H, Ninomiya I, Kitagawa H, et al. Angiotensin II induces tumor progression and fibrosis in intrahepatic cholangiocarcinoma through an interaction with hepatic stellate cells. Int J Oncol. 2010;37:1251–1259. doi: 10.3892/ijo_00000776. [DOI] [PubMed] [Google Scholar]
- 46.Anandanadesan R, Gong Q, Chipitsyna G, Witkiewicz A, Yeo CJ, Arafat HA. Angiotensin II induces vascular endothelial growth factor in pancreatic cancer cells through an angiotensin II type 1 receptor and ERK1/2 signaling. J Gastrointest Surg. 2008;12:57–66. doi: 10.1007/s11605-007-0403-9. [DOI] [PubMed] [Google Scholar]
- 47.Neo JH, Malcontenti-Wilson C, Muralidharan V, Christophi C. Effect of ACE inhibitors and angiotensin II receptor antagonists in a mouse model of colorectal cancer liver metastases. J Gastroenterol Hepatol. 2007;22:577–584. doi: 10.1111/j.1440-1746.2006.04797.x. [DOI] [PubMed] [Google Scholar]
- 48.Yasumatsu R, Nakashima T, Masuda M, Ito A, Kuratomi Y, Nakagawa T, Komune S. Effects of the angiotensin-I converting enzyme inhibitor perindopril on tumor growth and angiogenesis in head and neck squamous cell carcinoma cells. J Cancer Res Clin Oncol. 2004;130:567–573. doi: 10.1007/s00432-004-0582-7. [DOI] [PubMed] [Google Scholar]
- 49.Lever AF, Hole DJ, Gillis CR, McCallum IR, McInnes GT, MacKinnon PL, Meredith PA, Murray LS, Reid JL, Robertson JW. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet. 1998;352:179–184. doi: 10.1016/S0140-6736(98)03228-0. [DOI] [PubMed] [Google Scholar]
- 50.Meier CR, Derby LE, Jick SS, Jick H. Angiotensin-converting enzyme inhibitors, calcium channel blockers, and breast cancer. Arch Intern Med. 2000;160:349–353. doi: 10.1001/archinte.160.3.349. [DOI] [PubMed] [Google Scholar]
- 51.Friis S, Sorensen HT, Mellemkjaer L, McLaughlin JK, Nielsen GL, Blot WJ, Olsen JH. Angiotensin-converting enzyme inhibitors and the risk of cancer: a population-based cohort study in Denmark. Cancer. 2001;92:2462–2470. doi: 10.1002/1097-0142(20011101)92:9<2462::AID-CNCR1596>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 52.Lindholm LH, Anderson H, Ekbom T, Hansson L, Lanke J, Dahlof B, de Faire U, Forsen K, Hedner T, Linjer E, et al. Relation between drug treatment and cancer in hypertensives in the Swedish trial in old patients with hypertension 2: a 5-year, prospective, randomised, controlled trial. Lancet. 2001;358:539–544. doi: 10.1016/S0140-6736(01)05704-X. [DOI] [PubMed] [Google Scholar]
- 53.Sjoberg T, Garcia Rodriguez LA, Lindblad M. Angiotensin-converting enzyme inhibitors and risk of esophageal and gastric cancer: a nested case-control study. Clin Gastroenterol Hepatol. 2007;5(1160–1166):e1161. doi: 10.1016/j.cgh.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 54.Ronquist G, Rodriguez LA, Ruigomez A, Johansson S, Wallander MA, Frithz G, Svardsudd K. Association between captopril, other antihypertensive drugs and risk of prostate cancer. Prostate. 2004;58:50–56. doi: 10.1002/pros.10294. [DOI] [PubMed] [Google Scholar]
- 55.Sipahi I, Debanne SM, Rowland DY, Simon DI, Fang JC. Angiotensin-receptor blockade and risk of cancer: meta-analysis of randomised controlled trials. Lancet Oncol. 2010;11:627–636. doi: 10.1016/S1470-2045(10)70106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pasternak B, Svanstrom H, Callreus T, Melbye M, Hviid A. Use of angiotensin receptor blockers and the risk of cancer. Circulation. 2011;123:1729–1736. doi: 10.1161/CIRCULATIONAHA.110.007336. [DOI] [PubMed] [Google Scholar]
- 57.ARB Trialists Collaboration Effects of telmisartan, irbesartan, valsartan, candesartan, and losartan on cancers in 15 trials enrolling 138,769 individuals. J Hypertens. 2011;29:623–635. doi: 10.1097/HJH.0b013e328344a7de. [DOI] [PubMed] [Google Scholar]
- 58.Shiratori O. Growth inhibitory effect of cardiac glycosides and aglycones on neoplastic cells: in vitro and in vivo studies. Gan. 1967;58:521–528. [PubMed] [Google Scholar]
- 59.Avila J, Lecuona E, Morales M, Soriano A, Alonso T, Martín-Vasallo P. Opposite expression pattern of the human Na, K-ATPase β1 isoform in stomach and colon adenocarcinomasa. Ann NY Acad Sci. 1997;834:653–655. doi: 10.1111/j.1749-6632.1997.tb52341.x. [DOI] [PubMed] [Google Scholar]
- 60.Espineda C, Seligson DB, James Ball W, Rao J, Palotie A, Horvath S, Huang Y, Shi T, Rajasekaran AK. Analysis of the Na, K-ATPase α-and β-subunit expression profiles of bladder cancer using tissue microarrays. Cancer. 2003;97:1859–1868. doi: 10.1002/cncr.11267. [DOI] [PubMed] [Google Scholar]
- 61.Rajasekaran SA, Ball WJ, Bander NH, Liu H, Pardee JD, Rajasekaran AK. Reduced expression of beta-subunit of Na, K-ATPase in human clear-cell renal cell carcinoma. J Urol. 1999;162:574–580. doi: 10.1016/S0022-5347(05)68629-6. [DOI] [PubMed] [Google Scholar]
- 62.Kawazoe N, Aiuchi T, Masuda Y, Nakajo S, Nakaya K. Induction of apoptosis by bufalin in human tumor cells is associated with a change of intracellular concentration of Na+ ions. J Biochem. 1999;126:278–286. doi: 10.1093/oxfordjournals.jbchem.a022446. [DOI] [PubMed] [Google Scholar]
- 63.Raghavendra PB, Sreenivasan Y, Ramesh GT, Manna SK. Cardiac glycoside induces cell death via FasL by activating calcineurin and NF-AT, but apoptosis initially proceeds through activation of caspases. Apoptosis. 2007;12:307–318. doi: 10.1007/s10495-006-0626-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Manna SK, Sah NK, Newman RA, Cisneros A, Aggarwal BB. Oleandrin suppresses activation of nuclear transcription factor-kappaB, activator protein-1, and c-Jun NH2-terminal kinase. Cancer Res. 2000;60:3838–3847. [PubMed] [Google Scholar]
- 65.Bielawski K, Winnicka K, Bielawska A. Inhibition of DNA topoisomerases I and II, and growth inhibition of breast cancer MCF-7 cells by ouabain, digoxin and proscillaridin A. Biol Pharm Bull. 2006;29:1493–1497. doi: 10.1248/bpb.29.1493. [DOI] [PubMed] [Google Scholar]
- 66.Chen J-Q, Contreras RG, Wang R, Fernandez SV, Shoshani L, Russo IH, Cereijido M, Russo J. Sodium/potasium ATPase (Na + , K + -ATPase) and ouabain/related cardiac glycosides: a new paradigm for development of anti-breast cancer drugs? Breast Cancer Res Treat. 2006;96:1–15. doi: 10.1007/s10549-005-9053-3. [DOI] [PubMed] [Google Scholar]
- 67.Nesher M, Shpolansky U, Rosen H, Lichtstein D. The digitalis-like steroid hormones: new mechanisms of action and biological significance. Life Sci. 2007;80:2093–2107. doi: 10.1016/j.lfs.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 68.Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol-Cell Physiol. 2007;293:C509–C536. doi: 10.1152/ajpcell.00098.2007. [DOI] [PubMed] [Google Scholar]
- 69.Stenkvist B, Bengtsson E, Eriksson O, Holmquist J, Nordin B, Westman-Naeser S. Cardiac glycosides and breast cancer. Lancet. 1979;1:563. doi: 10.1016/S0140-6736(79)90996-6. [DOI] [PubMed] [Google Scholar]
- 70.Stenkvist B, Bengtsson E, Dahlqvist B, Eriksson O, Jarkrans T, Nordin B. Cardiac glycosides and breast cancer, revisited. N Engl J Med. 1982;306:484. [PubMed] [Google Scholar]
- 71.Stenkvist B. Is digitalis a therapy for breast carcinoma? Oncol Rep. 1999;6:493–496. [PubMed] [Google Scholar]
- 72.Haux J, Klepp O, Spigset O, Tretli S. Digitoxin medication and cancer; case control and internal dose-response studies. BMC Cancer. 2001;1:11. doi: 10.1186/1471-2407-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ahern TP, Lash TL, Sorensen HT, Pedersen L. Digoxin treatment is associated with an increased incidence of breast cancer: a population-based case-control study. Breast Cancer Res. 2008;10:R102. doi: 10.1186/bcr2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Biggar RJ, Wohlfahrt J, Oudin A, Hjuler T, Melbye M. Digoxin use and the risk of breast cancer in women. J Clin Oncol. 2011;29:2165–2170. doi: 10.1200/JCO.2010.32.8146. [DOI] [PubMed] [Google Scholar]
- 75.Biggar RJ, Wohlfahrt J, Melbye M. Digoxin use and the risk of cancers of the corpus uteri, ovary and cervix. Int J Cancer. 2012;131:716–721. doi: 10.1002/ijc.26424. [DOI] [PubMed] [Google Scholar]
- 76.Platz EA, Yegnasubramanian S, Liu JO, Chong CR, Shim JS, Kenfield SA, Stampfer MJ, Willett WC, Giovannucci E, Nelson WG. A novel two-stage, transdisciplinary study identifies digoxin as a possible drug for prostate cancer treatment. Cancer Discov. 2011;1:68–77. doi: 10.1158/2159-8274.CD-10-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Demierre M-F, Higgins PDR, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer. 2005;5:930–942. doi: 10.1038/nrc1751. [DOI] [PubMed] [Google Scholar]
- 78.Ogunwobi OO, Beales IL. Statins inhibit proliferation and induce apoptosis in Barrett’s esophageal adenocarcinoma cells. Am J Gastroenterol. 2008;103:825–837. doi: 10.1111/j.1572-0241.2007.01773.x. [DOI] [PubMed] [Google Scholar]
- 79.Kang S, Kim ES, Moon A. Simvastatin and lovastatin inhibit breast cell invasion induced by H-Ras. Oncol Rep. 2009;21:1317–1322. doi: 10.3892/or_00000357. [DOI] [PubMed] [Google Scholar]
- 80.Denoyelle C, Vasse M, Korner M, Mishal Z, Ganne F, Vannier JP, Soria J, Soria C. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: an in vitro study. Carcinogenesis. 2001;22:1139–1148. doi: 10.1093/carcin/22.8.1139. [DOI] [PubMed] [Google Scholar]
- 81.Graaf MR, Beiderbeck AB, Egberts AC, Richel DJ, Guchelaar H-J. The risk of cancer in users of statins. J Clin Oncol. 2004;22:2388–2394. doi: 10.1200/JCO.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 82.Blais L, Desgagné A, LeLorier J. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors and the risk of cancer: a nested case-control study. Arch Intern Med. 2000;160:2363–2368. doi: 10.1001/archinte.160.15.2363. [DOI] [PubMed] [Google Scholar]
- 83.Bansal D, Undela K, D’Cruz S, Schifano F. Statin use and risk of prostate cancer: a meta-analysis of observational studies. PLoS ONE. 2012;7:e46691. doi: 10.1371/journal.pone.0046691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Singh S, Singh AG, Singh PP, Murad MH, Iyer PG. Statins are associated with reduced risk of esophageal cancer, particularly in patients with Barrett’s esophagus: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2013;11:620–629. doi: 10.1016/j.cgh.2012.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu X-D, Zeng K, Xue F-Q, Chen J-H, Chen Y-Q. Statins are associated with reduced risk of gastric cancer: a meta-analysis. Eur J Clin Pharmacol. 2013;69:1855–1860. doi: 10.1007/s00228-013-1547-z. [DOI] [PubMed] [Google Scholar]
- 86.Emberson JR, Kearney PM, Blackwell L, Newman C, Reith C, Bhala N, Holland L, Peto R, Keech A, Collins R, et al. Lack of effect of lowering LDL cholesterol on cancer: meta-analysis of individual data from 175,000 people in 27 randomised trials of statin therapy. PLoS ONE. 2012;7:e29849. doi: 10.1371/journal.pone.0029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tan M, Song X, Zhang G, Peng A, Li X, Li M, Liu Y, Wang C. Statins and the risk of lung cancer: a meta-analysis. PLoS ONE. 2013;8:e57349. doi: 10.1371/journal.pone.0057349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li X, Wu X, Chen Q. Statin use is not associated with reduced risk of skin cancer: a meta-analysis. Br J Cancer. 2014;110:802–807. doi: 10.1038/bjc.2013.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kornblau SM, Banker DE, Stirewalt D, Shen D, Lemker E, Verstovsek S, Estrov Z, Faderl S, Cortes J, Beran M. Blockade of adaptive defensive changes in cholesterol uptake and synthesis in AML by the addition of pravastatin to idarubicin + high-dose Ara-C: a phase 1 study. Blood. 2007;109:2999–3006. doi: 10.1182/blood-2006-08-044446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Graf H, Jüngst C, Straub G, Dogan S, Hoffmann R-T, Jakobs T, Reiser M, Waggershauser T, Helmberger T, Walter A. Chemoembolization combined with pravastatin improves survival in patients with hepatocellular carcinoma. Digestion. 2008;78:34–38. doi: 10.1159/000156702. [DOI] [PubMed] [Google Scholar]
- 91.Leaute-Labreze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taieb A. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649–2651. doi: 10.1056/NEJMc0708819. [DOI] [PubMed] [Google Scholar]
- 92.Sans V, de la Roque ED, Berge J, Grenier N, Boralevi F, Mazereeuw-Hautier J, Lipsker D, Dupuis E, Ezzedine K, Vergnes P, et al. Propranolol for severe infantile hemangiomas: follow-up report. Pediatrics. 2009;124:e423–e431. doi: 10.1542/peds.2008-3458. [DOI] [PubMed] [Google Scholar]
- 93.Bertrand J, McCuaig C, Dubois J, Hatami A, Ondrejchak S, Powell J. Propranolol versus prednisone in the treatment of infantile hemangiomas: a retrospective comparative study. Pediatr Dermatol. 2011;28:649–654. doi: 10.1111/j.1525-1470.2011.01551.x. [DOI] [PubMed] [Google Scholar]
- 94.Izadpanah A, Izadpanah A, Kanevsky J, Belzile E, Schwarz K. Propranolol versus Corticosteroids in the treatment of infantile hemangioma. Plast Reconstr Surg. 2013;131:601–613. doi: 10.1097/PRS.0b013e31827c6fab. [DOI] [PubMed] [Google Scholar]
- 95.Leaute-Labreze C, Dumas de la Roque E, Nacka F, Abouelfath A, Grenier N, Rebola M, Ezzedine K, Moore N. Double-blind randomized pilot trial evaluating the efficacy of oral propranolol on infantile haemangiomas in infants <4 months of age. Br J Dermatol. 2013;169:181–183. doi: 10.1111/bjd.12217. [DOI] [PubMed] [Google Scholar]
- 96.Hogeling M, Adams S, Wargon O. A randomized controlled trial of propranolol for infantile hemangiomas. Pediatrics. 2011;128:e259–e266. doi: 10.1542/peds.2010-0029. [DOI] [PubMed] [Google Scholar]
- 97.Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, Guibaud L, Baselga E, Posiunas G, Phillips RJ, Caceres H, Lopez Gutierrez JC, Ballona R, et al. A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med. 2015;372:735–746. doi: 10.1056/NEJMoa1404710. [DOI] [PubMed] [Google Scholar]
- 98.Rao BS, Das DG, Taraknath VR, Sarma Y. A double blind controlled study of propranolol and cyproheptadine in migraine prophylaxis. Neurol India. 2000;48:223–226. [PubMed] [Google Scholar]
- 99.Diener HC, Hartung E, Chrubasik J, Evers S, Schoenen J, Eikermann A, Latta G, Hauke W. A comparative study of oral acetylsalicyclic acid and metoprolol for the prophylactic treatment of migraine. A randomized, controlled, double-blind, parallel group phase III study. Cephalalgia. 2001;21:120–128. doi: 10.1046/j.1468-2982.2001.00168.x. [DOI] [PubMed] [Google Scholar]
- 100.Silberstein SD, Holland S, Freitag F, Dodick DW, Argoff C, Ashman E. Evidence-based guideline update: pharmacologic treatment for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology. 2012;78:1337–1345. doi: 10.1212/WNL.0b013e3182535d20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garcia-Tsao G, Lim JK. Management and treatment of patients with cirrhosis and portal hypertension: recommendations from the Department of Veterans Affairs Hepatitis C Resource Center Program and the National Hepatitis C Program. Am J Gastroenterol. 2009;104:1802–1829. doi: 10.1038/ajg.2009.191. [DOI] [PubMed] [Google Scholar]
- 102.Hillon P, Lebrec D, Munoz C, Jungers M, Goldfarb G, Benhamou JP. Comparison of the effects of a cardioselective and a nonselective beta-blocker on portal hypertension in patients with cirrhosis. Hepatology. 1982;2:528–531. doi: 10.1002/hep.1840020503. [DOI] [PubMed] [Google Scholar]
- 103.Westaby D, Melia WM, Macdougall BR, Hegarty JE, Gimson AE, Williams R. B1 selective adrenoreceptor blockade for the long term management of variceal bleeding. A prospective randomised trial to compare oral metoprolol with injection sclerotherapy in cirrhosis. Gut. 1985;26:421–425. doi: 10.1136/gut.26.4.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bosch J. Carvedilol for portal hypertension in patients with cirrhosis. Hepatology. 2010;51:2214–2218. doi: 10.1002/hep.23689. [DOI] [PubMed] [Google Scholar]
- 105.Bernard B, Lebrec D, Mathurin P, Opolon P, Poynard T. Beta-adrenergic antagonists in the prevention of gastrointestinal rebleeding in patients with cirrhosis: a meta-analysis. Hepatology. 1997;25:63–70. doi: 10.1002/hep.510250112. [DOI] [PubMed] [Google Scholar]
- 106.Gluud LL, Langholz E, Krag A. Meta-analysis: isosorbide-mononitrate alone or with either beta-blockers or endoscopic therapy for the management of oesophageal varices. Aliment Pharmacol Ther. 2010;32:859–871. doi: 10.1111/j.1365-2036.2010.04418.x. [DOI] [PubMed] [Google Scholar]
- 107.Thiele M, Krag A, Rohde U, Gluud LL. Meta-analysis: banding ligation and medical interventions for the prevention of rebleeding from oesophageal varices. Aliment Pharmacol Ther. 2012;35:1155–1165. doi: 10.1111/j.1365-2036.2012.05074.x. [DOI] [PubMed] [Google Scholar]
- 108.Poynard T, Cales P, Pasta L, Ideo G, Pascal JP, Pagliaro L, Lebrec D. Beta-adrenergic-antagonist drugs in the prevention of gastrointestinal bleeding in patients with cirrhosis and esophageal varices. An analysis of data and prognostic factors in 589 patients from four randomized clinical trials. Franco-Italian Multicenter Study Group. N Engl J Med. 1991;324:1532–1538. doi: 10.1056/NEJM199105303242202. [DOI] [PubMed] [Google Scholar]
- 109.de Franchis R. Expanding consensus in portal hypertension: report of the Baveno VI Consensus Workshop: Stratifying risk and individualizing care for portal hypertension. J Hepatol. 2015;63:743–752. doi: 10.1016/j.jhep.2015.05.022. [DOI] [PubMed] [Google Scholar]
- 110.Groszmann RJ, Garcia-Tsao G, Bosch J, Grace ND, Burroughs AK, Planas R, Escorsell A, Garcia-Pagan JC, Patch D, Matloff DS, et al. Beta-blockers to prevent gastroesophageal varices in patients with cirrhosis. N Engl J Med. 2005;353:2254–2261. doi: 10.1056/NEJMoa044456. [DOI] [PubMed] [Google Scholar]
- 111.Serste T, Melot C, Francoz C, Durand F, Rautou PE, Valla D, Moreau R, Lebrec D. Deleterious effects of beta-blockers on survival in patients with cirrhosis and refractory ascites. Hepatology. 2010;52:1017–1022. doi: 10.1002/hep.23775. [DOI] [PubMed] [Google Scholar]
- 112.Moore RE, Smith CK, 2nd, Bailey CS, Voelkel EF, Tashjian AH., Jr Characterization of beta-adrenergic receptors on rat and human osteoblast-like cells and demonstration that beta-receptor agonists can stimulate bone resorption in organ culture. Bone Miner. 1993;23:301–315. doi: 10.1016/S0169-6009(08)80105-5. [DOI] [PubMed] [Google Scholar]
- 113.Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, Armstrong D, Ducy P, Karsenty G. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111:305–317. doi: 10.1016/S0092-8674(02)01049-8. [DOI] [PubMed] [Google Scholar]
- 114.Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, Shen J, Vinson C, Rueger JM, Karsenty G. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100:197–207. doi: 10.1016/S0092-8674(00)81558-5. [DOI] [PubMed] [Google Scholar]
- 115.Elefteriou F, Ahn JD, Takeda S, Starbuck M, Yang X, Liu X, Kondo H, Richards WG, Bannon TW, Noda M, et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434:514–520. doi: 10.1038/nature03398. [DOI] [PubMed] [Google Scholar]
- 116.Yang S, Nguyen ND, Eisman JA, Nguyen TV. Association between beta-blockers and fracture risk: a Bayesian meta-analysis. Bone. 2012;51:969–974. doi: 10.1016/j.bone.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 117.Song HJ, Lee J, Kim Y-J, Jung S-Y, Kim HJ, Choi N-K, Park B-J. β1 selectivity of β-blockers and reduced risk of fractures in elderly hypertension patients. Bone. 2012;51:1008–1015. doi: 10.1016/j.bone.2012.08.126. [DOI] [PubMed] [Google Scholar]
- 118.Toulis KA, Hemming K, Stergianos S, Nirantharakumar K, Bilezikian JP. β-adrenergic receptor antagonists and fracture risk: a meta-analysis of selectivity, gender, and site-specific effects. Osteoporos Int. 2013;25:121–129. doi: 10.1007/s00198-013-2498-z. [DOI] [PubMed] [Google Scholar]
- 119.Gribbin J, Hubbard R, Gladman JRF, Smith C, Lewis S. Risk of falls associated with antihypertensive medication: population-based case-control study. Age Ageing. 2010;39:592–597. doi: 10.1093/ageing/afq092. [DOI] [PubMed] [Google Scholar]
- 120.Zang G. Antihypertensive drugs and the risk of fall injuries: a systematic review and meta-analysis. J Int Med Res. 2013;41:1408–1417. doi: 10.1177/0300060513497562. [DOI] [PubMed] [Google Scholar]
- 121.Lipsitz LA, Habtemariam D, Gagnon M, Iloputaife I, Sorond F, Tchalla AE, Dantoine TF, Travison TG. Reexamining the effect of antihypertensive medications on falls in old age. Hypertension. 2015;66:183–189. doi: 10.1161/HYPERTENSIONAHA.115.05513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 123.Dietz HC. TGF-beta in the pathogenesis and prevention of disease: a matter of aneurysmic proportions. J Clin Invest. 2010;120:403–407. doi: 10.1172/JCI42014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473:308–316. doi: 10.1038/nature10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC., 3rd Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pees C, Laccone F, Hagl M, Debrauwer V, Moser E, Michel-Behnke I. Usefulness of losartan on the size of the ascending aorta in an unselected cohort of children, adolescents, and young adults with Marfan syndrome. Am J Cardiol. 2013;112:1477–1483. doi: 10.1016/j.amjcard.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 128.Groenink M, den Hartog AW, Franken R, Radonic T, de Waard V, Timmermans J, Scholte AJ, van den Berg MP, Spijkerboer AM, Marquering HA, et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J. 2013;34:3491–3500. doi: 10.1093/eurheartj/eht334. [DOI] [PubMed] [Google Scholar]
- 129.Lacro RV, Dietz HC, Sleeper LA, Yetman AT, Bradley TJ, Colan SD, Pearson GD, Tierney ES, Levine JC, Atz AM, et al. Atenolol versus Losartan in children and young adults with Marfan’s syndrome. N Engl J Med. 2014;371:2061–2071. doi: 10.1056/NEJMoa1404731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhou Q, Liao JK. Pleiotropic effects of statins: basic research and clinical perspectives. Circul J. 2010;74:818. doi: 10.1253/circj.CJ-10-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lindenauer PK, Pekow P, Wang K, Gutierrez B, Benjamin EM. Lipid-lowering therapy and in-hospital mortality following major noncardiac surgery. JAMA. 2004;291:2092–2099. doi: 10.1001/jama.291.17.2092. [DOI] [PubMed] [Google Scholar]
- 132.Hindler K, Shaw AD, Samuels J, Fulton S, Collard CD, Riedel B. Improved postoperative outcomes associated with preoperative statin therapy. J Am Soc Anesthesiol. 2006;105:1260–1272. doi: 10.1097/00000542-200612000-00027. [DOI] [PubMed] [Google Scholar]
- 133.Desai H, Aronow WS, Ahn C, Gandhi K, Amin H, Lai HM, Tsai FS, Sharma M, Babu S. Incidence of perioperative myocardial infarction and of 2-year mortality in 577 elderly patients undergoing noncardiac vascular surgery treated with and without statins. Arch Gerontol Geriatr. 2010;51:149–151. doi: 10.1016/j.archger.2009.09.042. [DOI] [PubMed] [Google Scholar]
- 134.Lau WC, Froehlich JB, Jewell ES, Montgomery DG, Eng KM, Shields TA, Henke PK, Eagle KA. Impact of adding aspirin to Beta-blocker and statin in high-risk patients undergoing major vascular surgery. Ann Vasc Surg. 2013;27:537–545. doi: 10.1016/j.avsg.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 135.Durazzo AE, Machado FS, Ikeoka DT, De Bernoche C, Monachini MC, Puech-Leão P, Caramelli B. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg. 2004;39:967–975. doi: 10.1016/j.jvs.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 136.Kristensen SD, Knuuti J, Saraste A, Anker S, Bøtker HE, De Hert S, Ford I, Gonzalez-Juanatey JR, Gorenek B, Heyndrickx GR: 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management. Eur Heart J. 2014:ehu282.
- 137.Fleisher LA, Fleischmann KE, Auerbach AD, Barnason SA, Beckman JA, Bozkurt B, Davila-Roman VG, Gerhard-Herman MD, Holly TA, Kane GC. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;64:e77–e137. doi: 10.1016/j.jacc.2014.07.944. [DOI] [PubMed] [Google Scholar]
- 138.Tumlin J, Stacul F, Adam A, Becker CR, Davidson C, Lameire N, McCullough PA. Pathophysiology of contrast-induced nephropathy. Am J Cardiol. 2006;98:14–20. doi: 10.1016/j.amjcard.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 139.John S, Schneider MP, Delles C, Jacobi J, Schmieder RE. Lipid-independent effects of statins on endothelial function and bioavailability of nitric oxide in hypercholesterolemic patients. Am Heart J. 2005;149:473. doi: 10.1016/j.ahj.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 140.Ridker PM, Rifai N, Clearfield M, Downs JR, Weis SE, Miles JS, Gotto AM., Jr Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med. 2001;344:1959–1965. doi: 10.1056/NEJM200106283442601. [DOI] [PubMed] [Google Scholar]
- 141.Wagner AH, Köhler T, Rückschloss U, Just I, Hecker M. Improvement of nitric oxide–dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol. 2000;20:61–69. doi: 10.1161/01.ATV.20.1.61. [DOI] [PubMed] [Google Scholar]
- 142.Gueler F, Rong S, Park J-K, Fiebeler A, Menne J, Elger M, Mueller DN, Hampich F, Dechend R, Kunter U. Postischemic acute renal failure is reduced by short-term statin treatment in a rat model. J Am Soc Nephrol. 2002;13:2288–2298. doi: 10.1097/01.ASN.0000026609.45827.3D. [DOI] [PubMed] [Google Scholar]
- 143.Li Y, Liu Y, Fu L, Mei C, Dai B. Efficacy of short-term high-dose statin in preventing contrast-induced nephropathy: a meta-analysis of seven randomized controlled trials. PLoS ONE. 2012;7:e34450. doi: 10.1371/journal.pone.0034450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Quintavalle C, Fiore D, De Micco F, Visconti G, Focaccio A, Golia B, Ricciardelli B, Donnarumma E, Bianco A, Zabatta MA. Impact of a high loading dose of atorvastatin on contrast-induced acute kidney injury. Circulation. 2012;126:3008–3016. doi: 10.1161/CIRCULATIONAHA.112.103317. [DOI] [PubMed] [Google Scholar]
- 145.Leoncini M, Toso A, Maioli M, Tropeano F, Villani S, Bellandi F. Early high-dose rosuvastatin for contrast-induced nephropathy prevention in acute coronary syndrome. J Am Coll Cardiol. 2014;63:71–79. doi: 10.1016/j.jacc.2013.04.105. [DOI] [PubMed] [Google Scholar]
- 146.Devine BL, Fife R, Trust PM. Minoxidil for severe hypertension after failure of other hypotensive drugs. Br Med J. 1977;2:667–669. doi: 10.1136/bmj.2.6088.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jacobs D, Buttigieg CF. Minoxidil experience in Australia 1974–1980. Med J Aust. 1981;1:477–478. doi: 10.5694/j.1326-5377.1981.tb135742.x. [DOI] [PubMed] [Google Scholar]
- 148.Kreindler TG. Topical minoxidil in early androgenetic alopecia. J Am Acad Dermatol. 1987;16:718–724. doi: 10.1016/S0190-9622(87)70093-0. [DOI] [PubMed] [Google Scholar]
- 149.Wester RC, Maibach HI, Guy RH, Novak E. Minoxidil stimulates cutaneous blood flow in human balding scalps: pharmacodynamics measured by laser Doppler velocimetry and photopulse plethysmography. J Invest Dermatol. 1984;82:515–517. doi: 10.1111/1523-1747.ep12261084. [DOI] [PubMed] [Google Scholar]
- 150.Lachgar S, Charveron M, Gall Y, Bonafe JL. Minoxidil upregulates the expression of vascular endothelial growth factor in human hair dermal papilla cells. Br J Dermatol. 1998;138:407–411. doi: 10.1046/j.1365-2133.1998.02115.x. [DOI] [PubMed] [Google Scholar]
- 151.Buhl AE, Waldon DJ, Conrad SJ, Mulholland MJ, Shull KL, Kubicek MF, Johnson GA, Brunden MN, Stefanski KJ, Stehle RG, et al. Potassium channel conductance: a mechanism affecting hair growth both in vitro and in vivo. J Invest Dermatol. 1992;98:315–319. doi: 10.1111/1523-1747.ep12499788. [DOI] [PubMed] [Google Scholar]
- 152.Roberts JL. Androgenetic alopecia: treatment results with topical minoxidil. J Am Acad Dermatol. 1987;16:705–710. doi: 10.1016/S0190-9622(87)70091-7. [DOI] [PubMed] [Google Scholar]
- 153.Olsen EA, Weiner MS, Amara IA, DeLong ER. Five-year follow-up of men with androgenetic alopecia treated with topical minoxidil. J Am Acad Dermatol. 1990;22:643–646. doi: 10.1016/0190-9622(90)70089-Z. [DOI] [PubMed] [Google Scholar]
- 154.Price VH, Menefee E, Strauss PC. Changes in hair weight and hair count in men with androgenetic alopecia, after application of 5% and 2% topical minoxidil, placebo, or no treatment. J Am Acad Dermatol. 1999;41:717–721. doi: 10.1016/S0190-9622(99)70006-X. [DOI] [PubMed] [Google Scholar]
- 155.Fryzek JP, Poulsen AH, Lipworth L, Pedersen L, Nørgaard M, McLaughlin JK, Friis S. A cohort study of antihypertensive medication use and breast cancer among Danish women. Breast Cancer Res Treat. 2006;97:231–236. doi: 10.1007/s10549-005-9091-x. [DOI] [PubMed] [Google Scholar]
- 156.Assimes TL, Elstein E, Langleben A, Suissa S. Long-term use of antihypertensive drugs and risk of cancer. Pharmacoepidemiol Drug Saf. 2008;17:1039–1049. doi: 10.1002/pds.1656. [DOI] [PubMed] [Google Scholar]
- 157.Powe DG, Voss MJ, Zänker KS, Habashy HO, Green AR, Ellis IO, Entschladen F. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget. 2010;1:628–638. doi: 10.18632/oncotarget.197. [DOI] [PMC free article] [PubMed] [Google Scholar]