

Abstract
Infiltration of activated lymphocytes into the central nervous system (CNS) is potentially harmful by damaging resident cells through release of cytokines. Among these is IFN-γ that is secreted by activated natural killer (NK) cells and T lymphocytes and can exert a cytotoxic effect on resident glial populations including oligodendrocytes. Here we show that treatment of mouse oligodendrocyte progenitor cell (OPC)-enriched cultures with IFN-γ resulted in a dose-dependent increase in apoptosis. IFN-γ-induced apoptosis is mediated, in part, through induction of the CXC chemokine ligand 10 (CXCL10; IP-10) from cultured OPCs. Treatment of OPCs with CXCL10 resulted in cell death in a concentration-dependent manner and IFN-γ-treatment of CXCL10−/− OPCs resulted in >50% reduction in cell death. Further, treatment of CXCR3−/− OPC cultures with either IFN-γ or CXCL10 resulted in reduced cell death supporting an important role for CXCL10 signaling in IFN-γ-mediated OPC apoptosis. Data is also provided demonstrating that signaling through CXCR2 protects either IFN-γ or CXCL10-treated OPC cultures from apoptosis and this effect is abolished in CXCR2−/− OPCs. CXCR2-mediated protection from apoptosis is associated with impaired cleavage of caspase 3 and elevated expression of the anti-apoptotic protein Bcl-2. These findings demonstrate a previously unappreciated role for CXCL10 in contributing to neuropathology by promoting oligodendrocyte apoptosis and emphasize the potential relevance in targeting CXCL10 in treating human demyelinating diseases including multiple sclerosis (MS).
Keywords: oligodendrocyte progenitors, chemokines, apoptosis, cytokines
INTRODUCTION
Interferon-γ (IFN-γ) is a pleiotropic cytokine released by activated natural killer (NK) cells and antigen-sensitized T lymphocytes following recognition of cognate antigen presented by the appropriate MHC molecule. While secretion of IFN-γ can aid in host defense in response to viral infection of the CNS by enhancing antigen presentation via elevated MHC expression and exerting direct anti-viral effects (Malone et al., 2008; Parra et al., 1999) this cytokine is also directly toxic to glial cells of the oligodendrocyte lineage. In particular, exposure of cultured oligodendrocyte progenitor cells (OPCs) to IFN-γ restricts proliferation and differentiation as well as triggering apoptosis (Baerwald and Popko, 1998; Balabanov et al., 2006; Chew et al., 2005; Grinspan et al., 1993, 1996; Horiuchi et al., 2006; Lin et al., 2008; Vartanian et al., 1995; Wang et al., 2010). Moreover, overexpression of IFN-γ within the CNS of transgenic mice results in severe behavioral deficits associated with deleterious consequences on oligodendrocytes that correlate with hypomyelination (LaFerla et al., 2000; Lin et al., 2006). Mechanisms by which IFN-γ mediates injury include STAT/MEK-ERK signaling pathways as well as endoplasmic reticulum (ER) stress (Baerwald and Popko, 1998; Horiuchi et al., 2006; Lin and Popko, 2009; Lin et al., 2006, 2007, 2008; Wang et al., 2010).
In addition to understanding how cytokines such as IFN-γ promote loss of OPCs/oligodendrocytes, it is also imperative to evaluate how these cells are protected from the damaging effects of IFN-γ signaling. Failed remyelination by OPCs is a characteristic feature of MS and may be associated with impaired OPC function and/or death of these cells (Chang et al., 2002). Further, during chronic inflammatory diseases such as MS, OPCs/oligodendrocytes are exposed to numerous inflammatory cytokines/chemokines that create a hostile and damaging environment. Therefore, it is reasonable to assume that these cells have developed mechanism(s) by which to dampen the toxic effect of exposure to harmful factors secreted into the microenvironment by inflammatory cells. IFN-γ induces transcription of numerous genes including inflammatory chemokines including the CXC chemokine ligand 10 (CXCL10). CXCL10 is a potent chemoattractant for activated Th1 cells and NK cells by binding to the receptor CXCR3. More CXCL10 signaling through CXCR3 has recently been shown to promote neuronal apoptosis indicating that CXCL10 may also contribute to neuropathology by killing resident cells of the CNS (Sui et al., 2004, 2006; van Marle et al., 2007; Zhang et al., 2010).
The current study examines the mechanisms by which IFN-γ mediates apoptosis of cultured OPCs. Evidence is provided that IFN-γ induces CXCL10 expression in cultured stem cell-derived OPCs and contributes to apoptosis through a caspase-dependent mechanism. OPC cultures derived from either CXCL10−/− or CXCR3−/− mice exhibited reduced sensitivity to IFN-γ-induced apoptosis. Moreover, signaling through the CXC chemokine receptor 2 (CXCR2) via engagement with ligand CXCL1 restricts both IFN-γ and CXCL10-mediated apoptosis associated with limiting cleavage of caspase 3 and sustaining Bcl2 expression. Importantly, incubation of CXCL1 with CXCR2−/− OPC cultures challenged with either IFN-γ or CXCL10 did not inhibit cleavage of caspase 3 or protect against apoptosis. These findings are relevant to the development of novel therapies designed to increase OPC survival and enhancing remyelination.
MATERIALS AND METHODS
Cell Culture and Immunofluorescence
Striatum from postnatal day 1 mice were dissected, triturated, and cultured as previously described (Hardison et al., 2006; Totoiu et al., 2004; Whitman et al., 2009). To assess culture purity, OPC cultures were plated on MatriGel coated imaging slides and 4-day differentiated cultures were washed in HBSS, fixed in 4% paraformaldehyde (Fisher Scientific) for 30 min, and blocked with 10% normal goat serum (NGS, Vector Laboratories, Burlingame, CA) 1 hr at room temperature. Primary antibodies were added serially overnight: rabbit anti-mouse GalC galactocerebroside 1:100 (Millipore, CA), rabbit anti-mouse NG2 chondroitin sulfate proteoglycan 1:200 (Millipore, CA), rabbit anti-mouse Map2 1:700 (Millipore, CA), rabbit anti-mouse GFAP rabbit 1:500 (Abcam), rat anti-mouse F4/80 1:100 (AbD Serotec) and mouse anti-mouse oligodendrocyte marker O1 1:100 (Sigma) or blocking solution (negative control, 10% NGS in PBS). Slides were rinsed three times with PBS and appropriate fluorescent-conjugated goat secondary antibodies (Alexa 488 nm and 594 nm, 1:500 dilution in 10% NGS; Invitrogen) were applied and incubated for 1 hr at room temperature. Slides were rinsed three times in PBS, and nuclear staining was conducted through inclusion of Vectashield Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA). Slides were analyzed using an Olympus FV1000 confocal microscope. Z-stacks from confocal images as well as cell quantification were generated using Volocity Image analysis software (Improvision). The frequency of immunopositive cells was determined by dividing the total number of immunopositive cells by the total number of DAPI-positive cells in three images from each chamber, and averaging the results from two different chambers per marker obtained from three separate experiments. Only immunopositive cells with a DAPI-positive nucleus were counted. OPC cultures were grown on imaging slides for the entire time of the treatments for a total of 9 days.
Mice
C57BL/6 mice (on the H-2b background) were purchased from the National Cancer Institute (Frederick, MD). Additional mouse strains used for OPC cultures included CXCL10−/−, CXCR2−/−, CXCR3−/− mice (all on the C57BL/6, H-2b background) and were bred in the University of California, Irvine animal facility. The animal protocols and procedures used for these studies were reviewed and approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
Cell Death Assays
For MTT assay, cells were cultured in 96-well plates with the addition of treatments, or media as a control and cytotoxicity was measured using the cell titer assay (Promega, Madison, WI) according to manufacturer’s instructions. The formazan formed from MTT in actively metabolizing cells was measured at an optical density of 570 nm and absorbance values were used to calculate the percentages of cell death relative to control. To identify apoptosis in experimental OPC primary cultures, a TMR red In Situ Oligo Ligation Apoptosis Detection kit (Roche) was used on fixed cells. Immumofluorescence was performed by using an Olympus Fluoview 1000 confocal microscope and imaging analysis were carried out using Volocity 3D software (Improvision). For cell quantification, each treatment was carried out in two chambers and three fields of each chamber were examined. Results are expressed as mean ± SD of at least three different experiments in each time point, and used for statistical analysis.
Western Blotting
Primary cultures were processed by western blotting as previously described (Hosking et al., 2010). Antibodies used: rabbit anti-total caspase 3 (1:1,000), rabbit anti-cleaved caspase 3 (1:1,000), rabbit anti-PARP (1:1,000; Cell Signaling, MA), goat anti-Bcl-2 (1:5,000; R&D System, MN), rabbit anti-CXCR2 (1:2,000; (Hosking et al., 2010)), rabbit anti-CXCR3 (1:500; Abcam) and mouse monoclonal actin (1:5,000; Millipore, MA). Immune complexes were detected using appropriate peroxidase-conjugated secondary antibodies (1:25,000; Jackson ImmunoResearch Laboratory) and then exposed to a chemiluminescent reagent (Super-Signal West-Femto, Pierce). Densitometric analysis was performed within the linear range with Image J (NIH) and normalized to actin levels. Results were normalized to respective control conditions.
Semiquantitative Real-Time PCR
Total cDNA experimental was generated as previously described (Hosking et al., 2010). Real-time Taqman analysis for Bcl-2, Bcl-xL, Bax, CXCL1, and GAPDH was performed using a BioRad (Hercules, CA) iCycler with defined primers. Amplicon expression was normalized to GAPDH. All primers were purchased from Invitrogen (Carlsbad, CA) and an iQ Supermix (BioRad) was used for all reactions. Assay conditions were as follows: a 10 min initial denaturation at 95°C, and 45 cycles of 30 sec at 95°C and 1 min at 60°C, for Bcl-2, Bcl-xL, and Bax and 1 min at 58°C for CXCL1. Data were analyzed with BioRad iCycler iQ5 and quantified with the Relative Expression Software Tool (Pfaffl, 2001).
Recombinant Mouse Cytokines/Chemokines
Primary OPC cultures were treated with recombinant mouse cytokines/chemokines including IFN-γ (10, 50, and 100 U/mL, Cell Science, Canton MA), CXCL10 (0.1, 1 and 10 ng/mL, Peprotech, Rocky Hill, NJ), CXCL1 and CXCL2 (10 ng/mL, Peprotech) for an extended period of 6 days.
ELISA
Assessment of CXCL9, CXCL10, CXCL1, and CXCL2 in the supernatants of treated as well as untreated control OPC cultures was determined with mouse CXCL9 (MIG), CXCL10 (IP-10), CXCL1 (KC), and CXCL2 (MIP-2) DuoSet sandwich ELISA kit (R&D Systems, Minneapolis, MN) according to manufacturer specifications and results are presented as pg/mL.
Statistical Analysis
All data is presented as average ± SD. Statistically significant differences were assessed by either unpaired Student’s t-test or one-way ANOVA, and p values less than 0.01 were considered significant.
RESULTS
NG2+O1− OPCs are Sensitive to IFN-γ-Induced Apoptosis
OPC-enriched cultures were differentiated from primary mouse neural progenitor cells (NPCs) as previously described (Hardison et al., 2006; Totoiu et al., 2004; Whitman et al., 2009) and analyzed by immunocytochemical staining carried out with stage-specific markers. Quantification of fluorescent staining performed on confocal images showed more than 90% of cells immunoreactive to NG2, and less than 10% were O1 positive immediately following differentiation. NG2 staining revealed bipolar cells characteristic of immature OPCs (Fig. 1A). By day 6 postdifferentiation, the frequency of NG2-positive cells decreased while O1 expression increased (Fig. 1B,C) suggesting maturation of cells. Indeed, the cellular morphology reveals many of the cells exhibit a branched morphology characteristic of late OPCs or immature oligodendrocytes (Fig. 1B). Under these culture conditions, GFAP-positive cells can be detected although at very low frequency and expression of F4/80 was not found (not shown). Cell cultures were exposed to IFN-γ at day 6 following differentiation to evaluate susceptibility to potential toxic effects. Using an MTT assay to assess cell viability, we demonstrated that OPC-enriched cultures displayed increased cell death when incubated with IFN-γ for 6 days and this response was dose-dependent (Fig. 2A). Approximately 40% of cultured cells were TUNEL-positive suggesting cells were undergoing apoptosis in response to IFN-γ treatment (Fig. 2B). Immunophenotyping of cultures reveals a predominant NG2+O1− bipolar population at the beginning of IFN-γ treatment (Fig. 2C). However, by day 6 post-IFN-γ treatment there was an ~30% drop in NG2+O1− cells suggesting these cells are susceptible to IFN-γ-induced apoptosis (Fig. 2D,G). In contrast, NG2−O1+ cells numbers did not change dramatically following IFN-γ-treatment suggesting these cells are resistant to IFN-γ-mediated death (not shown). Indeed, dual staining for TUNEL in combination with GalC revealed that GalC-positive cells (representing immature oligodendrocytes) were protected from IFN-γ-induced apoptosis compared with media-treated controls (Fig. 2E,F). Thus, NG2+O1−-enriched cultures treated with IFN-γ resulted in death consistent with previous findings (Baerwald and Popko, 1998; Balabanov et al., 2006; Chew et al., 2005; Horiuchi et al., 2006; Lin et al., 2008; Vartanian et al., 1995; Wang et al., 2010). Moreover, these observations are consistent with previous studies demonstrating differences in susceptibility to IFN-γ-induced apoptosis between OPCs and more mature cells of the oligodendrocyte lineage (Baerwald and Popko, 1998; Vartanian et al., 1995).
Fig. 1.

Differentiation of neural progenitor cultures. (A) NSCs from striatal tissues of P1 C57/B6 litters were differentiated and subsequently stained with antibodies reactive to either mouse NG2 or O1. (B) After 6 days, both NG2 and O1 cells were detected by immunofluorescent staining. In (A) and (B), nuclei were stained with DAPI (blue). (C) Frequency of NG2+O1− and NG2−O1+ cells at days 0 and 6 postdifferentiation. Data represent the percentage of cells staining positive for either antigen and presented as average ± SD from two independent experiments. **Statistical significance for P values ≤0.001. For (A) and (B), scale bar = 40 μm.
Fig. 2.

IFN-γ-induced apoptosis of OPC-enriched cultures. (A) Cell cultures were exposed to IFN-γ in increasing concentrations (10 U/mL, 50 U/mL, and 100 U/mL) and cell viability was measured at day 2, 4, and 6 following IFN-γ treatment by MTT assay as described in Materials and Methods. Values are compared with the media and expressed as the mean ± SD of three independent experiments performed in eight replicate wells per assay condition; treatment with IFN-γ at both 50 and 100 U/mL resulted in a significant (*P < 0.01, **P < 0.001, ***P < 0.0001, ****P < 0.00001) increase in cell death compared with treatment with IFN-γ at 10 U/mL. (B) Quantification of TUNEL-positive cells indicates that a significant (***P < 0.0001) increase in TUNEL staining in IFN-γ-treated cultures compared with media following 6 days. Data are expressed as mean percentages ± SD of apoptotic nuclei as described in Materials and Methods. Representative immunofluorescent staining reveals numerous NG2-positive cells with few O1-positive cells at the time of IFN-γ (100 U/mL) treatment (C) and 6 days post-treatment (D). Scale bar = 40 μm for panels (C) and (D). Representative confocal staining of GalC+ cells (green) treated with media alone (E) or IFN-γ (F) and stained for TUNEL-reactivity (red) as well as DAPI (blue nuclei) after 6 days of treatment; scale bar = 30 μm. (G) Quantification of NG2+O1− cells following 6-day treatment with either media, IFN-γ (100 U/mL), CXCL10 (10 ng/mL), IFN-γ + CXCL1 (10 ng/mL), and CXCL10 + CXCL1 (10 ng/mL). Data are presented as average ± SD and represent the results of two independent experiments performed in triplicate (n = 6 per experimental conditions); **P ≤ 0.001.
CXCL10 Promotes Apoptosis of OPC Cultures
Dose-dependent treatment of OPC cultures with IFN-γ resulted in elevated CXCL10 levels within supernatants (5,000–35,000 pg/mL) indicating that these cells are activated in response to IFN-γ treatment (Fig. 3A). In contrast, comparatively little (<80 pg/mL) of CXCL9 was detected in response to IFN-γ treatment (Fig. 3B). We next tested whether CXCL10 was capable of promoting cell death in OPCs primary cultures. Exposure of OPC-enriched glial cultures to CXCL10 for 6 days resulted in a dose-dependent decrease in cell survival (Fig. 3C). Moreover, inclusion of CXCL10 (10 ng/mL) was associated with increased cell death as determined by MTT assay (Fig. 3D) and TUNEL staining (Fig. 3E). Both IFN-γ and CXCL10 treatments resulted in similar level of TUNEL-positive cells (~ 40%) suggesting that IFN-γ-induced expression of CXCL10 is an important signaling pathway involved in cell death. IFN-γ treatment of OPC-enriched cultures derived from CXCL10−/− mice reduced cell death as determined both by MTT (Fig. 4A) and TUNEL staining (Fig. 4B) when compared with IFN-γ-treated OPC cultures generated from CXCL10+/+ mice. However, treatment of CXCL10−/− cells with CXCL10 (10 ng/mL) increased MTT staining (Fig. 4C) associated with elevated TUNEL-reactivity (Fig. 4D) indicating CXCL10−/− OPCs remained sensitive to CXCL10 treatment. These data indicate that one mechanism by which IFN-γ evokes apoptosis in OPC-enriched cultures is through inducing CXCL10 expression which functions in an autocrine and paracrine manner to induce apoptosis.
Fig. 3.

CXCL10 treatment results in OPC apoptosis. (A) Secreted CXCL10 protein levels in supernatant from OPC cultures treated with IFN-γ (10 U/mL, 50 U/mL, and 100 U/mL—48 h) were measured by ELISA. (B) Secreted CXCL9 protein in supernatant following treatment with IFN-g (100 U/mL—48 h). For (A) and (B), results represent the mean ± SD of three independent experiments carried out in triplicate. (C) OPC cultures were incubated with increasing concentrations of CXCL10 (0.1–10 ng/mL) for 2, 4, and 6 days. Cell viability was measured by trypan blue exclusion and results are presented as mean ± SD of three independent experiments performed in triplicate. Treatment with 10 ng/mL CXCL10 resulted in a significant increase (**P < 0.001) in cell death at days 4 and 6 when compared with media-treated cultures. (D) OPC cultures were exposed to CXCL10 (10 ng/mL) for 2, 4, and 6 days and cell death determined by MTT reduction. Such treatment resulted in a significant increase in cell death at days 4 (*P < 0.01) and 6 (***P < 0.0001) compared with media-treated cultures. Values are expressed as the mean ± SD of three independent experiments performed in duplicate. (E) Treatment of OPC cultures for 6 days with CXCL10 (10 ng/mL) showed a significant increase (***P < 0.0001) in TUNEL positive cells when compared with untreated cultures. Values are expressed as average ± SD. Data was obtained from three imaging slide fields and represents three independent experiments performed using duplicate wells (n = 6 for each experimental condition).
Fig. 4.
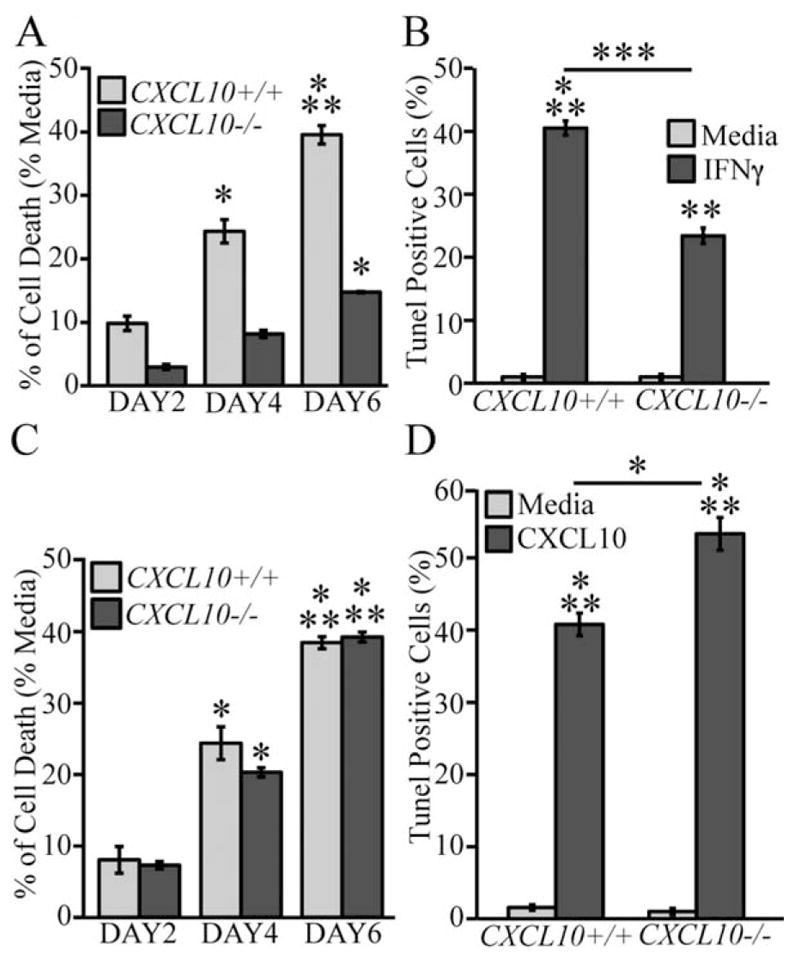
IFN-γ and CXCL10 treatment promotes OPC death. MTT assay showing the cytotoxic effects of either IFN-γ (100 U/mL) (A) or CXCL10 (10 ng/mL) (C) on OPC cultures derived from either WT or CXCL10−/− mice. Quantification of TUNEL-positive cells following treatment of OPC cultures from WT and CXCL10−/− mice with either IFN-γ (B) or CXCL10 (D) after 6 days of incubation. In (A) and (C), values are compared with the media and expressed as the mean ± SD of three independent experiments performed in eight replicate wells per assay condition and statistical significance is indicated by * for P < 0.01, and *** when P < 0.0001 (n = 3 separate experiments). For (B) and (D), data are derived from quantification of random confocal images of n = 3 indipendent experiments performed in duplicate and are presented as average ± SD. *P ≤ 0.01, **P ≤ 0.001, and ***P ≤ 0.0001.
CXCL10 Promotes Apoptosis Through Signaling Through CXCR3
Western blotting of proteins isolated from OPC-enriched cultures revealed detectable CXCR3 expression (Fig. 5A). Subsequently, we next evaluated if OPC cultures derived from CXCR3−/− mice were sensitive to either IFN-γ or CXCL10-mediated apoptosis. Treatment of CXCR3−/− cultures with either IFN-γ or CXCL10 resulted in muted cell death as determined by both MTT measurement (Fig. 5B) and TUNEL staining (Fig. 5C) when compared with treatment of CXCR3+/+ cultures. Notably, these findings also revealed that IFN-γ-treatment of CXCR3−/− cultures resulted in significantly greater cell death compared with CXCL10-treatment. These observations indicate that CXCL10 functions to evoke cell death through its receptor CXCR3, and also indicate that additional IFN-γ-inducible factors also can participate in apoptosis.
Fig. 5.
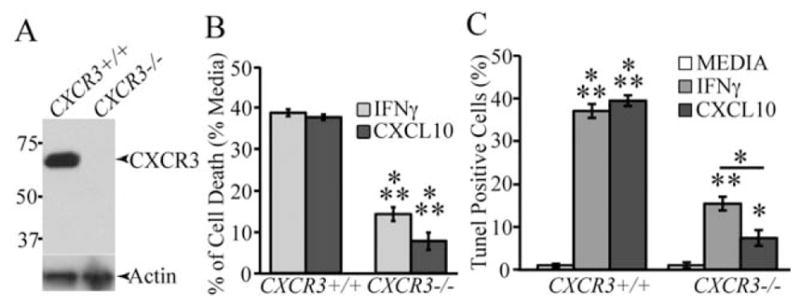
CXCR3 deficiency protects OPCs from IFN-γ and CXCL10-mediated cell death. (A) Western blotting of proteins isolated from OPC-enriched cultures obtained from either CXCR3+/+ or CXCR3−/− mice confirms that CXCR3 is expressed in wild-type cultures. A signal corresponding to ~72 kDa is detected in wild-type cultures and consistent with detection of dimerized protein that is detected with this antibody (manufacturer specifications) while signal is absent in knockout cultures. (B) MTT assay showing cell death following 6 days of treatment of CXCR3−/− or WT OPC cultures with either IFN-γ or CXCL10. Cell death is significantly (***P < 0.0001; n = 3 different experiments) reduced in CXCR3−/− cultures compared with WT cultures. (C) TUNEL quantification reveals the percentage of TUNEL-positive OPCs derived from WT and CXCR3−/− mice after 6 days of IFN-γ or CXCL10 treatment. Incubation of CXCR3−/− cultures with either IFN-γ or CXCL10 led to a significant (*P < 0.01, **P < 0.001, n = 3) reduction in Tunel staining compared with WT mice. For (C), data represents three separate experiments, performed in duplicate, and are presented as average ± SD; *P ≤ 0.01, **P < 0.001, ***P < 0.0001.
CXCL1 Signaling Through CXCR2 Protects OPCs from IFN-γ/CXCL10-Induced Apoptosis
We have recently shown that CXCL1 treatment of OPC-enriched cultures infected with the neurotropic JHM strain of mouse hepatitis virus (JHMV) protects from viral-induced apoptosis by signaling through the receptor CXCR2 (Hosking et al., 2010). Cultured OPC-enriched cultures constitutively express CXCR2 as determined by western blotting (Fig. 6A). To determine if CXCL1 protects OPC cultures from apoptosis following treatment with either IFN-γ or CXCL10 we added CXCL1 to the treatments and evaluated cell death. Inclusion of CXCL1 (10 ng/mL) to OPC cultures inhibited either IFN-γ or CXCL10-mediated death as determined by MTT assay (Fig. 6B) and TUNEL staining (Fig. 6C) when compared with stimulated cultures treated with vehicle alone. Phenotyping cell cultures demonstrated increased protection of NG2+O1− cells from either IFN-γ or CXCL10-induced death through addition of CXCL1 (Fig. 2G). Inclusion of CXCL1 to CXCR2−/− OPC cultures treated with either IFN-γ or CXCL10 resulted in no rescue from apoptosis highlighting the importance of CXCR2 signaling in protecting cells (Fig. 6D,E). Finally, the addition of CXCL2, another ligand for CXCR2, resulted in only marginal protection from either IFN-γ or CXCL10-induced death in comparison to CXCL1 indicating differential responses of cultured OPCs to CXCR2 ligands with regards to protection (Fig. 6B).
Fig. 6.

CXCR2 signaling protects OPC cultures from IFN-γ and CXCL10-mediated apoptosis. (A) Western blotting confirmed CXCR2 expression OPC-enriched cultures isolated from wild-type mice. The predominant signal is detected at ~70 kDa suggesting dimerization of CXCR2 which is consistent with previous studies (Martinez Munoz et al., 2009; Trettel et al., 2003). Less predominant CXCR2 signal is detected at ~50 and 42 kDa while no signal is detected in protein isolates obtained from OPC-enriched cultures derived from CXCR2−/− mice. (B) Inclusion of the CXCR2 ligand CXCL1 (10 ng/mL) protects OPC cultures from death following a 6-day incubation with either IFN-γ or CXCL10 as determined by MTT assay. Data are expressed as average ± SD of three different experiments performed in eight replicate wells per assay condition, for ***P < 0.0001, n = 3, and for *P < 0.01, n = 2 independent experiments. (C) Quantification of TUNEL staining revealed that CXCL1 protects OPCs from either IFN-γ or CXCL10 mediated apoptosis. In contrast, (D) CXCL1 did not rescue CXCR2−/− OPC cultures from death following 6-day treatment with either IFN-γ or CXCL10 as determined by MTT assay: values represent average ± SD of three different experiments performed in 12 replicate wells per assay condition. Significance was assessed by two-tail Student’s t-test and ***P ≤ 0.0001 (n = 3 independent experimental conditions) when compared with experimental cultures treated with CXCL1. (E) TUNEL staining on CXCR2−/− OPCs showed that CXCL1 co-treatments with IFN-γ or CXCL10 respectively, resulted in significant (***P < 0.0001, n = 3) increase in cell death compared with media-treated cultures. Data in (C–E) are presented as average± SD and represent three independent experiments performed in triplicate.
CXCL1-mediated protection is associated with blocking cleavage of caspase 3 accompanied by enhanced Bcl-2 expression. The active form of caspase-3 (molecular weight 17 kDa) was detected in IFN-γ and CXCL10 treated cultures at day 6 post-treatment (Fig. 7A) and this correlated with increased apoptosis. Addition of CXCL1 to OPC-enriched cultures exposed to either IFN-γ or CXCL10 resulted in undetectable levels of cleaved caspase 3 compared with untreated activated cultures (Fig. 7A) and this was associated with CXCL1-mediated protection from apoptosis. Exposure of OPC cultures derived from CXCR2−/− mice to either IFN-γ or CXCL10 resulted in cleavage of caspase 3 and inclusion of CXCL1 did not diminish levels of the activated form of this protein (Fig. 7B). These data indicate that the tonic effect of CXCL1-mediated protection from IFN-γ/CXCL10-mediated apoptosis is associated with prevention of cleavage of caspase 3. We next interrogated the effects of CXCL1 protection with expression of members of the Bcl-2 gene family. Treatment of OPC-enriched cultures with either IFN-γ or CXCL10 for 6 days resulted in diminished expression of transcripts encoding the anti-apoptotic Bcl-xl and Bcl2 family members while transcript levels were elevated for the pro-apoptotic Bax gene under these conditions (Fig. 7C). Addition of CXCL1 to either IFN-γ or CXCL10-treated cultures dampened Bax transcript levels whereas there was a marked increase in both Bcl-xl and Bcl-2 (Fig. 7C). CXCL1 treatment alone showed increased expression of both Bcl-xl and Bcl-2 when compared with media cultures (Fig. 7C). Western blotting confirmed that Bcl-2 protein levels were diminished in OPC cultures treated with either IFN-γ or CXCL10 yet inclusion of CXCL1 resulted in significantly increased Bcl-2 expression supporting transcript analysis (Fig. 7D,E). In contrast, CXCL1 treatment of CXCR2-deficient OPC cultures treated with either IFN-γ or CXCL10 did not elevate intracellular levels of Bcl-2 (Fig. 7D,E). Treatment of OPC-enriched cultures with either IFN-γ or CXCL10 did not increase expression of CXCL1 at either the transcriptional (Fig. 8A) or protein (Fig. 8B) level. However, addition of CXCL1 to cultures increased expression of CXCL1 and this was sustained following treatment with either IFN-γ or CXCL10 (Fig. 8A,B). In contrast, we could not detect CXCL2 protein following treatment (not shown). These findings indicate that CXCL1 treatment enhances CXCL1 expression at both the transcriptional and protein. In addition, our studies reveal that the lack of CXCR2 expression plays an important role in CXCL1 protection and establishes the involvement of CXCR2 signaling in modulating intracellular anti-apoptotic pathways in oligodendrocyte-lineage cells.
Fig. 7.

CXCR2 signaling prevents cleavage of caspase 3 and induces Bcl-2 up-regulation. Both WT (A) and CXCR2−/− (B) OPC cultures were treated with either IFN-γ (100 U/mL) or CXCL10 (10 ng/mL) alone or in combination with CXCL1 (10 ng/mL) for 6 days and cleavage of caspase 3 determined by Western blotting. While IFN-γ and CXCL10 treatment in WT cultures resulted in caspase 3 cleavage, the addition of CXCL1 blocked cleavage. In contrast, CXCL1 did not block cleavage of caspase 3 following either IFN-γ or CXCL10 treatment of CXCR2−/− cell cultures. (C) OPC cultures from WT mice were treated with either IFN-γ (100 U/mL) or CXCL10 (10 ng/mL) alone or in combination with CXCL1 (10 ng/mL) for 6 days and semiquantitative PCR analysis performed to measure transcripts encoding Bcl-2, Bcl-xl, and Bax. Treatment with either IFN-γ or CXCL10 alone resulted in reduced expression of both Bcl-2 and Bcl-xl yet transcripts encoding Bax were elevated. Values were determined by fold-increase in transcript levels compared with media-treated cultures and expressed as average of three experiments (n = 3) ±SD, statistical significance is indicated by * and ** for P values ≤ 0.01 and ≤ 0.001, respectively. (D) Representative Western blot analysis of Bcl-2 expression in WT and CXCR2−/− cultures following 6 days of culture under indicated experimental conditions. (E) Quantification of Bcl-2 levels within experimental cultures of OPCs from WT and CXCR2−/− mice. Treatment with CXCL1 led to a significant reduction (**P < 0.001) in Bcl-2 levels following treatment with either IFN-γ or CXCL10. Data in (D) and (E) represent three independent experiments and data in (E) are presented as average ± SD.
Fig. 8.

OPC-enriched cultures express CXCL1. (A) Total RNA was isolated from three different preparations of cells at day 6 following experimental treatments, and analyzed by semiquantitative real time PCR, for the expression of CXL1 using sequence specific primer. Data represent the fold induction relative to the media treated cultures. (B) Supernatants from same cultures in (A) were collected at day 6 and expression of CXCL1 was determined by ELISA. Data are expressed as mean of values ± SD of three different experiments.
DISCUSSION
Data presented in this study supports and extends earlier work by others demonstrating that IFN-γ treatment of OPCs induces apoptosis (Baerwald and Popko, 1998; Balabanov et al., 2006; Horiuchi et al., 2006; Lin et al., 2006, 2008; Vartanian et al., 1995; Wang et al., 2010). Susceptibility to IFN-γ-induced apoptosis was related to the maturation state of cells following differentiation with OPCs exhibiting greater sensitivity to IFN-γ compared with immature oligodendroytes and this is consistent with earlier findings (Baerwald and Popko, 1998; Vartanian, 1995). These findings provide new data indicating that one mechanism by which IFN-γ evokes apoptosis is through a CXCL10-dependent mechanism by signaling through CXCR3. OPC-mediated apoptosis following treatment with either IFN-γ or CXCL10 was associated with the presence of cleaved caspase 3 and diminished expression of the anti-apoptotic protein Bcl2. Importantly, signaling through CXCR2 protects OPCs from both IFN-γ and CXCL10-mediated apoptosis and this is associated with diminished cleavage of caspase 3 and elevated expression of Bcl-2. These findings further highlight the importance of chemokines in contributing to the neuropathology of inflammatory diseases of the CNS and demonstrate how chemokine signaling may also protect cells of an oligodendrocyte lineage from damage/death.
CXCL10 is a member of the non-ELR CXC chemokine family that is expressed by numerous cell types following exposure to IFN-γ and early studies indicated that CXCL10 expression was linked with many T cell-mediated inflammatory diseases (Lee et al., 2009; Luster, 1998). More recently, CXCL10 has been shown to contribute to tissue pathology by promoting apoptosis of targeted cell populations. Studies examining the effects of viral infection of neurons have demonstrated that CXCL10 signaling through CXCR3 expressed on neurons promotes apoptosis. CXCL10 is highly expressed within the brains of patients with human immunodeficiency virus-associated dementia. Both CXCL10 and CXCR3 were detected in neurons in rhesus macaques infected with simian immunodeficiency virus (Sui et al., 2004). Moreover, treatment of human fetal brain cultures with either simian human immunodeficiency virus (SHIV) or gp120 resulted in CXCL10 expression in neurons and treatment of cultured neurons with CXCL10 resulted in apoptosis that was associated with increased expression of cleaved caspase 3 (Sui et al., 2004, 2006). More recently, Klein and colleagues revealed that elevated levels of CXCL10 within the brain following experimental infection of mice with West Nile Virus (WNV) may induce neuronal apoptosis (Klein et al., 2005; Zhang et al., 2008, 2010). WNV-infected neurons express TNF-α that signals through TNFR1 resulting in decreased expression of CXCR3 that correlated with muted CXCL10-mediated calcium transients and delayed caspase 3 activation (Zhang et al., 2010). Importantly, loss of CXCR3 signaling through either genetic silencing or TNF-α treatment blocked neuronal apoptosis following in vitro infection of neurons with WNV (Zhang et al., 2010). Other models of inflammatory disease have also offered the possibility of CXCL10 signaling in contributing to apoptosis. Therapeutic neutralization of CXCL10 in a model of spinal cord injury reduces neuroinflammation as well as apoptosis that is associated with diminished neuronal loss (Gonzalez et al., 2007). A role for CXCL10 in the pathogenesis of pancreatitis by destruction of acinar cells has also been suggested (Singh et al., 2010a). Treatment of acinar cells with CXCL10 resulted in induction of apoptosis through signaling by CXCR3 and this was associated with increased expression of cleaved caspase 3 (Singh et al., 2010a). Collectively, these findings provide a compelling argument that CXCL10 signaling is a potent inducer of apoptosis and this provides an additional mechanism by which this chemokine may contribute to tissue pathology during chronic inflammatory disease.
We have provided new evidence that CXCL10 is important in contributing to apoptosis of OPCs using an in vitro model system. IFN-γ treatment of numerous cell types results in rapid expression of CXCL10, therefore, increased levels of CXCL10 in OPC-enriched cultures was not surprising. It is important to emphasize that we did not determine cell type(s) responsible for CXCL10 secretion in following IFN-γ treatment. Although treatment of mature cultures of oligodendrocytes with IFN-γ resulted in the production of a number of chemokines including CXCL10 (Balabanov et al., 2007), we do not know if OPCs are the primary source of CXCL10 following IFN-γ treatment. Additionally, astrocytes are very sensitive to IFN-γ-treatment with regards to production of CXCL10 therefore it is possible that the bulk of CXCL10 present within IFN-γ-treated cultures derives from the small population of astrocytes. Nevertheless, the findings reported in this study that apoptosis was reduced in IFN-γ treated CXCL10−/− and CXCR3−/− cultures highlights a potential role for CXCL10 in contributing to OPC death. It is important to note that genetic silencing of either CXCL10 or CXCR3 did not result in complete protection from IFN-γ-induced death. Indeed, apoptosis in IFN-γ-treated CXCL10−/− and CXCR3−/− cultures was reduced by ~ 50% to 60% compared with WT cultures suggesting additional mechanisms are employed in IFN-γ-mediated death. Compared with CXCL10 production in response to IFN-γ-production, very little CXCL9 was produced suggesting a potentially limited role in contributing to protection. CXCL11, the most potent CXCR3 ligand, is a pseudo-gene in B6 mice, so not relevant for this study. Our findings argue that local expression of CXCL10 within white matter tracts may contribute to demyelinating diseases by contributing to damage/death of cells of an oligodendrocyte lineage by signaling through CXCR3. This idea is supported by the demonstration that blocking of CXCL10 signaling in mice persistently infected with the neurotropic JHM strain of mouse hepatitis virus (MHV) results in extensive remyelination suggesting that myelin-producing cells of the CNS are protected (Liu et al., 2001). It is intriguing to speculate that OPCs are more sensitive to IFN-γ/CXCL10 death as these cells are considered critical during periods of remyelination as well as earlier findings arguing that susceptibility to IFN-γ-mediated death is dependent upon the developmental stage of oligodendrocyte lineage cells (Baerwald and Popko, 1998; Balabanov et al., 2007; Horiuchi et al., 2006). The mechanisms by which CXCL10 contribute to OPC apoptosis are not well understood at this time and are a focus of intense investigation. Presumably, similar mechanisms are employed in CXCL10-mediated apoptosis as are following IFN-γ treatment. Both STAT1 and IRF-1 have been shown to function as key components of the signaling pathway mediating OPC-death (Wang et al., 2010). Similarly, simultaneous activation of the STAT pathway by IFN-γ and the ERK pathway by exogenous factors has also been shown to play a role in OPC apoptosis (Horiuchi et al., 2006). We are currently examining whether these signaling pathways are evoked in response to CXCL10 treatment of OPC cultures.
The demonstration that CXCR2 signaling protects OPC cultures from both IFN-γ and CXCL10-mediated apoptosis provides potentially important insight into how oligodendrocyte lineage cells protect themselves from death during chronic neuroinflammation. Importantly, these findings support and extend earlier work from our laboratory showing that blocking CXCR2 signaling in a model of viral-induced demyelination resulted in delayed recovery in clinical disease that was associated with increased demyelination associated with apoptosis of both mature oligodendrocytes and OPCs without affecting either neuroinflammation or CNS viral burden (Hosking et al., 2010). While this study also showed that CXCR2 signaling protected OPC-enriched cultures from apoptosis following viral infection, the present work indicates that the protective effect of CXCR2 is not restricted to a viral model of neurologic disease. Rather, engagement of CXCR2 initiates protection in response to exposure to inflammatory factors IFN-γ and CXCL10 that are commonly detected during periods of neuroinflammation in animal models of disease. Inclusion of CXCL1 functioned in a tonic manner to protect OPCs from either IFN-γ or CXCL10-induced apoptosis and this was associated with impaired cleavage of caspase 3 and increased Bcl-2 expression. Our findings also argue for a more important role for CXCL1 in protection compared with CXCL2, another CXCR2 ligand. Whether this relates to differential effects in induction of anti-apoptotic proteins and/or modulation in caspase 3 cleavage is unknown at this time and under current investigation. Early studies examining the effects of chemokine signaling on cell function revealed a protective effect for CXCR2 in preventing apoptosis. For example, inhibition of CXCR2 signaling through use of a selective antagonist blocked ligand-mediated inhibition of neutrophil apoptosis (Glynn et al., 2002). Human endothelial cells express CXCR2 and incubation with the ligand IL-8 enhances proliferation and capillary tube organization as well as inhibiting apoptosis via enhance expression of anti-apoptotic genes including Bcl-xL and Bcl-2 (Li et al., 2003). In addition, an important role for CXCR2 in regulating tumor cell biology has also been reported. Expression of CXCR2 has been detected on melanoma cells (Singh et al., 2010b) and ovarian cancer lines (Yang et al., 2010). Expression of CXCR2 on cancer cells was associated with protection from apoptosis while genetic knock down of CXCR2 resulted in increased cell death associated with modulation of anti-apoptotic proteins (Singh et al., 2010a; Yang et al., 2010). CXCR2 has been shown to protect cultured astrocytes from Fas-induced death and toxic insult (Saas et al., 2002; Wang et al., 2006, 2010). Moreover, CXCL1/CXCR2 signaling is involved in the proliferation of human fetal OPCs (Filipovic and Zecevic, 2008).
Our findings reveal insight into potentially important mechanisms contributing to demyelinating disease including multiple sclerosis (MS). Specifically, these data show that IFN-γ evokes apoptosis in OPCs through CXCL10 signaling to CXCR3. Therefore, chronic CXCL10 expression within the CNS may not only attract activated CXCR3-expressing lymphocytes but also contribute to OPC death/apoptosis. CXCR3 expression has been detected on both immature and mature oligodendrocytes and elevated expression in was observed in tissues from MS patients, thus highlighting the possibility of responding to CXCL10 (Omari et al., 2005). As OPCs are considered critical in promoting remyelination, a decrease in numbers via IFN-γ/CXCL10 mediated death may contribute to remyelination failure often observed in the majority of MS patients (Franklin, 2002). These findings highlight that therapeutic strategies targeting CXCL10 may be beneficial in treating human demyelinating diseases including MS. In addition, approaches designed to enhance CXCR2 signaling on OPCs may potentiate the remyelination potential of these cells as well as limit myelin loss.
Acknowledgments
The authors gratefully thank Leslie Kirby for experimental assistance. This work was supported by NIH grants R01NS41249 (T.E.L.) and R01NS32151 (R.M.R.).
References
- Baerwald KD, Popko B. Developing and mature oligodendrocytes respond differently to the immune cytokine interferon-gamma. J Neurosci Res. 1998;52:230–239. doi: 10.1002/(SICI)1097-4547(19980415)52:2<230::AID-JNR11>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Balabanov R, Strand K, Goswami R, McMahon E, Begolka W, Miller SD, Popko B. Interferon-gamma-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J Neurosci. 2007;27:2013–2024. doi: 10.1523/JNEUROSCI.4689-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balabanov R, Strand K, Kemper A, Lee JY, Popko B. Suppressor of cytokine signaling 1 expression protects oligodendrocytes from the deleterious effects of interferon-gamma. J Neurosci. 2006;26:5143–5152. doi: 10.1523/JNEUROSCI.0737-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2002;346:165–173. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- Chew LJ, King WC, Kennedy A, Gallo V. Interferon-gamma inhibits cell cycle exit in differentiating oligodendrocyte progenitor cells. Glia. 2005;52:127–143. doi: 10.1002/glia.20232. [DOI] [PubMed] [Google Scholar]
- Filipovic R, Zecevic N. The effect of CXCL1 on human fetal oligodendrocyte progenitor cells. Glia. 2008;56:1–15. doi: 10.1002/glia.20582. [DOI] [PubMed] [Google Scholar]
- Franklin RJ. Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci. 2002;3:705–714. doi: 10.1038/nrn917. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn PC, Henney E, Hall IP. The selective CXCR2 antagonist SB272844 blocks interleukin-8 and growth-related oncogene-alpha-mediated inhibition of spontaneous neutrophil apoptosis. Pulm Pharmacol Ther. 2002;15:103–110. doi: 10.1006/pupt.2001.0323. [DOI] [PubMed] [Google Scholar]
- Gonzalez R, Hickey MJ, Espinosa JM, Nistor G, Lane TE, Keirstead HS. Therapeutic neutralization of CXCL10 decreases secondary degeneration and functional deficit after spinal cord injury in mice. Regen Med. 2007;2:771–783. doi: 10.2217/17460751.2.5.771. [DOI] [PubMed] [Google Scholar]
- Grinspan JB, Reeves MF, Coulaloglou MJ, Nathanson D, Pleasure D. Re-entry into the cell cycle is required for bFGF-induced oligodendroglial dedifferentiation and survival. J Neurosci Res. 1996;46:456–464. doi: 10.1002/(SICI)1097-4547(19961115)46:4<456::AID-JNR7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Grinspan JB, Stern JL, Franceschini B, Pleasure D. Trophic effects of basic fibroblast growth factor (bFGF) on differentiated oligodendroglia: A mechanism for regeneration of the oligodendroglial lineage. J Neurosci Res. 1993;36:672–680. doi: 10.1002/jnr.490360608. [DOI] [PubMed] [Google Scholar]
- Hardison JL, Nistor G, Gonzalez R, Keirstead HS, Lane TE. Transplantation of glial-committed progenitor cells into a viral model of multiple sclerosis induces remyelination in the absence of an attenuated inflammatory response. Exp Neurol. 2006;197:420–429. doi: 10.1016/j.expneurol.2005.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M, Itoh A, Pleasure D, Itoh T. MEK-ERK signaling is involved in interferon-gamma-induced death of oligodendroglial progenitor cells. J Biol Chem. 2006;281:20095–20106. doi: 10.1074/jbc.M603179200. [DOI] [PubMed] [Google Scholar]
- Hosking MP, Tirotta E, Ransohoff RM, Lane TE. CXCR2 signaling protects oligodendrocytes and restricts demyelination in a mouse model of viral-induced demyelination. PLoS One. 2010;5:e11340. doi: 10.1371/journal.pone.0011340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RS, Izikson L, Means T, Gibson HD, Lin E, Sobel RA, Weiner HL, Luster AD. IFN-inducible protein 10/CXC chemokine ligand 10-independent induction of experimental autoimmune encephalomyelitis. J Immunol. 2004;172:550–559. doi: 10.4049/jimmunol.172.1.550. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Sugarman MC, Lane TE, Leissring MA. Regional hypomyelination and dysplasia in transgenic mice with astrocyte-directed expression of interferon-gamma. J Mol Neurosci. 2000;15:45–59. doi: 10.1385/JMN:15:1:45. [DOI] [PubMed] [Google Scholar]
- Lee EY, Lee ZH, Song YW. CXCL10 and autoimmune diseases. Autoimmun Rev. 2009;8:379–383. doi: 10.1016/j.autrev.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170:3369–3376. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, Popko B. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest. 2007;117:448–456. doi: 10.1172/JCI29571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Kemper A, Dupree JL, Harding HP, Ron D, Popko B. Interferon-gamma inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain. 2006;129:1306–1318. doi: 10.1093/brain/awl044. [DOI] [PubMed] [Google Scholar]
- Lin W, Kunkler PE, Harding HP, Ron D, Kraig RP, Popko B. Enhanced integrated stress response promotes myelinating oligodendrocyte survival in response to interferon-gamma. Am J Pathol. 2008;173:1508–1517. doi: 10.2353/ajpath.2008.080449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Popko B. Endoplasmic reticulum stress in disorders of myelinating cells. Nat Neurosci. 2009;12:379–385. doi: 10.1038/nn.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MT, Keirstead HS, Lane TE. Neutralization of the chemokine CXCL10 reduces inflammatory cell invasion and demyelination and improves neurological function in a viral model of multiple sclerosis. J Immunol. 2001;167:4091–4097. doi: 10.4049/jimmunol.167.7.4091. [DOI] [PubMed] [Google Scholar]
- Luster AD. Chemokines–chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- Malone KE, Stohlman SA, Ramakrishna C, Macklin W, Bergmann CC. Induction of class I antigen processing components in oligodendroglia and microglia during viral encephalomyelitis. Glia. 2008;56:426–435. doi: 10.1002/glia.20625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Munoz L, Lucas P, Navarro G, Checa AI, Franco R, Martinez AC, Rodriguez-Frade JM, Mellado M. Dynamic regulation of CXCR1 and CXCR2 homo- and heterodimers. J Immunol. 2009;183:7337–7346. doi: 10.4049/jimmunol.0901802. [DOI] [PubMed] [Google Scholar]
- Omari KM, John GR, Sealfon SC, Raine CS. CXC chemokine receptors on human oligodendrocytes: Implications for multiple sclerosis. Brain. 2005;128:1003–1015. doi: 10.1093/brain/awh479. [DOI] [PubMed] [Google Scholar]
- Parra B, Hinton DR, Marten NW, Bergmann CC, Lin MT, Yang CS, Stohlman SA. IFN-gamma is required for viral clearance from central nervous system oligodendroglia. J Immunol. 1999;162:1641–1647. [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu K, D’Avila A, Heist EK, Rosenberg SP, Chou J, Yang M, Singh JP. Simultaneous electrical and mechanical mapping using 3D cardiac mapping system: Novel approach for optimal cardiac resynchronization therapy. J Cardiovasc Electrophysiol. 2010;21:219–222. doi: 10.1111/j.1540-8167.2009.01663.x. [DOI] [PubMed] [Google Scholar]
- Saas P, Walker PR, Quiquerez AL, Chalmers DE, Arrighi JF, Lienard A, Boucraut J, Dietrich PY. A self-defence mechanism of astrocytes against Fas-mediated death involving interleukin-8 and CXCR2. Neuroreport. 2002;13:1921–1924. doi: 10.1097/00001756-200210280-00018. [DOI] [PubMed] [Google Scholar]
- Singh L, Arora SK, Bakshi DK, Majumdar S, Wig JD. Potential role of CXCL10 in the induction of cell injury and mitochondrial dysfunction. Int J Exp Pathol. 2010a;91:210–223. doi: 10.1111/j.1365-2613.2009.00697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Sadanandam A, Varney ML, Nannuru KC, Singh RK. Small interfering RNA-mediated CXCR1 or CXCR2 knock-down inhibits melanoma tumor growth and invasion. Int J Cancer. 2010b;126:328–336. doi: 10.1002/ijc.24714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles LN, Liu MT, Kane JAC, Lane TE. CXCL10 and trafficking of virus-specific T cells during coronavirus demyelination. Autoimmunity. 2009;42:484–489. doi: 10.1080/08916930902810708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Potula R, Dhillon N, Pinson D, Li S, Nath A, Anderson C, Turchan J, Kolson D, Narayan O, Buch S. Neuronal apoptosis is mediated by CXCL10 overexpression in simian human immuno-deficiency virus encephalitis. Am J Pathol. 2004;164:1557–1566. doi: 10.1016/S0002-9440(10)63714-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Stehno-Bittel L, Li S, Loganathan R, Dhillon NK, Pinson D, Nath A, Kolson D, Narayan O, Buch S. CXCL10-induced cell death in neurons: Role of calcium dysregulation. Eur J Neurosci. 2006;23:957–964. doi: 10.1111/j.1460-9568.2006.04631.x. [DOI] [PubMed] [Google Scholar]
- Totoiu MO, Nistor GI, Lane TE, Keirstead HS. Remyelination, axonal sparing, and locomotor recovery following transplantation of glial-committed progenitor cells into the MHV model of multiple sclerosis. Exp Neurol. 2004;187:254–265. doi: 10.1016/j.expneurol.2004.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Di Bartolomeo S, Lauro C, Catalano M, Ciotti MT, Limatola C. Ligand-independent CXCR2 dimerization. J Biol Chem. 2003;278:40980–40988. doi: 10.1074/jbc.M306815200. [DOI] [PubMed] [Google Scholar]
- van Marle G, Antony J, Ostermann H, Dunham C, Hunt T, Halliday W, Maingat F, Urbanowski MD, Hobman T, Peeling J, Power C. West Nile virus-induced neuroinflammation: Glial infection and capsid protein-mediated neurovirulence. J Virol. 2007;81:10933–10949. doi: 10.1128/JVI.02422-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian T, Li Y, Zhao M, Stefansson K. Interferon-gamma-induced oligodendrocyte cell death: Implications for the pathogenesis of multiple sclerosis. Mol Med. 1995;1:732–743. [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Luo W, Stricker R, Reiser G. Protease-activated receptor-1 protects rat astrocytes from apoptotic cell death via JNK-mediated release of the chemokine GRO/CINC-1. J Neurochem. 2006;98:1046–1060. doi: 10.1111/j.1471-4159.2006.03950.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ren Z, Tao D, Tilwalli S, Goswami R, Balabanov R. STAT1/IRF-1 signaling pathway mediates the injurious effect of interferon-gamma on oligodendrocyte progenitor cells. Glia. 2010;58:195–208. doi: 10.1002/glia.20912. [DOI] [PubMed] [Google Scholar]
- Whitman L, Zhou H, Perlman S, Lane TE. IFN-gamma-mediated suppression of coronavirus replication in glial-committed progenitor cells. Virology. 2009;384:209–215. doi: 10.1016/j.virol.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik WJ, Swoveland P, Zhang X, Vanguri P. Chronic intrathecal infusion of phosphorothioate or phosphodiester antisense oligonucleotides against cytokine responsive gene-2/IP-10 in experimental allergic encephalomyelitis of lewis rat. J Pharmacol Exp Ther. 1996;278:404–410. [PubMed] [Google Scholar]
- Yang G, Rosen DG, Liu G, Yang F, Guo X, Xiao X, Xue F, Mercado-Uribe I, Huang J, Lin SH, Mills GB, Liu J. CXCR2 promotes ovarian cancer growth through dysregulated cell cycle, diminished apoptosis, and enhanced angiogenesis. Clin Cancer Res. 2010;16:3875–3886. doi: 10.1158/1078-0432.CCR-10-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Chan YK, Lu B, Diamond MS, Klein RS. CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J Immunol. 2008;180:2641–2649. doi: 10.4049/jimmunol.180.4.2641. [DOI] [PubMed] [Google Scholar]
- Zhang B, Patel J, Croyle M, Diamond MS, Klein RS. TNF-alpha-dependent regulation of CXCR3 expression modulates neuronal survival during West Nile virus encephalitis. J Neuroimmunol. 2010;224:28–38. doi: 10.1016/j.jneuroim.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]